

Abstract
Purpose of Review:
In this article, we summarize the current literature supporting metabolic and redox signaling pathways as important mechanisms underlying T cell activation in the context of hypertension.
Recent findings:
T cell immunometabolism undergoes dramatic remodeling in order to meet the demands of T cell activation, differentiation, and proliferation. Recent evidence demonstrates that the T cell oxidation-reduction (redox) system also undergoes significant changes upon activation, which can itself modulate metabolic processes and T cell function. Dysregulation of these signaling pathways can lead to aberrant T cell activation and inappropriate ROS production, both of which are linked to pathological conditions like hypertension.
Summary:
While the contribution of T cells to the progression of hypertension has been thoroughly investigated, how T cell metabolism and redox signaling changes, both separately and together, is an area of study that remains largely untouched. This review presents evidence from our own laboratory as well as others to highlight the importance of these two mechanisms in the study of hypertension.
Keywords: hypertension, immunometabolism, redox signaling, T cells, immune cells
Introduction
With the most recent update from the American College of Cardiology/American Heart Association now defining Stage 1 high blood pressure as having a systolic ≥130 mmHg over a diastolic ≥80 mmHg, the prevalence of hypertension among adults in the United States has jumped substantially to 46% [1]. Hypertension is the primary modifiable risk factor for the development of cardiovascular, renal, and cerebrovascular disease, making it the leading cause for mortality worldwide [2–4]. Despite advancements in research and available therapeutics, only 26% of hypertensive patients taking current standard-of-care medication achieve blood pressure control to recommended levels [5], increasing their risk of kidney, heart and vascular diseases [1, 6, 7]. Therefore, there is considerable need to understand the complex mechanisms underlying this significant human health crisis. While the study of the immunological component of hypertension, specifically T cells, has been a highly active area of investigation for the last 15 years, this review will focus exclusively on changes to immunometabolism and redox signaling as drivers of T cell function.
The History of T cells in Hypertension
The important contribution of T cells to the pathology of hypertension dates back to the 1970s, where thymus grafting into nude mice restored DOCA-salt-induced chronic hypertension and vascular disease similarly seen in wild-type mice [8]. This initial observation was followed by a 1980 study where normotensive recipient rats intravenously received splenic cells from hypertensive donors (either DOCA-salt or renal hypertensive) and developed elevated arterial hypertension compared to recipients receiving splenic cells from normotensive donors [9]. The field reemerged with the work by Rodríguez-Iturbe et al in 2002 where treatment with the immunosuppressant mycophenolate mofetil (MMF) normalized systemic hypertension in genetically hypertensive rats [10]. Interest in the role of T cells in hypertension was further rejuvenated in 2007 by Guzik et al, demonstrating that knockout of recombination-activating gene 1 (Rag1−/−) and the lack of T and B cells protected mice from the full development of angiotensin II- and DOCA-salt-induced hypertension [11]. More importantly, adoptive transfer of T cells restored the development of hypertension in Rag1−/− mice. This seminal paper ignited the entire field to explore the role of immunity and T cell signaling over the last 15 years [12–19].
Our laboratory was the first to elucidate the immunological component of salt-sensitive hypertension utilizing the Dahl Salt-Sensitive (SS) rat model. Salt-sensitive hypertension is a form of hypertension that is accompanied by particularly high morbidity and mortality [20, 21], and salt-sensitive hypertensive subjects have a 3-fold higher risk of developing cardiovascular diseases. The Dahl SS rat develops elevated blood pressure and renal end organ damage in response to a high salt diet [22–24], which importantly parallels the disease conditions observed in salt-sensitive hypertensive humans [25–29]. Treatment of SS rats with the immunosuppressive agent MMF reduced high salt-induced blood pressure and albuminuria, indicating the important role of the immune system in conferring salt-sensitivity [30]. It was observed that increasing dietary salt intake promoted T cell infiltration into the kidneys of SS rats, particularly around blood vessels in the renal cortex and vascular bundles in the renal medulla [31]. The lack of functional T and B cells via the genetic deletion of the Rag1 gene on the Dahl SS background resulted in blunted blood pressure, renal injury, and renal immune cell infiltration [32]. The specific contribution of T cells was demonstrated through deletion of the CD247 gene (encodes for the CD3 ζ chain of the T cell receptor) in the SS rat, which ablated CD3+ T cells, resulting in significantly reduced blood pressure and albuminuria in response to high salt challenge [33]. This provided evidence that functional T cells are specifically required for the full development of salt-sensitive hypertension. More recently, our laboratory has solidified the role of T cells in this salt-induced hypertensive response via adoptive transfer of wild-type SS splenocytes back into T cell-deficient SSCD247−/− rats [34], where the specific reconstitution and enrichment of CD4+ T helper cells led to the exacerbation of salt-sensitive hypertension and renal damage compared to control SSCD247−/− rats receiving PBS. This was the first demonstration of its kind in the Dahl SS rat.
