

Abstract
Cholangiocarcinoma (CCA) is a highly malignant cancer of the biliary tree. Although studies have implicated enhancer of Zeste homolog 2 (EZH2) in CCA growth, the role of EZH2 in CCA development has not been investigated, and the mechanism for EZH2-regulated gene expression in CCA remains to be further defined. The current study used a mouse model of CCA induced by hydrodynamic tail vein injection of Notch1 intracellular domain and myristoylated-AKT plasmids. Mice with liver-specific EZH2 knockout displayed reduced CCA development. In a xenograft model, EZH2 knockdown significantly decreased CCA progression. Administration of the EZH2 inhibitor GSK126 decreased CCA tumor burden in mice. Accordingly, EZH2 depletion or inhibition reduced the growth and colony formation capability of CCA cells. Analysis of high-throughput data identified a set of 12 tumor-inhibiting genes as targets of EZH2 in CCA. The experimental results suggest that EZH2 may down-regulate these tumor-inhibiting genes through methylation of lysine 27 on histone H3 (H3K27) in the gene louses and through regulation of specific miRNAs. High mobility group box 1 was shown to facilitate the methyltransferase activity of EZH2, which is implicated in the regulation of CCA cell growth. The study shows that EZH2 promotes CCA development and progression through a complicated regulatory network involving tumor-inhibiting genes, miRNAs, and high mobility group box 1, which support targeting EZH2 as a potentially effective strategy for CCA treatment.
Cholangiocarcinoma (CCA) is a highly malignant cancer of the biliary tree with poor prognosis.1, 2, 3, 4 Although the incidence rate and mortality of CCA are rising worldwide, currently there is no effective chemoprevention or systemic therapeutic option.1,2,5,6 Recent studies have shown the importance of aberrant histone epigenetic modification in CCA carcinogenesis and development.7, 8, 9, 10 Thus, a better understanding of the effect and underlying mechanism of histone modification in CCA may provide new therapeutic implications for CCA treatment.
Histone methylation is the modification of certain amino acids in a histone protein by the addition of methyl groups. Aberrant histone methylation induces tumor suppressor gene silencing and chromosomal instability, which results in cancer initiation and progression.11 Notably, enhancer of Zeste homolog 2 (EZH2), the core subunit of polycomb repressive complex 2 (PRC2), is a histone methyltransferase catalyzing the mono-, di-, and tri-methylation of lysine 27 on histone H3 (H3K27), a histone mark associated with compacted chromatin and repressed transcription.12 EZH2 overexpression or gain-of-function mutations, as well as its oncogenic role via affecting the expression of various target genes, have been documented in multiple types of cancers,13 including CCA.2,8,14, 15, 16, 17
Several lines of evidence have shown that the expression of EZH2 is typically up-regulated in CCA and negatively correlated with the prognosis of CCA in patients.8,14, 15, 16, 17 Preclinical data have shown that combined treatment with an EZH2 inhibitor and gemcitabine synergistically inhibits CCA cell growth.7 However, the role of EZH2 in CCA development has not been investigated, and the mechanism for EZH2-regulated gene expression in CCA remains to be further defined.
The current study provides novel evidence for an important role of EZH2 in CCA development. The experimental results show that EZH2 promotes CCA development and progression through a regulatory network involving tumor-inhibiting genes, miRNAs, and high mobility group box 1 (HMGB1). The data indicate that EZH2 down-regulates a set of 12 tumor-inhibiting genes through methylation of H3K27 in the gene louses and through regulation of specific miRNAs. Moreover, HMGB1 facilitates the methyltransferase activity of EZH2 via protein–protein interaction in CCA cells. These results provide further evidence for targeting EZH2 and related signaling pathways in CCA treatment.
Materials and Methods
Reagents
The EZH2 inhibitor GSK126 was purchased from Selleck Chemicals (Houston, TX). Captisol was purchased from AbMole BioScience (Houston, TX). siRNAs against human or mouse OSGIN1, PAX3, CDKN1A, and GAS1 were purchased from Integrated DNA Technologies (Coralville, IA). siRNA against mouse Hmgb1 was purchased from Thermo Fisher Scientific (Waltham, MA). Scramble control siRNA was purchased from Qiagen (Valencia, CA). Antibodies specific for EZH2 (#5246), Sox9 (#82630), and normal IgG were purchased from Cell Signaling Technology (Beverly, MA). Antibodies specific for H3K27me3 (#ab6002), HMGB1 (#ab18256), and Ki-67 (#ab15580) were purchased from Abcam (Cambridge, MA).
Induction of CCA in Mice
EZH2flox/flox (B6;129S1-EZH2tm2Sho/J) mice and albumin (Alb)-Cre (B6.Cg-Tg(Alb-cre) 21Mgn/J) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Liver-specific EZH2 knockout (EZH2-LKO) mice were generated by crossing EZH2flox/flox and Alb-Cre mice. Mice were kept at 22°C under a 12-hour light/dark cycle and were given ad libitum access to food and water. CCA was induced in 6- to 8-week–old EZH2-LKO mice and wild-type (WT) control mice by hydrodynamic tail vein (HDTV) injection of plasmids, including pT3-EF1aH-myc-NICD1 (15 μg per mouse), pT3-EF1aH-myr-AKT-HA (5 μg per mouse), and pCMV(CAT)T7-SB100 (1.25 μg per mouse) diluted in 2 mL saline; the plasmid mixtures were injected into the mouse tail vein in 5 to 7 seconds. Use of this method resulted in successful expression of exogenous gene in 40% to 50% of hepatocytes with one injection. Age matched Cre-negative littermates were used as WT controls.
All the mouse experimental procedures, as well as all the breeding and handling of mice, were approved by the Institutional Animal Care and Use Committee of Tulane University.
Treatment of CCA-Bearing Mice with the EZH2 Inhibitor GSK126
For this purpose, CCAs were induced in 8-week–old WT FVB mice by HDTV injection of transposase-based plasmids expressing Notch1 intercellular domain (NICD) and AKT. Seven days postinjection, the mice were randomized into two groups (n = 6 per group) and treated with GSK126 (150 mg/kg) or vehicle (20% Captisol) via i.p. injection every 2 days for 4 weeks. After the mice were euthanized, the livers were collected, and the liver/body weight ratios were calculated.
Xenograft Model of CCA
Five-week–old immunocompromised SCID mice (NOD.Cg-Prkdc scid/J) were purchased from The Jackson Laboratory. Stable EZH2-depleted or control CCLP1 cell suspensions were mixed with high-concentration BD Matrigel Matrix (BD Biosciences, Franklin Lakes, NJ) at a 1:1 ratio; 10 μL cell mixture (1 × 106 cells) was inoculated directly into the liver of each SCID mouse (n = 6 per group) using a Hamilton syringe (model 75NSYR; Hamilton Co., Reno, NV). The mice were observed for 12 weeks to monitor tumor development. After the mice were euthanized, the mouse livers were dissected surgically, and the ratios of liver/body weight were calculated.
Culture of CCA Cells
Human CCA cells (CCLP1) were cultured in Dulbecco’s modified Eagle’s medium (ATCC, Manassas, VA) supplemented with 10% fetal bovine serum (MilliporeSigma, St. Louis, MO) and Antibiotic-Antimycotic (100 U/mL penicillin, 100 μg/mL of streptomycin, and 0.25 μg/mL of Gibco Amphotericin B; Thermo Fisher Scientific). The cells were incubated in a humidified 5% carbon dioxide incubator at 37°C. Cells within 10 passages were used for experiments. The CCLP1 cell line was most recently authenticated by short tandem repeat DNA profiling analysis at ATCC in October 2021 and was excluded as having Mycoplasma contamination using the LookOut Mycoplasma PCR Detection Kit (Sigma-Aldrich) regularly. To knock down EZH2, CCLP1 cells were transfected with shEZH2-1 (5′-GAGATTATTTCTCAAGATG-3′), shEZH2-2 (5′-GAGGGAAAGTGTATGATAA-3′), or control shRNA plasmids, followed by selection in medium containing 1 μg/mL of Puromycin (Thermo Fisher Scientific) 48 hours after transfection.
