

ABSTRACT
Ubiquitin regulatory X domain-containing proteins (UBXN) might be involved in diverse cellular processes. However, their in vivo physiological functions remain largely elusive. We recently showed that UBXN3B positively regulated stimulator-of-interferon-genes (STING)-mediated innate immune responses to DNA viruses. Herein, we reported the essential role of UBXN3B in the control of infection and immunopathogenesis of two arthritogenic RNA viruses, Chikungunya (CHIKV) and O’nyong’nyong (ONNV) viruses. Ubxn3b deficient (Ubxn3b−/−) mice presented higher viral loads, more severe foot swelling and immune infiltrates, and slower clearance of viruses and resolution of inflammation than the Ubxn3b+/+ littermates. While the serum cytokine levels were intact, the virus-specific immunoglobulin G and neutralizing antibody levels were lower in the Ubxn3b−/− mice. The Ubxn3b−/− mice had more neutrophils and macrophages, but much fewer B cells in the ipsilateral feet. Of note, this immune dysregulation was also observed in the spleens and blood of uninfected Ubxn3b−/− mice. UBXN3B restricted CHIKV replication in a cell-intrinsic manner but independent of type I IFN signaling. These results demonstrated a dual role of UBXN3B in the maintenance of immune homeostasis and control of RNA virus replication.
KEYWORDS: UBXN, alphavirus, Chikungunya virus, arthritogenic, hematopoiesis
INTRODUCTION
Alphavirus is a genus of the Togaviridae family of positive sense, single-stranded RNA viruses. These viruses are primarily mosquito-borne and pose a public health threat worldwide, particularly in tropical/subtropical regions. Several of them, including Chikungunya (CHIKV), O’nyong’nyong (ONNV), and Ross River viruses (RRV), are known to elicit arthritis in humans. Of note, CHIKV causes acute and chronic crippling arthralgia and long-term neurological disorders. CHIKV was first discovered in 1952, in Tanzania and was relatively silent for several decades. However, since 2005 CHIKV has re-emerged and caused several large outbreaks in Africa, Asia, and South America (1). CHIKV appeared on the Caribbean Islands in 2013 and has spread rapidly throughout Central and South America since then, resulting in ~3 million human infections and ~500 deaths (data were from the Pan America Health Organization). CHIKV infection in humans can be divided into two phases, with an acute viremic stage for approximately 1 week and a chronic phase without active viral replication but sustained inflammation for several months to years. Both phases are typical of elevated levels of interferons (IFN) and inflammatory mediators, massive immune infiltrates primarily including macrophages and monocytes but also neutrophils (acute phase only), dendritic cells, NK cells, and lymphocytes (2–7). Approximately 50% of CHIKV-infected patients suffer from rheumatic manifestations, with ~5% of the victims suffering from chronic inflammatory rheumatism (1, 8). However, there are no approved vaccines or specific antiviral drugs. This is partly due to a poor understanding of the protective and detrimental immune responses elicited by CHIKV. In mouse models, CHIKV infection results in brief viremia for usually 5 to 7 days, which is controlled primarily by the type I interferon (IFN) response (6, 9–11). However, viral clearance depends on virus-specific antibody responses (12–19). When inoculated directly into a mouse foot, CHIKV induces the first peak of foot swelling characteristic of edema at 2 to 3 days postinfection followed by a second peak with massive immune infiltrates at 6 to 8 days postinfection (10, 20–26).
Ubiquitin regulatory X (UBX) domain-containing proteins (UBXN) are putative cofactors of a highly conserved AAA ATPase, p97/VCP, which participates in diverse cellular processes such as homotypic membrane fusion, endoplasmic reticulum (ER) associated degradation (ERAD) of misfolded proteins, cell cycle, etc. (27). UBXNs could also be adaptors interfacing E3 ubiquitin ligases and their substrates (28, 29). We and other research groups recently showed that several UBXNs regulate the immune system including viral RNA-sensing RIG-I (retinoic acid-inducible gene 1) like receptor-mitochondrial antiviral viral signaling (RLR-MAVS) (30–32), Nuclear factor-κB (NF-κB) signaling (33, 34), Janus kinase (JAK)-signal transducer and activator of transcription proteins (STAT) signaling (35), and mitophagy (36), etc. However, the in vivo physiological functions of UBXNs remain poorly understood. To this end, with a mouse model we recently found that UBXN3B controlled DNA virus infection by positively regulating the viral DNA-sensing, cGAS (cyclic di-GMP-AMP synthase)-STING (stimulator-of-interferon-genes) signaling and innate immune responses (37). Of note, STING is critical to the control of CHIKV infection and arthritis pathogenesis likely independently of the type I IFN response (38). Therefore, we asked if UBXN3B also played an important role in the control of CHIKV pathogenesis in mice. Our results showed that Ubxn3b deficient (Ubxn3b−/−) mice were more prone to CHIKV/ONNV infection than Ubxn3b+/+ littermates, which was characterized by higher viral loads, more severe foot swelling and immune infiltrates, slower clearance of viruses, and resolution of inflammation. The levels of serum type I interferons (IFNs) and inflammatory mediators were similar or higher, while the serum virus-specific immunoglobulin G and neutralizing antibody levels were lower in Ubxn3b−/−. Mechanistically, UBXN3B was essential for the maintenance of immune cell homeostasis during viral infection and in steady-state. UBXN3B also controlled CHIKV replication in a cell-intrinsic but in a type I IFN-independent manner.
RESULTS
An essential role of UBXN3B in the control of CHIKV/ONNV pathogenesis.
We recently showed that UBXN3B controls DNA virus infection via STING (37), and STING is critical to the control of CHIKV infection and arthritis pathogenesis in mice (38). However, the UBXN3B function in RNA virus infection has not been studied in depth. To this end, we employed a tamoxifen-inducible Cre/LoxP model because UBXN3B was essential for embryonic development. We directly injected CHIKV into one footpad to induce swelling and acute arthritis in Ubxn3b+/+ (corn-oil treated Cre+ Ubxn3bflox/flox), Ubxn3b−/− (tamoxifen-treated Cre+ Ubxn3bflox/flox), and Ubxn3bflox/flox (tamoxifen-treated Cre negative Ubxn3bflox/flox, a control to exclude any effect of tamoxifen on foot swelling/viral replication) littermates. Both the Ubxn3b+/+ and Ubxn3bflox/flox mice showed progressive footpad swelling, which peaked at 6 days postinfection (pi) and resolved rapidly. In contrast, the Ubxn3b−/− mice presented more severe and persistent foot swelling at 4 days pi through 14 days pi (Fig. 1A and B). Histopathological analyses by hematoxylin and eosin (H&E) staining confirmed more immune infiltrates in the muscle and synovial cavity of Ubxn3b−/− than those of Ubxn3b+/+ littermates at 8 and 16 days pi (Fig. 1C to E). Similar results were observed with infection of another arthritogenic alphavirus, O’nyong’nyong (ONNV) at 12 days pi (Fig. 1E and G). Moreover, the CHIKV viremia at 2 and 4 days pi, and viral loads in the ipsilateral feet of Ubxn3b−/− mice at 4 and 8 days pi were significantly higher than those in Ubxn3b+/+ and Ubxn3bflox/flox littermates (Fig. 2). Of note, the infectious virions were cleared from the blood by 4 days and the feet by 8 days pi in the Ubxn3b+/+ while it remained high in the Ubxn3b−/− mice (Fig. 2B and D), suggesting a critical role of UBXN3B in the resolution of viral infection.
FIG 1.

