

Abstract
Since its discovery in the 1960 s, doxorubicin (DOX) has constantly elicited the broadest spectrum of cancerocidal activity against human cancers. However, cardiotoxicity caused by DOX directly as well as its metabolites is a great source of concern over the continuous use of DOX in chemotherapy. While the exact mechanism of DOX-induced cardiotoxicity is yet to be completely understood, recent studies indicate oxidative stress, inflammation, and several forms of cell death as key pathogenic mechanisms that underpin the etiology of doxorubicin-induced cardiotoxicity (DIC). Notably, these key mechanistic events are believed to be negatively regulated by 3,4-dihydroxybenzoic acid or protocatechuic acid (PCA)—a plant-based phytochemical with proven anti-oxidant, anti-inflammatory, and anti-apoptotic properties. Here, we review the experimental findings detailing the potential ameliorative effects of PCA under exposure to DOX. We also discuss molecular insights into the pathophysiology of DIC, highlighting the potential intervention points where the use of PCA as a veritable chemoprotective agent may ameliorate DOX-induced cardiotoxicities as well as toxicities due to other anticancer drugs like cisplatin. While we acknowledge that controlled oral administration of PCA during chemotherapy may be insufficient to eliminate all toxicities due to DOX treatment, we propose that the ability of PCA to block oxidative stress, attenuate inflammation, and abrogate several forms of cardiomyocyte cell death underlines its great promise in the amelioration of DIC.
Keywords: Doxorubicin (DOX), DOX metabolism, Doxorubicin-induced cardiotoxicity (DIC), Protocatechuic acid (PCA), Chemoprotective agent
Graphical Abstract
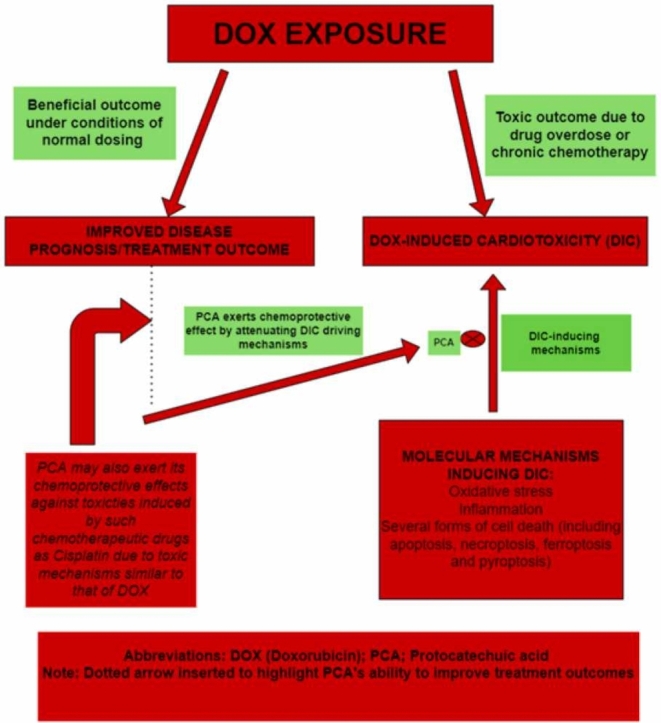
Highlights
-
•
Doxorubicin is a popular, broad-spectrum antineoplastic drug that has been in use for over 60 years.
-
•
DOX-induced cardiotoxicity (DIC) has threatened its continued use in chemotherapy.
-
•
Identified cardio-toxic mechanisms include inflammation, oxidative stress, and several forms of cell death (apoptosis, necroptosis, pyroptosis, and ferroptosis).
-
•
Protocatechuic acid (PCA)—an anthocyanin-derived phenolic compound, has been shown to negatively regulate these cardiotoxic processes (chemoprotection), hence its promise in the amelioration of DIC.
-
•
PCA may also exert chemoprotective effects against toxicities of other anticancer drugs such as cisplatin, for which experimental evidence has indicated oxidative stress and inflammation as key drivers.
1. Introduction
Cardiotoxicity due to doxorubicin (DOX) remains the most important public health concern questioning its continuous use in cancer chemotherapy. While markers of cardiotoxic injuries have been reported with the use of DOX alone, a much higher magnitude of cardiac damage, over 13 times more, has been observed following the metabolism of DOX into the highly toxic metabolite, doxorubicinol (DOXol) [1]. Aside from the adverse effects associated with DOXol, there is also the challenge of poor selectivity of the drug. While the use of nanoparticles—principally liposomal DOX (LD), and Pegylated DOX (PGD), PGD have improved selectivity, they are insufficient to completely prevent cardiac toxicity [2], [3], [4].
Although the exact mechanism of doxorubicin-induced cardiotoxicity (DIC) is far from being resolved, many studies have identified oxidative stress and several forms of cardiomyocyte cell death such as apoptosis, pyroptosis, necroptosis, and ferroptosis as key molecular drivers for the initiation and stabilization of DIC [5], [6], [7], [8], [9], [10], [11]. Stimulation of pro-oxidant mechanisms and inhibition of antioxidant enzyme activity have been recently identified as the fundamental molecular mechanisms underlying DIC [6], [8], [9], [12]. Moreover, once accumulated in the myocardium, DOX begins to activate cell-death pathways such as ferroptosis, necroptosis, and apoptosis which manifest in the clinical cardiotoxicity [8], [11].
Collectively, the non-selectivity of DOX, in addition to the associated oxidative stress and cell death in host cells following DOX exposure, elicits several layers of cellular perturbations, which ultimately results in the initiation of DIC while also lowering the therapeutic index of the drug and increasing morbidity and mortality. Given these challenges and the concomitant adverse effects associated with the use of the drug, serious consideration is currently being given to co-administration with potent chemoprotective agents such as PCA during chemotherapy to not only improve treatment outcomes, but also mitigate developing toxicities as well as prevent the onset of the life-threatening cardiomyopathy post-therapy [13], [14], [15], [16], [17].
The antioxidant and pharmacological activities of PCA have been extensively elucidated both in vivo and in vitro. PCA has also been compared to its precursor molecule, protocatechuic aldehyde (PLA)—the parent anthocyanin compound from which PCA is directly generated [18], [19]. Findings not only demonstrate the antioxidant, anti-cell death, and other pharmacological properties of PCA in rats but these properties of PCA, when compared with those of PLA, were also found to be significantly higher [18], [19]. Interestingly, the pharmacological properties of PCA, including its metal-chelating properties, show that these DIC events are negatively regulated by PCA [19]. Thus, while further investigation is warranted to firmly establish the clinical safety and reliability of PCA in chemotherapy, its proven anti-oxidant, anti-inflammatory, anti-cell death, and overall safety index at a therapeutic dose of 50–150 mg/kg [19], [20], demonstrates that PCA can be used as an effective chemopreventive intervention for the abrogation of the key molecular events underpinning clinically detectable DIC.
2. Protocatechuic acid: an overview
2.1. A brief elucidation
PCA (3,4-dihydrobenzoic acid), as shown in Fig. 1, is a phenolic compound widely found in many edible fruits, vegetables, and medicinal plants, especially Hibiscus sabdariffa L. (Hs, roselle; Malvaceae) [21], [22]. The PCA powder, is gray in color, decolorizes in air, and has melting and boiling points of 221 and 410 degrees, respectively [16], [23]. Additionally, PCA is incompatible with strong oxidizing agents and bases and requires the utmost careful handling as it is irritable to the eyes, lungs, and skin. PCA is only sparingly dissolved in water (1:50) but highly soluble in alcohol and ether in addition to corn oil, which is principally used for PCA dissolution in the experimental research [16], [23].
Fig. 1.
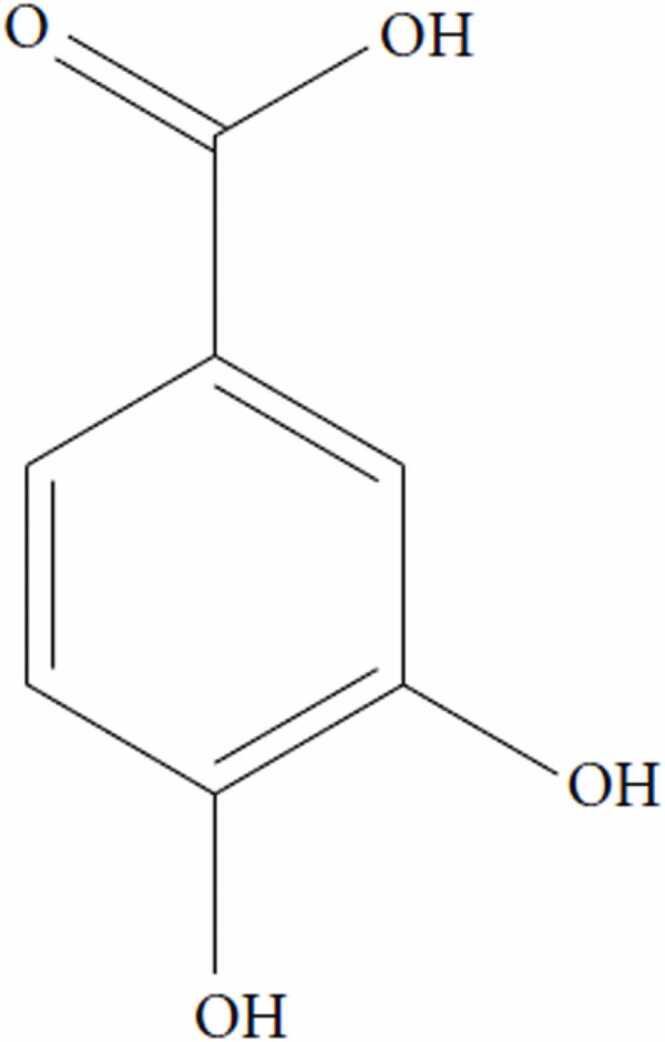
Chemical structure of protocatechuic acid (PCA).
