

Abstract
Chemoresistance remains a major obstacle to successful treatment of triple negative breast cancer (TNBC). Identification of druggable vulnerabilities is an important aim for TNBC therapy. Here, we report that SERCA2 expression correlates with TNBC progression in human patients, which promotes TNBC cell proliferation, migration and chemoresistance. Mechanistically, SERCA2 interacts with LC3B via LIR motif, facilitating WIPI2-independent autophagosome formation to induce autophagy. Autophagy-mediated SERCA2 degradation induces SERCA2 transactivation through a Ca2+/CaMKK/CREB-1 feedback. Moreover, we found that SERCA2-targeting small molecule RL71 enhances SERCA2–LC3B interaction and induces excessive autophagic cell death. The increase in SERCA2 expression predisposes TNBC cells to RL71-induced autophagic cell death in vitro and in vivo. This study elucidates a mechanism by which TNBC cells maintain their high autophagy activity to induce chemoresistance, and suggests increased SERCA2 expression as a druggable vulnerability for TNBC.
Key words: TNBC, SERCA2, Autophagy, LC3B, Chemoresistance, Druggable, Vulnerability
Graphical abstract
Increased SERCA2 expression enhances autophagy activity to induce chemoresistance, and might be a druggable vulnerability for TNBC therapy.
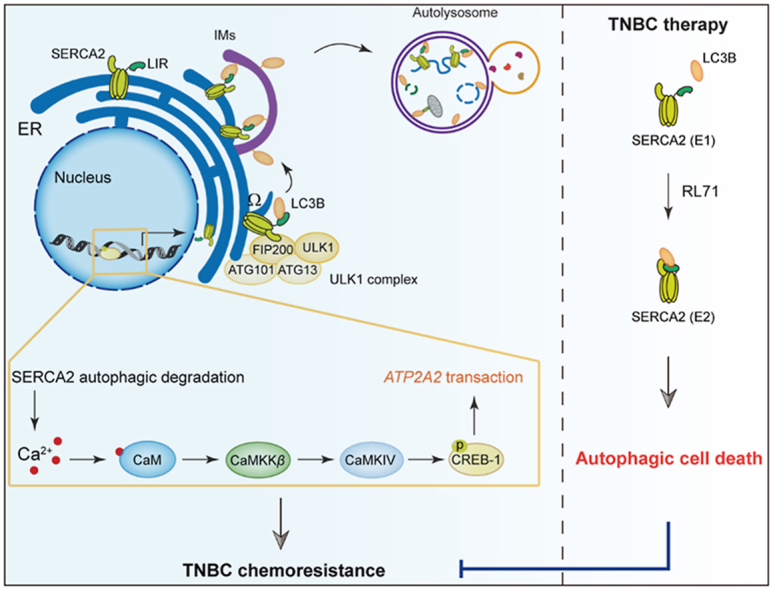
1. Introduction
Autophagy is a “double-edged sword” and possesses both tumor-suppressive and tumor-promoting functions, which are strongly dependent on the context of tumorigenesis1,2. Although autophagy is believed to have antitumoral potential before the onset of tumorigenesis, it frequently shifts to protumoral mechanisms, enabling the survival of tumor cells and conferring resistance to lethal attacks ‘executed’ by chemotherapy and radiotherapy in advanced cancer stages. Accordingly, many types of advanced cancers exhibit high autophagic activity3, 4, 5. Breast cancer is a malignant tumor characterized by disordered autophagy regulation6. Recent studies have demonstrated that in the TNBC subtype with the most aggressive phenotype, the expression of the autophagy-related marker LC3 is the highest among breast cancers7. High expression of LC3B has been associated with tumor progression and poor outcome in TNBC8, indicative of constitutive activation of basal autophagy and its protumoral function. Chemoresistance is a major obstacle in TNBC patients who mostly rely on chemotherapy9. However, the mechanism by which robust autophagic activity is sustained and consequently contributes to chemoresistance in TNBC and whether this sustained autophagy can be successfully targeted for therapeutic benefit remain to be elucidated.
Sarco/endoplasmic reticulum calcium-ATPases (SERCAs) act as Ca2+-ATPases that transfer Ca2+ from the cytosol to the lumen of the sarcoplasmic/endoplasmic reticulum (ER)10. SERCA-dependent calcium transport is the only calcium uptake mechanism in the ER. The levels of SERCA expression undergo significant changes during tumorigenesis. Our group and colleagues11, 12, 13 have reported that elevated expression of SERCA2, one isoform of SERCA, is closely related to the malignancy of different types of cancers.
In this study, we found that SERCA2 expression correlates with TNBC malignant progression, promoting cell proliferation, migration and chemoresistance. Further studies revealed that the ER-localized transmembrane protein SERCA2 directly interacted with LC3B and facilitated WIPI2-independent autophagosome formation to induce autophagy. Intriguingly, autophagy-mediated SERCA2 degradation triggered a transient increase in intracellular Ca2+ level and subsequently induced SERCA2 transactivation through a Ca2+/CaMKK/CREB-1 feedback. To therapeutically exploit these findings, we found that a small-molecule, RL71, has the potential to enhance the SERCA2–LC3B interaction and that overexpression of SERCA2 renders TNBC cells highly sensitive to RL71-induced autophagic cell death in vitro and in vivo. Our findings, for the first time, reveal increased SERCA2 expression as a novel druggable vulnerability in TNBC therapy.
2. Materials and methods
2.1. Reagents and antibodies
Paclitaxel (PTX, S1150), carboplatin (S1215), 5-fluorouracil (5-Fu, S1209), bafilomycin A1 (Baf A1, S1413), rapamycin (S1039), STO-609 (S8274), BAPTA (S7534) and compound C (S7840) were purchased from Selleck (Houston, TX, USA). Methyl thiazolyl tetrazolium (MTT, M5655), 4-phenyl butyric acid (4-PBA, P21005), thapsigargin (TG, T9033), actinomycin D (A1410) and crystal violet (C3886) were purchased from Sigma–Aldrich (St. Louis, MO, USA). RL71 (>97% purity, HPLC) was synthesized as described previously14. Anti-LC3B (3868), anti-SERCA2 (9580), anti-WIPI2 (8567), anti-calnexin (2679) antibodies and Autophagy Induction (ULK1 Complex) Antibody Sampler Kit (46486) were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-SERCA2 (sc-376235), anti-CREB-1 (sc-377,154), anti-p-CREB-1 (sc-81486), anti-COX4 (sc-376731), anti-Ki-67 (sc-23900) and anti-β-actin (sc-47778) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-SQSTM1/p62 antibody (ab101266) was purchased from Abcam (Cambridge, UK). Anti-ATG5 (10181-2-AP), ATG7 (10088-2-AP), anti-GFP (66002-1-Ig), anti-FLAG (20543-1-AP), anti-Lamin B1 (12987-1-AP), anti-SERCA1 (22361-1-AP) and anti-SERCA3 (13619-1-AP) antibodies were purchased from Proteintech Group (Chicago, IL, USA). Earle's Balanced Salt Solution (EBSS), MitoTracker Green FM (M7514), ER-Tracker Red (E34250), Alexa Fluor 488 goat anti-mouse IgG (R37114) and Alexa Fluor 594 goat anti-rabbit IgG (R11037) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). GST and GST-TRAF6 proteins were synthesized and purified by GenScript (Nanjing, China). GST-LC3B (BML-UW1155) was purchased from Enzo Life Sciences, Inc. (Farmingdale, NY, USA).
2.2. Cell culture
Human TNBC cell lines MDA-MB-231, SUM1315, MDA-MB-468 and HEK293T cells were purchased from the Cell Bank Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). All the cells were cultured in DMEM medium (Life Technologies, Grand Island, NY, USA) supplemented with 10% FBS (GIBCO, Grand Island, NY, USA), 100 U/mL penicillin, and 100 mg/mL streptomycin and incubated at 37 °C in a humidified atmosphere containing 5% CO2.
2.3. Mice
Female BALB/c nude mice (6–8 weeks old) were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). All animal experiments were performed in compliance with the guidelines (Ministry of Science and Technology of China, 2006) and relevant ethical regulations of Nanjing University. All efforts were made to minimize animal sufferings and the number of animals used.