UBXN3B was critical for the control of CHIKV/ONNV arthritis. Sex-and-age matched littermates were administered 1 × 105 PFU of CHIKV or 5 × 105 of ONNV subcutaneously in one footpad of Ubxn3b+/+ (corn-oil treated Cre+ Ubxn3bflox/flox), Ubxn3b−/− (tamoxifen-treated Cre+ Ubxn3bflox/flox), and Ubxn3bflox/flox (tamoxifen-treated Cre negative Ubxn3bflox/flox, a control to exclude any effect of tamoxifen on foot swelling/viral replication) littermates. (A) Representative images of foot swelling on day 8 postinfection (pi) (n = 3 animals per group). The red arrows indicate the foot inoculated with CHIKV. (B) Percent increase in the footpad dimensions of infected mice over before infection (baseline). Data point, mean ± SEM (n = 3 mice per genotype). (C to E) Representative micrographs of hematoxylin and eosin staining of ankle joints (C) on day 16 (N = 10 Ubxn3b+/+, 5 Ubxn3b−/−animals), (D) on day 8 (N = 3/group) post-CHIKV pi, and (E) on day 12 after ONNV infection (N = 7 Ubxn3b+/+, N = 6 Ubxn3b−/−animals). Objective lens, 4× and 20×. The lower plots (C to E) are the magnified (20×) portions of the squares in the upper plots. (F and G) The arbitrary scores of foot inflammation and damage using scales of 1 to 5, with 5 representing the worst disease presentation. Each symbol is one mouse. Error bar, mean ± SEM; *, P < 0.05; **, P < 0.01 (two-tailed Student's t test). The results are representative of 2 independent experiments.
FIG 2.
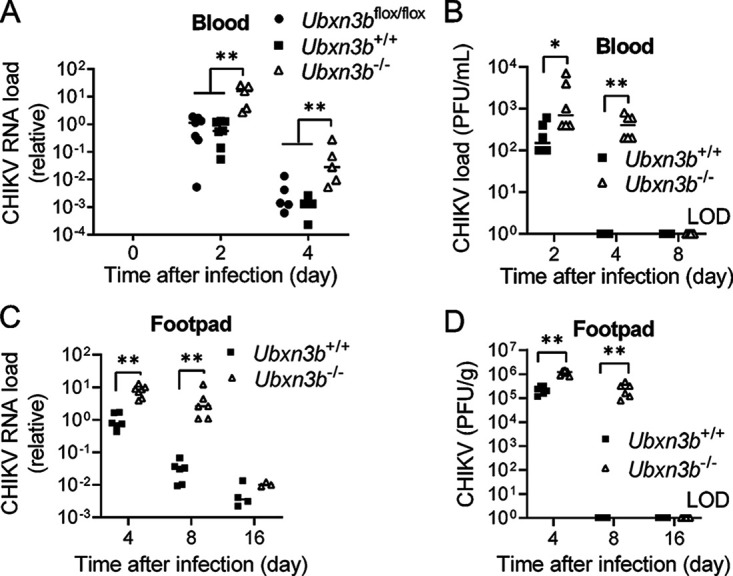
UBXN3B was essential for the control of CHIKV infection in mice. Sex-and-age matched littermates were administered 1 × 105 PFU of CHIKV subcutaneously in one footpad. (A and C) Quantitative RT-PCR (qPCR) quantification of CHIKV loads in (A) whole blood and (C) feet. (B and D) Viral titers in the (B) sera (plaque forming units [PFU/mL]) and (D) feet (PFU/gram tissue). Each symbol is one mouse, N = 6/group in (B and D). The short horizontal line is the median of the result. *, P < 0.05; **, P < 0.01 (two-tailed Student's t test). LOD, limit of detection. The results are representative of 2 independent experiments.
Acute CHIKV infection was associated with a systemic IFN and inflammatory response. Next, we measured the concentrations of serum innate immune molecules, including early antiviral type I IFNs (IFN-α) and type II IFN (IFN-γ), and those inflammatory mediators associated with severe arthritis in humans such as tumor necrosis factor (TNF-α), interleukin 6 (IL-6), IL-10, C-X-C motif chemokine ligand 10 (CXCL10), IL-1β, and granulocyte-macrophage colony-stimulating factor (GM-CSF). We noted that most cytokine levels were upregulated following CHIKV infection, though with different kinetics, except for IL-1β and GM-CSF (Fig. 3). The levels of CXCL10, TNF-α, IL-6, IL-10, CCL2, and CCL3 were higher in the Ubxn3b−/− than those in Ubxn3b+/+ mice at one or several time points even after viremia had been cleared at 8 days pi through 16 days pi (Fig. 3). The IFN-γ level peaked on day 6 in the Ubxn3b+/+ mice, and this peak shifted to day 8 in the Ubxn3b−/− animals, while the IFN-α level was similar between the Ubxn3b−/− and Ubxn3b+/+ mice (Fig. 3). During ONNV infection, we observed that the CXCL10 and IFN-α levels were higher in the Ubxn3b−/− than those in Ubxn3b+/+ littermates at several time points, and TNF-α at 12 days pi (Fig. 4). The differences between CHIKV and ONNV infection were likely due to different infection kinetics. We noted that ONNV infection caused more moderate foot swelling than CHIKV infection. Nonetheless, both the CHIKV and ONNV data demonstrated that cytokine responses were still intact or even higher in the Ubxn3b−/− mice.
FIG 3.

Cytokine responses were intact during CHIKV infection. Sex-and-age matched littermates were administered 1 × 105 PFU of CHIKV subcutaneously in one footpad. The serum cytokines, chemokines, and growth factors were quantitated by a bead-based multiplex ELISA, except for CCL2 and CCL3, which were quantified by individual ELISA. Each symbol is one mouse. The short horizontal line is the median of the result. *, P < 0.05; **, P < 0.01 (two-tailed Student's t test). The results are representative of 2 independent experiments.
FIG 4.

Cytokine responses were intact during ONNV infection. Sex-and-age matched littermates were administered 5 × 105 PFU of ONNV subcutaneously in one footpad. Serum cytokines, chemokines, and growth factors were quantitated by a bead-based multiplex ELISA. Each symbol is one mouse. The short horizontal line is the median of the result. *, P < 0.05; **, P < 0.01 (two-tailed Student's t test). The results are representative of 2 independent experiments.
An essential role of UBXN3B in the maintenance of immune cell homeostasis.
Because immune infiltration into muscles and joints was a hallmark of CHIKV arthritis, by H&E staining we confirmed that there were much more immune cells in the CHIKV-inoculated Ubxn3b−/− than Ubxn3b+/+ feet (Fig. 1C). We then resolved the different immune compartments in the feet inoculated with CHIKV by flow cytometry. We observed higher ratios and numbers of neutrophils and macrophages, but fewer B and CD4 T cells in the Ubxn3b−/− than Ubxn3b+/+ feet on day 7 pi. Notably, the B cell counts were reduced by ~5-fold. These differences were observed in other CHIKV-target immune organs, e.g., blood and spleens. In the spleen, both CD4 and CD8 T cell populations were also reduced by ~3-fold (Fig. 5A and B). Of note, the neutrophil-to-lymphocyte (N/L) ratios were much higher in all the tissues of the Ubxn3b−/− mice (Fig. 5C). These data suggested that UBXN3B signaling was essential for not only viral clearance but also immune cell homeostasis. We then asked if this dysregulated immune system in the Ubxn3b−/− mice was induced by CHIKV or constitutive. To this end, we analyzed the steady-state immune compartments. The total immune cell numbers in the feet of both uninfected Ubxn3b−/− and Ubxn3b+/+ mice were low with macrophages (mostly resident macrophages) being the dominant immune population, and there were no significant differences in either the myeloid or lymphoid compartments (Fig. 6). The ratios and counts of neutrophils and macrophages were much higher (4- to 7-fold), while B cells were much lower (3- to 10-fold) in the spleens and blood of Ubxn3b−/− than those in Ubxn3b+/+ animals (Fig. 6). The T cell counts were lower in the spleens of Ubxn3b−/− than those in Ubxn3b+/+ mice. After infection, the numbers of total immune cells and individual compartments in both Ubxn3b+/+ and Ubxn3b−/− feet increased significantly compared to the uninfected Ubxn3b+/+ and Ubxn3b−/− mice, respectively. While the changes (post/prior CHIKV) in neutrophil, B, and T cell counts were comparable between the Ubxn3b+/+ and Ubxn3b−/− mice, the change in macrophage in the Ubxn3b−/− mice was 2.5 times as many as in the Ubxn3b+/+ mice. Nonetheless, the absolute counts of neutrophils and macrophages were significantly greater, while the B cell number was significantly smaller in the infected-Ubxn3b−/− than infected-Ubxn3b+/+ mice (Fig. 5). These data demonstrated that UBXN3B was essential for immune cell homeostasis during steady-state and CHIKV infection.
FIG 5.