Therefore, it is well-established that T cells are critical to the development of hypertension, but what remains largely unknown are the specific signaling mechanisms that occur within these infiltrating T cells that promote disease. Additional recent work from our laboratory indicates that T cell immunometabolic and redox signaling mechanisms may be critical for the full development of salt-sensitive hypertension [35, 36]. Metabolic pathways and redox reactions are at the core of all biological processes and the control of T cell activation and function via these two mechanisms will be the focus of this brief review.
Immunometabolism in T cells
Immunometabolism is defined as the intracellular metabolic pathways that determine the fate and function of immune cells. Technological advances in our ability to precisely define changes in these processes via highly sensitive transcriptomic and metabolomic approaches have reinvigorated the study of metabolism by immunologists, giving rise to this field over the last decade. The study of immunometabolism specifically in T cells has been wonderfully and extensively reviewed [37, 38]. T cells require highly adaptive metabolic processes in order to maintain the continuous cycling between quiescent and activated states. Depending on the state, T cells rely primarily on two main metabolic processes to generate energy: oxidative phosphorylation (OXPHOS) and glycolysis. In the quiescent state, mature naïve T cells utilize OXPHOS for the generation of ATP [39] and critically depend upon the cytokine IL-7 for glucose uptake and the homeostatic proliferation, maintenance, and survival of naïve T cells [40]. Upon activation via T cell receptor ligation and costimulation, T cells undergo significant metabolic reprogramming characterized by aerobic glycolysis to support anabolic processes. Despite the oxygen-rich environment and less efficient ATP production compared to OXPHOS, aerobic glycolysis importantly produces critical intermediates required for nucleotide, amino acid, and reducing metabolite biosynthesis [41]. This T cell metabolic reprogramming and energy switch is central to the biosynthesis, cell growth, differentiation, and proliferation necessary to carry out its effector functions. Once the stimulus is removed and pathogen cleared, a small subset of these activated T cells will survive and shift back to a quiescent memory state, once again relying heavily on OXPHOS metabolism. However, memory T cells are metabolically reprogrammed with greater mitochondrial mass and substantially more spare respiratory capacity compared to naïve T cells to allow for their rapid recall and response to a reintroduced pathogen [42].
Exploration down to this level of T cell metabolism and activation in the context of disease is largely lacking in the field of hypertension. We have systematically investigated, at the transcriptomic level, how the T cell specifically changes in response to diet, salt, and location within a whole animal model of salt-sensitive hypertension. SS rats maintained on a whole grain, plant-based diet were considerably protected from the development of hypertension and renal damage compared to SS rats maintained on a casein, animal protein-based diet, despite being challenged to same high salt diet [43]. This interestingly corresponded with lesser immune cell infiltration into the kidneys of the rats fed the grain-based diet, and given our specific interest in T cells [31–33], we sought to transcriptionally phenotype T cells from both the periphery and kidney utilizing an RNAseq approach [35]. Our first major observation was the clear and remarkable activation of genes when comparing T cells from the periphery versus those infiltrating the kidney. Independent of diet or salt, over 50% of all annotated genes were found to be significantly differentially expressed between blood and kidney, indicative of a metabolic switch or ‘turning on’ from a quiescent to activated state upon recruitment to the site of injury. Second, the renal T cells isolated from the disease-prone SS rats fed the casein, animal-based diet were characterized by changes in genes associated with a distinctive proinflammatory phenotype (Figure 1). Ingenuity and KEGG Pathway Analysis revealed an overrepresentation of canonical pathways specifically related to T cell receptor signaling, T helper cell signaling, and the Th2 pathway. While this would seem expected, when looking at the significant pathways in the protected grain-fed SS rats, we did not simply observe an opposing downregulation of inflammatory genes. Instead, renal T cells isolated from the protected SS rats fed the grain, plant-based diet revealed a metabolically suppressed profile in response to high salt, with significant downregulation of pathways related to nucleotide, amino acid, and protein metabolism (Figure 1). While these results are of course only reflective of T cell metabolism at the transcript level, it is clear that gene activation in T cells is separate and distinct between these two hypertensive phenotypes, and whether these changes are cause or consequence of the disease remains to be elucidated. Determination of how these alterations to the T cell transcriptome translate to changes in metabolites, metabolic activity, and ultimately effector function of T cells is a current area of active investigation in our laboratory.
