

Summary
Triphenylphosphonium (TPP+) conjugated compounds selectively target cancer cells by exploiting their hyperpolarized mitochondrial membrane potential. To date, studies have focused on modifying either the linker or the cargo of TPP+-conjugated compounds. Here, we investigated the biological effects of direct modification to TPP+ to improve the efficacy and detection of mito-metformin (MMe), a TPP+-conjugated probe we have shown to have promising preclinical efficacy against solid cancer cells. We designed, synthesized, and tested trifluoromethyl and methoxy MMe analogs (pCF3-MMe, mCF3-MMe, and pMeO-MMe) against multiple distinct human cancer cells. pCF3-MMe showed enhanced selectivity toward cancer cells compared to MMe, while retaining the same signaling mechanism. Importantly, pCF3-MMe allowed quantitative monitoring of cellular accumulation via 19F-NMR in vitro and in vivo. Furthermore, adding trifluoromethyl groups to TPP+ reduced toxicity in vivo while retaining anti-tumor efficacy, opening an avenue to de-risk these next-generation TPP+-conjugated compounds.
Subject areas: Pharmaceutical science, chemistry, biological sciences, biochemistry
Graphical abstract

Highlights
-
•
Adding fluorine to triphenylphosphonium cation increases cancer cell selectivity
-
•
Fluorination retains the anti-proliferative activity of mitochondria-targeted metformin
-
•
Fluorinated mitochondria-targeted metformin has a half-life of nearly 2 h
-
•
Fluorinated mitochondria-targeted metformin has reduced toxicity in vivo
Pharmaceutical science; Chemistry; Biological sciences; Biochemistry
Introduction
Increases in the mitochondrial membrane potential are a recognized characteristic in multiple types of solid and liquid cancers.1,2,3,4 It remains unclear if changes in membrane potential are causal events or secondary events in cancer progression. The molecular mechanisms underlying mitochondrial membrane hyperpolarization remain enigmatic but have been experimentally exploited to selectively target cancer cells instead of normal cells. For example, rhodamine-123, mitochondria-penetrating peptides, Szeto-Schiller peptides, and triphenylphosphonium cation (TPP+)-conjugated compounds have all been synthesized to selectively target cancer cells.5,6,7 To date, TPP+-conjugated compounds have demonstrated the most promising in vitro and in vivo data.8,9,10,11,12,13 TPP+-conjugated compounds consist of the mitochondria-targeting TPP+ moiety, a linker typically consisting of an aliphatic carbon chain, and a functional cargo.12,13,14,15 TPP+-conjugated compounds were used to uncover the mechanism of coupling oxidative phosphorylation to mitochondrial membrane potential and, subsequently, to understand the role of oxidative metabolism in cancer.16 Moreover, early efforts by Murphy et al. focused on using the antioxidant Mito-Q, a TPP+-conjugated synthetic analog of coenzyme Q10, ubiquinone, to understand the role of mitochondrial redox recycling in disease progression.17,18 For over a decade, TPP+-conjugated compounds have been examined as tumor-selective anti-cancer agents, including mitochondria-targeted-3-carboxy-proxyl nitroxide, mitochondria-targeted-magnolol, mitochondria-targeted-metformin (MMe) and others. Each demonstrated broad anti-tumor efficacy by inhibiting complex I of the electron transport chain and activating energy-sensing signaling pathways.8,9,11,19,20 These TPP+-conjugated mitochondria-targeted compounds are examples of the functional consequences of changing the cargo. Changes in the carbon chain linker may skew TPP+-conjugated compounds to target complex III over complex I.19 While previous efforts have focused on modifying the aliphatic linker or the cargo, very few studies have investigated the functional and biological consequences of modifying the TPP+ moiety.21
The conventional wisdom underlying the use of TPP+ was that the large ionic radius, diffuse positive charge, and lipophilic nature allowed conjugated compounds to cross lipid bilayers and accumulate in the more negative inside of mitochondria.13,14,15 Most structural modifications to TPP+-conjugated agents have focused on modifying the length of the linker connecting TPP+ to cargo to fine-tune lipophilicity and localization of the mitochondria-targeted compound. A recent report has shown that TPP+ modifications can be employed to dissociate mitochondrial localization from functional effects on mitochondrial respiration.21 Notably, this report demonstrated a strong role for fluorine modifications in enhancing mitochondrial localization. Fluorine has different physical and chemical properties from other elements, including bond length, van der Waals radius, van der Waals volume, and electronegativity.22 Fluorine’s small van der Waals radius, 1.46 Å, can easily sterically replace hydrogen with a van der Waals radius of 1.2 Å.23 Unlike other halogens, non-polarizable fluorine does not participate in halogen bonding. Finally, the introduction of fluorine allows fine-tuning of the electrostatic properties of the compound based on the number and regiochemical placement of the fluorine atoms.24 The 19F isotope has 100% natural abundance and a +1/2-spin, resulting in an easily detectable NMR-active nucleus. As fluorinated compounds are scarce in nature, the introduction of 19F to a compound enables the use of 19F NMR detection of fluorinated TPP+-conjugated compounds with minimal noise resulting from endogenous molecules. Thus, fluorine substitution may have the added benefit of increasing the detectability of mitochondria-targeted compounds using 19F NMR or 18F positron emission tomography.
Here, we rationally redesigned and tested analogs of MMe, selected for its promising preclinical results, by adding electron-withdrawing trifluoromethyl groups in the para and meta positions of the TPP+ aryl rings (pCF3-MMe and mCF3-MMe). These analogs have reduced electron density on the phosphorus atom of the TPP group, which in turn improved the cell-type selectivity for cancer over normal cells. Importantly, fluorinating the TPP+ rings had the added benefit of in vivo detection and quantification via 19F-NMR. We also synthesized an electron-rich MMe analog containing para-methoxy aryl rings, pMeO-MMe, that allowed direct comparison of the effect of electron-withdrawing and electron-donating groups on the electron density of the phosphorus atom and the subsequent biological effects of TPP+-conjugated compounds. These data are the first to demonstrate that the addition of trifluoromethyl groups to the TPP+ moiety enhances selectivity for cancer cells, allows reproducible measurement of the accumulation of TPP+-conjugated compounds within tumor and non-tumor tissues, and improves the overall safety profile of TPP+-conjugated compounds while maintaining potent anti-tumor efficacy in vitro and in vivo.
Results
Design of next-generation mitochondria-targeted agents by direct triphenylphosphonium modification
Cationic lipophilic functional groups such as TPP+ move across biological membranes, particularly the mitochondrial membrane, without transporters.15 This phenomenon was previously exploited to selectively deliver metformin into cancer cells to enhance its anti-tumor efficacy and mitigate potential off-tumor side effects.9,10,11 Here, we fine-tuned the partial positive charge on the phosphorus center of MMe in a stepwise fashion by adding substituents to the aryl rings of the TPP+ moiety. Strong electron-withdrawing trifluoromethyl substituents were added to the meta or para positions of the TPP rings, mCF3-MMe, and pCF3-MMe, respectively (Figure S1). Trifluoromethyl substitution at the meta position increases the positive charge on the phosphorus center by withdrawing electrons through the aryl rings inductively. Trifluoromethyl substitution at the para position withdraws electrons both inductively and through resonance effects. Thus, the synthesis of each molecule enabled us to test the impact of gradients of increasing partial positive charge on the phosphorus center of the TPP+ moiety for enhanced accumulation in the mitochondria (Figures 1C and 1D). By comparison, the partial positive charge at the phosphorus center of MMe was decreased by adding para-methoxy substituents to the TPP aryl rings affording pMeO-MMe (Figure S1). Methoxy substituents at the para position of the phenyl rings increase electron density throughout the conjugated π-system via resonance, resulting in decreased partial positive charge at the phosphorus center (Figure 1A). Ultimately, these three novel MMe analogs allowed for a controlled decrease in electron density on the phosphorus atom: pMeO-MMe > MMe ≫ mCF3-MMe > pCF3-MMe (Figure 1). With these molecules in hand, we compared the biological effects produced by varying the cationic character of the phosphorus center compared to the parent MMe compound.