UBXN3B was essential for immune cell hemostasis during CHIKV infection. Sex-and-age matched littermates were administered 1 × 105 PFU of CHIKV subcutaneously in one footpad. On day 7 after infection, various immune cell compartments in the foot, spleen, and blood were analyzed by flow cytometry. (A) The percentage relative to CD45+ cells, (B) the counts of each immune cell type, and (C) the neutrophil-to-lymphocyte ratios (N/L). Neu, neutrophil; Mac, macrophage; DC, dendritic cell. The red/green arrows at the bottom indicate an increase/decrease in the Ubxn3b−/− compared to Ubxn3b+/+ mice. Bar, mean ± SEM; N = 6. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (two-tailed Student's t test). The results are representative of 2 independent experiments.
FIG 6.

UBXN3B was essential for steady-state immune cell hemostasis. Various immune cell compartments in the foot, spleen, and blood were analyzed by flow cytometry. (A) The percentage relative to CD45+ cells, and (B) counts of each immune cell type. Neu, neutrophil; Mac, macrophage; DC, dendritic cell. Each symbol is one mouse. Bar, mean ± SEM; N = 4. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (two-tailed Student's t test). The results are representative of 2 independent experiments.
Because B lymphocytes were significantly reduced in the Ubxn3b−/− mice, we asked if this deficiency influenced the production of virus-specific antibodies. Indeed, the concentrations of serum anti-CHIKV/ONNV immunoglobulin G (IgG) were lower in the Ubxn3b−/− than those in Ubxn3b+/+ mice (Fig. 7A). Moreover, the CHIKV-neutralizing antibody levels in the Ubxn3b−/− sera were also reduced. The fold-dilution for neutralizing 50% infectious virions (NT50) was 7595 for the Ubxn3b+/+ sera and 1595 for Ubxn3b−/− sera (day 16 pi), suggesting that the titers of CHIKV-neutralizing antibodies in the Ubxn3b−/− sera were reduced by ~3.7-fold (Fig. 7B).
FIG 7.

Virus specific-IgG production was impaired in Ubxn3b−/− mice. Sex-and-age matched littermates were administered 1 × 105 PFU of CHIKV or 5 × 105 of ONNV subcutaneously in one footpad. (A) The serum IgG levels were quantitated by ELISA and presented as optical density at 450 nm (OD450). Each symbol is one animal. The small horizontal line is the median of the result. (B) Percent plaque reduction of day 16 CHIKV sera over a preimmune serum. NT50, neutralizing titer of 50%. Preimmune sera assay for day 16 sera. *, P < 0.05; ***, P < 0.001 (two-tailed Student's t test). The results are representative of 2 independent experiments.
A cell-intrinsic antiviral function of UBXN3B.
The above-mentioned data demonstrated a deficiency in the B cell compartment and IgG production in the Ubxn3b−/− mice, which may underlie delayed viral clearance. However, the Ubxn3b−/− mice presented much higher CHIKV viremia as early as at 2 days pi with an intact type, I IFN response (Fig. 2A and B) before the adaptive immune response was activated. We then asked if UBXN3B played a cell-intrinsic antiviral role. To this end, we infected bone marrow-derive dendritic cells (BM-DCs) over 48 h. Skin DCs were one of the early targets of mosquito-transmitted CHIKV in nature and may help CHIKV disseminate systemically. Although CHIKV replication was not productive in DCs, the viral RNA loads in the Ubxn3b−/− were ~2-fold higher than those in the Ubxn3b+/+ cells at 48 h pi (Fig. 8A). The mRNA levels of Ifna, Tnfa, and Oas1a (an interferon-induced gene) in the Ubxn3b−/− cells trended slightly higher than those in the Ubxn3b+/+ cells (Fig. 8B). We confirmed the critical role of UBXN3B in the control of CHIKV replication in a highly permissive cell type, primary mouse embryonic fibroblast (MEF) (Fig. 8C) (37). These results suggested that UBXN3B restricted CHIKV replication independently of type I IFNs. To corroborate this, we examined the role of UBXN3B in CHIKV infection when type I IFN signaling was absent. We used an anti-IFNAR1 antibody to block the type I IFN receptor signaling. We noted that the viral loads in the anti-IFNAR1 antibody-treated cells were higher than those in control IgG-treated cells, and induction of interferon-stimulated genes (Oas1a, Isg15) was completely abolished suggesting an efficient blockade of IFNAR1 (Fig. 8D). Nonetheless, the viral loads in Ubxn3b−/− cells were still higher than those in Ubxn3b+/+ cells in either situation (Fig. 8E).
FIG 8.

UBXN3B restricted CHIKV infection in an IFN-I-independent manner. (A and B) Bone marrow-derived mouse dendritic cells were infected with CHIKV/ZIKV at a multiplicity of infection (MOI) of 1. Quantitative RT-PCR (qPCR) quantification of (A) viral RNA and (B) immune gene mRNA. Each symbol is one mouse. The small horizontal line is the median of the result. (C) Viral titers (PFU/mL) in the supernatants of mouse embryonic fibroblast (MEFs) infected with CHIKV (MOI = 0.5). Bar, mean ± SEM (N = 3). qPCR quantification of (D) ISG mRNA and (E) CHIKV RNA. MEFs were treated with an anti-IFNAR1 antibody or control IgG (8 μg/mL) for 2 h and then infected with CHIKV (MOI = 0.5) in the presence of antibodies for 24 h (N = 6 biological repeats). *, P < 0.05; **, P < 0.01 (two-tailed Student's t test). The results are representative of 3 independent experiments.
DISCUSSION
Although UBXN3B has been implicated in lipid metabolism (39–46), protein degradation (47, 48), mRNA stability (49), protein translocation (50), and stress granule disassembly (51), its in vivo physiological functions remain largely elusive. With a mouse model, we recently demonstrated that UBXN3B regulates STING-mediated innate immunity to a DNA virus (37). In this study, we discovered a critical role of UBXN3B in the control of RNA virus replication and pathogenesis. Similar to Sting deficiency (38), Ubxn3b deletion led to elevated viremia and exacerbated foot swelling during CHIKV and ONNV infection. However, the cytokine expression remained largely intact or even higher in the Ubxn3b−/− mice and DCs. Moreover, viral replication was still enhanced in Ubxn3b−/− cells when type I IFN signaling was simultaneously blocked. These results suggested that UBXN3B signaling restricted CHIKV and ONNV replication in a cell-intrinsic manner but independently of type I IFNs. However, both STING and UBXN3B are known to contribute to the induction of type I IFNs by some RNA viruses, including vesicular stomatitis virus (VSV) and Sendai virus (SeV) (37, 52). This discrepancy is likely because VSV and SeV solely depend on RIG-I (53), which can crosstalk with STING to induce type I IFNs (54). CHIKV and ONNV are less reliant on RIG-I and can activate melanoma differentiation-associated protein 5 (MDA5) for a sufficient type I IFN response (55). Mechanistically, STING is known to restrict the translation of RNA viruses (56). Because both UBXN3B and STING localize to the endoplasmic reticulum (ER), it is reasonable to speculate that UBXN3B works together with STING to inhibit RNA virus translation.
Notably, Ubxn3b deficiency increased myeloid immune cell (neutrophil, macrophage) numbers, while a reduction in B lymphocytes in the feet, spleens, and blood during CHIKV infection. Neutrophil and B cell ratios were strikingly different between the Ubxn3b−/− and Ubxn3b+/+ mice. Neutrophils infiltrate joints rapidly after CHIKV infection and seem to contribute to acute CHIKV arthritis pathogenesis (57), while B cells are essential for the control of CHIKV replication and arthritis (58). Macrophages constituted the largest immune cell population in feet following CHIKV infection, and its number was moderately higher in the Ubxn3b−/− mice. These activated macrophages could be the main cellular reservoir for CHIKV persistence during the late stages of infection and contribute to sustained inflammation (24, 59). On the other hand, heightened immunopathology and tissue damage persisted in the Ubxn3b−/− mice even after CHIKV clearance (16 days pi), suggesting that UBXN3B controls CHIKV immunopathogenesis separately from viral replication. Indeed, chronic CHIKV arthritis pathogenesis can progress without active viral replication (60). Thus, more myeloid, and fewer B cells may account for increased viral replication, delayed viral clearance, and exacerbated, and prolonged inflammation in the Ubxn3b−/− mice. Strikingly, this aberrant immune system was also observed in the uninfected Ubxn3b−/− mice, suggesting an important role of UBXN3B in constitutive hematopoietic homeostasis. This new function is irrelevant to STING as Sting-deficient mice have normal immune cell compartments (61). Instead, UBXN3B deficiency might resemble the aging immune state, which is characteristic of low-grade inflammation mainly driven by macrophages and immunosenescence (62).