2.4. Immunohistochemistry (IHC) and histologic analysis
The tissue microarrays of human TNBC (BR487c) were purchased from US Biomax, Inc. (Rockville, MD, USA). A tissue microarray containing 77 paired TNBC tissues (Cat. No. BRC1601) was purchased from the Shanghai Superbiotek Pharmaceutical Technology Co., Ltd. (Shanghai, China). Paraffin-embedded sections were stained with the antibody against SERCA2 and then determined using Real Envision Detection Kit (GK500705, GeneTech, Shanghai, China) according to the manufacturer's instructions. IHC scores for human breast cancer with different malignancy were evaluated as described previously12. Immunostaining of Ki-67 and p62 in the tumor tissues from mouse models was performed using a Real Envision Detection Kit according to the manufacturer's instructions. Paraffin-embedded sections of lung tissues from mice were stained with hematoxylin and eosin (H&E) using standard protocols. All data analysis was performed blindly.
2.5. Plasmids, transfection and RNA interference
FLAG-pcDNA3.1, FLAG-SERCA2 and FLAG-SERCA2 (H278A + K876A) plasmid were obtained as previously described14. FLAG-SERCA2 mutLIR (EFDEL LIR motif substituted by five alanine residues) was constructed by site-directed mutagenesis. Human cDNA encoding SERCA2 was inserted into pEGFP-C1 vector or mCherry-EGFP labeled pBABE-puro vector to obtain EGFP-SERCA2 or mCherry-EGFP-SERCA2. The mCherry-EGFP-LC3 plasmid (22418) and mCherry-DFCP1 plasmid (86746) were purchased from Addgene (Cambridge, MA, USA). pEGFP-C1 plasmid (VYC0084) and pEGFP-C1-LC3 plasmid (VXY0543) were from Yingrun Biotechnologies Inc. (Changsha, China). Transfection of plasmids was carried out with Lipofectamine 3000 (Thermo Fisher Scientific, L3000015). The lentivirus encoding SERCA2 was generated by Obio Technology Corp., Ltd. (Shanghai, China). Cells were infected with the lentivirus. Stably SERCA2-overexpressing cells were selected with 5 μg/mL puromycin and maintained in the culture medium with 0.5 mg/mL puromycin.
For RNA interference, cells were transfection with control or siRNA oligos using Lipofectamine RNAi MAX (ThermoFisher Scientific, 13778100). The resulting cells were cultured for 24–48 h prior to analysis. CREB-1 siRNA (sc-29281) was purchased from Santa Cruz Biotechnology. WIPI2 siRNA sequences were 5′-GGACCGGGUACUUCGGGAA-3′. Luciferase siRNA was used as controls. The sequences of ATG5, ATG7, SERCA2 and luciferase siRNAs was described as previously15.
2.6. Colony formation assay
Cells were transfected with FLAG-pcDNA3.1 or FLAG-SERCA2 plasmids for 24 h and then plated on 6-well plates (1000 cells/well). After 12 days of culture, cells were fixed by 4% paraformaldehyde for 15 min, washed twice with PBS and stained with 1% crystal violet solution for 15 min. After washing out crystal violet solution, plates were air-dried before photography.
2.7. Wound-healing assay
Wound-healing assay was performed as previously described16.
2.8. Cell viability assay
To assess the effects of SERCA2 on drug sensitivity, cells were transfected with FLAG-pcDNA3.1 or FLAG-SERCA2 plasmids, or control or SERCA2 siRNA for 24 h. Then the resulting cells were treated with various concentrations of paclitaxel, carboplatin and 5-fuorouracil for 48 h. Cell viability was determined using the MTT assay as previously described15.
2.9. Western blot, co-immunoprecipitation and transmission electron microscopy
The protocols for Western blot, co-immunoprecipitation and transmission electron microscopy have been reported previously15.
2.10. Immunofluorescence
SUM1315 cells on coverslips were fixed with 4% paraformaldehyde for 30 min at room temperature. The cells were permeabilized with PBS containing 0.1% Triton X-100 for 30 min, blocked with 3% BSA for 1 h and then incubated with the primary antibodies at 4 °C overnight. After washing three times with PBS, the cells were incubated with Alexa Fluor-conjugated secondary antibodies for 2 h. The nucleus was stained with DAPI (Beyotime, Shanghai, C1006) for 2 min. After staining, the cells were prepared for microscopic analysis. Fluorescent images were acquired using the FluoView TM FV1000 Confocal Microscope (Olympus, Tokyo, Japan) and analyzed by the Olympus Fluview Ver 1.7 b viewer (Olympus).
2.11. Flow cytometry
Cells were stained with ER-Tracker Red or MitoTracker Green FM for 30 min at 37 °C. Then the cells were washed and resuspended in a FACS buffer (1 × PBS, 1 mmol/L EDTA, 25 mmol/L HEPES, pH 7.3–7.5). The ER mass or mitochondria mass were analyzed with a FACSCalibur Flow Cytometer (Becton Dickinson, San Jose, CA, USA) by using FlowJo software.
2.12. GST pull-down
HEK293T cells were transfected with the indicated plasmids and lysed with lysis buffer (P0013, Beyotime, Shanghai, China). Lysates were cleared by centrifugation at 12,000 × g for 10 min. GST, GST-LC3B or GST-TRAF6 fusion proteins were incubated with glutathione–agarose (Sigma–Aldrich) for 4 h at 4 °C. After removing the supernatant, the lysates were incubated with GST-fusion-protein-loaded agarose for 4 h. The agarose was then washed three times in lysis buffer. The bound proteins were eluted and analyzed by Western blot.
2.13. Real-time RT-PCR and chromatin immunoprecipitation (ChIP)
Real-time RT-PCR was performed as previously described16. The primer sequences used in this study were: ATP2A2 forward, 5′-CGCTACCTCATCTCGTCCA-3′; ATP2A2 reverse, 5′-TCGGGTATGGGGATTCAA-3′. GADPH forward, 5′-AACAGCGACACCCACTCCTC-3′; GADPH reverse, 5′-GGAGGGGAGATTCAGTGTGGT-3′.
ChIP assay was performed using Pierce Magnetic ChIP Kit (ThermoFisher Scientific, 26157) according to the manufacturer's instructions. Anti-CREB-1 antibody was used for immunoprecipitation. The enrichment of CREB-1 at the promoter region of ATP2A2 was analyzed by real-time RT-PCR using the appropriated promoter primers. Relative quantification of target was normalized to input control. The primer sequences used in ChIP-PCR were as follows: ATP2A2 forward, 5′-TACATGCCAACGTCCACTCC-3′; ATP2A2 reverse, 5′-GGCTGGGCTTCAGGACTTAA-3′.
2.14. Molecular dynamics stimulation
The best pose in the largest cluster was extracted as a starting structure for MD simulation. The system with phospholipid bilayer was built by Charmm-GUI. The system was first minimized by 5000 steps of steepest and 5000 steps of conjugate gradient, and then heated to the target temperature of 300 K for a period of 20 ps in constant pressure periodic boundary conditions. The system was subjected to equilibrate the system by 20 ns of constant pressure and temperature (NPT) with time step of 1 fs, which was followed by 20 ns of production simulation performed in the same conditions. A cutoff of 10 A was used for non-bonded interactions and long-range electrostatic interactions were calculated with the Particle Mesh Ewald (PME) method. The MD simulation results were analyzed using the MDTraj and PyMol.
2.15. In vivo experiments
Two murine cancer models were used to examine the efficacy of RL71. In one model, SUM1315 cells (1 × 107 cells in 100 mL PBS) stably expressing vectors or SERCA2 (SERCA2-OE) were subcutaneously injected into the right flank of female nude mice. After 7 days, the tumor-bearing mice (an average size of 50 mm3) were randomly divided into three groups (n = 6 mice per group). Olive oil (vehicle control), RL71 (2 mg/kg/day) or PTX (10 mg/kg/7 days) were administered for 14 days by intraperitoneal injection. For another, MDA-MB-231 cells (2 × 106 cells in 20 mL PBS) stably expressing SERCA2 (SERCA2-OE) were injected into the fourth mammary fat pad in the lower abdomen of female nude mice. After 14 days, the tumor-bearing mice were treated as described as above. Tumor volumes were determined by caliper measurement every 2 days, and calculated using Eq. (1):
| V = 0.5236 × L1 × (L2)2 | (1) |
where L1 is the longer and L2 is the shorter tumor axis. The mice were sacrificed on Day 15. Tumor and lung tissues were collected, followed by photography and measurement. All animal experiments were performed twice independently.
2.16. Statistical analysis
Data are expressed as the means ± standard deviation (SD). All statistical analyses were performed using the GraphPad Prism 5.0 software (San Diego, CA, USA). Statistical significance was calculated using two-tailed Student's t-tests for comparisons between two groups and one-way ANOVA with post hoc Tukey HSD (Honestly Significant Difference) test using correction for comparisons among more than two groups. Post hoc tests were run only if F achieved P < 0.05 and there was no significant variance inhomogeneity. A probability level of 0.05 was chosen for statistical significance.