In a 2012 investigation, involving the use of Liquid Chromatography with Tandem Mass Spectroscopy (LC-MS-MS), the pharmacokinetics activity of PCA was characterized in mice. Following an oral administration of 50 mg/kg in addition to reaching a plasma peak of 73.6 μM in 5 min, PCA was reported to be rapidly absorbed with a half-life of 2.9 min [24]. Moreover, the characterization of Hs. extracts show that in addition to the presence of PCA, Hs extracts also possess organic acids (hydroxycitric acid and hibiscus acid) and anthocyanins (delphinidin-3-sambubioside and cyanidin-3-sambubioside) as well as flavonoids, which are likely to be responsible for the anti-oxidant, anti-inflammatory, anti-nephrotoxic, hepatoprotective, anti-aging, p53-upregulating, anti-apoptotic, antibacterial, and antidiabetic properties that have been amply elucidated by several studies [21], [25], [26], [27], [28], [29]. Notably, the flavonoids and polyphenols in PCA, have been associated with conferring protection against brain diseases, ulcerative colitis, cancers, diabetes, and a host of other non-communicable diseases [30], [31], [32], [33], [34]. Given the central role of inflammation in tumor initiation as well as the role of oxidative stress and cell death in the initiation, maintenance, and exacerbation of DIC, the role of PCA may indeed expand beyond simply ameliorating cardiotoxicity, to include suppressing tumor progression during chemotherapy [33], [35], [36].
2.2. Comparing PCA with other chemoprotective agents
Extensive work has been done on the mechanism of action and potential applications of chemoprotective agents other than PCA [13], [37], [38], [39], [40], [41], [42], [43], [44], [45], [46], [47], [48], [49], [50], [51], [52], [53], [54], [55], [56]. Table 1 below provides a comparison of the chemoprotective effects of some agents with those of PCA.
Table 1.
A comparison of PCA with other chemoprotective agents.
| Major Biological Sources | Prominent Mechanisms of Action | Other Chemoprotective Agents | Major Biological Sources | Prominent Mechanisms of Action | |
|---|---|---|---|---|---|
| PCA | Actinidia arguta (Kiwi berry) | Antioxidant activity [57] | Curcumin | Tumeric | Suppresses tumor promotion via its inhibition of NF-kB, AP1 and Cox2, β − Catenin activities [13], [37] |
| Morus alba (mulberry leaves) | Antioxidant and immunomodulatory activities [58] | Capsaicin | Chilli Pepper | Anti-tumor activity via the blockage of PMA- TNF-α induced Ap1 activation in mouse skin and cultured human leukemia HL-60 cells [38] |
|
| Hibiscus sabdariffa (Roselle) | Nephroprotective activity [19]) | [6]-Gingerol | Ginger | Suppresses tumor progression via the inhibition of epidermal growth-induced Ap1 activation [59] | |
| Hibiscus sabdariffa | Anti-hypertensive, hepatoprotective and anti-inflammatory activities [19] | Resveratrol | Grapes | Anticancer and cardioprotective activity (French paradox) through the downregulation of the activity of the Mmp-9, Cox2, PMA-induced activation of Ap1 as well as NF-kB activity [39], [40], [41], [42] | |
| Ginkgo biloba leaves (ginkgo) | Antioxidant activity [19] | Indole-3-carbinol | Cabbage | Anti-tumor activity, principally via the suppression of β − Catenin activity [43] | |
| Prunus amygdalus (almond) | Antioxidant activity [19] | Epigallocatechin-3-gallate | Green tea | Exerts antioxidant activity via the transcriptional activation of phase II detoxifying gene expression, strongly activated all three MAPKs (ERK, JNK and p38) [44] | |
| Euterpe oleracea (Acai berry) | Anti-inflammatory, antioxidant, antiproliferative, neuroprotective and cardioprotective activities [19], [60] | Diallyl sulfide | Garlic | Induces carcinogen detoxification and antioxidant genes, via the abrogation of KEAP1 repression of NRF2 [45], [47], [48], [49], [50], [51], [52], [53], [54], [55], [56] |
2.3. Toxicity profile
Although there is a surfeit of experimental data regarding the beneficial roles of PCA, especially in regards to the attenuation of oxidative stress, inflammation, hepatotoxicity, nephrotoxicity, ulcer, aging, diabetes, and various forms of cell death, PCA has also been associated with adverse outcomes following overdose [19], [26], [28], [61], [62]. At an oral dose of 500 mg/kg, PCA has been reported to cause depletions in hepatic and renal GSH levels in mice, albeit, but no mortality [19], [62]. Also, The LD50 of PCA was found to be 800 mg/kg by i.p. route and 3.5 g/kg by i.v. route in mice [19]. Furthermore, sub-chronic mice exposure to 0.1% PCA in drinking water for a period of 60 days, cause slight but significant hepatotoxic and nephrotoxic effects in mice, underlined by enhanced activity of plasma alanine transferase and urea level, respectively. Nonetheless, a dose of at least 500 mg/kg PCA administration does not directly equate to the induction of toxicities. Kakkar and Bais, for example, also reported that aqueous extract of PCA in a daily dose of 500 mg/kg BW/day decreased the oxidative stress levels, prevented cellular degeneration and necrosis of the renal tissues, and also decreased serum urea and creatinine levels while significantly increasing GSH and SOD levels. Given this nuanced outcome with PCA administration at a dose of 500 mg/kg, available literature exhaustively supports the suggestion that overall safety with PCA administration—that is, significant non-toxic outcomes may only be guaranteed at a dose of between 50 and 150 mg/kg, for which no toxic outcomes have been reported till date [19], [20].
2.4. Traditional use of PCA-containing plants
Many plants including Hibiscus sabdariffa L. (Hs, roselle; Malvaceae), Eupenicillium parvum, Ceratonia silique (especially its Carob pod aqueous extract, CPAE), Boswellia dalzielii, Ginkgo biloba L. (ginkgo), Agaricusbisporus and Phellinus linteus (Mushrooms), have been studied for both their pharmacological and traditional importance [19]. However, the traditional use of Hibiscus sabdariffa (Roselle), is the most widely studied [21]. For example, the fresh and dried calyces of Hibiscus sabdariffa L. (Hs, roselle; Malvaceae) known as cHs, have been used traditionally in herbal drinks, as a food as well as in hot and cold beverages, fermented drinks, and in wine-making [21]. From its use in making ‘’Cacody tea’’ in Egypt[63] and a drink known as ‘’Zoborodo’’ in Nigeria and ‘’Karkade’’ in Sudan [64], the PCA-containing Hibiscus plant, has also been locally applied to meet traditional culinary needs. Interestingly, in Mexico, ‘’Zoborodo’’ and ‘’Karkade’’ are slightly modified in a process that involves the boiling of fleshy calyces with sugar to produce a new kind of drink called Jamaica or ‘‘agua de Jamaica’’ or ‘‘t é de Jamaica’’. Moreover, in Sudan, fresh or dried cHs calyces are eaten garnished with onions or groundnuts or eaten directly as vegetables in Malaysia [65].
2.5. Application in clinical trials
PCA has also been applied in clinical settings, although progress in this area is still largely minimal. Relinqing granules (RLQ) granules are used alone or in combination with other antibacterial agents in the treatment of urological disorders. In a clinical study involving 12 healthy patients to study the pharmacokinetics of RLQ in humans and the potential of RLQ-perpetrated interactions on transporters, investigators concluded that both garlic acid (GA) and protocatechuic acid (PCA) given their appreciable modulation of transporters (especially OAT1 transporter), could serve as pharmacokinetic (PK)-markers in RLQ-related pharmacokinetic and drug interaction studies [66]. Thus, by serving as a PK-marker for RLQ, PCA (and GA) via their modulatory activities on the organic anion transporter protein 1 (OAT1), can synergistically increase the assessment of RLQ in the brain to modulate the centers of the brain involved in controlling functionality and homeostasis within the urological system. Also, PCA has been used in clinical settings as a topical antimicrobial agent for the amelioration of surgical site infection—essentially for surgical skin antisepsis. Using the Kirby-Bauer method of disc diffusion to investigate the in vitro antimicrobial activity and comparative effectiveness of PCA and 7 related compounds against surgical site infection (SSI) pathogens, Jalali et al. concluded that PCA demonstrated laboratory efficacy against pathogens implicated in SSI, including drug-resistant organisms and thus may be a novel antisepsis agent in the mitigation of the health burdens associated with SSI and bacteria resistance [67].
3. Metabolism of doxorubicin: a summary
Doxorubicin metabolism, even though required for its eliciting of its antineoplastic effects, also generates such toxic metabolites as DOXol, doxorubicin deoxyaglycone, and doxorubicinol hydroxyaglycone. [12], [68]. Using high and low DOX:Protein ratio as a marker for measuring the efficiency of each metabolic route, low DOX:Protein ratio was shown to be associated with the generation of doxorubicin deoxyagylcone and doxorubicinol hydroxyagylcone. Additionally, the DOX:Protein ratios were also associated with acute cardiotoxicity, which relative to chronic toxicity, is mild [12]. Conversely, high DOX:Protein ratio, which is associated with the generation of DOXol (itself reported to be 13 times more toxic than the parent molecule, DOX), is responsible for the chronic, life-threatening cardiomyopathy, following the cessation of DOX chemotherapy [12]. Although cardiac dysfunction and electrophysiologic indices of oxidative stress were found to be attenuated following catalase overexpression and antioxidant administration in transgenic mice [69], [70], [71], [72], such protective effects were found to wane after chronic DOX administration [73]. Interestingly, the administration of iron-chelating agents such as bis-dioxi-piperazine dexrazoxane, rescued the myocardium from observed DOX-induced toxicities, conferring a significant amount of cardiac protection [73], [74]. Notably, PCA, given its widely reported metal (including iron)-chelating activities, can effectively sequester the surfeit of iron generated during DOX-induced ferroptosis [6], [11] as well as effectively abrogate DOXol-induced oxidative stress, caused by both acute and chronic toxicities [12]. The pathway for the generation of DOX metabolites is illustrated in Fig. 2.
Fig. 2.