In humans, severe CHIKV arthritis is associated with elevated serum cytokine/chemokine levels, such as IL-1β, IL-6, TNF-α, CXCL10, etc. (63). Indeed, we noted that the serum CXCL10, IL-6 and TNF-α levels were higher in the Ubxn3b−/− than those in Ubxn3b+/+ mice, consistent with more severe arthritic pathology in the Ubxn3b−/− mice. Notably, the CXCL10 levels remained higher even after the viremia clearance at 8 to 16 days after CHIKV or ONNV infection, which is correlated with the prolonged foot inflammation in the Ubxn3b−/− mice. CXCL10 is a chemoattractant for monocytes/macrophages/T cells/natural killer cells/DCs and is associated with severe CHIKV arthritis in humans (64). CXCL10 may contribute to alphaviral arthritis pathogenesis by recruiting monocytes/macrophages to the alphavirus-infected mouse feet (65). IL-1β is another established biomarker of CHIKV arthritis severity in humans (66, 67). We also observed an increase in the serum CCL2 and CCL3 levels in the Ubxn3b−/− mice. CCL2 and CCL3 are chemoattractants expressed primarily by monocytes and macrophages and essential regulators of neutrophil and macrophage recruitment and function. These data agree with the increase of neutrophil and macrophage populations in the Ubxn3b−/− mice. However, we did not detect a significant increase in the serum IL-1β levels throughout CHIKV infection (1 to 16 days) compared with prior infection in all the mice. This might be not surprising because CHIKV infection is much less productive in adult wild-type mice than in humans. However, some studies have demonstrated the induction of IL-1β expression by CHIKV in mice. One study showed that the Il1b mRNA expression was upregulated in the ipsilateral feet of adult mice following CHIKV infection (~104 PFU). Another study with 4-week-old mice infected with 103 PFU of CHIKV showed that the IL-1β protein levels in the ipsilateral feet were upregulated too (68). In neonatal mice (8 to 9 day-old), the serum IL-1β levels were upregulated following CHIKV infection (1 × 105 to 2 × 105 PFU) (69, 70). In contrast, no significant serum IL-1β protein was detected during CHIKV infection (1 × 104 PFU) in adult mice (71). These studies demonstrated that, although CHIKV infection in mice upregulated IL-1β expression locally in the ipsilateral feet, whether it can increase the systemic (serum) IL-1β protein level was probably reliant on the animal models and infection conditions of choice.
In summary, our studies suggested that UBXN3B restricted CHIKV/ONNV infection and immunopathogenesis by dual mechanisms, including STING signaling and immune cell homeostasis. The mouse UBXN3B function may be potentially recapitulated in humans because the UBXN3B protein is highly conserved in mammals (98% identity between human and rodent protein). Immediate future work will pinpoint the role of UBXN3B in hematopoietic homeostasis, particularly B cell development.
MATERIALS AND METHODS
Mouse models.
The mouse line with the exon 1 of Ubxn3b flanked by two LoxP sites (Ubxn3bflox/flox) was generated via homologous recombination by Fujimoto at Nagoya University (43). The homozygous Ubxn3bflox/flox was then crossed with homozygous tamoxifen-inducible Cre recombinase-estrogen receptor T2 mice (Jackson Laboratories, stock number 008463) to generate Cre+ Ubxn3bflox/flox littermates. To induce Ubxn3b deletion, >6-weeks old mice were injected with 100 μL of tamoxifen (10 mg/ mL in corn oil) (Sigma, number T5648) via intraperitoneal (IP) every 2 days for a total duration of 8 days (4 doses). Successful deletion of Ubxn3b was confirmed in our recent study (32). Half of Cre+ Ubxn3bflox/flox litters were treated with tamoxifen and designated Ubxn3b−/−. The other half were treated with corn oil only and designated Ubxn3b+/+. Mice were allowed to purge tamoxifen for at least 4 weeks before any infection or analyses were performed. The animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Connecticut Health Center and Yale University.
Antibodies, cell lines, and viruses.
The goat anti-mouse IFNAR1 (number AF30369) was purchased from R&D Systems (Minneapolis, MN 55413, USA), rabbit anti-GAPDH (Clone D16H11, number 5174), anti-β-Actin (Clone 13E5, number 4970) and STING (Clone D2P2F, number 13647) were from Cell Signaling Technology (Danvers, MA, USA). Human embryonic kidney 293 cells transformed with T antigen of SV40 (HEK293T, number CRL-3216) and Vero cells (monkey kidney epithelial cells, number CCL-81) were purchased from American Type Culture Collection (ATCC) (Manassas, VA, USA). These cell lines were not listed in the database of commonly misidentified cell lines maintained by ICLAC. Cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics/antimycotics (Life Technologies, Grand Island, NY, USA). We routinely added MycoZAP (Lonza Group, Basel, Switzerland) to cell cultures to prevent mycoplasma contamination.
The CHIKV French La Reunion strain LR2006-OPY1 was a kind gift of the Connecticut Agricultural Experiment Station in New Haven, CT, USA. The CHIKV vaccine strain 181, clone 25 was recovered from a plasmid and used for in vitro infection and enzyme-linked immunosorbent assay (ELISA) (72, 73). The ONNV UgMP30 strain (NR-51661) was provided by BEI Resources. All these viruses were propagated in Vero cells.
Differentiation of bone marrow DCs, isolation and treatment of embryonic fibroblasts with 4-OH tamoxifen.
Bone marrow-derived DCs were induced from bone marrow cells with 10 ng/mL murine GM-CSF (PeproTech) in RPMI 1640 medium containing 10% (volume/volume [vol/vol]) FBS (Invitrogen, Carlsbad, CA, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen) for 6 to 8 days at 37°C and 5% CO2 (37). Mature BM-DCs were cultured in RPMI 1640 medium overnight and then washed once with the prewarmed fresh medium before use.
Pregnant Cre+ Ubxn3bflox/flox females were euthanized on days 12 to 14 of gestation. Embryos were decapitated and eviscerated and then digested with trypsin for 10 min at 37°C rotating. Fibroblasts were filtered through a 100 μM filter, cultured in RPMI 1640 medium (Life Technologies, NY, USA), and supplemented with 10% FBS and antibiotics/antimycotics. One-half of the Cre+ Ubxn3bflox/flox embryonic fibroblasts (MEFs) were treated with 2 μM 4-hydroxyl (OH) tamoxifen for 4 to 5 days to generate the Ubxn3b−/− cells. The other half was treated with solvent (dimethyl sulfoxide, DMSO), resulting in Ubxn3b+/+. After induction, the cells were further passaged three times in RPMI medium without tamoxifen.
Plaque-forming assay.
Quantification of infectious viral particles in sera, cell culture supernatants, and homogenized tissues were performed on Vero cell monolayers (74). Briefly, viral samples (appropriately diluted) were incubated with confluent Vero cells (6-well plate) at 37°C for 2 h. The inoculum was then removed and replaced with 2 mL of DMEM complete medium with 1% SeaPlaque agarose (catalog number 50100, Lonza). The cells were incubated at 37°C, 5% CO2 for 3 days, and on the fourth day, the cells were stained with Neutral Red (Sigma-Aldrich) overnight.
Mouse infection and monitoring.
About 1× 105 to 5 × 105 plague-forming units (PFU) of CHIKV LR2006-OPY1 or ONNV were inoculated at the ventral side of a hind foot subcutaneously. The thickness and width of the perimetatarsal area of the hind foot (inoculated with the virus) were measured using a precision metric caliper. The foot dimension was calculated as width × thickness, and the results were expressed as the percent increase in the foot dimension after infection (Dn) over its baseline before infection (D0), using the formula: 100% × (Dn-D0)/D0.
Tissue histology.
