

ABSTRACT
Candida albicans is an opportunistic human fungal pathogen that causes invasive infections in immunocompromised individuals. Despite the high anticandidal activity among the echinocandins (ECNs), a first-line therapy, resistance remains an issue. Furthermore, many clinical isolates display decreased ECN susceptibility, a physiological state which is thought to lead to resistance. Determining the factors that can decrease susceptibility is of high importance. We searched for such factors genome-wide by comparing the transcriptional profiles of five mutants that acquired decreased caspofungin susceptibility in vitro in the absence of canonical FKS1 resistance mutations. The mutants were derived from two genetic backgrounds and arose due to independent mutational events, some with monosomic chromosome 5 (Ch5). We found that the mutants exhibit common transcriptional changes. In particular, all mutants upregulate five genes from Ch2 in concert. Knockout experiments show that all five genes positively influence caspofungin and anidulafungin susceptibility and play a role in regulating the cell wall mannan and glucan contents. The functions of three of these genes, orf19.1766, orf19.6867, and orf19.5833, were previously unknown, and our work expands the known functions of LEU42 and PR26. Importantly, orf19.1766 and LEU42 have no human orthologues. Our results provide important clues as to basic mechanisms of survival in the presence of ECNs while identifying new genes controlling ECN susceptibility and revealing new targets for the development of novel antifungal drugs.
KEYWORDS: Candida albicans, echinocandin susceptibility, drug adaptation, RNA profiling, drug tolerance
INTRODUCTION
Recent reviews have highlighted the worldwide increase in fungal infections, including Candida species, which are emerging as a major problem. Indeed, Candida albicans is the most common cause of systemic candidiasis, along with infections by non-albicans Candida species, increasingly displaying resistance to antifungals (1–5). C. albicans is a unicellular budding fungus that lives as part of the normal human gut and genital microflora but is an important opportunistic infectious agent in immunocompromised individuals (6). Because of their low toxicity and high efficacy, especially against isolates that are resistant to azole drugs, compounds from the echinocandin (ECN) class are now recommended “frontline” treatments. These drugs inhibit glucan synthase and kill C. albicans cells by disrupting cell wall biosynthesis. Although overall resistance to ECNs remains relatively low, the increased use of ECNs is associated with increased resistance to ECNs in C. albicans as well as some non-albicans Candida species (6–10). ECN resistance is currently recognized as being due to a single known mechanism involving mutations in the FKS1 (orf19.2929) gene, which encodes a catalytic subunit of the 1,3-β-glucan synthase complex, in various Candida species and the FKS1 gene or the orthologous FKS2 gene (orf19.3269) in Candida glabrata (11).
However, it has now become clear that C. albicans possesses mechanisms independent of FKS1 mutations that can decrease susceptibility to ECNs, although these mechanisms do not confer clinical resistance (12). In this regard, positive regulators for which gene depletion increases ECN susceptibility have been reported in C. albicans and the related species C. glabrata, including genes as diverse as heat shock protein Hsp90 acting via calcineurin; mitogen-activated protein (MAP) kinase Mkc1 acting in concert with calcineurin and Hsp90; the transcription factor CAS5; the chaperonin complex chaperonin containing TCP-1 (CCT); CHS genes for chitin synthases; the YCK2 stress kinase; the cell wall factors YPS1, YPK2, and SLT2; mitochondrial complexes I and IV; as well as the posttranscriptional regulator SSD1 (13–20). In addition, we identified negative regulators for which gene depletion decreases ECN susceptibility in C. albicans, including the chitinase CHT2, the glucanosyltransferase PGA4, and a putative transcription factor, CSU51 (12).
Genes that can decrease drug susceptibility are now considered to be of critical importance as they can provide a window of opportunity for the appearance of FKS1 mutations and thus can govern the evolution of ECN resistance, as has been demonstrated by Singh-Babak and colleagues using C. glabrata (21). Despite the importance of genes modulating ECN susceptibility and a substantial collective effort by several laboratories to identify such genes, they remain largely unexplored.
In this work, we took a novel approach by initiating a genome-wide search for genes that influence ECN susceptibility and can contribute to the adaptation of C. albicans to ECNs. We used mutants with decreased caspofungin susceptibility in the absence of FKS1 mutations that arose on plates supplemented with lethal amounts of caspofungin. Differentially expressed genes were identified in isogenic parent-mutant pairs and compared among the adapted mutants. Our strategy identified a set of five genes on chromosome 2 (Ch2) that exhibit consistency in transcriptional changes among caspofungin-adapted mutants and individually contribute to ECN susceptibility characteristics.
RESULTS
Genome-wide expression changes in five caspofungin-adapted mutants.
We used RNA sequencing (RNA-seq) to prepare genome-wide transcriptional profiles of five representative mutants that arose due to distinct mutational events on plates supplemented with lethal amounts of caspofungin and independently acquired decreased susceptibility to caspofungin in the absence of FKS1 mutations (22). Of these strains, JMC160-2-5 and JMC200-2-5 are normal diploids, whereas JMC200-3-4, SMC60-2-5, and SMC60-3-4 lack one Ch5, a condition known to confer adaptation to caspofungin (22) (see Table 1 for the origins, relationships, and properties of the adapted mutants). The chromosome complements of all adapted mutants were extensively characterized previously (22).
TABLE 1.
C. albicans parental strains and caspofungin-adapted mutantsa used in the study
| Strain | Description | Phenotype | Source and/or reference |
|---|---|---|---|
| SC5314 | Parental strain; diploid | Susceptible to caspofungin | A. D. Johnson laboratory; 48 |
| SMC60-2-5 | Same as SC5314 but with Ch5 monosomy; MTLa | Adapted to caspofungin | 22 |
| SMC60-3-4 | Same as SC5314 but with Ch5 monosomy; MTLa | Adapted to caspofungin | 22 |
| JRCT1 | Parental strain; diploid | Adapted to caspofungin | 22 |
| JMC200-3-4 | Same as JRCT1 but with Ch5 monosomy; MTLα | Adapted to caspofungin | 22 |
| JMC160-2-5 | Same as JRCT1, but see the phenotype | Adapted to caspofungin | 22 |
| JMC200-2-5 | Same as JRCT1, but see the phenotype | Adapted to caspofungin | 22 |
| DPL225 | Parental strain; no FKS1 mutation | Elevated caspofungin MIC | 24 |
We performed stringent differential expression analysis by DESeq2 (at a false discovery rate [FDR] of <0.05) (see Data Set S1 in the supplemental material) and present differentially expressed genes having FDR values of <0.05 for each mutant by volcano plots that allow tracking genes of interest across the mutants (Fig. 1). Of the 6,018 predicted genes in C. albicans, we find a relatively large number of differentially expressed genes in each mutant, ranging from 14% to 41% of genes across the mutants. Most expression changes, as determined by the mutant/parent ratio, were within a ±4-fold range in all mutants. Importantly, we identified a downregulation of CHT2 (orf19.3895) in all mutants (Fig. 1; Data Sets S1 and S2), thus validating our statistical analysis. CHT2, which encodes glycosylphosphatidylinositol (GPI)-linked chitinase, was previously demonstrated to serve as a negative regulator of ECN susceptibility (23).