Diagram of Doxorubicin metabolism and generated metabolites. Four major metabolites of Doxorubicin (DOX) metabolism, including doxorubicinol (DOXol), doxorubicin aglycone, doxorubicin deoxyglycone, and doxorubicin hydroxyaglycone. Although several reports have highlighted a role for DOX treatment in the induction of arrhythmogenic events, the mechanistic basis for this phenomenon or which DOX metabolite may be involved, is not clearly understood, hence the denotation ‘???’.
4. Potential chemoprotective properties of PCA under DOX treatment
Cancer chemoprevention is collectively defined as the use of natural, synthetic, biological, or chemical agents in the inhibition, blocking, suppression, or prevention of cancer progression to an invasive tumor or full-blown metastasis [75], [76], [77], [78], [79]. In line with this definition, chemopreventive agents are categorized as ‘blocking agents’, when their mechanism of action is to inhibit the initiation of tumorigenesis and as ‘suppressing agents’ when they function to abrogate tumor promotion/progression [75], [80]. Recent investigations identified oxidative stress and programmed cell death as key mechanisms underlying the DOX-induced acute cardiotoxicity [11]. Additionally, several studies have also found that inflammation is a strong factor predisposing cells to the development of cancer. Indeed, inflammation has been underscored as underpinning all the stages of tumorigenesis as well as DIC [81], [82], [83], [84]. Furthermore, inflammation, like carcinogens, UV light, and variable radiation, can induce mutations and/or epigenetic alterations in genes and signaling pathways involved in tumor suppression, principally in the Tp53 gene [81].
Moreover, evidence from chronic intestinal inflammation shows a steady accumulation of inflammation-induced mutations in the Tp53 gene and other cancer-suppressing genes in the intestinal epithelium [85], [86], [87], [88]. Additional finding along this line, also shows that the accumulation of inflammation-induced mutations alone, devoid of other extrinsic carcinogens, is sufficient for the induction of epithelial carcinogenesis [89]. It is noteworthy, however, to mention that at the basis of these inflammation-induced genotoxic insults, is oxidative stress—especially from macrophages and neutrophils, which generate huge amounts of reactive oxygen species (ROS) and reactive nitrogen species (RNI) which in turn, induces the mutations that result in cardiac cell death [81]. To induce its chemoprotective effect following DOX exposure, PCA being a potently antioxidant, exerts its free-radical scavenging effects on the generated ROS/RNIs, thereby abrogating cardiomyocyte cell death via inflammation-induced DNA damage [5], [8], [11], [19], [81].
Decreased activity of inflammatory markers such as interleukin-6, tumor necrosis factor-α (TNF-α), myeloid peroxidase, and nitric oxide have also been reported in tissues of streptozotocin-induced diabetic adult male mice following PCA treatment, thus further demonstrating PCA as a potent tumor suppressive agent [26], [77]. Taken together, the chemopreventive properties of PCA relevant to its potential ameliorative effects against DIC, are its antioxidant, metal-chelating effects (especially in ferroptosis), anti-cell death (apoptosis, pyroptosis and necroptosis), anti-inflammatory properties, and p53-upregulatory effects [11], [19], [26]. The latter, up-regulation of p53 expression, was observed in gastric adenocarcinoma cells (AGS) exposed to a combined treatment of PCA (500 mM) and 5-fluorouracil, 5-FU (10 μM), leading to the upregulation of p53 expression and downregulation of Bcl-2 proteins [90]. A summary sketch of the molecular processes underpinning DIC and the potential ameliorative function of PCA administration is provided in Fig. 3.
Fig. 3.

Summarization of DIC mechanisms and potential ameliorative role for PCA. Generation of DOX-semiquinone via an NADPH oxidase 2 (NOX2)-catalyzed single electron reduction is the common point for the induction of molecular toxicities such as oxidative stress, inflammation, DNA fragmentation/adduction, and cell deaths of several forms (apoptosis, pyroptosis, and necroptosis) in cardiomyocytes. Left unchecked or unregulated, these processes collectively result in DIC. Interestingly, PCA, due to its established antioxidant, anti-inflammatory, anti-cell death, and DNA-protecting activities abrogates the induction of molecular toxicities due to DOX-quinone generation thereby rescuing cardiac cells from DOX-induced injuries.
4.1. Anti-cell death property of PCA under DOX treatment
4.1.1. Apoptosis
Apoptosis—an automated programmed cell death pathway devoid of inflammation in surrounding tissues—is by far the most dominant form of cell death that occurs in the DIC [8]. Evidence from human stem cell-derived cardiomyocytes, highlights increased sensitivity of cardiac cells to DOX treatment under conditions of increased topoisomerase 2β (Top2β) gene expression. In contrast, cardiomyocyte-specific deletion of the Top2β gene was found to positively correlate with decreased apoptosis, resulting in the attenuation of DOX-induced cardiotoxicities [91], [92]. Essentially, a high Top2β gene expression in cardiomyocytes increases their sensitivity to DOX treatment, which in turn, causes the induction of DNA damage, p53 phosphorylation, and apoptosis via a process undergirded by the formation of the rate-limiting DOX-Top2β-DNA complex [11], [91], [92]. While the direct effect of PCA on Top2β expression is unknown, the DNA damage, p53 phosphorylation, and apoptotic cell death that ensues are underpinned by oxidative stress, which PCA has been widely reported to effectively abrogate [26], [93].
Other common forms of DOX-induced death of cardiomyocytes include necroptosis, pyroptosis, and ferroptosis [11]. Although beneficial to the cell under physiological conditions due to its role in the normal development of cardiomyocytes and the establishment of cardiac homeostasis, apoptosis becomes dysregulated and cardiotoxic following DOX-treatment, ultimately inducing cell death, genome instability, and clinically detectable DIC [8]. Notably, DOX-induced apoptosis could be via any of the two canonical pathways—intrinsic and extrinsic pathways.
The induction of the intrinsic pathway following DOX treatment is crucially accompanied by mitochondrial outer membrane permeabilization (MOMP). This represents a loss in the mitochondrial membrane potential and is commonly referred to as the ‘point-of-no return’ along the intrinsic pathway of the apoptosis [8], [94]. Once established, the MOMP, triggers the release of cytochrome c (Cyt c), an intracellular mitochondrial protein that mediates the trafficking of electrons between complex III and complex IV into the cytosol. The following release into the cytosol, Cyt c, triggers the oligomerization of apoptosis protease activating factor-1 protein (Apaf-1), which in turn, generates the apoptosome. Inactive caspase-9 dimers are recruited, cleaved, and activated using energy from ATP hydrolysis. Caspase-9 once generated, activates the executioner caspases 3/7. To drive the final step of induction of apoptosis, caspase 3 (not caspase-7), is translocated into the nucleus to initiate the induction of apoptosis. In the nucleus, caspase 3 stimulates the release and activation of caspase-3-activated DNase (CAD), by cleaving the inhibitor of CAD (ICAD), which results in a DNA fragmentation [8]. Studies have also shown that while not translocated into the nucleus (remains in the cytoplasm), caspase-7 is also able to mediate the nucleus-located inhibition of ICAD, stressing the heterogeneous localization of ICAD [8], [95].
Poly (ADP-ribose) polymerase, PARP, activity is often activated as an endogenous response mechanism to DNA damage. However, under conditions of DOX-induced apoptosis, PARP polymerization activity is inactivated by caspase-3, allowing for the potentiation of DNA damage and genomic instability [96]. Interestingly, apoptosis via the intrinsic pathway can also occur in a caspase-independent manner.
Following the loss of the mitochondrial membrane potential due to DOX treatment, mitochondria-bound pro-apoptotic factors such as the BAX and BAK proteins, apoptosis-inducing factor (AIF), and endonuclease G (EndoG), are released into the cytosol and then translocated to the nucleus to drive apoptosis [8]. Unlike AIF which lacks DNase activity and is thus unable to mediate oligonucleosomal DNA fragmentation, EndoG having both DNase and RNase activity mediates the cleavage of high order chromatin into high resolution melting (HRM) DNA fragments [97], [98]. Together, this aberrant apoptosis induced by DOX treatment accelerates the death of cardiomyocytes, which manifests clinically as DIC (Fig. 4).
Fig. 4.

Schematic representation of DOX-induced intrinsic apoptosis, highlighting the potential attenuating effects of PCA. DOX following its entry into the myocardium and inducing MOMP can cause DNA fragmentation, leading to apoptosis in a caspase-dependent or -independent manner. While the former process is directly brought about by the executioner caspase-3, which mediates the inhibitory phosphorylation of Poly ADP-ribose polymerase (PARP) and Caspase-activated DNase enzyme (ICAD), the latter process is mediated by the pro-apoptotic factors like BAX and BAK as well as by endonuclease G (EndoG) and apoptosis-inducing factor (AIF). Notably, while the caspase-3 suppressing effects of PCA have been widely reported in both in vivo and in vitro studies, very few findings observe the effects of PCA on such DNA-damage driving factors as AIF and EndoG.
In contrast to the intrinsic pathway, induction of the extrinsic pathway following DOX exposure, is via the overstimulation of the death-signaling receptors—Fas, TNF receptor superfamily 1 A (TNFR1A), TNF-related apoptosis-inducing ligand receptor 10a (TRAIL 1), and TNF-related apoptosis-inducing ligand receptor 10b (TRAIL 2), all of which rely on binding of the complementary ligands [99], [100]. Interestingly, while cardiac cells demonstrate resistance to Fas-induced apoptosis under normal physiological conditions, available literature shows that DOX-exposure induces extrinsic apoptosis in the myocardium in a Fas-dependent manner [99], [101]. To execute DOX-induced cardiotoxic effects following the formation of the associated death receptor-ligand complexes, a dynamic multi-protein structure known as the death-inducing signaling complex I and II (DISC) is formed to positively regulate the activation of pro-caspase-8 to caspase-8. Activated caspase-8 once formed, catalyze the proteolytic maturation and cleavage (activation) of the executioner caspases-3/6/7, which in turn, mediates apoptosis of cardiomyocytes through DNA damage [99]. Additionally, DOX administration mediates the reduction of XIAP (X-linked inhibitor of apoptosis protein) and cFLIPS (cellular FLICE-like inhibitory proteins), both of which otherwise inhibit apoptosis and concomitantly enhance the activity of TRAIL 10a and 10b to trigger the induction, progression, and potentiation of cardiomyocyte apoptosis.