Tissues were fixed in 4% paraformaldehyde (PFA), embedded in paraffin, cut into 4 μM-thick sections, immobilized to glass slides, decalcified, and processed for hematoxylin and eosin staining. Arbitrary arthritic disease scores (on a 1 to 5 scale with 1 being the slightest and 5 the worst) were assessed using a combination of histological parameters, including exudation of fibrin and inflammatory cells into the joints, alteration in the thickness of tendons or ligament sheaths, and hypertrophy and hyperlexia of the synovium in a double-blinded manner (75).
Reverse transcription and quantitative PCR.
Up to 1 × 106 cells or 10 mg of tissue was collected in 350 μL of lysis buffer (Qiagen RNeasy Minikit). RNA was extracted following the Qiagen RNeasy manufacturer’s instructions. Reverse transcription of RNA into complementary DNA (cDNA) was performed using the BIO-RAD iScript cDNA Synthesis kit. Quantitative PCR (qPCR) was performed with gene-specific primers and SYBR green PCR master mix. Results were calculated using the –ΔΔCt method and a housekeeping gene, beta-actin, as an internal control. The qPCR primers were reported in our previous studies (38, 65).
Fluorescence-activated cell sorting.
FACS was performed according to our published study (65). Mouse tissues were minced with a fine scissor and digested in 4 mL of digestion medium (20 mg/mL collagenase IV [Sigma-Aldrich, St. Louis, MO, USA], 5 U/mL dispase [StemCell, Cambridge, MA, USA], and 50 mg/mL DNase I mix [Qiagen, Germantown, MD, USA] in complete RPMI 1640 medium) at 37°C for 4 h. The lysate was filtrated with a 40 μm cell strainer. Cells were then pelleted down by centrifugation at 500 × g for 5 min. The red blood cells in the cell pellet were lysed three times with a lysis buffer (catalog number 420301 from Biolegend, San Diego, CA, USA). Cells were suspended in FACS buffer and stained for 30 min at 4°C with the desired antibody cocktails (Biolegend, San Diego, CA, US) of APC-Fire 750-anti CD11b (catalog number 101261, clone M1/70), Alexa Fluor 700-anti Ly-6G (catalog number 127621, clone 1A8), Brilliant Violet 421-anti CD11c (catalog number 117343, clone N418), PerCP-Cy5.5-anti MHC II (catalog number 107625, clone M5/114.15.2), PE-anti Tetherin (PCDA1) (catalog number 12703, clone 10C2), Brilliant Violet 510-anti F4/80 (catalog number 123135, clone BM8), APC-anti CD68 (catalog number 137007, clone FA-11), PE-Dazzle 594-anti CD3 epsilon (catalog number 100347, clone145 to 2C11), Brilliant Violet 711-anti CD4 (catalog number 100557, clone RM4-5), Brilliant Violet 570-anti CD8a (catalog number 100739, clone53 to 6.7), Brilliant Violet 650 anti-CD161 (NK1.1) (catalog number 108735, clone PK136), Zombie UV (catalog number 423107), PE-Cy7-anti CD45 (catalog number 103113, clone 30-F11), TruStain FcX-anti CD16/32 (catalog number 101319, clone 93), Brilliant Violet 421-anti CD45R (B220) (catalog number 103239, clone RA3-6B2), and Alexa Fluor 700-anti CD19 (catalog number 115527, clone 6D5). After staining and washing, the cells were fixed with 4% PFA and analyzed on a Becton, Dickinson FACS ARIA II, CyAn advanced digital processor (ADP). Data were analyzed using the FlowJo software. Among CD45+ cells, CD11b+ Ly6G+ cells were classified as neutrophils, Ly6G− CD11b+ F4/80+ as macrophages, Ly6G− CD11b+ CD115+ as monocytes, Ly6G− CD11c+ MHC II+ as dendritic cells (DC), CD3+ as total T cells, CD3+ CD4+ as CD4 T cells, CD3+ CD8+ as CD8 T cells, CD19+ as B cells.
Multiplex enzyme-linked immunosorbent assay.
We used a LEGENDPlex (Biolegend, San Diego, CA 92121, USA) bead-based immunoassay to quantify serum cytokine concentrations. The procedures were the same as in the product manual. Briefly, the supernatants or standards were mixed with antibody-coated microbeads in a filter-bottom microplate and incubated at room temperature for 2h with vigorous shaking at 500 rpm. After the removal of unbound analytes and two washes, 25 μL of detection antibodies were added to each well, and the plate was incubated at room temperature for 1 h with vigorous shaking at 500 rpm. Next, 25 μL of SA-PE reagent was then added directly to each well, and the plate was incubated at room temperature for 30 min with vigorous shaking at 500 rpm. The beads were washed twice with wash buffer and then transferred to a microcentrifuge tube. The beads were fixed with 4% PFA for 15 min and resuspended in the assay buffer. The beads were run through a BIORAD ZE5 and the concentrations of analytes were calculated with the standards included in the assay kit using the LEGENDPlex software. The CCL2 (number 88-7391-22) and CCL3 (number 88-56013-22) ELISA kits were purchased from Invitrogen and performed according to the product manual exactly (Waltham, MA, USA).
Quantification of IgG by ELISA.
Next, ~1 × 106 viral particles in coating buffer (0.05 M carbonate-bicarbonate, pH 9.6) were coated to a 96-well microplate at 4°C overnight. The plate was washed once with wash solution (50 mM Tris, 0.14 M NaCl, 0.05% Tween 20, pH 8.0), and blocked with 4% bovine serum albumin at room temperature for 2 h. Next, 100 μL of each diluted serum (500-fold) was added to a well and incubated at room temperature for 1 h, then washed three times. Then, 100 μL of diluted horseradish peroxidase-conjugated goat anti-mouse IgG was added to each well and incubated at room temperature for 1 h. After stringency wash, 100 μL of substrate 3,3′,5,5′-tetramethylbenzidine (TMB) was added to each well and incubated at room temperature for5 to 30min for color development and terminated by 100 μL of 0.16 M sulfuric acid. The absorption at wavelength 450 nm (A450) was read on a Cytation 1 plate reader (BioTek, Winooski, VT, USA).
Plaque neutralizing assay.
The preimmune and anti-CHIKV mouse sera were heated at 56°C for 20 min and serially diluted in phosphate-buffered saline (PBS). One hundred 20 PFU of CHIKV were mixed with 100 μL of diluted sera, and the mix was incubated at 37°C for 1 h. The mix and fetal bovine serum (FBS)-free DMEM medium was added to Vero cells (see description of the plaque-forming assay for preparation of Vero cells) in a 6-well plate and incubated at 37°C for 4 h. The inoculum was then removed and replaced with 2 mL of DMEM complete medium with 1% SeaPlaque agarose (catalog number 50100, Lonza). The cells were incubated at 37°C, 5% CO2 for 3 days, and on the fourth day, the cells were stained with Neutral Red (Sigma-Aldrich) overnight. The dilutions and percent plaque reduction (over control serum) were plotted for polynomial equations, which were used to calculate the neutralizing titer at which viral plaques were reduced by 50% (NT50).
Data availability.
The sample sizes chosen for our animal experiments in this study were estimated according to our prior experience in similar sets of experiments and power analysis calculations (http://isogenic.info/html/power_analysis.html). All animals were included, and no method of randomization was applied. All data were analyzed with GraphPad Prism software by nonparametric or nonparametric two-tailed Student's t test depending on the data distribution. P ≤ 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
T.G. designed and performed most of the experimental procedures and data analyses. D.Y. and T.L. contributed to some of the experimental procedures and/or provided technical support. P.W. conceived and oversaw the study. J.G.C. helped to improve the writing. T.G. and P.W. wrote the paper and all the authors reviewed and/or modified the manuscript.
This project was funded in part by National Institutes of Health grants to P. W., R01AI132526, and R21AI155820.
We have no financial or nonfinancial interest to disclose.
Contributor Information
Penghua Wang, Email: pewang@uchc.edu.
James Weger-Lucarelli, Virginia Tech.