To establish mouse EZH2-floxed CCA cells, EZH2flox/flox mice were euthanized 5 weeks after HDTV injection; the tumor tissues were then collected and rinsed with phosphate-buffered saline and minced into small pieces. After digestion with 0.1% type IV collagenase (Sigma-Aldrich) for 30 minutes at 37°C, disaggregated cell suspension was obtained by filtering through a 40 μm cell strainer. The filtrate was diluted with F12 medium (GIBCO BRL, Gaithersburg, MD) and centrifuged at 1000 rpm at 4°C for 5 minutes. The cell pellet was re-suspended in F12 medium and layered onto density gradients of 70%, 50%, and 40% iso-osmotic Percoll in F12 medium (from bottom to top). The Percoll gradients were centrifuged at 4500 rpm at 4°C for 30 minutes, and then cell fractions in 40% Percoll layer were collected. After washing with F12 medium three times, the collected cells were seeded into a 10-cm tissue culture dish (1 million cells per dish) with F12 medium supplied with 10% fetal bovine serum and incubated at 37°C in a humidified atmosphere with 5% carbon dioxide. The medium was changed twice a week. Possible fibroblast contamination was eliminated by differential attachment characteristics of fibroblast versus cancer cells. Two lines of EZH2-floxed CCA cells were established (#22891 and #22893), and cells were subjected to subculture while they reached approximately 80% confluence. For EZH2 depletion, Cre-expressing adenovirus or control adenovirus (Vector Biolab, Malvern, PA) was added to the cell cultures with a multiplicity of infection of 100 for 72 to 96 hours.
Cell Proliferation and Colony Formation Assays
For cell proliferation assay by WST-1 reagent, CCA cells (2 × 103 per well) with indicated treatment were seeded in 96-well plates at least in triplicate. At the indicated time point, the culture medium was removed, and the cells were incubated with 100 μL of serum-free medium containing 10 μL of WST-1 reagents (Roche, Indianapolis, IN) for 1 to 2 hours at 37°C. The absorbance of each sample was measured at 450 nm by using an ELISA plate reader.
For cell proliferation assay by cell counting, 1 × 105 CCA cells with or without EZH2 depletion were seeded in each well of the six-well plates. On the following day, the cells were transfected with indicated siRNA or scramble control siRNA. Seventy-two hours after siRNA transfection, an aliquot of the cells was stained by Trypan blue (1:1 ratio) to exclude dead cells, and the viable cells were counted by using an automated cell counter (Invitrogen, Carlsbad, CA).
For colony formation assays, CCA cells (2 × 103) with indicated treatments were plated in 10-cm dishes and cultured for 14 days to allow colony formation. The colonies were fixed with 100% methanol at room temperature for 10 minutes followed by 0.1% crystal violet staining for 20 minutes.
Transwell Cell Invasion Assay
Transwell assays were performed by using Corning BioCoat Matrigel Invasion Chambers (Tewksbury, MA) following the manufacturer’s instructions. Then, 500 μL cell suspensions (5 × 104 cells) in 1% fetal bovine serum media were loaded onto the upper chamber and 10% fetal bovine serum media to the lower chamber; the chambers were incubated for 22 hours in a 37°C humidified incubator with 5% carbon dioxide. Noninvading cells were removed by using a cotton swab. The invading cells on the lower surface of the membrane were fixed in methanol and stained with 0.5% crystal violet. Invading cells were counted in 10 randomly selected fields under an optical microscope.
RNA-Sequencing and Chromatin Immunoprecipitation Sequencing
Total RNAs from mouse CCA cells (#22891) treated with Cre-recombinase–expressing adenovirus (Ad-Cre) or control adenovirus were isolated with the RNeasy Mini Kit (Qiagen) following the manufacturer’s instructions. RNA samples were then processed for preparation of small RNA and mRNA sequencing libraries; the libraries were sequenced on a HiSeq2000 instrument (Illumina, San Diego, CA). RNA-sequencing (RNA-Seq) data were aligned to the mouse mm9 reference genome assembly using the TopHat (v1.4.1)-Cufflinks (v2.2.1) pipeline (https://tophat.com). Differential gene expression was analyzed by using the edgeR package (https://bioconductor.org/packages/release/bioc/html/edgeR.html).
Chromatin immunoprecipitation assays were performed in mouse CCA cells by using a SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology) according to the manufacturer’s instructions. After immunoprecipitation, the protein–DNA cross-links were reversed, and the DNA was purified and sent to GENEWIZ Company (South Plainfield, NJ) for sequencing on a HiSeq configuration. Sequence reads for each sample were mapped to the mouse mm10 reference genome. EaSeq version 1.04 software was used for data visualization.18
Immunoprecipitation and Mass Spectrometry Assay
CCA cells (1 × 107) were lysed in 1 mL Pierce IP Lysis Buffer (Thermo Fisher Scientific) with phosphatase and protease inhibitors. Then, 500 μL cell lysates were used for immunoprecipitation with specific antibodies. In brief, cell lysate was precleared with 30 μL protein A/G Magnetic beads (Thermo Fisher Scientific) by rotation at 4°C for 1 hour. The precleared supernatants were incubated with 2 μg antibodies by rotation at 4°C overnight and then with the addition of 100 μL A/G Magnetic beads (MilliporeSigma) at 4°C for 4 more hours. The samples were collected by magnetic stand, followed by washing five times with a beads wash solution (50 mmol/L Tris-HCl [pH 7.6], 150 mmol/L NaCl, 1 mmol/L EDTA, and 0.1% NP-40) and washing three times with phosphate-buffered saline. The protein/beads mixtures were sent to the Biomolecular/Proteomics Mass Spectrometry Facility at the University of California San Diego (San Diego, CA) for analysis. For immunoblotting, the bead complexes were suspended in 30 μL SDS-PAGE sample loading buffer and subjected to SDS-PAGE.
Histone Methyltransferase Activity Assay
EZH2 enzymatic activity was measured by using an ELISA-based EpiQuik Histone Methyltransferase Activity/Inhibition Assay Kit (H3-K27; EpiGentek, Farmingdale, NY). Briefly, nuclear proteins of CCLP1 cells with or without HMGB1 depletion by siRNA (Thermo Fisher Scientific) were extracted and incubated with substrate and assay buffer for 1 hour, followed by anti-H3K27me3 incubation for 1 hour. Then, horseradish peroxidase–conjugated detection antibody and developing solution were added for color development. Absorbance measurements were performed on a microplate reader at 450 nm.
Immunohistochemistry
Formalin-fixed tissue slides (4 μm thick) were deparaffinized in xylene, hydrated in ethanol, and subjected to heat retrieval at 100°C for 20 minutes in citrate buffer (pH 6.0). After blocking, the slides were incubated subsequently with primary antibodies at 4°C overnight and horseradish peroxidase–conjugated secondary antibodies at room temperature for 1 hour. After washing, the slides were subjected to 3,3′- diaminobenzidine for chromogenic development.
Western Blot Analysis
The tissue samples were homogenized in NP-40 lysis buffer, and the cultured cells were lysed in radioimmuno-precipitation assay buffer. The homogenates were centrifuged at 12,000 × g for 20 minutes at 4°C to collect the supernatants. After being denatured, equal amounts of protein were separated on SDS-PAGE gel and then transferred onto the nitrocellulose membrane (Bio-Rad, Hercules, CA). The milk-blocked blots were incubated with different primary antibodies at 4°C overnight. After three washings with phosphate-buffered saline with Tween20, blots were incubated with IRDye 800CW or 680LT conjugated secondary antibodies (LI-COR Biosciences, Lincoln, NE) for 1 hour at room temperature and scanned by using the LI-COR Odyssey Imaging system.
Statistical Analysis
Data are presented as means ± SD from a minimum of three replicates. Differences between groups were evaluated by using SPSS version 19.0 (IBM Corp., Armonk, NY) with one-way analysis of variance, two-tailed t-test, Mann-Whitney U test, χ2 test, or repeated measures analysis of generalized linear model when applicable. Kaplan-Meier survival and log-rank tests were used for mortality analysis. P < 0.05 was considered statistically significant.
