

Abstract
Formalin-fixed, paraffin-embedded (FFPE) tissues are banked in large repositories to cost-effectively preserve valuable specimens for later study. With the rapid growth of spatial proteomics, FFPE tissues can serve as a more accessible alternative to more commonly used frozen tissues. However, extracting proteins from FFPE tissues is challenging due to cross-links formed between proteins and formaldehyde. Here, we have adapted the nanoPOTS sample processing workflow, which was previously applied to single cells and fresh-frozen tissues, to profile protein expression from FFPE tissues. Following the optimization of extraction solvents, times, and temperatures, we identified an average of 1312 and 3184 high-confidence master proteins from 10 μm thick FFPE-preserved mouse liver tissue squares having lateral dimensions of 50 and 200 μm, respectively. The observed proteome coverage for FFPE tissues was on average 88% of that achieved for similar fresh-frozen tissues. We also characterized the performance of our fully automated sample preparation and analysis workflow, termed autoPOTS, for FFPE spatial proteomics. This modified nanodroplet processing in one pot for trace samples (nanoPOTS) and fully automated processing in one pot for trace sample (autoPOTS) workflows provides the greatest coverage reported to date for high-resolution spatial proteomics applied to FFPE tissues. Data are available via ProteomeXchange with identifier PXD029729.
Keywords: spatial proteomics, mass spectrometry, FFPE, nanodroplet, autoPOTS, LCM
Graphical Abstract
INTRODUCTION
Biological tissues exhibit phenotypic heterogeneity and plasticity arising from differences in cell type, developmental stage, microenvironment, and stochastic biochemical expression.1 The need to better understand the spatial organization and heterogeneity of protein expression during tissue development and disease progression is critical for developing new therapies and gaining a deeper understanding of biological systems.2 While a small number of proteins may be studied with (sub)cellular resolution using, e.g., immunohistochemistry,3 imaging mass cytometry,4 and matrix-assisted laser desorption/ionization (MALDI) imaging,5,6 insufficient measurement sensitivity has limited the application of mass spectrometry (MS) to the global profiling of protein expression with high spatial resolution and depth of coverage.
To fill this gap, laser capture microdissection (LCM) has been combined with MS-based proteomics to spatially profile protein expression in tissues. While initial couplings analyzed relatively large tissue amounts comprising many thousands of cells,7–9 sample input requirements have decreased dramatically in recent years due to advances in efficient sample processing and increasingly sensitive liquid chromatography (LC)-MS. For example, Xu et al. coupled LCM to a spin-tip-based sample processing workflow termed SISPROT,10 which contained strong cation-exchange beads and a C18 disk within a spin-tip. Protein extraction, reduction, alkylation, and digestion were carried out within the tip, and the authors identified ~500 proteins from 0.1 mm2 tissue samples. Davis et al. implemented a series of sample preparation optimizations with respect to sample collection, selection of extraction buffers, and digestion conditions. Using the single-pot, solid-phase-enhanced (SP3) sample processing method, approximately 1500 proteins were identified from 10 μm thick brain tissue sections with a 0.06 mm2 area.11 Our group has also focused on improving the sensitivity of MS-based proteomics for low-input samples including single cells12–16 and trace tissue samples17–23 through improvements in sample preparation, separations, ionization, and mass spectrometry. To minimize losses during sample preparation, we developed nanodroplet processing in one pot for trace samples (nanoPOTS),17 which miniaturizes processing volumes to the nanoliter scale to minimize surface exposure and transfer losses while enhancing reaction kinetics. We also developed the automated transfer of laser-microdissected samples from histology slides to nanowells19 and have analyzed trace tissues from a variety of biological systems.17–23 For example, we quantified protein expression across mouse uterine tissues, achieving a depth of coverage of >2000 protein groups at a 100 μm spatial resolution.22 This was a first-of-its-kind example of in-depth, high-resolution proteome tissue mapping. Additionally, with the added sensitivity of field asymmetric ion mobility spectrometry (FAIMS), we quantified >1000 protein groups from individual motor neurons and interneurons excised by LCM from human spinal tissues.15
In the single-cell and near-single-cell proteomic analyses of laser-microdissected tissues described above, the samples were obtained from fresh-frozen specimens. Fresh tissue preservation by flash freezing can be considered a gold standard for biomarker discovery and omics profiling due to its ability to maintain biomolecules in their native state. However, because frozen tissues degrade quickly at room temperature, and due to the high cost of storage at required low temperatures, there is a limited collection of fresh-frozen samples available for research purposes.24 Alternatively, tissues may be fixed in formalin and embedded in paraffin wax for long-term preservation.25 These formalin-fixed, paraffin-embedded (FFPE) tissues preserve the morphological and structural properties of proteins and can be stored for decades at room temperature.26 Because of the routine FFPE preservation of tissues in clinical settings and the low cost of room-temperature storage, there are many large repositories of FFPE tissues with associated clinical data for patients, including histology reports, treatments, and responses to treatment. However, while the extensive formaldehyde-mediated cross-links that form between proteins, DNA, and RNA contribute to the long-term preservation of these tissues, they also impact the ability to retrieve biomolecules for further study.27 Despite these challenges, in-depth proteomic analyses using microdissected FFPE tissues have been demonstrated. In 2011, Wiśniewski et al. used filter-aided sample preparation (FASP) and LC fractionation to identify up to 4400 proteins from FFPE tissues containing ~20,000 cells.28 Further decreases in sample size were reported by Longuespée et al., who developed an improved sample preparation workflow to identify ~1400 proteins from ~0.4 mm2 tissue areas containing ~2700 cells.29 This approach enabled FFPE tissues to be used in various studies for the discovery of potential biomarkers and classification of tumors.30,31 Griesser and colleagues profiled 1–3 mm2 sections of human substantia nigra tissues to achieve a depth of proteome coverage of 5600 proteins using SP3 sample preparation combined with an isobaric labeling workflow.32 MALDI-based small-molecule imaging has recently guided the selection of tissue regions of interest for microdissection and proteome profiling.33,34 While FFPE tissue regions as small as 0.1 mm2 were analyzed, proteome coverage was limited to ~200 protein groups at that size scale.33 To render FFPE tissues suitable for high-spatial-resolution proteome profiling approaching the single-cell level, methods for efficient protein extraction are needed.
Here, we have applied the nanoPOTS sample processing workflow and ultrasensitive LC-MS/MS analysis to in-depth proteome profiling of 10 μm thick FFPE tissues as small as 50 μm × 50 μm. Following a comparison of various extraction solvents, times, and temperatures, and under the most favorable conditions, we respectively identified an average of 1312 and 3184 high-confidence master proteins from 0.0025 mm2 (50 μm × 50 μm) and 0.04 mm2 (200 μm × 200 μm) FFPE-preserved mouse liver tissue sections. This was on average 88% of the proteome coverage achieved for frozen tissues across three different tissue sizes. We also utilized our fully automated autoPOTS35 sample preparation and analysis workflow using only commercial instrumentation and observed only a modest reduction in proteome coverage for various tissue sizes relative to nanoPOTS (Figure 1). This work significantly extends spatial proteomics of FFPE tissues toward the single-cell level.
Figure 1.