Substantial parts of both the intrinsic and extrinsic pathways of apoptosis following DOX exposure terminate with the generation of the executioner caspase-3, whose activity PCA has been widely reported to effectively abrogate [19], [26], [102]. Thus, because PCA can rescue cell death from caspase-3-induced apoptosis, its administration as an adjuvant during chemotherapy at the safe oral dose of 100 mg/kg represents a unique therapeutic intervention for the amelioration of DOX-induced cardiotoxicities in the chemotherapy [19]. Further, it can attenuate the occurrence of life-threatening cardiomyopathy that may occur during a long post-chemotherapeutic period [12].
4.1.2. Pyroptosis
Pyroptosis is the inflammatory form of programmed cell death (apoptosis), which occurs upon infection with intracellular pathogens. Although generally occurring as a result of the inflammatory response to microbes, pyroptosis has been identified as one of the major cell death forms associated with DIC [103], [104]. By upregulating the expression of BH3-only protein Bcl-2/adenovirus E1B19-kDa-interacting protein 3 (Bnip3) in myocytes, it triggers the activation of caspase-3, cleavage of Gasdermin E (GSDME), membrane rupture, and pyroptosis in cardiomyocytes [105], [106]. Further investigations showed that either knockdown of GSDME with siRNA or complete deletion of caspase-3 prevented approximately 50% of DOX-induced pyroptosis in the macrophages [107]. Notably, the involvement of caspase-3 expression in the mediation of DOX-induced pyroprosis in macrophages and the well-reported negative regulation of caspase-3 by PCA highlights the role of PCA in rescuing cardiac cells from DOX-induced pyrotosis during chemotherapy [19], [26], [102].
DOX treatment was also found to trigger the activation of Toll-like receptor 4 (TLR4) while also augmenting the formation of NLRP3 (NACHT, LRR, and PYD domains-containing protein 3) inflammasomes to activate caspase-1 and GSDMD, which ultimately mediates induction of pyroptosis [108]. Furthermore, these studies also found that the activation of NLRP3, following DOX exposure, occurred in a TLR4-independent but ROS-dependent manner. In the context of a role as a potential attenuator of DOX-induced pyroptosis, PCA exerts its antioxidant function by scavenging the ROS and/or free radicals upon which the activation of NLRP3 depends. PCA is also able to block the activation of caspase-1 and GSDMD which are directly responsible for pyroptotic cardiac cell death [26], [108], [109].
Available literature shows that the overexpression of NOX1/NOX4 following DOX treatment triggers mitochondrial fission and promotes the subsequent activation of NLRP3 in a ROS-dependent manner, thereby mediating the onset of pyroptosis in the heart [108], [110]. Moreover, NLRP3- and caspase-1-deficient mice were found to be resistant to DOX-induced pyroptotic cell death, suggesting a critical role for NLRP3 inflammasomes and caspase-1 in DOX-induced pyroptotic cell death [110]. Although additional studies are required to either confirm or refute these findings [110], it holds an enormous therapeutic or chemoprotective potential as it stresses the centrality of NLRP3 inflammasomes activation—an ROS-dependent process, in the onset of DOX-induced pyroptosis. This ROS-driven induction of pyroptosis highlights a chemopreventive potential (abrogation of pyroptosis) for PCA as an established anti-oxidant [19], [26]. Collectively, the potential ameliorative effects of PCA on DOX-induced pyroptosis are schematically summarized in Fig. 5.
Fig. 5.

Schematic illustration of the crosstalk between DOX-induced pyroptosis and apoptosis and the potential ameliorative function of PCA. The schematic illustration shows the crosstalk between DOX-induced pyroptosis and DOX-induced apoptosis. The left side of the schematic illustration shows the formation of NLRP3 inflammation, a process that is heavily ROS-dependent and TLR4 (Dox-activated toll-like receptor)-independent. Formation of NLRP3 allows for its activation of caspase-1 which could directly induce pyroptosis by the activation of Gasdermin D (GSDMD) or by activation of caspase-3 in the absence of GSDMD (Taabazuing et al., 2017). The right side of the schematic illustration shows a caspase-3-dependent induction of apoptosis following DOX treatment. Notably, introduction of PCA, either to scavenge ROS or attenuate Caspase-3 activity, ultimately results in limiting both pyroptosis and apoptosis and ipso facto, DIC.
5. Antioxidant property of PCA and DOX treatment
Oxidative stress is defined as the imbalance in the production/generation of free radicals or ROS/RNI at a rate that exceeds the scavenging or protective activity of the body’s endogenous antioxidant mechanisms e.g., superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GSH-PX), and blockers of lipid peroxidation [18], [26], [111], [112]. To exert its anti-oxidative effects and confer protection against oxidative stress, PCA up-regulates the activities of endogenous antioxidant systems and attenuates pro-oxidant systems [19]. As a result, the free radicals that induce apoptosis, DNA damage, and oxidative stress, following DOX exposure, can be effectively scavenged. Moreover, the reduction of pro-oxidant enzyme systems like those of xanthine oxidase (XOD), peroxisomes, and NADP oxidase (NOX) as well as the reduction of the level of malondialdehydes (MDAs) which are a marker for lipid peroxidation by PCA, further underlines the potential chemoprotective function of PCA in attenuating DIC [8], [18], [113], [114], [115], [116]. Notably, of the seven members of the NOX family, NOX2 and NOX4 are predominantly expressed in the mammalian heart [117]. While normally remaining inactive on the cell membrane of cardiomyocytes, NOX2 is readily activated following DOX treatment to mediate the reduction of DOX (to semiquinone). This in turn generates super-oxide anion radicals and hydrogen peroxide, both of which are effectively scavenged, sequestered, and converted into less toxic moieties by the endogenous antioxidant mechanisms, which are upregulated by PCA [26], [118].
DOX treatment, asides from being associated with glutathione (GSH) depletion, also potently induces a form of oxidative stress called ferroptosis, which results in iron overload in the cardiomyocytes and corresponding cell death [8], [11]. By elevating the expression of transferrin (Tf) and its receptor, transferrin receptor (TrF) through its induction of a mutation in human homeostatic iron protein (HFE), DOX hyper-activates the entry of iron into the cardiomyocytes inducing ferroptosis [5], [8]. Because reduced glutathione (GSH) is an essential repository for intracellular cysteine and also plays a crucial role in redox homeostasis, both enzymatic anti-oxidant systems (SOD, CAT, GPXs, and GSTs) and non-enzymatic anti-oxidant systems (alpha-tocopherol/vitamin E), are directly inhibited by DOX-induced GSH depletion, increasing the vulnerability of the cell to oxidative stress following DOX treatment [119]. Moreover, DOX-induced GSH depletion has also been reported to induce a dysregulation in the activity of nicotinamide adenine dinucleotide phosphate (NADPH), which under normal physiological conditions plays a critical role in the conversion of oxidized glutathione (GSSG) to reduced glutathione (GSH) via a process catalyzed by the enzyme glutathione reductase [8].
Under conditions of DOX exposure, NADPH switches from being beneficial to being deleterious, as it is recruited by NADPH oxidases (NOXs) to serve as a substrate in reactions that ultimately result in ROS generation [120], [121]. Generation of ROS, with the concomitant depletion of GSH, results in the downregulation of both the enzymatic and non-enzymatic antioxidant systems, allowing the build-up of the oxidative stress [122]. Furthermore, the DOX-Fe complexes formed, due to the iron-chelating ability of DOX, potentiates oxidative stress and accompanying oxidative damage to cardiac cells. Specifically, iron overload results in the generation of superoxide anion radical, O2-; hydrogen peroxide, H2O2; and malondialdehydes (MDAs) into more toxic ROS entities, like hydroxyl radicals (-OH) [123], [124]. Interestingly, the metal-chelating and metal radical-scavenging properties of PCA have been reported in vitro using the positive controls Trolox and Butylated Hydroxy Toluene (BHT) [19], [125], [126]. Notably, the metal-chelating capacity of PCA on Fe2+ and Cu2+ as well as its ability to scavenge DPPH radicals was observed to be dose-dependent and significantly higher than those of the positive controls [126]. Various antioxidant assays were used to demonstrate the metal-chelating as well as superoxide anion radical-scavenging and hydroxyl radical-scavenging powers of PCA concerning positive controls [19], [125]. When compared to the positive control, Trolox, PCA shows much more effective antioxidant activity in vitro in both lipid and aqueous media, indicating the possibility of PCA as a safer and more effective antioxidant relative to established or standard anti-oxidants [19]. Altogether, PCA, with widely reported free radical scavenging activities, has the potential to sequester toxic ROS moieties, thereby attenuating the severity of the DIC [18], [19], [26], [93].
6. Anti-inflammatory properties of PCA and DOX-treatment
The induction of inflammatory responses following DOX treatment occurs through the regulation of a variety of downstream inflammatory effectors such as the binding of TNF-α to TNFR1, impairment of nuclear factor kappa beta (NF-κβ), and inhibition of the Janus kinase (JAK)/signal transducer and activator of transcription (STAT3) pathway [101]. To begin this complex inflammatory process, TNF-α binds to TNFR1, which causes the assembly of the cytoplasmic complex II—consisting of the receptor-interacting protein-1 (RIPK1), Caspase-8, and complex Fas-associated via death domain (FADD) [101], [127]. The cytoplasmic complex II once formed, triggers the phosphorylation and oligomerization of the receptor-interacting protein kinases 1/3 (RIPK1/3) at the serine/threonine residues, while also simultaneously inhibiting caspase-8, a process which ultimately initiates the induction of necroptosis (programmed necrosis) via the phosphorylation of mixed lineage kinase domain-like protein (MLKL) [101], [127].
DOX treatment also impairs NF-κβ. NF-κβ impairment is achieved by two key mechanisms: the upregulation of interleukin 1 beta (IL-1β)/interleukin 6 (IL-6) and the disruption of the Ikappa B kinase complex (IKK) which consists of IKKα, IKKβ, and NF-κβ essential modulator (NEMO) [99], [100]. Furthermore, the inhibition of the JAK/STAT3 signaling pathway inhibits the transcription of anti-apoptotic or in this context, pro-survival genes (such as the BCL-2 gene), which further worsens cardiac outcomes due to sustained death of cardiac cells by both apoptosis and necroptosis (Fig. 6).