Results
Analysis of EZH2 in Human CCA Tissues
Analysis of CCA patient data sets GSE76297,19 GSE26566,20 and GSE10794321 (NIH, https://www.ncbi.nlm.nih.gov/geo, last accessed September 29, 2022) indicated that EZH2 mRNA levels in CCA tissues were significantly higher than those in non-tumorous tissues (Figure 1A). Further survival analysis of patients with available clinical data from GSE107943 and TCGA-CHOL data sets (National Cancer Institute, https://www.cancer.gov/tcga, last accessed September 29, 2022) revealed that CCA patients with high EZH2 mRNA levels (more than the median of each cohort) exhibited significantly worse disease-free survival compared with patients with low EZH2 mRNA levels (less than the median of each cohort) (Figure 1B).
Figure 1.

Liver-specific deletion of enhancer of Zeste homolog 2 (EZH2) inhibits development of cholangiocarcinoma (CCA) in mice. A: EZH2 expression in human CCAs and non-tumorous tissues from Gene Expression Omnibus data sets GSE76297 (containing 91 CCAs and 92 adjacent nontumor samples) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76297), GSE26566 (containing 104 CCAs, 59 matched nontumor samples, as well as six normal intrahepatic bile duct samples) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE26566), and GSE107943 (containing 30 CCAs and 27 matched nontumor samples) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE107943). B: Kaplan-Meier survival and log-rank test showing the disease-free survival periods of patients with CCA in TCGA-CHOL and GSE107943 data sets. Patients were assigned to EZH2 high or EZH2 low groups with the median EZH2 level of each cohort as cutoff. C and D: Intrahepatic CCAs in liver-specific EZH2 knockout (EZH2-LKO) mice and control mice (C57BL/6 background) induced by hydrodynamic injection of Notch1 intercellular domain (NICD), AKT, and Sleeping Beauty (SB) transposase plasmids. Gross images of livers of each group at 5 weeks postinjection are shown in C, and the liver/body weight ratios of these mice are shown in D. E: Kaplan-Meier survival and log-rank test of EZH2-LKO and littermate control (wild-type [WT]) mice injected with NICD/AKT/SB plasmids (n = 12 per group). F and G: Hematoxylin and eosin (H&E) and immunohistochemistry stains (F), and Western blot analysis for indicated molecules (G) in the liver/tumor tissues from NICD/AKT/SB–injected mice and noninjected control mice (as shown in C). Data are expressed as means ± SD. ∗P < 0.05. Scale bars = 100 μm. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Depletion of EZH2 Prevents the Development of CCA in Mice
Mice with liver-specific knockout of EZH2 (EZH2-LKO) were used to determine the role of EZH2 in CCA development. The EZH2-LKO mice were generated by crossing EZH2flox/flox mice and Alb-Cre mice. The animals were subjected to a CCA development protocol as previously described,22 in which CCA was induced by HDTV injection of Sleeping Beauty (SB) transposase-based plasmids expressing Notch1 intercellular domain (NICD) and myristoylated-AKT. In this system, 8-week–old EZH2-LKO mice and their littermate WT controls were subjected to HDTV injection of NICD/AKT/SB plasmids. As shown in Figure 1C, whereas HDTV injection of NICD/AKT/SB plasmids induced the development of CCA in WT mice, the EZH2-LKO mice under the same HDTV injection protocol developed fewer and smaller tumor nodules in the liver. Accordingly, the EZH2-LKO mice displayed significantly lower liver/body weight ratios compared with their littermate controls under the CCA induction protocol (Figure 1D). The overall survival period of the EZH2-LKO mice was significantly longer than that of control mice (Figure 1E). Immunohistochemistry and immunoblotting analyses of the liver and CCA tissues showed successful deletion of EZH2 in EZH2-LKO mice and verified overexpression of NICD and AKT in mice receiving HDTV injection of NICD/AKT/SB plasmids (Figure 1, F and G). Immunohistochemistry and immunofluorescence analyses indicated that EZH2 is predominantly expressed in CCA cells but not in non-tumorous liver parenchymal cells (Figure 1F and Supplemental Figures S1 and S2). These results show that EZH2 enhances CCA development in mice.
EZH2 Silences a Panel of Tumor-Inhibiting Genes in CCA
To further investigate the mechanism of EZH2 in CCA, two lines of EZH2-floxed CCA cells (termed #22891 and #22893) were established from NICD/AKT/SB–induced CCA in EZH2flox/flox mice, as outlined in Figure 2A. As shown in Figure 2B, transduction of the EZH2-floxed CCA cells with Ad-Cre virus successfully depleted EZH2 and consequently decreased H3K27me3 levels as shown by Western blot analysis (Figure 2B). Depletion of EZH2 by Ad-Cre in EZH2-floxed CCA cells significantly decreased cell growth and reduced colony formation capabilities (Figure 2, C and D). The aforementioned CCA cells with or without EZH2 deletion were then processed for RNA-Seq analysis to identify EZH2 downstream targets. To this end, RNA-Seq was performed by using total RNA isolated from EZH2-floxed CCA cells infected with Ad-Cre or control virus for 72 hours. This approach led to the identification of 839 up-regulated genes (>1.7 fold) (Supplemental Table S1) in EZH2-depleted CCA cells versus control cells.
Figure 2.

Enhancer of Zeste homolog 2 (EZH2) deletion inhibits mouse cholangiocarcinoma (CCA) cell growth and up-regulates the expression of tumor-inhibiting genes. A: Schematic illustration for establishment of EZH2-floxed CCA cells from mouse CCA tissues. B: Western blot analysis showing the levels of EZH2 and H3K27me3 in mouse #22891 and #22893 CCA cells with or without EZH2 deletion (infected by Ad-Cre or control virus [Ad-Con] for 3 days). C: The proliferation of mouse EZH2-deleted or control CCA cells as determined by WST-1 assays. D: Representative images (left) and quantification (right) of colony formation assays of mouse CCA cells with or without EZH2 deletion. E: Gene Ontology analysis of genes upregulated ≥1.7-fold (P < 0.001) in EZH2-deleted #22891 cells versus control cells. F: Heatmap illustrating the expression of the 12 identified tumor-inhibiting genes in patients with CCA from the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76297; accession number GSE76297). G: Quantitative RT-PCR analysis to determine the levels of the 12 tumor-inhibiting genes in EZH2-depleted and control CCA cells. H and I: EZH2-depleted human CCA cells (CCLP1) (H) and mouse #22891 CCA cells (I) were transfected with siRNAs targeting OSGIN1, PAX3, CDKN1A, or GAS1; 72 hours posttransfection, the cells were counted on an automatic cell counter after being stained with Trypan blue. ∗P < 0.05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Because the role of EZH2 in cancer is largely attributable to the silencing of tumor-inhibiting genes, the current analysis focused on genes that were up-regulated in EZH2-depleted CCA cells. These genes were enriched in various biologic processes, including cell adhesion, growth, migration, angiogenesis, and differentiation (Figure 2E), as indicated by gene ontology analysis. Among them, 18 genes were identified as being involved in the process of negative regulation of cell growth and migration (Supplemental Table S2) and 92 genes in the process of cell differentiation (Supplemental Table S3). Gene validation in patient data sets and identified a panel of 12 genes whose expression levels were significantly decreased in patient CCA tissues versus matched nontumor tissues. These genes included Pax3, Wnt11, Wt1, Sfrp1, Ptprv, Osgin1, Il1rn, Cdkn1a, Itga7, Bbc3, Sulf1, and Gas1 (Figure 2F). Among them, Pax3, Wt1, and Sfrp1 are known to be implicated in cell differentiation as well as in negative regulation of tumor cell growth. The effect of EZH2 on the expression of the aforementioned 12-gene panel was further verified by quantitative RT-PCR analysis of CCA cells with or without EZH2 deletion (Figure 2G). Finally, four of the aforementioned genes (OSGIN1, PAX3, CDKN1A, and GAS1) were selected for further rescue experiments to document their tumor-inhibitory functions. Knockdown of these genes individually partially rescued EZH2 depletion–induced inhibition of CCA cell growth (Figure 2, H and I).