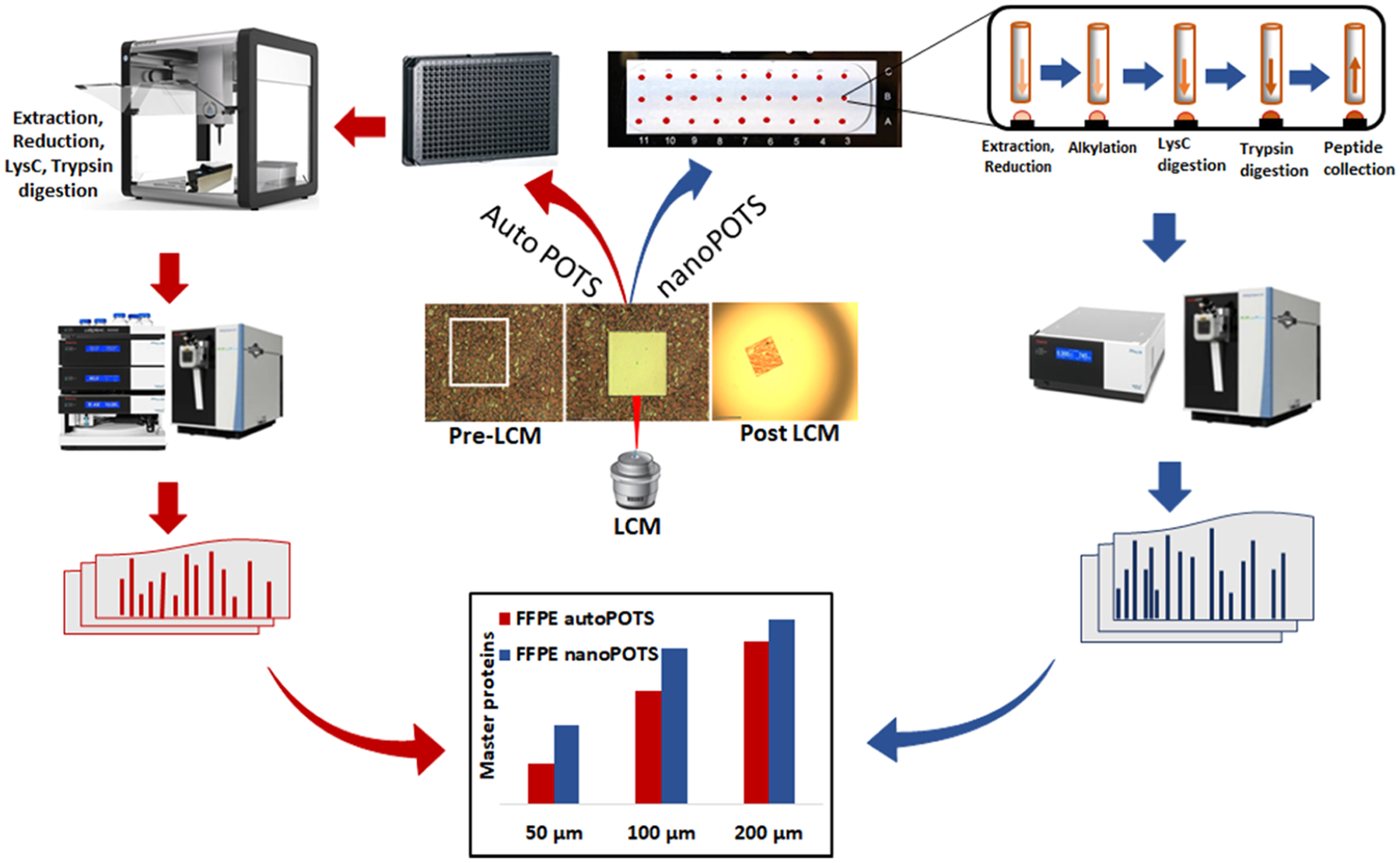
Overview depicting the processing and analysis of microdissected FFPE tissues using either the nanoPOTS (blue arrows) or autoPOTS (red arrows) workflows. For autoPOTS, samples are transferred to a 384-well plate, prepared in an automated fashion using the Opentrons OT-2 liquid handler and analyzed by LC-MS using a commercial autosampler. For nanoPOTS, the excised tissue samples are transferred to custom nanowell chips, prepared using an in-house-developed robotic platform and manually injected for LC-MS analysis.
MATERIALS AND METHODS
Materials
Dithiothreitol (DTT), iodoacetamide (IAA), formic acid (LC-MS grade), Na2HPO4, and NaH2PO4 were purchased from Thermo Fisher Scientific (Waltham, MA). Ammonium bicarbonate (ABC) buffer (50 mM) was freshly prepared from a 500 mM stock solution. LC-MS grade water and acetonitrile were purchased from Honeywell (Charlotte, NC). Trypsin gold and rLys-C (MS grade) were from Promega (Madison, WI). Scott’s tap water substitute bluing reagent was from Ricca Chemical (Arlington, TX). Ethanol was purchased from Decon Laboratories, King of Prussia, PA, and paraffin wax was from Blended Waxes, Inc. (Oshkosh, WI). Other chemicals were from Sigma-Aldrich (St. Louis, MO). Neutral buffered formalin (pH 6.8–7.0) was prepared by combining 25 mL (37–40% w/w) of formaldehyde with 225 mL of purified water, 1.0 g of NaH2PO4, and 1.625 g of anhydrous Na2HPO4. Carboxymethylcellulose solution (CMC; 2.5% w/v in water) was prepared for embedding fresh samples prior to freezing. A stock solution of n-dodecyl-β-d-maltoside (DDM, Sigma-Aldrich), a nonionic surfactant used for protein extraction and solubilization,36 was prepared at 1% (w/v) in water and further diluted for use, as described below. Nanowell chips were fabricated as described previously.17
Tissue Preparation
All protocols involving animal subjects were approved by the Institutional Animal Care and Use Committee of Brigham Young University. A C57BL6 mouse, purchased from Jackson Laboratory (Bar Harbor, Maine), was housed and fed under standard protocols in the BYU Life Sciences Vivarium. The mouse was anesthetized with carbon dioxide and euthanized by cardiac puncture. The liver was rapidly removed and bisected into two halves in a sagittal plane to produce two mirror images. One half was placed on dry ice and then immersed in a 2.5% carboxymethylcellulose (CMC) embedding solution for 1 min at 4 °C prior to freezing at −80 °C for storage. CMC serves as an MS-compatible substitute for the more commonly used optimal cutting temperature (OCT) embedding compound. The other half of the mouse liver was immersed in a 10% neutral buffered formalin solution for 48 h. To dehydrate and paraffin-embed the fixed liver tissue, it was first immersed in 70% ethanol for 1 h, 80% ethanol twice for 30 min, 100% ethanol twice for 30 min, xylene twice for 1 h, and melted paraffin wax at 55 °C twice for 90 min. The FFPE-preserved liver sample was then stored at room temperature until further use. A cryostat (TN50, Tanner Scientific, Sarasota, FL) was set to −13 °C and used to section the fresh-frozen tissue to a thickness of 10 μm. The tissue sections were deposited onto poly(ethylene naphthalate) (PEN) membrane slides (Carl Zeiss Microscopy, Oberkochen, Germany) and stored at −80 °C. A tabletop microtome was used to cut FFPE tissues to the same 10 μm thickness; these were placed on PEN membrane slides and stored at room temperature. Tissue sections of both types were stained using hematoxylin and eosin. Briefly, fresh-frozen tissues were fixed in ice-cold 70% ethanol for 30 s, rehydrated with deionized water for 30 s, and stained in Mayer’s hematoxylin solution (10% v/v in water) for 10 s. Excess stain was rinsed with water, and the slides were immersed in a bluing solution for 10 s. The tissues were then dehydrated using 70% ethanol for 15 s followed by dipping in eosin (20% v/v in ethanol) for 5 s. Further dehydration was accomplished by dipping in 95% ethanol twice for 30 s followed by 100% ethanol twice for 30 s. The FFPE tissue sections were dewaxed by immersing in xylene twice for 1 min followed by rehydration in 100% ethanol for 15 s, 95% ethanol for 15 s, 70% ethanol for 15 s, and deionized water for 10 s. Once rehydrated, the tissues were stained in hematoxylin for 5 s, rinsed in deionized water for 10 s, and dehydrated in 70% ethanol for 10 s. The tissue sections were counterstained with a quick dip in eosin followed by dehydration in 95% ethanol for 15 s, 100% ethanol for 15 s, and xylene for 10 s. After processing and staining the fresh-frozen and FFPE sections, they were dried in a vacuum desiccator for 10 min.