Fig. 6.

Doxorubicin-induced inflammatory signaling and the potential suppressive effect of PCA. DOX exposure upregulates the expression of tumor necrosis factor alpha, TNF-α which binds to its receptor, the TNF-α receptor 1, TNFR1. The ligand-receptor complex allows for the activation of TRADD (TNFα receptor activated death domain), also known as Apo2L, as well as the assembly of complex II—consisting of the Fas-associated Death domain, FADD, Caspase-8, Casp-8, and Receptor-interacting protein-1, RIPK1. Within this complex, Casp-8 is activated by autophosphorylation, allowing it to exercise its dual functionality as an inducer of apoptosis (via the activation of caspase 3) as well as an inhibitor of necroptosis (via the blockade of the activity of necroptosis mediators RIPKI/RIPK3). Interestingly, DOX treatment, not only blocks the activity of caspase-8, releasing RIPKI/RIPK3 from its inhibitory effect, DOX treatment also initiates necroptosis via the induction of an activation phosphorylation of the mixed lineage kinase domain-like protein, MLKL. Collectively, by suppressing TNF-α, IL6, IL-1β expressions, PCA impairs the relevant ligand-receptor interactions needed to the complex signaling process underpinning the induction of toxicities and ipso facto DIC by necroptosis.
It is instructive to point out that inflammation due to DOX treatment, also induces cardiac cell death, principally by the induction of both necroptosis and apoptosis, both of which in turn, have being reported by various studies to be negatively regulated by PCA [19], [22], [26], [93]. In brief, the ‘co-conspiration’ of inflammation and oxidative stress to induce cell death via DNA damage under conditions of DOX exposure and the abundance of laboratory-based evidence underscoring the anti-oxidant, anti-inflammatory, and anti-apoptotic properties of PCA; PCA represents an effective chemopreventive intervention in blocking, suppressing, and attenuating the many cardiotoxicities associated with DOX chemotherapy. Therefore, it may ultimately result in increasing the therapeutic index of DOX therapy for the treatment of cancer.
7. Chemoprotection beyond DOX-induced toxicities
Importantly, the chemoprotective properties of PCA, including its anti-inflammatory and cardio-protective properties—the latter being achieved through its abrogation of various forms of cell death of cardiomyocytes, are not specific for doxorubicin-induced toxicities, although, certain limitations exist. In the case of cisplatin, for example, where mechanisms of toxicity are known (principally oxidative stress and inflammation), PCA has a clear potential of attenuating cisplatin-induced toxicities—hepatotoxic and gastrointestinal toxicity [20], [128]. However, for a drug such as 5-FU, where toxicities could be due to genetic factors such as a deficiency in the DYDP (dihydropyrimidine dehydrogenenase) gene or cardio-toxicities due to an interplay of various pathophysiological processes such as myocardial inflammation, coronary spasm and increased endothelial thrombogenicity [129], the outcome of PCA use is not known and may yield very minimal chemoprotection, if any. Because DOX toxicity is associated with its induction of oxidative stress, inflammation, and various forms of cell death including apoptosis, necrosis, necroptosis, and/or Ferroptosis, all of which PCA negatively regulates, PCA-conferred chemoprotection against DOX-induced toxicities, is more significant when compared to other cancer drugs whose mechanism of toxicity is either not fully understood or whose mechanism of toxicity PCA can only minimally attenuates.
Also, because different anticancer drugs have different mechanisms of toxicity—some established and well-known and others nuanced and still yet to be fully understood—the chemoprotective effect of PCA would be dependent on whether the toxic mechanism of the drug is known or on when there is experimental evidence supporting the negatively regulation or attenuation of identified toxic mechanisms by PCA. In the case of cisplatin mentioned above, very recent investigations have shown, that suppressing serum MDA and NO levels, decreases the expressions of IL-6 and TNF-α and NF-kB p65 subunit and iNOS, as well as significantly lowers the levels of apoptotic (annexin-V, Bax, caspase-3) biomarkers, while concurrently raising serum GSH and SOD levels, PCA attenuates cisplatin-induced hepatotoxicity[20]. Moreover, because oxidative stress, inflammation, and apoptosis have been proven to play critical roles in the pathogenesis of cisplatin-induced hepatotoxicity [20], and because all three toxic mechanisms have been demonstrated by a broad spectrum of experimental investigations to be negatively regulated by PCA, that remains a veritable rescuing agent for the attenuation of the harmful effects associated with the use of anti-cancer drugs, such as cisplatin. In a related study, Dioscin—a steroidal saponin with potential anti-cancer, anti-oxidative and anti-inflammatory properties, was reported to ameliorate cisplatin-induced intestinal toxicity by attenuating oxidative stress and inflammation, thereby, improving the therapeutic efficacy of the drug. Identified dioscin-driven mechanisms include induction of the Nrf2-HO-1 pathway and amelioration of intestinal oxidative stress, inhibition of both NLRP3 inflammasome activation and inflammatory factor expression, and finally, promotion of Wnt3A/β-catenin signaling to relieve cisplatin-induced proliferation inhibition [128]. Notably, since PCA also exerts an inhibitory effect on the NLRP3 inflammasome activation [19], it has the capacity of replicating the ameliorative effect of dioscin against cisplatin-induced gastrointestinal toxicity if it were to be replaced with dioscin. Nonetheless, for some anti-cancer drugs such as 5-FU, whose toxic mechanisms are yet to be fully dissected and for which reported cardiotoxic effects are due to the contribution of various pathophysiological mechanisms such as myocardial inflammation, coronary spasm, and increased endothelial thrombogenicity [129], caution must be taken in the application of PCA.
8. Limitations and future perspectives
Findings from several studies have dissociated the tumouricidal activity of DOX from observed cardiotoxicity, stressing a role for the metabolites generated following DOX metabolism in the induction of the cardiotoxicity [8], [11], [12], [70]. DOX has also been reported to induce destabilization of the electrophysiology of cardiac cells, albeit a dearth of commensurate evidence elucidating the mechanistic basis for this phenomenon [130]. Interestingly, a possible role for DOXol in the perturbation of cardiac electrophysiology has also been identified in vitro [70]. Briefly, DOXol was not only more potent than DOX in impairing both systolic and diastolic heart function, but it was also more effective in inhibiting the activity of pumps such as the Ca2+ pump of the sarcoplasmic reticulum, Na+/K+ pump of the sarcolemma, and the FOF1 proton pump of the mitochondria, all of which play critical roles in the induction and stabilization of myocardial electric homeostasis [70]. Moreover, these findings stress a potential role for DOX, especially in a DOXol-dependent manner, in the alteration of the electrophysiology of cardiac cells and the induction of arrhythmias including more lethal arrhythmic events like complete heart block and ventricular fibrillation [12], [130], [131], [132], [133].
While PCA can negatively regulate oxidative stress, inflammation and several other forms of cell death as elucidated in this review [19], [26], [102], these chemoprotective properties of PCA do not readily demonstrate possible anti-arrhythmogenic effects. This is especially problematic because the mechanistic basis of DOX-induced arrhythmia is far from being comprehensively understood [130]. Given this limitation of the current review, that is, uncertainties around the possible effect of PCA in limiting the molecular mechanisms underpinning arrhythmia, future investigations on this subject are warranted. Investigators may want to focus on designing experiments in appropriate models to assess a potential role for PCA in the stabilization of all stages of cardiac electric activity from direct effects on single cell ionic currents to the whole animal telemetry [130]. Whether this proposed ‘anti-arrhythmogenic effects of PCA rely on its ability to negatively regulate the adverse effects of DOXol, remains to be determined. Consequently, it may signal the discovery of new research paradigms toward a more comprehensive understanding of DOX-induced cardio-electric alterations and the potential for novel ameliorative/chemoprotective strategies.
9. Conclusion
Oxidative stress, inflammation, apoptosis, as well as other forms of cell death such as pyroptosis, necroptosis, and ferroptosis, have been identified as the molecular mechanisms of DIC. Interestingly, the antioxidant, anti-inflammatory, anti-apoptotic, anti-cell death, and chemoprotective properties of PCA have been amply underlined by several laboratory-based investigations, indicating a potential role for PCA in the attenuation of cardiotoxicities during DOX chemotherapy as well as in the mitigation of the more life-threatening cardiomyopathy after treatment cessation. Moreover, PCA may also exert chemoprotective effects against toxicities induced by anticancer drugs such as cisplatin since reported cases of cisplatin-induced hepatotoxicity and gastrointestinal toxicities, are principally driven by oxidative stress and inflammation. Although much is still unknown about the exact molecular mechanisms by which PCA abrogates pathways such as the death receptor-based extrinsic apoptotic, the caspase-independent, and the pro-apoptotic factors-driven intrinsic pathways; PCA, at a dose from 50 to 150 mg/kg, still represents a safe and effective intervention. Further experimental and clinical studies are warranted to elucidate these potentials of PCA and to possibly establish its usage as a veritable chemo-protective adjuvant in the amelioration of DIC both during and after chemotherapy.
Funding
This work was supported by the Ministry of Science and Technology of China (grant number G2022200008L), National Natural Science Foundation of China, NSFC (grant number. 3217090070), Natural Science Foundation of Anhui Province, China, NSFA (grant Number. 2108085MC89) and the Chinese government scholarship council, CSC.
CRediT authorship contribution statement
EO conceptualized the idea for the review manuscript and SH mentored and guided its development. EO wrote the first draft of the manuscript, including the design of original diagrams as provided. EO, IA, MK, EF, and SH edited and revised several drafts of the manuscript. EO and XG contributed to the proofreading of the final draft as well as to the design, formatting, and standardization of the final diagrams. XG, particularly oversaw the alignment, standardization, and harmonization of cited references. EO, IA, XG, MK, EF, and SH approved the final draft of this manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors would like to acknowledge the financial support from the Ministry of Science and Technology of China, the National Natural Science Foundation of China, the Natural Science Foundation of Anhui Province, China, and the Chinese government scholarship council, through the Division of Life Science, University of Science and Technology of China, USTC. The authors also especially acknowledge the contribution of Ms. Chuyun Yan in interfacing with relevant commercial vendors during the processing of manuscript diagrams.