Nisha K. Duggal, Virginia Tech
REFERENCES
- 1.Silva LA, Dermody TS. 2017. Chikungunya virus: epidemiology, replication, disease mechanisms, and prospective intervention strategies. J Clin Invest 127:737–749. doi: 10.1172/JCI84417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sourisseau M, Schilte C, Casartelli N, Trouillet C, Guivel-Benhassine F, Rudnicka D, Sol-Foulon N, Le Roux K, Prevost MC, Fsihi H, Frenkiel MP, Blanchet F, Afonso PV, Ceccaldi PE, Ozden S, Gessain A, Schuffenecker I, Verhasselt B, Zamborlini A, Saib A, Rey FA, Arenzana-Seisdedos F, Despres P, Michault A, Albert ML, Schwartz O. 2007. Characterization of reemerging chikungunya virus. PLoS Pathog 3:e89. doi: 10.1371/journal.ppat.0030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teng TS, Kam YW, Lee B, Hapuarachchi HC, Wimal A, Ng LC, Ng LF. 2015. A systematic meta-analysis of immune signatures in patients with acute chikungunya virus infection. J Infect Dis 211:1925–1935. doi: 10.1093/infdis/jiv049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruiz Silva M, van der Ende-Metselaar H, Mulder HL, Smit JM, Rodenhuis-Zybert IA. 2016. Mechanism and role of MCP-1 upregulation upon chikungunya virus infection in human peripheral blood mononuclear cells. Sci Rep 6:32288. doi: 10.1038/srep32288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Her Z, Malleret B, Chan M, Ong EK, Wong SC, Kwek DJ, Tolou H, Lin RT, Tambyah PA, Renia L, Ng LF. 2010. Active infection of human blood monocytes by chikungunya virus triggers an innate immune response. J Immunol 184:5903–5913. doi: 10.4049/jimmunol.0904181. [DOI] [PubMed] [Google Scholar]
- 6.Schilte C, Couderc T, Chretien F, Sourisseau M, Gangneux N, Guivel-Benhassine F, Kraxner A, Tschopp J, Higgs S, Michault A, Arenzana-Seisdedos F, Colonna M, Peduto L, Schwartz O, Lecuit M, Albert ML. 2010. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J Exp Med 207:429–442. doi: 10.1084/jem.20090851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wauquier N, Becquart P, Nkoghe D, Padilla C, Ndjoyi-Mbiguino A, Leroy EM. 2011. The acute phase of chikungunya virus infection in humans is associated with strong innate immunity and T CD8 cell activation. J Infect Dis 204:115–123. doi: 10.1093/infdis/jiq006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Javelle E, Ribera A, Degasne I, Gauzere BA, Marimoutou C, Simon F. 2015. Specific management of post-chikungunya rheumatic disorders: a retrospective study of 159 cases in Reunion Island from 2006-2012. PLoS Negl Trop Dis 9:e0003603. doi: 10.1371/journal.pntd.0003603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rudd PA, Wilson J, Gardner J, Larcher T, Babarit C, Le TT, Anraku I, Kumagai Y, Loo YM, Gale M, Jr., Akira S, Khromykh AA, Suhrbier A. 2012. Interferon response factors 3 and 7 protect against chikungunya virus hemorrhagic fever and shock. J Virol 86:9888–9898. doi: 10.1128/JVI.00956-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poo YS, Rudd PA, Gardner J, Wilson JA, Larcher T, Colle MA, Le TT, Nakaya HI, Warrilow D, Allcock R, Bielefeldt-Ohmann H, Schroder WA, Khromykh AA, Lopez JA, Suhrbier A. 2014. Multiple immune factors are involved in controlling acute and chronic chikungunya virus infection. PLoS Negl Trop Dis 8:e3354. doi: 10.1371/journal.pntd.0003354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Couderc T, Chretien F, Schilte C, Disson O, Brigitte M, Guivel-Benhassine F, Touret Y, Barau G, Cayet N, Schuffenecker I, Despres P, Arenzana-Seisdedos F, Michault A, Albert ML, Lecuit M. 2008. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog 4:e29. doi: 10.1371/journal.ppat.0040029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akahata W, Yang ZY, Andersen H, Sun S, Holdaway HA, Kong WP, Lewis MG, Higgs S, Rossmann MG, Rao S, Nabel GJ. 2010. A virus-like particle vaccine for epidemic Chikungunya virus protects nonhuman primates against infection. Nat Med 16:334–338. doi: 10.1038/nm.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fric J, Bertin-Maghit S, Wang CI, Nardin A, Warter L. 2013. Use of human monoclonal antibodies to treat Chikungunya virus infection. J Infect Dis 207:319–322. doi: 10.1093/infdis/jis674. [DOI] [PubMed] [Google Scholar]
- 14.Goh LY, Hobson-Peters J, Prow NA, Gardner J, Bielefeldt-Ohmann H, Pyke AT, Suhrbier A, Hall RA. 2013. Neutralizing monoclonal antibodies to the E2 protein of chikungunya virus protects against disease in a mouse model. Clin Immunol 149:487–497. doi: 10.1016/j.clim.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 15.Metz SW, Gardner J, Geertsema C, Le TT, Goh L, Vlak JM, Suhrbier A, Pijlman GP. 2013. Effective chikungunya virus-like particle vaccine produced in insect cells. PLoS Negl Trop Dis 7:e2124. doi: 10.1371/journal.pntd.0002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pal P, Dowd KA, Brien JD, Edeling MA, Gorlatov S, Johnson S, Lee I, Akahata W, Nabel GJ, Richter MK, Smit JM, Fremont DH, Pierson TC, Heise MT, Diamond MS. 2013. Development of a highly protective combination monoclonal antibody therapy against Chikungunya virus. PLoS Pathog 9:e1003312. doi: 10.1371/journal.ppat.1003312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prow TW, Chen X, Prow NA, Fernando GJ, Tan CS, Raphael AP, Chang D, Ruutu MP, Jenkins DW, Pyke A, Crichton ML, Raphaelli K, Goh LY, Frazer IH, Roberts MS, Gardner J, Khromykh AA, Suhrbier A, Hall RA, Kendall MA. 2010. Nanopatch-targeted skin vaccination against West Nile Virus and Chikungunya virus in mice. Small 6:1776–1784. doi: 10.1002/smll.201000331. [DOI] [PubMed] [Google Scholar]
- 18.Selvarajah S, Sexton NR, Kahle KM, Fong RH, Mattia KA, Gardner J, Lu K, Liss NM, Salvador B, Tucker DF, Barnes T, Mabila M, Zhou X, Rossini G, Rucker JB, Sanders DA, Suhrbier A, Sambri V, Michault A, Muench MO, Doranz BJ, Simmons G. 2013. A neutralizing monoclonal antibody targeting the acid-sensitive region in chikungunya virus E2 protects from disease. PLoS Negl Trop Dis 7:e2423. doi: 10.1371/journal.pntd.0002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang D, Suhrbier A, Penn-Nicholson A, Woraratanadharm J, Gardner J, Luo M, Le TT, Anraku I, Sakalian M, Einfeld D, Dong JY. 2011. A complex adenovirus vaccine against chikungunya virus provides complete protection against viraemia and arthritis. Vaccine 29:2803–2809. doi: 10.1016/j.vaccine.2011.01.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fox JM, Diamond MS. 2016. Immune-Mediated Protection and Pathogenesis of Chikungunya Virus. J Immunol 197:4210–4218. doi: 10.4049/jimmunol.1601426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teo TH, Lum FM, Lee WW, Ng LF. 2012. Mouse models for Chikungunya virus: deciphering immune mechanisms responsible for disease and pathology. Immunol Res 53:136–147. doi: 10.1007/s12026-012-8266-x. [DOI] [PubMed] [Google Scholar]
- 22.Teo TH, Lum FM, Claser C, Lulla V, Lulla A, Merits A, Renia L, Ng LF. 2013. A pathogenic role for CD4+ T cells during Chikungunya virus infection in mice. J Immunol 190:259–269. doi: 10.4049/jimmunol.1202177. [DOI] [PubMed] [Google Scholar]
- 23.Gasque P, Couderc T, Lecuit M, Roques P, Ng LF. 2015. Chikungunya virus pathogenesis and immunity. Vector Borne Zoonotic Dis 15:241–249. doi: 10.1089/vbz.2014.1710. [DOI] [PubMed] [Google Scholar]
