

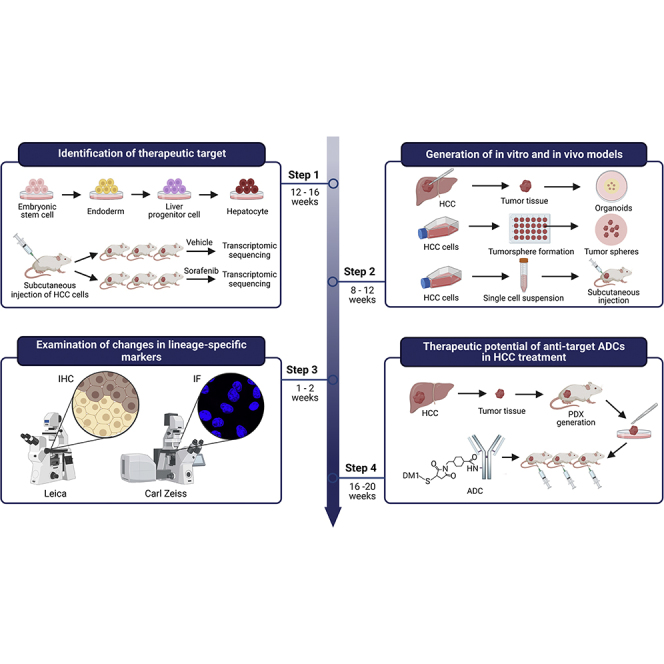
Summary
Here, we present a detailed protocol for the identification of potential oncofetal targets for hepatocellular carcinoma (HCC) patients through a hepatocyte differentiation model and a sorafenib refractory cell-line-derived xenograft model. We describe the procedures of tumor sphere formation, organoid generation, and subcutaneous tumor formation for functional studies. We then detail the procedures of immunohistochemistry and immunofluorescence for examination of changes in lineage-specific markers. Finally, we describe the development of antibody-based therapeutics targeting tumor lineage plasticity in HCC.
For complete details on the use and execution of this protocol, please refer to Kong et al. (2021).1
Subject areas: Cancer, Health Sciences, Model Organisms, Organoids
Graphical abstract
Highlights
-
•
Generation of hepatocyte differentiation model and sorafenib refractory CDX model
-
•
Generating organoids and PDX models from human HCC biopsies
-
•
Development of ADCs against potential oncofetal targets
-
•
This protocol is applicable to other genes of interest in HCC treatment
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Here, we present a detailed protocol for the identification of potential oncofetal targets for hepatocellular carcinoma (HCC) patients through a hepatocyte differentiation model and a sorafenib refractory cell-line-derived xenograft model. We describe the procedures of tumor sphere formation, organoid generation, and subcutaneous tumor formation for functional studies. We then detail the procedures of immunohistochemistry and immunofluorescence for examination of changes in lineage-specific markers. Finally, we describe the development of antibody-based therapeutics targeting tumor lineage plasticity in HCC.
Before you begin
In this protocol, we use the CLDN6 gene as an example to describe the steps of screening potential therapeutic targets with functional potential.
Institutional permissions
The animal experiments must acquire ethical approval and institutional permissions and follow the guidelines of the institutional animal care and use committee.
Human biopsies should be obtained from patients who provide informed consent, and research ethics approval is needed.
Laboratory preparation
-
1.
All experiments involving cell lines, organoids and mice must be performed under sterile conditions.
-
2.
Prepare all solutions as indicated in the materials and equipment section before starting the experiments.
Establishment of stable cell lines
Timing: 4–8 weeks
The lentivirus for CLDN6 overexpression or knockdown was obtained from IGEbio (∼1 × 108 TU/mL, Guangzhou, China).
-
3.Before the experiment:
-
a.Thaw the virus on ice. Depending on usage, consider quantitative redistribution of the virus aliquot into different tubes.Note: One cycle of freeze-thawing of the virus will lead to a loss of over 20% of the viral titer. Avoid repeating freeze-thawing. Virus aliquots need to be stored in a −80°C freezer or in liquid nitrogen.Note: The right antibiotics need to be chosen according to the vector system. The concentration that kills all uninfected negative cells is used for the selection concentration.
-
b.Use a GFP-expressing control lentivirus to determine the optimal multiplicity of infection (MOI) in the target cells.Note: The MOI refers to the number of viruses that can infect each cell. MOI=1 refers to infection of 1 transduction unit (TU) virus per cell.
 CRITICAL: Lentivirus-based vectors have biological risk potential. It is strongly recommended to handle lentivirus according to the requirements of Biosafety Level 2 (BSL-2).
CRITICAL: Lentivirus-based vectors have biological risk potential. It is strongly recommended to handle lentivirus according to the requirements of Biosafety Level 2 (BSL-2).
-
a.
-
4.
Plate 5 × 104 CRL-8024 or HepG2 cells in 6-well plates to achieve 30%–50% confluence after 18–20 h of culture.
-
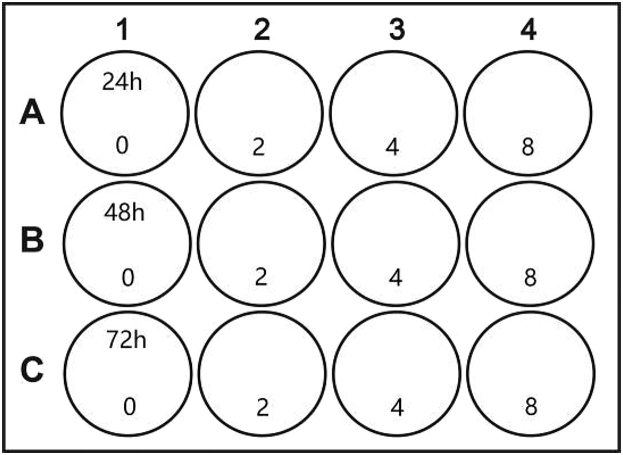
5.Thaw the single-use virus on ice. Place an appropriate amount of virus with the desired MOI in a proper amount of culture medium and mix it gently.Note: To maximize the efficiency of infection, only the minimum volume of media to cover the surface of the plate should be used.Note: If the cells are susceptible to infection, start infecting cells with an MOI between 1 and 10. A higher MOI may be required for some cell lines.Optional: The addition of polybrene will enhance lentiviral transduction. Overexposure to polybrene (> 12 h) might be toxic to some cell lines.Optimization of the polybrene conditions:
-
a.Make a stock of 8 mg/mL of polybrene in sterilized ddH2O.
-
b.Label the 12-well plate as follows:
-
c.A total of 1 × 105 cells/mL should be diluted in cell culture medium.
-
d.Prepare the following virus solution:
-
i.3 mL medium + 30 μL GFP-expressing control lentivirus (10 MOI).
-
ii.3 mL medium + 30 μL GFP-expressing control lentivirus + 1.5 μL polybrene (final =2 μg/mL).
-
iii.3 mL medium + 30 μL GFP-expressing control lentivirus + 3 μL polybrene (final =4 μg/mL).
-
iv.3 mL medium + 30 μL GFP-expressing control lentivirus + 6 μL polybrene (final =8 μg/mL).Note: The final concentration of polybrene can be changed.
-
i.
-
e.Add 1 milliliter of cell suspension and 1 mL of virus solution to the corresponding labeled wells.
-
f.Gently rotate the plate and place it in a cell culture incubator for 6–8 h.
-
g.Replace the medium with fresh, complete medium and return the cells to the incubator.Note: The complete medium varies depending on the cell type and application. Here, we used basic DMEM (1×). Store the medium at 4°C for up to 6 weeks.
-
h.The fluorescence intensity of GFP should be inspected under a microscope at the indicated times.
-
a.
-
6.
Discard the old culture medium and add lentivirus-containing medium to the cells.

Note: Leave an additional well to serve as a negative control.
-
7.
Gently rotate the place and place it in a cell culture incubator for 6–8 h.
-
8.
Replace the medium with fresh, complete medium and incubate the cells for 48–72 h to engineer cells with the targeted genes.
-
9.
Replace the medium with fresh, complete medium containing the appropriate concentration of antibiotics every 2–3 d until the negative control cells all die. Infected cells will grow under antibiotic selection.
-
10.
Digest the cells with trypsin, load a hemocytometer to count the number of cells, dilute the cells to 5 cells/mL with culture medium containing the selective antibiotics.
-
11.
Add 200 μL of diluted cells per well into a 96-well plate and then put it back into the cell culture incubator.
-
12.
The plate should be checked every day, and the wells that only form one colony should be marked.
-
13.
Upon reaching 30–40 cells per colony, remove the culture medium and wash the cells once with 1×PBS.
Note: Depending on the cell type, the time for colony formation varies from 1–3 weeks.
-
14.
Incubate the colonies with TrypLE until they detach from the surface. Add 100 μL of complete medium to dilute the TrypLE. Carefully resuspend the cells and place them into a 6-well plate for a further week of cell growth. At least 10 colonies per virus should be collected.
-
15.
When the confluency of the cells reaches ∼80%, harvest half of the cells to perform RT-qPCR and western blotting for validation, and culture the remaining cells in a T75 cell culture flask.
-
16.
When the confluency of the cells reaches ∼80%, use 1 mL of trypsin to detach the cells and then add 9 mL of complete medium to neutralize the trypsin. The split ratio varies depending on the doubling time and the intended use of the cells.