Depletion of EZH2 by shRNA in Human CCA Cells Restores the Expression of Tumor-Inhibiting Genes and Inhibits the Growth of Tumor Cells, in Vitro and in Vivo
To further determine the role of EZH2 in CCA, human CCA cells (CCLP1) with stable depletion of EZH2 were established by shRNA plasmid transfection. Depletion of EZH2 protein was confirmed by Western blot analysis (Figure 3A). EZH2 depletion led to a remarkable decrease in cell proliferation, colony formation, and invasion/migration (Figure 3, B-D). Quantitative RT-PCR analysis was performed to determine the level of tumor-inhibiting genes in CCLP1 cells with or without EZH2 depletion. Consistent with the effect of EZH2 on the expression of tumor-inhibiting genes as documented in patient CCA samples and mouse CCA cells, mRNA levels of the aforementioned 12 tumor-inhibiting genes in human CCA cells were also significantly increased after knockdown of EZH2 (Figure 3E). CCPL1 cells stably transfected with EZH2 shRNA or control vectors were next inoculated into the livers of SICD mice, and the animals were observed for 12 weeks after tumor cell inoculation. EZH2 knockdown significantly decreased tumor size and liver/body weight ratios (Figure 3E). Hematoxylin and eosin stain and immunostain for SOX9 indicated that the livers inoculated with EZH2-depleted cells developed smaller intrahepatic tumor lesions compared with livers inoculated with control vector cells (Figure 3F). Immunohistochemistry for the proliferation marker Ki-67 showed decreased nuclear staining in EZH2-depleted tumor cells (Figure 3G). Together, these results indicate that depletion of EZH2 in human CCA cells restored the expression of tumor-inhibiting genes and inhibited the growth of tumor cells in vitro and in vivo.
Figure 3.

Enhancer of Zeste homolog 2 (EZH2) depletion inhibits human cholangiocarcinoma (CCA) cell growth and restores the expression of tumor-inhibiting genes. A: Knockdown efficiency of EZH2 by shRNA in Human CCA cells (CCLP1) was determined by immunoblotting. B: The proliferation of EZH2-depleted and control CCLP1 cells was determined by using WST-1 assays. C: Representative images (upper) and quantification (lower) of colony formation assays of CCLP1 cells with or without EZH2 depletion. D: Representative images (upper) and quantification (lower) of transwell invasion assay of CCLP1 cells with or without EZH2 depletion. E: Quantitative RT-PCR analyses of the 12 tumor-inhibiting genes in CCLP1 cells with or without EZH depletion by shRNA. F: Gross liver images (upper) and liver/body weight ratio (lower) of SCID mice at 12 weeks post–intrahepatic inoculation of EZH2-depleted or control CCLP1 cells (1 × 106 per mouse, n = 6 for each group). G: Representative images of hematoxylin and eosin (H&E) and immunohistochemistry stains of SRY-box transcription factor 9 (SOX9) and Ki-67 in the liver tissues shown in F; high-power image of Ki-67 stain in right panel. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001. Scale bars: 100 μm (D); 200 μm (G). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
The EZH2 Inhibitor, GSK126, Inhibits CCA Growth and Restores the Expression of Tumor-Inhibiting Genes in CCA Cells
Because GSK126 is a highly selective small molecular inhibitor of EZH2,23 the effects of GSK126 on CCA growth were evaluated next. The efficacy of GSK126 on CCLP1 cell growth was first examined in vitro. Successful reduction of H3K27 trimethylation by GSK126 treatment was confirmed by immunoblotting analysis (Figure 4A). GSK126 treatment significantly reduced CCLP1 cell proliferation (Figure 4B). To assess the extent of GSK126 treatment on the expression of tumor-inhibiting genes, quantitative RT-PCR was performed by using total RNA isolated from CCLP1 cells treated by GSK126 or vehicle control (dimethyl sulfoxide). As shown in Figure 4C, GSK126 treatment significantly increased the expression of the 12 tumor-inhibiting genes. These in vitro studies were followed by further experiments to assess the efficacy of GSK126 on CCA development in mice. WT mice were subjected to HDTV injection of NICD/AKT/SB plasmid solution. Seven days postinjection, the mice were randomized into two groups and treated with GSK126 (150 mg/kg) or vehicle control (20% Captisol) via i.p. injection every 2 days for 4 weeks. The mice were euthanized 24 hours after the final treatment. As shown in Figure 4D, GSK126 treatment significantly decreased liver tumor burden, with the average liver/body weight ratio for GSK126 group much lower compared with that of the control group. Histologic analysis revealed that GSK126 treatment significantly decreased CCA tumor lesions and inhibited tumor cell proliferation (Figure 4E). Together, these results show that the EZH2 inhibitor GSK126 inhibits CCA cell growth and restores the expression of tumor-inhibiting genes in CCA cells.
Figure 4.

Treatment with GSK126 inhibits cholangiocarcinoma (CCA) growth in vitro and in mice. A: Immunoblotting for enhancer of Zeste homolog 2 (EZH2) and H3K27me3 levels in human CCA cells (CCLP1) treated with GSK126 or vehicle control for 24 hours. B: WST-1 assays of CCLP1 cells treated with GSK126 or vehicle control. C: The mRNA levels of the 12 tumor-inhibiting genes in CCLP1 cells with or without GSK126 treatments were measured by using quantitative RT-PCR. D: CCAs in wild-type FVB mice were induced by hydrodynamic tail vein injection of Notch1 intercellular domain/AKT/Sleeping Beauty plasmids. Seven days postinjection, mice were treated with GSK126 (150 mg/kg) or 20% Captisol (vehicle control) via i.p. injection every 2 days for 4 weeks (n = 6 per group). Representative livers from each group of mice are shown in the left panel. The liver/body weight ratios were calculated and are shown in the right panel. E: Representative hematoxylin and eosin (H&E) and immunohistochemistry stains of SOX9 and Ki-67 in the liver/tumor tissues shown in D; high-power image of Ki-67 stain in the right panel. ∗P < 0.05, ∗∗P < 0.01. Scale bars = 200 μm. DMSO, dimethyl sulfoxide.
Both EZH2 deletion and GSK126 treatment led to increased expression of tumor-inhibiting genes, although the extent of the increase for individual genes varied. While the exact reason(s) for the observed variations in fold change is not clear, possible explanations may include different approaches of EZH2 inhibition, the selection of EZH2 shRNA-transfected cells, or the concentration of GSK126 used in the experiments.
EZH2 Regulates Tumor-Inhibiting Genes in CCA Cells via Histone Methylation and miRNAs
To delineate the mechanism underlying EZH2 regulation of tumor-inhibiting genes in CCA, chromatin immunoprecipitation assays with anti-EZH2 and anti-H3K27me3 were performed followed by high-throughput sequencing in CCA cells. The enrichment peaks by anti-EZH2 and anti-H3K27me3 are shown in Figure 5A. Of the 12 tumor-inhibiting genes, five (Pax3, Wnt11, Wt1, Sfrp1 and Ptprv) were found to be significantly enriched both by anti-EZH2 and by anti-H3K27me3, which suggests that the expression of these five genes may be directly regulated by EZH2-mediated H3K27 methylation (Figure 5B). Conversely, the remaining seven genes (Osgin1, Il1rn, Cdkn1a, Itga7, Bbc3, Sulf1, and Gas1) were not enriched by anti-H3K27me3 or anti-EZH2. Given that deregulation of miRNAs is known to be involved in CCA development,8,24,25 small-RNA sequencing was performed to identify miRNAs that may mediate EZH2 regulation of tumor-inhibiting genes in CCA. Small-RNA sequencing results showed that 99 miRNAs were down-regulated (>1.5 fold) (Supplemental Table S4) in CCA cells with EZH2 depletion. miRNA target prediction tool TargetScan26 identified 36 miRNAs with the potential to target the tumor-inhibiting panel of 12 genes (Table 1). These results suggest that EZH2 may down-regulate tumor-inhibiting genes in CCA cells via methylation of H327 in the DNA locus of these genes or via regulation of specific miRNAs.
Figure 5.