Open Access
The funders support publishing this article under the ‘Gold open access’ option, Under a CC BY license.
Handling Editor: Dr. Lawrence Lash
Contributor Information
Ebenezer O. Farombi, Email: olatunde_farombi@yahoo.com.
Shuxin Han, Email: hsx66@ustc.edu.cn.
Data availability
Data will be made available on request.
References
- 1.Zeng X., et al. Pharmacokinetics and cardiotoxicity of doxorubicin and its secondary alcohol metabolite in rats. Biomed. Pharmacother. 2019;116 doi: 10.1016/j.biopha.2019.108964. [DOI] [PubMed] [Google Scholar]
- 2.Kanwal U., et al. Advances in nano-delivery systems for doxorubicin: an updated insight. J. Drug Target. 2018;26(4):296–310. doi: 10.1080/1061186X.2017.1380655. [DOI] [PubMed] [Google Scholar]
- 3.Makwana V., et al. Liposomal doxorubicin as targeted delivery platform: current trends in surface functionalization. Int. J. Pharm. 2021;593 doi: 10.1016/j.ijpharm.2020.120117. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y., Castro Bravo K.M., Liu J. Targeted liposomal drug delivery: a nanoscience and biophysical perspective. Nanoscale Horiz. 2021;6(2):78–94. doi: 10.1039/d0nh00605j. [DOI] [PubMed] [Google Scholar]
- 5.Rawat P.S., et al. Doxorubicin-induced cardiotoxicity: an update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021;139 doi: 10.1016/j.biopha.2021.111708. [DOI] [PubMed] [Google Scholar]
- 6.Fang X., et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA. 2019;116(7):2672–2680. doi: 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang X., et al. FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death Differ. 2020;27(2):540–555. doi: 10.1038/s41418-019-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sangweni N.F., et al. Molecular insights into the pathophysiology of doxorubicin-induced cardiotoxicity: a graphical representation. Arch. Toxicol. 2022;96(6):1541–1550. doi: 10.1007/s00204-022-03262-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuzu M., et al. Morin attenuates doxorubicin-induced heart and brain damage by reducing oxidative stress, inflammation and apoptosis. Biomed. Pharmacother. 2018;106:443–453. doi: 10.1016/j.biopha.2018.06.161. [DOI] [PubMed] [Google Scholar]
- 10.Tedesco L., et al. A special amino-acid formula tailored to boosting cell respiration prevents mitochondrial dysfunction and oxidative stress caused by doxorubicin in mouse cardiomyocytes. Nutrients. 2020;12(2) doi: 10.3390/nu12020282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kong C.Y., et al. Underlying the mechanisms of doxorubicin-induced acute cardiotoxicity: oxidative stress and cell death. Int. J. Biol. Sci. 2022;18(2):760–770. doi: 10.7150/ijbs.65258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Licata S., et al. Doxorubicin metabolism and toxicity in human myocardium: role of cytoplasmic deglycosidation and carbonyl reduction. Chem. Res. Toxicol. 2000;13(5):414–420. doi: 10.1021/tx000013q. [DOI] [PubMed] [Google Scholar]
- 13.Surh Y.J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer. 2003;3(10):768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 14.Mori H., et al. Chemoprevention by naturally occurring and synthetic agents in oral, liver, and large bowel carcinogenesis. J. Cell Biochem. Suppl. 1997;27:35–41. [PubMed] [Google Scholar]
- 15.Greenwald P. The future of cancer prevention. Semin. Oncol. Nurs. 2005;21(4):296–298. doi: 10.1016/j.soncn.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 16.Masella R., et al. Protocatechuic acid and human disease prevention: biological activities and molecular mechanisms. Curr. Med. Chem. 2012;19(18):2901–2917. doi: 10.2174/092986712800672102. [DOI] [PubMed] [Google Scholar]
- 17.Lin J.K. Cancer chemoprevention by tea polyphenols through modulating signal transduction pathways. Arch. Pharm. Res. 2002;25(5):561–571. doi: 10.1007/BF02976924. [DOI] [PubMed] [Google Scholar]
- 18.Zhang S., et al. Antioxidant effects of protocatechuic acid and protocatechuic aldehyde: old wine in a new bottle. Evid. Based Complement. Altern. Med. 2021;2021 doi: 10.1155/2021/6139308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kakkar S., Bais S. A review on protocatechuic Acid and its pharmacological potential. ISRN Pharmacol. 2014;2014 doi: 10.1155/2014/952943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Habib S.A., et al. The protective effect of protocatechuic acid on hepatotoxicity induced by cisplatin in mice. Life Sci. 2021;277 doi: 10.1016/j.lfs.2021.119485. [DOI] [PubMed] [Google Scholar]
- 21.Da-Costa-Rocha I., et al. Hibiscus sabdariffa L. - a phytochemical and pharmacological review. Food Chem. 2014;165:424–443. doi: 10.1016/j.foodchem.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 22.Adedara I.A., et al. Impact of prepubertal exposure to dietary protocatechuic acid on the hypothalamic-pituitary-testicular axis in rats. Chem. Biol. Interact. 2018;290:99–109. doi: 10.1016/j.cbi.2018.05.013. [DOI] [PubMed] [Google Scholar]
- 23.Szumilo J. [Protocatechuic acid in cancer prevention] Post. Hig. Med. Dosw. 2005;59:608–615. [PubMed] [Google Scholar]
- 24.Chen W., et al. Pharmacokinetics of protocatechuic acid in mouse and its quantification in human plasma using LC-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012;908:39–44. doi: 10.1016/j.jchromb.2012.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ajiboye T.O., et al. Antioxidant and drug detoxification potentials of Hibiscus sabdariffa anthocyanin extract. Drug Chem. Toxicol. 2011;34(2):109–115. doi: 10.3109/01480545.2010.536767. [DOI] [PubMed] [Google Scholar]
- 26.Adedara I.A., et al. Dietary protocatechuic acid abrogates male reproductive dysfunction in streptozotocin-induced diabetic rats via suppression of oxidative damage, inflammation and caspase-3 activity. Eur. J. Pharmacol. 2019;849:30–42. doi: 10.1016/j.ejphar.2019.01.033. [DOI] [PubMed] [Google Scholar]
- 27.Ali B.H., et al. Effect of Hibiscus sabdariffa and its anthocyanins on some reproductive aspects in rats. Nat. Prod. Commun. 2012;7(1):41–44. [PubMed] [Google Scholar]
- 28.Farombi E.O., Fakoya A. Free radical scavenging and antigenotoxic activities of natural phenolic compounds in dried flowers of Hibiscus sabdariffa L. Mol. Nutr. Food Res. 2005;49(12):1120–1128. doi: 10.1002/mnfr.200500084. [DOI] [PubMed] [Google Scholar]
- 29.Ali B.H., Al Wabel N., Blunden G. Phytochemical, pharmacological and toxicological aspects of Hibiscus sabdariffa L.: a review. Phytother. Res. 2005;19(5):369–375. doi: 10.1002/ptr.1628. [DOI] [PubMed] [Google Scholar]
- 30.Farombi E.O., et al. Dietary protocatechuic acid ameliorates dextran sulphate sodium-induced ulcerative colitis and hepatotoxicity in rats. Food Funct. 2016;7(2):913–921. doi: 10.1039/c5fo01228g. [DOI] [PubMed] [Google Scholar]
- 31.Gao J.W., et al. DJ-1-Mediated protective effect of protocatechuic aldehyde against oxidative stress in SH-SY5Y cells. J. Pharmacol. Sci. 2011;115(1):36–44. doi: 10.1254/jphs.10271fp. [DOI] [PubMed] [Google Scholar]
- 32.Tseng T.H., et al. Induction of apoptosis by hibiscus protocatechuic acid in human leukemia cells via reduction of retinoblastoma (RB) phosphorylation and Bcl-2 expression. Biochem. Pharmacol. 2000;60(3):307–315. doi: 10.1016/s0006-2952(00)00322-1. [DOI] [PubMed] [Google Scholar]
- 33.Tseng T.H., et al. Inhibitory effect of Hibiscus protocatechuic acid on tumor promotion in mouse skin. Cancer Lett. 1998;126(2):199–207. doi: 10.1016/s0304-3835(98)00010-x. [DOI] [PubMed] [Google Scholar]
- 34.Zhong Z., et al. Protocatechuic aldehyde mitigates hydrogen peroxide-triggered PC12 cell damage by down-regulating MEG3. Artif. Cells Nanomed. Biotechnol. 2020;48(1):602–609. doi: 10.1080/21691401.2020.1725535. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka T., et al. Chemoprevention of colon carcinogenesis by the natural product of a simple phenolic compound protocatechuic acid: suppressing effects on tumor development and biomarkers expression of colon tumorigenesis. Cancer Res. 1993;53(17):3908–3913. [PubMed] [Google Scholar]
- 36.Hirose Y., et al. Chemoprevention of urinary bladder carcinogenesis by the natural phenolic compound protocatechuic acid in rats. Carcinogenesis. 1995;16(10):2337–2342. doi: 10.1093/carcin/16.10.2337. [DOI] [PubMed] [Google Scholar]
- 37.Mahmoud N.N., et al. Plant phenolics decrease intestinal tumors in an animal model of familial adenomatous polyposis. Carcinogenesis. 2000;21(5):921–927. doi: 10.1093/carcin/21.5.921. [DOI] [PubMed] [Google Scholar]
- 38.Han S.S., et al. Suppression of phorbol ester-induced NF-kappaB activation by capsaicin in cultured human promyelocytic leukemia cells. Arch. Pharm. Res. 2002;25(4):475–479. doi: 10.1007/BF02976605. [DOI] [PubMed] [Google Scholar]
- 39.Subbaramaiah K., et al. Resveratrol inhibits cyclooxygenase-2 transcription in human mammary epithelial cells. Ann. N. Y. Acad. Sci. 1999;889:214–223. doi: 10.1111/j.1749-6632.1999.tb08737.x. [DOI] [PubMed] [Google Scholar]
- 40.Subbaramaiah K., et al. Resveratrol inhibits cyclooxygenase-2 transcription and activity in phorbol ester-treated human mammary epithelial cells. J. Biol. Chem. 1998;273(34):21875–21882. doi: 10.1074/jbc.273.34.21875. [DOI] [PubMed] [Google Scholar]
- 41.Mouria M., et al. Food-derived polyphenols inhibit pancreatic cancer growth through mitochondrial cytochrome C release and apoptosis. Int. J. Cancer. 2002;98(5):761–769. doi: 10.1002/ijc.10202. [DOI] [PubMed] [Google Scholar]