Note: For a 1:2 split, transfer 5 mL of the cell suspension to a new T75 flask and add complete medium. For a 1:5 split, transfer 2 mL of the cell suspension to a new T75 flask and add complete medium. For a 1:10 split, transfer 1 mL of cell suspension to a new T75 flask and add complete medium.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-CLDN6 (1:100 dilution) | Affinity Biosciences | AF5213 |
| Mouse monoclonal anti-CLDN6 (0.25 μg/106 cells) | R&D Systems | MAB3656 |
| CLDN6-specific monoclonal antibodies (1 mg/mL) | Affinity Biosciences | N/A |
| Normal mouse IgG (1 mg/mL) | Santa Cruz Biotechnology | sc-2025 |
| Normal rabbit IgG (1:100 dilution) | R&D Systems | AB-105-C |
| Goat Anti-Rabbit IgG H&L (Alexa Fluor 488) (1:500 dilution) | Abcam | ab150077 |
| Goat Anti-Mouse IgG H&L (Alexa Fluor 488) (1:500 dilution) | Abcam | ab150113 |
| Goat Anti-Rabbit IgG H&L (Alexa Fluor 594) (1:500 dilution) | Abcam | ab150080 |
| Goat Anti-Mouse IgG H&L (Alexa Fluor 594) (1:500 dilution) | Abcam | ab150116 |
| Donkey Anti-Rabbit IgG H&L (Alexa Fluor 647) (1:500 dilution) | Abcam | ab150075 |
| Goat Anti-Mouse IgG H&L (Alexa Fluor 647) (1:500 dilution) | Abcam | ab150115 |
| Bacterial and virus strains | ||
| pLenti-CMV-GFP-3FLAG-PGK-Puro | Obio Technology (Shanghai) Corp., Ltd. | N/A |
| pLenti-CMV-MCS-3FLAG-PGK-Puro | Obio Technology (Shanghai) Corp., Ltd. | N/A |
| pLenti-CMV-CLDN6-3FLAG-PGK-Puro | Obio Technology (Shanghai) Corp., Ltd. | N/A |
| pcDNA3.0-shRNA-Control | IGE Biotechnology (Guangzhou) Co., Ltd. | N/A |
| pcDNA3.0-shRNA-CLDN6 | IGE Biotechnology (Guangzhou) Co., Ltd. | N/A |
| Biological samples | ||
| HCC patient-derived organoids | (Broutier et al.2) | N/A |
| Surgical specimen of tumor removed from HCC patients | Affiliated Cancer Hospital and Institute of Guangzhou Medical University | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Activin A | R&D Systems | 338-AC |
| Advanced DMEM/F-12 | Thermo Fisher Scientific | 12634028 |
| A8301 (TGFβ inhibitor) | Tocris Bioscience | 2939 |
| B27 Supplement (50×), minus vitamin A | Thermo Fisher Scientific | 12587010 |
| B-27 Supplement (50×), serum free | Thermo Fisher Scientific | 17504044 |
| Bovine serum albumin (BSA) | R&D Systems | 5217/100G |
| Collagenase D | Merck | 11088858001 |
| Cultrex Reduced Growth Factor BME, Type 2 PathClear (BME2) |
Merck | 3533-005-02 |
| 2-Mercaptoethanol | Thermo Fisher Scientific | 21985023 |
| DNaseI | Merck | DN25 |
| DMEM basic (1×) | Thermo Fisher Scientific | C11995500BT |
| DMEM, high glucose, GlutaMAX, pyruvate | Thermo Fisher Scientific | 10569010 |
| Dexamethasone | Merck | D4902 |
| DMSO | Merck | C6164 |
| Earle’s balanced salt solution (EBSS) | Thermo Fisher Scientific | 24010043 |
| Ellman’s reagent | MedChemExpress | HY-15915 |
| Fetal bovine serum (FBS) | Thermo Fisher Scientific | 10099141C |
| Forskolin | Tocris Bioscience | 1099 |
| GlutaMAX supplement | Thermo Fisher Scientific | 35050061 |
| HEPES, 1 M | Thermo Fisher Scientific | 15630080 |
| Insulin | Merck | I2643 |
| Keratinocyte growth factor | R&D Systems | 251-KG |
| KnockOut DMEM | Thermo Fisher Scientific | 10829018 |
| KnockOut DMEM/F-12 | Thermo Fisher Scientific | 12660012 |
| KnockOut serum replacement | Thermo Fisher Scientific | 10828010 |
| L-Glutamine (200 mM) | Thermo Fisher Scientific | 25030149 |
| [Leu15]-gastrin I human | Merck | G9145 |
| Madin-Darby bovine kidney maintenance medium | Merck | 14581C |
| MEM non-essential amino acids solution (100×) | Thermo Fisher Scientific | 11140050 |
| Mertansine (DM1) | MedChemExpress | HY-19792 |
| N2 supplement (100×) | Thermo Fisher Scientific | 17502048 |
| N-acetylcysteine | Merck | A0737-5MG |
| Nicotinamide | Merck | N0636 |
| Oncostatin M | R&D Systems | 295-OM |
| Paraformaldehyde (PFA), 4% in PBS | Thermo Fisher Scientific | AAJ61899AP |
| 1×PBS, pH 7.4 | Thermo Fisher Scientific | 10010023 |
| Penicillin/streptomycin (10,000 U/mL) | Thermo Fisher Scientific | 15140122 |
| Pentobarbital sodium | R&D Systems | 4579/50 |
| pHrodo™ iFL dye | Invitrogen | P36011 |
| Potassium bromide | Aladdin | P116276 |
| RPMI 1640 | Thermo Fisher Scientific | 11875119 |
| Recombinant human EGF | Peprotech | AF-100-15 |
| Recombinant human FGF-basic | Peprotech | 100-18B |
| Recombinant human FGF 4 | Peprotech | 100-31 |
| Recombinant human FGF10 | Peprotech | 100-26 |
| Recombinant human HGF | Peprotech | 100-39 |
| Rho kinase inhibitor Y-27632 dihydrochloride | Merck | Y0503 |
| RNAlater® RNA stabilization solution | Thermo Fisher Scientific | AM7020 |
| R-spodin1-conditioned medium | (Broutier et al.3) | N/A |
| Serum replacement solution | Peprotech | SR-100 |
| Sorafenib tosylate | AbMole | M1827 |
| Succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-car boxylate (SMCC) | AAT Bioquest | 4501 |
| TrypLE Express | Thermo Fisher Scientific | 12605028 |
| Trypsin-EDTA (0.05%) | Thermo Fisher Scientific | 25300120 |
| TO type biological tablet transparent agent (TO) | Beijing Solarbio Science & Technology | CB44716046 |
| Wnt-3a | R&D Systems | 5036-WN |
| Critical commercial assays | ||
| GTVisionTM Ⅲ Detection System/Mo&Rb | GTVision | GK500710 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | 23225 |
| Quick-RNA Miniprep Kit | Zymo Research | R1054 |
| Experimental models: Cell lines | ||
| Human embryonic stem cells (hESCs) | (Liu et al.4) | N/A |
| HepG2 | ATCC | HB-8065 |
| PLC/PRF/5 | ATCC | CRL-8024 |
| Experimental models: Organisms/strains | ||
| Mouse model: BALB/cNj-Foxn1nu/Gpt (Female, 4–6 weeks old) | GemPharmatech, China |
D000521 |
| Mouse model: NOD/ShiLtJGpt-Prkdcem26Dd52II2rgem26Cd22/Gpt (Female, 4–6 weeks old) | GemPharmatech, China |
T001475 |
| Software and algorithms | ||
| GraphPad | Prism, San Diego, USA | Version 8.0. https://www.graphpad.com |
| LAS X | Leica | Version 3.4.2 https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| Other | ||
| Cell strainer size 70 μm | Merck | CLS431751 |
| Forceps | N/A | N/A |
| Surgical scissors | N/A | N/A |
| 2.0 mL Microtubes | Merck | AXYMCT200CS |
| Corning® Costar® Ultra-Low Attachment 24-Well Plate | Merck | CLS3473 |
| Nunc™ 15 mL Conical Sterile Polypropylene Centrifuge Tubes | Thermo Fisher Scientific | 339651 |
| Nunc™ 50 mL Conical Sterile Polypropylene Centrifuge Tubes | Thermo Fisher Scientific | 339653 |
| Dialysis sacks | Merck | D6066 |
| Falcon® Round Bottom Polystyrene Tubes | Corning | 352052 |
| Aperio CS2 slide scanner | Leica | N/A |
| Leica DM6 B Fluorescence Motorized Microscope | Leica | N/A |
| ZEISS LSM 980 Confocal Laser Scanning Microscope | Carl Zeiss Microscopy | N/A |
| Cary 60 UV-Vis Spectrophotometer | Agilent | N/A |
| Zetasizer Nano ZS (DLS) | Malvern, UK | N/A |
| FTIR TENSOR27 Spectrophotometer | Bruker, Germany | N/A |
Materials and equipment
Complete medium
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM basic (1×) | N/A | 445 mL |
| Penicillin/Streptomycin (100×) |
1% | 5 mL |
| FBS | 10% | 50 mL |
| Total | N/A | 500 mL |
Note: Prepare in a sterile environment. Store the medium at 4°C for up to 6 weeks.
hESCs culture medium
| Reagent | Final concentration | Amount |
|---|---|---|
| KnockOut DMEM/F-12 | N/A | 41.41 mL |
| KnockOut Serum Replacement |
15% | 7.5 mL |
| Non-Essential Amino Acids Solution (100×) |
1% | 0.5 mL |
| L-Glutamine (200 mM) | 2 mM | 0.5 mL |
| 2-Mercaptoethanol (55 mM) | 0.1 mM | 91 μL |
| Recombinant human FGF-basic (100 μg/mL) |
4 ng/mL | 2 μL |
| Total | N/A | 50 mL |
Note: Prepare in a sterile environment. Store the medium at 4°C for up to 2 weeks.
hESCs feeder free culture medium
| Reagent | Final concentration | Amount |
|---|---|---|
| RPMI 1640 | N/A | 49.38 mL |
| Activin A (10 μg/mL) | 100 ng/mL | 0.5 mL |
| Wnt3a (10 μg/mL) | 25 ng/mL | 125 μL |
| Total | N/A | 50 mL |
Note: Prepare in a sterile environment. Store the medium at 4°C for up to 2 weeks.