Chromatin immunoprecipitation (ChIP)-sequencing identifies the association of enhancer of Zeste homolog 2 (EZH2) and H3K27 to tumor-inhibiting genes in cholangiocarcinoma cells. A: ChIP assays were performed in #22891 cells, followed by high-throughput sequencing. Heatmap clustering of ChIP-sequencing profiles indicating EZH2-binding peaks enriched by anti-EZH2, anti-H3K27me3, and IgG are shown. B: The ChIP-sequencing peaks enriched by anti-EZH2 (top), anti-H3K27me3 (middle) and IgG (bottom) in the locus of Pax3, Wnt11, Wt1, Sfrp1, and Ptprv genes.
Table 1.
miRNAs Targeting the Tumor-Inhibiting Panel of 12 Genes
| Gene | miRNA |
|---|---|
| Pax3 | miR-704, miR-1927 |
| Wnt11 | miR-1938, miR-3966 |
| Wt1 | miR-1931, miR-1949, miR-704 |
| Sfrp1 | miR-1938, miR-378b, miR-1927, miR-5125, miR-6240 |
| Ptprv | miR-7069 |
| Osgin1 | miR-344c, miR-326, miR-7647, miR-6975, miR-6992, miR-5627, miR-3077, miR-6913, miR-7030, miR-5626 |
| Il1rn | miR-1931, miR-709 |
| Cdkn1a | miR-6538 |
| Itga7 | miR-1938, miR-5119 |
| Bbc3 | miR-1938, miR-2137, miR-709, miR-5126 |
| Sulf1 | miR-378b, miR-6240 |
| Gas1 | miR-1969, miR-3470b |
HMGB1 Facilitates EZH2-Mediated H3K27 Methylation
Because EZH2 is also known to regulate cell functions through direct interaction with other proteins27, 28, 29, 30, 31, 32, 33 to form a multiprotein complex, a mass spectrometry–based proteomics approach was used to identify proteins associated with EZH2 protein in CCA cells. Specifically, immunoprecipitation was performed in tandem with mass spectrometry assays to identify the physically interacting proteins of EZH2 in CCLP1 cells. This approach led to the identification of 187 proteins precipitated by anti-EZH2 antibody (Supplemental Table S5). Among those potential EZH2-binding proteins, HMGB1 was identified with high coverage and thus was selected for further investigation. Subsequent immunoprecipitation and immunoblotting analysis confirmed the interaction between EZH2 and HMGB1 in both human and mouse CCA cells (Figure 6A).
Figure 6.

High mobility group box 1 (HMGB1) facilitates enhancer of Zeste homolog 2 (EZH2) methyltransferase activity via protein–protein interaction. A: Cholangiocarcinoma (CCA) cell extracts were subjected to immunoprecipitation (IP) using anti-EZH2 antibody or isotype IgG followed by Western blot analysis. B: Mouse CCA cells were transfected with HMGB1-targeting siRNA or scramble control. Seventy-two hours posttransfection, nuclear extracts were collected and subjected to EZH2 methyltransferase activity assays using H3K27 as substrates. C: Detection of EZH2 and HMGB1 by immunoblotting in mouse CCA cells with EZH2 deletion (by Ad-Cre virus infection) (upper panel) or with HMGB1 knockdown (by siRNA transfection) (lower panel). D: HMGB1 expression levels in human CCAs and non-tumorous tissues of Gene Expression Omnibus data sets GSE76297 and GSE107943. E: HMGB1 levels in CCAs from wild-type mouse at 5 weeks post–Notch1 intercellular domain (NICD)/AKT/Sleeping Beauty (SB) plasmids injection. Liver tissues from noninjected mice were used as controls. F: Correlation of EZH2 and HMGB1 mRNA levels in patient tissues from data sets GSE76297 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76297) or GSE107943 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE107943). G: Schematic illustration of the mechanisms of EZH2 in CCA. ∗P < 0.05.
Histone methyltransferase activity assays were performed to examine whether HMGB1 might affect the methyltransferase activity of EZH2. For this purpose, CCA cells were transfected with specific siRNA to knock down HMGB1. As shown in Figure 6B, HMGB1 knockdown by siRNA significantly decreased the EZH2 methyltransferase activity (as reflected by decreased production of H3K27me3). However, the level of EZH2 protein was not altered by HMGB1 depletion, and vice versa (Figure 6C). These results suggest that HMGB1 may facilitate EZH2 methyltransferase activity through a protein–protein interaction. Data mining from patients with CCA and mouse CCA tissue analysis indicated that HMGB1 expression was significantly increased in both human and mouse CCA tissues compared with respective liver/non-tumorous tissues (Figure 6, D and E). Furthermore, levels of EZH2 and HMGB1 in patient CCA tissues were positively correlated (Figure 6F). These findings suggest that HMGB1 may enhance the tumor-promoting effect of EZH2 in CCA.
Collectively, the results presented in this study show that EZH2 promotes CCA growth through silencing a panel of tumor-inhibiting genes via methylating histone H3K27 in the gene locus or through regulation of specific miRNAs to target tumor-inhibiting genes. The results also indicate that the tumor-promoting effect of EZH2 in CCA could be further enhanced by HMGB1 (Figure 6G).
Discussion
EZH2, the enzymatic subunit of PRC2, canonically catalyzes the methylation reactions of H3K27 at target DNA locus for gene silencing.12,13,34 The association of EZH2 overexpression with aggressive and advanced CCA is well-documented.8,35, 36, 37 However, the role of EZH2 in CCA development has not been previously investigated, and the target genes of EZH2 in CCA, as well as the underlying regulatory mechanisms, remain to be defined. The current study used a mouse model of CCA development induced by HDTV injection of NICD/AKT/SB plasmids. This approach combines HDTV delivery and transposase-mediated somatic integration of oncogenes (activated NICD1 and AKT) into the hepatocyte genome, which is known to effectively initiate hepatocyte transformation leading to rapid development of CCA.22 This model indicated that EZH2 depletion efficiently prevents CCA development. To our knowledge, this is the first study establishing EZH2 as a pro-oncogenic molecule in the development of CCA.
This study established EZH2-floxed CCA cells isolated from EZH2flox/flox mice, which allows efficient deletion of EZH2 in CCA cells. Comparison of RNA-Seq data from CCA cells with or without Ad-Cre–mediated EZH2 deletion in conjunction with the analysis of human CCA tissues identified a panel of 12 genes (Pax3, Wnt11, Wt1, Sfrp1, Ptprv, Osgin1, Il1rn, Cdkn1a, Itga7, Bbc3, Sulf1, and Gas1) as downstream targets of EZH2 in CCA cells. The identified genes were further validated by quantitative RT-PCR analysis of CCA cells with EZH2 depletion and with EZH2 inhibitor treatment. All of the aforementioned genes are known to be involved in pathways related to negative regulation of cell proliferation and migration, with tumor-inhibiting functions documented in multiple cancer types.38, 39, 40, 41, 42 For example, Osign1 has been identified as a tumor suppressor in human hepatocellular carcinoma38; PAX3, a transcription factor involved in cell proliferation and survival, is a tumor-suppressor in thyroid cancer39,40; and CDKN1A has been reported as a tumor suppressor in CCA.41 GAS1 induces cell arrest and apoptosis, and acts as a tumor suppressor in multiple cancers.42 The current study provided novel evidence of these genes being regulated by EZH2 in CCA cells. The functional impact of representative genes in EZH2-mediated CCA cell growth is further corroborated by the observations that knockdown of Cdkn1a, Osgin1, Pax3, or Gas1 in EZH2-depleted CCA cells partially reverses the inhibiting effects of EZH2 depletion on CCA cell proliferation.
To explore the mechanisms by which EZH2 regulates the expression of tumor-inhibiting genes in CCA cells, chromatin immunoprecipitation assays with anti-EZH2 or anti-H3K27me3 were performed, followed by high-throughput sequencing in CCA cells. EZH2 was shown to induce hypermethylation status of H3K27 in the locus of five of the 12 tumor-inhibiting genes (Pax3, Wnt11, Wt1, Sfrp1, and Ptprv) in CCA cells, suggesting that the expression of these five genes is likely regulated by EZH2-mediated H3K27 methylation. Additionally, small-RNA sequencing analysis identified 36 EZH2-regulated miRNAs that target the aforementioned 12 tumor-inhibiting genes in CCA cells. These results, along with the RNA-Seq and chromatin immunoprecipitation sequencing data, suggest that EZH2 may promote CCA growth through mechanisms that include down-regulation of tumor-inhibiting genes via H327 methylation as well as miRNAs.
