

Abstract
Upregulation of NADPH oxidases (NOXs) in cancer cells leads to chronic increase in intracellular reactive oxygen species (ROS) and adaptation to a high ROS level for cell survival and, thereby, low sensitivity to radiotherapy. To overcome resistance to radiotherapy, we have developed a bioactive and CD44 targeted hyaluronic acid nanoparticle encapsulated with an NOX inhibitor, GKT831 (HANP/GKT831). We found that HANP/GKT831 had stronger inhibitory effects on ROS generation and cell proliferation than that of GKT831 alone in cancer cells. Systemic delivery of HANP/GKT831 led to the targeted accumulation in breast cancer patient derived xenograft (PDX) tumors in nude mice. Importantly, the combination of systemic delivery of HANP/GKT831 with a low dose of local radiotherapy significantly enhanced tumor growth inhibition in breast cancer PDX models. Our results showed that HANP/GKT831 primed tumor cells to radiation-induced DNA damage and cell death by downregulation of DNA repair function and oncogenic signal pathways.
Keywords: Redox imbalance, reactive oxygen species, hyaluronic acid nanoparticle (HANP), radiotherapy resistance, NADPH oxidases, GKT831, targeted cancer therapy
Graphical Abstract
About 50% of cancer patients with solid tumors received radiotherapy alone or in combination.1 Radiotherapy induces DNA damage in cancer cells directly by high-energy X-ray-induced DNA double-strand breaks and indirectly through ionizing water molecules to produce reactive oxygen species (ROS).2,3 Extensive DNA damage activates the DNA damage response, cell cycle checkpoint, and apoptotic cell death.3,4 During tumorigenesis, genetic mutations, alternations in signal pathways, and changes in the tumor microenvironment confer resistance to radiation therapy in human cancers.1,4 Clinical significance of effective cancer therapeutic approaches for overcoming resistance to radiotherapy has attracted extensive investigations into tumor responses to radiotherapy, resistant mechanisms, and radiosensitizing agents.4-7 Increasing evidence showed that tumor hypoxia, increased DNA repair, and upregulation of a survival pathway contribute to a poor response to radiotherapy.4-6,8-11
Inorganic nanoparticles synthesized from high atomic number materials produce secondary electrons after absorbing X-ray energy and directly cause DNA damage.12-15 Nanoparticle-induced production of ROS and oxidative stress in tumor cells further enhance the response to radiation. The radiosensitizing effect of gold nanoparticles has been demonstrated in mouse tumor models.12,13 Nanodroplets carrying oxygen and gold nanoparticles enhanced ROS production and DNA damage and improved the efficacy of radiotherapy in a mouse mammary tumor model.16 Currently, hafnium oxide (NBTXR3) and gadolinium (AGuIX) nanoparticles have been approved for clinical trials in cancer patients to enhance the effect of radiation therapy.14,15 Several chemotherapeutic drugs sensitize tumor cells to radiotherapy by induction of DNA damage, inhibition of DNA repair, and ROS production.6,17 PEGylated (PEG = poly(ethylene glycol)) liposomes carrying doxorubicin and cisplatin enhanced the therapeutic effect in head and neck cancer xenografts.18 A combination of nanoparticle–camptothecin conjugates with radiation produced a strong chemoradiotherapy effect by inhibiting DNA repair.19 Nanometal–Organic frameworks incorporated with a mitochondria targeting agent, ruthenium, that generated high levels of hydroxyl radicals and singlet oxygen upon radiation, enhanced the effect of radiodynamic therapy in mouse colon cancer models.20
Redox status in normal cells is tightly controlled to keep a balance between the production and neutralization of ROS. A significant increase in intracellular ROS can activate cell death.21,22 Radiotherapy induces cell death by ROS and DNA damage-induced stress response.21,22 Current approaches for enhancing the effect of radiotherapy mostly rely on increasing ROS, enhancing DNA damage, and reducing DNA repair.4-6,9-16,18-20,23,24 However, tumor cells have increased cell stress response and ROS production.4-6,9-11 Chronic oxidative stress and upregulation of intracellular ROS induce a redox imbalance in tumor cells that alters signal pathways that regulate cell proliferation, survival, and metabolism, enabling tumor cells to adapt to a high level of oxidative stress for cell survival that is insensitive to ROS-induced cell death.21,22,25,26
The presence of a hypoxic tumor microenvironment limits ROS production for induction of cell death.11 Thus, those tumor cells are resistant to therapeutic agents with their effects mediated by ROS induction. Furthermore, tumor cells developed resistant mechanisms in DNA repairing, ROS scavenging, tumor metabolism, and cell death/survival pathways.1,2,4,9 Therefore, the development of cancer therapeutics that act upon ROS-dependent survival signals, enhance DNA damage, and downregulate DNA repair in cancer cells hold great promise for overcoming radioresistance in human cancer.
NADPH oxidases (NOXs) and mitochondrial respiration are the major sources of intracellular ROS.27 NOXs catalyze the transfer of electrons from molecular oxygen across biological membranes.27,28 Among the NOX family members, NOX1 is responsible for an elevated ROS level in 80% of human breast cancers.29 Additionally, overexpression of NOX4 in normal breast epithelial cells potentiated tumorigenic transformation, and its overexpression in breast cancer cells further increased tumorigenicity.30,31 However, inhibition of NOX1 or NOX4 could reduce cancer cell proliferation and suppress tumor growth in vivo.32 The antitumor effect was mediated by inhibition of ROS and modulation of tumor metabolism.33 Moreover, NOX1 and NOX4 are critical mediators involved in the hypoxia-induced chemo- and radio-resistance via activation of hypoxia-inducible transcription factor alpha (HIF-α).34 Previous reports showed that upregulation of NOX1 and/or NOX4 enabled tumor cells to better cope with the redox stress by activation of MAPK, PI3K, NF-κB, and JAK/Stat pathways and by metabolic reprogramming, leading to low sensitivity to chemotherapeutics and radiation.28,32-34 Increasing evidence supports a paradoxical approach for overcoming radioresistance by inhibiting the ROS signal that plays an important role in tumor cell survival and stress responses.21,22,26,28,32,33 The potential for inhibition of NOX signal pathways in overcoming radioresistance is supported by study results from cancer cells and animal tumor models.14,16,25,28,32,33,35 Antitumor effects of small-molecule NOX inhibitors have been demonstrated in animal tumor models.32,35 Setanaxib or GKT831 is a dual NOX1 and NOX4 inhibitor35 with a minimal effect on NOX 2, 3, or 5.32,36 Upregulation of NOX4 is also the major driver of inflammation-induced fibrosis and tumor-associated fibroblasts.35-37 The effect of GKT831 on the inhibition of inflammatory fibrosis has been demonstrated in mouse models and human patients. Currently, GKT831 is the only Food and Drug Administration (FDA) approved NOX inhibitor in phase II/III clinical trials for pulmonary and kidney fibrosis and primary biliary cholangitis.36,37 It has been shown that upregulation of NOX4 in tumor-associated fibroblasts of human cancer tissues is associated with poor survival.35 Downregulation of NOX4 using siRNA or GKT831 inhibited tumor growth in a mouse lung cancer model.35
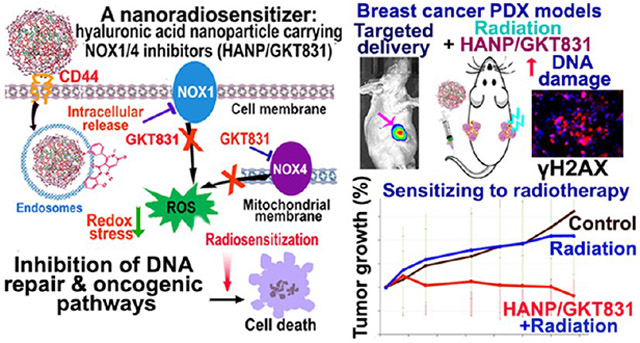
In this study, we developed a hyaluronic acid nanoparticle (HANP) encapsulated with GKT831 (HANP/GKT831) and evaluated its effect on tumor cells in vitro and breast cancer patient derived xenograft (PDX) models in vivo. Hyaluronic acid (HA) is a polysaccharide synthesized by cells as a high molecular weight form (HMW-HA, 1000–8000 kDa) and then degraded into low molecular weight fragments (LMW-HA, 20–250 kDa) by hyaluronidase II (Hyal 2) on the cell membrane. LMW-HA then interacts with CD44 receptor on the cell surface to be internalized into endosomes for degradation by hyaluronidase I (Hyal 1).38,39 Conjugation of a hydrophobic molecule, such as 5β-cholanic acid (5β-CA), to LMW-HAs induces self-assembling into a reel of thread-like nanoparticles (HANPs), with a hydrophilic surface and hydrophobic caves within for drug encapsulation.40,41 Results of our study showed that HANP-encapsulated GKT831 had a stronger inhibitory effect on ROS generation and cell growth in human cancer cell lines than that of GKT831 alone. In breast cancer PDX models, systemic administrations of HANP/GKT831 significantly enhanced the therapeutic effect of radiation on the PDX tumors.
RESULTS
Levels of NOX1 and NOX4 Were Upregulated in Human Breast Cancer Tissues.
To determine whether targeting the redox imbalance in tumor cells by inhibiting NOX pathway is a valid approach for the development of a cancer therapeutic agent, we examined the levels of NOX1 and NOX4 in representative human breast cancer and normal tissues. We found a low level of NOX1 expression in the normal breast ducts, but the expression level increased 1.9–2.1-fold in triple negative breast cancer (TNBC) and estrogen receptor positive (ER+) tumors (Figure 1A). Normal breast ducts had a very low level of NOX4. However, the level of NOX4 upregulated 3.6–5.2-fold in TNBC and ER+ primary and metastatic tumors compared to the normal breast ductal epithelial cells (Figure 1A).
Figure 1.

Levels of NOX1 and NOX4 expression are upregulated in human breast cancer tissues, and radiation further increased NOX4 expression. Normal breast tissues were obtained from the adjacent normal tissue areas from surgically resected breast. Breast I, II, and VI tumors were triple negative breast cancer (TNBC), Breast VII (P), and (LM) were ER+ breast cancer tissues obtained from surgically resected primary (P) and lymph node metastases (LM). Breast IX was ER+ breast cancer. Frozen tissue sections were used for immunofluorescence labeling of anti-NOX1 and NOX4 antibodies. (A) Detection of the levels of NOX1 and NOX4 in breast normal and cancer tissues. The mean fluorescence intensity was quantified from 4 to 10 images using the NIH ImageJ. (B) Effect of radiation on NOX1 and NOX4 expression. Frozen tissue sections of a residual tumor of Breast VII PDX model following 10 Gy radiation were examined. Western blot used cell lysates from MCF-7 cells following 4 h of 5 Gy irradiation. The mean relative level of NOX4 over β-actin from three blots is shown.
Radiation Increased the Level of NOX4 in Breast PDX Tumors and Cancer Cells.
To assess the effect of radiation on the levels of NOX1 and NOX4 expression, we analyzed residual tumors of the breast VII PDX model after radiotherapy. We found that radiotherapy-resistant tumors had a slightly increased level of NOX1. However, the level of NOX4 in the resistant tumors was 2.34-fold higher than that in the control tumor (Figure 1B). Results of a western blot analysis also showed a twofold increase in the level of NOX4 protein in cell lysates of MCF-7 human breast cancer cells 4 h after 5 Gy of radiation (Figure 1B). Thus, inhibition of the level and function of NOX1 and NOX4 offers an opportunity to modulate the elevated ROS and enhance the effect of radiotherapy on resistant cancer cells.
Characterization of Biophysical and Biological Activities of HANP/GKT831 In Vitro.