3. Results
3.1. SERCA2 expression correlates with TNBC progression and induces chemoresistance
We initially examined SERCA2 protein expression in a tissue array of TNBC. IHC analysis of tumor tissues from human TNBC patients in different stages showed that elevated SERCA2 expression was associated with the degree of malignancy (Fig. 1A). When the relationship between the SERCA2 level and survival rate in 77 TNBC patients was compared, we found that higher ATP2A2 expression correlated with poor prognosis for TNBC progression (Fig. 1B and Supporting Information Table S1). Moreover, overexpression of SERCA2 increased colony formation activity in multiple TNBC cell lines, including MDA-MB-231, SUM1315 and MDA-MB-468 (Fig. 1C). Wound-healing assay showed that SERCA2 overexpression promoted cell migration in SUM1315 cells (Fig. 1D). In contrast, silencing SERCA2 decreased colony formation in those three TNBC cell lines and inhibited cell migration in SUM1315 cells (Supporting Information Fig. S1A–S1E). Given that chemoresistance is a major obstacle in TNBC treatment, we assessed the role of SERCA2 in drug sensitivity. Of note, overexpression of SERCA2 markedly reduced the sensitivity of TNBC cells to standard-of-care chemotherapy agents, including paclitaxel (PTX), carboplatin and 5-fuorouracil (Fig. 1E and Fig. S1F). In contrast, silencing SERCA2 augmented their chemosensitivity (Fig. 1F and Fig. S1G). Because P53 is an important player in chemoresistance and p53 mutation frequently occurs in TNBC cells17,18, we examined the effects of SERCA2 expression on luminal breast cancer cells ZR75-1 and MCF-7 harboring non-mutant p53 to rule out the relevance of mutant p53 in SERCA2-mediated chemoresistance. In accordance with our data in the multiple TNBC cell lines with p53 mutation, overexpression of SERCA2 increased colony formation (Supporting Information Fig. S2A and S2B), and SERCA2 levels determined the chemosensitivity in ZR75-1 and MCF-7 cells (Fig. S2C and S2D). Together, our data implicate that SERCA2 may be critical for TNBC progression and chemoresistance.
Figure 1.

Highly expressed SERCA2 promotes TNBC progression and induces chemoresistance. (A) Immunohistochemistry (IHC) to detect SERCA2 protein levels in human cancer normal adjacent tissues (NAT) and TNBC tissues in different clinical stages. Left panel, representative images. Scale bar, 50 μm. Right panel, IHC scores. The variance was analyzed with one-way ANOVA. (B) Correlation of SERCA2 protein levels with overall survival in TNBC patients. (i) IHC to detect SERCA2 protein levels. Scale bar, 100 μm. (ii) The number of patients used in the analysis of SERCA2 expression. (iii) The relevance of SERCA2 expression with prognosis of TNBC patients was analysis. (C) Colony formation assay using the indicated TNBC cells transfected with FLAG-pcDNA3.1 or FLAG-SERCA2 plasmids. The quantification results are shown (right panel). (D) Wound-healing assay using SUM1315 cells transiently expressing FLAG-pcDNA3.1 or FLAG-SERCA2. The wound-healing areas were calculated (right panel). (E, F) MTT assay to assess the sensitivity of the indicated TNBC cells transfected with (E) FLAG-pcDNA3.1 or FLAG-SERCA2 plasmids or transfected with (F) NC-siRNA or siRNA targeting SERCA2 to paclitaxel (PTX). Data are shown as the mean ± SD of at least three independent experiments. ∗∗∗P < 0.001.
3.2. SERCA2 modulates autophagic activity by interacting with LC3B in TNBC
To determine the mechanism underlying SERCA2-mediated TNBC progression and chemoresistance, we performed gene set enrichment analysis in ATP2A2high and ATP2A2low breast cancers. Our analysis characterized increased expression of autophagy-associated genes in ATP2A2high tumors (Fig. 2A). In accordance with this finding, degradation of p62 and LC3B lipidation induced by EBSS starvation, was markedly enhanced in SERCA2-overexpressing SUM1315 cells compared with the cells transfected with vectors. The change in autophagic flux was inhibited by the addition of the autophagy inhibitor bafilomycin A1 (Baf A1) (Fig. 2B). When autophagy was induced by rapamycin, SERCA2 overexpression augmented the degradation of p62 and LC3B lipidation (Supporting Information Fig. S3A). Such changes in autophagic flux were also inhibited by Baf A1. In contrast, autophagic flux was not affected in SERCA2-silenced SUM1315 cells upon either EBSS starvation or rapamycin treatment (Fig. 2C and Fig. S3B). Autophagosome formation and LC3 turnover were further examined by transfecting SERCA2-overexpressing or SERCA2-silenced SUM1315 cells with mCherry-EGFP-LC3 plasmids. Under basal conditions, more mCherry-positive and EGFP-negative puncta were observed in SERCA2-overexpressing cells than in control or SERCA2-silenced cells, indicating higher basal autophagic activity owing to the higher stability of mCherry in the acidic milieu (Fig. 2D)19. After EBSS starvation, the increase in the number of LC3 puncta and mCherry-LC3 dots was more prominent in SERCA2-overexpressing cells than in control cells, whereas only a few mCherry-LC3 dots were observed in SERCA2-silenced cells. Similarly, transmission electron microscopy confirmed that autolysosomes were more abundant in SERCA2-overexpressing SUM1315 cells than in controls under basal conditions, an effect that was more prominent upon EBSS starvation (Fig. 2E). In contrast, autolysosome accumulation was negligibly detected in SERCA2-silenced cells under either basal or starvation conditions. Additionally, transient overexpression of SERCA2 significantly reduced the mass of both ER and mitochondria upon EBSS starvation, which was reversed by the addition of Baf A1 (Supporting Information Fig. S4A–S4D). SERCA2 overexpression also induced remarkable collapse of the mitochondrial membrane potential in SUM1315 cells under either basal or starvation conditions (Fig. S4E). These data suggest that SERCA2 could induce removal of both ER (ER-phagy) and mitochondria (mitophagy) via autophagy. Moreover, pharmacological or genetic autophagy inhibition reversed the insensitivity of SERCA2-overexpressing SUM1315 cells to PTX after treatment with Baf A1 and siRNA against ATG5 or ATG7, respectively (Fig. 2F and G).
Figure 2.

SERCA2 facilitates autophagy in TNBC cells. (A) Gene Set Enrichment Analysis of significantly altered pathways between ATP2A2high and ATP2A2low breast cancer based on the dataset GES21653 from GEO database. Gene sets were obtained and analyzed against Gene Ontology datasets. (B) Western blot analysis of indicated proteins in SUM1315 cells transiently expressing FLAG-pcDNA3.1 or FLAG-SERCA2, which were pretreated with or without 100 nmol/L bafilomycin A1 (Baf A1) for 2 h and then treated with EBSS for 12 h. β-Actin was used as a loading control. The densitometry of the immunoblots was performed with image J software and is presented in the histograms (right panel). The data are shown as the mean ± SD of three independent experiments. (C) Western blot analysis of indicated proteins in SUM1315 cells with siRNA-mediated knockdown of SERCA2, which were pretreated with or without 100 nmol/L Baf A1 for 2 h and then treated with EBSS rapamycin for 12 h. (D) SERCA2-overexpressing or -silenced SUM1315 cells were transfected with the mCherry-EGFP-LC3 plasmid. The number of LC3 puncta and mCherry-LC3 dots as well as the proportion of mCherry-positive but EGFP-negative (red) LC3 puncta were quantified under basal conditions or after 4 h of EBSS starvation (lower panel). n = 200 puncta. Scale bars, 5 μm; insets, 2 μm. (E) Transmission electron microscopy of SERCA2-overexpressing or -silenced SUM1315 cells under basal conditions or after 4 h of EBSS starvation. Autolysosomes were indicated by arrowheads. N, nucleus; Scale bars, 500 nm. (F, G) MTT assay to assess the sensitivity of the SUM1315 cells expressing FLAG-pcDNA3.1 or FLAG-SERCA2 to PTX, which were pretreated with or without 100 nmol/L Baf A1 for 2 h (F) or cotransfected with the indicated siRNAs (G). Data are shown as the mean ± SD of at least three independent experiments. ∗P < 0.05, ∗∗∗P < 0.001.