- 42.Banerjee S., Bueso-Ramos C., Aggarwal B.B. Suppression of 7,12-dimethylbenz(a)anthracene-induced mammary carcinogenesis in rats by resveratrol: role of nuclear factor-kappaB, cyclooxygenase 2, and matrix metalloprotease 9. Cancer Res. 2002;62(17):4945–4954. [PubMed] [Google Scholar]
- 43.Blum C.A., et al. beta-Catenin mutation in rat colon tumors initiated by 1,2-dimethylhydrazine and 2-amino-3-methylimidazo[4,5-f]quinoline, and the effect of post-initiation treatment with chlorophyllin and indole-3-carbinol. Carcinogenesis. 2001;22(2):315–320. doi: 10.1093/carcin/22.2.315. [DOI] [PubMed] [Google Scholar]
- 44.Chen C., et al. Activation of antioxidant-response element (ARE), mitogen-activated protein kinases (MAPKs) and caspases by major green tea polyphenol components during cell survival and death. Arch. Pharm. Res. 2000;23(6):605–612. doi: 10.1007/BF02975249. [DOI] [PubMed] [Google Scholar]
- 45.Itoh K., et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997;236(2):313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 46.Ramos-Gomez M., et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA. 2001;98(6):3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kwak M.K., et al. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003;278(10):8135–8145. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- 48.Chan K., Han X.D., Kan Y.W. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc. Natl. Acad. Sci. USA. 2001;98(8):4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McMahon M., et al. The Cap'n'Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001;61(8):3299–3307. [PubMed] [Google Scholar]
- 50.Cho H.Y., et al. Role of NRF2 in protection against hyperoxic lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2002;26(2):175–182. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- 51.Chanas S.A., et al. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem. J. 2002;365(Pt 2):405–416. doi: 10.1042/BJ20020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thimmulappa R.K., et al. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62(18):5196–5203. [PubMed] [Google Scholar]
- 53.Enomoto A., et al. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol. Sci. 2001;59(1):169–177. doi: 10.1093/toxsci/59.1.169. [DOI] [PubMed] [Google Scholar]
- 54.Ishii T., et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000;275(21):16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 55.Chan K., Kan Y.W. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc. Natl. Acad. Sci. USA. 1999;96(22):12731–12736. doi: 10.1073/pnas.96.22.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Itoh K., et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13(1):76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang J., et al. Phenolics profile and antioxidant activity analysis of kiwi berry (Actinidia arguta) flesh and peel extracts from four regions in China. Front. Plant Sci. 2021;12 doi: 10.3389/fpls.2021.689038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang B., Luo H. Effects of mulberry leaf silage on antioxidant and immunomodulatory activity and rumen bacterial community of lambs. BMC Microbiol. 2021;21(1):250. doi: 10.1186/s12866-021-02311-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bode A.M., et al. Inhibition of epidermal growth factor-induced cell transformation and activator protein 1 activation by [6]-gingerol. Cancer Res. 2001;61(3):850–853. [PubMed] [Google Scholar]
- 60.MN A.L., Mellor I.R., Carter W.G. A preliminary assessment of the nutraceutical potential of acai berry (Euterpe sp.) as a potential natural treatment for Alzheimer's disease. Molecules. 2022;27(15) doi: 10.3390/molecules27154891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shin S., et al. Anti-skin aging properties of protocatechuic acid in vitro and in vivo. J. Cosmet. Dermatol. 2020;19(4):977–984. doi: 10.1111/jocd.13086. [DOI] [PubMed] [Google Scholar]
- 62.Nakamura Y., Torikai K., Ohigashi H. Toxic dose of a simple phenolic antioxidant, protocatechuic acid, attenuates the glutathione level in ICR mouse liver and kidney. J. Agric. Food Chem. 2001;49(11):5674–5678. doi: 10.1021/jf0106594. [DOI] [PubMed] [Google Scholar]
- 63.Kochhar S.L. Macmillan Publishers Ltd; London, UK: 1986. Tropical Crops: A Textbook of Economic Botany. [Google Scholar]
- 64.Gibbon D., Pain A. Crops of the Drier Regions of the Tropics. first ed. English Language Book Society; Longman, England: 1985. [Google Scholar]
- 65.Ismail A., Ikram E.H.K., Nazri H.S.M. Roselle (Hibiscus sabdariffa L.) seeds nutritional composition protein quality and health benefits. Food. 2008;2(1):1–16. [Google Scholar]
- 66.Li Z., et al. Pharmacokinetics of gallic acid and protocatechuic acid in humans after dosing with Relinqing (RLQ) and the potential for RLQ-perpetrated drug-drug interactions on organic anion transporter (OAT) 1/3. Pharm. Biol. 2021;59(1):757–768. doi: 10.1080/13880209.2021.1934039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jalali O., et al. Protocatechuic acid as a topical antimicrobial for surgical skin antisepsis: preclinical investigations. JB JS Open Access. 2020;5(3) doi: 10.2106/JBJS.OA.19.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Forrest G.L., et al. Genomic sequence and expression of a cloned human carbonyl reductase gene with daunorubicin reductase activity. Mol. Pharmacol. 1991;40(4):502–507. [PubMed] [Google Scholar]
- 69.Herman E.H., et al. Comparison of the effectiveness of (+/-)-1,2-bis(3,5-dioxopiperazinyl-1-yl)propane (ICRF-187) and N-acetylcysteine in preventing chronic doxorubicin cardiotoxicity in beagles. Cancer Res. 1985;45(1):276–281. [PubMed] [Google Scholar]
- 70.Olson R.D., et al. Doxorubicin cardiotoxicity may be caused by its metabolite, doxorubicinol. Proc. Natl. Acad. Sci. USA. 1988;85(10):3585–3589. doi: 10.1073/pnas.85.10.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Siveski-Iliskovic N., et al. Probucol protects against adriamycin cardiomyopathy without interfering with its antitumor effect. Circulation. 1995;91(1):10–15. doi: 10.1161/01.cir.91.1.10. [DOI] [PubMed] [Google Scholar]
- 72.Kang Y.J., Chen Y., Epstein P.N. Suppression of doxorubicin cardiotoxicity by overexpression of catalase in the heart of transgenic mice. J. Biol. Chem. 1996;271(21):12610–12616. doi: 10.1074/jbc.271.21.12610. [DOI] [PubMed] [Google Scholar]
- 73.Weiss R.B. The anthracyclines: will we ever find a better doxorubicin? Semin. Oncol. 1992;19(6):670–686. [PubMed] [Google Scholar]
- 74.Minotti G., Cairo G., Monti E. Role of iron in anthracycline cardiotoxicity: new tunes for an old song? FASEB J. 1999;13(2):199–212. [PubMed] [Google Scholar]
- 75.Morse M.A., Stoner G.D. Cancer chemoprevention: principles and prospects. Carcinogenesis. 1993;14(9):1737–1746. doi: 10.1093/carcin/14.9.1737. [DOI] [PubMed] [Google Scholar]
- 76.Tsao A.S., Kim E.S., Hong W.K. Chemoprevention of cancer. CA Cancer J. Clin. 2004;54(3):150–180. doi: 10.3322/canjclin.54.3.150. [DOI] [PubMed] [Google Scholar]
- 77.Wattenberg L.W. Chemoprevention of cancer. Cancer Res. 1985;45(1):1–8. [PubMed] [Google Scholar]
- 78.Yilmaz S., et al. The effect of antioxidants in Ehrlich Ascites Cancer. Cell Mol. Biol. 2021;67(2):20–24. doi: 10.14715/cmb/2021.67.2.4. [DOI] [PubMed] [Google Scholar]
- 79.Farabegoli F., Pinheiro M. Epigallocatechin-3-Gallate delivery in lipid-based nanoparticles: potentiality and perspectives for future applications in cancer chemoprevention and therapy. Front. Pharmacol. 2022;13 doi: 10.3389/fphar.2022.809706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Manson M.M., et al. Blocking and suppressing mechanisms of chemoprevention by dietary constituents. Toxicol. Lett. 2000;112–113:499–505. doi: 10.1016/s0378-4274(99)00211-8. [DOI] [PubMed] [Google Scholar]
- 81.Greten F.R., Grivennikov S.I. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. 2019;51(1):27–41. doi: 10.1016/j.immuni.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arthur J.C., et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338(6103):120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Binnewies M., et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018;24(5):541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Albrengues J., et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361:6409. doi: 10.1126/science.aao4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Canli O., et al. Myeloid cell-derived reactive oxygen species induce epithelial mutagenesis. Cancer Cell. 2017;32(6):869–883. doi: 10.1016/j.ccell.2017.11.004. e5. [DOI] [PubMed] [Google Scholar]
- 86.Chang W.C., et al. Loss of p53 enhances the induction of colitis-associated neoplasia by dextran sulfate sodium. Carcinogenesis. 2007;28(11):2375–2381. doi: 10.1093/carcin/bgm134. [DOI] [PubMed] [Google Scholar]