To develop an effective therapeutic agent inhibiting the NOX-ROS pathway, we produced a bioactive and biocompatible HANP encapsulated with GKT831, a dual NOX1/NOX4 inhibitor,32,36 using an established protocol42 (Figure 2A). The hydrophobic GKT831 molecules were loaded into HANPs with a drug loading efficiency of 90.07 ± 3.15%, and a high load capacity of 18% of the nanoparticle weight, determined by a high-performance liquid chromatography (HPLC) analysis. A dynamic light scattering analysis determined the hydrodynamic size of HANP at 187 ± 18 nm and HANP/GKT831 at 204 ± 15 nm in diameter (Figure 2B), which are consistent with transmission electron microscopy (TEM) images (Figure 2B inset and Figure S1). The stability of HANP/GKT831 in different buffers was evaluated by monitoring nanoparticle sizes for two weeks. As shown in Figure S2, we did not find apparent changes in the dispersion and diameters of HANP/GKT831 during the study. There were no obvious morphology changes after storage of HANP or HANP/GKT831 at 4 °C for one month (Figure S1).
Figure 2.

Production and characterization of HANP/GKT831. (A) 200 kDa HA (1) was conjugated with 5β-cholanic acid to form HA-CA (2). GKT831 was encapsulated into HA-CA by self-assembling, resulting in HANP/GKT831 (3). (B) Hydrodynamic size of HANP (180 nm) and HANP/GKT831 (204 nm). (inset) TEM image of a HANP/GKT831. (C) Quantification of the level of intracellular ROS by DCFDA assay. Breast cancer MCF-7 cells treated with various concentrations of HANP/GKT831 or GKT831. DCFDA assay was performed 24 h following treatment. Fluorescence intensity (FI, a.u.) of treated cells represents the level of ROS, mostly H2O2. The mean fluorescence intensity of four repeat samples is shown. (D) GKT831 release profile from HANP/GKT831 in the absence and presence of hyaluronidase (HAase). (E, F) SRB cell proliferation assay. IC50 of HANP/GKT831: Breast VII PDX-derived cell line (IC50 = 0.45 μM), MCF-7 cell line (IC50 = 2 μM). IC50 of GKT831: Breast VII PDX tumor cell line (6 μM) and MCF-7 cell line (>16 μM, not reached). Neither HANP/GKT831 nor GKT831 inhibited proliferation of normal HDF (G).
Next, the uptake of near-infrared (NIR) 830 dye-labeled HANP/GKT831 in tumor cells was determined in the Breast VII PDX-derived tumor and breast cancer MCF7 cell lines. Following 4 h of incubation of NIR 830-HANP-GKT831 with tumor cells, high levels of NIR fluorescence signals were observed in both cancer cells (Figure S3A). To determine whether the HANP carrier increases the efficacy of drug delivery, the amount of GKT831 in MCF7 cells was quantified using HPLC. HANP/GKT831-treated cells had 3.6 times higher level of GKT831 than that in cells treated with free GKT831 (Figure S3B,C). Then, we examined the effect of HANP/GKT831 on the level of ROS production in cancer cells using a diacetyldichlorofluorescein (DCFH-DA) ROS assay43 to measure intracellular H2O2, which was generated directly from NOX4 activity and indirectly from NOX1-induced O2·− that was converted to H2O2 by superoxide dismutase.27,28 Twenty-four hours following HANP/GKT831 treatment, the level of ROS was significantly reduced in MCF-7 cells (Figure 2C). In comparison with the no-treatment control, there was a 50% inhibition of ROS in cells treated with 1 μM of GKT831 equiv HANP/GKT831. Higher concentrations (8–16 μM) of HANP/GKT831 resulted in 73–75% ROS inhibition. However, without an HANP formulation, 1–16 μM of GKT831 led to 21–27% ROS inhibition (Figure 2C). These results indicated that the HANP formulation of GKT831 is critical for its effect on inhibition of ROS production in cancer cells.
Next, we determined the release profile of GKT831 from HANP/GKT831 in vitro (Figure 2D). The cumulative release of GKT831 from the HANP/GKT831 complex under an acidic condition (pH = 5.0) was calculated as 68.6 ± 10.9% (w/w) in the presence of native hyaluronidase, and 56.0 ± 7.4% (w/w) of GKT831 was found without hyaluronidase in the first 6 h. 82.4 ± 7.1% of GKT831 was released from HANP/GKT831 in the presence of hyaluronidase after 20 h of incubation, which is about 1.7-fold higher than that of GKT831 in the absence of hyaluronidase, suggesting this GKT831 release is enhanced by hyaluronidase. Because native hyaluronidase is active under acidic conditions in the endosomes/lysosomes in cells, CD44 targeted internalization into tumor cells ensures the release of GKT831 in tumor cells but not in the blood circulation and normal organs and, thereby, reduces systemic side effects in vivo.
HANP/GKT831 Inhibited the Growth of Cancer Cells but Not Normal Fibroblasts.
We investigated the effect of HANP/GKT831 on the proliferation/cytotoxicity of representative human cancer cell lines, including ER+ breast cancer MCF-7 and Breast VII PDX-derived tumor cell lines, and pancreatic cancer PANC II PDX-derived tumor cell line. A normal human dermal fibroblast cell line (HDF) was used as a control. A dose-dependent inhibition of tumor cell growth was observed in all three tumor cell lines following HANP/GKT831 treatment using GKT equivalent doses from 0.125 to 16 μM (Figure 2E-G). The half-maximal inhibitory concentration (IC50) of HANP/GKT831 was 0.45 μM in Breast VII PDX tumor cells and 2.0 μM in MCF-7 cells. The inhibitory effect of HANP/GKT831 was also detected in the PANC II PDX tumor cells with a low IC50 of 0.38 μM (Figure S4). However, conventional GKT831 had a weak activity on tumor cells. The IC50 of GKT831 was 9- and 13-fold higher than that of HANP/GKT831 in PANC II and Breast VII tumor cells, respectively. MCF-7 cells had a relatively lower sensitivity to HANP/GKT831. GKT831 failed to reach the IC50 value even at a high concentration of 16 μM. One of the differences among these three cell lines is that the MCF-7 cell line has a wild type p53,44 but the breast and pancreatic PDX tumors have a high percentage of tumor cells with mutant p53 proteins. Furthermore, neither GKT831 nor HANP/GKT831 showed a significant effect on the proliferation of normal HDF cells.
To determine whether HANP/GKT831 induced tumor cell death is mediated by apoptosis, the Breast VII PDX tumor cells were treated with HANP, free GKT831, and HANP/GKT831 for 72 h, and the apoptotic cells were quantified by using an Annexin V/7AAD apoptosis assay and flow cytometry. As shown in Figure S5, the HANP/GKT831 treatment induced 27.4% of apoptotic cells. Including the late-stage apoptosis/necrotic cells resulted in a total of 44.6% of tumor cells death. However, a free GKT treatment led to 9.1% of the apoptotic cell death. HANP carrier-treated cells had 12% of the apoptotic cells. Collectively, our results suggest that HANP/GKT831 has the potential for the development of an NOX-ROS targeted cancer therapeutic agent that inhibits proliferation and induces apoptosis in tumor cells.
HANP/GKT831 Efficiently Accumulated in Breast PDX Tumors Following Systemic Administration.
The efficiency of targeted delivery and the intratumoral and systemic distribution of HANP/GKT831 were examined in representative breast cancer PDX models. We found that both primary cancer and PDX tumor tissues expressed a high level of CD44 and had a large number of fibroblast active protein positive (FAP+) cells (Figure 3A). Most FAP+ stromal fibroblasts also expressed an intermediate level of CD44 (Figure 3A). Following an intravenous (i.v.) injection of NIR 830 dye-labeled HANP/GKT831 into nude mice bearing orthotopic Breast VII PDX tumors for 24 h, whole-body fluorescent imaging detected a strong signal in tumors (Figure 3B). Ex vivo imaging of collected tumors and normal organs showed NIR signal in tumors. Most normal organs had a very low signal, except the liver (Figures 3B and S6). Since HAs are cleared out from the blood through hyaluronan receptor-mediated endocytosis by the liver sinusoidal endothelial cells and Kupffer cells,38,39 detection of a high level of signal in the liver is likely due to uptake and clearance of HANPs in the liver.
Figure 3.

Targeted delivery of HANPs in orthotopic Breast VII PDX tumors following a systemic administration. (A) Dual immunofluorescence labeling. A high level of CD44 was detected in Breast VII patient cancer tissues and PDX tumors (green, yellow arrows). Tumor-associated fibroblasts identified by an anti-FAP antibody (red) also express CD44 (white arrows). (B) NIR optical imaging of targeted delivery. NIR 830 dye-labeled HANP (0.1 mg) was injected via the tail vein into nude mice bearing orthotopic PDX tumors for 24 h. A high level of NIR signal was detected in the tumor (pink arrow). Ex vivo imaging showed the accumulation of NIR 830-HANPs in the tumor (pink arrow) and liver (blue arrow). (C) NIR fluorescence microscopy of intratumoral distribution of NIR 830-HANPs (red) and immunofluorescence labeling of CD31+ tumor endothelial cells (green). A high level of HANPs was detected in the necrotic and avascular tumor center (white arrows). (D) NIR 830-HANPs were colocalized with CD44+ and CK18+ tumor cells. Many of NIR 830-HANPs were not detected in CD68+ macrophages (white arrows). (E) Distribution of HANP/GKT831 in the liver and their location in relationship to CD68+ Kupffer cells.
Examinations of intratumoral distribution of HANPs by immunofluorescence labeling and fluorescence microscopy revealed a high level of NIR 830-HANP/GKT831 in tumor tissues. An intermediate level of NIR 830-HANPs was detected in the stroma near tumor vessels (CD31+) (Figure 3C). Strong NIR signals were detected in the tumor cell nests and even in the avascular tumor area and necrotic center (Figure 3C). Importantly, colocalization of HANPs with CD44+ and CK18+ tumor cells was detected (Figure 3D). Although nonspecific uptake by intratumoral macrophages was commonly found in many types of nanoparticles, the majority of the NIR 830-HANP positive cells were not colocalized with CD68+ macrophages, suggesting that HANPs had a low level of nonspecific uptake by tumor-associated macrophages (Figure 3D). Furthermore, an intermediate level of NIR 830-HANP/GKT831 signal was detected in the liver (Figure 3E). However, most of the scattered HANP-containing cells were not colocalized with CD68+ and CD163+ Kupffer cells, which is consistent with the clearance of the majority of HAs by the liver sinusoidal endothelial cells (Figures 3E and S6B).
Systemic Delivery of HANP/GKT831 Significantly Enhanced Therapeutic Responses of Therapy-Resistant Breast Cancers to a Low-Dose Radiation Therapy.
Breast VII and IX PDX tumors were derived from ER+ tumor tissues that were resistant to multiple chemotherapy drugs. Breast PDX tumors were implanted in the right and left mammary fat pads of each mouse (Figure 4A). Twenty-four hours following i.v. delivery of HANP/GKT831, 2 Gy of radiation was applied to one side of the PDX tumors (Figure 4A). The above procedure was repeated weekly for five treatments for a total of 10 Gy of radiation. Although the treatment started when Breast VII PDX tumor sizes were large (~200 mm3), the combination of HANP/GKT831 with radiation significantly inhibited tumor growth, which led to 18% of tumor regression compared to the starting tumor volume. However, the tumor volume in the no-treatment control group increased 261% compared to its initial tumor size, which represented a 3.19-fold increase in the tumor volume compared to the HANP/GKT831+radiotherapy (RT) treated tumors (Figure 4B). There was 68.7% of tumor growth inhibition in the HANP/GKT831+RT group compared to the no-treatment control. In contrast, the PDX tumors showed a poor response to single therapies using GKT831, HANP/GKT831, radiation, or GKT831+RT. RT alone results in 19.9% of tumor growth inhibition, while GKT 831 or HANP/GKT831 inhibited tumor growth for 7.5% or 8.8% compared to the no-treatment control. The GKT831+RT treatment had 13.4% of tumor growth inhibition, which was not statistically significant (p = 0.55). Furthermore, HANP/GKT831+RT treated mice had 60.9% or 63.8% of tumor growth inhibition compared with the mice treated with radiation alone or GKT831+RT (Figure 4B upper).
Figure 4.