Endoderm induction medium 1
| Reagent | Final concentration | Amount |
|---|---|---|
| KnockOut DMEM | N/A | 48.95 mL |
| Keratinocyte growth factor(25 μg/mL) | 25 ng/mL | 50 μL |
| FBS | 2% | 1 mL |
| Total | N/A | 50 mL |
Note: Prepare in a sterile environment. Store the medium at 4°C for up to 1 week.
Endoderm induction medium 2
| Reagent | Final concentration | Amount |
|---|---|---|
| KnockOut DMEM | N/A | 38.66 mL |
| Serum Replacement Solution(100×) | 20% | 10 mL |
| L-Glutamine (200 mM) | 1 mM | 250 μL |
| Non-Essential Amino Acids Solution (100×) | 1% | 0.5 mL |
| 2-Mercaptoethanol (55 mM) | 0.1 mM | 91 μL |
| DMSO | 1% | 0.5 mL |
| Total | N/A | 50 mL |
Note: Prepare in a sterile environment. Store the medium at 4°C for up to 1 week.
Passage medium
| Reagent | Final concentration | Amount |
|---|---|---|
| RPMI 1640 | N/A | 48.49 mL |
| GlutaMAX (100×) | 1% | 0.5 mL |
| KnockOut SerumReplacement | 2% | 1 mL |
| Recombinant human HGF(100 μg/mL) | 10 ng/mL | 5 μL |
| Recombinant human FGF-4 (100 μg/mL) | 10 ng/mL | 5 μL |
| Total | N/A | 50 mL |
Note: Prepare in a sterile environment. Store the medium at 4°C for up to 2 weeks.
Liver progenitor induction medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Madin-Darby bovine kidney maintenance medium | N/A | 49.44 mL |
| GlutaMAX (100×) | 1% | 0.5 mL |
| BSA (500 mg/mL) | 0.5 mg/mL | 50 μL |
| Recombinant human HGF(100 μg/mL) | 10 ng/mL | 5 μL |
| Recombinant human FGF-4(100 μg/mL) | 10 ng/mL | 5 μL |
| Total | N/A | 50 mL |
Note: Prepare in a sterile environment. Store the medium at 4°C for up to 2 weeks.
Mature medium
| Reagent | Final concentration | Amount |
|---|---|---|
| KnockOut DMEM | N/A | 44.40 mL |
| FBS | 10% | 5 mL |
| Recombinant human HGF (100 μg/mL) |
10 ng/mL | 5 μL |
| Oncostatin M (10 μg/mL) | 20 ng/mL | 0.1 mL |
| Dexamethasone (50 μM) | 0.5 μM | 0.5 mL |
| Total | N/A | 50 mL |
Note: Prepare in a sterile environment. Store the medium at 4°C for up to 2 weeks.
Digestion solution
| Reagent | Final concentration | Amount |
|---|---|---|
| EBSS | N/A | 9.8 mL |
| Collagenase D (250 mg/mL) | 2.5 mg/mL | 0.1 mL |
| DNasel (10 mg/mL) | 0.1 mg/mL | 0.1 mL |
| Total | N/A | 10 mL |
Note: Freshly prepare in a sterile environment and use it immediately.
Basal medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Advanced DMEM/F-12 | N/A | 485 mL |
| Penicillin/streptomycin (100×) |
1% | 5 mL |
| GlutaMAX (100×) | 1% | 5 mL |
| HEPES (1 M) | 10 mM | 5 mL |
| Total | N/A | 500 mL |
Note: Prepare in a sterile environment. Store the medium at 4°C for up to 1 month.
Wash medium
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM (high glucose, GlutaMAX and pyruvate) | N/A | 490 mL |
| Penicillin/streptomycin (100×) | 1% | 5 mL |
| FBS | 1% | 5 mL |
| Total | N/A | 500 mL |
Note: Prepare in a sterile environment. Store the medium at 4°C for up to 1 month.
Isolation medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Advanced DMEM/F-12 | N/A | 46.14 mL |
| Penicillin/Streptomycin (100×) | 1% | 0.5 mL |
| GlutaMAX (100×) | 1% | 0.5 mL |
| HEPES (1 M) | 10 mM | 0.5 mL |
| B27 supplement (without vitamin A) (50×) |
2% | 1 mL |
| N2 supplement (100×) | 1% | 0.5 mL |
| N-acetylcysteine (625 mM) | 1.25 mM | 0.1 mL |
| Nicotinamide (1 M) | 10 mM | 0.5 mL |
| [Leu15]-gastrin I human (10 μM) | 10 nM | 50 μL |
| Recombinant human EGF (100 μg/mL) | 50 ng/mL | 25 μL |
| Recombinant human FGF10 (100 μg/mL) | 100 ng/mL | 50 μL |
| Recombinant human HGF (50 μg/mL) | 25 ng/mL | 25 μL |
| Forskolin (25 mM) | 10 μM | 20 μL |
| A8301 (12.5 mM) | 5 μM | 20 μL |
| Dexamethasone (3 μM) | 3 nM | 50 μL |
| Y-27632 (25 mM) | 10 μM | 20 μL |
| Total | N/A | 50 mL |
Note: Prepare in a sterile environment. Store the medium at 4°C for up to 2 weeks.
Expansion medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Advanced DMEM/F-12 | N/A | 41.21 mL |
| Penicillin/Streptomycin (100×) | 1% | 0.5 mL |
| GlutaMAX (100×) | 1% | 0.5 mL |
| HEPES (1 M) | 10 mM | 0.5 mL |
| R-spodin1-conditioned medium | 10% | 5 mL |
| B27 supplement (without vitamin A) (50×) |
2% | 1 mL |
| N2 supplement (100×) | 1% | 0.5 mL |
| N-acetylcysteine (625 mM) | 1.25 mM | 0.1 mL |
| Nicotinamide (1 M) | 10 mM | 0.5 mL |
| [Leu15]-gastrin I human (10 μM) | 10 nM | 50 μL |
| Recombinant human EGF (100 μg/mL) | 50 ng/mL | 25 μL |
| Recombinant human FGF10 (100 μg/mL) | 100 ng/mL | 50 μL |
| Recombinant human HGF (50 μg/mL) | 25 ng/mL | 25 μL |
| Forskolin (25 mM) | 10 μM | 20 μL |
| A8301 (12.5 mM) | 5 μM | 20 μL |
| Total | N/A | 50 mL |
Note: Prepare in a sterile environment. Store the medium at 4°C for up to 2 weeks.
Tumorsphere culture medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Advanced DMEM/F-12 | N/A | 24.22 mL |
| Recombinant human EGF (100 μg/mL) |
20 ng/mL | 5 μL |
| Recombinant human FGF-basic (100 μg/mL) |
10 ng/mL | 2.5 μL |
| Insulin (4 mg/mL) | 4 μg/mL | 25 μL |
| Penicillin/Streptomycin (100×) |
1% | 250 μL |
| B27 supplement (50×) | 2% | 0.5 mL |
| Total | N/A | 25 mL |
Note: Prepare in a sterile environment. Store the medium at 4°C for up to 2 weeks.
Step-by-step method details
Identification of potential oncofetal targets for HCC patients refractory to chemotherapy
This section describes the procedures for establishing models to profile differentially expressed genes that facilitate the identification of potential therapeutic targets for HCC patients who are refractory to therapeutic drugs.
-
1.Generation of hepatocyte differentiation model.
-
a.Culture of hESCs on mitotically inactivated human or mouse embryonic fibroblasts (HEFs or MEFs) feeder layer at a density of ∼2,500 cells per cm2 in hESCs culture medium.Note: The hESCs culture medium can be stored at 4°C for up to 2 weeks.Note: Cells were cultured at 37°C in a humidified chamber with 5% CO2. HEFs or MEFs may take up to a week to reach confluence, and the medium may be changed daily to increase the growth rate of the HEFs or MEFs. Routinely passage the hESCs every 6 or 7 d.Note: Differentiation will occur in some hESC colonies. Pasteur pipettes can be used to routinely aspirate the differentiated colonies.
-
b.Use a minimal volume of warm 0.05% trypsin-EDTA to passage hESCs on 6 cm2 dishes coated with a feeder-free system until the confluence reaches 50%–70%.
-
c.Inducing hESCs to differentiate into hepatic endoderm.
-
i.Replace the medium with hESCs feeder free culture medium (Store at 4°C for up to 2 weeks) for 3–5 d, changing the medium every other day.
-
ii.Aspirate the medium and add endoderm induction medium 1 (Store at 4°C for up to 1 week) for 2 d.
-
iii.Change the medium to endoderm induction medium 2 (Store at 4°C for up to 1 week) for 4–7 d.
-
i.
-
d.Inducing hepatic endoderm into liver progenitor cells.