Figure 1.

Kidney T cell transcriptome analysis of differentially expressed genes via Ingenuity Pathway Analysis (IPA) or Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis showed enrichment of pro-inflammatory pathways related to T cell signaling in disease-prone SS rats fed an animal-based diet. Conversely, kidney T cell derived from disease-protected SS rats fed a plant-based diet revealed enrichment of gene pathways primarily related to metabolic processes. Figure redrawn with permission from Abais-Battad et al [35].
Redox Signaling in T cells
The generation of reactive oxygen species (ROS) in cells were once considered to be a purely detrimental occurrence, since free radicals are known to induce irreversible damage to DNA, proteins, and lipids. However, it is now widely accepted and demonstrated that controlled levels of ROS have very specific roles in normal cellular signaling, whereas uncontrolled ROS production or ‘oxidative stress’ is correlated with a number of pathological conditions, including hypertension [44]. While studies examining the contribution of endogenously produced ROS in T cells are relatively new [45], its homeostatic role in T cell activation, metabolism, and function is clear. In addition to ROS being unavoidably generated from OXPHOS, another major source of ROS in T cells comes from NADPH oxidase (NOX), with T cells being shown to express NOX2 [46, 47]. Jackson et al provided early evidence that phagocytic NOX2 is activated upon T cell receptor (TCR) stimulation, and the generation of this appropriate, low-level ROS in T cells from both NOX activity as well as mitochondrial OXPHOS enhances, sustains, and regulates TCR signaling [47, 48] to promote the metabolic rewiring necessary for the controlled switch between the quiescent and effector states. If left unchecked by antioxidant systems, continued and excessive ROS production results in defective metabolic rewiring, aberrant T cell function and response [49, 50], and ultimately pathological conditions. While studies utilizing a systemic knock-out approach of various NOX2 subunits (p47phox, gp91phox) certainly demonstrated that NADPH oxidase-derived ROS is critical for T cell polarization and skewing the Th response [47, 51], a major issue is the inability to dissect whether these results are direct effects of NOX2 loss within T cells or are global, non-T cell effects due to loss of NOX2 throughout the entire host.
It is well-documented that the inappropriate production of ROS is associated with the development of hypertension and cardiovascular disease [44, 52], and in the Dahl SS rat, increased production of superoxide via NADPH oxidase in the renal medulla is linked to the progression of salt-sensitive hypertension and renal damage [53, 54]. While the null mutation of the p67phox gene on the Dahl SS background (SSp67phox−/−) was the first evidence that ROS specifically derived from the NOX2 isoform of NADPH oxidase is critical for the development of salt-sensitive hypertension [55], these studies are again limited by the use of a systemic p67phox knockout. With the infiltration of immune cells into target organs considered to be central to the development of hypertension [22] and with NOX2 known to be highly expressed on immune cells [56], it is conceivable that these immune cells serve as a critical source of ROS contributing to disease progression. Therefore, our laboratory first began the investigation of NOX2 in immune cells by determining whether infiltrating T cells isolated from the SS rat kidneys express NOX2 subunits. Indeed, these renal T cells highly express p67phox, gp91phox, p22phox, and p47phox, especially upon exposure to high salt, indicating the presence of the molecular machinery necessary for NOX2-derived ROS production [46]. To importantly dissociate the contribution of ROS from immune cells versus the renal parenchyma, we utilized a total body irradiation/bone marrow transfer approach to transfer functional, ROS-producing SS wild-type bone marrow cells into irradiated SSp67phox−/− recipients to generate a chimeric rat with the ability to produce NOX2-derived ROS only in cells of the hematopoietic lineage [36]. This technique addresses the major issue of dissecting the effect of global versus immune cell NOX2 function. We discovered that the production of NOX2-derived ROS solely in cells of hematopoietic origin (immune cells), but absent in the remainder of the host, was sufficient to drive and exacerbate hypertension and renal damage, highlighting the fundamental role of immune cell ROS in the pathogenesis of salt-sensitivity (Figure 2). While the further determination of the specific immune cell type remains the current focus of our investigation, these results demonstrate the importance of the exact cellular localization and source of ROS production, which becomes critical when developing effective antioxidant therapies.