Determination of therapeutic efficacy of HANP/GKT831 without or with in combination with radiation therapy in orthotopic breast PDX tumor models. (A) Treatment protocol. Nude mice bearing two orthotopic breast PDX tumors on the right and left mammary fat pads received 5 mg/kg of GKT831 equiv dose of HANP/GKT831 via the tail vein injections once per week for five weeks. 2 Gy of radiation was given to the right-side tumor 24 h after each HANP/GKT831 injection, while the left-side tumors were shielded. (B) (left) Relative tumor growth curves following different treatments in Breast VII and Breast IX PDX tumor models in nude mice. (right) PDX-tumor images of treatment groups. Student’s t-test: *p < 0.05, **p < 0.01, ***p < 0.005. Breast VII PDX: No-treatment control vs HANP/GKT831+RT: p = 0.002; HANP/GKT831+RT vs RT: p = 0.004 or vs GKT831+RT: p = 0.047. Breast IX PDX: No treatment control vs RT: p = 0.39; HANP/GKT831+RT vs no treatment: p = 0.0085 or vs RT: p = 0.015. No-treatment control vs HANP/GKT831: p = 0.07, or vs GKT831+RT: p = 0.03. n = 5 mice/group. (C) Determination of systemic toxicity by monitoring body weights.
Breast IX PDX tumors had a modest response to 10 Gy of radiation that led to a 31% reduction in tumor growth compared to the no-treatment control. However, HANP/GKT831+ RT inhibited tumor growth for 73.9% or 81.9% compared to radiation alone or the no-treatment control (Figure 4B lower). GKT831 alone only slowed the tumor growth inhibition by 3.7%. HANP/GKT831 alone showed 40% of tumor growth inhibition, but the antitumor effect is not statistically significant compared with the control. In this PDX tumor, the GKT831+RT treatment also resulted in a significant tumor growth inhibition of 44.2% (Figure 4B, lower). The result of our study showed that HANP/GKT831 or HANP/GKT831+RT treatment at the current therapeutic dose did not have apparent systemic toxicity, since there was no body weight change in mice of all experimental groups during the treatment (Figure 4C).
Evaluation of Therapeutic Responses in Tumor Tissues Using Histological and Immunofluorescence Analyses.
Breast PDX tumors collected 5 d after the last treatment were analyzed for Ki67+ proliferating cells. Breast VII PDX tumor was an aggressive tumor with 83.2 ± 10.1% of proliferating cells. Following five weekly combination therapies of HANP/GKT831+RT, Ki67+ proliferating cells significantly decreased to 20.0 ± 3.4% (Figure 5A). Although the tumor volumes in mice treated with GTK831, RT, or GKT+RT only reduce slightly, the treatments had inhibited cell proliferation with 51.9 ± 6.5%, 53.7 ± 9.0%, and 40.5 ± 6.7% of Ki67+ proliferating cells, respectively (Figure 5A). HANP/GKT831 alone reduced Ki67+ cells to 34.0 ± 4.4% (Figure 5A). Furthermore, using an antiactive caspase-3 antibody, we found the highest level of the apoptotic cells in HANP/GKT831+RT-treated tumors (52.3 ± 13.8%), while significantly lower levels of apoptotic cells were detected in the control groups, including the nontreated control (3.8 ± 3.3%), GKT831 alone (3.3 ± 2.2%), GKT831+RT (15.9 ± 4.0%), HANP/GKT831 (4.9 ± 3.6%), and radiation alone (8.2 ± 3.8%). The tumor suppressor gene, p53, plays an important role in DNA repair, cell cycle, and apoptosis. 2,3 Since wild type p53 protein has a short half-life, detection of the p53 protein in tumor tissue sections has been considered as an indication of the accumulation of mutant p53 proteins.45 Immunofluorescence labeling showed 60% of p53+ cells in Breast VII PDX tumor tissues without treatment (Figure 5C). Radiation, GKT831, or GKT831+RT treatments slightly increased p53+ cells. However, HANP/GKT831 or HANP/GKT831+RT treatment significantly reduced the percentage of p53+ cells in tumors to 17 and 21%, respectively, suggesting that tumor cells with a mutant p53 were more sensitive to HANP/GKT831 (Figure 5C). Breast cancer cells expressing a high level of CD44 and a low level of CD24 (CD44+++/CD24−/+) have been defined as cancer stem-like cells.46 The biological significance of cancer stem-like cells in tumor progression and therapy resistance has been demonstrated in many types of human cancers.47 We found that Breast VII PDX tumors had a high level of CD44+++/CD24−/+ cancer stem-like cells. HANP/GKT831 treatment alone slightly reduced CD44+++/CD24∓ cells and switched to mostly CD44+++/CD24+2 tumor cells. Radiation or GTK831+RT treatment also reduced the percentage of CD44+++ cancer cells, but residual tumor cells retained a CD44+++/CD24−/+ stem-like phenotype (Figure 5D). However, HANP/GKT831+RT markedly reduced the level of CD44+++ tumor cells, and residual tumor cells expressed a high level of CD24, a noncancer stem cell population (Figure 5D).
Figure 5.

Histological analysis of the effect of HANP/GKT831 and HANP/GKT831+RT on Breast PDX tumors. Frozen tissue sections of Breast VII PDX tumors collected 5 d after the last treatment were examined by immunofluorescence labeling. (A) Ki67+ proliferating cells. The bar figure showed the percentage of Ki67+ cells of Hoechst 33342 nuclear staining cells. (B) Induction of apoptotic cell death, shown as an increased level of active caspase-3 (green). (C) Inhibition of the percentage of mutant p53 tumor cells in tumors. (D) Effect of treatments on the levels of CD44 and CD24 expression in tumors. Since the levels of CD44 or CD24 expression in tumor cells, rather than the number of CD44 or CD24 positive cells, reflect the stem-like property of tumor cells, the mean fluorescence intensity of CD44 or CD24 of 6–8 images is shown. Upper images: dual CD44 (green) and CD24 (red) labeling. Lower images: CD24 labeling only. NIH ImageJ was used to quantify all images obtained under a 20× lens. The mean percentage of positive cells per image field from 4 to 6 images is shown for each group in (A–C). Student’s t-test: experimental groups vs No-treatment control, *p < 0.05; ** p < 0.01, ***p < 0.005.
HANP/GKT831 Induced DNA Damage and Increased the Level of γH2AX in Tumors.
Detection of γH2AX in cells has been used as a specific and sensitive molecular marker for double-strand DNA damage.48 Radiation alone in the Breast VII PDX tumors slightly increased the level of γH2AX by 1.6-fold in tumor cells. GKT831, GKT831+RT, or HANP/GKT831 treatment further increased the levels of γH2AX in tumors to 2.0-, 2.4-, or 2.6-fold, respectively (Figure 6A). Furthermore, the highest level of γH2AX was detected in tumors treated with HANP/GKT831+RT, and there was a 4.3-fold increase compared to the no-treatment control (Figure 6A). The effect of HANP/GKT831+RT treatment on increasing the level of γH2AX was further confirmed by a western blot analysis of cell lysates obtained from treated MCF-7 cancer cells (Figure 6B). Radiation or HANP/GKT831 increased the protein level of γH2AX by 1.5-fold. HANP/GKT831+RT showed 2.4- to 3-fold increases in the level of γH2AX in tumor cells (Figure 6B).
Figure 6.

HANP/GKT831 treatment downregulated the expression levels of DNA repair genes and enhanced radiation-induced DNA damage. (A) Levels of γH2AX in Breast VII PDX tumors detected by immunofluorescence labeling. The mean fluorescence intensity of 5–10 images from each treatment is shown. (B) Western blot analysis of the levels of γH2AX protein in breast cancer MCF-7 cells treated with RT alone, 0.5 or 1 μM of HANP/GKT831, without or with 5 Gy RT. The mean relative level of γH2AX protein is shown as the ratio of intensity of γH2AX vs β-actin from four repeat blots. (C) (left) Levels of expression of DNA repair genes in MCF-7 cells after HANP/GKT831 treatment for 24 h and RT for 4 h. (right) RNA-seq analysis of Breast VII PDX tumors obtained 5 d after the last treatment. The levels of expression of DNA repair genes are shown as Log2 decrease or increase compared to the no-treatment control. The mean value obtained from two PDX tumors in each group is shown.
To elucidate the mechanism by which HANP/GKT831 enhanced the response to radiation in tumor cells, gene expression profiles of MCF-7 cells were analyzed by Nanostring. Results showed that all genes in the DNA repair pathway were downregulated dramatically for 2- to over 32-fold after an HANP/GTK831 or HANP/GKT831+RT treatment (Figure 6C). HANP/GTK831, without or with in combination with radiation, had a similar level of inhibition of DNA repair genes, suggesting that HANP/GTK831 played a major role in blocking DNA repair function. Radiation had a minimal effect on the expression of those genes. Additionally, HANP/GKT831 decreased the level of p53 gene expression in MCF-7 cells that have a wild type p53 gene. Next, the levels of gene expression profiles of the Breast VII PDX tumors collected 5 d after five weekly treatments were examined by RNA-seq analysis. Consistent with the result observed in breast cancer cells in vitro 4 h after RT, the level of γH2AX gene expression increased in HANP/GKT831 or HANP/GKT831+RT-treated tumors (Figure 6C). The levels of many DNA repair genes were decreased mostly in HANP/GKT831- or HANP/GKT831+RT-treated tumors (Figure 6C). Since the DNA damage response occurs early after radiation, the levels of downregulation of DNA repair genes in tumors 5 d after the last HANP/GKT831 treatment were not as strong as that in the treated MCF-7 cells. However, radiation slightly increased the levels of expression of some DNA repair genes in the tumor (Figure 6C). Therefore, results of this study suggest that HANP/GKT831 treatment inhibited DNA repair function in tumor cells, which primed sensitivity of tumor cells to DNA damaging agents, such as radiation.
HANP/GKT831 Treatment Modulated Tumor Stromal Fibroblasts and Macrophages.
The NOX4-ROS signal plays an important role in activation and proliferation of tumor-associated fibroblasts.35 We found that GKT831 treatment significantly reduced the level of FAP+ and alpha-smooth muscle actin (α-SMA) + fibroblasts in tumor tissues, suggesting that GKT831 was able to enter the tumor stroma to reach a concentration that inhibited fibroblasts, especially FAP + fibroblasts (Figure 7A). HANP/GKT831 had a similar inhibitory effect on FAP+ and α-SMA + fibroblasts in tumors. However, a marked inhibition of active fibroblasts by GKT831 or HANP/GKT831 did not have a significant effect on the tumor growth in the Breast VII PDX model. Although radiation therapy modestly increased the levels of both FAP+ and α-SMA+ fibroblasts in tumor tissues, the combination of RT with GKT831 or HANP/GKT831 could significantly inhibit the levels of both types of fibroblasts (Figure 7A).
Figure 7.

Effect of HANP/GKT831 treatment on tumor stromal cells. Frozen tissue sections of Breast VII PDX tumors after different treatments were examined for the changes in tumor stromal fibroblasts and macrophages using immunofluorescence labeling. (A) FAP+ and α-SMA+ stromal fibroblasts. The mean of the total FAP+ or α-SMA+ fluorescent signal of four images per treatment is shown. (B) Tumor-associated macrophages. CD68 is a pan–macrophage biomarker. CD163+ is a biomarker for M2-like macrophages. The number of CD68+ or CD163+ macrophages in each fluorescence image was quantified to determine changes in the macrophage populations after treatment. The mean number of positive cells per image field under a 20× lens of 5–6 images are shown. * p < 0.05, **p < 0.01, *** p < 0.005.
Tumor-associated macrophages are involved in aggressive biology, tumor invasion, and metastasis and are resistant to therapy.49 Our results showed that GKT831+RT-, HANP/GKT831-, or HANP/GKT831+RT-treated tumors had significantly reduced levels of CD68+ macrophages in tumors (Figure 7B). Importantly, the numbers of CD163+ M2-like macrophages49,50 were significantly decreased following treatment with HANP/GKT831 or HANP/GKT831+RT (Figure 7B). Radiation also inhibited CD163+ macrophages in tumors (Figure 7B).
Bioactive HANP/GKT831 Sensitized Tumor Cells to Radiation by Inhibition of Oncogenic Signaling Pathways.