EZH2 is known to regulate cellular functions through mechanisms independent of H3K27 methylation in various cell types.27, 28, 29, 30, 31, 32, 33 For example, multiple lines of evidence have shown that EZH2 functions as a transcription activator in cancers.27 EZH2 is known to interact with ERα and β-catenin to regulate c-Myc and cyclin D1 expression in breast cancer.28 EZH2 has also been found to increase the expression of multiple Wnt pathway genes via interacting with β-catenin in epithelial hyperplasia and colon cancer.29,30 In addition, EZH2 reportedly serves as a transcriptional co-activator with androgen receptor in castration-resistant prostate cancer cells,31 and as a co-activator of cyclin D1 transcription in T-cell lymphoma.33 EZH2 can also enhance the activities of certain transcription factors through lysine methylation, as shown in methylation of multiple lysine residues in STAT3 leading to complete activation of STAT3 in glioblastoma.32 The results presented in the current study indicate that EZH2 in CCA cells regulates the levels of multiple miRNAs, which may provide an explanation for H3K27me3-independent functions of EZH2.
Another novel aspect of this study is the identification of a functional connection between HMGB1 and EZH2 in CCA. HMGB1 mediates a broad range of physiological and pathologic processes in the liver, including cell proliferation, autophagy, inflammation, and immunomodulation.43, 44, 45 Targeting HMGB1 constitutes a favorable therapeutic strategy for inflammatory diseases and cancer.46 With respect to CCA, previous studies on HMGB1 have primarily focused on its clinical and prognostic values in patients,47,48 whereas the potential function of HMGB1 in CCA has not been investigated. The current study indicated a positive corelation between the mRNA levels of EZH2 and HMGB1 in patient CCA tissues. The interaction between EZH2 and HMGB1 proteins in CCA cells was identified by mass spectrometry and confirmed by immunoprecipitation and immunoblotting analysis. Further histone methyltransferase activity assays showed that HMGB1 knockdown decreased EZH2 methyltransferase activity. Given that the level of EZH2 protein was not altered by HMGB1 depletion (or vice versa), we reason that HMGB1 may regulate EZH2 methyltransferase activity through protein–protein interaction. Because EZH2 methyltransferase activity is known to be regulated allosterically,49 it is possible that HMGB1 may bind to EZH2 and enhance its enzyme activity through allosteric regulation. The precise mechanism for HMGB1-mediated regulation of EZH2 activity remains to be delineated in future studies. In this context, the results presented in the current study suggest a possible synergistic action of HMGB1 on EZH2 methyltransferase activity through direct protein–protein interaction, which may reflect their cancer-promoting effects in CCA. Whether the interaction between EZH2 and HMGB1 can be targeted for CCA treatment remains to be determined.
The current study used a mouse model of CCA induced by NICD/AKT to assess the therapeutic efficacy of the EZH2 inhibitor, GSK126. GSK126 treatment significantly decreased the development of CCA in mice. These findings provide additional preclinical evidence for utilization of GSK126 in CCA treatment. A potential pitfall for the NICD/AKT–induced CCA model is the short disease course of CCA, which prevents assessment of treatment efficacy at a later time point when the disease has progressed. Further clinical studies are needed to determine whether GSK126 can be used as an effective therapeutic agent for the treatment of patients with CCA.
The current studies indicated that EZH2 knockout was more effective than GSK126 treatment in reducing the CCA tumor burden. For the EZH2 knockout model, the liver/body weight ratio was decreased from 0.178 to 0.114, reflecting an approximately 35% reduction of tumor burden. For the GSK126 treatment model, the liver/body weight ratio was reduced from 0.289 to 0.231, indicating an approximately 20% reduction of tumor burden. However, because the experiments for EZH2 knockout and GSK126 treatment were performed in mice with different genetic backgrounds, it is challenging to accurately compare the tumor-inhibiting efficiency of those two different methodologies.
In summary, this study presents important evidence for the role of EZH2 in CCA development and tumor progression. By using complementary approaches of CCA models, RNA-Seq, miRNA-sequencing, proteomics, and biochemical analyses, the study dissected an important regulatory network critical in EZH2-mediated CCA growth. The current results showed that EZH2 promotes CCA growth through silencing a panel of tumor-inhibiting genes via methylating histone H3K27 in the gene locus or through regulation of specific miRNAs to target tumor-inhibiting genes. Furthermore, they revealed a novel regulatory mechanism for regulation of EZH2 by HMGB1 in CCA. The experimental findings provide further evidence in support of targeting EZH2 and related signaling mechanism as a potentially effective strategy for CCA treatment.
Footnotes
Supported by the NIH grants CA102325, CA219541, and CA226281, and Department of Defense grant CA180361.
J.Z. and W.C. contributed equally to this article.
Disclosures: None declared.
Supplemental material for this article can be found at http://doi.org/10.1016/j.ajpath.2022.08.008.
Contributor Information
Jinqiang Zhang, Email: jzhang@tulane.edu.
Tong Wu, Email: twu@tulane.edu.
Supplemental Data
Supplemental Figure S1.
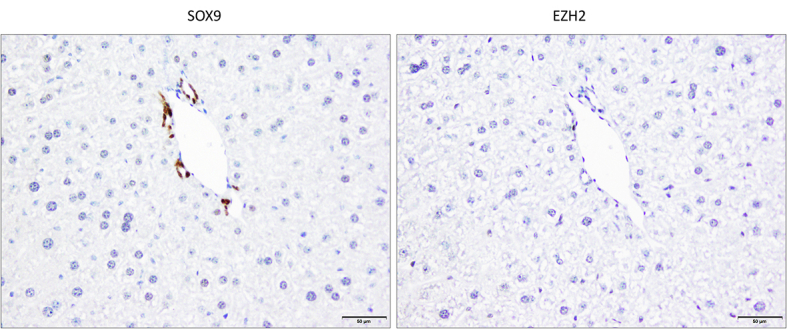
Representative immunohistochemical staining for enhancer of Zeste homolog 2 (EZH2) and SRY-box transcription factor 9 (SOX9) in serial sections of liver tissues from un-injected control wild-type mice. The interlobular bile duct epithelium (staining with the biliary marker SOX9) (left panel) is negative for EZH2 (right panel). The nonneoplastic hepatocytes also show no significant staining for EZH2. Scale bar = 50 μm.
Supplemental Figure S2.
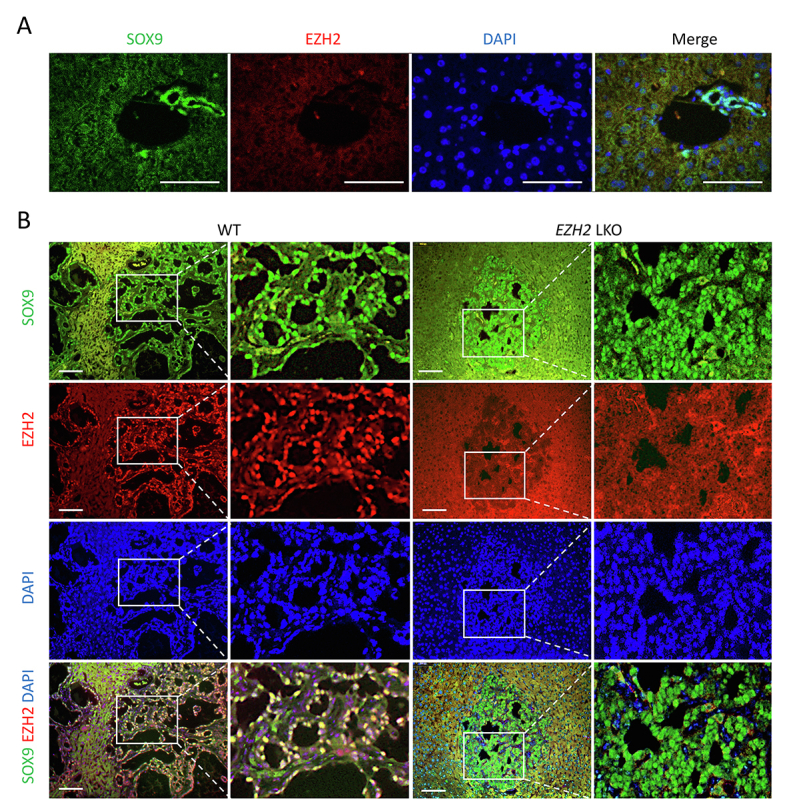
A: Co-immunofluorescence staining of enhancer of Zeste homolog 2 (EZH2) and the biliary marker SRY-box transcription factor 9 (SOX9) in liver tissues from un-injected wild-type (WT) control. B: Co-immunofluorescence staining of EZH2 and the biliary marker SOX9 in liver tissues from Notch1 intercellular domain/AKT/Sleeping Beauty–injected mice. The EZH2 signal is detected in the cholangiocarcinoma cells but not in the non-tumorous liver parenchymal cells. Note the background autofluorescence in the normal liver region. Scale bars: 50 μm (A); 100 μm (B).