-
i.Passage hepatic endoderm cells with 0.05% trypsin-EDTA at a ratio of 1:3 or 1:6 and then plate them onto collagen I-coated dishes in passage medium (Store at 4°C for up to 2 weeks) for 2 d.Note: Stop digestion as soon as the cells have detached from the culture surface, which usually takes 1–5 min.
-
ii.Change the medium to liver progenitor induction medium (Store at 4°C for up to 2 weeks) for 10 d. Replace the medium every other day.
-
i.
-
e.Inducing liver progenitor cells to differentiate into hepatocyte-like cells.Replace the medium with mature medium (Store at 4°C for up to 2 weeks) for 7 more days.
-
f.Harvest the cells from each differentiation phase, following the manufacturer’s instructions for the Quick-RNA Miniprep Kit from Zymo Research to extract RNA.
-
g.Send the RNA for transcriptome sequencing (Sangon Biotech, China) to profile the genes that are active in hESCs and progressively decrease in expression throughout hepatocyte differentiation (Figure 1).
-
a.
-
2.Generation of a sorafenib refractory cell line-derived xenograft (CDX) model.Note: Sorafenib is the most widely used first-line drug for treating advanced HCC.
-
a.When the confluency of CRL-8024 or HepG2 cells reaches ∼80% in a 25 cm2 cell culture flask, remove the medium and wash the cells twice with 3 mL of PBS.
-
b.Aspirate the PBS and add 500 μL of trypsin and incubate the flask at 37°C until the cells detach.
-
c.Stop the dissociation by adding at least 3 volumes of medium containing 10% FBS.
-
d.Pellet the cells by centrifugation at 1,000× rpm (86 × g) for 5 min.
-
e.Discard the supernatant and wash the cells with 5 mL sterile PBS by centrifugation at 1,000× rpm (86 × g) for 5 min.
-
f.Discard the supernatant and gently resuspend the cells in 5 mL of sterile PBS, and take a 10 μL aliquot for counting.Optional: place a 10 μL aliquot of cells in a 200 μL sterile Eppendorf tube with 10 μL of 0.4% trypan blue, mix well, and load into a hemocytometer immediately to count the number of cells. The formula “Number of viable cells × 104 × dilution factor = cells/mL” is used to calculate the number of viable cells.Note: Avoid counting dark blue cells, which are dead.
-
g.Resuspend the cells in PBS at least 1 × 106 cells/100 μL for labeling.Note: The cell number required depends on the tumor cell type, usually 1–10 million cells.Optional: Thaw Matrigel on ice and add an equal volume of Matrigel to the cells and mix by pipetting up and down.Note: Matrigel has proven to be extremely useful in establishing xenografts from cells that are difficult to xenograft in the absence of Matrigel. Matrigel will solidify at room temperature (∼20°C–25°C).Note: Use a prechilled 1 mL insulin syringe. The syringes containing the cell/Matrigel mixture need to be placed on ice prior to injection to avoid solidification of the Matrigel.
-
h.Inject a total of 200 μL of the cell/Matrigel mixture into the right dorsal flank of immune-deficient mice subcutaneously.Note: Mice are considered juvenile from 4 weeks to 6 weeks old.
-
i.Divide the mice bearing 100–150 mm3 tumors into two groups: the solvent group and the sorafenib (60 mg/kg) group, with 5–10 mice per group.
-
j.Perform intraperitoneal injection of the treatment or solvent thrice per week (Monday, Wednesday and Friday).Note: Typically, inject into the mouse’s lower right quadrant of the abdomen to prevent damage to the abdominal organs.
-
k.Assess the tumor volume with a caliper every 3 days and calculate it according to the formula V=0.5 × L × W2.
-
l.If residual tumors appeared, euthanize the mice by using CO2 inhalation for 5 min.Note: The relative tumor volume (RTV) is calculated by the following formula: RTV = (tumor volume now)/(tumor volume on the day initiating the treatment). The tumor size will slowly increase with sorafenib treatment and then may start to grow quickly after some time. The tumor growth inhibition ratio (TGI) is calculated via the following formula: TGI(%) = [1-(RTV of the sorafenib group)/(RTV of the control group)] × 100%. TGI (%) drops by over 5% for 3 consecutive days after drug treatment are regarded as residual tumors, which may harbor potential drug refractory populations (e.g., for the PLC-8024 (2 × 106 cells/mouse)-generated CDX model, it takes approximately 3 weeks to develop residual tumors when treated with 60 mg/kg sorafenib) (Figure 2).
-
m.With sterile scissors and forceps, the subcutaneous tumor is separated from the surrounding skin. The tumors are minced into small pieces (2–5 mm) using sterile scissors or a scalpel. The tissues are divided into three parts for RNA extraction, protein extraction and fixation.
-
n.Soak the tissues for RNA extraction from step m in RNAlater® RNA Stabilization Solution and store at −20°C until RNA extraction.
-
o.Extract the RNA following the manufacturer’s instructions for the Quick-RNA Miniprep Kit from Zymo Research.
-
p.Send the RNA for transcriptome sequencing (Sangon Biotech, China) to profile the genes that are active in the residual tumors.Note: Sequencing depths between 20 M and 50 M reads per sample are recommended.
-
q.The tissues from step m need to be stored at −80°C or in liquid nitrogen until protein extraction.
-
r.Fix the tissues from step m in fixative solution for 1 day, and substitute 70% ethanol for the fixative solution for long-term storage at 4°C.
-
a.
Figure 1.

Establishment of an in vitro hepatocyte differentiation model
hESCs were induced to differentiate along hepatic lineages into adult hepatocytes. Cells from different developmental stages were selected for transcriptomic sequencing.
Figure 2.

PLC-8024 cells (2 × 106 cells/mouse) were subcutaneously injected into nude mice
The mice were randomly divided into two groups with different setups: treatment with PBS or sorafenib (60 mg/kg, thrice per week) (n=3). Tumor volumes were measured thrice per week.
(A) The tumor volume was calculated by V=0.5 × L × W2. Data are shown as the mean ±SD. ∗p < 0.05.
(B) Relative tumor volume change was estimated by TGI (%). The red dot indicates residual tumors.
Generation of in vitro and in vivo models
This section describes the procedures for establishing in vitro and in vivo models to explore the functional roles of potential oncofetal targets in HCC.
-
3.In vitro three-dimensional (3D) culture of HCC tissues.
-
a.Collect samples from patients who have not received any previous local or systemic treatment before the operation.CRITICAL: High-quality fresh-tissue samples are key for establishing successful organoids. Process the samples as soon as possible (i.e., within 1–4 h of surgical resection) to preserve the tissue viability.
-
b.Prewarm 24-well ultralow attachment surface cell culture plates at 37°C for 1 h to overnight (12–16 h). Thaw the frozen BME2 on ice until ready for use.
-
c.Dissect the tissues (0.25–1 cm3). In a laminar flow cabinet, place the samples in a 100 mm culture dish on ice and mince the tissue into small pieces (∼0.5 mm3) using sterile scissors and a scalpel.
-
d.Warm the digestion solution to 37°C prior to use.Note: Freshly prepare the digestion solution in a sterile environment and use it immediately.
-
e.Use a 3 mL pasteurized pipette to transfer small pieces into a 15 mL centrifuge tube containing 10 mL of ice-cold wash medium, gently wash with a pasteurized pipette, and then settle the tissue pieces for 1–2 min. Remove ∼7.5 mL of supernatant, including any blood cells and floating pieces of fat. Repeat this step twice.Note: The wash medium can be stored at 4°C for up to 1 month.
-
f.Carefully aspirate the remaining wash medium and add 5 mL of prewarmed digestion solution.
-
g.Rotate the tube at 37°C for digestion.
-
h.After initial digestion for 30 min, place 10 μL of the solution in a hemocytometer and observe under an inverted microscope to check for the presence of single cells. If few single cells are visible, continue rotating the tube at 37°C for digestion and check with a hemocytometer every 10 min to avoid overdigestion.Note: Stop the reaction when in contains 80%–100% single cells.
-
i.Stop the digestion by adding cold wash medium up to 15 mL, gently wash with a pasteurized pipette and filter through a 70 μm cell strainer.
-
j.Continue to add ice-cold wash medium to wash any remaining small pieces in the 15 mL centrifuge tube and then filter them through the cell strainer to remove any cell clusters or debris up to a volume of 50 mL.
-
k.Spin the cells down at 300 × g for 5 min at 4°C. Carefully remove the supernatant with a glass or plastic pipette, resuspend the pellet by adding 15 mL of ice-cold wash medium.
-
l.Repeat step k.
-
m.Spin down at 300 × g for 5 min at 4°C. Carefully discard the supernatant and wash the pellet with 10 mL of basal medium (Store at 4°C for up to 1 month). Take 10 μL of the solution to count the number of cells. Centrifuge the required amount of cells at 300 × g for 5 min at 4°C.
-
n.Remove the supernatant and resuspend the cells in 50 μL BME2 in a 24-well plate and gently pipette up and down until the cells are totally resuspended.Note: BME2 will solidify at room temperature (∼20°C–25°C). Keep BME2 on ice during the process. One tissue sample can be split into approximately 8–16 wells of a 24-well plate.
-
o.Spot the mixture dome in the center of each well according to the chart below, and then move the plate carefully into a 37°C cell culture incubator until the BME2 solidifies (∼30 min).Note: We recommend 1,000–5,000 cells per organoid in each well of a 24-well plate. A high cell density and/or the spread of BME2 to the wall of the well may cause the cells to attach to the culture plate.
-
p.Add 500 μL of isolation medium (Store at 4°C for up to 2 weeks) and then return the plate to the 37°C cell culture incubator.Note: The volumes differ according to the different culture conditions, as described in the table below.