- 87.Hussain S.P., Hofseth L.J., Harris C.C. Radical causes of cancer. Nat. Rev. Cancer. 2003;3(4):276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 88.Robles A.I., et al. Whole-exome sequencing analyses of inflammatory bowel disease-associated colorectal cancers. Gastroenterology. 2016;150(4):931–943. doi: 10.1053/j.gastro.2015.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meira L.B., et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Invest. 2008;118(7):2516–2525. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Motamedi Z., et al. Combined effects of protocatechuic acid and 5-fluorouracil on p53 gene expression and apoptosis in gastric adenocarcinoma cells. Turk. J. Pharm. Sci. 2020;17(6):578–585. doi: 10.4274/tjps.galenos.2019.69335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang S., et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012;18(11):1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 92.Cui N., et al. Doxorubicin-induced cardiotoxicity is maturation dependent due to the shift from topoisomerase IIalpha to IIbeta in human stem cell derived cardiomyocytes. J. Cell Mol. Med. 2019;23(7):4627–4639. doi: 10.1111/jcmm.14346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee W.J., Lee S.H. Protocatechuic acid protects hepatocytes against hydrogen peroxide-induced oxidative stress. Curr. Res. Food Sci. 2022;5:222–227. doi: 10.1016/j.crfs.2022.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chang L.K., et al. Mitochondrial involvement in the point of no return in neuronal apoptosis. Biochimie. 2002;84(2–3):223–231. doi: 10.1016/s0300-9084(02)01372-x. [DOI] [PubMed] [Google Scholar]
- 95.Dadsena S., Zollo C., Garcia-Saez A.J. Mechanisms of mitochondrial cell death. Biochem. Soc. Trans. 2021;49(2):663–674. doi: 10.1042/BST20200522. [DOI] [PubMed] [Google Scholar]
- 96.Park H.J., et al. The PARP inhibitor olaparib potentiates the effect of the DNA damaging agent doxorubicin in osteosarcoma. J. Exp. Clin. Cancer Res. 2018;37(1):107. doi: 10.1186/s13046-018-0772-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kitazumi I., Tsukahara M. Regulation of DNA fragmentation: the role of caspases and phosphorylation. FEBS J. 2011;278(3):427–441. doi: 10.1111/j.1742-4658.2010.07975.x. [DOI] [PubMed] [Google Scholar]
- 98.Artus C., et al. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. EMBO J. 2010;29(9):1585–1599. doi: 10.1038/emboj.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Galluzzi L., et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kroemer G., et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16(1):3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Font-Belmonte E., et al. Post-ischemic salubrinal administration reduces necroptosis in a rat model of global cerebral ischemia. J. Neurochem. 2019;151(6):777–794. doi: 10.1111/jnc.14789. [DOI] [PubMed] [Google Scholar]
- 102.Li Z., et al. Neuroprotective effects of protocatechuic acid on sodium arsenate induced toxicity in mice: role of oxidative stress, inflammation, and apoptosis. Chem. Biol. Interact. 2021;337 doi: 10.1016/j.cbi.2021.109392. [DOI] [PubMed] [Google Scholar]
- 103.Zhang L., et al. Calycosin alleviates doxorubicin-induced cardiotoxicity and pyroptosis by inhibiting NLRP3 inflammasome activation. Oxid. Med. Cell Longev. 2022;2022 doi: 10.1155/2022/1733834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hou X., et al. USP47-mediated deubiquitination and stabilization of TCEA3 attenuates pyroptosis and apoptosis of colorectal cancer cells induced by chemotherapeutic doxorubicin. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.713322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zheng X., et al. Bnip3 mediates doxorubicin-induced cardiomyocyte pyroptosis via caspase-3/GSDME. Life Sci. 2020;242 doi: 10.1016/j.lfs.2019.117186. [DOI] [PubMed] [Google Scholar]
- 106.Burton T.R., Gibson S.B. The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ. 2009;16(4):515–523. doi: 10.1038/cdd.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mai F.Y., et al. Caspase-3-mediated GSDME activation contributes to cisplatin- and doxorubicin-induced secondary necrosis in mouse macrophages. Cell Prolif. 2019;52(5) doi: 10.1111/cpr.12663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tavakoli Dargani Z., Singla D.K. Embryonic stem cell-derived exosomes inhibit doxorubicin-induced TLR4-NLRP3-mediated cell death-pyroptosis. Am. J. Physiol. Heart Circ. Physiol. 2019;317(2):H460–H471. doi: 10.1152/ajpheart.00056.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sagulenko V., et al. Caspase-1 is an apical caspase leading to caspase-3 cleavage in the AIM2 inflammasome response, independent of Caspase-8. J. Mol. Biol. 2018;430(2):238–247. doi: 10.1016/j.jmb.2017.10.028. [DOI] [PubMed] [Google Scholar]
- 110.Zeng C., et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. 2020;34 doi: 10.1016/j.redox.2020.101523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Park J., et al. Daily injection of melatonin inhibits insulin resistance induced by chronic mealtime shift. Physiol. Rep. 2022;10(6) doi: 10.14814/phy2.15227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shi X., et al. Oxidative stress, vascular endothelium, and the pathology of neurodegeneration in retina. Antioxidants. 2022;11(3) doi: 10.3390/antiox11030543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mukhopadhyay P., et al. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am. J. Physiol. Heart Circ. Physiol. 2009;296(5):H1466–H1483. doi: 10.1152/ajpheart.00795.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Krishnamurthy B., et al. Febuxostat ameliorates doxorubicin-induced cardiotoxicity in rats. Chem. Biol. Interact. 2015;237:96–103. doi: 10.1016/j.cbi.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 115.Ashtar M., et al. The roles of ROS generation in RANKL-induced osteoclastogenesis: suppressive effects of Febuxostat. Cancers. 2020;12(4) doi: 10.3390/cancers12040929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Moruno-Manchon J.F., et al. Peroxisomes contribute to oxidative stress in neurons during doxorubicin-based chemotherapy. Mol. Cell Neurosci. 2018;86:65–71. doi: 10.1016/j.mcn.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tayeh Z., Ofir R. Asteriscus graveolens extract in combination with cisplatin/etoposide/doxorubicin suppresses lymphoma cell growth through induction of Caspase-3 dependent apoptosis. Int. J. Mol. Sci. 2018;19(8) doi: 10.3390/ijms19082219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McLaughlin D., et al. Signalling mechanisms underlying doxorubicin and Nox2 NADPH oxidase-induced cardiomyopathy: involvement of mitofusin-2. Br. J. Pharmacol. 2017;174(21):3677–3695. doi: 10.1111/bph.13773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dukhande V.V., et al. Reduced glutathione regenerating enzymes undergo developmental decline and sexual dimorphism in the rat cerebral cortex. Brain Res. 2009;1286:19–24. doi: 10.1016/j.brainres.2009.05.029. [DOI] [PubMed] [Google Scholar]
- 120.Xiao W., et al. NAD(H) and NADP(H) redox couples and cellular energy metabolism. Antioxid. Redox Signal. 2018;28(3):251–272. doi: 10.1089/ars.2017.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Brandes R.P., Weissmann N., Schroder K. Nox family NADPH oxidases: molecular mechanisms of activation. Free Radic. Biol. Med. 2014;76:208–226. doi: 10.1016/j.freeradbiomed.2014.07.046. [DOI] [PubMed] [Google Scholar]
- 122.Efentakis P., et al. Levosimendan prevents doxorubicin-induced cardiotoxicity in time- and dose-dependent manner: implications for inotropy. Cardiovasc. Res. 2020;116(3):576–591. doi: 10.1093/cvr/cvz163. [DOI] [PubMed] [Google Scholar]
- 123.Hayyan M., Hashim M.A., AlNashef I.M. Superoxide ion: generation and chemical implications. Chem. Rev. 2016;116(5):3029–3085. doi: 10.1021/acs.chemrev.5b00407. [DOI] [PubMed] [Google Scholar]
- 124.Tang J., et al. Selective hydrogen peroxide conversion tailored by surface, interface, and device engineering. Joule. 2021;5(6):1432–1461. [Google Scholar]
- 125.Saito S., Kawabata J. Effects of electron-withdrawing substituents on DPPH radical scavenging reactions of protocatechuic acid and its analogues in alcoholic solvents. Tetrahedron. 2005;61(34):8101–8108. [Google Scholar]
- 126.Aslam Bhatti H., et al. Protocatecheuic acid underlies the antioxidant activity exhibited by Illicium verum fruit. J. Anal. Pharm. Res. 2017;6(3) [Google Scholar]
- 127.Font-Belmonte E., Gonzalez-Rodriguez P., Fernandez-Lopez A. Necroptosis in global cerebral ischemia: a role for endoplasmic reticulum stress. Neural Regen. Res. 2020;15(3):455–456. doi: 10.4103/1673-5374.266054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jin S., et al. Dioscin ameliorates cisplatin-induced intestinal toxicity by mitigating oxidative stress and inflammation. Int. Immunopharmacol. 2022;111 doi: 10.1016/j.intimp.2022.109111. [DOI] [PubMed] [Google Scholar]
- 129.Papanastasopoulos P., Stebbing J. Molecular basis of 5-fluorouracil-related toxicity: lessons from clinical practice. Anticancer Res. 2014;34(4):1531–1535. [PubMed] [Google Scholar]
- 130.Benjanuwattra J., et al. Doxorubicin and its proarrhythmic effects: a comprehensive review of the evidence from experimental and clinical studies. Pharmacol. Res. 2020;151 doi: 10.1016/j.phrs.2019.104542. [DOI] [PubMed] [Google Scholar]
- 131.Larsen R.L., et al. Electrocardiographic changes and arrhythmias after cancer therapy in children and young adults. Am. J. Cardiol. 1992;70(1):73–77. doi: 10.1016/0002-9149(92)91393-i. [DOI] [PubMed] [Google Scholar]
- 132.Raabe N.K., Storstein L. Cardiac arrhythmias in patients with small cell lung cancer and cardiac disease before, during and after doxorubicin administration. An evaluation of acute cardiotoxicity by continuous 24-hour Holter monitoring. Acta Oncol. 1991;30(7):843–846. doi: 10.3109/02841869109091832. [DOI] [PubMed] [Google Scholar]
- 133.Kilickap S., et al. Early and late arrhythmogenic effects of doxorubicin. South Med. J. 2007;100(3):262–265. doi: 10.1097/01.smj.0000257382.89910.fe. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.