Since SERCA2 is an ER-resident transmembrane protein and might be involved in selective autophagy, we hypothesized that SERCA2 may serve as an autophagy receptor. Intriguingly, a bioinformatics analysis revealed the presence of a conserved LC3-interacting region (LIR) motif in SERCA family members (-EFDEL-, Fig. 3A). GST fusion of LC3B, but not unrelated TRAF6, specifically pulled down FLAG-SERCA2 from HEK293T cell lysates (Fig. 3B). An immunoprecipitation (IP) assay confirmed that endogenous and overexpressed SERCA2 interacted with LC3 (Fig. 3C and D), and exogenous LC3 precipitated SERCA2 from cell lysates (Fig. 3E). Similar results were obtained for SERCA1 and SERCA3 (Supporting Information Fig. S5). Substitution of amino acids in this LIR motif with alanine residues (EFDEL/AAAAA, mutLIR) completely abolished the interaction of SERCA2 with LC3 (Fig. 3F and G). Additionally, an immunofluorescence analysis confirmed the increased colocalization of SERCA2 with LC3B after EBSS starvation, which did not occur in the case of SERCA2 with mutant LIR (Fig. 3H). And the number of cells with LIR-mutant SERCA2/LC3B dual positive foci was much lower than that of cells with wide-type SERCA2 2/LC3B dual positive foci under basal condition, suggesting that the SERCA2/LC3B interaction via the LIR motif accounts for high basal autophagic activity in SERCA2-overexpressing cells. The LIR-mutant SERCA2 failed to further enhance autophagy after EBSS starvation, as shown by the less reduction in degradation of p62 compared with wide-type SERCA2 control (Fig. 3I). These data thus show a direct interaction of SERCA2 and LC3B via the LIR motif, which is required for SERCA2-mediated autophagy.
Figure 3.

SERCA2 interacts with LC3B. (A) Domain architecture of SERCA and alignment of the LC3-interacting region (LIR) motif. Black, LIR. (B) GST pull-down assays with GST-fused LC3B or TRAF6 to overexpressed FLAG-SERCA2 from HEK293T cell lysates. (C) Co-immunoprecipitation (IP) of SERCA2 with endogenous LC3B in SUM1315 cells. (D) Co-IP of FLAG-SERCA2 with EGFP-LC3 in transfected HEK293T cells. (E) Co-IP of EGFP-LC3 with FLAG-SERCA2. (F) GST pull-down assays with GST-fused LC3B to overexpressed FLAG-SERCA2 or FLAG-SERCA2 mutLIR. (G) Co-IP of EGFP-LC3 with FLAG-SERCA2 or FLAG-SERCA2 mutLIR in transfected HEK293T cells. (H) Co-localization of FLAG-SERCA2 or FLAG-SERCA2 mutLIR with LC3B in SUM1315 cells treated with or without EBSS for 4 h. Right panel, representative images. Scale bar, 5 μm. Left panel, Quantification of SERCA2 co-localization with LC3B. (I) Western blot analysis of indicated proteins in SUM1315 cells transiently transfected with the indicated plasmids, which were pretreated with or without 100 nmol/L Baf A1 for 2 h and then treated with EBSS for 12 h. Data are shown as the mean ± SD of at least three independent experiments. ∗P < 0.05, ∗∗∗P < 0.001.
3.3. The SERCA2–LC3B interaction facilitates WIPI2-independent autophagosome formation
Next, we investigated the step at which SERCA2 acts in the autophagy pathway. Phagophore nucleation is an initial process of autophagosome formation during which a subset of autophagy proteins, such as the ULK1/FIP200 complex, are located on the ER and trigger the formation of PI(3)P-enriched ER subdomains that are termed “omegasomes” and marked by the ER-localized PI3P-binding protein DFCP12,20,21. Co-IP assays showed that overexpressed SERCA2 could interact with all of the ULK1 complex members, ULK1, FIP200, ATG13 and ATG101 (Fig. 4A). Moreover, exogenously expressed SERCA2 was detectable in the form of a small number of foci upon autophagy initiation (Fig. 4B). Strong colocalization of DFCP1 with SERCA2 was observed, suggesting that SERCA2 may be involved in the formation of omegasomes. In mammalian cells, omegasomes act as platforms for phagophore expansion and are associated with the isolation membrane (IM), which is labeled by LC322,23. We found that a co-immunoprecipitating complex consisting of endogenous LC3B and ULK1 complex members was abundant in the presence of overexpressed SERCA2, and its level was profoundly reduced in the presence of LIR-mutant SERCA2 (Fig. 4C). Because WIPI2 is also associated with both omegasomes and IM and is required for the formation of autophagosomes24, we silenced WIPI2 using siRNA to determine the role of SERCA2. Knockdown of WIPI2 profoundly reduced the degradation rate of p62 in the starved cells, which, intriguingly, was reversed by overexpressed SERCA2 (Fig. 4D). The mCherry-EGFP-LC3 reporter also showed that simultaneous overexpression of SERCA2 restored LC3 degradation in WIPI2-silenced cells under starvation conditions (Supporting Information Fig. S6). In addition, LC3 puncta appeared to be closely associated with FIP200 in starved control cells (Fig. 4E). Silencing either SERCA2 or WIPI2 facilitated dissociation of FIP200 from the LC3 puncta. However, simultaneous overexpression of SERCA2 led to the resumed colocalization of LC3 puncta with FIP200 in WIPI2-silenced cells, suggesting that in addition to WIPI2, SERCA2 can function as an alternative bridge to tether omegasomes to the IM through interaction with the ULK/FIP200 complex and LC3B. As autophagosomes close, they detach from the ER. To exclude the possibility that SERCA2 interferes with the dissociation of IM-ER contacts after autophagosome maturation, we used thapsigargin (TG), which blocks autophagosome fusion with lysosomes. LC3 puncta in TG-treated cells are reported to closely associate with the ER and lead to the failure of autophagosomes to completely close25. After 4 h of EBSS starvation, DFCP1-LC3 colocalization was much weaker in the cells without TG treatment than in TG-treated cells regardless of the presence of overexpressed SERCA2 (Fig. 4F).
Figure 4.

SERCA2 tethers the ER with the isolation membrane through interaction with the ULK1/FIP200 complex and LC3B. (A) Co-IP of FLAG-SERCA2 with the ULK1/FIP200 complex members in transfected SUM1315 cells. (B) Co-localization of DFCP1 with SERCA2 in SUM1315 cells treated with or without EBSS for 1 h. Left panel, Representative images. Scale bar, 5 μm. Right panel, quantification of co-localization. n = 200 puncta. (C) Co-IP of LC3B with the ULK1/FIP200 complex members in SUM1315 cells transiently transfected with the indicated plasmids. (D) Western blot analysis of indicated proteins in SUM1315 cells transfected with the indicated siRNA or plasmids, which were then treated with EBSS for 12 h. (E) Co-localization of FIP200 with LC3 in SUM1315 cells transfected with the indicated siRNA or plasmids, which were then treated with EBSS for 1 h. Upper panel, representative images. Scale bars, 5 μm; insets, 2 μm. Lower panel, quantification of co-localization. n = 200 puncta. (F) Co-localization of DFCP1 with LC3 in SUM1315 cells transfected with the indicated plasmids, which were then treated with EBSS for 4 h in the absence or presence of 100 nmol/L thapsigargin (TG). Upper panel, representative images. Scale bars, 5 μm; insets, 2 μm. Lower panel, quantification of co-localization. n = 200 puncta. Data are shown as the mean ± SD of at least three independent experiments. ∗P < 0.05, ∗∗∗P < 0.001.
3.4. SERCA2 gene expression is inducible via autophagy
To dissect the fate of SERCA2 after autolysosome formation, we transfected SUM1315 cells with mCherry-EGFP-SERCA2 plasmids. More mCherry-positive and EGFP-negative puncta were observed after the cells were starved for 1 h compared with the cells cultured under basal conditions (Fig. 5A). Flow cytometry analysis confirmed that, similar to those of LC3, the fluorescence intensities of EGFP-SERCA2 were reduced, whereas those of mCherry-SERCA2 were increased upon EBSS starvation (Supporting Information Fig. S7). Starvation-induced EGFP-SERCA2 degradation was reversed by blocking lysosome function with Baf A1 but not by the proteasome inhibitor MG132. These data suggest that SERCA2 is rapidly degraded by lysosomes via autophagy.
Figure 5.