FIG 1.

Volcano plots of differentially expressed genes, as determined by the mutant/parent ratio, according to DESeq2 analysis of RNA-seq profiling of caspofungin-adapted mutants SMC60-2-5, SMC60-3-4, JMC200-3-4, JMC160-2-5, and JMC200-2-5. Included here are only differentially expressed genes having FDR values of <0.05. Statistical significance (FDR value) is plotted against the expression change (log2 fold) in mutant strains versus the respective parental strains. Red and blue dots represent up- and downregulated genes, respectively. Red and blue arrows indicate the total numbers of up- and downregulated genes, respectively. Black dots represent ECS1, ECS2, ECS3, PR26, and LEU42 of Ch2 as well as the control gene CHT2, as indicated.
We found that 62 differentially expressed genes were common across all adapted mutants (Table 2 and Fig. 2; Data Set S2). The statistical significance of the overlap between every set of differentially expressed genes was assessed using a pairwise hypergeometric test, and every pairwise comparison returned P values of less than 0.05, indicating that the overlaps are nonrandom and, hence, unique and significant.
TABLE 2.
Numbers of differentially expressed genes that are in common in caspofungin-adapted mutants JMC200-3-4, JMC160-2-5, JMC200-2-5, SMC60-2-5, and SMC60-3-4a
| Mutant | No. of differentially expressed genes | No. of genes in common |
|---|---|---|
| JRCT1 genetic background | ||
| JMC200-3-4 | 864 | 240 |
| JMC160-2-5 | 2,112 | |
| JMC200-2-5 | 1,632 | |
| SC5314 genetic background | ||
| SMC60-2-5 | 1,292 | 703 |
| SMC60-3-4 | 2,441 |
There were 62 genes in common across all mutants.
FIG 2.
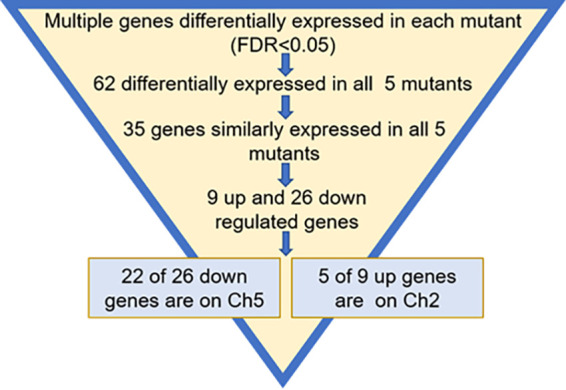
Schematic of the search for genes that change expression similarly in all mutants and choice of genes for validation.
Most importantly, of the 62 total genes shared by all five mutants, 35 genes exhibited similar changes in all mutants, with 26 genes being downregulated and 9 being upregulated (Fig. 2). Strikingly, there is a strong bias in the distribution of these genes over two chromosomes, with 22 out of 26 downregulated genes residing on Ch5 and 5 out of 9 upregulated genes residing on Ch2. The remaining 8 genes are distributed over various chromosomes.
We used Gene Ontology (GO) Slim Mapper with the specific term “Candida-GO slim: Process” to analyze the 35 genes exhibiting similar expression changes in each caspofungin-adapted mutant. Genes mapped to the following categories: 11 genes for the RNA metabolic process, 9 genes for the regulation of biological processes, 8 genes for ribosome biogenesis, 6 genes for transport, 5 genes for the response to stress, and 4 genes for biological processes and the response to chemicals (Fig. S1). Next, we used Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis for the 16 annotated genes within the total set of 35 genes. We found no significant enrichment of any pathway.
Validation of the importance of Ch2 genes for ECN susceptibility.
We set about validating the five Ch2 genes that are simultaneously upregulated in all caspofungin-adapted mutants (Fig. 2). We initially prepared independent deletions of each gene in the clinically resistant strain DPL15, which harbors the FKS1 mutation F641S/F641S. We observed no change in the growth of these deletion mutants in the presence of echinocandins (data not shown). We next prepared deletion mutants in the clinical isolate DPL225 (24), which serves as a model for an adapted mutant as it harbors no FKS1 mutation but exhibits an elevated MIC for caspofungin of ~0.50 μg/mL (Fig. 3). We prepared mutants in which both copies of orf19.1766, orf19.6867, or LEU4 (orf19.1375) were independently deleted, but we were able to obtain only mutants in which one copy of PR26 (orf19.5793) or orf19.5833 was deleted. Consistent with the inability to prepare null mutants, PR26 is listed as an essential gene in the Candida Genome Database (CGD) (http://www.candidagenome.org/), and we conclude that orf19.5833 is essential as well. We prepared three biological replicates of mutants for each gene and tested the growth of all deletion mutants versus the parental DPL225 strain in the presence of the ECN drug caspofungin or anidulafungin.
FIG 3.

Single-deletion mutants exhibit growth defects in the presence of ECNs. The heat maps represent the growth of various C. albicans strains by a broth microdilution assay. CAS and ANI refer to caspofungin and anidulafungin, respectively. Shown are parental strain DPL225; null mutants independently lacking ECS1, ECS2, or LEU42; and heterozygous null mutants lacking one copy of PR26 or ECS3. Also shown are three pairwise deletion mutants lacking ECS1 ECS2, ECS2 LEU42, and ECS1 LEU42 (essential genes are excluded). Names, genotypes of strains, and MIC50s are indicated on the right. The assay was conducted according to CLSI methods in RPMI 1640 medium with 2% glucose. The assay included a maximum caspofungin concentration of 0.5 μg/mL, a maximum anidulafungin concentration of 0.015 μg/mL, and 2-fold serial dilutions. A total of 103 cells were inoculated into each well in either triplicates or quadruplicates (technical replicates), and the tray was incubated at 35°C for 24 h. Control wells without the drug or without cells were included. The no-cell control was used to subtract the background. The no-drug control was used for normalization. The color bar for percent growth is presented below each panel. Note the elevated caspofungin MIC of DPL225.