Plate Number of domes per well Matrigel volume per dome (μL) Medium volume per well (mL) 48-well plate 1 25 0.25 24-well plate 1 50 0.5 6-well plate 8 50 3 -
q.Refresh the isolation medium twice a week.
-
r.After 2–3 weeks for HCC-derived organoids, change the isolation medium to organoid expansion medium (Store at 4°C for up to 2 weeks).
-
s.Change the expansion medium twice a week.
-
t.Harvest or passage the organoids when they develop dense cultures or large structures (exceeding 20% diameter of the dome).Note: Passage the cultures when the dome tends to detach from the plate.
-
u.Passaging organoids.
-
i.Prewarm a 24-well cell culture plate at 37°C for 1 h to overnight (12–16 h). Thaw the frozen BME2 on ice until ready for use.Note: Organoids are generally passaged at a 1:4–1:6 ratio.
-
ii.Remove the culture medium, add 500 μL of cold basal medium to each well, and break the dome apart by scraping and pipetting using a 1,000 μL pipette.
-
iii.Place a 15 mL centrifuge tube on ice and transfer the suspension from 3 wells into one tube.
-
iv.Continue to add cold basal medium to ∼13–14 mL, incubate on ice for 20 min, and keep pipetting the cell pellet multiple times to break it up. The incubation and pipetting need to be repeated if residual Matrigel is still present.Note: To better dissolve the BME2, do not combine more than 3 wells for a 24-well plate or 6 wells for a 48-well plate together at one time.
-
v.When the Matrigel has completely dissolved and a clear cell pellet is observable, centrifuge the tube at 4°C and 200 × g for 5 min.
-
vi.Aspirate the basal medium as much as possible without disturbing the pellet. Resuspend the pellet in 1 mL of prewarmed TrypLE, mixed by pipetting up and down multiple times, incubate at 37°C, and then check the digestion solution under a bright-field microscope every 2 min.Note: Overdigestion will lead to decreased cell viability.
-
vii.Add 10 mL of cold basal medium to inactivate the digestion when the majority of the organoid fragments turn into single cells. Filter the solution through a 70 μm cell strainer.
-
viii.Centrifuge the cells at 4°C 300 × g for 5 min.
-
ix.Resuspend the cell pellet in expansion medium. Mix the cells gently and take 10 μL of the organoid solution to count the number of cells. Transfer the required amount of cell suspension to another 15 mL conical tube and spin it down.Note: Domes could be seeded with 1,000–5,000 live cells. The seeding density should be optimized for each donor.
-
x.Generate the organoid domes according to steps n to o.
-
xi.Place the plate in the hood, add 500 μL of expansion medium containing the required amount of lentivirus or drug, and then carefully move the plate into the 37°C cell culture incubator (Figure 3).Note: Comparison of the characteristics of the organoids cultured with isolation medium or expansion medium.
-
i.
-
a.
| Organoids | Cultured in isolation medium (2–3 weeks) | Cultured in expansion medium (1–2 weeks) |
|---|---|---|
| Shape | Clusters | Spherical or oval |
| Diameter | 100–300 μm | 100–300 μm |
| Growth | Slower | Faster |
| Components | Tumor cells, normal liver cells | Tumor cells |
-
4.Sphere formation assay.
-
a.See Generation of the sorafenib refractory cell line-derived xenograft (CDX) model from step a to e.
-
b.Take the required number of CLDN6-overexpressed or -knockdown HCC cells and dilute the cells into a 24-well ultralow attachment plate at a final concentration of 2,000 cells/mL in tumorsphere culture medium.Note: The density of cells may vary with cell type.
-
c.Seed a total of 250 μL of cell mixture into each well (500 cells per well).Note: The density of the cells will vary by cell type.
-
d.Check the spheres and replenish with 30 μL of tumorsphere culture medium (Store at 4°C for up to 2 weeks) every second day.
-
e.After 3–10 days of incubation, once the spheres are generated, transfer the spheres into a 1.5 mL sterile Eppendorf tube.
-
f.Settle the cells for 10–15 min.
-
g.Aspirate as much of the supernatant as possible with no disturbance of the cells.
-
h.Add a few tens of microliters of TrypLE into the tube, pipette up and down several times to break the spheres apart.Note: If using trypsin, neutralize with trypsin inhibitor.
-
i.Centrifuge the cells at 1,000× rpm (86 × g) for 5 min and aspirate the supernatant.
-
j.Resuspend the cells in tumorsphere culture medium.
-
k.Seed the cells in 24-well ultralow attachment plates at the desired density to form next-generation spheres.
-
a.
-
5.In vivo CDX model.
-
a.Remove the medium from the CLDN6-overexpressing CRL-8024 cells (control, CLDN6) or CLDN6 knockdown HepG2 cells (shControl, shCLDN6) and wash the cells twice with 3 mL of PBS in a 25 cm2 cell culture flask.
-
b.See Generation of sorafenib refractory cell line-derived xenograft (CDX) model from steps b to h.
-
c.Assess the tumor volumes thrice per week (Monday, Wednesday and Friday) and calculate it with the formula V=0.5 × L × W2.
-
d.Euthanize the mice by using CO2 inhalation for 5 min.
-
e.See Generation of the sorafenib refractory cell line-derived xenograft (CDX) model from steps m to n, q to r.
-
a.
Figure 3.

Representative images of HCC organoids cultured under isolation and expansion conditions at different time points
Scale bar = 250 μm.
Examination of changes in the lineage-specific markers
Immunohistochemistry and immunofluorescence are useful tools to detect the expression and localization of liver progenitor cell surface markers, biliary markers and hepatic markers associated with lineage plasticity at the protein level.
-
6.Immunohistochemistry staining.
-
a.Tissue block preparation and deparaffinization.
-
i.Fix freshly harvested tissues (thickness<3 mm) in cassettes by immersion in 4% PFA fixative solution for 24 h at 4°C.
-
ii.Wash the tissues in running tap water for 30 min to 1 h.
-
iii.Place the cassettes into 70% ethanol for 24 h.
 Pause point: Tissues can be stored in 70% ethanol for a longer time.
Pause point: Tissues can be stored in 70% ethanol for a longer time. -
iv.Turn the paraffin wax machine on to melt the paraffin prior to embedding.Note: Paraffin melts at 58°C.
-
v.Dehydrate the tissues according to the following procedure:
Step Solution Time (min) 1 80% Ethanol 60–120 2 90% Ethanol 45–60 3 95% Ethanol 30–45 4 100% Ethanol 30 4 100% Ethanol 30 5 TO 30 6 TO 30 7 Paraffin Wax 30–60 8 Paraffin Wax 30–60 Note: Xylene or histoclear can be substituted for TO. -
vi.Fill an embedding mold with paraffin wax and place the tissues in the molten paraffin.
-
vii.After the paraffin solidifies, remove the embedded construct from the mold and store it at 4°C until ready for sectioning.
-
viii.Use a rotary microtome to cut 4–5 μm thick tissue sections at room temperature (∼20°C–25°C).Note: Place the blocks on ice before cutting.
-
ix.Float the sections in a 40°C–45°C warm water bath containing distilled water.
-
x.Place an adhesion microscope slide into the water bath and mount each section on an individual slide.
-
xi.Air dry the sections for 12–18 h at room temperature (∼20°C–25°C).
-
xii.Store the sections at room temperature (∼20°C–25°C) or 2°C–8°C for long-term storage.
-
i.
-
b.Rehydration.
-
i.Place the slides in an electric thermostatic drying oven at 60°C–65°C for 1–3 h.
-
ii.Immerse the slides in TO three times for 10 min each.
-
iii.Immerse the slides in 100% ethanol twice for 5 min each.
-
iv.Immerse the slides in 95% ethanol twice for 5 min each.
-
v.Immerse the slides in 90% ethanol for 5 min.
-
vi.Immerse the slides in 80% ethanol for 5 min.
-
vii.Immerse the slides in 70% ethanol for 5 min.
-
viii.Gently rinse the slides in distilled water for 5 min.Caution: TO is a nontoxic clearing agent that replaces xylene. The odor of TO is a stimulus, and the rehydration process should be conducted in a fume hood.
-
i.
-
c.Antigen retrieval.
-
i.Prepare 1 liter of 10 mM sodium citrate buffer, pH 6.0, in distilled water and then pour it into a pressure cooker.
-
ii.Immerse the slides in the sodium citrate buffer and maintain it at 120°C for 5 min in the pressure cooker.
-
iii.Remove the cooker from the hot plate and allow the slides to naturally cool to room temperature (∼20°C–25°C) before proceeding to the next step.Note: Check the antibody datasheet for individual antigen retrieval buffers and manipulation. The optimal heating time should be adjusted by the user.
-
iv.Rinse the slides in distilled water and then wash them twice with 1×PBS for 5 min each.
-
i.
-
d.Immunostaining.
-
i.Drain and carefully remove any excess solution around the tissue sections. Create a hydrophobic barrier around the tissue using a barrier pen.
-
ii.If an HRP-conjugated primary antibody is to be applied, block any endogenous peroxidase with 3% hydrogen peroxide for 15 min, and then rinse the slides three times with 1×PBS for 5 min each.
-
iii.After draining and carefully wiping away any excess solution around the tissue sections, block the specimens in ready-to-use normal goat serum for 30 min at room temperature (∼20°C–25°C).Note: The blocking solution should be selected according to the primary antibody datasheet.
-
iv.Drain any excess solution, and incubate each section with the diluted primary antibody in a humidified chamber overnight (12–16 h) at 4°C.Note: Refer to the antibody datasheet for the antibody diluent, dilution ratio and working conditions.