SERCA2 autophagic degradation leads to its transcriptional activation. (A) SUM1315 cells were transfected with the mCherry-EGFP-SERCA2 plasmids. The mCherry-positive but EGFP-negative (red) SERCA2 puncta were quantified under basal conditions or after 1 h of EBSS starvation (lower panel). n = 200 puncta. Scale bars, 5 μm. (B) Western blot analysis of SERCA2 and p62 in SUM1315 cells treated with EBSS for the indicated time. (C) Real-time quantitative PCR analysis of ATP2A2 in SUM1315 cells treated with EBSS for the indicated time. GAPDH was used as an internal control. (D, E) SUM1315 cells were pretreated with or without 25 μmol/L STO-609 for 2 h and then treated with EBSS for 12 h. Western blot analysis of (D) SERCA2 and (E) phosphorylated cAMP response element-binding protein-1 (CREB-1) and CREB-1. (F) Diagram of the location of CREB-1 binding site in the human ATP2A2 promoter region. (G) ChIP assays using an antibody against CREB-1, followed by real-time quantitative PCR with primers designed for the CREB-1 binding site in the ATP2A2 promoter region. (H, I) The (H) mRNA and (I) protein levels of SERCA2 expression in SUM1315 cells with siRNA-mediated knockdown of CREB-1, which were treated with EBSS for 12 h. (J, K) The (J) mRNA and (K) protein levels of SERCA2 expression in SUM1315 cells, which were treated with EBSS in the absence or presence of 0.5 μmol/L actinomycin D for 12 h. Data are shown as the mean ± SD of at least three independent experiments. ∗P < 0.05, ∗∗P < 0.005, ∗∗∗P < 0.001.
Despite these findings, a time-dependent increase in both the protein and mRNA levels of SERCA2 was observed in the three TNBC cell lines during EBSS starvation (Fig. 5B and C, Supporting Information Fig. S8A–S8B). As a control, the protein levels of p62 gradually decreased. SERCA2 is a ubiquitous Ca2+ pump26. It was hypothesized that autophagy-mediated SERCA2 degradation would upregulate cytosolic Ca2+ levels, subsequently leading to SERCA2 transactivation. Indeed, the specific Ca2+ chelator BAPTA completely inhibited the increase in SERCA2 expression after EBSS starvation (Fig. S8C). Neither the ER stress inhibitor 4-phenyl butyric acid (4-PBA) nor the AMPK inhibitor compound C had an effect on the upregulation of SERCA2, ruling out the possibility that Ca2+-associated ER stress or AMPK signaling pathways were the cause of SERCA2 upregulation (Fig. S8D and S8E)27,28. Instead, we discovered that STO-609, an inhibitor of Ca2+/calmodulin-dependent protein kinase (CaMKK), eliminated SERCA2 upregulation under starvation conditions (Fig. 5D and Fig. S8F). It had been previously reported that upon a transient increase in intracellular Ca2+, CaMKK activates CaMKIV and renders the enzyme capable of phosphorylating transcriptional nuclear proteins such as the cAMP response element-binding protein (CREB)29. Correspondingly, the level of CREB-1 phosphorylation was increased, reaching a peak after 4 h of EBSS starvation (Fig. S8G). STO-609 inhibited the phosphorylation of CREB-1 under starvation conditions (Fig. 5E). To investigate whether phosphorylated CREB-1 can transactivate SERCA2, we performed sequence analysis of the ATP2A2 gene. In the promoter regions of ATP2A2 gene, there is a CREB-1-binding site, as shown in Fig. 5F. ChIP assays with an anti-CREB-1 antibody showed that EBSS starvation significantly enhanced the recruitment of CREB-1 to the ATP2A2 promoter regions (Fig. 5G). Moreover, silencing CREB-1 suppressed the increase in SERCA2 expression at both the mRNA and protein levels after EBSS starvation (Fig. 5H and I). When we blocked transcription using actinomycin D, SERCA2 upregulation was also inhibited under EBSS starvation (Fig. 5J and K). These data suggest that due to SERCA2 autophagic degradation, a transient increase in intracellular Ca2+ provides a feedback “CaMKK–CaMKIV–CREB-1 axis”, possibly accounting for the maintenance of high levels of SERCA2 in advanced stages of TNBC.
3.5. Small-molecule RL71 is a novel enhancer of the SERCA2–LC3B interaction
To test whether increased SERCA2 expression, which correlates with TNBC progression and chemoresistance, is a druggable target for TNBC therapy, we sought to find potent small molecules to target SERCA2 at high levels. Our previous studies showed that RL71, a second-generation curcumin analog, targets SERCA2 and induces excessive autophagic cell death in TNBC cells14,15. Surprisingly, a co-IP assay showed that RL71 enhanced the interaction of endogenous and exogenous SERCA2 with LC3B (Fig. 6A and B), and reciprocal LC3 immunoprecipitation confirmed the increased binding of SERCA2 to LC3 in the presence of RL71 (Fig. 6C). RL71 showed no effect on SERCA2 expression at either mRNA or protein levels in MDA-MB-231 cells (Supporting Information Fig. S9). Further, we found that RL71 treatment augmented the degradation of p62 and LC3B lipidation in SUM1315 cells transfected with wild-type SERCA2 in a time-dependent manner, whereas RL71 failed to affect autophagic flux in the cells transfected with SERCA2 harboring mutant LIR (Fig. 6D), suggesting that the LIR motif is critical for RL71-induced autophagy. To explore the mode of action of RL71 to enhance the SERCA2–LC3 interaction, we treated SUM1315 cells with RL71 in the presence of 4-PBA or BAPTA; neither of these inhibitors had an effect on the interaction (Supporting Information Fig. S10), ruling out the possible involvement of Ca2+-associated signaling. RL71 directly binds to SERCA2 in the E2 state at a cleft on the luminal side of the ER, and the amino acids Lys 876 and His 278 are critical for its specific binding14. Next, we examined the effect of SERCA2 harboring His278 A and Lys876 A mutations on its interaction with LC3B. As shown in Fig. 6E, the mutations markedly impaired the binding of SERCA2 to LC3B, implying that the conformation change in SERCA2 upon RL71 binding might affect the SERCA2–LC3 interaction. Thus, we carried out molecular dynamic simulations and found that the binding of RL71 induced the conformational change of SERCA2 from the E1 to the E2 state, and simultaneously, three cytoplasmic domains (N, P and A domains) gathered to form a single headpiece (Fig. 6F and Supporting Information Fig. S11A)14,30. The LIR motif locates in the cytoplasmic P domain. The headpiece in the E2 state reduced the flexibility of SERCA2 and facilitated the stable binding of LC3 with the lowest estimated free energy for binding (−29.71 ± −8.33 kcal/mol, Fig. 6G and Fig. S11B). These findings suggest that the small-molecule RL71 can be used as a novel autophagy inducer by enhancing the SERCA2–LC3B interaction via directly binding to SERCA2.
Figure 6.

The small-molecule RL71 promotes SERCA2–LC3B interaction. (A) Co-IP of SERCA2 with endogenous LC3B in SUM1315 cells 6 h after the treatment of 2 μmol/L RL71. (B) Co-IP of FLAG-SERCA2 with endogenous LC3B and (C) Co-IP of EGFP–LC3 with endogenous SERCA2 in the transfected SUM1315 cells 6 h after the treatment of 2 μmol/L RL71. (D) Western blot analysis of p62 and LC3B in SUM1315 cells, which were transfected with the indicated plasmids and then treated with 2 μmol/L RL71 for 0–12 h. (E) Co-IP of FLAG-SERCA2 or its mutant with endogenous LC3B in SUM1315 cells, which were transfected with the indicated plasmids and then treated with 2 μmol/L RL71 for 6 h. (F) Ribbon representation of SERCA2 in the LC3-bound E1 form and that in E2 form bound with both LC3 and RL71. The cytoplasmic part of SERCA2 consists of N domain (yellow), P domain (blue) and A domain (purple)30. LIR (red) in P domain interacts with LC3 (green). RL71 (dark purple) binds to SERCA2 at the cleft on the luminal side of the ER14. TM, transmembrane. (G) Cartoon representation of SERCA2 in E1 or E2 form bound with LC3 (gray). Red indicates high flexibility and green indicates low flexibility. Data are shown as the mean ± SD of at least three independent experiments. ∗P < 0.05, ∗∗P < 0.005, ∗∗∗P < 0.001.