Laser Microdissection
A glass nanowell chip was thoroughly cleaned, air-dried, and prefilled with 80 nL of dimethylsulfoxide (DMSO) using the nanoPOTS nanoliter liquid handling system and placed in a freezer at −20 °C to reduce evaporation. A PALM MicroBeam LCM system (Carl Zeiss) was used for microdissection of small regions of interest from tissue sections, as described previously.19 The nanowell chip was fixed on a microscope slide adapter (Slide Collector 48, Carl Zeiss) and mounted on the robotic arm of the LCM system. To excise square regions of interest having edge lengths of either 100 or 200 μm, both fresh and FFPE mouse liver samples were marked and cut using a 10× objective, an energy of 52, and a focus of 91. For 50 μm square tissue regions, a cutting energy of 48 and a focus of 76 were used with a 20× objective. The cut tissues were then catapulted into DMSO-containing nanowells with a laser-pressure catapulting (LPC) energy of 18 and a focus of 15. Following the transfer of the tissue samples to the nanoPOTS chips, they were then stored at −20 °C for up to a week until being further processed.
Sample Preparation
For frozen tissues, following LCM and tissue transfer to the DMSO-containing nanowells, our previously developed protocol was followed.17 Briefly, 100 nL of protein extraction buffer (containing 0.1% DDM and 5 mM DTT in 50 mM ABC) was dispensed into each nanowell and incubated at 70 °C for 1 h. The nanowells were again allowed to air-dry, after which proteins were alkylated with 150 nL of 30 mM IAA for 30 min in the dark. A two-step digestion was carried out at 37 °C with 50 nL of Lys-C (0.1 ng/nL) and 50 nL of trypsin (0.2 ng/nL) for 3 and 10 h, respectively. The digestion was quenched with the addition of 50 nL of 0.1% trifluoroacetic acid (TFA), and peptide solution was collected into a capillary tube (4 cm long, 200 μm id). The capillary was then sealed with parafilm M at both ends and stored at −20 °C until LC-MS/MS analysis.
For FFPE tissues, the same procedure was followed but with variations in the protein extraction step evaluated for improved proteome coverage, as described in the Results and Discussion section. These included incubating with the same extraction buffer but varying the extraction time from 60 to 180 min and the temperature from 60 to 90 °C. In addition, an alternate extraction buffer consisting of 50% trifluoroethanol (TFE) in 50 mM ABC buffer and 5 mM DTT was evaluated, which was incubated at 90 °C for 90 min. AutoPOTS sample preparation was performed using an Opentrons OT-2 liquid handler (Brooklyn, NY) following a previously reported protocol.35 Specific parameters for all liquid handling steps are shown in Figure S1.
NanoLC-MS/MS Analysis
For nanoPOTS-prepared samples, nanoLC-MS/MS analysis was performed manually, as described previously.17 The solid-phase extraction (SPE or trapping) column (100 μm i.d., 5 cm long) and analytical column (30 μm i.d., 50 cm long) were packed in-house with Dr. Maisch 120 Å C18-AQ 3 μm particles. AutoPOTS-prepared samples were analyzed using an Ultimate 3000 TPL autosampler (Thermo) modified with a 10-port valve.35 The dimensions and media used for the SPE and analytical columns matched those used to analyze the nanoPOTS-prepared samples. For peptide separation, the percentage of mobile phase B (MP B, 0.1% FA in acetonitrile) was increased from 2 to 8% for 3 min, followed by a 100 min linear gradient from 8 to 25% MP B and a 20 min gradient from 25 to 45% MP B. The column was then washed to elute DDM and other hydrophobic species with a linear gradient from 45 to 90% MP B for 20 min followed by a decrease to 2% MP B for 2 min and an increase to 90% MP B for another 5 min. The column was then re-equilibrated for 30 min with 2% mobile phase B. Mass spectra were acquired on a Thermo Orbitrap Exploris 480 mass spectrometer equipped with a nanospray flex source. The electrospray voltage on the 10 μm i.d. chemically etched nanospray emitter (Micromics Technologies, Spanish Fork, UT) was set to 2.2 kV, and the temperature of the ion transfer tube was 200 °C. For MS1 acquisition, the orbitrap resolution was set to 120,000 full width at half-maximum (FWHM) at m/z 200 with a normalized automatic gain control (AGC) target of 100% (1E6). The maximum injection time was set to 100 ms. For tissue squares having edge lengths of 200 and 100 μm, the precursor intensity threshold for tandem mass spectrometry (MS2) fragmentation was set to 8000 and the selected charge states were +2 to +7. The dynamic exclusion duration was set to 30 s, and the time between master scans was 1.5 s. The MS2 Orbitrap resolution was set to 30,000, the maximum injection time was set to 54 ms, and the normalized collision energy was 30%. For 50 μm tissue squares, the dynamic exclusion duration was increased to 60 s, the time between master scans was 3 s, and the orbitrap resolution for MS2 was increased to 120,000 (m/z 200) with a maximum injection time of 500 ms, while other parameters remained the same. For the LC-MS/MS analysis of autoPOTS-prepared samples, the separation conditions were the same as for the manual analysis of nanoPOTS-prepared samples but were fully automated, as described previously.35
Data Analysis
Raw files were processed using Proteome Discoverer Software 2.5 (Thermo). The mouse proteome database was downloaded from UniProtKB on June 6th, 2019. A two-step database search was performed using Sequest HT and INFERYS. The MS/MS spectra were searched for fully tryptic peptides with N-terminal protein acetylation, methionine oxidation, and N-terminal methionine loss acetylation set as variable modifications and carbamidomethylation of cysteine residues as a fixed modification. The precursor and fragment mass tolerances were set to 5 ppm and 0.02 Da, respectively. The match between runs (MBR) feature detection (referred to as Feature Mapping in the software) matched features having a normalized retention time shift of up to 0.7 min and a mass tolerance of 5 ppm. The precursor ion quantifier node was used to calculate protein abundances based on the top three distinct peptides from each protein. The peptides and proteins were filtered with a maximum false discovery rate (FDR) of 0.01. To compare frozen and FFPE samples, global scaling normalization was achieved using scaling coefficients calculated as the ratio of protein abundance to the median protein abundance measured for each grouping (FFPE and frozen). Both unique and razor peptides were selected for protein quantification. Other unmentioned parameters were used as default.