-
v.Gently rinse the slides three times with 1×PBS for 5 min each.
-
vi.Drain and shake off any excess solution. Incubate each section with diluted secondary antibody in a humidified chamber for 1 h at room temperature (∼20°C–25°C).
-
vii.Gently rinse the slides three times with 1×PBS for 5 min each.CRITICAL: Maintain tissue hydration at all times during the immunostaining steps.
-
i.
-
e.Detection.
-
i.Calculate the total volume of DAB-peroxidase substrate solution required to cover all tissue sections. Apply freshly prepared DAB substrate to the tissue sections and incubate at room temperature (∼20°C–25°C).Caution: DAB is a potent carcinogen. Gloves, laboratory coats, and safety goggles need to be used to prevent contact.Note: Approximately 100–200 μL of DAB solution is required per slide.
-
ii.Monitor the staining under a light microscope. When suitable staining develops, place the slide in distilled water.
-
iii.Rinse the slides three times with distilled water for 5 min each.
-
iv.Counterstain the slides with hematoxylin for 10–30 s.
-
v.Rinse the slides in running tap water for 5–10 min.
-
i.
-
f.Dehydration.
-
i.Immerse the slides in 70% ethanol for 5 min.
-
ii.Immerse the slides in 80% ethanol for 5 min.
-
iii.Immerse the slides in 90% ethanol for 5 min.
-
iv.Immerse the slides in 95% ethanol twice for 5 min each.
-
v.Immerse the slides in 100% ethanol twice for 5 min each.
-
vi.Immerse the slides in TO three times for 5 min each.
-
vii.Dry the slides for 1–2 h at room temperature (∼20°C–25°C).
-
viii.Add a drop of mounting media to the tissue sections on the slide and top with a coverslip.Note: Remove air bubbles between the coverslip and slide.
-
ix.Dry the slides overnight (12–16 h) at room temperature (∼20°C–25°C).
-
x.Visualize staining of the tissue under a bright-field microscope.
-
i.
-
a.
-
7.Immunofluorescence staining.
-
a.See Immunohistochemistry staining for steps a to c.
-
b.Rinse the slides gently three times with 1×PBS for 5 min each.
-
c.Block the specimens in ready-to-use normal goat serum for 30 min at room temperature (∼20°C–25°C).Note: Select the blocking solutions according to the primary antibody specification.
-
d.While blocking, dilute the primary antibody in the correct buffer.
-
e.Aspirate the blocking solution and incubate the sections with diluted primary antibody in a humidified chamber overnight (12–16 h) at 4°C.
-
f.Rinse the slides with 1×PBS three times for 5 min each.
-
g.Dilute the fluorescence-conjugated secondary antibody in 1×PBS following the antibody’s information sheet. Incubate the sections with the secondary antibodies for 1–2 h at room temperature (∼20°C–25°C) in the dark.
-
h.Rinse the slides in 1×PBS three times for 5 min each time protected from the light.
-
i.Stain the specimens with 0.5–10 μg/mL DAPI for 5–10 min protected from the light.
-
j.Rinse the slides in 1×PBS three times for 5 min each time protected from the light.
-
k.Add a drop of mounting media to the tissue sections on the slide and top with a coverslip protected from the light.Note: Remove air bubbles between the coverslip and slide.
-
l.Measure the fluorescence immediately or store the slides at 4°C in the dark for long-term storage.Note: The fluorescence intensity will be weakened with time.
-
a.
Development of antibody-drug conjugates for HCC treatment
The previous sections describe the procedures for the development of in vitro and in vivo models to explore the functional roles of potential targets in HCC. We then detail the procedures of immunohistochemistry and immunofluorescence for investigation of changes in the expression of lineage-specific markers. As the studied candidate CLDN6 is a transmembrane oncofetal protein, this section describes the design and development of antibody-based therapeutics to precisely target CLDN6+HCC cells.
-
8.
Generation of antibody-drug conjugates (ADCs).
To illustrate this protocol, we used CLDN6-DM1-ADC as an example (Figure 4).-
a.Preparation.
-
i.Extract information of CLDN6-specific amino acid sequences using Expasy (https://web.expasy.org/protparam/).
-
ii.Calculate the amount of available moles of primary amines via the formula m/M(1+n1+n2+n3+n4). The variable m represents the mass of the protein, M represents the molecular weight of the protein, and n1, n2, n3, and n4 represent the total number of asparagine, glutamine, lysine, and arginine residues in the amino acid sequences, respectively.Note: The maximum drug loading depends on the number of free amino groups on the antibody.
-
iii.Design the immunogen using AbDesigner (https://esbl.nhlbi.nih.gov/AbDesigner/).
-
iv.CLDN6-specific monoclonal antibodies were generated by immunizing BALB/c mice with gene-specific extracellular peptides at Affinity Biosciences Co., Ltd. IgG was used as a negative control.
-
v.Dissolve 1 mg of the linker species SMCC in 500 μL DMSO to prepare a 0.5 mg/mL stock solution.Note: Succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) is a heterobifunctional crosslinker containing N-hydroxy succinimide-ester and maleimide, which react with amines and thiols, respectively.
-
vi.Prepare 1 mg/mL of antibody solution in 1×PBS (pH 7.4), and the final concentration should be no less than 1 mg/mL.
-
i.
-
b.Antibody-drug conjugate reaction.
-
i.Place 2 mL of antibody solution in a Florence flask.
-
ii.Couple the antibody with SMCC (1 μL, 0.5 mg/mL) shaking at 100 r/min at 4°C for 4–6 h.Note: A sufficient amount of Antibody-NH2 could avoid yield losses in the subsequent reactions. If the antibody has any free sulfhydryls, protect them with benzyl chloride before the reaction, and reduce the disulfide bonds using 2-mercaptoethylamine. HCl (β-MEA) after the reaction.
-
iii.Dialyze the product after the reaction with 1×PBS (pH 7.4) at 4°C for 12 h to remove the free SMCC and byproducts. Change the PBS every 3 h.
-
iv.Place the antibody-SMCC product in a 15 mL centrifuge tube. The volume can be determined according to the graduated scale on the centrifuge tube.
-
v.Calculate the concentration as the ratio of the total initial antibody mass to the volume.
-
vi.Combine the drug DM1 and the intermediate antibody SMCC in the desired molar ratio and mix by stirring at 100 r/min for 4–6 h at 4°C.Note: Determine the optimal solvent (e.g., ethanol, acetone or water) for the chemical.Note: Longer incubation times do not negatively affect conjugation, and conjugations can also be left overnight (12–16 h).
-
vii.Calculate the thiols in the protein with Elman’s reagent. Prepare a tube containing 100 μL of Elman’s reagent and 10 μL of antibody-SMCC-DM1 reaction buffer, and incubate the mixture at room temperature (∼20°C–25°C) for 15 min.
-
viii.Measure the absorbance of each sample with a NanoDrop spectrophotometer at 412 nm. Continue to the next step if the OD value >0.15; if 0.05<OD<0.15, replenish the reaction with additional drugs to meet the conjugation requirements. If OD<0.05, the antibody-SMCC reaction should be performed again.Note: Elman's reagent is specifically used for quantitating free sulfhydryl groups. When it reacts with sulfhydryls, the reaction buffer turns yellow. The color of the reaction buffer will not change when sulfhydryls are converted into dimeric or multimeric oxidized forms.
-
ix.Dialyze the product after the reaction with 1×PBS (pH 7.4) at 4°C for 12 h to remove the free DM1. Change the PBS every 3 h.Note: Avoid using buffers containing primary amines (e.g., Tris or glycine) and sulfhydryl groups during the conjugation as they will react with SMCC. If necessary, dialyze or desalt the sample into an appropriate buffer, such as PBS.
-
x.Collect the dialysate and lyophilize it in a vacuum freeze dryer. Quantify the protein samples with BCA according to the manufacturer’s protocol.
-
i.
-
c.Determination of the antibody conjugation efficiency.
-
i.Prepare 5 mg/mL of chemical drug solution, apply a UV-vis spectrophotometer with a full wavelength scan to determine the maximum absorption peak of the drug.
-
ii.Make two fold serial dilutions (dilution factor of two) with a range of concentrations from 5 mg/mL to 0.05 mg/mL. Measure the absorption values of the different drug concentrations at specific wavelengths, and use the wavelength corresponding to the maximum absorption peak to draw a standard curve of absorbance versus solution concentration.Note: Overconcentration of the drug should be avoided. The range of concentrations should be 0.05 mg/mL to 5 mg/mL. The drug concentration increases with the extent of absorption, and the maximum (peak) absorption is determined.
-
iii.Calculate the molecular mole according to the Formula n = m/M. Repeat substeps b-x and c-i, ii to quantify the protein samples with BCA three times to calculate the average number of drug molecules conjugated to an antibody.
-
iv.Dissolve two milligrams of lyophilized powder from each group (e.g., IgG-DM1, CLDN6-DM1) in 2 mL of double-distilled water and filter it through a 0.45 μm membrane. Detect the particle size and zeta potential of the filtrates using dynamic light scattering (DLS) with a Zetasizer Nano ZS.
-
v.Dry FT-IR grade potassium bromide powder at 110°C for 4 h and then mix it according to a mass ratio of 100:1 with lyophilized powder from each group (SMCC, Ab, Ab-SMCC, DM1, Ab-DM1).