3.6. Increased SERCA2 predisposes TNBC cells to RL71-induced autophagic cell death
Next, we tested the cytotoxic effects of RL71 on SUM1315 cells expressing SERCA2 at different levels. Overexpression of SERCA2 rendered the cells more sensitive to RL71, whereas silencing SERCA2 impaired this responsiveness (Fig. 7A and B). This finding was in profound contrast with the response to the standard-of-care agents described above. When combined with Baf A1, RL71-induced cell death was remarkably reduced regardless of SERCA2 levels and LIR mutation in SERCA2 attenuated the sensitivity to RL71 (Fig. 7C), which suggested that RL71 induced autophagic cell death depending on the SERCA2–LC3 interaction. Moreover, Western blotting showed that upon RL71 treatment, LC3B lipidation and the degradation of p62 were more evident in the cells overexpressing SERCA2 than in the controls (Fig. 7D). In contrast, no change in autophagic flux was observed in the case of SERCA2 silence (Fig. 7E). In addition, a co-IP assay showed that RL71 enhanced the interaction of LC3 with several organelle-specific proteins, including the mitochondrial marker Cox 4, ER marker calnexin and nuclear lamina protein lamin B1 (Fig. 7F). Immunofluorescence analyses confirmed the elevated colocalization of LC3 with ER tracker and MitoTracker upon RL71 treatment (Fig. 7G and H). These data suggest that RL71 could promote the delivery of organelles or their fragments to autolysosomes depending on the SERCA2–LC3 interaction and facilitate their lysosomal degradation, eventually leading to excessive autophagic cell death.
Figure 7.

RL71 shows potent cytotoxicity on TNBC cells with high SERCA2 expression by inducing excessive autophagic cell death. (A, B) MTT assay to assess the sensitivity of SUM1315 cells transfected with (A) FLAG-pcDNA3.1 or FLAG-SERCA2 plasmids or transfected with (B) NC-siRNA or SERCA2 siRNA to RL71. (C) MTT assay to assess the sensitivity of SUM1315 cells transfected with the indicated plasmids to RL71 in the absence or presence of 100 nmol/L Baf A1. (D, E) Western blot analysis of LC3B and p62 in SUM1315 cells, which were transfected with (D) FLAG-pcDNA3.1 or FLAG-SERCA2 plasmids or transfected with (E) NC-siRNA or SERCA2 siRNA, and then treated with 2 μmol/L RL71 for 24 h. (F) Co-IP of EGFP–LC3 with the indicated proteins in the transfected SUM1315 cells after treatment with 2 μmol/L RL71 for 6 h. (G, H) Co-localization of LC3 with (G) ER tracker or (H) Mitotracker in the transfected SUM1315 cells after treatment with 2 μmol/L RL71 for 6 h. Left panel, representative images. Scale bars, 5 μm. Right panel, quantification of co-localization. n = 200 puncta. Data are shown as the mean ± SD of at least three independent experiments. ∗P < 0.05, ∗∗P < 0.005, ∗∗∗P < 0.001.
To determine the therapeutic potential of RL71 in highly malignant TNBC, which shows increased SERCA2 expression during cancer progression, we transduced SUM1315 cells with lentivirus to stably overexpress SERCA2 (Supporting Information Fig. S12A) and then injected these cells subcutaneously into the right flank of nude mice. Compared with the control group mice transplanted with wild-type tumor cells, the overexpression of SERCA2 rendered the xenograft tumors to be faster growing and increasingly malignant, as evidenced by the increase in tumor volume, weight and extensive pulmonary metastasis (Fig. 8A–C). Treatment with 10 mg/kg PTX inhibited tumor development to a level comparable to 2 mg/kg RL71 treatment in the mice transplanted with wild-type tumor cells. Of note, in the SERCA2-overexpressing group, the tumor became resistant to PTX treatment, whereas RL71 treatment robustly inhibited tumor progression. IHC of tumor tissues showed that treatment with RL71, but not PTX, greatly reduced Ki-67 expression in both the control and SERCA2-overexpressing groups (Fig. 8D). Furthermore, positive immunostaining for p62 showed that the RL71-treated tumors exhibited a substantial decrease in p62-positive cells compared with the olive oil-treated controls (Fig. 8E). Western blot analysis also showed a reduction in p62 and LC3B lipidation in the tumors growing in RL71-treated mice compared with those growing in the mice treated with olive oil or PTX (Fig. 8F). In addition, we assayed the therapeutic potential of RL71 in an orthotopic transplant model using MDA-MB-231 cells stably overexpressing SERCA2 (Fig. S12B). Similarly, a reduction in tumor volume and weight was observed only in the RL71-treated mice compared with the olive oil-treated control mice (Fig. 8G and H). p62 reduction and LC3B lipidation were confirmed in the tumors in RL71-treated mice (Fig. 8I). These data suggest that RL71 may be a promising inducer of excessive autophagic cell death against highly malignant and PTX-resistant TNBC with high SERCA2 expression.
Figure 8.

High expression of SERCA2 renders TNBC tumor preferentially sensitive to RL71 treatment. (A–F) Female nude mice were inoculated with SUM1315 cells stably expressing vectors or SERCA2 (SERCA2-OE) into the right flank, and administered with olive oil, RL71 (2 mg/kg/day) or PTX (10 mg/kg/7 days) 7 days later. The mice (n = 6/group) were sacrificed on Day 15 after administration. And tumors and lung tissues were collected, followed by photography and measurement. (A) The tumor growth curves. Left panel: representative images of the tumors (n = 6). (B) Tumor weight. (C) The proportion of mice with lung metastases in each group. Upper panel: representative images of lung section stained with H&E. Black arrowheads indicate metastatic foci in the lungs. Scale bar: 100 μm. (D) IHC for Ki-67 in tumor tissues. Lower panel: quantification of IHC for Ki-67. Scale bar: 100 μm. (E) IHC for p62 in tumor tissues. Lower panel: quantification of IHC for p62. Scale bar: 100 μm. (F) Western blot analysis of LC3B and p62 in tumor tissues. (G–I) Female nude mice were inoculated with MDA-MB-231 cells stably expressing SERCA2 into the mammary fat pad. After 14 days, the mice (n = 6/group) were treated as described above. (G) The tumor growth curves. (H) Tumor weight. (I) Western blot analysis of LC3B and p62 in tumor tissues. Animal experiments were performed twice independently. Data are shown as the mean ± SD of at least three independent experiments. #P < 0.05, ∗P < 0.05, ∗∗P < 0.005, ∗∗∗P < 0.001.
4. Discussion
Chemotherapy is the primary established treatment option for patients with both early-stage and advanced-stage TNBC31. Despite an initial good response to chemotherapy, patients with TNBC have a much poorer prognosis than patients with other breast cancer subtypes, largely due to the inherently aggressive clinical behavior of TNBC, unsustained response to conventional chemotherapies, and lack of targeted therapies. Therefore, the identification of novel druggable vulnerabilities in TNBC is an important aim. Herein, we found that SERCA2 expression correlated with poor prognosis in TNBC patients. Overexpressed SERCA2 promoted the proliferation, invasion and chemoresistance of TNBC cells by inducing autophagy. More importantly, we found that the small-molecule RL71 could be used to take advantage of SERCA2-mediated autophagy and facilitate cell switching from autophagic survival to autophagic death. TNBC cells with high SERCA2 expression showed preferential sensitivity to RL71-induced excessive autophagic cell death in vitro and in vivo. Our data reveal increased SERCA2 expression as a potential druggable vulnerability for TNBC therapy.
As ER calcium pumps, SERCAs have been accepted as a therapeutic target for cancer treatment, especially drug-resistant cancers32,33. In this study, we delineated a role for SERCA2 as a key regulator of autophagy. SERCA2 can increase starvation-induced autophagic flux in TNBC cells through direct interaction with LC3B via its LIR motif. Mutations in the LIR motif led to the loss of SERCA2-mediated autophagy. Autophagy has been previously implicated in cancer resistance mechanisms because of its cytoprotective properties34, 35, 36. Indeed, in our study, overexpression of SERCA2 rendered TNBC cells chemoresistant to standard-of-care agents, an effect that was reversed by SERCA2 silencing. In the case of pharmacological or genetic autophagy inhibition, the insensitivity of SERCA2-overexpressing TNBC cells to chemotherapy drug PTX was markedly impaired. Thus, SERCA2-mediated autophagy enables the survival of TNBC cells and may be critical for chemoresistance in TNBC patients.