Broth microdilution assays (see Materials and Methods) consistently showed the diminished growth of all deletion mutants in the presence of ECNs for all biological replicates (Fig. 3; Fig. S2). Of note, the lack of one copy of orf19.5833, which is denoted ECS3 in the figures, produced less growth in the presence of anidulafungin but not caspofungin (MIC50 of 0.0078 μg/mL versus 0.0156 μg/mL for the parental strain), whereas the lack of one copy of PR26 produced less growth in the presence of caspofungin but not anidulafungin (MIC50 of 0.250 μg/mL versus 0.500 μg/mL for the parental strain) (Fig. 3). The growth rates of the deletion strains in rich universal medium were the same or differed only slightly compared to those of the parental strain (Fig. S3). In summary, broth microdilution assays demonstrated that orf19.1766, orf19.6867, orf19.5833, LEU42, and PR26 act as positive regulators of ECN susceptibility. We therefore refer to orf19.1766, orf19.6867, and orf19.5833 as ECS1 (echinocandin susceptibility), ECS2, and ECS3, respectively.
Ch2 genes that are simultaneously upregulated in caspofungin-adapted mutants are important for the cell wall.
In order to address the function of the above-described Ch2 genes, we tested the survival of deletion mutants independently lacking one or both copies of ECS1, ECS2, ECS3, LEU42, or PR26 in the presence of chemical agents affecting various metabolic or signaling pathways, including tunicamycin (endoplasmic reticulum stress), geldanamycin and radicicol (inhibition of Hsp90), cyclosporine or FK506 (calcineurin pathway), H2O2 (oxidative stress), aureobasidin A (sphingolipid biosynthesis), hygromycin B (protein synthesis), and caffeine (antifungal activity). None of these tests showed significant changes in the growth of the deletion mutants versus the parental strain DPL225 (data not shown).
Spot assays were conducted to test for growth in the presence of the cell wall stressors calcofluor white and Congo red, the cell membrane disruptor SDS, or Zymolyase, an enzyme mixture that lyses the C. albicans cell wall (25, 26). Remarkably, the growth of the mutants was not affected by calcofluor white, Congo red, or SDS (data not shown). However, in the presence of Zymolyase, cells of all deletion mutants were lysed significantly faster than cells of the parental strain (Fig. 4, left). As essential activities of Zymolyase include β-1,3-glucan laminaripentao-hydrolase activity and β-1,3-glucanase activity (25), these data indicate that the cell wall glucan is more exposed in the deletion mutants.
FIG 4.
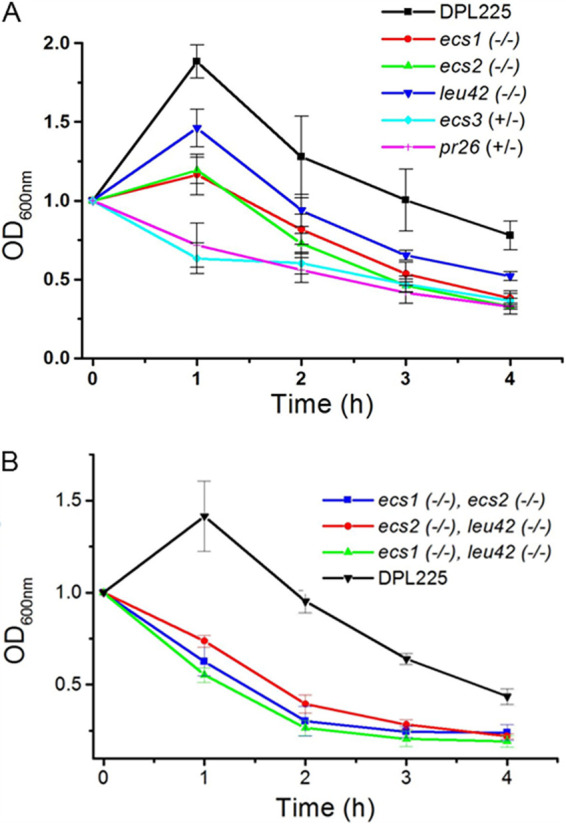
Survival of single-deletion (A) and double-deletion (B) mutants and parental strain DPL225 in the presence of Zymolyase. For better comparison, the optical density at the 0-h time point was scaled to 1 in all strains. The means and standard deviations of the optical density at 600 nm (OD600) values of each strain from four technical replicates are plotted against the time of incubation with Zymolyase. (A) DPL225, ecs1−/−, ecs2−/−, leu42−/−, ecs3+/−, and pr26+/−. Student’s t test was performed between the parental strain and each mutant strain at the 1-, 2-, 3-, and 4-h time points. P values for each time point ranged from 0.0000023 to 0.01, except for leu42 at 2 h, where the P value was 0.05. (B) DPL225, ecs1−/− ecs2−/−, ecs2−/− leu42−/−, and ecs1−/− leu41−/−. Student’s t test was performed between the parental strain and each double-deletion mutant at the 1-, 2-, 3-, and 4-h time points. P values for each time point were <0.001.
In order to obtain further information regarding cell wall changes in the deletion mutants, we measured the levels of glucans, chitin, and mannans, three major cell wall components. As presented in Fig. 5A, left, the bulk level of glucans increased in all deletion mutants from 10% to 80%. Taken together with no change in resistance to calcofluor white, Congo red, or SDS, this result is indicative of an increase in glucan exposure (27). Such exposure can be caused by a reduction in mannan/mannoprotein (28). Indeed, mannan levels are reduced to ~80% of the parental level in all deletion mutants except the ECS1 deletion mutant (Fig. 5C). The ecs1−/− mutant also showed a slight increase in chitin, while the remaining mutants had slightly decreased chitin levels, ranging from 1% to 10% (Fig. 5B). We conclude that ECS1, ECS2, ECS3, LEU42, or PR26 is involved in regulating cell wall components. However, the mechanism(s) governing cooperative changes in the levels of glucan, mannans, and chitin is yet to be elucidated.
FIG 5.

Deletion mutants exhibited distinct altered levels of cell wall glucans, chitin, and mannans. The left panels show parental strain DPL225; independent null mutants lacking ECS1, ECS2, or LEU42; and independent heterozygous null mutants lacking one copy of ECS3 or PR26, whereas the right panels show parental strain DPL225 and null mutants lacking both copies of ECS1 ECS2, ECS2 LEU42, or ECS1 LEU42. The experiments were performed on two biological replicates with four technical replicates for glucans and mannans, whereas one biological replicate with four technical replicates was used for chitin. The amount of glucans and chitin in the parental strain is set as 100%. The asterisks indicate a P value of <0.01 (**) or <0.001 (***), determined using Student’s t test.
In an effort to further characterize upregulated Ch2 genes, three double-null mutants, ecs1−/− ecs2−/−, ecs2−/− leu42−/−, and ecs1−/− leu42−/− (essential genes were excluded), were prepared in the parental strain DPL225. Drug susceptibility assays conducted with caspofungin and anidulafungin surprisingly showed a lack of sensitivity or reversed MIC phenotypes with the double-null mutants (Fig. 3). We next addressed the conditions of the cell walls of these mutants. Assays for cell lysis by Zymolyase as well as for measuring the levels of chitin or glucans showed overall similarities between single- and double-deletion mutants (Fig. 4 and Fig. 5A and B). However, mannans were increased in the double-deletion mutants, opposite of the decrease in the single-deletion mutants (Fig. 5C). One simple explanation for this effect is that deleting one gene has relatively modest effects on the cell wall while deleting two genes has more severe effects that then upregulate some alternative compensating process activated only under extreme circumstances.