Protein accession numbers of the filtered proteins for both fresh-frozen and FFPE tissues having at least one unique peptide were compiled into a list. The list was then loaded into the jvenn software37 and plotted using the VennDiagramPlotter (https://omics.pnl.gov/software/venn-diagram-plotter). Pearson correlations were generated as follows. High-confidence master proteins for both fresh-frozen and FFPE samples having at least two unique peptides were loaded into Perseus (v1.6.5.0).38 Normalized abundances were log10-transformed and filtered such that proteins having valid values detected in 70% of a sample group were retained. The median values were determined for each sample type, and Pearson correlations were plotted for the two sample types.
DATA AVAILABILITY
Mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with data set identifier PXD029729.
RESULTS AND DISCUSSION
Effective protein extraction is a critical step in sample preparation for any proteomics analysis, and studies have reported that incubation at an elevated temperature can improve protein extraction for FFPE tissues.39–41 We first compared proteome coverage between 200 μm square frozen and FFPE mouse liver tissue sections using the nanoPOTS workflow in a solvent containing 0.1% DDM with incubation at 70 °C for 60 min. These general conditions have been used extensively for frozen tissues.17–20,22,23 As shown in Figure 2 and the first two rows of Table 1 (frozen and FFPE 1), these extraction conditions yielded reductions of 51 and 35% in peptide and proteome coverage (excluding match between run values), respectively, for the FFPE samples relative to frozen tissues, indicating poor suitability of these conditions for FFPE. To improve protein extraction from FFPE tissues, we explored other extraction solvents, temperatures, and incubation times. These conditions, along with the observed proteome coverage, are listed as FFPE 2 – FFPE 4 in Table 1.
Figure 2.
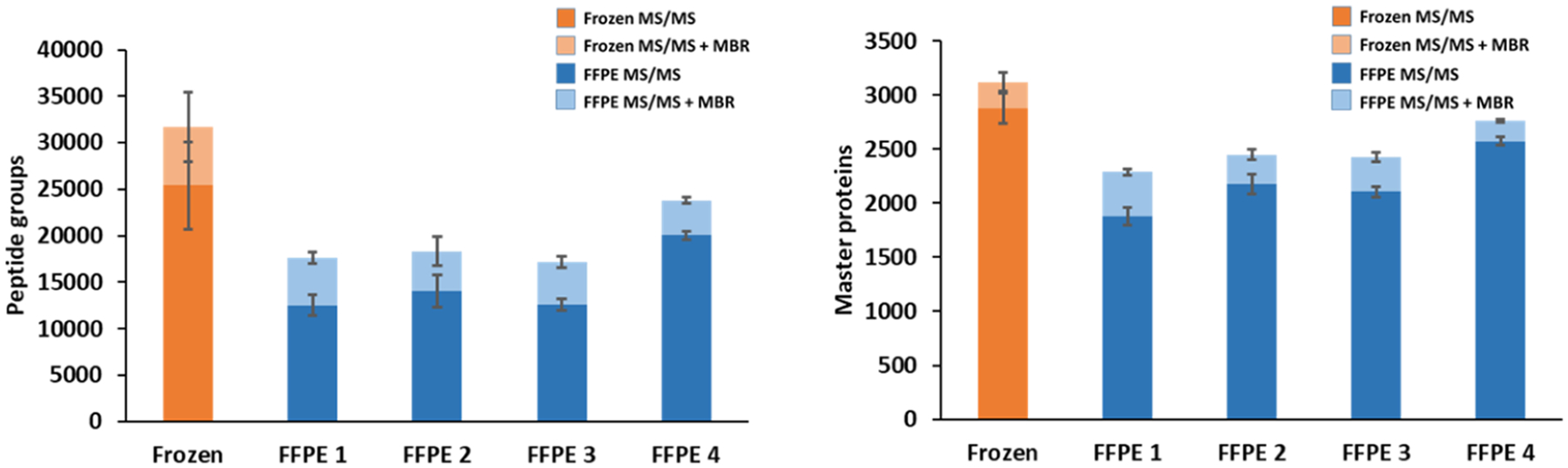
Peptide groups (left) and master proteins (right) identified from 200 μm × 200 μm tissue samples. Specific extraction conditions are shown in Figure 1. N = 4 for each condition.
Table 1.
Protein Extraction Conditions, Peptides, and Proteome Coverages for Both Fresh-Frozen and FFPE Tissue Samplesa
| tissue sample | extraction conditions | peptide ID (with MBR in parentheses) | protein ID (with MBR in parentheses) | % reduction in peptide coverage relative to frozen (MBR in parentheses) | % reduction in protein coverage relative to frozen (MBR in parentheses) |
|---|---|---|---|---|---|
| Frozen | 0.1% DDM, 70 °C, 60 min | 25,500 ± 4700 | 2900 ± 140 | 0 | 0 |
| (31,700 ± 3700) | (3260 ± 100) | (0) | (0) | ||
| FFPE 1 | 0.1% DDM, 70 °C, 60 min | 12,500 ± 1100 | 1900 ± 100 | 51 | 35 |
| (17,700 ± 600) | (2300 ± 30) | (44) | (28) | ||
| FFPE 2 | 0.1% DDM, 70 °C, 180 min | 14,000 ± 1700 | 2200 ± 100 | 45 | 25 |
| (18,300 ± 1600) | (2500 ± 60) | (42) | (23) | ||
| FFPE 3 | 50% TFE, 90 °C, 90 min | 12,600 ± 600 | 2100 ± 50 | 51 | 27 |
| (17,200 ± 600) | (2500 ± 50) | (45) | (23) | ||
| FFPE 4 | 0.1% DDM, 90 °C, 90 min | 20,100 ± 400 | 2600 ± 40 | 21 | 11 |
| (23,800 ± 300) | (2800 ± 20) | (25) | (13) |
Values show MS/MS acquisition and match between runs (MBRs) in the table. N = 4 for each condition.
FFPE tissue incubation with 0.1% DDM at 90 °C for 90 min (FFPE 4 in Table 1) yielded the highest level of coverage when compared with other extraction conditions (Figure 2 and Table 1). Fresh-frozen tissues were also analyzed using all conditions shown in Table 1, but our standard conditions of extraction in 0.1% DDM at 70 °C for 60 min provided greater proteome coverage for frozen tissues (data not shown). TFE has been reported previously to be an effective solvent for protein extraction.11,42,43 In our experiments, the less favorable performance of TFE (FFPE 3 in Table 1) may be due to its higher volatility leading to rapid evaporation in our nanoliter preparation volumes. As such, we utilized extraction with 0.1% DDM at 90 °C for 90 min (FFPE 4 in Table 1) for the subsequent analysis of FFPE tissues, as these conditions provided the highest proteome coverage and were compatible with nanoliter sample processing.
A Pearson correlation plot of protein intensities for fresh-frozen and FFPE tissues is shown in Figure 3. A modest correlation coefficient of 0.77 for proteins identified with at least two unique peptides indicates significant differences in observed intensities for the two methods of tissue storage. For reference, R2 values of 0.97 and 0.98 were obtained for replicate analyses of fresh frozen and FFPE tissue samples, respectively (Figure S2), indicating that the relatively low correlation between fresh-frozen and FFPE tissues was not due to technical variability but rather to differences in storage methods. A Venn diagram was also plotted between fresh-frozen and FFPE tissues (Figure 3). A total of 3110 and 2677 proteins were identified for fresh-frozen and FFPE tissues, respectively, with 2464 proteins (74%) overlapping between the two storage conditions. Gene ontology analysis as provided by PD 2.5 classified identified proteins according to cellular compartment, molecular function, and biological processes (Figure S3). A close agreement was observed between fresh-frozen and FFPE tissues in all cases, indicating that any biases were not specific to proteins of a given cellular component or process.