Figure 1.

Impact of aryl ring substitutions on the electron density of MMe
(A–D) Chemical structures (top), stick model (middle), and electron density maps (bottom) of MMe (B) and the analogs pMeO-MMe (A), mCF3-MMe (C), and pCF3-MMe (D). The chemical stick model displays the position, bonds, and predicted three-dimensional orientation of the individual atoms within each compound. The heatmap depicts the range of electron density for all four compounds. Red indicates increased electron density (decreased partial positive charge); blue indicates decreased electron density (increased partial positive charge). The arrows and white and black markers denote the range of electron density seen for MMe.
Modifying triphenylphosphonium increases selectivity while maintaining anti-tumor efficacy
We previously reported broad anti-tumorigenic effects of TPP+-conjugated compounds, with MMe consistently 100- to 1000-fold more efficacious on pancreatic, breast, and colon cancers compared to the FDA-approved biguanide compound metformin.8,10,11 Here, we explored the anti-tumorigenic effects of the newly synthesized trifluoromethyl and methoxy MMe analogs, pCF3-MMe, mCF3-MMe, and pMeO-MMe, on a battery of murine (KPC1242) and human (Panc-1, AsPC-1) pancreatic, colonic (HCT-116), breast (MDA-MB-231), and ovarian (OVCAR-4, OVCAR-8) cancer cell lines. To investigate tumor cell selectivity, human pancreatic nestin-expressing cells (HPNE) were used as a surrogate for normal epithelial cells, and human pancreatic stellate cells (HPSC) were studied as a non-transformed stromal cell type. Cells were exposed to escalating concentrations of MMe, pCF3-MMe, mCF3-MMe, and pMeO-MMe (Figures 2A–2D) and percent confluency was measured as a surrogate for daily proliferation. Growth curves and photomicrographs were used to calculate IC50 values at either 96 or 120 h (Figures 2A–2D) and confirmed that cell confluency was decreased in a concentration-dependent manner in each of the cancer cells treated with MMe (Figure S2), pCF3-MMe (Figure S3), and mCF3-MMe (Figure S4). Examination of pMeO-MMe in human pancreatic cancer cells also revealed a concentration-dependent decrease in confluence (Figure S5).
Figure 2.

Growth inhibitory effects of MMe, pCF3-MMe, and mCF3-MMe on pancreatic, ovarian, breast, and colon cancers and non-transformed normal epithelial cells
(A–E) Logarithmic dose-response fit curves of KPC1242, Panc-1, AsPC-1, HPSC, and HPNE confluence in response to treatment with MMe (A) or pCF3-MMe (C), or OVCAR-8, OVCAR-4, MDA-MB-231, and HCT-116 confluence in response to treatment with MMe (B) or pCF3-MMe (D). Densitometric quantification (E) in KPC1242 cells treated with MMe (0.7 μM) or pCF3-MMe (3 μM). Representative images of levels of Ki-67 (green) were recorded using the IncuCyte of KPC1242 treated with MMe (0.7 μM) or pCF3-MMe (3 μM), followed by the quantification of the total integrated intensity of Ki-67. Values are mean ± SD, n = 3-4, each experiment completed in technical duplicates or triplicates. IC50 values were calculated from confluence curves on the final day of the study as shown in Figures S2–S6. Statistical differences in panel E were determined using Student’s t-test (∗, p < 0.05; ∗∗, p < 0.01).
To test the off-target effects of the refined MMe analogs on normal non-transformed cells, the growth of HPNE cells (Figures S6A, S6D, and S6G), used as a surrogate for normal pancreatic ductal epithelium, or normal stromal HPSC cells (Figures S6B, S6E, and S6H) were measured in increasing concentrations of MMe, pCF3-MMe, or mCF3-MMe and compared to the vehicle control. Normal, non-transformed cells were more than a log-unit less sensitive to MMe, pCF3-MMe, and mCF3-MMe compared to cancer cells (Figures S5A–S5H). Logarithmic fitting of growth curves revealed strong growth inhibitory effects of MMe (Figures 2A–2C), pCF3-MMe (Figures 2D–2F), and mCF3-MMe (Figures S4A and S4B). Moreover, logarithmic fitting of growth curves demonstrated that pCF3-MMe had the least toxic effects toward non-transformed HPNE and HPSC-1 cells compared to the parent MMe and the mCF3-MMe analog (Figures 2A, 2C, and S4A).
Next, we determined the direct effects of MMe or pCF3-MMe treatment on cellular proliferation by measuring Ki-67 levels; increased Ki-67 expression is associated with increased cellular growth. KPC1242 cells treated with MMe or pCF3-MMe demonstrated a significant 20-30% reduction in Ki-67 levels after 24-h treatment (Figure 2E). Subsequent experiments focused on pCF3-MMe as it demonstrated the highest preclinical potential with efficacy toward cancer cells and decreased toxicity toward normal cells.
pCF3-MMe maintains bioenergetic metabolism inhibitory effects of mito-metformin
Both the parent compound metformin and mitochondria-targeted compound MMe inhibit complex I-mediated oxygen consumption and activate energy-sensing signaling pathways.9 To determine if the newly synthesized variants retain this functional specificity, we next determined if pCF3-MMe effectively impacts mitochondrial function and/or induces bioenergetic stress. KPC1242 cells were treated for 24 h with increasing concentrations of MMe or pCF3-MMe, and oxygen consumption rate (OCR) was measured as a surrogate for oxidative phosphorylation-linked mitochondrial respiration.25,26 The Seahorse Cell Mito Stress Test, which provides a quantitative assessment of mitochondrial function that includes basal respiration, ATP-linked respiration, maximal and reserve capacities, and non-mitochondrial respiration, revealed a concentration-dependent decrease in basal OCR, ATP-linked respiration, and maximal respiratory capacity in MMe (Figure 3A) and pCF3-MMe (Figure 3E) treated cells. Next, we evaluated if the observed decrease in OCR with pCF3-MMe corresponded to an inhibition of mitochondrial respiratory complex I. In agreement with our previous reports,9,11 MMe and pCF3-MMe decreased complex I-derived OCR in the KPC1242 cell line in a concentration-dependent manner. This effect was attenuated by supplementing succinate, a complex II substrate, allowing bypass of complex I,27,28 which suggests the mechanism for OCR inhibition is indeed through complex I for both MMe and pCF3-MMe. Moreover, logarithmic fitting revealed potent basal OCR inhibition (Figures 3C and 3G) and complex I inhibition at IC50 values comparable to growth inhibition (Figures 3D and 3H).
Figure 3.