Properties of proteins encoded by the five simultaneously upregulated Ch2 genes.
In an effort to better understand the functions of proteins that are encoded by the five simultaneously upregulated genes on Ch2, we pooled available information from the CGD. Despite coordinated regulation, the proteins are quite diverse, as inferred from amino acid sequence similarity and size, ranging from 129 amino acids (aa) to 571 aa. Multiple-sequence alignments failed to show common motifs or domains.
ECS1 has approximately 25% amino acid sequence identity with glycosylhydrolase from Bacteroides thetaiotaomicron VPI-5482. The protein possibly has a role in the hydrolysis of glycosidic bonds from complex polysaccharides in the cell wall (mannan, glucan, or chitin) of C. albicans. ECS2 has 38% amino acid sequence identity with a cytochrome b5-like fold from Arabidopsis thaliana. The protein possibly has a role in redox reactions in the electron transport chain in mitochondria. ECS3 has 73% amino acid sequence identity with ubiquitin binding motifs from Ufd1 of Saccharomyces cerevisiae. The protein possibly has a role in the ubiquitin-dependent protein catabolic process. Leu42p has a putative role in catalysis as an α-isopropylmalate synthase in the leucine biosynthesis pathway. Pr26p has significant amino acid sequence similarity with the 26S proteasome regulatory subunits from Saccharomyces cerevisiae and Homo sapiens and might have a role in proteasome-mediated protein degradation.
DISCUSSION
Several dozen clinical isolates of Candida species have been reported to display a wide range of increased MIC values for ECNs without canonical FKS1 mutations for resistance, including some at or below the MIC breakpoints (9, 29–31). Candida mutants with decreased susceptibility to ECNs can also be easily generated in vitro on agar plates supplemented with caspofungin (22, 32). These mutants typically exhibit 2- to 8-fold increases in MICs but lack critical FKS1 resistance mutations, similar to clinical isolates with increased MICs.
In an effort to understand factors that determine elevated MICs, we analyzed the genome-wide transcriptional profiles of five mutants that acquired increased MICs for caspofungin in the absence of FKS1 mutations (22). Each adapted mutant contained multiple genes exhibiting statistically significant expression changes, up to as much as 41% of the total genes, with ~50% of these genes having unknown functions. Large amounts of differentially expressed genes were also found in other studies in which Candida cells were treated with micafungin for 24 h or tunicamycin for 1 h (13, 33). We find that 62 differentially expressed genes were shared across all adapted mutants, with 35 exhibiting changes in the same direction. This is despite the findings that the adapted mutants were each independently derived from two genetic backgrounds and that 3 of 5 mutants harbored monosomic Ch5 ploidy. Statistical analyses indicated that this pattern of expression is by far not random but, instead, unique and significant (see Results). Multiple shared genes imply that similar pathways might be involved in adaptation to ECNs among the mutants.
We noted an apparent nonrandom genomic distribution of the 35 commonly regulated genes. Of these, 22 (~85%) of 26 downregulated genes reside on Ch5, while over half of the 9 upregulated genes reside on Ch2. This selective distribution is in accordance with the previously demonstrated role of Ch5 and Ch2 ploidy in the regulation of caspofungin susceptibility. The loss of one Ch5 decreases susceptibility due to the wholesale downregulation of Ch5 genes, including negative regulators of susceptibility (23), while the duplication of one Ch2 to generate trisomy upregulates a still-to-be-identified positive effector(s) residing on this chromosome (33).
The validation of the ECN phenotypes of the 22 genes that are downregulated on Ch5 is under way and will be reported elsewhere. Here, we report that the five genes that we identified as ECS1, ECS2, ECS3, LEU42, and PR26 that are simultaneously upregulated from Ch2 across all caspofungin-adapted mutants encode positive regulators of ECN susceptibility. PR26 has also been identified as a positive regulator of ECN susceptibility in a screen of the GRACE mutant collection (15). This provides additional support for the proposed role of PR26 in this work. It is of interest that ECS1 and LEU42 have no human orthologues.
Analyses of the deletion strains show that the above-described Ch2 genes contribute to the regulation of mannans, glucans, and chitin, three major cell wall components. We find that deletions of each of the genes have distinct effects on each of these components, which is reminiscent of glucan masking (see Results). The involvement of Ch2 genes in the control of the cell wall is consistent with the results of a previous study that determined that the cell wall, a target of ECN drugs, is remodeled in adapted mutants (22). Interestingly, combinations of deletions of the nonessential ECS1, ECS2, and LEU42 genes do not exhibit additive effects with regard to mannan and glucan contents, likely reflecting the individual genes’ regulation of diverse pathways leading to decreases in ECN susceptibility. However, further research is needed to better understand how the concerted upregulation of these genes contributes to the control of the cell wall and, subsequently, drug adaptation.
The simultaneous upregulation of multiple genes on normal disomic Ch2 is indicative of yet-to-be-elucidated epigenetic mechanisms that may holistically control the genes without a change in the gene copy number. Similarly, epigenetic regulation is likely also responsible for the simultaneous downregulation of multiple genes on disomic Ch5, as observed in two caspofungin-adapted mutants. Indeed, in a previous study, we found chromosome-wide increases in histone acetylation in adapted mutants exhibiting Ch5 monosomy, presumably associated with compensation for the dosage of genes not related to drug susceptibility (34). However, detailing the epigenetic mechanisms that control differentially expressed genes on disomic Ch5 and play a role in drug susceptibility remains a subject for future studies.
The transcriptome analysis presented here has inevitable limitations due to combining a stringent statistical analysis (DESeq2) with the requirement to consider only candidate genes with only increases in expression in all adapted mutants on Ch2 for validation and characterization. This approach obviously excluded upregulated genes on other chromosomes, genes exhibiting expression changes in opposite directions among adapted mutants, as well as genes that changed in some but not all mutants. An ability of these genes to modulate drug susceptibility might be obscured by, for example, alterations in the DNA sequence (inside or outside coding regions), epigenetic changes, some unspecified changes in the genetic background of an individual adapted mutant, as well as the culturing method. These genes can still be important factors and will be investigated in future studies.
Our study identifies the activation of similar pathways in independent caspofungin-adapted mutants with increased MICs. A part of the shared survival strategy is the concerted upregulation of five genes from Ch2, three of them with previously unknown functions, that contribute to the regulation of cell wall components. Elucidating the regulatory circuits that affect such holistic gene regulation in response to ECN challenge is a major goal of future work.