Figure 3.
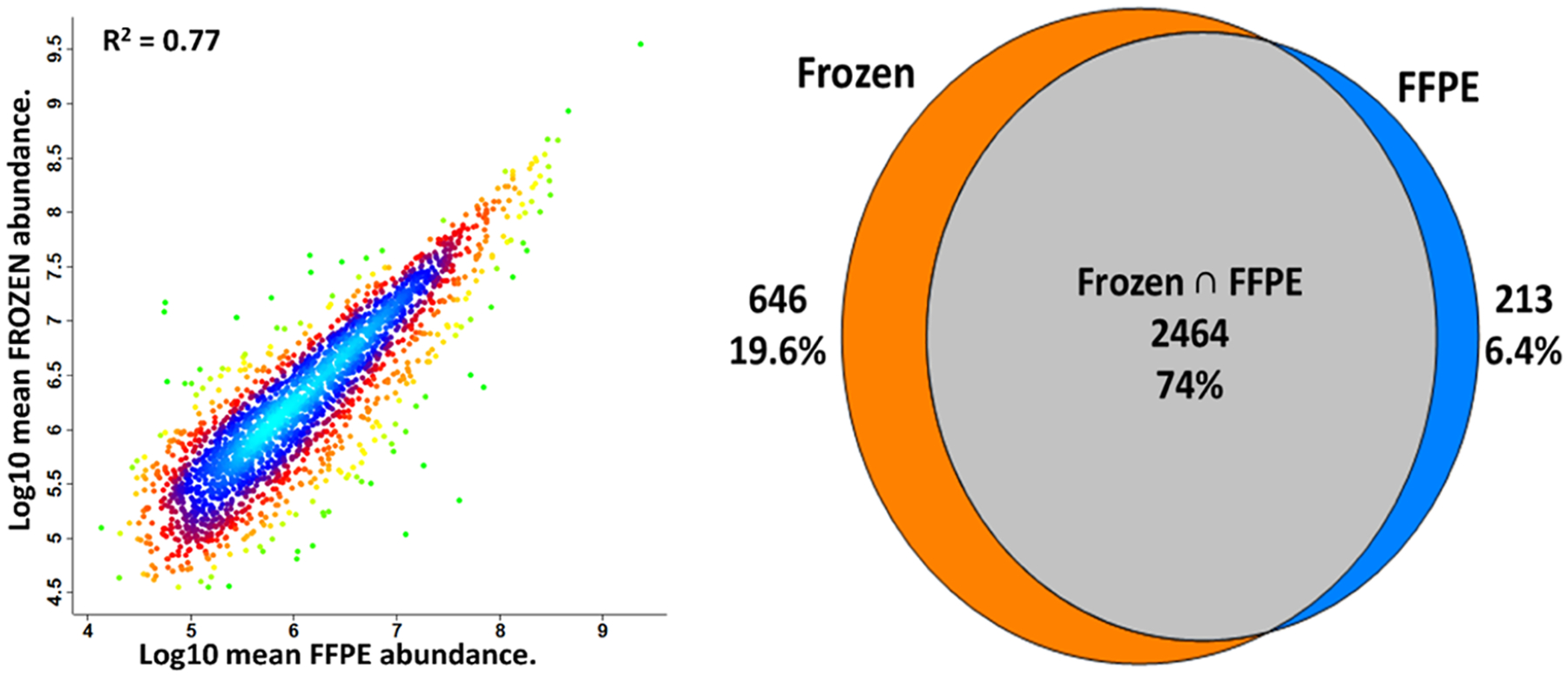
Comparison between frozen and FFPE mouse liver tissues obtained from the same mouse. (Left) Pearson correlation plot between fresh-frozen and FFPE tissue types. (Right) Venn diagram showing the overlap of high-confidence master proteins between fresh-frozen and FFPE tissues. Only proteins identified in all four replicates, including those identified with MBR, are included.
We then studied whether any biases were observed against hydrophilic or hydrophobic peptides during the preparation of FFPE tissues using our optimized method. Using GRAVY index scoring of identified peptides based on the average hydrophobicity of constituent amino acids,44 we found little to no reduction in hydrophobic species and that the reduced peptide coverage for the FFPE tissue was reflected in the identification of fewer hydrophilic species (Figure S4). Given that formaldehyde forms cross-links with hydrophilic amino acids,45 this provided preliminary evidence that protein extraction is adequate, but that a significant degree of cross-linking remains following extraction, resulting in the loss of those cross-linked peptides.
With appropriate extraction conditions, the modified nanoPOTS protocol enables reproducible, in-depth proteome profiling of microdissected FFPE tissues. However, this workflow as performed here is semiautomated and requires expertise in manufacturing nanowell chips, skill in using the robotic system for sample preparation, and delicate manual injection of samples for LC-MS analysis. These requirements limit adoption by other laboratories. To address this, our lab recently developed a fully automated sample processing workflow, termed “autoPOTS”,35 which uses a low-cost commercial pipetting robot (Opentrons OT-2). This open-source liquid handler prepares samples in low-microliter volumes within 384-well plates for a fully automated workflow and facile interfacing with commercial autosamplers. Despite a modest reduction in sensitivity relative to nanoPOTS, autoPOTS can reliably quantify proteins in low-input samples including single cells.35 We therefore evaluated the performance of this fully automated workflow for the global profiling of FFPE tissues. We sampled different sizes of tissue sections in the range of 50 μm × 50 μm to 200 μm × 200 μm from both fresh-frozen and FFPE-preserved mouse liver using both nanoPOTS and autoPOTS. As shown in Figure 4 and the corresponding Table S1, nanoPOTS consistently achieved greater coverage for frozen tissues relative to FFPE, and the use of autoPOTS resulted in an average 30% reduction in proteome coverage for FFPE tissues relative to nanoPOTS when MBR identifications were excluded. Importantly, the reduction in coverage for autoPOTS was 42% for 50 μm tissue squares and only 23% for 200 μm squares, indicating that with the smallest samples, a nanodroplet sample processing workflow is most critical. When employing MBR (feature mapping in PD 2.5), the reduction in coverage for autoPOTS averaged just 19%, indicating that some of the lost peptides provide a sufficient MS1 signal to still be identified. The modest reduction in coverage for autoPOTS relative to nanoPOTS, particularly for larger samples, indicates that it is suitable for many applications involving the low-input profiling of FFPE tissues.
Figure 4.