Mechanistically, we demonstrated that the ER-localized transmembrane protein SERCA2 is involved in autophagosome formation, including phagophore nucleation and expansion. SERCA2 can tether the ER to the IM through its interaction with both the ULK1 complex and LC3B. Although previous studies reported that SERCA2 activity is required for the regulation of ER–IM contact21, our results for the first time demonstrated that the LIR motif of SERCA2 was also indispensable for ER–IM contact during autophagy, as evidenced by the fact that LIR mutation led to the blockade of the interaction between LC3B and the ULK1 complex. Despite SERCA2-mediated tethering, the dissociation of the IM from the ER was not disrupted after autophagosome maturation, since overexpressed SERCA2 failed to maintain its strengthening effect on the contact between DFCP1-labeled omegasomes and LC3-labeled IMs after 4 h of EBSS starvation. Additionally, regardless of the function of the PI(3)P effector WIPI2 to tether ER-IM contacts21,24, simultaneous overexpression of SERCA2 restored the autophagic progress in WIPI2-scilenced cells. These findings suggest that, in addition to WIPI2, SERCA2 can tether the ER to the IM, highlighting the action of SERCA2 as a scaffold-like protein during autophagy.
Although pharmacological inhibition of autophagy has been proposed as a strategy to enhance the efficacy of cancer therapies, multiple clinical trials with cancer patients have been largely disappointing2,37,38. Especially in TNBC, some studies demonstrate that the restriction of autophagy promotes tumor survival39,40. Cotzomi-Ortega et al.39 reported that autophagy inhibition in TNBC cells induced the secretion of macrophage migration inhibitory factor to promote tumor malignancy via immunomodulation. In contrast, recent studies have highlighted a beneficial role for autophagy activation in cancer therapies14,41, 42, 43. High activation of autophagy can be a cause of cell death44,45. A growing body of evidence suggests that some cancer cells respond to cytotoxic drugs due to the induction of autophagic cell death46, 47, 48, 49. In the present study, we provided evidence that the SERCA2 gene is inducible in TNBC cells, possibly accounting for the persistence of high SERCA2 levels in this disease. SERCA2 autophagic degradation induces a transient increase in cytosolic Ca2+ levels, which stimulates its own transactivation through Ca2+/CaMKK/CREB-1 feedback. Because of its sustained high expression and close association with autophagy, we reasoned that SERCA2 may be utilized in TNBC therapy. Intriguingly, we found that the small-molecule RL71 directly bound SERCA2 to enhance the SERCA2–LC3B interaction by changing the conformation of SERCA2 from the E1 to E2 state, culminating in the induction of excessive autophagic cell death. Although autophagy is required for the survival of TNBC cells50, it is feasible that a further enhancement of autophagy activation through targeting the increased SERCA2 can predispose TNBC cells to death.
Autophagic cell death could occur in conditions of prolonged stress and consequently high activation of autophagy, which could cause an excessive protein and organelle turnover that overwhelm the capacity of the cell51. In fact, RL71 treatment increased the marked degradation of p62 in SERCA2-overexpressing TNBC cells than in the controls. Co-IP and immunofluorescence analyses also showed that RL71 promoted the delivery of organelles or their fragments to autolysosomes and facilitated their lysosomal degradation. To date, the molecular machinery that distinguishes autophagy-dependent cell death from autophagic cell survival remains to be established44,52. Our findings suggest that modifying the SERCA2–LC3B interaction might contribute to the regulation of autophagy-dependent cell death.
5. Conclusions
This study elucidates a mechanism by which TNBC cells maintain their high autophagic activity to induce chemoresistance and suggests increased SERCA2 expression as a druggable vulnerability in TNBC.
Acknowledgments
We gratefully acknowledge Dr Yi Fan and Dr Yanqing Gong from University of Pennsylvania, Perelman School of Medicine for critical reading of the manuscript. This study was funded by National Natural Science Foundation of China (Nos. 21937005 and 81974504), Natural Science Foundation of Jiangsu Province (No. BK 20191251, China), the Fundamental Research Funds for the Central Universities (China) and National Key R&D Program of China (No. 2017YFA0506000).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2022.05.009.
Contributor Information
Yan Shen, Email: shenyan@nju.edu.cn.
Qiang Xu, Email: molpharm@163.com.
Author contributions
Qiang Xu and Yan Shen conceived and led the project. Yan Shen, Qiang Xu and Minmin Fan wrote the paper. Yan Shen designed and coordinated the study. Jian Gao, Lin Zhou performed the human sample studies. Minmin Fan, Jian Gao, Wenwen Xue, Yixuan Wang and Jingwei Chen performed mechanism-related studies. Minmin Fan, Wuhao Li, Ying Yu and Bo Liu contributed to the design and performed the pharmacology studies.
Conflicts of interest
The authors declare no conflicts of interest.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Zhou Y., Rucker E.B., 3rd, Zhou B.P. Autophagy regulation in the development and treatment of breast cancer. Acta Biochim Biophys Sin. 2016;48:60–74. doi: 10.1093/abbs/gmv119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dikic I., Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–364. doi: 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 3.Guo J.Y., Chen H.Y., Mathew R., Fan J., Strohecker A.M., Karsli-Uzunbas G., et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuwen D., Ma Y., Wang D., Gao J., Li X., Xue W., et al. Prognostic role of circulating exosomal miR-425-3p for the response of NSCLC to platinum-based chemotherapy. Cancer Epidemiol Biomarkers Prev. 2019;28:163–173. doi: 10.1158/1055-9965.EPI-18-0569. [DOI] [PubMed] [Google Scholar]
- 5.Perera R.M., Stoykova S., Nicolay B.N., Ross K.N., Fitamant J., Boukhali M., et al. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–365. doi: 10.1038/nature14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou X., Yue G.G., Tsui S.K., Pu J., Fung K.P., Lau C.B. Elaborating the role of natural products on the regulation of autophagy and their potentials in breast cancer therapy. Curr Cancer Drug Targets. 2018;18:239–255. doi: 10.2174/1568009617666170330124819. [DOI] [PubMed] [Google Scholar]
- 7.Choi J., Jung W., Koo J.S. Expression of autophagy-related markers beclin-1, light chain 3A, light chain 3B and p62 according to the molecular subtype of breast cancer. Histopathology. 2013;62:275–286. doi: 10.1111/his.12002. [DOI] [PubMed] [Google Scholar]
- 8.Zhao H., Yang M., Zhao J., Wang J., Zhang Y., Zhang Q. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med Oncol. 2013;30:475. doi: 10.1007/s12032-013-0475-1. [DOI] [PubMed] [Google Scholar]
- 9.Saatci O., Kaymak A., Raza U., Ersan P.G., Akbulut O., Banister C.E., et al. Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nat Commun. 2020;11:2416. doi: 10.1038/s41467-020-16199-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arbabian A., Brouland J.P., Gelebart P., Kovacs T., Bobe R., Enouf J., et al. Endoplasmic reticulum calcium pumps and cancer. Biofactors. 2011;37:139–149. doi: 10.1002/biof.142. [DOI] [PubMed] [Google Scholar]
- 11.Chung F.Y., Lin S.R., Lu C.Y., Yeh C.S., Chen F.M., Hsieh J.S., et al. Sarco/endoplasmic reticulum calcium-ATPase 2 expression as a tumor marker in colorectal cancer. Am J Surg Pathol. 2006;30:969–974. doi: 10.1097/00000478-200608000-00006. [DOI] [PubMed] [Google Scholar]
- 12.Li W., Ouyang Z., Zhang Q., Wang L., Shen Y., Wu X., et al. SBF-1 exerts strong anticervical cancer effect through inducing endoplasmic reticulum stress-associated cell death via targeting sarco/endoplasmic reticulum Ca2+-ATPase 2. Cell Death Dis. 2014;5 doi: 10.1038/cddis.2014.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L., Wang L., Song R., Shen Y., Sun Y., Gu Y., et al. Targeting sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 2 by curcumin induces ER stress-associated apoptosis for treating human liposarcoma. Mol Cancer Therapeut. 2011;10:461–471. doi: 10.1158/1535-7163.MCT-10-0812. [DOI] [PubMed] [Google Scholar]