pCF3-MMe inhibits complex I derived mitochondrial respiration
(A and E) Representative mitochondrial stress tests on the KPC1242 pancreatic cancer cell line preceded by 24 h treatment with MMe (A) or pCF3-MMe (E). Injections of oligomycin (1 μg/mL), 2,4-dinitrophenol (50 μM), rotenone (1 μM), and antimycin A (10 μM) were injected as indicated by the arrows.
(B and F) Representative complex I-derived oxygen consumption rate (OCR) after 24 h treatment of increasing concentrations of MMe (B) or pCF3-MMe (F) followed by rotenone to inhibit complex I and succinate to bypass complex I inhibition.
(C and G) Logarithmic dose-response curves of basal oxygen consumption rate in response to increasing concentrations of MMe (C) or pCF3-MMe (G) in KPC1242 cells.
(D and H) Logarithmic dose-response curves of complex I-derived oxygen consumption rate in response to increasing concentrations of MMe (D) or pCF3-MMe (H) in KPC1242 cells. Treated cells were normalized to vehicle (C, G, and D). Values are mean ± SD from 3 biological replicates completed in technical quadruplicate.
TPP-conjugated compounds effectively target the mitochondria of cancer cells.1,9,15,17,18,29,30 Thus, we tested and compared the efficacy of pCF3-MMe to target the mitochondria of cancer cells compared to normal cells using tetramethylrhodamine ethyl ester perchlorate (TMRE), a cell-permeant, positively charged dye that accumulates in active mitochondria, and is released into the cytosol upon mitochondrial damage.31,32 Differences in TMRE retention are often used to measure mitochondrial damage.31,32 As expected,9,21 and shown in Figures 4A–4D, MMe significantly reduced TMRE accumulation within the mitochondria of AsPC-1 measured decreased fluorescence; similarly, pCF3-MMe significantly reduced TMRE accumulation within the mitochondria of AsPC-1 (Figures 4A and 4C). Although not significant, pCF3-MMe demonstrated a stronger trend toward decreased TMRE accumulation compared to MMe. Consistent with their selectivity toward cancer cells, both MMe and pCF3-MMe did not affect TMRE accumulation within the HPSC cell line (Figures 4B and 4D).
Figure 4.

pCF3-MMe selectively depolarizes mitochondria and activates bioenergetic signaling in cancer cells
(A–G) AsPC-1 and HPSC cells were serum-starved overnight, loaded with tetramethylrhodamine ethyl ester (TMRE), and treated for 1 h with the indicated concentrations of MMe or pCF3-MMe or, as the vehicle control, DMSO. Mitochondrial TMRE retention was measured by flow cytometry. Representative histograms of n = 3 of TMRE fluorescence in AsPC-1 (A) and HPSC (B) in response to treatment. Normalized Mean fluorescence intensity of TMRE in treated AsPC-1 (C) shows the depolarization of mitochondrial membrane in response to treatment and a lack of depolarization in the HPSC cells (D). Total cellular ATP levels were analyzed using a luminescent ATP detection assay of KPC1242 cells treated with MMe (0.7 μM) or pCF3-MMe (3 μM), and changes in ATP levels were assayed (E). Representative immunoblot analyses of phosphorylated and total AMPK levels (F) and densitometric quantification (G) in KPC1242 cells treated with MMe (0.7 μM) or pCF3-MMe (3 μM). Representative images of levels of Ki-67 (green) were recorded using the IncuCyte of KPC1242 treated with MMe (0.7 μM) or pCF3-MMe (3 μM), followed by the quantification of the total integrated intensity of Ki-67. Values are mean ± SD from 3 biological replicates. Statistical differences in panels A, C, and E were determined using Student’s t-test (∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001, ns: not significant).
The functional consequence of inhibiting mitochondria complex I and subsequently oxidative phosphorylation is a reduction of overall ATP levels in the cell.33 We previously reported the ability of MMe to reduce ATP levels in numerous cancer cell types.9,11 Thus, we explored if pCF3-MMe retained similar properties. KPC1242 cells were treated with MMe or pCF3-MMe for 24 h at concentrations that inhibit cellular growth and complex I-derived OCR (0.7 μM MMe or 3 μM pCF3-MMe). Levels of ATP were significantly reduced in both MMe and pCF3-MMe treated cells, consistent with the inhibition of metabolic respiration (Figure 4E). Cells, including cancer cells, must maintain a high ATP:ADP ratio to maintain cellular functions and survival.34 Moreover, AMP levels rise whenever the ATP:ADP ratio decreases, and an increase in the AMP:ATP ratio signals an energetic crisis in the cell. This energetic crisis is sensed by the master regulator AMP-Activated Protein Kinase (AMPK), which is activated through Thr172 phosphorylation and controls many signaling pathways ranging from energy metabolism to cellular growth.34 Therefore, we evaluated if the reduction in ATP levels induced by MMe or pCF3-MMe is sufficient to activate AMPK. KPC1242 cells treated with 0.7 μM MMe or 3 μM pCF3-MMe, previously demonstrated to decrease ATP levels (Figure 4E), showed a significant increase in AMPK activation as quantified by increased Thr172 phosphorylation (Figures 4F and 4G).
Detection of pCF3-MMe in vitro and in vivo
As natural organic compounds containing fluorine are rare in living organisms, we specifically chose trifluoromethyl groups when designing next-generation MMe analogs.35 We aimed to leverage the nine chemically equivalent fluorine atoms in the trifluoromethyl groups of pCF3-MMe using 19F-NMR. To detect cellular accumulation of pCF3-MMe in vitro, KPC1242 cells were incubated with 1 mg pCF3-MMe or vehicle (DMSO) in growth media for 2 h, followed by the removal of the media, compound extraction, and NMR analysis (Figure S7A). Four extraction conditions were tested: 8:2 MeOH:H2O, 9:1 CHCl3:MeOH, CH2Cl2, and 1:1 MeCN:H2O. MeOH:H2O (8:2) provided the most efficient extraction, evidenced by the strongest signal observed in 19F-NMR (−61.6 ppm) among all tested conditions (Figures S7B–S7E). An additional peak was observed at −73.4 ppm in both the vehicle and pCF3-MMe treated groups that likely originated from tissue culture plastics as removing the fluorinated plasticizer-containing research consumables completely abolished the signal (Figures S7not shown). To assess in vivo detection of pCF3-MMe, mice engrafted with KPC1242 cells were administered 1 mg/mouse pCF3-MMe for 2 h, followed by tumor harvest and compound extraction (Figure S8A). NMR analysis demonstrated the consistent detection of pCF3-MMe, albeit with slightly different efficacy for each of the organic solvents tested (Figure S8). Much like extraction from cell culture, 8:2 MeOH:H2O extraction was the most efficient for tissues (Figures S8B–S8E).
To validate 19F-NMR as a viable detection approach, we analyzed the persistence of pCF3-MMe in tumors. KPC1242 engrafted mice were administered 1 mg/mouse pCF3-MMe starting 12 days after implantation. Mice were sacrificed, and tumors were extracted with 8:2 MeOH:H2O 30 min, 2, 4, 6, or 24 h after treatment (Figure 5A). 19F-NMR revealed measurable levels of pCF3-MMe within the tumors 24 h after injection (Figure 5B). We next designed a quantitative NMR approach to measure the in vivo persistence of pCF3-MMe. First, serial dilutions of known concentrations of pCF3-MMe were prepared, and the intensity of the NMR peaks at a set scan number (n = 64) was used to create a standard curve. Next, a one-phase decay least-squares fit equation was generated to calculate the amount of pCF3-MMe at each time point using the standardized scan number. Nonlinear fit curve analysis revealed that pCF3-MMe had a t1/2 of 1.9 h (114 min) (Figure 5C). As an initial study of its pharmacodynamics, quantitative 19F-NMR indicated that pCF3-MMe was undetectable in the blood and liver 2 h after injection (Figure S9) and was barely detectable above the signal-to-noise ratio at 6 h (not shown). Taken together, these data reveal that the modification to TPP+, particularly by adding chemically equivalent trifluoromethyl groups, allows ease of detection and monitoring of TPP+-conjugated compounds.