We further investigated molecular mechanisms of HANP/GKT831 enhanced responses to radiotherapy by an analysis of gene expression profiles of breast cancer cells in vitro and PDX tumors following different treatments. Following treatment of MCF-7 cells with a cytotoxic dose (4 μM of GKT831 equiv) of HANP/GKT831 for 24 h, and then RT for 4 h, RNAs were isolated from cells for a gene expression analysis using Nanostring. We found that HANP/GKT831 or HANP/GKT831+RT treatment significantly downregulated the levels of many genes in the oncogenic pathways that are associated with cell proliferation/cycle, apoptosis, survival, and resistance to chemo- and radiotherapy samples (Figure 8). For example, the levels of signal molecule genes that are associated with resistance to radiation, such as KRAS, EGFR, IGF-1R, PI3K/AKT/mTOR, MAPK, Notch, TGF-β, and STAT,4,5,7,51 were markedly decreased in the treated tumor cells (Figure 8A-D,F,H). Overexpression of TTK protein kinase that regulates cell division has been shown to correlate with early tumor recurrence after radiotherapy. Inhibition of TTK impaired homologous recombination of DNA, and it radiosensitizes breast cancer to radiotherapy.52 Interestingly, HANP/GKT831 treatment decreased the level of TTK gene expression by 30-fold (Figure 8C). On the other hand, there was a fourfold increase in the level of the suppressor of cytokine signaling (SOCS1) gene, which inhibits STAT3 to enhance radiation-induced DNA damage and improve response to radiotherapy53 (Figure 8H). HANP/GKT831 or HANP/GKT831+RT induced fourfold increases in the expression level of TNF, an apoptosis-inducing gene.54,55 Furthermore, the expression levels of cell cycle regulating genes, including cyclins (CCNA, B, D, E), CDC25 (A, B, C), CDK (4, 6), and CHEK (1, 2), were significantly inhibited (Figure 8C). However, the level of the growth arrest and DNA-damage-inducible protein (GADD45A/G) that is associated with an increased response to radiotherapy in human cancers56 was elevated 8- to 16-fold in HANP/GKT831 or HANP/GKT831+RT treated tumor cells compared to the no-treatment control (Figure 8C). Additionally, the ability of targeted delivery in CD44 expressing tumor cells resulted in downregulation of cancer stemness signals in the WNT pathway (Figure 8E). HANP/GKT831 and HANP/GKT831+RT also decreased the expression levels of genes controlling epigenetic regulation of gene transcription that are associated with an aggressive tumor biology, such as the DNA methyl transferases (DNMTs) and histone deacetylases (HDACs) (Figure 8G). Interestingly, many genes regulating stromal fibroblasts and extracellular matrix proteins were downregulated in HANP/GKT831- or HANP/GKT831+RT-treated cells (Figure 8I). Results of the above gene expression analysis in tumor cells in vitro revealed HANP/GKT831 or RT-induced signal changes at the early stage of the treatment.
Figure 8.

Gene expression profiles of breast cancer MCF-7 cells after treatment in vitro. RNA samples isolated from MCF-7 cells after treatment with HANP/GKT831 for 24 h and 5 Gy of RT for 4 h were analyzed by Nanostring using the nCounter PanCancer Pathways Panel. Changes in the levels of representative genes in key signal pathways are shown as log2 increase or decrease compared to the values of the no-treatment control. (A) Oncogenic genes. (B) PI3k/AKT/MAPK. (C) Cell cycle/proliferation. (D) TGF-β. (E) WNT. (F) Notch. (G) Transcription regulation. (H) JAK/State. (I) Stroma/matrix. (*) Upregulation is associated with tumor growth inhibition.
To determine changes in the regulation of gene expression in residual breast PDX tumors 5 d after we completed five weekly treatments, RNAs isolated from the PDX tumors were analyzed by RNaseq. In the PDX tumors that showed a good response, the residual tumors were small and had increased tumor stroma. Results of gene expression profiles were different from those detected in culture tumor cells 24 h after HANP/GKT831 and 4 h following RT treatment. We found that the levels of expression of many genes that promote tumor growth and therapy resistance were downregulated in tumors treated with HANP/GKT831+RT. The expression levels of several representative genes that promote tumor cell proliferation and survival, such as EGFR, PI3K (PIK3R3), MAPK3, STAT1, high mobility group A2 (HMGA2), lysophosphatidic Acid Receptor (LPAR1), and cyclin D (CCND2), were decreased in tumors treated with HANP/GKT831+RT57,58 (Figure 9A,B). HANP/GKT831+RT treatment increased levels of cyclin-dependent kinase inhibitors (CDKN1A, 2A, 2B) that inhibit cell proliferation, but GKT831 decreased the expression levels of those genes (Figure 9B). Importantly, HANP/GKT831+RT treatment downregulated the expression of genes associated with cancer stem cells and drug and radiation resistance, such as WNT, CD44, TWIST2,59 and ABCA2 (Figure 9C). Additionally, the combination therapy of either GKT831+RT or HANP/GKT831+RT showed a strong inhibitory effect on the expression of genes that regulate tumor stromal fibroblasts and extracellular matrix, such as FGF, FGFR, hyaluronan and proteoglycan link protein 3 (HAPLN3),60 and laminin subunit α S (LAMA5) (Figure 9D). The above treatments also decreased the levels of expression of the Chitinase 3-like 1 (CHI3L1) gene, which is associated with tumor metastasis and immune suppression in the tumor stroma61 (Figure 9D). Furthermore, the combination therapy significantly downregulated the expression levels of proinflammatory and tumor-promoting genes, such as COX2, S100 (AB, A9), and TGFBI (Figure 9D). Although in vivo efficacy studies were conducted in immunodeficient nude mice bearing PDX tumors, the combination therapy of GKT831+RT or HANP/GKT831+RT downregulated the levels of proinflammatory, tumor promoting, and immunosuppressive cytokines, such as CXCL1, CXCL2, CXCL3, and IL1A, or cytokine receptors (IL1R, IL22R, IL6R and CSF3R). The lysyl oxidase (LOX) family of proteins modify stromal collagen and elastin to enhance tumor cell invasion.62 HANP/GKT831 and in combination with RT decreased the level of LOX gene expression (Figure 9E). HANP/GKT831+RT or GKT831+RT treatment increased the levels of cytokines and toll-like receptors (TLRs) that increase immune cell infiltration and activity and activation of tumor immune response, including CXCL10, CXCL14, IL23A, TRAF3IP3, TLR1, and TLR6 (Figure 9E).63,64 Although the effect of immunoactivating cytokines and molecules did not have a significant contribution to the antitumor effect in breast PDX tumors, it is likely that the ability of the activation of an immune response should enhance the overall therapeutic response in immune-competent animal tumor models and in cancer patients. Therefore, the results of a gene expression analysis support the effect of HANP/GKT831 on simultaneous downregulation of multiple cell-signal pathways that are associated with resistance to radiation therapy.
Figure 9.

Gene expression profiling of Breast VII PDX tumors after treatment. RNAs isolated from Breast VII PDX tumors collected from in vivo studies 5 d after completing a five weekly treatment schedule were analyzed by RNA-seq. The mean expression value of two PDX tumors in each group is shown as log2 increase or decrease compared to the no-treatment control. (A) Tumor-promoting genes. (B) Cell proliferation/apoptosis. (C) Stem cell and drug resistance. (D) Stroma and inflammation. (E) Cytokines and immune regulatory molecules. (*) Upregulation is associated with tumor growth inhibition. Cyan boxed: tumor-promoting and immune-suppressive cytokines and genes. Pink boxed: cytokines and genes with antitumor and immunoactivating effects.
Discussion.
The development of cancer therapeutics that target the altered signal pathways in human cancer cells offers an opportunity for improvement of therapeutic efficacy. Extensive efforts have been devoted to the development of therapeutic agents, including nanoparticle-based radiosensitizers, with the ability to induce ROS, which leads to DNA damage, oxidation of cellular proteins and lipids, or redox stress-induced cell death.10,12-16,20,24 Although such approaches could enhance therapeutic responses in some tumor cells, there is a need to develop approaches to improve therapeutic efficacy in tumor cells that have adapted to survive under a high level of intracellular ROS (redox imbalance) developed during tumorigenesis or after chemo- and radiotherapy. Furthermore, tumor hypoxia limits the therapeutic effect of ROS-inducing agents. Thus, therapeutic agents that effectively downregulate the level of intracellular ROS can block the critical survival signal for tumor cells with a redox imbalance and convert radiosensitivity in hypoxic tumor cells. Since many human cancers have a higher level of intracellular ROS than normal tissues,21,22,65 inhibition of ROS by targeting signal molecules that regulate ROS production in tumor cells should have a significant impact on survival signals in tumor cells but not on normal cells. In human tumor cells, mitochondria and NOX are the major contributors of intracellular ROS.21,22,25,66 The levels of NOX1 and/or NOX4 are upregulated in many types of human cancers.27,28,30,33-35,67 Our results showed breast cancer tissues obtained from drug-resistant tumor tissues have high levels of NOX1 and/or NOX4 expression, including both primary and metastatic tumor lesions, but not in normal breast ducts. The level of NOX4 was further upregulated in radiotherapy-resistant tumors. Those results validated the significance of inhibition of the NOX-ROS signal for the development of radiosensitizers and cancer therapeutic agents. In this study, we have developed a CD44 targeted nanoparticle carrying a dual NOX1/NOX4 inhibitor (HANP/GKT831) for overcoming resistance to radiotherapy in human cancer (Figure 10). Using two breast cancer PDX models with different responses to radiation, we demonstrated that systemic delivery of HANP/GKT831 led to the accumulation of the nanoparticles in tumors and resulted in a significant enhancement of therapeutic responses to low-dose radiotherapy in both PDX tumor models that showed an intermediate response or resistance to radiation therapy.
Figure 10.

Mechanisms of the effect of HANP/GKT831 on inhibition of NOX-ROS and oncogenic signal pathways and enhancement of response to radiation therapy. The levels of NOX1 and NOX4 are upregulated in human cancer cells that are resistant to chemo- and radiotherapy. NOX1 on the plasma membrane and endosome generates superoxide anion (O2•−), and NOX4 locates in the mitochondria and nuclear and endoplasmic reticulum membrane to generate intracellular hydrogen peroxide (H2O2). Chronic upregulation of intracellular ROS in tumor cells alters signal pathways that regulate cell proliferation, survival, invasion, and inflammation, which contribute to aggressive tumor biology and adaptation to a high level of ROS. This redox imbalance reduces sensitivity of tumor cells to ROS-activated cell death and a poor response to radiotherapy. Systemic administrations of HANP/GKT831 lead to targeted delivery into tumors and CD44 receptor-mediated internalization into endosomes/lysosomes of tumor cells for intracellular GKT831 release. GKT831 inhibits activity of NOX1 and NOX4 and reduces the ROS generation. The redox stress in tumor cells results in downregulation of DNA repair genes that primes tumor cells to radiation-induced DNA damage. Significant inhibition of oncogenic and survival signals, including oncogenes, growth factor receptors, PI3K/AKT, WNT, and cell cycle genes as well as TGF-β and JAK/Stat genes, further sensitizes tumor cells to cell death and improves the response to radiation.