Figure 2.
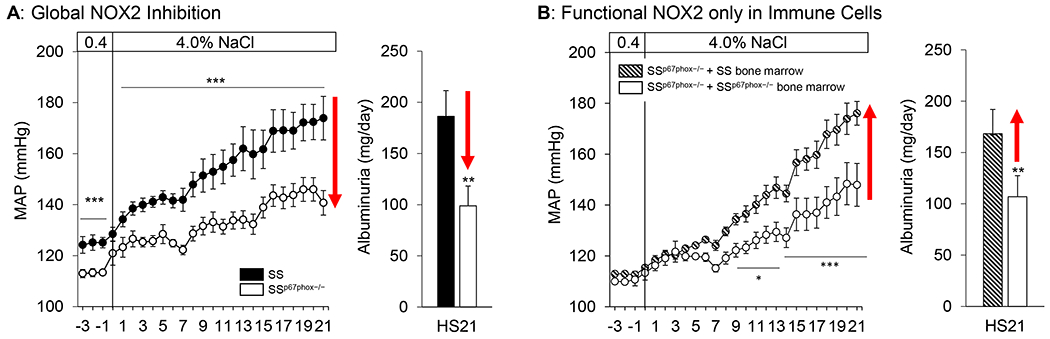
Global genetic deletion of the p67phox subunit of NOX2 attenuates salt-induced hypertension and renal damage in SSp67phox−/− rats (A). To delineate the contribution of free radical production from immune cells versus that of parenchymal cells, we generated a chimeric rat where SSp67phox−/− rats received SS bone marrow and p67phox would only be expressed in cells of hematopoietic origin (immune cells). Shown in (B), intact p67phox and functional NOX2 in immune cells was sufficient to restore hypertension and renal damage to SSp67phox−/− rats lacking p67phox in the parenchyma. *p<0.05, **p<0.005, ***p<0.001 SSp67phox−/− vs SS. Data redrawn with permission from Abais-Battad et al [36]. Reprinted from Free Radical Biology and Medicine, Vol. 146, Justine M. Abais-Battad, Hayley Lund, John Henry Dasinger, Daniel J. Fehrenbach, Allen W. Cowley, and David L. Mattson, NOX2-derived reactive oxygen species in immune cells exacerbates salt-sensitive hypertension, 333-339, 2020, with permission from Elsevier
Therapeutic Implications of Immunometabolic Modulators and Antioxidants
The extensive beneficial effects of a plant-based diet to human health and disease are recognized not only within the scientific community but the general public as well. What is less recognized are the specific plant components and underlying mechanisms driving this protection in humans. A few clinical studies have attempted to address the effect of plant-based or Mediterranean diets on immunity and T cell function, but have provided contradictory results. Adults at high risk of cardiovascular disease saw a significant anti-inflammatory effect with a 5-year Mediterranean diet intervention, to include a reduction in adhesion molecule expression on circulating T cells [27440261]. In other studies, a vegetarian or vegan diet had no effect on number or function of circulating T cells, specifically [2072829, 32147197]. One major limitation among these human studies is the use of circulating peripheral blood mononuclear cells to assess T cell function; these cells may not accurately reflect the inflammatory milieu that exists in target organs, which may account for the discrepancy in results between these studies. As demonstrated in our work, circulating T cells display a drastically different transcriptional profile compared to T cells found in the kidney, highlighting the importance of cellular localization when performing these analyses on immune cell function and metabolism.
In terms of targeting redox signaling, the use of antioxidants for treatment have held great promise in numerous pre-clinical animal models; however, there have been underwhelming effects of antioxidants in human trials [22419320, 18474268] due to short trial duration and poor antioxidant bioavailability [19694341, 30121293]. The studies discussed in this review again demonstrate the importance of cellular localization as well as source of ROS production, which becomes critical when determining targets for potential therapeutics. In light of this work, perhaps taking a more targeted approach to deliver high enough concentrations of antioxidants to the cells specifically responsible for producing deleterious ROS would lead to more efficacious therapies and ultimately better outcomes in humans. While no such therapies currently exist to specifically target T cell metabolism or ROS production, this is a future area of drug development with great potential.