Figure 5.

Quantification of pCF3-MMe persistence within the tumor microenvironment
(A) Tumor engraftment and treatment schedule. KPC1242 murine pancreatic cancer engrafted mice were treated with an intra-tumoral injection of 1 mg pCF3-MMe.
(B) Following treatment, pCF3-MMe was extracted using 8:2 MeOH:H2O at 30 min, 2, 4, 6, or 24 h and levels of pCF3-MMe at each time point were quantified using quantitative NMR.
(C) Nonlinear fit curve analysis of pCF3-MMe levels was used to generate t1/2. Values are mean ± SEM from 1 to 4 biological replicates.
pCF3-MMe attenuates tumor progression in vivo with superior tolerability to mito-metformin
With the ability to detect pCF3-MMe in vitro and in vivo in hand, along with anti-tumor properties and decreased toxicity toward non-transformed cells in culture, we next compared the tolerability of pCF3-MMe in mice. Non-tumor-bearing mice received an intra-peritoneal injection with decreasing amounts (1 mg, 250, 100, 50, or 25 μg) of unsubstituted MMe or pCF3-MMe. Additional mice were treated with 1 mg/mouse metformin as a treatment control (Figure 6A). Surprisingly, a single intra-peritoneal injection of 250 μg or 1 mg MMe to non-tumor-bearing mice resulted in significant and rapid mortality (Figures 6B and 6C). In stark contrast, pCF3-MMe demonstrated vastly superior tolerability, with 100% of mice injected with 1 mg of the fluorinated analog surviving (Figures 6B and 6C). Metformin demonstrated a similar safety profile as pCF3-MMe.
Figure 6.

pCF3-MMe inhibits tumor growth with enhanced biosafety relative to MMe
(A–I) C57BL/6J mice were treated with 1 mg MMe, 1 mg pCF3-MMe, 1 mg MMe, 250 μg MMe, 100 μg MMe, 50 μg MMe, or 25 μg MMe for 6 h (A) or 5 days (D). Kaplan-Meier survival curve of mice treated for 6 h (B) or 5 days (E), n = 3-5. Numbers of mice surviving over time after a single injection (C) or 5 repeated doses (F). Significance was determined using a Mantel-Cox test (∗, p < 0.05; ∗∗, p < 0.01). KPC1242 murine pancreatic cancer engrafted mice were treated with 1 mg pCF3-MMe or vehicle 5 days on and 2 days off (G). Tumors isolated from vehicle and treated mice were pictured (H). Tumor volume was measured ex vivo from vehicle and pCF3-MMe-treated mice (I). Values are mean ± SD. Significant differences were determined by Student’s t-test (∗, p < 0.05).
Given the strong safety shown from a single injection, we next determined if mice tolerated repeated injection using our published treatment schedule by injecting non-tumor-bearing mice daily for 5 consecutive days with doses ranging from as little as 25 μg/mouse to as much as 1 mg/mouse20 (Figure 6D). Survival curves indicate that while each of the treated mice tolerated a 1 mg dose of pCF3-MMe, parallel groups succumbed to 4-fold lower doses of un-fluorinated MMe (Figures 6E and 6F). Having shown that 1 mg/mouse pCF3-MMe was well tolerated in tumor-free animals, we next tested if pCF3-MMe could inhibit tumor growth in vivo. Mice were implanted with murine cancer cells and injected 17 days after engraftment with 1 mg/mouse pCF3-MMe (Figure 6G). Consistent with its anti-proliferative effects in culture, pCF3-MMe significantly decreased tumor size and volume (Figures 6H and 6I). Taken together, trifluoromethylation of TPP+ improves the reproducible detection, quantitative measurement, and safety profile of TPP+-conjugated agents while retaining potent anti-tumor effects in vivo.
Discussion
Cancer cell mitochondrial dysfunction and resulting membrane hyperpolarization have previously been exploited to study and deliver diagnostic and therapeutic agents.1,17,18,30,36 While no underlying mechanism has been identified, this property is routinely exploited to selectively detect and target cancer in vitro and in vivo.1,8,9,10,11,12,17,18,19,20,30,36,37 TPP+-conjugated agents take advantage of this hyperpolarization to accumulate in cancer cells and have shown the most promising results in preclinical testing. However, early drug development efforts using TPP+ have focused on modifying the aliphatic linker or the cargo.12,19 Moreover, the development of TPP+-conjugated compounds has been hindered by the lack of sensitive and unbiased approaches to easily detect TPP+-conjugated compounds and monitor their distribution in animal models.12 Herein, TPP+-conjugated MMe was rationally redesigned by directly modifying the TPP+ moiety to enhance the partial positive charge on the phosphorus atom of the TPP+ moiety. Using cell culture and preclinical models, we demonstrate that trifluoromethyl-modified MMe has improved selectivity and tolerability with the added benefit of sensitive and quantitative detection of fluorinated MMe in vivo. These data illustrate that harnessing the electron density of the phosphorus atom in the TPP+ provides novel insight into the biological effects of modifying the TPP+ moiety.
The large ionic radius of the unsubstituted TPP+, mainly due to the delocalized cationic charge on the phosphorus atom, likely plays a role in the ability of TPP+ to cross the cellular membrane and target the more negative inside membrane potential of mitochondria.17 Thus, any substitution on the aryl rings of TPP+ will likely impact the ability of these compounds to penetrate the membrane of cancer cells successfully and selectively.17 A previous report investigated some of the chemical properties of aryl substitution on TPP+, lacking the aliphatic carbon chain linker and a cargo, on normal non-transformed C2C12 myoblast cells.21 However, gaps in this report included examining the roles of TPP+ aryl substitution on the anti-tumor efficacy when a functional cargo is attached and assessing the preclinical potential of aryl-substituted TPP+ compounds. Here, we modified the three TPP+ aryl rings to determine if we could improve the mitochondrial localization and detectability of these reagents. The three novel compounds described, pCF3-MMe, mCF3-MMe, and pMeO-MMe, had distinct anti-tumor properties. The addition of trifluoromethyl groups in the para position on the TPP+ moiety, pCF3-MMe, was the most optimal modification, with only a slight change in efficacy compared to MMe in several types of cancer. Interestingly, the strongest effect was observed in cell selectivity, with pCF3-MMe and mCF3-MMe demonstrating a nearly 10-fold reduction in toxicity to normal cells compared to MMe. These data are consistent with a prior report demonstrating that pCF3-TPP+, a molecule lacking the aliphatic carbon chain linker and bioactive cargo, had no impact on OCR or proliferation of a non-transformed murine myoblast cell line.21 In contrast to the fluorinated compound, pMeO-MMe exhibited lower efficacy in pancreas cancer cells, a diminishment we speculate reflects superior selectivity of pCF3-MMe toward cancer cells, compared to the electron-donating methoxy groups in pMeO-MMe. Alternatively, the differences in the IC50 curves between individual TPP-modified compounds or between the varying cancer cell lines may reflect the ability of each compound to activate discrete cellular pathways in different types of cancer cells. Indeed, we have previously shown that shortening the carbon linker between TPP and the bioactive moiety modulates the inhibitory effect of mitochondria-targeted agents.38,39
Fine-tuning the TPP+ aryl rings with trifluoromethyl substituents increased lipophilicity while methoxy decreased lipophilicity as measured by the octanol/water partition coefficient (cLogP) pCF3-MMe. These data suggest a potential mechanism for pCF3-MMe’s improved ability to cross the lipid bilayer and preferentially accumulate in mitochondria membranes. The increase in the partition coefficient of the lipophilic cation may be associated with a greater accumulation of the compound on the matrix side of mitochondria, consistent with the decreased retention of TMRE in pCF3-MMe, relative to pMeO-MMe, treated cells. While the ratio of the membrane-bound and unbound pools of the compound is defined by the partition constant, the total accumulation at equilibrium is heavily controlled by the membrane potential, as predicted by the Nernst equation, with correction due to membrane/cytosol or matrix partitioning. Thus, changes in the partition coefficient and the electron density may work cooperatively to enhance pCF3-MMe selectivity toward cancer cells.