References
- 1.Banales J.M., Marin J.J.G., Lamarca A., Rodrigues P.M., Khan S.A., Roberts L.R., Cardinale V., Carpino G., Andersen J.B., Braconi C., Calvisi D.F., Perugorria M.J., Fabris L., Boulter L., Macias R.I.R., Gaudio E., Alvaro D., Gradilone S.A., Strazzabosco M., Marzioni M., Coulouarn C., Fouassier L., Raggi C., Invernizzi P., Mertens J.C., Moncsek A., Rizvi S., Heimbach J., Koerkamp B.G., Bruix J., Forner A., Bridgewater J., Valle J.W., Gores G.J. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17:557–588. doi: 10.1038/s41575-020-0310-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rizvi S., Khan S.A., Hallemeier C.L., Kelley R.K., Gores G.J. Cholangiocarcinoma—evolving concepts and therapeutic strategies. Nat Rev Clin Oncol. 2018;15:95–111. doi: 10.1038/nrclinonc.2017.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fabris L., Sato K., Alpini G., Strazzabosco M. The tumor microenvironment in cholangiocarcinoma progression. Hepatology. 2021;73(Suppl 1):75–85. doi: 10.1002/hep.31410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brindley P.J., Bachini M., Ilyas S.I., Khan S.A., Loukas A., Sirica A.E., Teh B.T., Wongkham S., Gores G.J. Cholangiocarcinoma. Nat Rev Dis Primers. 2021;7:65. doi: 10.1038/s41572-021-00300-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan S.A., Tavolari S., Brandi G. Cholangiocarcinoma: epidemiology and risk factors. Liver Int. 2019;39(Suppl 1):19–31. doi: 10.1111/liv.14095. [DOI] [PubMed] [Google Scholar]
- 6.Razumilava N., Gores G.J. Classification, diagnosis, and management of cholangiocarcinoma. Clin Gastroenterol Hepatol. 2013;11:13–21.e1. doi: 10.1016/j.cgh.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakagawa S., Sakamoto Y., Okabe H., Hayashi H., Hashimoto D., Yokoyama N., Tokunaga R., Sakamoto K., Kuroki H., Mima K., Beppu T., Baba H. Epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A inhibits the growth of cholangiocarcinoma cells. Oncol Rep. 2014;31:983–988. doi: 10.3892/or.2013.2922. [DOI] [PubMed] [Google Scholar]
- 8.Kwon H., Song K., Han C., Zhang J., Lu L., Chen W., Wu T. Epigenetic silencing of miRNA-34a in human cholangiocarcinoma via EZH2 and DNA methylation: impact on regulation of notch pathway. Am J Pathol. 2017;187:2288–2299. doi: 10.1016/j.ajpath.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Rourke C.J., Munoz-Garrido P., Aguayo E.L., Andersen J.B. Epigenome dysregulation in cholangiocarcinoma. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1423–1434. doi: 10.1016/j.bbadis.2017.06.014. [DOI] [PubMed] [Google Scholar]
- 10.Ma W., Han C., Zhang J., Song K., Chen W., Kwon H., Wu T. The histone methyltransferase G9a promotes cholangiocarcinogenesis through regulation of the hippo pathway kinase LATS2 and YAP signaling pathway. Hepatology. 2020;72:1283–1297. doi: 10.1002/hep.31141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Audia J.E., Campbell R.M. Histone modifications and cancer. Cold Spring Harb Perspect Biol. 2016;8:a019521. doi: 10.1101/cshperspect.a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greer E.L., Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim K.H., Roberts C.W.M. Targeting EZH2 in cancer. Nat Med. 2016;22:128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wasenang W., Puapairoj A., Settasatian C., Proungvitaya S., Limpaiboon T. Overexpression of polycomb repressive complex 2 key components EZH2/SUZ12/EED as an unfavorable prognostic marker in cholangiocarcinoma. Pathol Res Pract. 2019;215:152451. doi: 10.1016/j.prp.2019.152451. [DOI] [PubMed] [Google Scholar]
- 15.Nakagawa S., Okabe H., Ouchi M., Tokunaga R., Umezaki N., Higashi T., Kaida T., Arima K., Kitano Y., Kuroki H., Mima K., Nitta H., Imai K., Hashimoto D., Yamashita Y.-I., Chikamoto A., Baba H. Enhancer of zeste homolog 2 (EZH2) regulates tumor angiogenesis and predicts recurrence and prognosis of intrahepatic cholangiocarcinoma. HPB (Oxford) 2018;20:939–948. doi: 10.1016/j.hpb.2018.03.018. [DOI] [PubMed] [Google Scholar]
- 16.Nakagawa S., Okabe H., Sakamoto Y., Hayashi H., Hashimoto D., Yokoyama N., Sakamoto K., Kuroki H., Mima K., Nitta H., Imai K., Chikamoto A., Watanabe M., Beppu T., Baba H. Enhancer of zeste homolog 2 (EZH2) promotes progression of cholangiocarcinoma cells by regulating cell cycle and apoptosis. Ann Surg Oncol. 2013;20(Suppl 3):S667–S675. doi: 10.1245/s10434-013-3135-y. [DOI] [PubMed] [Google Scholar]
- 17.Tang B., Du J., Li Y., Tang F., Wang Z., He S. EZH2 elevates the proliferation of human cholangiocarcinoma cells through the downregulation of RUNX3. Med Oncol. 2014;31:271. doi: 10.1007/s12032-014-0271-6. [DOI] [PubMed] [Google Scholar]
- 18.Lerdrup M., Johansen J.V., Agrawal-Singh S., Hansen K. An interactive environment for agile analysis and visualization of ChIP-sequencing data. Nat Struct Mol Biol. 2016;23:349–357. doi: 10.1038/nsmb.3180. [DOI] [PubMed] [Google Scholar]
- 19.Chaisaingmongkol J., Budhu A., Dang H., Rabibhadana S., Pupacdi B., Kwon S.M., et al. Common molecular subtypes among Asian hepatocellular carcinoma and cholangiocarcinoma. Cancer Cell. 2017;32:57–70.e3. doi: 10.1016/j.ccell.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersen J.B., Spee B., Blechacz B.R., Avital I., Komuta M., Barbour A., Conner E.A., Gillen M.C., Roskams T., Roberts L.R., Factor V.M., Thorgeirsson S.S. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology. 2012;142:1021–1031.e15. doi: 10.1053/j.gastro.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahn K.S., O’Brien D., Kang Y.N., Mounajjed T., Kim Y.H., Kim T.-S., Kocher J.-P.A., Allotey L.K., Borad M.J., Roberts L.R., Kang K.J. Prognostic subclass of intrahepatic cholangiocarcinoma by integrative molecular-clinical analysis and potential targeted approach. Hepatol Int. 2019;13:490–500. doi: 10.1007/s12072-019-09954-3. [DOI] [PubMed] [Google Scholar]
- 22.Fan B., Malato Y., Calvisi D.F., Naqvi S., Razumilava N., Ribback S., Gores G.J., Dombrowski F., Evert M., Chen X., Willenbring H. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest. 2012;122:2911–2915. doi: 10.1172/JCI63212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morera L., Lübbert M., Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin Epigenetics. 2016;8:57. doi: 10.1186/s13148-016-0223-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki H., Maruyama R., Yamamoto E., Kai M. Epigenetic alteration and microRNA dysregulation in cancer. Front Genet. 2013;4:258. doi: 10.3389/fgene.2013.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salati M., Braconi C. Noncoding RNA in cholangiocarcinoma. Semin Liver Dis. 2019;39:13–25. doi: 10.1055/s-0038-1676097. [DOI] [PubMed] [Google Scholar]