Our data add to a growing body of evidence pointing to the complexity of mechanisms responding to the action of toxic agents. This work expands our understanding of genes that modulate ECN susceptibility. Identifying new genes controlling ECN susceptibility helps to reveal new drug targets for the development of much-needed novel antifungal drugs.
MATERIALS AND METHODS
Strains, plasmids, and primers.
We used five C. albicans caspofungin-adapted mutants that arose due to independent mutational events from two genetic backgrounds, the reference strain SC5314 and the clinical isolate JRCT1 (Table 1). The chromosome conditions of all strains were extensively characterized previously (22) and also confirmed by DNA sequencing (DNA-seq), which will be presented elsewhere.
The plasmids and primers used in this study are presented in Table S1 in the supplemental material.
Maintenance and growth of strains and media.
Cells were maintained, stored, and grown using our standardized approach that prevents the induction of chromosome instability, as previously described (35). This approach favors maintaining cells that represent a major fraction of the population of cells (36). Briefly, cells were stored at −70°C. When needed, cells from a −70°C stock were streaked for independent colonies onto yeast extract-peptone-dextrose (YPD) plates and incubated at 37°C until young colonies with a size of approximately 1 × 105 to 3 × 105 cells/colony grew up. Young colonies were collected, a proper dilution in sterile water was prepared with the aid of a hemacytometer, approximately 3,000 CFU were plated onto each plate, and plates were incubated until young colonies appeared.
Cells were stored in a 25% (vol/vol) glycerol solution at −70°C to interrupt metabolism and routinely grown at 37°C.
YPD medium (1% yeast extract, 2% peptone, 2% dextrose), synthetic dextrose (SD) medium (0.67% yeast nitrogen base without amino acids, 2% dextrose), and synthetic medium with sorbitol as the sole carbon source (0.67% yeast nitrogen base without amino acids, 2% sorbitol) were previously described (37, 38). RPMI 1640 medium (Sigma, St. Louis, MO, USA) was supplemented with 2% glucose. In order to prepare solid medium, 2% (wt/vol) agar was added. Nourseothricin at 150 μg/mL (Jena Bioscience GmbH, Jena, Germany), uridine at 50 μg/mL (Sigma-Aldrich, St. Louis, MO, USA), caspofungin (Merck Sharp & Dohme Corp., Kenilworth, NJ, USA), or anidulafungin (Pfizer Inc., New York, NY, USA) was added when needed. We also used Zymolyase 100T (U.S. Biological, Swampscott, MA, USA), calcofluor white (MP Biomedicals, Solon, OH, USA), Congo red (MP Biomedicals, Solon, OH, USA), SDS (Sigma-Aldrich, St. Louis, MO, USA), tunicamycin (Sigma-Aldrich, St. Louis, MO, USA), geldanamycin (MedChemExpress LLC, Monmouth Junction, NJ, USA), radicicol (Adipogen Corp., San Diego, CA, USA), cyclosporine (TCI, Portland, OR, USA), FK506 (Research Products International, Mt. Prospect, IL, USA), H2O2 (Ward’s Science, Rochester, NY, USA), aureobasidin A (TaKaRa, Mountain View, CA, USA), hygromycin B (MedChemExpress LLC, Monmouth Junction, NJ, USA), and caffeine (Sigma-Aldrich, St. Louis, MO, USA).
RNA sequencing and analysis.
Cells from a −70°C stock were streaked for independent colonies onto YPD plates and incubated at 37°C until young colonies with a size of approximately 1 × 105 to 3 × 105 cells/colony grew up. Young colonies were collected, a proper dilution in sterile water was prepared with the aid of a hemacytometer, approximately 3,000 CFU were plated onto each plate with sorbitol medium to prevent catabolite repression of glucose, and plates were incubated until young colonies appeared. Sorbitol plates were used to grow batches of independent cultures for each strain, and total RNA was prepared from three independent batches for each strain, as described previously (38). The RNA concentration was determined with the NanoDrop 1000 spectrophotometer (NanoDrop, Wilmington, DE), and RNA quality was assessed with the Agilent (Santa Clara, CA) Bioanalyzer at the Genomics Research Center of the University of Rochester. Transcription profiling from the RNA obtained from each batch was performed by RNA-seq using an Illumina HiSeq 2500 sequencer at the Genomics Research Center of the University of Rochester, as described previously (38).
Sequences were aligned against the C. albicans SC5314 genome ASM18296v3 using the Splice Transcript Alignment to a Reference (STAR) algorithm (39), counted with HTSeq (40), and normalized for total counts (counts per million [CPM]). We used a nonspecific filtering strategy to remove genes with low expression values. Differential expression was assessed by DESeq2 (41) to identify genes with significant differences in mean expression (FDR of <0.05).
Mutants were analyzed versus their parental strains (see Table 1 for the relationships between strains).
Two strains monosomic for Ch5, SMC60-2-5 and JMC200-3-4, were previously used by us to address the dosage compensation effect on a single Ch5 (38). In this previous publication, only the Ch5 transcriptome was analyzed using the CuffDiff2 method. In this work, the entire transcriptomes of these two strains were reanalyzed with the more stringent DESeq2 method.
Gene deletions by the transient CRISPR method.
We used the transient CRISPR-Cas9 method allowing the deletion of both copies of the target genes in C. albicans with a single transformation (42). The Candida codon-optimized CAS9 gene and the target-specific single guide RNA (sgRNA) were PCR amplified from plasmid pV1093. The homologous repair template having NAT1 was PCR amplified from plasmid pJK863 with primers 80 bp up- and downstream of the target gene. The CAS9 expression cassette, the sgRNA expression cassette, and the homologous repair templates at a ratio of 1 μg/1 μg/3 μg were used to transform C. albicans cells by lithium acetate transformation (23). NATR or HYGR transformants were selected on YPD plates supplemented with 150 μg/mL of nourseothricin or 600 μg/mL of hygromycin B. The correct target gene deletion was verified by PCR amplification using the 5′-flanking region and either the gene of interest (GI), NATR, or HYGR as well as by the amplification of the entire GI, NATR, or HYGR using 5′- and 3′-flanking regions (Fig. S4).
Broth microdilution assay to determine MICs.
We employed a broth microdilution assay according to the CLSI document M27-A3 broth microdilution method for yeasts (43), with some modifications. Briefly, the assay was conducted with RPMI 1640 medium buffered to pH 7.0 with 0.165 M morpholinepropanesulfonic acid (MOPS) supplemented with 2% glucose in a total volume of 200 μL. A series of 2-fold dilutions of caspofungin or anidulafungin was prepared directly in 96-well, flat-bottom, polystyrene microtiter plates. A total of 103 cells in a total volume of 100 μL were inoculated into each well in triplicates or quadruplicates (technical replicates) and incubated at 35°C for 24 h. The parental strain was included on each plate. Control wells without cells were used to subtract the background. A no-drug control well was used for normalization. The turbidities were measured with a Spark multimode microplate reader (Tecan, Zurich, Switzerland) at 600 nm. Normalized readings were generated in Microsoft Excel and presented as heat maps.