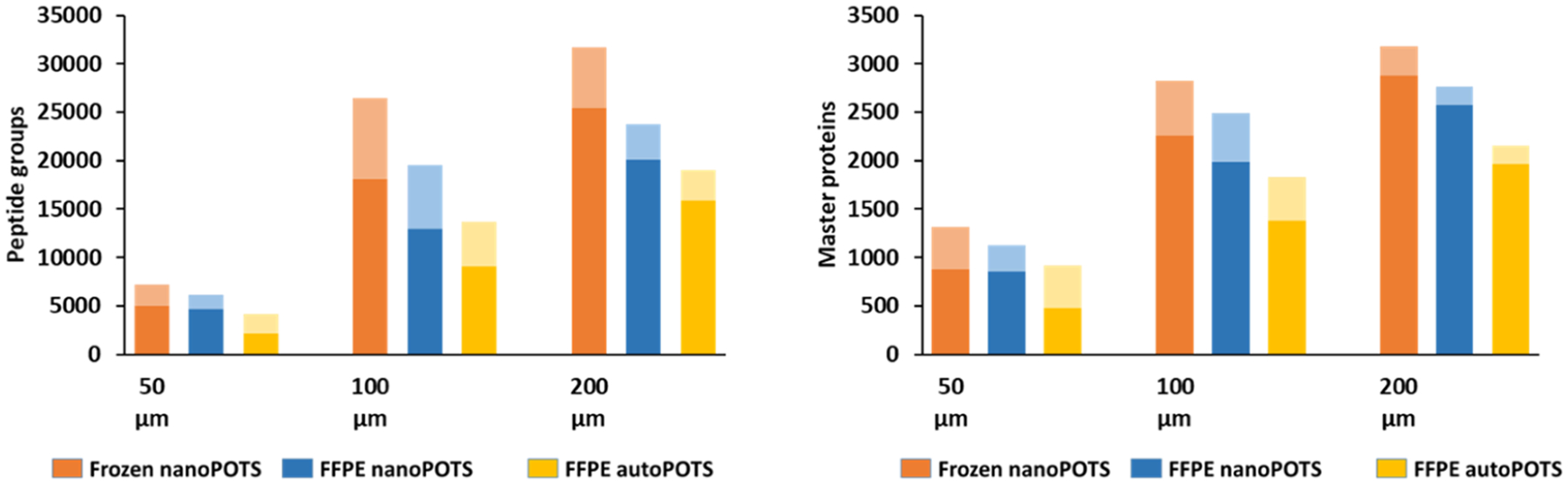
Number of identified peptide groups (Left) and high-confidence master proteins (Right) for the nanoPOTS analysis of frozen and FFPE-preserved mouse liver tissue squares having lateral dimensions of 50–200 μm, as well as autoPOTS analysis of the FFPE tissue sections of the same dimensions. The lighter shading in each bar indicates additional identifications made using MBR. Standard deviations for each condition (N = 4) are provided in Table S1.
CONCLUSIONS
There is limited availability of fresh-frozen tissues for translational medicine due to costs of storage and changes in morphology over extended periods. FFPE tissues are stored very cost-effectively at room temperature for up to decades, and large repositories exist in many hospitals and research facilities. These specimens are generally banked with extensive metadata and are essential for biomedical research. We have adapted the nanoPOTS workflow to increase proteome coverage for trace amounts of laser-microdissected FFPE tissues. A loss of primarily hydrophilic peptides in FFPE reduced proteome coverage by 10–15% relative to frozen tissues, and these losses were presumably due to remaining cross-links.
In terms of best practices for the proteome profiling of trace FFPE tissues, we recommend extracting protein at 90 °C for 90 min in the presence of the nonionic surfactant DDM (condition 4 in Table 1), which resulted in greater proteome coverage than other tested conditions. As demonstrated in this work, both the more sensitive nanoPOTS workflow and the fully automated autoPOTS workflow are compatible with the processing and analysis of FFPE tissues, and both approaches provide a greater level of proteome coverage than previously been reported for tissues as small as 50 μm × 50 μm × 10 μm (corresponding to the protein content of 10 cells having a radius of ~8 μm). As observed from our autoPOTS results, the use of the more sensitive nanoPOTS workflow is best used for a smaller region of interest in the 50 μm × 50 μm range for better protein identification, while regions of interest in the 200 μm × 200 μm range and above may be analyzed using the autoPOTS sample workflow as a lower cost and less technically demanding alternative with only a modest reduction in proteome coverage.
To apply the platform to study differential protein expression within FFPE-preserved clinical tissues, there are additional considerations that should be addressed during experimental design. For example, the number of replicate measurements within a tissue and across subjects will depend on a variety of factors that are specific to a given study. Additionally, as with all low-input proteomics, there is a clear tradeoff between the amount of sample analyzed and the achievable proteome coverage. A compromise must therefore be reached between molecular depth and spatial resolution, which will depend on the size scale of features of interest and the relative abundance of proteins of greatest interest. However, the results presented here lead to specific recommendations for designing a spatial proteomics study. That is, using nanoPOTS sample processing or a similar low-volume 1-step preparation method, sample voxels as small as 10 μm × 50 μm × 50 μm may reasonably be analyzed, provided that proteins of biological interest in the study are among the ~1000 most abundant proteins in the sample. The ~2500 most abundant proteins may be studied in 10 μm thick tissue squares of 100 μm edge length, and 200 μm squares can reasonably access the ~3000 most abundant proteins. Of course, improvements to low-input proteomics workflows are ongoing, and further developments in sample preparation, separations, ionization, MS acquisition, and data analysis will enable greater proteome coverage at a higher spatial resolution. Ideally, these measurements will be achieved at a much higher throughput relative to the current state of the art. While the field of MS-based spatial proteomics is in its early stages, this work establishes the capability of profiling FFPE tissues with high spatial resolution and molecular depth.
Supplementary Material
ACKNOWLEDGMENTS
The research reported in this publication was supported by the National Cancer Institute and the National Institute of General Medical Sciences of the National Institutes of Health under award numbers R33CA225248 and R01GM138931. A.J.N., S.A.M., T.T., and R.T.K. received funding from R33CA225248. A.J.N., R.H.C., K.G.I.W., N.B.A., Y.L., S.M.J., and R.T.K. received funding from R01GM138931. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.2c00409.
AutoPOTS sample preparation workflow using the Opentrons OT-2 (Figure S1); representative Pearson correlation plots for replicate analyses of 200 μm × 200 μm sections of FFPE and frozen tissues (Figure S2); gene ontology plots with respect to cellular component, molecular function, and biological process for proteins identified in frozen and FFPE tissues (Figure S3); GRAVY score showing the relative hydrophobicity of unique peptides from 200 μm tissue squares (Figure S4); and proteome coverage achieved for 50, 100, and 200 μm fresh-frozen and FFPE tissue squares (Table S1) (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jproteome.2c00409
The authors declare the following competing financial interest(s): RTK has a financial interest in MicrOmics Technologies, which provided nanoelectrospray emitters for this study.
Contributor Information
Andikan J. Nwosu, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States
Santosh A. Misal, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States
Thy Truong, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States.
Richard H. Carson, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States
Kei G. I. Webber, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States
Nathaniel B. Axtell, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States
Yiran Liang, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States.
S. Madisyn Johnston, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States.
Kenneth L. Virgin, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States
Ethan G. Smith, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States
George V. Thomas, Knight Cancer Center, Oregon Health & Science University, Portland, Oregon 97239, United States
Terry Morgan, Department of Pathology, Oregon Health & Science University, Portland, Oregon 97239, United States.