Conclusions
In this review, we have briefly summarized the recent evidence regarding the role of metabolic and redox signaling in the determination of T cell function, and its implications for pathological conditions like hypertension, specifically salt-sensitivity. However, the reality is that immunometabolism and redox mechanisms are highly interdependent and tightly interwoven with one another, where metabolic reprogramming can alter ROS and vice versa, ROS can affect immune cell function via direct effects on metabolic enzymes and metabolites. In order to understand the complexities that occur during pathological conditions, thorough investigation of these redox-metabolism interactions in the regulation of immune function remains a critical area of future research.
Funding
The authors’ work is supported by National Institutes of Health grants HL137748 and HL116264, American Heart Association grant 19CDA34660184, and the Georgia Research Alliance.
Conflict of Interest
David L. Mattson reports grants from National Institutes of Health, grants from American Heart Association, other from Georgia Research Alliance, during the conduct of the study.
Justine M. Abais-Battad reports grants from American Heart Association, grants from National Institute of Health, during the conduct of the study.
Footnotes
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Whelton PK, Carey RM, Aronow WS, Casey DE Jr., Collins KJ, Dennison Himmelfarb C et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):e13–e115. doi: 10.1161/HYP.0000000000000065. [DOI] [PubMed] [Google Scholar]
- 2.Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365(9455):217–23. doi: 10.1016/S0140-6736(05)17741-1. [DOI] [PubMed] [Google Scholar]
- 3.Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2224–60. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poulter NR, Prabhakaran D, Caulfield M. Hypertension. Lancet. 2015;386(9995):801–12. doi: 10.1016/S0140-6736(14)61468-9. [DOI] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention (CDC). Hypertension Cascade: Hypertension Prevalence, Treatment and Control Estimates Among US Adults Aged 18 Years and Older Applying the Criteria From the American College of Cardiology and American Heart Association’s 2017 Hypertension Guideline—NHANES 2015–2018. Atlanta, GA: US Department of Health and Human Services; 2021. [Google Scholar]
- 6.Muntner P, Carey RM, Gidding S, Jones DW, Taler SJ, Wright JT, Jr. et al. Potential US Population Impact of the 2017 ACC/AHA High Blood Pressure Guideline. Circulation. 2018;137(2):109–18. doi: 10.1161/CIRCULATIONAHA.117.032582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Primatesta P, Brookes M, Poulter NR. Improved hypertension management and control: results from the health survey for England 1998. Hypertension. 2001;38(4):827–32. [PubMed] [Google Scholar]
- 8.Svendsen UG. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta Pathol Microbiol Scand A. 1976;84(6):523–8. doi: 10.1111/j.1699-0463.1976.tb00150.x. [DOI] [PubMed] [Google Scholar]
- 9.Olsen F Transfer of arterial hypertension by splenic cells from DOCA-salt hypertensive and renal hypertensive rats to normotensive recipients. Acta Pathol Microbiol Scand C. 1980;88(1):1–5. doi: 10.1111/j.1699-0463.1980.tb00065.x. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez-Iturbe B, Quiroz Y, Nava M, Bonet L, Chavez M, Herrera-Acosta J et al. Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol Renal Physiol. 2002;282(2):F191–201. doi: 10.1152/ajprenal.0197.2001. [DOI] [PubMed] [Google Scholar]
- 11.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204(10):2449–60. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55(2):500–7. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF et al. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension. 2011;57(3):469–76. doi: 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- 14.Tipton AJ, Baban B, Sullivan JC. Female spontaneously hypertensive rats have greater renal anti-inflammatory T lymphocyte infiltration than males. Am J Physiol Regul Integr Comp Physiol. 2012;303(4):R359–67. doi: 10.1152/ajpregu.00246.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji H, Zheng W, Li X, Liu J, Wu X, Zhang MA et al. Sex-specific T-cell regulation of angiotensin II-dependent hypertension. Hypertension. 