- 24.Hoarau JJ, Jaffar Bandjee MC, Krejbich Trotot P, Das T, Li-Pat-Yuen G, Dassa B, Denizot M, Guichard E, Ribera A, Henni T, Tallet F, Moiton MP, Gauzere BA, Bruniquet S, Jaffar Bandjee Z, Morbidelli P, Martigny G, Jolivet M, Gay F, Grandadam M, Tolou H, Vieillard V, Debre P, Autran B, Gasque P. 2010. Persistent chronic inflammation and infection by Chikungunya arthritogenic alphavirus in spite of a robust host immune response. J Immunol 184:5914–5927. doi: 10.4049/jimmunol.0900255. [DOI] [PubMed] [Google Scholar]
- 25.Petitdemange C, Becquart P, Wauquier N, Beziat V, Debre P, Leroy EM, Vieillard V. 2011. Unconventional repertoire profile is imprinted during acute chikungunya infection for natural killer cells polarization toward cytotoxicity. PLoS Pathog 7:e1002268. doi: 10.1371/journal.ppat.1002268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stoermer KA, Burrack A, Oko L, Montgomery SA, Borst LB, Gill RG, Morrison TE. 2012. Genetic ablation of arginase 1 in macrophages and neutrophils enhances clearance of an arthritogenic alphavirus. J Immunol 189:4047–4059. doi: 10.4049/jimmunol.1201240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuberth C, Buchberger A. 2008. UBX domain proteins: major regulators of the AAA ATPase Cdc48/p97. Cell Mol Life Sci 65:2360–2371. doi: 10.1007/s00018-008-8072-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P, Warren G. 1997. p47 is a cofactor for p97-mediated membrane fusion. Nature 388:75–78. doi: 10.1038/40411. [DOI] [PubMed] [Google Scholar]
- 29.Alexandru G, Graumann J, Smith GT, Kolawa NJ, Fang R, Deshaies RJ. 2008. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell 134:804–816. doi: 10.1016/j.cell.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang P, Yang L, Cheng G, Yang G, Xu Z, You F, Sun Q, Lin R, Fikrig E, Sutton RE. 2013. UBXN1 interferes with Rig-I-like receptor-mediated antiviral immune response by targeting MAVS. Cell Rep 3:1057–1070. doi: 10.1016/j.celrep.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim JH, Park ME, Nikapitiya C, Kim TH, Uddin MB, Lee HC, Kim E, Ma JY, Jung JU, Kim CJ, Lee JS. 2017. FAS-associated factor-1 positively regulates type I interferon response to RNA virus infection by targeting NLRX1. PLoS Pathog 13:e1006398. doi: 10.1371/journal.ppat.1006398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dai T, Wu L, Wang S, Wang J, Xie F, Zhang Z, Fang X, Li J, Fang P, Li F, Jin K, Dai J, Yang B, Zhou F, van Dam H, Cai D, Huang H, Zhang L. 2018. FAF1 regulates antiviral immunity by inhibiting MAVS but is antagonized by phosphorylation upon viral infection. Cell Host Microbe 24:776–790.e5. doi: 10.1016/j.chom.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Wang YB, Tan B, Mu R, Chang Y, Wu M, Tu HQ, Zhang YC, Guo SS, Qin XH, Li T, Li WH, Li AL, Zhang XM, Li HY. 2015. Ubiquitin-associated domain-containing UBX protein UBXN1 is a negative regulator of NF-kappaB signaling. J Biol Chem 290:10395–10405. doi: 10.1074/jbc.M114.631689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu Y, O'Boyle K, Auer J, Raju S, You F, Wang P, Fikrig E, Sutton RE. 2017. Multiple UBXN family members inhibit retrovirus and lentivirus production and canonical NFkappaBeta signaling by stabilizing IkappaBalpha. PLoS Pathog 13:e1006187. doi: 10.1371/journal.ppat.1006187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ketkar H, Harrison AG, Graziano VR, Geng T, Yang L, Vella AT, Wang P. 2021. UBX domain protein 6 positively regulates JAK-STAT1/2 signaling. J Immunol 206:2682–2691. doi: 10.4049/jimmunol.1901337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mengus C, Neutzner M, Bento A, Bippes CC, Kohler C, Decembrini S, Hausel J, Hemion C, Sironi L, Frank S, Scholl HPN, Neutzner A. 2022. VCP/p97 cofactor UBXN1/SAKS1 regulates mitophagy by modulating MFN2 removal from mitochondria. Autophagy 18:171–190. doi: 10.1080/15548627.2021.1922982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang L, Wang L, Ketkar H, Ma J, Yang G, Cui S, Geng T, Mordue DG, Fujimoto T, Cheng G, You F, Lin R, Fikrig E, Wang P. 2018. UBXN3B positively regulates STING-mediated antiviral immune responses. Nat Commun 9:2329. doi: 10.1038/s41467-018-04759-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geng T, Lin T, Yang D, Harrison AG, Vella AT, Fikrig E, Wang P. 2020. A critical role for STING signaling in limiting pathogenesis of Chikungunya virus. J Infect Dis 223:2186–2196. doi: 10.1093/infdis/jiaa694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JN, Kim H, Yao H, Chen Y, Weng K, Ye J. 2010. Identification of Ubxd8 protein as a sensor for unsaturated fatty acids and regulator of triglyceride synthesis. Proc Natl Acad Sci USA 107:21424–21429. doi: 10.1073/pnas.1011859107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olzmann JA, Richter CM, Kopito RR. 2013. Spatial regulation of UBXD8 and p97/VCP controls ATGL-mediated lipid droplet turnover. Proc Natl Acad Sci USA 110:1345–1350. doi: 10.1073/pnas.1213738110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang CW, Lee SC. 2012. The ubiquitin-like (UBX)-domain-containing protein Ubx2/Ubxd8 regulates lipid droplet homeostasis. J Cell Sci 125:2930–2939. doi: 10.1242/jcs.100230. [DOI] [PubMed] [Google Scholar]
- 42.Kim H, Zhang H, Meng D, Russell G, Lee JN, Ye J. 2013. UAS domain of Ubxd8 and FAF1 polymerizes upon interaction with long-chain unsaturated fatty acids. J Lipid Res 54:2144–2152. doi: 10.1194/jlr.M037218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Imai N, Suzuki M, Hayashi K, Ishigami M, Hirooka Y, Abe T, Shioi G, Goto H, Fujimoto T. 2015. Hepatocyte-specific depletion of UBXD8 induces periportal steatosis in mice fed a high-fat diet. PLoS One 10:e0127114. doi: 10.1371/journal.pone.0127114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohsaki Y, Cheng J, Fujita A, Tokumoto T, Fujimoto T. 2006. Cytoplasmic lipid droplets are sites of convergence of proteasomal and autophagic degradation of apolipoprotein B. Mol Biol Cell 17:2674–2683. doi: 10.1091/mbc.e05-07-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohsaki Y, Cheng J, Suzuki M, Fujita A, Fujimoto T. 2008. Lipid droplets are arrested in the ER membrane by tight binding of lipidated apolipoprotein B-100. J Cell Sci 121:2415–2422. doi: 10.1242/jcs.025452. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki M, Otsuka T, Ohsaki Y, Cheng J, Taniguchi T, Hashimoto H, Taniguchi H, Fujimoto T. 2012. Derlin-1 and UBXD8 are engaged in dislocation and degradation of lipidated ApoB-100 at lipid droplets. Mol Biol Cell 23:800–810. doi: 10.1091/mbc.E11-11-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia Y, Yan LH, Huang B, Liu M, Liu X, Huang C. 2014. Pathogenic mutation of UBQLN2 impairs its interaction with UBXD8 and disrupts endoplasmic reticulum-associated protein degradation. J Neurochem 129:99–106. doi: 10.1111/jnc.12606. [DOI] [PubMed] [Google Scholar]
- 48.Phan VT, Ding VW, Li F, Chalkley RJ, Burlingame A, McCormick F. 2010. The RasGAP proteins Ira2 and neurofibromin are negatively regulated by Gpb1 in yeast and ETEA in humans. Mol Cell Biol 30:2264–2279. doi: 10.1128/MCB.01450-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou HL, Geng C, Luo G, Lou H. 2013. The p97-UBXD8 complex destabilizes mRNA by promoting release of ubiquitinated HuR from mRNP. Genes Dev 27:1046–1058. doi: 10.1101/gad.215681.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Glinka T, Alter J, Braunstein I, Tzach L, Wei Sheng C, Geifman S, Edelmann MJ, Kessler BM, Stanhill A. 2014. Signal-peptide-mediated translocation is regulated by a p97-AIRAPL complex. Biochem J 457:253–261. doi: 10.1042/BJ20130710. [DOI] [PubMed] [Google Scholar]