The mechanism of action of pCF3-MMe remained intact relative to the unsubstituted MMe. As shown for the parental compound and several other TPP+-conjugated compounds, pCF3-MMe decreased mitochondrial respiration, inhibited complex I, decreased ATP levels, and activated AMPK signaling in a concentration-dependent manner.8,9,10,11,12,19,20,37 These data indicate the para trifluoromethyl substitution did not impact critical binding interactions or decrease the activity of the parent molecule. Importantly, while the para trifluoromethyl aryl substitutions did not interfere with the mechanism of action, this substitution improved the selectivity and tolerability of the molecules in vitro and in vivo. This partly reflects the increase in partial positive charge on the phosphorus atom of pCF3-MMe, which suggests better accumulation within tumor cells. Kulkarni and colleagues showed that using a pCF3-TPP+ substituted on the phenyl rings, in the absence of any conjugated cargo, did not prevent the modified TPP+ moiety from accumulating within the mitochondria of intact cells. Indeed, para-substitution of the TPP+ phenyl rings led to a higher mitochondrial accumulation of cargo than that mediated by unmodified TPP+ 21. Combined, these data demonstrate that the pCF3-TPP+ moiety is a very promising lead for developing mitochondria-targeting agents with increased safety profiles. Our results support those findings with the increased benefit that adding the bioactive cargo metformin to generate pCF3-MMe retained strong inhibitory effects on mitochondrial respiration. These data suggest that the metformin cargo conjugated to pCF3-TPP provided anti-proliferative impact on cancer cells.
Cumulatively, pCF3-MMe has significant other advantages over traditional unsubstituted TPP+-conjugated compounds. First, the detection of pCF3-MMe using 19F-NMR is greatly enhanced in vitro and in vivo. The ability to quantitative measurement via 19F-NMR is a marked improvement on the traditional detection of unsubstituted TPP+-conjugated compounds requiring a laborious process for detection using LC/MS requiring unique method development for each compound synthesized. Using 19F-NMR, the half-life of pCF3-MMe was determined to be ∼2 h, a level that closely resembles the pharmacodynamics of many anti-cancer agents.40 The inability to detect pCF3-MMe in the blood and liver at 30 -minutes or 6 h supports the idea that the compound is retained at the injection site. A caveat to that interpretation is that metformin is not metabolized and is instead excreted as an intact molecule in the urine.41 While reports suggest metformin has a half-life of ∼5 h, the compound is poorly absorbed, with large inter-individual variability in pharmacokinetics.42 In contrast to pCF3-MMe, pharmacokinetic and pharmacodynamic studies of MitoQ, a different, non-fluorinated, TPP+-conjugated compound, revealed rapid uptake followed by a sharp decline in the compound after administration, with a half-life of ∼30 min, with the molecule being excreted as an intact molecule in the urine.43 Yet another advantage is that pCF3-MMe demonstrated a significantly improved safety profile in vivo similar, with the fluorinated compound demonstrating comparable potency to the FDA-approved parent drug metformin. While additional studies are needed to evaluate the pharmacokinetics, pharmacodynamics, and safety profile of TPP+-conjugated metformin analogs, the work herein demonstrate that these newly designed compounds may be useful in completing rigorous pharmacology studies. Moreover, pCF3-MMe opens the door for the non-invasive monitoring of pCF3-TPP+ compounds with complementary techniques such as 19F-MRI and 18F-PET.
Limitations of the study
Our data indicate that TPP+-modified variants of MMe retain anti-tumor effects on cancer cell growth and mitochondrial respiration. While we demonstrated strong sensitivity for in vivo and in vitro detection using NMR spectroscopy, 18F positron emission tomography or other reliable approaches for in vivo quantification remain to be optimized. Similarly, the detection and quantification of fluorinated MMe variants within the heterogeneous cell populations of solid tumors have yet to be completed. Those studies would facilitate the validation of tumor selectivity by TPP+-modified compounds. Further chemical modifications, including fluorination at the ortho-carbon position, or saturation with additional fluorines have yet to be synthesized and tested.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC-conjugated monoclonal anti-Ki-67 (SolA15) | ThermoFisher | RRID: AB-11151330 |
| Rabbit anti-phospho-AMPKalpha | Cell Signaling Technologies | RRID:AB-330330 |
| Rabbit anti-total-AMPKalpha | Cell Signaling Technologies | RRID:AB-330331 |
| HRP-conjugated rabbit IgG | Cytiva, Amersham | RRID:AB-772191 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Gibco Life Technologies | Cat# 11965084 |
| RPMI 1640 Medium | Gibco Technologies | Cat# 11875119 |
| McCoy’s modified medium | Gibco Technologies | Cat# 16600082 |
| Fetal bovine serum | Omega | Cat# FB-12 |
| L-glutamine GlutaMAX | Gibco Technologies | Cat# 35050-061 |
| D-glucose | Gibco Technologies | Cat# J60067.EQE |
| M3-Base A | Incell | Cat# M300F-500 |
| Human EGF | Gibco | Cat# PHG0313 |
| Puromycin | ThermoFisher | Cat# A1113802 |
| Dulbecco’s-modified PBS | Gibco Life Technologies | Cat# 14190 |
| Paraformaldehyde | Sigma | Cat# 158127 |
| Triton X-100 | Fisher Scientific | Cat# 327371000 |
| Oligomycin | Sigma | Cat# O4876 |
| Dinitrophenol | Sigma | Cat# D198501 |
| Rotenone | Sigma | Cat# R8875 |
| Antimycin A | Sigma | Cat# A8674 |
| Protease inhibitor cocktail set III | Millipore Sigma | Cat# 539134 |
| Nonidet P-40 | ThermoFisher | Cat# 28324 |
| EDTA | Life Technologies | Cat# 15575-038 |
| Sodium fluoride | Sigma | Cat# S-1504 |
| SDS | ThermoFisher | Cat# BP166500 |
| Sodium orthovanadate | Sigma | Cat# S6508 |
| Beta-glycerol phosphate | Sigma | Cat#G9891 |
| N-(10-bromodecyl)phthalimide | Combi-Blocks, Inc | Cat#QI-5596 |
| anhydrous acetonitrile (CH3CN) | Sigma Aldrich | Cat # 271004 |