Biocompatible and biodegradable HANPs have biophysical properties that are favorable for drug delivery carriers.38-41 The structure of an HA polysaccharide chain has hydrophobic patches that prevent nonspecific interactions with proteins and cells.68,69 The antifouling, viscoelastic, and elasticity features of HAs offer advantages of nanodrug carriers with reduced protein corona and improved intratumoral penetration and distribution.38,39,69 Although a liposome is a commonly used nanodrug carrier, nontargeted liposome enters tumors via the enhanced permeability retention (EPR) effect. The majority of liposomes/drugs accumulate in the tumor stroma areas and release the payload drug in the stroma.70 However, HANPs can bind to CD44 receptor expressing cells, leading to the internalization into tumor cells to be degraded by Hyal1 and release drug molecules in tumor cells. Although Hyal 2 on the extracellular cell membrane can hydrolyze HAs, it is likely that HANPs produced from 200 kDa HAs have a good stability in the extracellular space or cell surface, since Hyal 2 only degrades high molecular weight HAs (>1000 kDa). Furthermore, degraded HA fragments have additional biological activities on tumor cells and the tumor stroma to enhance therapeutic response.38,39
Although nanoparticles with smaller sizes have been considered more suitable for intratumoral delivery and penetration, unlike many other metallic and polymeric nanoparticles,38,41,68,69 HANPs have good elasticity and intermediate intrinsic stiffness between flexible and semiflexible polymers. More importantly, the biological activity of HAs depends on their molecular weights (MWs). HANPs produced from hyaluronic acids with a MW around 200 kDa have biological properties that resemble native hyaluronic acids in the modulation of tumor stroma cells and migration inside the tumor stroma to move away from the tumor vessels and to reach the tumor center that contains necrotic and hypoxic tumor areas. Although HANPs produced from 200 kDa HAs are relatively large (~200 nm), multiple binding sites of HANPs to CD44 and RHAMM receptors on the tumor endothelial cells enhance delivery of HANP/drugs into tumors. Our results demonstrated intratumoral delivery of 200 nm HANPs in breast PDX tumors with dense tumor stroma and large avascular tumor areas following a systemic delivery. As shown in Figure 3C,D, HANPs delivered into CK18+ and CD44+ tumor cells in the avascular tumor areas and many of the HANP positive cells did not colocalize with CD68+ macrophages.
In addition to tumor, we recognized that systemic delivery of HANP/GKT831 also leads to accumulation in the liver. This could be attributed to the internalization of HANP/GKT831 by sinusoidal endothelial and Kupffer cells in the liver. Results of clinical trials demonstrated that oral doses of GKT831 at 800–1600 mg daily for 24 weeks (an equivalent oral dose of 164–328 mg/kg of GKT831 in mice) are safe and well-tolerated in patients with lung, kidney, and liver fibrosis. It has been shown that GKT831 treatment improved the liver function by reducing fibrosis and inflammation.71 Results of our preclinical studies in mice also showed that HANP/GKT831 at 5 mg/kg once per week did not induce apparent systemic toxicity. For future clinical translation, we expect that therapeutic dose of i.v. delivered HANPS/GKT831 will be lower than that of the blood GKT831 concentration of 800–1600 mg oral daily doses that have been used in clinical trials. However, extensive preclinical evaluations and future clinical trials will further determine the systemic effect of HANP/GKT831, especially on the liver function.
The nanotherapeutic agent developed in this study, HANP/GKT831, showed a strong inhibitory effect on ROS production and cell proliferation in human tumor cells in vitro. Results of in vivo efficacy studies in two ER+ breast cancer PDX models further supported the effect of HANP/GKT831 on the significant enhancement of the therapeutic response to radiotherapy in resistant tumors. Breast VII PDX tumors showed a poor response to radiotherapy, GKT831 or HANP/GKT831 alone, or GKT831+RT treatment. Only the combination of HANP/GKT831 with radiation overcame the therapy resistance and resulted in tumor regression in this highly resistant tumor model. Although Breast IX PDX tumor was derived from a doxorubicin, cyclophosphamide, and taxol-resistant residual tumor, it had intermediate responses to radiotherapy. Conventional GKT831 treatment was able to enhance the response to radiation. Because HANP/GKT831 induced reduction of intracellular ROS and redox stress, resulting in significant inhibition of oncogenic, cell proliferation, and survival signal pathways in tumor cells, our result showed that HANP/GKT831 treatment alone could inhibit the growth of Breast IX PDX tumors that were not highly resistant to therapies. However, HANP/GKT831-induced downregulation of DNA repair function could further enhance the therapeutic response to radiation therapy in this PDX model. Therefore, our results support the importance of HANP-formulated GKT831 in overcoming radioresistance in human tumor cells.
At present, molecular mechanisms by which HANP/GKT831 induces a stronger radiosensitization in resistant tumors than GKT831 have yet to be fully elucidated. It is likely that multiple mechanisms lead to overcoming radioresistance in tumor cells, including increased GKT831 delivery into tumor cells and efficient inhibition of intracellular ROS by HANP/GKT831, which results in a redox-stress in tumor cells with a redox imbalance, dependence on a high level of ROS to survive, and defective DNA repair function. CD44-mediated internalization of HANP/GKT831 into the endosomes and Hyal l-triggered drug release intracellularly may contribute to effective inhibition of NOX1 and NOX4 signals located in endosomes, endoplasmic reticulum, and mitochondrial membrane.30,33 Furthermore, biomaterial HAs, especially small molecular HAs, have biological activities that modulate signal pathways to potentiate the effect of GKT831 on tumor cells.38,39,41 A marked advantage of HANP-mediated delivery of GKT831 is selectively entering into CD44+ cells in tumors, including angiogenic endothelial, active stromal, and tumor cells. Following systemic delivery, HANPs enter tumors by both the EPR effect through the leaky vascular structures and active targeting of CD44 and RHAMM receptor expressing endothelial cells.40,41,72 In tumor tissues, HANP/GKT831 binds to CD44-expressing tumor cells, stromal fibroblasts, and tumor-associated macrophages. Interaction of HANPs with stromal fibroblasts and macrophages enhances retention of the nanoparticle/drug in tumors as well as modulates NOX-ROS signals in stromal cells. We observed a decrease in the levels of FAP+ and α-SMA + fibroblasts in the PDX tumors treated with GKT831 and HANP/GKT831, GKT831+RT, or HANP/GKT831+RT. In the tumor stroma, HANP/GKT831 also interacts with tumor-associated macrophages by actively targeting CD44. It is likely that inhibition of NOX1 and NOX4 activities by HANP/GKT831 does not significantly affect macrophage function, since NOX2 activity is involved in phagocytosis function.73 A recent study showed that inhibition of NOX4 in macrophages could upregulate NOX2 to enhance the pro-inflammatory effect of macrophages.74 Our results showed that HANP/GKT831 or HANP/GKT831+RT treatment significantly reduced the level of M2-like macrophages in tumors.
Therapy approaches that inhibit DNA repair could bypass radioresistant mechanisms in tumor cells due to tumor hypoxia and adaptation to a redox imbalance. Our results showed that HANP/GKT831 markedly inhibited the expression levels of DNA repair genes and induced accumulation of DNA damage. Combination therapy of HANP/GKT831 with radiation further enhanced radiation-induced DNA damage. Notably, the total radiation dose used in tumor-bearing mice was 10 Gy, which is a lower dose and less-intensive schedule compared to the standard radiotherapy in breast cancer using the dose of 2 Gy daily for a total dose of 45–60 Gy.75 Additionally, DNA damage activates the ATM-p53 signal pathway that promotes cell cycle arrest and DNA repair.76 Tumors with p53 mutations have a higher sensitivity to radiation therapy than the wild-type tumors.77 Breast VII PDX tumors contain a high percentage of mutant p53-expressing tumor cells. Following five weekly treatments, most tumor cells in HANP/GKT831+RT-treated residual tumors lacked detectable mutant p53 protein, suggesting that tumor cells with mutant p53 genes are more sensitive to the treatment.
Results of a gene expression analysis of breast cancer cells further delineated the effect of HANP/GKT831, without or with radiation, on cell signal pathways. HANP/GKT831 significantly downregulated oncogenic signals that are associated with resistance to radiation therapy, such as KRAS, EGFR, PI3K/AKT, MAPK, WNT, Stat, Notch, and TGF-β.7,51 The levels of gene expression regulating the DNA damage responses, such as DNA repair and cell cycle, were also inhibited.4,5,9 Inhibition of the growth factor receptor signals using antibodies or small-molecule drugs sensitizes tumor cells to radiotherapy in animal tumor models and in clinical trials in cancer patients.1,7 Most radiosensitization approaches targeting cell signals act upon one molecular target.7 Recently, therapeutic agents that target the DNA repair pathway, such ATM or ATR inhibitors, have attracted great attention. Those DNA repair inhibitors are in clinical trials and have shown enhanced therapeutic responses to radiation and chemotherapy drugs.7 Results of our study showed that HANP/GKT831 treatment inhibited the level of expression not only of ATM and ATR genes but also of many other DNA repair genes. Thus, such an approach should have advantages over single-agent inhibition using a small-molecule drug. Additionally, increasing evidence supports the effect of radiation-induced tumor stroma and immunological changes on promoting radioresistance.78 Targeting the tumor stromal environment has been shown to be a promising approach for enhancing a response to radiotherapy. It is likely that strong inhibitory effects of HANP/GKT831 on tumor-associated fibroblasts, M2 macrophages, inflammatory molecules, and protumor cytokines also contribute to the radiosensitization effect on breast PDX tumors.
The HANP/GKT831 nanoradiosensitizer developed in this study inhibited multiple signal pathways to effectively induce apoptotic cell death in tumor cells in vitro. Since our in vivo study used breast PDX models derived from multidrug-resistant tumors, the PDX tumors showed poor to modest responses to radiotherapy or HANP/GKT83 alone. However, HANP/GKT831 in combination with radiation further enhanced DNA damage and downregulated survival signals to overcome resistance to radiotherapy in those tumor cells. Currently, the downstream events of ROS inhibition by HANP/GKT831 that lead to the inhibition of the DNA repair and oncogenic signal pathways are under investigation. A recent in vitro study using human embryonic kidney 293 cells stably transfected with different NOX isoforms suggested that the effect of GKT831 on ROS inhibition might not be mediated by direct inhibition of the NOX4 activity.79 Therefore, further studies are required to determine whether HANP/GKT831 has different mechanisms of action in regulating ROS and cell-signal pathways compared with GKT831. Nevertheless, demonstration of a strong inhibitory effect of HANP/GKT831 on important cell-signal pathways that lead to increased DNA damage and sensitizing tumor cells to radiotherapy in this study should provide us with an effective therapeutic approach for overcoming radioresistance in human cancer.
CONCLUSIONS
In summary, we have developed a tumor-targeted and bioactive NOX nanoinhibitor by encapsulation of GKT831 into HANPs. Systemic delivery of HANP/GKT831 leads to significantly enhanced therapeutic responses to radiotherapy in resistant breast PDX tumors. The effect of HANP/GKT831 is likely mediated by downregulating DNA repair that primes tumor cells to radiation-induced DNA damage in tumor cells. The combination of HANP/GKT831 with radiation increased DNA damage and inhibited oncogenic and survival signals, which sensitize tumor cells to radiotherapy. Therefore, HANP/GKT831 is a promising nanotherapeutic agent for the development of combination therapies for the treatment of resistant human cancers.
METHODS/EXPERIMENTAL
Preparation of Hyaluronic Acid Nanoparticle Carrying GKT831 (HANP/GKT831).
HANP/GKT831 nanocomplexes were produced according to our established protocol with minor modifications42 (Figure 2A). Sodium hyaluronic acid (HA, 234 kDa) was purchased from Lifecore Biomedical Company. HA was first converted to the tetrabutylammonium (TBA) salt of HA according to previously reported methods. The HA-TBA was then chemically modified with 5β-cholanic acid (CA) in the presence of 1-ethyl-3(3-(dimethylamino) propyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) at 60 °C in dimethyl sulfoxide (DMSO) (Sigma-Aldrich) to produce HA-CA. After dialysis against methanol/ultrapure water (1:1, v/v) and then ultrapure water for 4–12 h, HA-CA was lyophilized and stored at 4 °C in the dark until further use. Next, 80 mg of HA-CA conjugates were dissolved in 14 mL of distilled water and subjected to a high-pressure homogenizer (D-3L, PhD Technology) for 5 min to form HA nanoparticles (HANPs). Hydrophobic GKT 831 was loaded to HANP via a high-pressure homogenizer at a ratio of 10 mg GKT831/40 mg of HANP. Thirty milligrams of GKT831 (MedKoo Biosciences #522357) dissolved in 1 mL of DMSO was added dropwise into the solution containing 120 mg of HANP and then circulated for an additional 5 min in the homogenizer. HANP/GKT831 nanocomplex was then dialyzed for 8 h in distilled water to remove free drugs and organic solvents. Finally, HANP/GKT831 was lyophilized into white powder and kept at 4 °C.
In Vitro Characterization of HANP/GKT831.