2014;64(3):573–82. doi: 10.1161/HYPERTENSIONAHA.114.03663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pollow DP, Uhrlaub J, Romero-Aleshire M, Sandberg K, Nikolich-Zugich J, Brooks HL et al. Sex differences in T-lymphocyte tissue infiltration and development of angiotensin II hypertension. Hypertension. 2014;64(2):384–90. doi: 10.1161/HYPERTENSIONAHA.114.03581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caillon A, Mian MOR, Fraulob-Aquino JC, Huo KG, Barhoumi T, Ouerd S et al. gammadelta T Cells Mediate Angiotensin II-Induced Hypertension and Vascular Injury. Circulation. 2017;135(22):2155–62. doi: 10.1161/CIRCULATIONAHA.116.027058. [DOI] [PubMed] [Google Scholar]
- 18.Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. 2017;551(7682):585–9. doi: 10.1038/nature24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drummond GR, Vinh A, Guzik TJ, Sobey CG. Immune mechanisms of hypertension. Nat Rev Immunol. 2019;19(8):517–32. doi: 10.1038/s41577-019-0160-5. [DOI] [PubMed] [Google Scholar]
- 20.Morimoto A, Uzu T, Fujii T, Nishimura M, Kuroda S, Nakamura S et al. Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet. 1997;350(9093):1734–7. doi: 10.1016/S0140-6736(97)05189-1. [DOI] [PubMed] [Google Scholar]
- 21.Weinberger MH, Fineberg NS, Fineberg SE, Weinberger M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension. 2001;37(2 Pt 2):429–32. doi: 10.1161/01.hyp.37.2.429. [DOI] [PubMed] [Google Scholar]
- 22.Mattson DL. Immune mechanisms of salt-sensitive hypertension and renal end-organ damage. Nat Rev Nephrol. 2019;15(5):290–300. doi: 10.1038/s41581-019-0121-z. [DOI] [PubMed] [Google Scholar]
- 23.Mattson DL. Infiltrating immune cells in the kidney in salt-sensitive hypertension and renal injury. Am J Physiol Renal Physiol. 2014;307(5):F499–508. doi: 10.1152/ajprenal.00258.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mattson DL, Dasinger JH, Abais-Battad JM. Amplification of Salt-Sensitive Hypertension and Kidney Damage by Immune Mechanisms. Am J Hypertens. 2021;34(1):3–14. doi: 10.1093/ajh/hpaa124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cowley AW Jr., Roman RJ. The role of the kidney in hypertension. JAMA. 1996;275(20):1581–9. [PubMed] [Google Scholar]
- 26.Bigazzi R, Bianchi S, Baldari D, Sgherri G, Baldari G, Campese VM. Microalbuminuria in salt-sensitive patients. A marker for renal and cardiovascular risk factors. Hypertension. 1994;23(2):195–9. [DOI] [PubMed] [Google Scholar]
- 27.Aviv A, Hollenberg NK, Weder A. Urinary potassium excretion and sodium sensitivity in blacks. Hypertension. 2004;43(4):707–13. doi: 10.1161/01.HYP.0000120155.48024.6f. [DOI] [PubMed] [Google Scholar]
- 28.Sullivan JM, Prewitt RL, Ratts TE. Sodium sensitivity in normotensive and borderline hypertensive humans. Am J Med Sci. 1988;295(4):370–7. doi: 10.1097/00000441-198804000-00025. [DOI] [PubMed] [Google Scholar]
- 29.Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension. 1996;27(3 Pt 2):481–90. [DOI] [PubMed] [Google Scholar]
- 30.Mattson DL, James L, Berdan EA, Meister CJ. Immune suppression attenuates hypertension and renal disease in the Dahl salt-sensitive rat. Hypertension. 2006;48(1):149–56. doi: 10.1161/01.HYP.0000228320.23697.29. [DOI] [PubMed] [Google Scholar]
- 31.De Miguel C, Das S, Lund H, Mattson DL. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2010;298(4):R1136–42. doi: 10.1152/ajpregu.00298.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol. 2013;304(6):R407–14. doi: 10.1152/ajpregu.00304.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rudemiller N, Lund H, Jacob HJ, Geurts AM, Mattson DL, PhysGen Knockout P. CD247 modulates blood pressure by altering T-lymphocyte infiltration in the kidney. Hypertension. 2014;63(3):559–64. doi: 10.1161/HYPERTENSIONAHA.113.02191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fehrenbach DJ, Dasinger JH, Lund H, Zemaj J, Mattson DL. Splenocyte transfer exacerbates salt-sensitive hypertension in rats. Exp Physiol. 2020;105(5):864–75. doi: 10.1113/EP088340. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This work demonstrates that adoptive transfer of splenocytes back into CD247−/− SS rats lacking T cells restores salt-sensitive hypertension and renal damage.