-
vi.Grind the mixtures to a powder and press them in a powder tablet pressing machine. Measure the FTIR spectra of the pressed pellet samples with a TESOR 27 spectrophotometer and analyze the functional groups in each component.Note: Lyophilization is required for the ADC products to improve their stability, and the products should be placed in a vacuum drying oven to remove moisture for long-term storage.
-
i.
-
a.
-
9.Examine the binding of the antibody-DM1 to the HCC cells.
-
a.Harvest the HCC cells (0.5 × 106–1 × 106), wash them with 1×PBS once and centrifuge at 300 × g for 5 min.Note: Cells are usually stained in Falcon® Round Bottom Polystyrene Tubes. Cells can also be stained in Eppendorf tubes.
-
b.Block the cell samples in ready-to-use normal goat serum (100 μL) for 30 min at room temperature (∼20°C–25°C) with gentle shaking 2–3 times.Note: Select appropriate blocking solutions according to the primary antibody specification.
-
c.While blocking, dilute the primary antibody in terms of the antibody datasheet.
-
d.Centrifuge the cell samples at 300 × g for 5 min and aspirate the blocking solution.
-
e.Incubate the cells with the diluted primary antibody at 4°C for 1 h.
-
f.Wash the samples with 1×PBS twice for 5 min each.
-
g.Dilute the fluorescence-conjugated secondary antibody in PBS as indicated in the antibody information sheet. Incubate the cell samples with diluted secondary antibody at 4°C for 1 h.
-
h.Wash the cell samples with 1×PBS twice for 5 min each.
-
i.Resuspend the cell samples in 0.5–1 mL of 1× PBS.
-
j.Run the stained cell samples immediately on a flow cytometer.Note: Perform steps g to j in the dark.
-
a.
-
10.Antibody internalization assay.
-
a.Place a sterile 12 mm round coverslip in each well of a 24-well plate.Note: 18 mm round coverslips for 12-well plates, 25 mm round coverslips for 6-well plates.Note: Sterilize coverslips by washing in absolute ethanol or placing under UV light in a cell-culture hood for no less than 60 min.
-
b.Harvest the target cells and calculate the cell concentration (see Generation of sorafenib treatment cell line-derived xenograft (CDX) model for steps a to f.
-
c.Transfer the required number of cells directly to the coverslips to achieve 35%–45% confluence Overnight (12–16 h).
-
d.On the next day, aspirate the cell medium and wash the cells twice with 1×PBS.
-
e.Incubate the cells with an appropriate amount of ADC and cell culture medium for the indicated time points. For example, HepG2 cells were seeded at 1 × 104 cells per well and then incubated with 20 ng/μL unconjugated anti-CLDN6 antibody or ADCs for 0 h, 1 h, 2 h, 4 h and 8 h.
-
f.After incubation, aspirate the medium and wash the cells three times with 1×PBS for 5–10 min each to remove any unbound antibody.
-
g.Aspirate the PBS and incubate the cells with anti-goat IgG (H&L) at 4°C for 1 h.
-
h.Rinse the cells three times with 1×PBS for 5 min each.
-
i.Stain the cell nucleus with 0.5–10 μg/mL DAPI for 5–10 min. Wash the cells three times with 1×PBS for 5 min each.
-
j.Fix the cells in 4% PFA for 20–30 min at room temperature (∼20°C–25°C).
-
k.Aspirate the fixative and rinse three times with 1×PBS for 5 min each time.
-
l.Add 10 μL of mounting media to each slide.
-
m.Utilize forceps to retrieve the coverslips from the 24-well plate. Dry the coverslip quickly on filter paper and place the coverslip cells face down on the slide.Note: After fixation, many biological samples become autofluorescent once completely dried.Note: Remove any air bubbles between the coverslip and slide.
-
n.Use a confocal microscope to take fluorescence images.Note: The manipulation from g to n must be protected from the light.Pause point: Prior to imaging, mounted slides can be stored at 4°C in the dark for at least 4 weeks.Pause point: If confocal microscopy imaging can be performed immediately, skip steps j and k.
-
a.
-
11.pH-sensitive internalization assay.
-
a.Dilute 500 μg of amine-reactive pHrodo™ iFL dye in 75 μL of anhydrous DMSO to prepare an ∼10 mM solution.
-
b.Seed cells in a 24-well plate according to the Antibody internalization assay from steps a to d.
-
c.On the next day, add 10–20× molar excess of pHrodo™ iFL dye to the appropriate amount of ADC in 0.1 M sodium bicarbonate buffer, pH 8.4.
-
d.Mix the ADC and pHrodo™ iFL dye gently and incubate for 10–60 min in the dark.
-
e.Add the ADC-dye mixture to the cell culture medium and then incubate for 2–4 h.Note: Sufficient incubation times will enable specific binding of the mixture to the cell surface targets.
-
f.After incubation, wash the cells three times with 1×PBS for 5–10 min each to remove any unbound antibody.Optional: Stain the cell nucleus with 0.5–10 μg/mL DAPI for 5–10 min in the dark.
-
g.Wash the cells three times with 1×PBS for 5 min each.
-
h.For mounting and image acquisition, refer to Antibody internalization assay from steps j to n.
-
a.
-
12.Patient-derived xenograft (PDX) establishment.
-
a.PDX models were established in 4- to 6-weeks-old male NCG mice (GemPharmatech, China). In a laminar flow cabinet, tumor samples were transferred from the section “In vitro three-dimensional (3D) culture of HCC tissues” in a 100 mm culture dish on ice, and the tissue was minced into small pieces (i.e., 2 mm × 2 mm, F0) using sterile scissors and a scalpel.
-
b.Add 10 mL of cold DMEM/F12 with penicillin-streptomycin to a 15 mL centrifuge tube and place the tissue pieces into the tube on ice until use.
-
c.Enter the mouse facility. Clean up the biological safety cabinet with 75% ethanol.
-
d.Place sterile paper towels on the work surface. Turn on the heating pad.
-
e.Record the body weight of each mouse. Intraperitoneally inject the proper volume of 1% pentobarbital sodium according to the body weight, as described in the table below.
Body weight (g) Volume (μL) 18 90 19 95 20 100 21 105 22 110 23 115 24 120 25 125 -
f.Ensure that anesthesia is complete. Punch the ear for identification and remove the fur from the dorsal region with an electric clipper.
-
g.Make a 0.5 cm incision halfway in the lower dorsal region of the mouse with scissors and curved forceps.
-
h.Lift the cut skin with curved forceps, and create a pocket with scissors by blunt dissection of the left lower back dorsal skin.
-
i.Use straight forceps to subcutaneously implant one piece of tumor into the pocket.
-
j.Sew up the cut using medical sutures. Clean the wound using sterile alcohol prep pads or alcohol-soaked cotton balls (Figure 5).
-
k.Place the mouse on a warm heating pad and monitor until fully mobile.
-
l.Return the mouse to its home cage. These mice in the first-generation phase are regarded as F1 mice.
-
m.After 4–10 weeks, when the tumor reaches ∼1.5 cm, another transplant should be prepared.Note: The number of implanted mice should be determined according to the experimental design.Note: See Methods video S1 for steps g to j.Methods video S1. A procedure for implanting tumor piecesThis video displays the procedure for establishing the PDX model. The tumor piece is implanted into the formed “pocket”, and then the incision is closed with sutures, related to substeps g to j in step 12.Download video file (13.3MB, mp4)
-
n.Euthanize the F1 mice by using CO2 inhalation for 5 min. Take the euthanized mouse into a sterilized biological safety cabinet.
-
o.Isolate the tumor and surrounding skin using sterile scissors and a scalpel.
-
p.Transfer the resected tumor to a 100 mm culture dish containing cold DMEM/F12 with penicillin-streptomycin.
-
q.Cut the tissues into equal pieces (i.e., 2 mm × 2 mm) using sterile scissors and a scalpel.Note: The engrafted tissues should be gathered into one place rather than scattering them to minimum the size difference before drug treatment.
-
r.Anesthetize the mouse as described in steps e to f.
-
s.Implant a subcutaneous piece of tumor into the left lower back region of the mouse as described in steps g to k. These mice in the second-generation phase are regarded as F2 mice.
-
t.Monitor the tumor growth. When the tumor volume reaches between 80 and 120 mm3, randomly divide the mice into two groups: IgG-DM1 (4 mg/kg) or CLDN6-2-DM1 (4 mg/kg), with 5–10 mice per group.
-
u.Perform intraperitoneal injection of the treatment three times per week (Figure 6).CRITICAL: Ensure that the injection site is correct to avoid the possibility of organ damage caused by injection. An intraperitoneal injection is typically given in the lower right or left quadrants of the abdomen, not in the middle. It is recommended that the needle be relatively short (e.g., 0.5 inches or less) to prevent puncturing the intestines or bowels.
-
v.Assess the tumor volumes every 3 days and calculate the volume according to the formula V=0.5 × L × W2.
-
w.When the difference in the tumor size between the groups reaches statistical significance, euthanize the mice and extract the tumors under sterile conditions. See Generation of sorafenib treatment cell line-derived xenograft (CDX) model for steps l to o, q to r.CRITICAL: The tumor size must not exceed 20 mm at the largest diameter in mice.
-
a.
Figure 4.

The process of ADC generation
The SMCC is first reacted with the antibody to produce an intermediate antibody-SMCC, which is further coupled with the drug to form the final ADC.
Figure 5.

Schema for the construction of PDX models
(A) Resection of the tumor masses.
(B) Anesthetization of the mice.
(C) Cut a wound in the lower dorsal flanks.
(D) Blunt dissection of the left lower back dorsal skin with scissors.
(E) Use forceps to subcutaneously implant one piece of the tumor away from the wound.