Zymolyase assay.
Cells were streaked from a −80°C stock onto YPD plates for independent colonies and incubated at 37°C for 19 h. Colonies were then collected, and cells were counted with a hemacytometer. The assay was conducted in sterile-suspension culture plates with 96 flat-bottomed wells (Greiner Bio-One, Monroe, NC, USA). A total of 7 × 106 cells were adjusted in 200 μL of 10 mM Tris-HCl (pH 7.5) containing 50 μg/mL of Zymolyase and incubated at 37°C for 4 h. The optical density was measured every 15 min with a Spark multimode microplate reader (Tecan, Zurich, Switzerland) at 600 nm. The optical density from the background, the no-cell control, was subtracted from raw data, and the reading for each strain at the 0-h time point was considered 1.
Determination of glucan content in the cell wall.
The level of bulk of glucans was determined by an aniline blue assay (44). Briefly, cells were streaked from a −80°C stock onto YPD plates for independent colonies and incubated at 37°C for 19 h. Colonies were then collected, and cells were counted with a hemacytometer. A total of 106 cells from each sample were harvested and washed twice with Tris-EDTA (TE) buffer (10 mM Tris-HCl, 1 mM EDTA [pH 8]). Cells were suspended in 500 μL of TE buffer. Next, 100 μL of 6 M NaOH was added, and the cells were incubated at 80°C for 30 min for glucan solubilization. A total of 2.1 mL of an aniline blue mixture (0.03% aniline blue, 0.18 M HCl, 0.49 M glycine-NaOH [pH 9.5]) was added to solubilized glucans. The samples were incubated at 50°C for 30 min and then at room temperature for 30 min. Fluorescence was measured in a plate reader (Spectra Max; Molecular Devices Corp.) at an excitation wavelength of 400 nm and an emission wavelength of 469 nm, with a cutoff of 455 nm.
Determination of chitin content in the cell wall.
The amount of chitin was determined by measuring the absorbance of glucosamine released by acid hydrolysis of the purified cell wall as described previously (45). Briefly, cells were inoculated from a −80°C stock into 10 mL of YPD and incubated at 37°C for 16 h with shaking at 220 rpm. Cells were harvested, washed with distilled water, suspended in sorbitol lysis buffer (1 M sorbitol, 0.1 M EDTA [pH 7.4]), and disrupted with 0.5-mm glass beads (product no. 11079105; BioSpec Products Inc.). The pellet was washed 5 times with 1 M NaCl. Cell wall proteins were extracted with SDS-mercaptoethanol extraction buffer (50 mM Tris, 2% SDS, 0.3 M mercaptoethanol) by incubation at 100°C for 10 min. The pellet was washed three times with distilled water. Cell wall pellets were air dried and weighed. Dry cell wall samples were incubated with 1 mL of 6 M HCl at 100°C for 17 h. HCl was evaporated by incubating the samples at 65°C for ~25 h. The acid hydrolysate was dissolved in 1 mL of distilled water. One hundred microliters of each sample was mixed with 100 μL of 1.5 M Na2CO3 in 4% acetyl acetone and incubated at 100°C for 20 min. Next, 700 μL of 96% ethanol and 100 μL of a p-dimethylaminobenzaldehyde solution in a 1:1 mixture of ethyl alcohol and concentrated HCl were added. The mixture was incubated at room temperature for 1 h. The absorbance at 520 nm was measured in a plate reader (Spectra Max; Molecular Devices Corp., Sunnyvale, CA). Glucosamine (Sigma-Aldrich, St. Louis, MO) was used to prepare the standard curve for chitin measurement. The chitin level was calculated as a percentage of the cell wall dry weight for each sample.
Determination of mannan content in the cell wall.
Mannan contents were determined with the alcian blue staining method (46). Briefly, cells were streaked from a −80°C stock onto YPD plates for independent colonies and incubated at 37°C for 19 h. The colonies were then collected, and cells were counted with a hemacytometer. Approximately 1.4 × 107 cells were collected and washed with distilled water. Cell pellets were suspended in 1 mL of 30 μg/mL of alcian blue in 0.02 M HCl (pH 3.0) and incubated at room temperature for 10 min with shaking. Cell suspensions were centrifuged, and the supernatants were collected. The absorbance at 620 nm was measured for 100 μL of the supernatants collected from each sample using a plate reader (Spectra Max; Molecular Devices Corp., Sunnyvale, CA). The alcian blue concentration was determined using a standard curve. Alcian blue binding per cell (picograms per cell) was calculated according to the formula x = [(u − v)/n] × 106, where x is the bound alcian blue (picograms per cell), u is the original alcian blue concentration (micrograms per milliliter), v is the final alcian blue concentration (micrograms per milliliter), and n is the number of cells stained.
Gene annotation and KEGG analysis.
GO annotation was performed on the differentially expressed genes to identify categories of biological processes using GO Slim Mapper at the CGD with the specific GO set name “Candida-GO slim: Process” (http://www.candidagenome.org). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed using DAVID database software. The Benjamini and Bonferroni procedures were used for multiple-testing correction for KEGG pathway analysis (47).
Data availability.
RNA-seq raw data for SC5314, JRCT1, SMC60-2-5, SMC60-3-4, JMC200-3-4, JMC160-2-5, JMC200-2-5 are available at Gene Expression Omnibus (GEO), https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE217094, with submission ID GSE217094.
ACKNOWLEDGMENTS
We thank Mark Dumont for stimulating discussions and intellectual input. We are grateful to David Perlin for intellectual input. We thank CGD curator Mark Skrzypek for helpful assistance. We thank Merck and Co., Inc., for the generous donation of caspofungin and Pfizer Inc., for the generous donation of anidulafungin via a compound transfer program.