Freeze-dried HANP and HANP/GKT831 samples were dissolved in H2O and homogenized in the high-pressure homogenizer. Nanoparticle sizes of HANP and HANP/GKT831 were determined using a dynamic light scattering (DLS) analysis (DelsaMax PRO, Beckman Coulter Life Sciences). The amount of GKT831 loading in HANP/GKT831 was determined by a reversed-phase high-performance liquid chromatography system (RP-HPLC, Dionex, Thermo Fisher Scientific) using a C18 column in a 10–65% linear gradient of an acetonitrile/water mixture (containing 0.1% trifluoroacetic acid) for 30 min at a flow rate of 1 mL/min. Absorption intensity at a wavelength of 254 nm was obtained. The loading content of GKT831 in HANP/GKT831 was calculated using the following equation: loading content (%) = WGKT831 loaded/WGKT831 input × 100.
Determination of Drug Release In Vitro.
GKT831 release from HANP/GKT831 was examined under a physiological temperature of 37 °C, in the presence of 10 mM hyaluronidase (Hyase, Sigma) at pH 5.0 using a dialysis bag (molecular weight cutoff (MWCO) 10 kDa, Spectrum Laboratories). 200 μL of solution in the dialysis system was harvested at different time points. The level of GKT831 in the solution was analyzed by an analytical HPLC (Thermo Fisher Scientific) using a reverse-phase C18 column. Amount of GKT831 released from HANP/GKT831 was calculated using a standard GKT831 curve (y = 2156.65x – 1.05, R2 = 0.9999). Experiments for all samples were performed three times at each pH value.
Cytotoxicity Assay.
The effect of HANP/GKT831 on human cancer cells was determined using Sulforhodamine B (SRB) Cell Proliferation assay (MP Biomedicals, LLC). The MCF-7 human breast cancer cell line (ATCC) is a representative estrogen receptor positive (ER+) breast cancer cell line that has a wild-type p53 gene.44 Breast PDX tumor-derived cell line (Breast VII) and pancreatic PDX tumor-derived cell line (PANC II) were established in our research lab from PDX tumors with a high percentage of tumor cells containing a mutant p53 gene, which was determined by RNA-seq of the PDX tumors and by immunofluorescence labeling of p53 protein in tumor tissues. Normal human dermal fibroblast cell line (HDF) was obtained from ATCC. Cells were cultured in 96-well plates for 24 h. A serial dilution of HANP/GKT831 and free GKT831 was then added to the culture wells for 72 h. Following an SRB assay, the optical density (OD) at a wavelength of 510 nm was measured using the Synergy H1 microplate reader (BioTek instruments).
Determination of ROS Production in Cells.
The level of ROS in viable cells was quantified using a Cellular ROS Assay Kit that contains 2′,7′-dichlorodihydrofluorescindiacetate (DCFH-DA, Thermo Fisher Scientific). DCFH-DA is cell-permeable probe that is hydrolyzed intracellularly to the DCFH carboxylate anion and then oxidized by ROS (H2O2) to a fluorescent DCF.43 Following treatment with GKT831 and HANP/GKT831 for 24 h, 1 μM DCFHDA was added into each culture well for 1 h. The fluorescence intensity was measured using a Synergy H1 microplate spectrometer and excitation/emission wavelengths (Ex 488/Em 525 nm).
Establishment of Orthotopic Human Breast Patient Derived Xenograft (PDX) Tumor Models in Nude Mice.
Breast cancer PDX models (Breast VII and Breast IX) were established in our lab from surgically resected breast cancer tissues following an approved Institutional Review Board protocol of Emory University. Selected tumor tissues were collected from the breast cancer patients treated with neoadjuvant chemotherapy and had large resistant residual tumors. The Breast VII patient had an ER+ invasive ductal carcinoma and disease progression during the combination chemotherapy of taxotere and cyclophosphosphamide. The Breast IX patient had an ER+ invasive ductal carcinoma that was resistant to the neoadjuvant chemotherapy of doxorubicin, cyclophosphamide, and taxol and then a 50 Gy radiotherapy. Small fragments (1–2 mm) of fresh cancer tissues were implanted into the mammary fat pad of athymic nude mice (8–10 weeks old, female) using a surgical procedure. Orthotopic PDX tumors grew to 10 to 15 mm in diameter in about 10 to 15 weeks. PDX tumors were removed after the mice were sacrificed. Fresh tumor fragments were then implanted into the mammary fat pad of nude mice for large-scale studies.
In Vivo Fluorescent Imaging of Targeted Delivery in PDX Tumors in Nude Mice.
Nude mice bearing orthotopic PDX tumors at a size of ~80 mm3 received an intravenous (i.v.) injection of NIR 830 dye-labeled HANPs by covalent conjugation via ethylenediamine. Whole body optical imaging was performed 24 h after the injection using the IVIS Spectrum In Vivo Imaging System (Ex/Em: 745/800 nm, PerkinElmer). After whole body imaging, breast PDX tumors and normal organs were collected for the acquisition of fluorescence signals. Fluorescence intensities from whole body and ex vivo images were analyzed and quantified using the software of the IVIS Spectrum In Vivo Imaging System.
In Vivo Antitumor Efficacy.
Nude mice bearing orthotopic Breast VII or Breast IX PDX tumors (in vivo passage 2) were randomized into experimental groups. Each nude mouse had two tumors implanted into the right and left mammary fat pads (Figure 4A). When the tumor reached sizes of ~250 mm3 (Breast VII) or ~120 mm3 (Breast IX), tumor-bearing mice received tail vein injections of conventional GKT831 or HANP/GKT831 at a GKT831 equiv dose of 5 mg/kg body weight once per week for a total of five treatments. Conventional GKT831 was first dissolved in DMSO and then mixed with kolliphor EL (Sigma-Aldrich). The mixture was further diluted in H2O before injection at a concentration of 1 mg/mL GKT831. The ratio of DMSO, kolliphor EL, and H2O was controlled as 1:2:7 (v/v). Twenty-four hours following the nanodrug injection, radiation therapy (X-RAD 320, Precision X-ray) at 2 Gy each treatment was applied on the right-side tumor once per week for five weeks, while the left side was shielded from the irradiation using a lead blocker. A total irradiation dose was 10 Gy. During the treatment, tumor size and mouse body weight were monitored once per week. Five days after the final treatment, mice were sacrificed, and tumors were collected for histological and molecular analysis.
Western Blot Analysis.
MCF-7 breast cancer cells were treated with HANP/GKT831 (0.5 and 1 μM of equivalent dose of GKT831) for 24 h and then received 5 Gy of irradiation. Cells were collected 4 h after irradiation and lysed in an immunoprecipitation assay buffer (Cell Signaling Technology). Protein extracts of cells were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then blotted onto poly(vinylidene difluoride) membranes. Blots were incubated with rabbit antihuman γH2AX antibody (sc-10790, Santa Cruz Biotechnology) and then horseradish peroxidase (HRP) conjugated goat antirabbit IgG antibody (Santa Cruz Biotechnology). Positive protein bands were detected using the G:BOX Systems Imaging System (Syngene).
Nanostring or RNA-seq Analysis of the Levels of Gene Expression in Tumor Cells and PDX Tumors.
Human breast cancer MCF-7 cells were treated with 4 μM GKT831 or HANP/GKT831 for 24 h and then 5 Gy of irradiation. Cells were collected 4 h after irradiation, an early time point allowing accurate detection of changes in the expression of DNA repair genes. RNA samples of cells were isolated using the SV Total RNA Isolation kit (Promega). The levels of gene expression were analyzed by NanoString and the nCounter Pan-cancer pathway panel (NanoString).
Gene expression profiling of Breast VII PDX tumors after treatment was examined by RNA-seq analysis. PDX tumors were collected from in vivo studies after five weekly treatments of 5 mg/kg of GKT831, HANP/GKT831, without or with in combination with 2 Gy of radiation (IR). Tumors were collected 5 d after the last therapy. RNAs were isolated from frozen tissue samples and analyzed by RNA-seq (Illumina HiSeq platform, GENEWIZ).
Immunofluorescence Labeling.
Frozen tissue sections (10 μm) of tumor and normal tissues were used for single or double immunofluorescence labeling. Anti-NOX1 (MBS9609001) and anti-NOX4 (MBS820230) antibodies were from MYBioSource. Goat antimouse CD44, rabbit antimouse CD24, rabbit antimouse Ki67 antibody, and goat antihuman fibroblast active protein (FAP) antibodies were purchased from Santa Cruz Biotechnology (sc7051, sc11406, sc15402, and sc71094, respectively). A rat antimouse CD31 antibody (ThermoFisher) was used to identify tumor blood vessels. A mouse monoclonal anti-CK18 antibody was from Sigma-Aldrich. A rabbit antihuman γH2AX antibody and a rabbit antihuman p53 antibody were from Cell Signaling. A rat antimouse CD68 antibody (Bio-Rad, MCA1957) and a rat antimouse CD163 antibody (155302, BioLegend) were used to identify macrophages. Alexa Fluor 488 dye or Alexa Fluor 555 dye-labeled secondary antibodies against corresponding species of primary antibodies (Thermo Fisher Scientific) were used to detect biomarker-positive cells following incubation with single or dual primary antibodies. Fluorescent images were taken using an inverted fluorescence microscope (BZ-X710 All-in-one, Keyence). To determine cell types containing NIR-830 dye labeled HANPs, NIR signals were also captured from tissue sections at the same imaging field using an NIR filter set (Ex/Em: 780/800 nm) in the fluorescence microscope. Fluorescence images were analyzed and quantified using the NIH ImageJ software.
Statistical Analysis.
All experimental results were presented as the mean ± standard deviation from more than three repeat samples. Statistical analysis was performed using a two-tailed student’s t-test. A statistically significant difference was defined as a p-value less than or equal to 0.05.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Emory Integrated Genomics Core for Gene expression analysis using NanoString nCounter FLEX, research funding support of NIH grant awards (U01CA19891301, R01EB020601-01, R01CA154129, and R43 CA246827), and Nancy Panoz Chair fund to L.Y.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.2c07440.
Procedures of the cell internalization and drug delivery of HANP/GKT831 in vitro; TEM images of HANP and HANP/GKT831; stability of HANP/GKT831; imaging and quantification of HANP/GKT831 in tumor cells; cell proliferation assay of HANP/GKT831 on pancreatic cancer PDX derived tumor cells; quantification of HANP/GKT831 induced apoptosis; biodistribution of HANP/GKT831 ex vivo (PDF)
The authors declare no competing financial interest.
Contributor Information
Lei Zhu, Department of Surgery and Winship Cancer Institute, Emory University School of Medicine, Atlanta 30322 Georgia, United States.
Yi Zhao, Department of Surgery and Winship Cancer Institute, Emory University School of Medicine, Atlanta 30322 Georgia, United States; Jiangxi University of Traditional Chinese Medicine, Nanchang 330004, China.
Tongrui Liu, Department of Surgery and Winship Cancer Institute, Emory University School of Medicine, Atlanta 30322 Georgia, United States.
Minglong Chen, Department of Surgery and Winship Cancer Institute, Emory University School of Medicine, Atlanta 30322 Georgia, United States; Department of Nuclear Medicine, China-Japan Union Hospital, Jilin University, Changchun 130033, China.
Wei Ping Qian, Department of Surgery and Winship Cancer Institute, Emory University School of Medicine, Atlanta 30322 Georgia, United States.
Binghua Jiang, Department of Pathology, Anatomy and Cell Biology, Thomas Jefferson University, Philadelphia 19107 Pennsylvania, United States.
Benjamin G. Barwick, Department of Hematology and Medical Oncology, Emory University School of Medicine, Atlanta 30322 Georgia, United States
Lumeng Zhang, Department of Surgery and Winship Cancer Institute, Emory University School of Medicine, Atlanta 30322 Georgia, United States; Department of Nuclear Medicine, China-Japan Union Hospital, Jilin University, Changchun 130033, China.
Toncred M Styblo, Department of Surgery and Winship Cancer Institute, Emory University School of Medicine, Atlanta 30322 Georgia, United States.