| N,N-dimethylformamide (DMF) | Sigma Aldrich | Cat# 227056 |
| methanol | Sigma Aldrich | Cat # 179337 |
| Dichloromethane (DCM) | Sigma Aldrich | Cat #D65100 |
| Triphenylphosphine | Oakwood | Cat # 037818 |
| Tris(4-trifluoromethyl)phosphine | Ambeed | Cat # A108436 |
| Tris(4-methoxyphenyl)phosphine | Ambeed | Cat # A354955 |
| Ethanol | Decon Labs | Cat # UN1987 |
| Hydrazine | Sigma Aldrich | Cat # 21555 |
| Diethyl ether | Sigma Aldrich | Cat # 673811 |
| n-butanol | Sigma Aldrich | Cat # 360465 |
| HCl | Sigma Aldrich | Cat # 294837 |
| cyanoguanidine | Sigma Aldrich | Cat #D76609 |
| Critical commercial assays | ||
| Luminescent ATP Detection Assay | Abcam | Cat# ab113849 |
| Experimental models: Cell lines | ||
| Panc-1 | ATCC | RRID:CVCL-0480 |
| MDA-MB-231 | ATCC | RRID:CVCL-0062 |
| AsPC1 | ATCC | RRID:CVCL-0152 |
| OVCAR-8 | ATCC | RRID:CVCL-1629 |
| OVCAR-4 | ATCC | RRID:CVCL-1627 |
| HCT-116 | ATCC | RRID:CVCL-0291 |
| HPNE | ATCC | RRID:CVCL-C466 |
| KPC1242 | David Tuveson | |
| HPSC | Rosa Hwang | PMCID: PMC2519173 |
| Experimental models: Organisms/strains | ||
| C57BL/6J | The Jackson Laboratories | RRID:IMSR-JAX:000,664 |
| Software and algorithms | ||
| GraphPad Prism v.9.2.0 | GraphPad Software, San Diego, CA | RRID:SCR_002798 |
| Alpha View Software v.3.5.0 | Protein Simple, Santa Clara, CA | |
| Schrodinger Computational Suite v.2021-1 | RRID:SCR_014879 | |
| ChemDraw Professional 18.0 | PerkinElmer | https://www.perkinelmer.com/category/chemdraw |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Michael Dwinell (mdwinell@mcw.edu).
Materials availability
Chemical modifications of mitochondria-targeted compounds, including those included in this report, are covered by a provisional patent [U.S. Application No. 63/285,374].
Compounds generated in this work will be provided following the completion of an institutional signed material transfer agreement.
Experimental model details
Cell culture
Murine pancreatic cancer cells KPC1242 (David Tuveson, Cold Spring Harbor), human pancreatic cancer cells, Panc-1 (RRID:CVCL-0480), human breast cancer cells MDA-MB-231 (RRID:CVCL-0062), and human pancreatic stellate cells HPSC (Rosa F. Hwang, MD Anderson Cancer Center)44 were all cultured in DMEM (Gibco ThermoFisher Scientific, Waltham, MA, Cat#11965084) containing 10% v/v fetal bovine serum (Omega Scientific, Inc., Tarzana, CA, Cat# FB-12). Human pancreatic cancer cells AsPC-1 (RRID:CVCL-0152), human ovarian cancer cells OVCAR-8 (RRID:CVCL-1629), and human ovarian cancer cells OVCAR-4 (RRID:CVCL-1627) were all cultured in RPMI media (Cat# 22400089) containing 10% v/v fetal bovine serum (Omega Scientific). Human colon cancer cells HCT-116 (RRID:CVCL-0291) were cultured in McCoy’s 5A Modified Medium (Gibco, Cat# 16600082) containing 10% v/v fetal bovine serum (Omega Scientific). Human pancreatic duct epithelial cells HPNE (RRID:CVCL-C466) were cultured in DMEM and 200 mM L-Glutamine and 7.5% w/v NaCO3 further supplemented with 15% v/v fetal bovine serum, M3-Base A (Incell, San Antonio, TX), 1M D-Glucose (Gibco Cat#J60067.EQE), 100 μg/mL human EGF (Gibco Cat# PHG0313) and 10 mg/mL puromycin (ThermoFisher, Cat#A1113802).
Animal models
All animal studies were approved by the Institutional Animal Care and Use Committees at the Medical College of Wisconsin. C57BL/6J mice (RRID: IMSR JAX:000,664) were purchased from The Jackson Laboratory (Bar Harbor, ME). Six-to eight-week-old male or female mice were randomly assigned to treatment or control groups and implanted subcutaneously in the right flank with 1 × 106 KPC1242 murine pancreatic cancer cells. Tumors were allowed to establish for 12-17 days before intra-tumoral treatment. Mice were randomly assigned to control or experimental treatment groups immediately before treatment by an investigator blinded to the ex vivo tumor measurements. Treated tumor-bearing mice received a single injection of compound resuspended in Dulbecco’s-modified PBS (Gibco Life Technologies, Cat#14190) in a final 50 μL volume directly to the tumor mass using a 1.5 mL syringe and 28-gauge × 0.5 cm needle (Franklin Lakes, NJ, Cat#329461). At study end tumors were excised, and volume (length × width × depth = mm3) measured postmortem using calipers. Tumors were then fixed in zinc formalin and cleared in 70% v/v ethanol, embedded in paraffin, and 7 μm sections stained for immunohistochemical analysis.
Method details
Cell growth and proliferation
Cells were plated to standard 96-well tissue culture plates at 4,000 cells/well. Mitochondria-targeted compounds were diluted in full serum medium and added to wells. Cell confluence was measured using an IncuCyte S3 every 2 h at 10× magnification. To measure proliferation, KPC1242 cells (2 × 104) were treated with MMe or pCF3-MMe for 24 h, fixed with 1% v/v PFA (Sigma, Cat#158127), permeabilized with 0.2% v/v Triton X-100 (Fisher, Cat# 327371000), and stained using FITC-conjugated Ki-67 antibody (ThermoFisher, Cat# 11-5698-82 [RRID: AB-11151330]). Image analyses were conducted using the IncuCyte S3 imaging software system.
Extracellular flux
Cells were plated to Seahorse XF96 cell culture microplates at 20,000 cells/well. Stock compounds in DMSO were diluted in RPMI (Gibco, Cat#11875119) medium to a final concentration in 100 mL volume, incubated at 37°C for 24 h, centrifuged at 200 × g for 2 min, and conditioned medium removed and replaced with serum-free RPMI (200 mL). Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured after injections of 1 mg/mL oligomycin (Sigma, Cat# O4876), 50 mM dinitrophenol (Sigma, Cat# D198501), 1 mM rotenone (Sigma, Cat# R8875), and 10 mM antimycin A (Sigma, Cat# A8674). Analysis was performed by normalizing OCR and ECAR readouts to vehicle controls. Complex I and II activity were analyzed after permeabilization of the cells.
ATP measurements
KPC1242 cells (2 × 104) were treated with MMe or pCF3-MMe for 24 h. Intracellular ATP levels were determined in cell lysates using a Luminescent ATP Detection Assay Kit per the manufacturer’s directions (Abcam, Cambridge, UK, Cat# ab113849).