John C. Price, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States
Ryan T. Kelly, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States
REFERENCES
- (1).Almendro V; Marusyk A; Polyak K Cellular Heterogeneity and Molecular Evolution in Cancer. Annu. Rev. Pathol.: Mech. Dis 2013, 8, 277–302. [DOI] [PubMed] [Google Scholar]
- (2).Lundberg E; Borner GHH Spatial Proteomics: A Powerful Discovery Tool for Cell Biology. Nat. Rev. Mol. Cell. Biol 2019, 20, 285–302. [DOI] [PubMed] [Google Scholar]
- (3).Uhlen M; Oksvold P; Fagerberg L; Lundberg E; Jonasson K; Forsberg M; Zwahlen M; Kampf C; Wester K; Hober S; Wernerus H; Björling L; Ponten F Towards a Knowledge-Based Human Protein Atlas. Nat. Biotechnol 2010, 28, 1248–1250. [DOI] [PubMed] [Google Scholar]
- (4).Giesen C; Wang HAO; Schapiro D; Zivanovic N; Jacobs A; Hattendorf B; Schüffler PJ; Grolimund D; Buhmann JM; Brandt S; Varga Z; Wild PJ; Günther D; Bodenmiller B Highly Multiplexed Imaging of Tumor Tissues with Subcellular Resolution by Mass Cytometry. Nat. Methods 2014, 11, 417–422. [DOI] [PubMed] [Google Scholar]
- (5).Yagnik G; Liu Z; Rothschild KJ; Lim MJ Highly Multiplexed Immunohistochemical MALDI-MS Imaging of Biomarkers In Tissues. J. Am. Soc. Mass Spectrom 2021, 32, 977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ryan DJ; Spraggins JM; Caprioli RM Protein Identification Strategies in MALDI imaging Mass Spectrometry: a Brief Review. Curr. Opin. Chem. Biol 2019, 48, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Johann DJ; Rodriguez-Canales J; Mukherjee S; Prieto DA; Hanson JC; Emmert-Buck M; Blonder J Approaching Solid Tumor Heterogeneity on a Cellular Basis by Tissue Proteomics Using Laser Capture Microdissection and Biological Mass Spectrometry. J. Proteome Res 2009, 8, 2310–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Satoskar AA; Shapiro JP; Bott CN; Song H; Nadasdy GM; Brodsky SV; Hebert LA; Birmingham DJ; Nadasdy T; Freitas MA; Rovin BH Characterization of Glomerular Diseases Using Proteomic Analysis of Laser Capture Microdissected Glomeruli. Mod. Pathol 2012, 25, 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liao L; Cheng D; Wang J; Duong DM; Losik TG; Gearing M; Rees HD; Lah JJ; Levey AI; Peng J Proteomic Characterization of Postmortem Amyloid Plaques Isolated by Laser Capture Microdissection. J. Biol. Chem 2004, 279, 37061–37068. [DOI] [PubMed] [Google Scholar]
- (10).Xu R; Tang J; Deng Q; He W; Sun X; Xia L; Chen Z; He L; You S; Hu J; et al. Spatial-Resolution Cell Type Proteome Profiling of Cancer Tissue by Fully Integrated Proteomics technology. Anal. Chem 2018, 90, 5879–5886. [DOI] [PubMed] [Google Scholar]
- (11).Davis S; Scott C; Ansorge O; Fischer R Development of a Sensitive, Scalable Method for Spatial, Cell-Type-Resolved Proteomics of the Human Brain. J. Proteome Res 2019, 18, 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhu Y; Clair G; Chrisler WB; Shen Y; Zhao R; Shukla AK; Moore RJ; Misra RS; Pryhuber GS; Smith RD; Ansong C; Kelly RT Proteomic Analysis of Single Mammalian Cells Enabled by Microfluidic Nanodroplet Sample Preparation and Ultrasensitive NanoLC-MS. Angew. Chem., Int. Ed 2018, 57, 12370–12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cong Y; Liang Y; Motamedchaboki K; Huguet R; Truong T; Zhao R; Shen Y; Lopez-Ferrer D; Zhu Y; Kelly RT Improved Single-Cell Proteome Coverage Using Narrow-Bore Packed NanoLC Columns and Ultrasensitive Mass Spectrometry. Anal. Chem 2020, 92, 2665–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Williams SM; Liyu AV; Tsai C-F; Moore RJ; Orton DJ; Chrisler WB; Gaffrey MJ; Liu T; Smith RD; Kelly RT; Pasa-Tolic L; Zhu Y Automated Coupling of Nanodroplet Sample Preparation with Liquid Chromatography-Mass Spectrometry for High-Throughput Single-Cell Proteomics. Anal. Chem 2020, 92, 10588–10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cong Y; Motamedchaboki K; Misal SA; Liang Y; Guise AJ; Truong T; Huguet R; Plowey ED; Zhu Y; Lopez-Ferrer D; Kelly RT Ultrasensitive Single-Cell Proteomics Workflow Identifies >1000 Protein Groups per Mammalian Cell. Chem. Sci 2021, 12, 1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Webber KGI; Truong T; Johnston SM; Zapata SE; Liang Y; Davis JM; Buttars AD; Smith FB; Jones HE; Mahoney AC; Carson RH; Nwosu AJ; Heninger JL; Liyu AV; Nordin GP; Zhu Y; Kelly RT Label-Free Profiling of up to 200 Single-Cell Proteomes per Day Using a Dual-Column Nanoflow Liquid Chromatography Platform. Anal. Chem 2022, 94, 6017–6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhu Y; Piehowski PD; Zhao R; Chen J; Shen Y; Moore RJ; Shukla AK; Petyuk VA; Campbell-Thompson M; Mathews CE; Smith RD; Qian W-J; Kelly RT Nanodroplet Processing Platform for Deep and Quantitative Proteome Profiling of 10–100 Mammalian Cells. Nat. Commun 2018, 9, No. 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dou M; Zhu Y; Liyu A; Liang Y; Chen J; Piehowski PD; Xu K; Zhao R; Moore RJ; Atkinson MA; Mathews CE; Qian W-J; Kelly RT Nanowell-Mediated Two-Dimensional Liquid Chromatography Enables Deep Proteome Profiling of <1000 Mammalian Cells. Chem. Sci 2018, 9, 6944–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhu Y; Dou M; Piehowski PD; Liang Y; Wang F; Chu RK; Chrisler WB; Smith JN; Schwarz KC; Shen Y; Shukla AK; Moore RJ; Smith RD; Qian WJ; Kelly RT Spatially Resolved Proteome Mapping of Laser Capture Microdissected Tissue with Automated Sample Transfer to Nanodroplets. Mol. Cell. Proteomics 2018, 17, 1864–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Liang Y; Zhu Y; Dou M; Xu K; Chu RK; Chrisler WB; Zhao R; Hixson KK; Kelly RT Spatially Resolved Proteome Profiling of <200 Cells from Tomato Fruit Pericarp by Integrating Laser-Capture Microdissection with Nanodroplet Sample Preparation. Anal. Chem 2018, 90, 11106–11114. [DOI] [PubMed] [Google Scholar]
- (21).Xu K; Liang Y; Piehowski PD; Dou M; Schwarz KC; Zhao R; Sontag RL; Moore RJ; Zhu Y; Kelly RT Benchtop-Compatible Sample Processing Workflow for Proteome Profiling of < 100 Mammalian Cells. Anal. Bioanal. Chem 2019, 411, 4587–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Piehowski PD; Zhu Y; Bramer LM; Stratton KG; Zhao R; Orton DJ; Moore RJ; Yuan J; Mitchell HD; Gao Y; Webb-Robertson BJM; Dey SK; Kelly RT; Burnum-Johnson KE Automated Mass Spectrometry Imaging of over 2000 Proteins from Tissue Sections at 100-μm Spatial Resolution. Nat. Commun 2020, 11, No. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Balasubramanian VK; Purvine SO; Liang Y; Kelly RT; Pasa-Tolic L; Chrisler WB; Blumwald E; Stewart CN Jr.; Zhu Y; Ahkami AH Cell-Type-Specific Proteomics Analysis of a Small Number of Plant Cells by Integrating Laser Capture Microdissection with a Nanodroplet Sample Processing Platform. Curr. Protoc 2021, 1, No. e153. [DOI] [PubMed] [Google Scholar]