Xiaoxian Li, Department of Pathology and Laboratory Medicine, Emory University School of Medicine, Atlanta 30322 Georgia, United States.
Lily Yang, Department of Surgery and Winship Cancer Institute, Emory University School of Medicine, Atlanta 30322 Georgia, United States.
REFERENCES
- (1).Schaue D; McBride WH Opportunities and Challenges of Radiotherapy for Treating Cancer. Nat. Rev. Clin Oncol 2015, 12, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Curtin NJ DNA Repair Dysregulation from Cancer Driver to Therapeutic Target. Nature Reviews Cancer 2012, 12, 801–817. [DOI] [PubMed] [Google Scholar]
- (3).Lord CJ; Ashworth A The DNA Damage Response and Cancer Therapy. Nature 2012, 481, 287–294. [DOI] [PubMed] [Google Scholar]
- (4).Pilie PG; Tang C; Mills GB; Yap TA State-of-the-Art Strategies for Targeting the DNA Damage Response in Cancer. Nat. Rev. Clin Oncol 2019, 16, 81–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pearl LH; Schierz AC; Ward SE; Al-Lazikani B; Pearl FMG Therapeutic Opportunities within the DNA Damage Response. Nature Reviews Cancer 2015, 15, 166–180. [DOI] [PubMed] [Google Scholar]
- (6).Lawrence TS; Blackstock AW; McGinn C The Mechanism of Action of Radiosensitization of Conventional Chemotherapeutic Agents. Semin Radiat Oncol 2003, 13, 13–21. [DOI] [PubMed] [Google Scholar]
- (7).Petroni G; Cantley LC; Santambrogio L; Formenti SC; Galluzzi L Radiotherapy as a Tool to Elicit Clinically Actionable Signalling Pathways in Cancer. Nature Reviews Clinical Oncology 2022, 19, 114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Delaney G; Jacob S; Featherstone C; Barton M The Role of Radiotherapy in Cancer Treatment - Estimating Optimal Utilization from a Review of Evidence-Based Clinical Guidelines. Cancer 2005, 104, 1129–1137. [DOI] [PubMed] [Google Scholar]
- (9).Morgan MA; Lawrence TS Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways. Clin. Cancer Res 2015, 21, 2898–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Tofilon PJ; Camphausen K Molecular Targets for Tumor Radiosensitization. Chem. Rev 2009, 109, 2974–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dewhirst MW A Potential Solution for Eliminating Hypoxia as a Cause for Radioresistance. Proc. Natl. Acad. Sci. U.S.A 2018, 115, 10548–10550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wang AZ; Tepper JE Nanotechnology in Radiation Oncology. J. Clin Oncol 2014, 32, 2879–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lee J; Chatterjee DK; Lee MH; Krishnan S Gold Nanoparticles in Breast Cancer Treatment: Promise and Potential Pitfalls. Cancer Lett. 2014, 347, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bonvalot S; Rutkowski PL; Thariat J; Carrere S; Ducassou A; Sunyach MP; Agoston P; Hong A; Mervoyer A; Rastrelli M; Moreno V; Li RK; Tiangco B; Herraez AC; Gronchi A; Mangel L; Sy-Ortin T; Hohenberger P; de Baere T; Le Cesne A; et al. Nbtxr3, a First-in-Class Radioenhancer Hafnium Oxide Nanoparticle, Plus Radiotherapy Versus Radiotherapy Alone in Patients with Locally Advanced Soft-Tissue Sarcoma (Act.In.Sarc): A Multicentre, Phase 2–3, Randomised, Controlled Trial. Lancet Oncol 2019, 20, 1148–1159. [DOI] [PubMed] [Google Scholar]
- (15).Bort G; Lux F; Dufort S; Cremillieux Y; Verry C; Tillement O Epr-Mediated Tumor Targeting Using Ultrasmall-Hybrid Nanoparticles: From Animal to Human with Theranostic Aguix Nanoparticles. Theranostics 2020, 10, 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Jiang W; Li Q; Xiao L; Dou J; Liu Y; Yu W; Ma Y; Li X; You YZ; Tong Z; Liu H; Liang H; Lu L; Xu X; Yao Y; Zhang G; Wang Y; Wang J Hierarchical Multiplexing Nanodroplets for Imaging-Guided Cancer Radiotherapy Via DNA Damage Enhancement and Concomitant DNA Repair Prevention. ACS Nano 2018, 12, 5684–5698. [DOI] [PubMed] [Google Scholar]
- (17).Bonner JA; Lawrence TS Doxorubicin Decreases the Repair of Radiation-Induced DNA Damage. Int. J. Radiat. Biol 1990, 57, 55–64. [DOI] [PubMed] [Google Scholar]
- (18).Harrington KJ; Rowlinson-Busza G; Syrigos KN; Vile RG; Uster PS; Peters AM; Stewart JS Pegylated Liposome-Encapsulated Doxorubicin and Cisplatin Enhance the Effect of Radiotherapy in a Tumor Xenograft Model. Clin. Cancer Res 2000, 6, 4939–4949. [PubMed] [Google Scholar]
- (19).Tian X; Nguyen M; Foote HP; Caster JM; Roche KC; Peters CG; et al. Crlx101, a Nanoparticle-Drug Conjugate Containing Camptothecin, Improves Rectal Cancer Chemoradiotherapy by Inhibiting DNA Repair and Hif1 Alpha (Vol 77, Pg 112, 2017). Cancer Res. 2017, 77, 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ni KY; Lan GX; Veroneau SS; Duan XP; Song Y; Lin WB Nanoscale Metal-Organic Frameworks for Mitochondria-Targeted Radiotherapy-Radiodynamic Therapy. Nat. Commun 2018, 9. DOI: 10.1038/s41467-018-06655-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nogueira V; Hay N Molecular Pathways: Reactive Oxygen Species Homeostasis in Cancer Cells and Implications for Cancer Therapy. Clin. Cancer Res 2013, 19, 4309–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Trachootham D; Alexandre J; Huang P Targeting Cancer Cells by Ros-Mediated Mechanisms: A Radical Therapeutic Approach? Nat. Rev. Drug Discov 2009, 8, 579–591. [DOI] [PubMed] [Google Scholar]
- (23).Tian X; Lara H; Wagner KT; Saripalli S; Hyder SN; Foote M; Sethi M; Wang EN; Caster JM; Zhang LZ; Wang AZ Improving DNA Double-Strand Repair Inhibitor Ku55933 Therapeutic Index in Cancer Radiotherapy Using Nanoparticle Drug Delivery. Nanoscale 2015, 7, 20211–20219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Benej M; Hong XQ; Vibhute S; Scott S; Wu JH; Graves E; Le QT; Koong AC; Giaccia AJ; Yu B; Chen SC; Papandreou I; Denko NC Papaverine and Its Derivatives Radiosensitize Solid Tumors by Inhibiting Mitochondrial Metabolism. Proc. Natl. Acad. Sci. U.S.A 2018, 115, 10756–10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Schieber M; Chandel NS Ros Function in Redox Signaling and Oxidative Stress. Curr. Biol 2014, 24, R453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Tong L; Chuang CC; Wu S; Zuo L Reactive Oxygen Species in Redox Cancer Therapy. Cancer Lett. 2015, 367, 18–25. [DOI] [PubMed] [Google Scholar]
- (27).Roy K; Wu Y; Meitzler JL; Juhasz A; Liu H; Jiang G; Lu J; Antony S; Doroshow JH Nadph Oxidases and Cancer. Clin Sci. (Lond) 2015, 128, 863–875. [DOI] [PubMed] [Google Scholar]
- (28).Mortezaee K; Goradel NH; Amini P; Shabeeb D; Musa AE; Najafi M; Farhood B Nadph Oxidase as a Target for Modulation of Radiation Response; Implications to Carcinogenesis and Radiotherapy. Current Molecular Pharmacology 2019, 12, 50–60. [DOI] [PubMed] [Google Scholar]
- (29).Desouki MM; Kulawiec M; Bansal S; Das G; Singh KK Cross Talk between Mitochondria and Superoxide Generating Nadph Oxidase in Breast and Ovarian Tumors. Cancer Biology & Therapy 2005, 4, 1367–1373. [DOI] [PubMed] [Google Scholar]
- (30).Graham KA; Kulawiec M; Owens KM; Li XR; Desouki MM; Chandra D; Singh KK Nadph Oxidase 4 Is an Oncoprotein Localized to Mitochondria. Cancer Biol. Ther 2010, 10, 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tobar N; Guerrero J; Smith PC; Martinez J Nox4-Dependent Ros Production by Stromal Mammary Cells Modulates Epithelial Mcf-7 Cell Migration. Br. J. Cancer 2010, 103, 1040–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Teixeira G; Szyndralewiez C; Molango S; Carnesecchi S; Heitz F; Wiesel P; Wood JM Therapeutic Potential of Nadph Oxidase 1/4 Inhibitors. Br. J. Pharmacol 2017, 174, 1647–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Shanmugasundaram K; Nayak BK; Friedrichs WE; Kaushik D; Rodriguez R; Block K Nox4 Functions as a Mitochondrial Energetic Sensor Coupling Cancer Metabolic Reprogramming to Drug Resistance. Nat. Commun 2017, 8. DOI: 10.1038/s41467-017-01106-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Zhan M; Wang H; Chen T; Chen W; Yang LH; He M; Xu SW; Wang J Nox1Mediates Chemoresistance Via Hif1 Alpha/Mdr1 Pathway in Gallbladder Cancer. Biochem. Biophys. Res. Commun 2015, 468, 79–85. [DOI] [PubMed] [Google Scholar]
- (35).Hanley CJ; Mellone M; Ford K; Thirdborough SM; Mellows T; Frampton SJ; Smith DM; Harden E; Szyndralewiez C; Bullock M; Noble F; Moutasim KA; King EV; Vijayanand P; Mirnezami AH; Underwood TJ; Ottensmeier CH; Thomas GJ Targeting the Myofibroblastic Cancer-Associated Fibroblast Phenotype through Inhibition of Nox4. JNCI-J. Natl. Cancer Inst 2018, 110, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Laleu B; Gaggini F; Orchard M; Fioraso-Cartier L; Cagnon L; Houngninou-Molango S; Gradia A; Duboux G; Merlot C; Heitz F; Szyndralewiez C; Page P First in Class, Potent, and Orally Bioavailable Nadph Oxidase Isoform 4 (Nox4) Inhibitors for the Treatment of Idiopathic Pulmonary Fibrosis. J. Med. Chem 2010, 53, 7715–7730. [DOI] [PubMed] [Google Scholar]
- (37).Dalekos G; Invernizzi P; Nevens F; Hans VV; Zigmond E; Andrade RJ; Ben Ari Z; Heneghan M; Huang J; Harrison S; Minuk G; Jorn S; Moreno C; Vierling J; Vincent C; Bowlus C; Lurie Y; Muratori L; Niro G; Hirschfield G; et al. Efficacy of Gkt831 in Patients with Primary Biliary Cholangitis and Inadequate Response to Ursodeoxycholic Acid: Interim Efficacy Results of a Phase 2 Clinical Trial. J. Hepatology 2019, 70, E1–E2. [Google Scholar]
- (38).Cowman MK; Lee HG; Schwertfeger KL; McCarthy JB; Turley EA The Content and Size of Hyaluronan in Biological Fluids and Tissues. Front. Immunol 2015, 6. DOI: 10.3389/fimmu.2015.00261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Tavianatou AG; Caon I; Franchi M; Piperigkou Z; Galesso D; Karamanos NK Hyaluronan: Molecular Size-Dependent Signaling and Biological Functions in Inflammation and Cancer. Febs Journal 2019, 286, 2883–2908. [DOI] [PubMed] [Google Scholar]
- (40).Choi KY; Min KH; Na JH; Choi K; Kim K; Park JH; Kwon IC; Jeong SY Self-Assembled Hyaluronic Acid Nanoparticles as a Potential Drug Carrier for Cancer Therapy: Synthesis, Characterization, and in Vivo Biodistribution. J. Mater. Chem 2009, 19, 4102–4107. [Google Scholar]