Immunoblotting
Cells were serum-starved for 24 h followed by compound treatment for 24 h before lysis in RIPA buffer composed of 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.25% v/v sodium deoxycholate, 1% v/v Nonidet P-40 (ThermoFisher, Cat#28324), 0.1% v/v SDS (Fisher, Cat#BP166500), 1 mM EDTA (Life Technologies, Cat#15575-038), 10 mM sodium orthovanadate (Sigma Cat#S6508), 40 mM β-glycerol phosphate (Sigma Cat# G9891), 20 mM sodium fluoride (Sigma, Cat#S1504), and protease inhibitors cocktail set III (Millipore Sigma, Cat#539134). Protein (10 μg) was loaded to each lane and separated by reducing SDS-PAGE. The separated proteins were electro-transferred to PVDF membranes and incubated with primary anti-phospho-AMPKα or anti-total AMPKα (Cell Signaling Technology #2531S [RRID:AB_330330] or #2532S [RRID:AB_330331], respectively) or anti-β-actin (Cell Signaling, Cat# 4970S [RRID:AB_2223172]). Primary antibodies were detected by incubating with secondary horseradish peroxidase-conjugated antibody (Cytiva, Amersham ECL rabbit IgG-HRP, Cat# NA9340 [RRID:AB_772191]). Densitometric quantification of phosphorylated, total or actin protein levels was measured using chemiluminescence and calculated using Alpha View Software (ProteinSimple, Santa Clara, CA).
Sample preparation and NMR analysis
Cells (1 × 107 cells/10 cm dish) were allowed to adhere overnight and then treated with varying concentrations of mitochondria-targeted compounds. Compounds were extracted using multiple methods and optimized for detection and quantification: A) Cells or tumors were placed in a dry ice ethanol bath followed by 80% v/v MeOH in water. Cells were placed on a dry ice ethanol bath on an orbital shaker for 20 min, and then cells were collected and centrifuged at 10,000 × g; B) Post-treatment cells were lifted with TrypLE (Gibco, Cat# 11558856) and centrifuged, then compound was extracted from the cells or tumors with 9:1 CHCl3:H2O, CH2Cl2, or 1:1 MeCN:H2O. Following extraction, solvents were evaporated using a Biotage V-10 Touch (Uppsala, Sweden), and before NMR acquisition, samples were resuspended in an equal volume of DMSO-d6.
NMR data collection
19F 1D-NMR spectra were collected on a Bruker Avance-III 500 MHz NMR spectrometer equipped with a 1H&19F/13C/15N–TCI cryoprobe. All 19F data were collected at 25°C using a 1D pulse sequence with a 30° flip-angle, 64 transients, and an automatic receiver gain. TopSpin 3.6.1 was used to process all spectra and integrate the peak intensity corresponding to the pCF3-MMe 19F signal at −61.6 ppm. To evaluate pCF3-MMe concentrations in biological samples, a standard curve was generated using pCF3-MMe concentrations of 0.25, 0.50, 0.75, 1.0, 1.25, and 1.5 mg/mL, and the resulting peak intensities were analyzed by linear regression. The standard curve was then used to calculate the amount of pCF3-MMe present per gram of tissue in the processed biological samples.
Electron density calculations
Computational calculations were performed with the Schrodinger Computational Suite v. 2021-1 (RRID:SCR_014879). Using the LigPrep workflow, all molecules were prepared in the OPLS4 force field at pH = 7. Using the Jaguar interface and default settings, electron density and electrostatic potential surfaces of the prepared molecules were calculated using B3LYP-D3 theory and the 6-31G∗∗ basis set. Color ramping (minimum and maximum) for the heatmap was normalized to unconjugated MMe.
General compound synthesis methods and equipment
All reagents and solvents were commercial grade and purified before use. Thin-layer chromatography was performed using glass-backed silica gel (250 μm) plates. UV light and/or iodine, potassium permanganate, and potassium iodoplatinate stains were used to visualize products. Medium pressure liquid chromatography was performed on a Biotage Isolera in conjunction with a Biotage Dalton 2000 using the conditions indicated. Nuclear magnetic resonance spectra (NMR) were acquired on a Bruker Avance-III-500 MHz spectrometer equipped with a TCI cryoprobe. 1H and 13C chemical shifts were measured relative to residual solvent peaks as an internal standard set to δ 7.26 and δ 77.0 (CDCl3), δ 2.50 and δ 39.5 (DMSO-d6), or δ 3.31 and δ 49.00 (CD3OD). Low-resolution mass spectra were recorded on a Biotage Dalton 2000 or Advion Expression Compact Mass Spectrometer using the indicated ionization method. High-resolution mass spectra were recorded at the Indiana University Mass Spectrometry Facility on a Thermo Scientific Orbitrap XL spectrometer using the indicated ionization method. Post-acquisition gain correction was applied using reserpine as a lock mass.
General procedure-a for the preparation of compounds 2a-d
N-(10-bromodecyl)phthalimide (1 equiv, Combi-Blocks, Inc., cat # QI-5596) was dissolved in anhydrous acetonitrile (CH3CN) (Sigma-Aldrich, cat # 271004-1L) or N,N-dimethylformide (DMF) (Sigma-Aldrich, cat# 227056-1L) (0.32 M) and placed under nitrogen. This was treated with the indicated phosphine (1 equiv). The reaction was then heated at reflux for CH3CN or in a pressure vessel at 140°C for DMF for 18-51 h. The reaction was then concentrated in vacuo to give crude 2a-d. The material was purified by silica column chromatography (0-30% v/v MeOH (Sigma- Aldrich, cat # 179337)/dichloromethane (DCM) (Sigma-Aldrich, cat #D65100)) to provide the title compound.
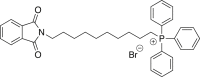 (2a) [10-(1,3-dioxo-2,3-dihydro-1H-isoindol-2-yl)decyl]triphenylphosphonium bromide
(2a) [10-(1,3-dioxo-2,3-dihydro-1H-isoindol-2-yl)decyl]triphenylphosphonium bromide
A mixture of 2.5 g (6.83 mmol) of N-(10-bromodecyl)phthalimide in CH3CN (21 mL) was refluxed with triphenylphosphine (Oakwood, cat # 037818) for 18 h. Flash chromatography gave 3.98 g (93%) of the title compound. Spectra match those previously reported.21
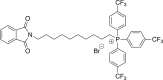 (2b) [10-(1,3-dioxo-2,3-dihydro-1H-isoindol-2-yl)decyl]tris[4-(trifluoromethyl)phenyl]-phosphonium bromide
(2b) [10-(1,3-dioxo-2,3-dihydro-1H-isoindol-2-yl)decyl]tris[4-(trifluoromethyl)phenyl]-phosphonium bromide
A mixture of 1.5 g (4.10 mmol) of N-(10-bromodecyl)phthalimide in DMF (12.6 mL) was heated with tris(4-trifluoromethyl)phosphine (1.91 g, 4.10 mmol, Ambeed, cat # A108436) for 51 h. Flash chromatography gave 2.59 g (76%) of the title compound. Spectra match those previously reported.21
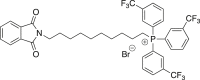 (2c) [10-(1,3-dioxo-2,3-dihydro-1H-isoindol-2-yl)decyl]tris[3-(trifluoromethyl)phenyl]-phosphonium bromide
(2c) [10-(1,3-dioxo-2,3-dihydro-1H-isoindol-2-yl)decyl]tris[3-(trifluoromethyl)phenyl]-phosphonium bromide
A mixture of 0.500 g (1.37 mmol) of N-(10-bromodecyl)phthalimide in DMF (12.6 mL) was heated with tris(3-trifluoromethyl)phosphine45 (0.637 g, 1.37 mmol) for 51 h. Flash chromatography gave 0.442 g (76%) of the title compound.