- 35.Abais-Battad JM, Alsheikh AJ, Pan X, Fehrenbach DJ, Dasinger JH, Lund H et al. Dietary Effects on Dahl Salt-Sensitive Hypertension, Renal Damage, and the T Lymphocyte Transcriptome. Hypertension. 2019;74(4):854–63. doi: 10.1161/HYPERTENSIONAHA.119.12927. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This study shows how dietary factors can influence the T cell transcriptome, characterized by either a pro-inflammatory or metabolically-suppressed state, which associates with susceptibility to salt-sensitive hypertension.
- 36.Abais-Battad JM, Lund H, Dasinger JH, Fehrenbach DJ, Cowley AW Jr., Mattson DL. NOX2-derived reactive oxygen species in immune cells exacerbates salt-sensitive hypertension. Free Radic Biol Med. 2020;146:333–9. doi: 10.1016/j.freeradbiomed.2019.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This study demonstrates that free radical production derived from NOX2 in immune cells promotes salt-sensitive hypertension.
- 37.Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212(9):1345–60. doi: 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moshfegh CM, Case AJ. The Redox-Metabolic Couple of T Lymphocytes: Potential Consequences for Hypertension. Antioxid Redox Signal. 2021;34(12):915–35. doi: 10.1089/ars.2020.8042. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This review extensively dissects the coupling of redox and metabolic effects on T cells in the regulation of adaptive immunity.
- 39.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5(11):844–52. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- 40.Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI et al. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci U S A. 2001;98(15):8732–7. doi: 10.1073/pnas.161126098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36(1):68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geurts AM, Mattson DL, Liu P, Cabacungan E, Skelton MM, Kurth TM et al. Maternal diet during gestation and lactation modifies the severity of salt-induced hypertension and renal injury in Dahl salt-sensitive rats. Hypertension. 2015;65(2):447–55. doi: 10.1161/HYPERTENSIONAHA.114.04179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cifuentes ME, Pagano PJ. Targeting reactive oxygen species in hypertension. Curr Opin Nephrol Hypertens. 2006;15(2):179–86. doi: 10.1097/01.mnh.0000214776.19233.68. [DOI] [PubMed] [Google Scholar]
- 45.Muri J, Kopf M. Redox regulation of immunometabolism. Nat Rev Immunol. 2021;21(6):363–81. doi: 10.1038/s41577-020-00478-8. [DOI] [PubMed] [Google Scholar]; ** This review summarizes the role of ROS and antioxidant systems in the control of T cell proliferation, survival, and function.
- 46.De Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol. 2011;300(3):F734–42. doi: 10.1152/ajprenal.00454.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol. 2004;5(8):818–27. doi: 10.1038/ni1096. [DOI] [PubMed] [Google Scholar]
- 48.Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38(2):225–36. doi: 10.1016/j.immuni.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity. 2017;46(4):675–89. doi: 10.1016/j.immuni.2017.03.019. [DOI] [PubMed] [Google Scholar]
- 50.Case AJ, McGill JL, Tygrett LT, Shirasawa T, Spitz DR, Waldschmidt TJ et al. Elevated mitochondrial superoxide disrupts normal T cell development, impairing adaptive immune responses to an influenza challenge. Free Radic Biol Med. 2011;50(3):448–58. doi: 10.1016/j.freeradbiomed.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tse HM, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD et al. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J Immunol. 2010;185(9):5247–58. doi: 10.4049/jimmunol.1001472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Araujo M, Wilcox CS. Oxidative stress in hypertension: role of the kidney. Antioxid Redox Signal. 2014;20(1):74–101. doi: 10.1089/ars.2013.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Makino A, Skelton MM, Zou AP, Roman RJ, Cowley AW Jr. Increased renal medullary oxidative stress produces hypertension. Hypertension. 2002;39(2 Pt 2):667–72. doi: 10.1161/hy0202.103469. [DOI] [PubMed] [Google Scholar]
- 54.Taylor NE, Glocka P, Liang M, Cowley AW Jr. NADPH oxidase in the renal medulla causes oxidative stress and contributes to salt-sensitive hypertension in Dahl S rats. Hypertension. 2006;47(4):692–8. doi: 10.1161/01.HYP.0000203161.02046.8d. [DOI] [PubMed] [Google Scholar]
- 55.Feng D, Yang C, Geurts AM, Kurth T, Liang M, Lazar J et al. Increased expression of NAD(P)H oxidase subunit p67(phox) in the renal medulla contributes to excess oxidative stress and salt-sensitive hypertension. Cell Metab. 2012;15(2):201–8. doi: 10.1016/j.cmet.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]