This work was supported by National Institutes of Health grants 1R21AI159877-01 to M.D.K., R01GM052426 to J.J.H., and R01AI110764 and R01AI141884 to E.R.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Heredia MY, Gunasekaran D, Ikeh MAC, Nobile CJ, Rauceo JM. 2020. Transcriptional regulation of the caspofungin-induced cell wall damage response in Candida albicans. Curr Genet 66:1059–1068. 10.1007/s00294-020-01105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee Y, Puumala E, Robbins N, Cowen LE. 2021. Antifungal drug resistance: molecular mechanisms in Candida albicans and beyond. Chem Rev 121:3390–3411. 10.1021/acs.chemrev.0c00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pristov KE, Ghannoum MA. 2019. Resistance of Candida to azoles and echinocandins worldwide. Clin Microbiol Infect 25:792–798. 10.1016/j.cmi.2019.03.028. [DOI] [PubMed] [Google Scholar]
- 4.Robbins N, Cowen LE. 2021. Antifungal drug resistance: deciphering the mechanisms governing multidrug resistance in the fungal pathogen Candida glabrata. Curr Biol 31:R1520–R1523. 10.1016/j.cub.2021.09.071. [DOI] [PubMed] [Google Scholar]
- 5.Whaley SG, Berkow EL, Rybak JM, Nishimoto AT, Barker KS, Rogers PD. 2016. Azole antifungal resistance in Candida albicans and emerging non-albicans Candida species. Front Microbiol 7:2173. 10.3389/fmicb.2016.02173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arendrup MC, Perlin DS. 2014. Echinocandin resistance: an emerging clinical problem? Curr Opin Infect Dis 27:484–492. 10.1097/QCO.0000000000000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Rubio R, Hernandez RY, Clear A, Healey KR, Shor E, Perlin DS. 2021. Critical assessment of cell wall integrity factors contributing to in vivo echinocandin tolerance and resistance in Candida glabrata. Front Microbiol 12:702779. 10.3389/fmicb.2021.702779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Healey KR, Perlin DS. 2018. Fungal resistance to echinocandins and the MDR phenomenon in Candida glabrata. J Fungi (Basel) 4:105. 10.3390/jof4030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsumoto E, Boyken L, Tendolkar S, McDanel J, Castanheira M, Pfaller M, Diekema D. 2014. Candidemia surveillance in Iowa: emergence of echinocandin resistance. Diagn Microbiol Infect Dis 79:205–208. 10.1016/j.diagmicrobio.2014.02.016. [DOI] [PubMed] [Google Scholar]
- 10.Coste AT, Kritikos A, Li J, Khanna N, Goldenberger D, Garzoni C, Zehnder C, Boggian K, Neofytos D, Riat A, Bachmann D, Sanglard D, Lamoth F, Fungal Infection Network of Switzerland (FUNGINOS) . 2020. Emerging echinocandin-resistant Candida albicans and glabrata in Switzerland. Infection 48:761–766. 10.1007/s15010-020-01475-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perlin DS. 2015. Mechanisms of echinocandin antifungal drug resistance. Ann N Y Acad Sci 1354:1–11. 10.1111/nyas.12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sah SK, Hayes JJ, Rustchenko E. 2021. The role of aneuploidy in the emergence of echinocandin resistance in human fungal pathogen Candida albicans. PLoS Pathog 17:e1009564. 10.1371/journal.ppat.1009564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia-Rubio R, Jimenez-Ortigosa C, DeGregorio L, Quinteros C, Shor E, Perlin DS. 2021. Multifactorial role of mitochondria in echinocandin tolerance revealed by transcriptome analysis of drug-tolerant cells. mBio 12:e01959-21. 10.1128/mBio.01959-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Healey KR, Paderu P, Hou X, Jimenez Ortigosa C, Bagley N, Patel B, Zhao Y, Perlin DS. 2020. Differential regulation of echinocandin targets Fks1 and Fks2 in Candida glabrata by the post-transcriptional regulator Ssd1. J Fungi (Basel) 6:143. 10.3390/jof6030143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caplan T, Polvi EJ, Xie JL, Buckhalter S, Leach MD, Robbins N, Cowen LE. 2018. Functional genomic screening reveals core modulators of echinocandin stress responses in Candida albicans. Cell Rep 23:2292–2298. 10.1016/j.celrep.2018.04.084. [DOI] [PubMed] [Google Scholar]
- 16.Caplan T, Lorente-Macías Á, Stogios PJ, Evdokimova E, Hyde S, Wellington MA, Liston S, Iyer KR, Puumala E, Shekhar-Guturja T, Robbins N, Savchenko A, Krysan DJ, Whitesell L, Zuercher WJ, Cowen LE. 2020. Overcoming fungal echinocandin resistance through inhibition of the non-essential stress kinase Yck2. Cell Chem Biol 27:269–282.e5. 10.1016/j.chembiol.2019.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LaFayette SL, Collins C, Zaas AK, Schell WA, Betancourt-Quiroz M, Gunatilaka AAL, Perfect JR, Cowen LE. 2010. PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog 6:e1001069. 10.1371/journal.ppat.1001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh SD, Robbins N, Zaas AK, Schell WA, Perfect JR, Cowen LE. 2009. Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog 5:e1000532. 10.1371/journal.ppat.1000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker LA, Gow NAR, Munro CA. 2013. Elevated chitin content reduces the susceptibility of Candida species to caspofungin. Antimicrob Agents Chemother 57:146–154. 10.1128/AAC.01486-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie JL, Qin L, Miao Z, Grys BT, Diaz JDLC, Ting K, Krieger JR, Tong J, Tan K, Leach MD, Ketela T, Moran MF, Krysan DJ, Boone C, Andrews BJ, Selmecki A, Ho Wong K, Robbins N, Cowen LE. 2017. The Candida albicans transcription factor Cas5 couples stress responses, drug resistance and cell cycle regulation. Nat Commun 8:499. 10.1038/s41467-017-00547-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh-Babak SD, Babak T, Diezmann S, Hill JA, Xie JL, Chen Y-L, Poutanen SM, Rennie RP, Heitman J, Cowen LE. 2012. Global analysis of the evolution and mechanism of echinocandin resistance in Candida glabrata. PLoS Pathog 8:e1002718. 10.1371/journal.ppat.1002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang F, Zhang L, Wakabayashi H, Myers J, Jiang Y, Cao Y, Jimenez-Ortigosa C, Perlin DS, Rustchenko E. 2017. Tolerance to caspofungin in Candida albicans is associated with at least three distinctive mechanisms that govern expression of FKS genes and cell wall remodeling. Antimicrob Agents Chemother 61:e00071-17. 10.1128/AAC.00071-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suwunnakorn S, Wakabayashi H, Rustchenko E. 2016. Chromosome 5 of human pathogen Candida albicans carries multiple genes for negative control of caspofungin and anidulafungin susceptibility. Antimicrob Agents Chemother 60:7457–7467. 10.1128/AAC.01888-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiménez-Ortigosa C, Paderu P, Motyl MR, Perlin DS. 2014. Enfumafungin derivative MK-3118 shows increased in vitro potency against clinical echinocandin-resistant Candida species and Aspergillus species isolates. Antimicrob Agents Chemother 58:1248–1251. 10.1128/AAC.02145-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitamura K, Yamamoto Y. 1972. Purification and properties of an enzyme, Zymolyase, which lyses viable yeast cells. Arch Biochem Biophys 153:403–406. 10.1016/0003-9861(72)90461-4. [DOI] [PubMed] [Google Scholar]