- 26.McGeary S.E., Lin K.S., Shi C.Y., Pham T.M., Bisaria N., Kelley G.M., Bartel D.P. The biochemical basis of microRNA targeting efficacy. Science. 2019;366:eaav1741. doi: 10.1126/science.aav1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamaguchi H., Hung M.-C. Regulation and role of EZH2 in cancer. Cancer Res Treat. 2014;46:209–222. doi: 10.4143/crt.2014.46.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi B., Liang J., Yang X., Wang Y., Zhao Y., Wu H., Sun L., Zhang Y., Chen Y., Li R., Zhang Y., Hong M., Shang Y. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol. 2007;27:5105–5119. doi: 10.1128/MCB.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jung H.-Y., Jun S., Lee M., Kim H.-C., Wang X., Ji H., McCrea P.D., Park J.-I. PAF and EZH2 induce Wnt/[beta]-catenin signaling hyperactivation. Mol Cell. 2013;52:193–205. doi: 10.1016/j.molcel.2013.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X., Gonzalez M.E., Toy K., Filzen T., Merajver S.D., Kleer C.G. Targeted overexpression of EZH2 in the mammary gland disrupts ductal morphogenesis and causes epithelial hyperplasia. Am J Pathol. 2009;175:1246–1254. doi: 10.2353/ajpath.2009.090042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu K., Wu Z.J., Groner A.C., He H.H., Cai C., Lis R.T., Wu X., Stack E.C., Loda M., Liu T., Xu H., Cato L., Thornton J.E., Gregory R.I., Morrissey C., Vessella R.L., Montironi R., Magi-Galluzzi C., Kantoff P.W., Balk S.P., Liu X.S., Brown M. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim E., Kim M., Woo D.-H., Shin Y., Shin J., Chang N., Oh Y.T., Kim H., Rheey J., Nakano I., Lee C., Joo K.M., Rich J.N., Nam D.-H., Lee J. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23:839–852. doi: 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan J., Ng S.-B., Tay J.L.-S., Lin B., Koh T.L., Tan J., Selvarajan V., Liu S.-C., Bi C., Wang S., Choo S.-N., Shimizu N., Huang G., Yu Q., Chng W.-J. EZH2 overexpression in natural killer/T-cell lymphoma confers growth advantage independently of histone methyltransferase activity. Blood. 2013;121:4512–4520. doi: 10.1182/blood-2012-08-450494. [DOI] [PubMed] [Google Scholar]
- 34.Kim J., Lee Y., Lu X., Song B., Fong K.-W., Cao Q., Licht J.D., Zhao J.C., Yu J. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 2018;25:2808–2820.e4. doi: 10.1016/j.celrep.2018.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang M., Yang H., Wan L., Wang Z., Wang H., Ge C., Liu Y., Hao Y., Zhang D., Shi G., Gong Y., Ni Y., Wang C., Zhang Y., Xi J., Wang S., Shi L., Zhang L., Yue W., Pei X., Liu B., Yan X. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J Hepatol. 2020;73:1118–1130. doi: 10.1016/j.jhep.2020.05.039. [DOI] [PubMed] [Google Scholar]
- 36.Sasaki M., Matsubara T., Yoneda N., Nomoto K., Tsuneyama K., Sato Y., Nakanuma Y. Overexpression of enhancer of zeste homolog 2 and MUC1 may be related to malignant behaviour in intraductal papillary neoplasm of the bile duct. Histopathology. 2013;62:446–457. doi: 10.1111/his.12016. [DOI] [PubMed] [Google Scholar]
- 37.Sasaki M., Ikeda H., Itatsu K., Yamaguchi J., Sawada S., Minato H., Ohta T., Nakanuma Y. The overexpression of polycomb group proteins Bmi1 and EZH2 is associated with the progression and aggressive biological behavior of hepatocellular carcinoma. Lab Invest. 2008;88:873–882. doi: 10.1038/labinvest.2008.52. [DOI] [PubMed] [Google Scholar]
- 38.Liu M., Li Y., Chen L., Chan T.H.M., Song Y., Fu L., Zeng T.-T., Dai Y.-D., Zhu Y.-H., Li Y., Chen J., Yuan Y.-F., Guan X.-Y. Allele-specific imbalance of oxidative stress-induced growth inhibitor 1 associates with progression of hepatocellular carcinoma. Gastroenterology. 2014;146:1084–1096. doi: 10.1053/j.gastro.2013.12.041. [DOI] [PubMed] [Google Scholar]
- 39.Boudjadi S., Chatterjee B., Sun W., Vemu P., Barr F.G. The expression and function of PAX3 in development and disease. Gene. 2018;666:145–157. doi: 10.1016/j.gene.2018.04.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu W., Sui F., Liu J., Wang M., Tian S., Ji M., Shi B., Hou P. PAX3 is a novel tumor suppressor by regulating the activities of major signaling pathways and transcription factor FOXO3a in thyroid cancer. Oncotarget. 2016;7:54744–54757. doi: 10.18632/oncotarget.10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu Y., Zhang M., Wang N., Li Q., Yang J., Yan S., He X., Ji G., Miao L. Epigenetic silencing of tumor suppressor gene CDKN1A by oncogenic long non-coding RNA SNHG1 in cholangiocarcinoma. Cell Death Dis. 2018;9:746. doi: 10.1038/s41419-018-0768-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Segovia J., Zarco N. Gas1 is a pleiotropic regulator of cellular functions: from embryonic development to molecular actions in cancer gene therapy. Mini Rev Med Chem. 2014;14:1139–1147. doi: 10.2174/1389557514666141127142301. [DOI] [PubMed] [Google Scholar]
- 43.Mendonça Gorgulho C., Murthy P., Liotta L., Espina V., Lotze M.T. Different measures of HMGB1 location in cancer immunology. Methods Enzymol. 2019;629:195–217. doi: 10.1016/bs.mie.2019.10.011. [DOI] [PubMed] [Google Scholar]
- 44.Tripathi A., Shrinet K., Kumar A. HMGB1 protein as a novel target for cancer. Toxicol Rep. 2019;6:253–261. doi: 10.1016/j.toxrep.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ugrinova I., Pasheva E. HMGB1 protein: a therapeutic target inside and outside the cell. Adv Protein Chem Struct Biol. 2017;107:37–76. doi: 10.1016/bs.apcsb.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 46.Xue J., Suarez J.S., Minaai M., Li S., Gaudino G., Pass H.I., Carbone M., Yang H. HMGB1 as a therapeutic target in disease. J Cell Physiol. 2021;236:3406–3419. doi: 10.1002/jcp.30125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu Y.-F., Liu Z.-L., Pan C., Yang X.-Q., Ning S.-L., Liu H.-D., Guo S., Yu J.-M., Zhang Z.-L. HMGB1 correlates with angiogenesis and poor prognosis of perihilar cholangiocarcinoma via elevating VEGFR2 of vessel endothelium. Oncogene. 2019;38:868–880. doi: 10.1038/s41388-018-0485-8. [DOI] [PubMed] [Google Scholar]
- 48.Xu Y.-F., Ge F.-J., Han B., Yang X.-Q., Su H., Zhao A.-C., Zhao M.-H., Yang Y.-B., Yang J. High-mobility group box 1 expression and lymph node metastasis in intrahepatic cholangiocarcinoma. World J Gastroenterol. 2015;21:3256–3265. doi: 10.3748/wjg.v21.i11.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee C.-H., Yu J.-R., Kumar S., Jin Y., LeRoy G., Bhanu N., Kaneko S., Garcia B.A., Hamilton A.D., Reinberg D. Allosteric activation dictates PRC2 activity independent of its recruitment to chromatin. Mol Cell. 2018;70:422–434.e6. doi: 10.1016/j.molcel.2018.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.