- 14.Yang B., Zhang M., Gao J., Li J., Fan L., Xiang G., et al. Small molecule RL71 targets SERCA2 at a novel site in the treatment of human colorectal cancer. Oncotarget. 2015;6:37613–37625. doi: 10.18632/oncotarget.6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao J., Fan M., Peng S., Zhang M., Xiang G., Li X., et al. Small-molecule RL71-triggered excessive autophagic cell death as a potential therapeutic strategy in triple-negative breast cancer. Cell Death Dis. 2017;8:e3049. doi: 10.1038/cddis.2017.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan M., Chen J., Gao J., Xue W., Wang Y., Li W., et al. Triggering a switch from basal- to luminal-like breast cancer subtype by the small-molecule diptoindonesin G via induction of GABARAPL1. Cell Death Dis. 2020;11:635. doi: 10.1038/s41419-020-02878-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Capaci V., Bascetta L., Fantuz M., Beznoussenko G.V., Sommaggio R., Cancila V., et al. Mutant p53 induces Golgi tubulo-vesiculation driving a prometastatic secretome. Nat Commun. 2020;11:3945. doi: 10.1038/s41467-020-17596-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi Y., Jin J., Ji W., Guan X. Therapeutic landscape in mutational triple negative breast cancer. Mol Cancer. 2018;17:99. doi: 10.1186/s12943-018-0850-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khaminets A., Heinrich T., Mari M., Grumati P., Huebner A.K., Akutsu M., et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522:354–358. doi: 10.1038/nature14498. [DOI] [PubMed] [Google Scholar]
- 20.Itakura E., Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–776. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Y.G., Chen Y., Miao G., Zhao H., Qu W., Li D., et al. The ER-localized transmembrane protein EPG-3/VMP1 regulates SERCA activity to control ER-isolation membrane contacts for autophagosome formation. Mol Cell. 2017;67:974–989. doi: 10.1016/j.molcel.2017.08.005. e6. [DOI] [PubMed] [Google Scholar]
- 22.Graef M., Friedman J.R., Graham C., Babu M., Nunnari J. ER exit sites are physical and functional core autophagosome biogenesis components. Mol Biol Cell. 2013;24:2918–2931. doi: 10.1091/mbc.E13-07-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Axe E.L., Walker S.A., Manifava M., Chandra P., Roderick H.L., Habermann A., et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polson H.E., de Lartigue J., Rigden D.J., Reedijk M., Urbe S., Clague M.J., et al. Mammalian Atg 18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6:506–522. doi: 10.4161/auto.6.4.11863. [DOI] [PubMed] [Google Scholar]
- 25.Ganley I.G., Wong P.M., Gammoh N., Jiang X. Distinct autophagosomal–lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell. 2011;42:731–743. doi: 10.1016/j.molcel.2011.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monteith G.R., McAndrew D., Faddy H.M., Roberts-Thomson S.J. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer. 2007;7:519–530. doi: 10.1038/nrc2171. [DOI] [PubMed] [Google Scholar]
- 27.Hoyer-Hansen M., Bastholm L., Szyniarowski P., Campanella M., Szabadkai G., Farkas T., et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 28.Orrenius S., Zhivotovsky B., Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 29.Lemrow S.M., Anderson K.A., Joseph J.D., Ribar T.J., Noeldner P.K., Means A.R. Catalytic activity is required for calcium/calmodulin-dependent protein kinase IV to enter the nucleus. J Biol Chem. 2004;279:11664–11671. doi: 10.1074/jbc.M312613200. [DOI] [PubMed] [Google Scholar]
- 30.Toyoshima C., Nomura H. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature. 2002;418:605–611. doi: 10.1038/nature00944. [DOI] [PubMed] [Google Scholar]
- 31.Bianchini G., Balko J.M., Mayer I.A., Sanders M.E., Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674–690. doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu S.W., Law B.Y.K., Qu S.L.Q., Hamdoun S., Chen J., Zhang W., et al. SERCA and P-glycoprotein inhibition and ATP depletion are necessary for celastrol-induced autophagic cell death and collateral sensitivity in multidrug-resistant tumor cells. Pharmacol Res. 2020;153:104660. doi: 10.1016/j.phrs.2020.104660. [DOI] [PubMed] [Google Scholar]
- 33.Roti G., Carlton A., Ross K.N., Markstein M., Pajcini K., Su A.H., et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell. 2013;23:390–405. doi: 10.1016/j.ccr.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu Y.L., Jahangiri A., Delay M., Aghi M.K. Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Cancer Res. 2012;72:4294–4299. doi: 10.1158/0008-5472.CAN-12-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janser F.A., Adams O., Butler V., Schlafli A.M., Dislich B., Seiler C.A., et al. Her2-targeted therapy induces autophagy in esophageal adenocarcinoma cells. Int J Mol Sci. 2018;19:3069. doi: 10.3390/ijms19103069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang L., Fu J., Wang S., Cen H., Zhang L., Mandukhail S.R., et al. MiR-142-3p enhances chemosensitivity of breast cancer cells and inhibits autophagy by targeting HMGB1. Acta Pharm Sin B. 2020;10:1036–1046. doi: 10.1016/j.apsb.2019.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Towers C.G., Thorburn A. Therapeutic targeting of autophagy. EBioMedicine. 2016;14:15–23. doi: 10.1016/j.ebiom.2016.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukhopadhyay S., Sinha N., Das D.N., Panda P.K., Naik P.P., Bhutia S.K. Clinical relevance of autophagic therapy in cancer: investigating the current trends, challenges, and future prospects. Crit Rev Clin Lab Sci. 2016;53:228–252. doi: 10.3109/10408363.2015.1135103. [DOI] [PubMed] [Google Scholar]
- 39.Cotzomi-Ortega I., Rosas-Cruz A., Ramirez-Ramirez D., Reyes-Leyva J., Rodriguez-Sosa M., Aguilar-Alonso P., et al. Autophagy inhibition induces the secretion of macrophage migration inhibitory factor (MIF) with autocrine and paracrine effects on the promotion of malignancy in breast cancer. Biology. 2020;9:20. doi: 10.3390/biology9010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maxfield K.E., Macion J., Vankayalapati H., Whitehurst A.W. SIK2 restricts autophagic flux to support triple-negative breast cancer survival. Mol Cell Biol. 2016;36:3048–3057. doi: 10.1128/MCB.00380-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galluzzi L., Bravo-San Pedro J.M., Demaria S., Formenti S.C., Kroemer G. Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol. 2017;14:247–258. doi: 10.1038/nrclinonc.2016.183. [DOI] [PubMed] [Google Scholar]
- 42.Xiang H., Zhang J., Lin C., Zhang L., Liu B., Ouyang L. Targeting autophagy-related protein kinases for potential therapeutic purpose. Acta Pharm Sin B. 2020;10:569–581. doi: 10.1016/j.apsb.2019.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng X., Li W., Xu H., Liu J., Ren L., Yang Y., et al. Sinomenine ester derivative inhibits glioblastoma by inducing mitochondria-dependent apoptosis and autophagy by PI3K/AKT/mTOR and AMPK/mTOR pathway. Acta Pharm Sin B. 2021;11:3465–3480. doi: 10.1016/j.apsb.2021.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Denton D., Kumar S. Autophagy-dependent cell death. Cell Death Differ. 2019;26:605–616. doi: 10.1038/s41418-018-0252-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Denton D., Nicolson S., Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19:87–95. doi: 10.1038/cdd.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong V.K., Li T., Law B.Y., Ma E.D., Yip N.C., Michelangeli F., et al. Saikosaponin-d, a novel SERCA inhibitor, induces autophagic cell death in apoptosis-defective cells. Cell Death Dis. 2013;4 doi: 10.1038/cddis.2013.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ouyang L., Zhang L., Fu L., Liu B. A small-molecule activator induces ULK1-modulating autophagy-associated cell death in triple negative breast cancer. Autophagy. 2017;13:777–778. doi: 10.1080/15548627.2017.1283470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laane E., Tamm K.P., Buentke E., Ito K., Kharaziha P., Oscarsson J., et al. Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death Differ. 2009;16:1018–1029. doi: 10.1038/cdd.2009.46. [DOI] [PubMed] [Google Scholar]
- 49.Zhang S., Zhang J., An Y., Zeng X., Qin Z., Zhao Y., et al. Multi-omics approaches identify SF3B3 and SIRT3 as candidate autophagic regulators and druggable targets in invasive breast carcinoma. Acta Pharm Sin B. 2021;11:1227–1245. doi: 10.1016/j.apsb.2020.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maycotte P., Gearheart C.M., Barnard R., Aryal S., Mulcahy Levy J.M., Fosmire S.P., et al. STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res. 2014;74:2579–2590. doi: 10.1158/0008-5472.CAN-13-3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marino G., Niso-Santano M., Baehrecke E.H., Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dasari S.K., Bialik S., Levin-Zaidman S., Levin-Salomon V., Merrill A.H., Jr., Futerman A.H., et al. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ. 2017;24:1288–1302. doi: 10.1038/cdd.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.