- (41).Misra S; Heldin P; Hascall VC; Karamanos NK; Skandalis SS; Markwald RR; Ghatak S Hyaluronan-Cd44 Interactions as Potential Targets for Cancer Therapy. Febs Journal 2011, 278, 1429–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zhang L; Gao S; Zhang F; Yang K; Ma Q; Zhu L Activatable Hyaluronic Acid Nanoparticle as a Theranostic Agent for Optical/Photoacoustic Image-Guided Photothermal Therapy. ACS Nano 2014, 8, 12250–12258. [DOI] [PubMed] [Google Scholar]
- (43).Aranda A; Sequedo L; Tolosa L; Quintas G; Burello E; Castell JV; Gombau L Dichloro-Dihydro-Fluorescein Diacetate (Dcfh-Da) Assay: A Quantitative Method for Oxidative Stress Assessment of Nanoparticle-Treated Cells. Toxicology in Vitro 2013, 27, 954–963. [DOI] [PubMed] [Google Scholar]
- (44).Negrini M; Sabbioni S; Haldar S; Possati L; Castagnoli A; Corallini A; Barbantibrodano G; Croce CM Tumor and Growth Suppression of Breast-Cancer Cells by Chromosome-17-Associated Functions. Cancer Res. 1994, 54, 1818–1824. [PubMed] [Google Scholar]
- (45).Allred DC; Clark GM; Elledge R; Fuqua SAW; Brown RW; Chamness GC; Osborne CK; Mcguire WL Association of P53 Protein Expression with Tumor-Cell Proliferation Rate and Clinical Outcome in Node-Negative Breast-Cancer. Journal of the National Cancer Institute 1993, 85, 200–206. [DOI] [PubMed] [Google Scholar]
- (46).Abraham BK; Fritz P; McClellan M; Hauptvogel P; Athelogou M; Brauch H Prevalence of Cd44(+)/Cd24(−/Low) Cells in Breast Cancer May Not Be Associated with Clinical Outcome but May Favor Distant Metastasis. Clin. Cancer Res 2005, 11, 1154–1159. [PubMed] [Google Scholar]
- (47).Baumann M; Krause M; Hill R Exploring the Role of Cancer Stem Cells in Radioresistance. Nature Reviews Cancer 2008, 8, 545–554. [DOI] [PubMed] [Google Scholar]
- (48).Ivashkevich A; Redon CE; Nakamura AJ; Martin RF; Martin OA Use of the Gamma-H2ax Assay to Monitor DNA Damage and Repair in Cancer Research. Cancer Letters 2012, 327, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Noy R; Pollard JW Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity 2014, 41, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Tang XQ Tumor-Associated Macrophages as Potential Diagnostic and Prognostic Biomarkers in Breast Cancer. Cancer Letters 2013, 332, 3–10. [DOI] [PubMed] [Google Scholar]
- (51).Wang M; Han J; Marcar L; Black J; Liu Q; Li XY; Nagulapalli K; Sequist LV; Mak RH; Benes CH; Hong TS; Gurtner K; Krause M; Baumann M; Kang JX; Whetstine JR; Willers H Radiation Resistance in Kras-Mutated Lung Cancer Is Enabled by Stem-Like Properties Mediated by an Osteopontin-Egfr Pathway. Cancer Res. 2017, 77, 2018–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Chandler BC; Moubadder L; Ritter CL; Liu ML; Cameron M; Wilder-Romans K; Zhang A; Pesch AM; Michmerhuizen AR; Hirsh N; Androsiglio M; Ward T; Olsen E; Niknafs YS; Merajver S; Thomas DG; Brown PH; Lawrence TS; Nyati S; Pierce LJ; et al. Ttk Inhibition Radiosensitizes Basal-Like Breast Cancer through Impaired Homologous Recombination. J. Clin. Invest 2019, 130, 958–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Sugase T; Takahashi T; Serada S; Fujimoto M; Hiramatsu K; Ohkawara T; Tanaka K; Miyazaki Y; Makino T; Kurokawa Y; Yamasaki M; Nakajima K; Kishimoto T; Mori M; Doki Y; Naka T Socs1 Gene Therapy Improves Radiosensitivity and Enhances Irradiation-Induced DNA Damage in Esophageal Squamous Cell Carcinoma. Cancer Res. 2017, 77, 6975–6986. [DOI] [PubMed] [Google Scholar]
- (54).Wu X; Wu MY; Jiang M; Zhi Q; Bian X; Xu MD; Gong FR; Hou J; Tao M; Shou LM; Duan W; Chen K; Shen M; Li W TNF-Alpha Sensitizes Chemotherapy and Radiotherapy against Breast Cancer Cells. Cancer Cell Int. 2017, 17, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Wu X; Wu MY; Jiang M; Zhi QM; Bian XJ; Xu MD; Gong FR; Hou J; Tao M; Shou LM; Duan WM; Chen K; Shen M; Li W TNF-Alpha Sensitizes Chemotherapy and Radiotherapy against Breast Cancer Cells. Cancer Cell Int. 2017, 17. DOI: 10.1186/s12935-017-0382-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Tront JS; Hoffman B; Liebermann DA Gadd45a Suppresses Ras-Driven Mammary Tumorigenesis by Activation of C-Jun Nh2-Terminal Kinase and P38 Stress Signaling Resulting in Apoptosis and Senescence. Cancer Res. 2006, 66, 8448–8454. [DOI] [PubMed] [Google Scholar]
- (57).Geraldo LHM; Spohr TCLD; do Amaral RF; da Fonseca ACC; Garcia C; Mendes FD; Freitas C; dosSantos MF; Lima FRS Role of Lysophosphatidic Acid and Its Receptors in Health and Disease: Novel Therapeutic Strategies. Signal Transduct. Target. Ther 2021, 6. DOI: 10.1038/s41392-020-00367-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Mansoori B; Duijf PHG; Mohammadi A; Najafi S; Roshani E; Shanehbandi D; Hajiasgharzadeh K; Shirjang S; Ditzel HJ; Kazemi T; Mokhtarzadeh A; Gjerstorff MF; Baradaran B Overexpression of Hmga2 in Breast Cancer Promotes Cell Proliferation, Migration, Invasion and Stemness. Expert Opinion on Therapeutic Targets 2020, 24, 255–265. [DOI] [PubMed] [Google Scholar]
- (59).Fang X; Cai Y; Liu J; Wang Z; Wu Q; Zhang Z; Yang CJ; Yuan L; Ouyang G Twist2 Contributes to Breast Cancer Progression by Promoting an Epithelial-Mesenchymal Transition and Cancer Stem-Like Cell Self-Renewal. Oncogene 2011, 30, 4707–4720. [DOI] [PubMed] [Google Scholar]
- (60).Kuo SJ; Chien SY; Lin C; Chan SE; Tsai HT; Chen DR Significant Elevation of Cldn16 and Hapln3 Gene Expression in Human Breast Cancer. Oncol. Rep 2010, 24, 759–766. [DOI] [PubMed] [Google Scholar]
- (61).Libreros S; Garcia-Areas R; Shibata Y; Carrio R; Torroella-Kouri M; Iragavarapu-Charyulu V Induction of Proinflammatory Mediators by Chi3l1 Is Reduced by Chitin Treatment: Decreased Tumor Metastasis in a Breast Cancer Model. Int. J. Cancer 2012, 131, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Saatci O; Kaymak A; Raza U; Ersan PG; Akbulut O; Banister CE; Sikirzhytski V; Tokat UM; Aykut G; Ansari SA; Dogan HT; Dogan M; Jandaghi P; Isik A; Gundogdu F; Kosemehmetoglu K; Dizdar O; Aksoy S; Akyol A; Uner A; et al. Targeting Lysyl Oxidase (Lox) Overcomes Chemotherapy Resistance in Triple Negative Breast Cancer. Nat. Commun 2020, 11. DOI: 10.1038/s41467-020-16199-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Propper DJ; Balkwill FR Harnessing Cytokines and Chemokines for Cancer Therapy. Nature Reviews Clinical Oncology 2022, 19, 237–253. [DOI] [PubMed] [Google Scholar]
- (64).Javaid N; Choi S Toll-Like Receptors from the Perspective of Cancer Treatment. Cancers 2020, 12, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Ray PD; Huang BW; Tsuji Y Reactive Oxygen Species (Ros) Homeostasis and Redox Regulation in Cellular Signaling. Cell Signal 2012, 24, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Yamamori T; Yasui H; Yamazumi M; Wada Y; Nakamura Y; Nakamura H; Inanami O Ionizing Radiation Induces Mitochondrial Reactive Oxygen Species Production Accompanied by Upregulation of Mitochondrial Electron Transport Chain Function and Mitochondrial Content under Control of the Cell Cycle Checkpoint. Free Radic Biol. Med 2012, 53, 260–270. [DOI] [PubMed] [Google Scholar]
- (67).Gurer-Orhan H; Ince E; Konyar D; Saso L; Suzen S The Role of Oxidative Stress Modulators in Breast Cancer. Curr. Med. Chem 2018, 25, 4084–4101. [DOI] [PubMed] [Google Scholar]
- (68).Monslow J; Govindaraju P; Pure E Hyaluronan - a Functional and Structural Sweet Spot in the Tissue Microenvironment. Front. Immunol 2015, 6. DOI: 10.3389/fimmu.2015.00231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Scott JE Secondary Structures in Hyaluronan Solutions - Chemical and Biological Implications. Ciba Foundation Symposia 1989, 143, 6–20. [DOI] [PubMed] [Google Scholar]
- (70).Liu D; Mori A; Huang L Role of Liposome Size and Res Blockade in Controlling Biodistribution and Tumor Uptake of Gm1-Containing Liposomes. Biochim. Biophys. Acta 1992, 1104, 95–101. [DOI] [PubMed] [Google Scholar]
- (71).Moehlen M Emerging Treatment Strategies for Patients with Primary Biliary Cholangitis. Gastroenterol Hepatol (N Y) 2021, 17, 598–601. [PMC free article] [PubMed] [Google Scholar]
- (72).Cao G; Savani RC; Fehrenbach M; Lyons C; Zhang L; Coukos G; Delisser HM Involvement of Endothelial Cd44 During in Vivo Angiogenesis. Am. J. Pathol 2006, 169, 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Feng MY; Jiang W; Kim BYS; Zhang CC; Fu YX; Weissman IL Phagocytosis Checkpoints as New Targets for Cancer Immunotherapy. Nature Reviews Cancer 2019, 19, 568–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Helfinger V; Palfi K; Weigert A; Schroder K The Nadph Oxidase Nox4 Controls Macrophage Polarization in an Nf Kappa B-Dependent Manner. Oxid. Med. Cell. Longev 2019, 2019. DOI: 10.1155/2019/3264858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Bartelink H; Maingon; Poortmans P Whole-Breast Irradiation with or without a Boost for Patients Treated with Breast-Conserving Surgery for Early Breast Cancer: 20-Year Follow-up of a Randomised Phase 3 Trial. Lancet Oncology 2015, 16, 47–56. [DOI] [PubMed] [Google Scholar]
- (76).Canman CE; Lim DS; Cimprich KA; Taya Y; Tamai K; Sakaguchi K; Appella E; Kastan MB; Siliciano JD Activation of the Atm Kinase by Ionizing Radiation and Phosphorylation of P53. Science 1998, 281, 1677–1679. [DOI] [PubMed] [Google Scholar]
- (77).El-Deiry WS The Role of P53 in Chemosensitivity and Radiosensitivity. Oncogene 2003, 22, 7486–7495. [DOI] [PubMed] [Google Scholar]
- (78).Barker HE; Paget JTE; Khan AA; Harrington KJ The Tumour Microenvironment after Radiotherapy: Mechanisms of Resistance and Recurrence. Nature Reviews Cancer 2015, 15, 409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Augsburger F; Filippova A; Rasti D; Seredenina T; Lam M; Maghzal G; Mahiout Z; Jansen-Durr P; Knaus UG; Doroshow J; Stocker R; Krause KH; Jaquet V Pharmacological Characterization of the Seven Human Nox Isoforms and Their Inhibitors. Redox Biol. 2019, 26. DOI: 10.1016/j.redox.2019.101272 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.