(F) The wound is sutured after the application of an alcohol-soaked cotton ball.
(G) Sterilization of the wound.
(H) The mouse is moved to a warm heating pad and monitored until fully mobile.
Figure 6.

Intraperitoneal administration of ADC
(A) The injection was performed in the mouse’s lower right quadrant of the abdomen.
(B and C) Proceeding with the injection when negative pressure is observed.
Expected outcomes
Increasing evidence, including our recent study, suggests an important link between tumor lineage and therapeutic resistance. Therefore, the purpose of this protocol is to provide essential and reliable methods to identify therapeutic targets for drug-tolerant HCC, unveil the changes in cell lineage triggered by candidate targets, uncover the functions in tumorigenicity and drug refractoriness, and further develop therapeutic antibodies specifically for candidates to precisely target drug-refractory HCC tumors.
We recently used this procedure to identify CLDN6 as a novel therapeutic target related to lineage plasticity in HCC. A de novo anti-CLDN6 monoclonal antibody conjugated with the cytotoxic agent DM1 was developed to precisely target CLDN6+ HCC cells with tumor lineage plasticity, which led to attenuation of the tolerance of HCC cells to sorafenib (See (Kong et al.1) for more details).
Limitations
Increasing evidence has implied the importance of the association between developmental signals and therapeutic resistance. Oncofetal proteins are principally active in embryonic development but silenced in the terminal differentiation stage. The hepatocyte differentiation model mimics liver development and could be applied to identify oncofetal targets in HCC. The CDX model was established to generate residual tumors after persistent drug treatment. Integrating the transcriptomic data of both models will help to provide candidate therapeutic targets. However, more databases, such as the GTEX and TCGA datasets, could be imported to screen for the candidate, as an ideal candidate is usually highly expressed in HCC but expressed at extremely low levels in normal organs. For example, our published CLDN6 was identified by evaluating the intersection of genes in the following 4 groups: Group 1 contained the genes that were highly expressed in the embryonic stage and progressively decreased in expression through the premature hepatocyte stage. Group 2 comprised the genes that had extremely low expression (TPM, transcript per million < 0.1) in mature hepatocytes. Group 3 comprised the genes that progressively increased in expression from distant normal liver (>1 cm from the tumor) to para-tumor liver (<1 cm from the tumor) to tumor tissues. Group 4 included the genes that were upregulated in the HCC residual tumors compared with their expression in the transplanted HCC cells. CLDN6 was the top potential therapeutic target based on the differences.
Tumor sphere formation and organoids are applied for in vitro functional studies. Tumor spheres are generated to assess the proliferation of the stem/progenitor cells residing in tumors. Organoids are stem cell-derived 3D constructs that recapitulate the complex and dynamic nature of primary tumors. However, the lack of an immune system impedes the in vitro evaluation of immune-based therapeutics in the organoid model.
Immunohistochemistry and immunofluorescence are useful tools to detect the expression and localization of liver progenitor cell surface markers, biliary markers and hepatic markers associated with lineage plasticity at the protein level. In addition, other useful techniques, such as western blotting and qPCR assays, could be combined to fully clarify the tumor lineage plasticity.
HCC is highly heterogeneous and plastic, and a single ADC may not function to eliminate all tumor cell populations. Some cancer cell populations may lose certain tumor antigens under therapeutic pressure and escape antibody recognition. The combination of ADCs with other therapeutic drugs may enhance antitumor efficacy during HCC treatment.
Troubleshooting
Problem 1
Low or no-yield production of organoids or the organoid lines grow slowly (In vitro 3D culture of HCC tissues steps).
Potential solution
Tissue digestion should be frequently checked under a microscope to avoid overdigestion. Confirm the cell pellet is obtained after dissociation. Appropriate medium components need to be selected, and fresh medium should be prepared to culture different sources of organoids. Change the medium frequently (2–3 times a week).
Problem 2
Tumorspheres become large cell aggregates or clumps (Sphere formation assay steps).
Potential solution
Begin with a single cell suspension at a lower cell density. The plate should remain flat and move as little as possible during incubation. When the tumorspheres are too large, the incubation time should be decreased.
Problem 3
Nonspecific signals, high background, a weak or no signal occurs in immunohistochemistry and immunofluorescence staining (Examination of changes in the lineage-specific markers steps).
Potential solution
Nonspecific signal: Try different antigen retrieval approaches for the antigen or tissue; reduce the antibody concentration and incubation time; and wash and incubate sections with 0.3% Triton X-100 to clean the tissues (immunohistochemistry/immunofluorescence).
High background: Try to test multiple concentrations of primary and secondary antibodies, prolong the blocking time or change the blocking solution, and increasing the wash times and numbers might reduce the background (immunohistochemistry/immunofluorescence).
Weak or no signal: Prevent overblocking the tissues. Increase the concentration of the antibody and incubation time. Permeabilize the cells with 0.3% Triton X-100 if formaldehyde was used to fix the tissues. Run a positive control to ensure that the conditions are effective. The primary and secondary antibodies should be compatible (immunohistochemistry/immunofluorescence). An enhanced DAB substrate system (immunohistochemistry) can be used. Reduce the laser power, adjust the cutoff points of the detectors and use different fluorophores with no obvious interference between emission spectra. Note that the fluorescence of endogenous GFP or similar fluorescent tags can be affected by fixation conditions (immunofluorescence).
Problem 4
The drug-loading efficiency is low (Generation of antibody-drug conjugates steps).
Potential solution
It is common for an antibody to have a drug load of 0–8 drugs. A molar excess of SMCC over the amount of antibody-lysine residue (-NH2) will result in a sufficient conjugated reaction. It is recommended to add an appropriate amount of SMCC to the antibody solution as follows:
-
•
Add 40- to 80-fold molar excess to protein samples < 1 mg/mL.
-
•
Add 20-fold molar excess to protein samples 1–4 mg/mL.
-
•
Add 5- to 10-fold molar excess to protein samples at 5–10 mg/mL.
Problem 5
The chemical does not harbor free sulfhydryl groups (Generation of antibody-drug conjugates steps).
Potential solution
If it is not a sulfhydryl-containing molecule, 2-iminothiolane·HCl (Traut’s Reagent) or N-succinimidyl S-acetylthioacetate (SATA) can be used to modify the primary amines into sulfhydryl groups.
Problem 6
Few or no PDX tumors formed (Patient-derived xenograft establishment steps).
Potential solution
Ensure implants with an appropriate size of fresh tissue (i.e., 2 mm × 2 mm) and remove necrotic areas of the tumor. Due to the heterogeneity of unknown features of the patient, some patient samples may not form PDX tumors.
Problem 7
Combination treatment has no obvious antitumor effect (Patient-derived xenograft establishment steps).
Potential solution
A dose and concentration adjustment might be necessary to increase the antitumor efficacy if the survival or weight does not change.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ming Liu (liuming@gzhmu.edu.cn).
Materials availability
This study did not generate unique reagents.
Acknowledgments
Graphical abstract is created with BioRender.com and quoted with permission from BioRender. This research was supported by the National Natural Science Foundation of China (82122048; 82003773).
Author contributions
M.M.L. designed the experiments, summarized the protocols, wrote the manuscript, and drew the figures. F.E.K. designed the experiments and provided technical suggestions. G.M.L. contributed to the generation of antibody-drug conjugate and drew Figure 4. Y.T.H. and J.K.L. assisted M.M.L. in some of the experiments and drew the figures. X.F.Z. and C.Y.Z. generated HCC organoids. X.Y.G. and N.F.M. reviewed and revised the manuscript. M.B.X. and M.L. designed the study and revised the manuscript. All authors reviewed and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101921.
Contributor Information
Mao-Bin Xie, Email: maobinxie@gzhmu.edu.cn.
Ming Liu, Email: liuming@gzhmu.edu.cn.
Data and code availability
This study did not include any new data or codes that are not presented in the published article.
References
- 1.Kong F.E., Li G.M., Tang Y.Q., Xi S.Y., Loong J.H.C., Li M.M., Li H.L., Cheng W., Zhu W.J., Mo J.Q., et al. Targeting tumor lineage plasticity in hepatocellular carcinoma using an anti-CLDN6 antibody-drug conjugate. Sci. Transl. Med. 2021;13:eabb6282. doi: 10.1126/scitranslmed.abb6282. [DOI] [PubMed] [Google Scholar]
- 2.Broutier L., Mastrogiovanni G., Verstegen M.M., Francies H.E., Gavarró L.M., Bradshaw C.R., Allen G.E., Arnes-Benito R., Sidorova O., Gaspersz M.P., et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017;23:1424–1435. doi: 10.1038/nm.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broutier L., Andersson-Rolf A., Hindley C.J., Boj S.F., Clevers H., Koo B.K., Huch M. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat. Protoc. 2016;11:1724–1743. doi: 10.1038/nprot.2016.097. [DOI] [PubMed] [Google Scholar]
- 4.Liu M., Yan Q., Sun Y., Nam Y., Hu L., Loong J.H., Ouyang Q., Zhang Y., Li H.L., Kong F.E., et al. A hepatocyte differentiation model reveals two subtypes of liver cancer with different oncofetal properties and therapeutic targets. Proc. Natl. Acad. Sci. USA. 2020;117:6103–6113. doi: 10.1073/pnas.1912146117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This video displays the procedure for establishing the PDX model. The tumor piece is implanted into the formed “pocket”, and then the incision is closed with sutures, related to substeps g to j in step 12.
Data Availability Statement
This study did not include any new data or codes that are not presented in the published article.