- (24).Sánchez-Navarro I; Gámez-Pozo A; González-Barón M; Pinto-Marín Á; Hardisson D; López R; Madero R; Cejas P; Mendiola M; Espinosa E; Vara JÁF Comparison of Gene Expression Profiling by Reverse Transcription Quantitative PCR between Fresh Frozen and Formalin-Fixed, Paraffin-Embedded Breast Cancer Tissues. Biotechniques 2010, 48, 389–397. [DOI] [PubMed] [Google Scholar]
- (25).Kokkat TJ; Patel MS; McGarvey D; LiVolsi VA; Baloch ZW Archived Formalin-Fixed Paraffin-Embedded (FFPE) Blocks: A Valuable Underexploited Resource for Extraction of DNA, RNA, and Protein. Biopreserv. Biobanking 2013, 11, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Piehowski PD; Petyuk VA; Sontag RL; Gritsenko MA; Weitz KK; Fillmore TL; Moon J; Makhlouf H; Chuaqui RF; Boja ES; Rodriguez H; Lee JSH; Smith RD; Carrick DM; Liu T; Rodland KD Residual Tissue Repositories as a Resource for Population-Based Cancer Proteomic Studies. Clin. Proteomics 2018, 15, No. 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Nirmalan NJ; Harnden P; Selby PJ; Banks RE Mining the Archival Formalin-Fixed Paraffin-Embedded Tissue Proteome: Opportunities and Challenges. Mol. Biosyst 2008, 4, 712–720. [DOI] [PubMed] [Google Scholar]
- (28).Wiśniewski JR; Ostasiewicz P; Mann M High Recovery FASP Applied to the Proteomic Analysis of Microdissected Formalin Fixed Paraffin Embedded Cancer Tissues Retrieves Known Colon Cancer Markers. J. Proteome Res 2011, 10, 3040–3049. [DOI] [PubMed] [Google Scholar]
- (29).Longuespée R; Alberts D; Pottier C; Smargiasso N; Mazzucchelli G; Baiwir D; Kriegsmann M; Herfs M; Kriegsmann J; Delvenne P; De Pauw E A Laser Microdissection-Based Workflow for FFPE Tissue Microproteomics: Important Considerations for Small Sample Processing. Methods 2016, 104, 154–162. [DOI] [PubMed] [Google Scholar]
- (30).Herfs M; Longuespée R; Quick CM; Roncarati P; Suarez-Carmona M; Hubert P; Lebeau A; Bruyere D; Mazzucchelli G; Smargiasso N; Baiwir D; Lai K; Dunn A; Obregon F; Yang EJ; De Pauw E; Crum CP; Delvenne P Proteomic Signatures Reveal a Dualistic and Clinically Relevant Classification of Anal Canal Carcinoma. J. Pathol 2017, 241, 522–533. [DOI] [PubMed] [Google Scholar]
- (31).Pottier C; Kriegsmann M; Alberts D; Smargiasso N; Baiwir D; Mazzucchelli G; Herfs M; Fresnais M; Casadonte R; Delvenne P; De Pauw E; Longuespée R Microproteomic Profiling of High-Grade Squamous Intraepithelial Lesion of the Cervix: Insight into Biological Mechanisms of Dysplasia and New Potential Diagnostic Markers. Proteomics: Clin. Appl 2019, 13, No. 1800052. [DOI] [PubMed] [Google Scholar]
- (32).Griesser E; Wyatt H; Have ST; Stierstorfer B; Lenter M; Lamond AI Quantitative Profiling of the Human Substantia Nigra Proteome from Laser-Capture Microdissected FFPE tissue. Mol. Cell. Proteomics 2020, 19, 839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Dewez F; Martin-Lorenzo M; Herfs M; Baiwir D; Mazzucchelli G; De Pauw E; Heeren RMA; Balluff B Precise Co-Registration of Mass Spectrometry Imaging, Histology, and Laser Microdissection-Based Omics. Anal. Bioanal. Chem 2019, 411, 5647–5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mezger STP; Mingels AMA; Bekers O; Heeren RMA; Cillero-Pastor B Mass Spectrometry Spatial-Omics on a Single Conductive Slide. Anal. Chem 2021, 93, 2527–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Liang Y; Acor H; McCown MA; Nwosu AJ; Boekweg H; Axtell NB; Truong T; Cong Y; Payne SH; Kelly RT Fully Automated Sample Processing and Analysis Workflow for Low-Input Proteome Profiling. Anal. Chem 2021, 93, 1658–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ellinger P; Kluth M; Stindt J; Smits SHJ; Schmitt L Detergent Screening and Purification of the Human Liver ABC transporters BSEP (ABCB11) and MDR3 (ABCB4) Expressed in the Yeast Pichia pastoris. PLoS One 2013, 8, No. e60620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bardou P; Mariette J; Escudié F; Djemiel C; Klopp C Jvenn: An Interactive Venn Diagram Viewer. BMC Bioinformatics 2014, 15, No. 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Tyanova S; Cox J Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol 2018, 1711, 133–148. [DOI] [PubMed] [Google Scholar]
- (39).Ikeda K; Monden T; Kanoh T; Tsujie M; Izawa H; Haba A; Ohnishi T; Sekimoto M; Tomita N; Shiozaki H; Monden M Extraction and Analysis of Diagnostically Useful Proteins from Formalin-Fixed, Paraffin-Embedded Tissue Sections. J. Histochem. Cytochem 1998, 46, 397–403. [DOI] [PubMed] [Google Scholar]
- (40).Fowler CB; Waybright TJ; Veenstra TD; O’Leary TJ; Mason JT Pressure-Assisted Protein Extraction: A Novel Method for Recovering Proteins from Archival Tissue for Proteomic Analysis. J. Proteome Res 2012, 11, 2602–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Azimzadeh O; Barjaktarovic Z; Aubele M; Calzada-Wack J; Sarioglu H; Atkinson MJ; Tapio S Formalin-Fixed Paraffin-Embedded (FFPE) Proteome Analysis Using Gel-Free and Gel-Based Proteomics. J. Proteome Res 2010, 9, 4710–4720. [DOI] [PubMed] [Google Scholar]
- (42).Coscia F; Doll S; Bech JM; Schweizer L; Mund A; Lengyel E; Lindebjerg J; Madsen GI; Moreira JM; Mann M A Streamlined Mass Spectrometry–Based Proteomics Workflow for Large-Scale FFPE Tissue Analysis. J. Pathol 2020, 251, 100–112. [DOI] [PubMed] [Google Scholar]
- (43).Wang H; Qian W-J; Mottaz HM; Clauss TRW; Anderson DJ; Moore RJ; Camp DG; Khan AH; Sforza DM; Pallavicini M; Smith DJ; Smith RD Development and Evaluation of a Micro- and Nanoscale Proteomic Sample Preparation Method. J. Proteome Res 2005, 4, 2397–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kyte J; Doolittle RF A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol 1982, 157, 105–132. [DOI] [PubMed] [Google Scholar]
- (45).O’Rourke MB; Padula MP Analysis of Formalin-Fixed, Paraffin-Embedded (FFPE) Tissue via Proteomic Techniques and Misconceptions of Antigen Retrieval. Biotechniques 2016, 60, 229–238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with data set identifier PXD029729.