- 51.Gwon Y, Maxwell BA, Kolaitis RM, Zhang P, Kim HJ, Taylor JP. 2021. Ubiquitination of G3BP1 mediates stress granule disassembly in a context-specific manner. Science 372:eabf6548. doi: 10.1126/science.abf6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 54.Ishikawa H, Ma Z, Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akhrymuk I, Frolov I, Frolova EI. 2016. Both RIG-I and MDA5 detect alphavirus replication in concentration-dependent mode. Virology 487:230–241. doi: 10.1016/j.virol.2015.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Franz KM, Neidermyer WJ, Tan YJ, Whelan SPJ, Kagan JC. 2018. STING-dependent translation inhibition restricts RNA virus replication. Proc Natl Acad Sci USA 115:E2058–E2067. doi: 10.1073/pnas.1716937115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Poo YS, Nakaya H, Gardner J, Larcher T, Schroder WA, Le TT, Major LD, Suhrbier A. 2014. CCR2 deficiency promotes exacerbated chronic erosive neutrophil-dominated chikungunya virus arthritis. J Virol 88:6862–6872. doi: 10.1128/JVI.03364-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lum FM, Teo TH, Lee WW, Kam YW, Renia L, Ng LF. 2013. An essential role of antibodies in the control of Chikungunya virus infection. J Immunol 190:6295–6302. doi: 10.4049/jimmunol.1300304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Labadie K, Larcher T, Joubert C, Mannioui A, Delache B, Brochard P, Guigand L, Dubreil L, Lebon P, Verrier B, de Lamballerie X, Suhrbier A, Cherel Y, Le Grand R, Roques P. 2010. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J Clin Invest 120:894–906. doi: 10.1172/JCI40104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chang AY, Martins KAO, Encinales L, Reid SP, Acuna M, Encinales C, Matranga CB, Pacheco N, Cure C, Shukla B, Ruiz Arteta T, Amdur R, Cazares LH, Gregory M, Ward MD, Porras A, Rico Mendoza A, Dong L, Kenny T, Brueggemann E, Downey LG, Kamalapathy P, Lichtenberger P, Falls O, Simon GL, Bethony JM, Firestein GS. 2018. Chikungunya arthritis mechanisms in the Americas: a cross-sectional analysis of chikungunya arthritis patients twenty-two months after infection demonstrating no detectable viral persistence in synovial fluid. Arthritis Rheumatol 70:585–593. doi: 10.1002/art.40383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McGuckin Wuertz K, Treuting PM, Hemann EA, Esser-Nobis K, Snyder AG, Graham JB, Daniels BP, Wilkins C, Snyder JM, Voss KM, Oberst A, Lund J, Gale M, Jr.. 2019. STING is required for host defense against neuropathological West Nile virus infection. PLoS Pathog 15:e1007899. doi: 10.1371/journal.ppat.1007899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haynes L. 2020. Aging of the immune system: research challenges to enhance the health span of older adults. Front Aging 1:602108. doi: 10.3389/fragi.2020.602108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ng LFP. 2017. Immunopathology of chikungunya virus infection: lessons learned from patients and animal models. Annu Rev Virol 4:413–427. doi: 10.1146/annurev-virology-101416-041808. [DOI] [PubMed] [Google Scholar]
- 64.Kelvin AA, Banner D, Silvi G, Moro ML, Spataro N, Gaibani P, Cavrini F, Pierro A, Rossini G, Cameron MJ, Bermejo-Martin JF, Paquette SG, Xu L, Danesh A, Farooqui A, Borghetto I, Kelvin DJ, Sambri V, Rubino S. 2011. Inflammatory cytokine expression is associated with chikungunya virus resolution and symptom severity. PLoS Negl Trop Dis 5:e1279. doi: 10.1371/journal.pntd.0001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lin T, Geng T, Harrison AG, Yang D, Vella AT, Fikrig E, Wang P. 2020. CXCL10 signaling contributes to the pathogenesis of arthritogenic alphaviruses. Viruses 12:1252. doi: 10.3390/v12111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chirathaworn C, Rianthavorn P, Wuttirattanakowit N, Poovorawan Y. 2010. Serum IL-18 and IL-18BP levels in patients with Chikungunya virus infection. Viral Immunol 23:113–117. doi: 10.1089/vim.2009.0077. [DOI] [PubMed] [Google Scholar]
- 67.Ng LF, Chow A, Sun YJ, Kwek DJ, Lim PL, Dimatatac F, Ng LC, Ooi EE, Choo KH, Her Z, Kourilsky P, Leo YS. 2009. IL-1beta, IL-6, and RANTES as biomarkers of Chikungunya severity. PLoS One 4:e4261. doi: 10.1371/journal.pone.0004261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cook LE, Locke MC, Young AR, Monte K, Hedberg ML, Shimak RM, Sheehan KCF, Veis DJ, Diamond MS, Lenschow DJ. 2019. Distinct roles of interferon alpha and beta in controlling chikungunya virus replication and modulating neutrophil-mediated inflammation. J Virol 94:e00841-19. doi: 10.1128/JVI.00841-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patil DR, Hundekar SL, Arankalle VA. 2012. Expression profile of immune response genes during acute myopathy induced by chikungunya virus in a mouse model. Microbes Infect 14:457–469. doi: 10.1016/j.micinf.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 70.Werneke SW, Schilte C, Rohatgi A, Monte KJ, Michault A, Arenzana-Seisdedos F, Vanlandingham DL, Higgs S, Fontanet A, Albert ML, Lenschow DJ. 2011. ISG15 is critical in the control of Chikungunya virus infection independent of UbE1L mediated conjugation. PLoS Pathog 7:e1002322. doi: 10.1371/journal.ppat.1002322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gardner J, Anraku I, Le TT, Larcher T, Major L, Roques P, Schroder WA, Higgs S, Suhrbier A. 2010. Chikungunya virus arthritis in adult wild-type mice. J Virol 84:8021–8032. doi: 10.1128/JVI.02603-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gorchakov R, Wang E, Leal G, Forrester NL, Plante K, Rossi SL, Partidos CD, Adams AP, Seymour RL, Weger J, Borland EM, Sherman MB, Powers AM, Osorio JE, Weaver SC. 2012. Attenuation of Chikungunya virus vaccine strain 181/clone 25 is determined by two amino acid substitutions in the E2 envelope glycoprotein. J Virol 86:6084–6096. doi: 10.1128/JVI.06449-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tretyakova I, Hearn J, Wang E, Weaver S, Pushko P. 2014. DNA vaccine initiates replication of live attenuated chikungunya virus in vitro and elicits protective immune response in mice. J Infect Dis 209:1882–1890. doi: 10.1093/infdis/jiu114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang P, Arjona A, Zhang Y, Sultana H, Dai J, Yang L, LeBlanc PM, Doiron K, Saleh M, Fikrig E. 2010. Caspase-12 controls West Nile virus infection via the viral RNA receptor RIG-I. Nat Immunol 11:912–919. doi: 10.1038/ni.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang L, Geng T, Yang G, Ma J, Wang L, Ketkar H, Yang D, Lin T, Hwang J, Zhu S, Wang Y, Dai J, You F, Cheng G, Vella AT, Flavell RA, Fikrig E, Wang P. 2020. Macrophage scavenger receptor 1 controls Chikungunya virus infection through autophagy in mice. Commun Biol 3:556. doi: 10.1038/s42003-020-01285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sample sizes chosen for our animal experiments in this study were estimated according to our prior experience in similar sets of experiments and power analysis calculations (http://isogenic.info/html/power_analysis.html). All animals were included, and no method of randomization was applied. All data were analyzed with GraphPad Prism software by nonparametric or nonparametric two-tailed Student's t test depending on the data distribution. P ≤ 0.05 was considered statistically significant.