- 26.Zlotnik H, Fernandez MP, Bowers B, Cabib E. 1984. Saccharomyces cerevisiae mannoproteins form an external cell wall layer that determines wall porosity. J Bacteriol 159:1018–1026. 10.1128/jb.159.3.1018-1026.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wheeler RT, Fink GR. 2006. A drug-sensitive genetic network masks fungi from the immune system. PLoS Pathog 2:e35. 10.1371/journal.ppat.0020035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li D, Williams D, Lowman D, Monteiro MA, Tan X, Kruppa M, Fonzi W, Roman E, Pla J, Calderone R. 2009. The Candida albicans histidine kinase Chk1p: signaling and cell wall mannan. Fungal Genet Biol 46:731–741. 10.1016/j.fgb.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Espinel-Ingroff A, Arendrup M, Cantón E, Cordoba S, Dannaoui E, García-Rodríguez J, Gonzalez GM, Govender NP, Martin-Mazuelos E, Lackner M, Lass-Flörl C, Linares Sicilia MJ, Rodriguez-Iglesias MA, Pelaez T, Shields RK, Garcia-Effron G, Guinea J, Sanguinetti M, Turnidge J. 2017. Multicenter study of method-dependent epidemiological cutoff values for detection of resistance in Candida spp. and Aspergillus spp. to amphotericin B and echinocandins for the Etest agar diffusion method. Antimicrob Agents Chemother 61:e01792-16. 10.1128/AAC.01792-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfaller MA, Messer SA, Woosley LN, Jones RN, Castanheira M. 2013. Echinocandin and triazole antifungal susceptibility profiles for clinical opportunistic yeast and mold isolates collected from 2010 to 2011: application of new CLSI clinical breakpoints and epidemiological cutoff values for characterization of geographic and temporal trends of antifungal resistance. J Clin Microbiol 51:2571–2581. 10.1128/JCM.00308-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfaller MA, Messer SA, Diekema DJ, Jones RN, Castanheira M. 2014. Use of micafungin as a surrogate marker to predict susceptibility and resistance to caspofungin among 3,764 clinical isolates of Candida by use of CLSI methods and interpretive criteria. J Clin Microbiol 52:108–114. 10.1128/JCM.02481-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Healey KR, Katiyar SK, Castanheira M, Pfaller MA, Edlind TD. 2011. Candida glabrata mutants demonstrating paradoxical reduced caspofungin susceptibility but increased micafungin susceptibility. Antimicrob Agents Chemother 55:3947–3949. 10.1128/AAC.00044-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang F, Gritsenko V, Slor Futterman Y, Gao L, Zhen C, Lu H, Jiang Y-Y, Berman J. 2021. Tunicamycin potentiates antifungal drug tolerance via aneuploidy in Candida albicans. mBio 12:e02272-21. 10.1128/mBio.02272-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wakabayashi H, Tucker C, Bethlendy G, Kravets A, Welle SL, Bulger M, Hayes JJ, Rustchenko E. 2017. NuA4 histone acetyltransferase activity is required for H4 acetylation on a dosage-compensated monosomic chromosome that confers resistance to fungal toxins. Epigenetics Chromatin 10:49. 10.1186/s13072-017-0156-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rustchenko-Bulgac EP. 1991. Variations of Candida albicans electrophoretic karyotypes. J Bacteriol 173:6586–6596. 10.1128/jb.173.20.6586-6596.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rustchenko E. 2007. Chromosome instability in Candida albicans. FEMS Yeast Res 7:2–11. 10.1111/j.1567-1364.2006.00150.x. [DOI] [PubMed] [Google Scholar]
- 37.Sherman F. 2002. Getting started with yeast. Methods Enzymol 350:3–41. 10.1016/s0076-6879(02)50954-x. [DOI] [PubMed] [Google Scholar]
- 38.Tucker C, Bhattacharya S, Wakabayashi H, Bellaousov S, Kravets A, Welle SL, Myers J, Hayes JJ, Bulger M, Rustchenko E. 2018. Transcriptional regulation on aneuploid chromosomes in diverse Candida albicans mutants. Sci Rep 8:1630. 10.1038/s41598-018-20106-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Min K, Ichikawa Y, Woolford CA, Mitchell AP. 2016. Candida albicans gene deletion with a transient CRISPR-Cas9 system. mSphere 1:e00130-16. 10.1128/mSphere.00130-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clinical and Laboratory Standards Institute. 2008. M27-A3 reference method for broth dilution antifungal susceptibility testing of yeasts: approved standard, 3rd ed. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 44.Suwunnakorn S, Wakabayashi H, Kordalewska M, Perlin DS, Rustchenko E. 2018. FKS2 and FKS3 genes of opportunistic human pathogen Candida albicans influence echinocandin susceptibility. Antimicrob Agents Chemother 62:e02299-17. 10.1128/AAC.02299-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Munro CA, Whitton RK, Bleddyn Hughes H, Rella M, Selvaggini S, Gow NAR. 2003. CHS8—a fourth chitin synthase gene of Candida albicans contributes to in vitro chitin synthase activity, but is dispensable for growth. Fungal Genet Biol 40:146–158. 10.1016/S1087-1845(03)00083-5. [DOI] [PubMed] [Google Scholar]
- 46.Hobson RP, Munro CA, Bates S, MacCallum DM, Cutler JE, Heinsbroek SEM, Brown GD, Odds FC, Gow NAR. 2004. Loss of cell wall mannosylphosphate in Candida albicans does not influence macrophage recognition. J Biol Chem 279:39628–39635. 10.1074/jbc.M405003200. [DOI] [PubMed] [Google Scholar]
- 47.Jiao X, Sherman BT, Huang DW, Stephens R, Baseler MW, Lane HC, Lempicki RA. 2012. DAVID-WS: a stateful Web service to facilitate gene/protein list analysis. Bioinformatics 28:1805–1806. 10.1093/bioinformatics/bts251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ahmad A, Kabir MA, Kravets A, Andaluz E, Larriba G, Rustchenko E. 2008. Chromosome instability and unusual features of some widely used strains of Candida albicans. Yeast 25:433–448. 10.1002/yea.1597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 and Fig. S1 to S4. Download aac.00977-22-s0001.pdf, PDF file, 0.5 MB (523KB, pdf)
Data Set S1. Download aac.00977-22-s0002.xlsx, XLSX file, 2.3 MB (2.3MB, xlsx)
Data Set S2. Download aac.00977-22-s0003.xlsx, XLSX file, 0.03 MB (27.8KB, xlsx)
Data Availability Statement
RNA-seq raw data for SC5314, JRCT1, SMC60-2-5, SMC60-3-4, JMC200-3-4, JMC160-2-5, JMC200-2-5 are available at Gene Expression Omnibus (GEO), https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE217094, with submission ID GSE217094.