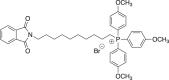 (2d) [10-(1,3-dioxo-2,3-dihydro-1H-isoindol-2-yl)decyl]tris(4-methoxyphenyl)phosphonium bromide
(2d) [10-(1,3-dioxo-2,3-dihydro-1H-isoindol-2-yl)decyl]tris(4-methoxyphenyl)phosphonium bromide
A mixture of 0.500 g (1.37 mmol) of N-(10-bromodecyl)phthalimide in CH3CN (4.2 mL) was refluxed with tris(4-methoxyphenyl)phosphine (Ambeed, cat # A354955) for 19 h. Flash chromatography gave 0.920 g (93%) of the title compound. 1H NMR (500 MHz, DMSO) δ 7.92–7.85 (m, 4H), 7.69 (dd, J = 10.3, 10.3 Hz, 6H), 7.32 (dd, J = 2.1, 8.8 Hz, 6H), 3.92 (s, 9H), 3.58 (t, J = 7.1 Hz, 2H), 1.59 (m, J = 7.1 Hz, 2H), 1.54–1.40 (m, 5H), 1.32–1.17 (m, 11H).
General Procedure-B for the preparation of compounds 4a-c
Compound 2a-c (1 equiv) was dissolved in EtOH (0.16 M, Decon Labs, inc. cat # UN1987) and placed under nitrogen. This was treated with hydrazine (1 equiv or 1.5 equiv, Sigma-Aldrich, cat # 21555). The reaction was then heated to reflux for 20 h. The reaction was then concentrated, and the byproduct was precipitated using EtOH/Et2O Sigma-Aldrich, cat # 673811) and removed by vacuum filtration to give crude intermediate 3a-d. The material was purified by silica column chromatography using amine-treated silica (0-20% v/v MeOH/DCM). The residue (1 equiv) was dissolved in n-BuOH (0.24 M, Sigma-Aldrich, cat # 360465) and placed under nitrogen. This was treated with N-[amino(methylsulfanyl)methylidene)guanidine hydroiodide46 (1.15 equiv). The reaction was heated to 130°C for 3 h and concentrated. The material was purified by silica column chromatography using amine-treated silica (1-20% v/v MeOH/DCM) to give the title compound.
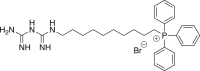 (4a, MMe) 10-[(carbamimidamidomethanimidoyl)amino]decyl-triphenylphosphonium bromide
(4a, MMe) 10-[(carbamimidamidomethanimidoyl)amino]decyl-triphenylphosphonium bromide
A mixture of 2a (1.5 g, 2.39 mmol) and hydrazine (0.074 mL, 2.39 mmol) was refluxed in ethanol (15 mL) for 20 h. Purification by silica column chromatography afforded the amine as an amorphous solid, which was carried forward without further manipulation. LRMS (+ESI) m/z [M−Br]+, Calculated for C28H37NP 418.3; Found 418.4. A mixture of 0.906 g (1.82 mmol) of (10-aminodecyl)triphenylphosphonium in n-BuOH (7.5 mL) was heated with N-[amino(methylsulfanyl)methylidene)guanidine hydroiodide (0.543 g, 2.09 mmol). Flash chromatography gave 0.367 g (35%) of the title compound as an amorphous solid. HRMS (+ESI) m/z: [M−Br]+ calculated for C30H41N5P 502.3094; Found 502.3094.
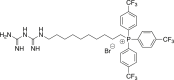 (4b, pCF3-MMe) (10-(3-carbamimidoylguanidino)decyl)tris(4-(trifluoromethyl)phenyl)phosphonium bromide
(4b, pCF3-MMe) (10-(3-carbamimidoylguanidino)decyl)tris(4-(trifluoromethyl)phenyl)phosphonium bromide
A mixture of 2b (2.59 g, 3.11 mmol) and hydrazine (0.145 mL, 4.66 mmol) was refluxed in ethanol (19 mL) for 20 h. Purification by silica column chromatography afforded the amine as an amorphous solid, which was carried forward without further manipulation. LRMS (+ESI) m/z [M−Br]+, Calculated for C31H34F9NP 622.2; Found 622.2. A mixture of 0.630 g (0.879 mmol) of (10-aminodecyl)tris[4-(trifluoromethyl)phenyl]phosphonium bromide in n-BuOH (3.7 mL) was heated with N-[amino(methylsulfanyl)methylidene)guanidine hydroiodide (0.268 g, 1.03 mmol). Flash chromatography gave 0.224 g (32%) of the title compound as an amorphous solid. 1H NMR (500 MHz, CD3OD) δ 8.06–7.73 (12H, m), 3.26–3.19 (2H, m), 3.12–3.05 (1H, m), 2.97–2.90 (4H, m), 2.51–2.40 (1H, m),1.55–1.29 (6H, m), 1.29–1.10 (10H, m). 19F NMR (470 MHz, CD3OD) δ −64.66. HRMS (+ESI) m/z: [M−Br]+ calculated for C33H38F9N5P 706.2716; Found 706.2722.
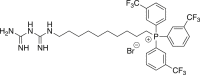 (4c, mCF3-MMe) (10-(3-carbamimidoylguanidino)decyl)tris(3-(trifluoromethyl)phenyl)phosphonium bromide
(4c, mCF3-MMe) (10-(3-carbamimidoylguanidino)decyl)tris(3-(trifluoromethyl)phenyl)phosphonium bromide
A mixture of 2c (0.4421 g, 0.531 mmol) and hydrazine (0.025 mL, 0.796 mmol) was refluxed in ethanol for 20 h. Purification by silica column chromatography afforded the amine as an amorphous solid, which was carried forward without further manipulation. LRMS (+ESI) m/z [M−Br]+, Calculated for C31H34F9NP 622.2; Found 622.2. A mixture of 0.069 g (0.098 mmol) of (10-aminodecyl)tris[3-(trifluoromethyl)phenyl]phosphonium bromide in n-BuOH (1 mL) was heated with N-[amino(methylsulfanyl)methylidene)guanidine hydroiodide (0.029 g, 0.113 mmol). Flash chromatography gave 0.006 g (7%) of the title compound as an amorphous solid. 1H NMR (500 MHz, DMSO-d6) δ 8.40–7.62 (6H, m), 6.71–6.53 (10H, m), 1.64–1.14 (16H, m). 19F NMR (470 MHz, CD3OD) δ −61.19. Significant 31P splitting complicated the proton spectrum. HRMS (+ESI) m/z: [M−Br]+ Calculated for C33H38F9N5P 706.2716; Found 706.2713.
 (4d, pMeO-MMe) [10-(2-carbamimidoylethanimidamido)decyl]tris(4-methoxyphenyl)phosphonium bromide
(4d, pMeO-MMe) [10-(2-carbamimidoylethanimidamido)decyl]tris(4-methoxyphenyl)phosphonium bromide
Compound 2days (0.920 g, 1.28 mmol) was dissolved in EtOH (8 mL) and placed under nitrogen. This was treated with hydrazine (0.040 mL, 1.28 mmol). The reaction was then heated to reflux for 20 h. The reaction was then concentrated, and the byproduct was precipitated using EtOH/Et2O and removed by vacuum filtration. The filtrate was concentrated in vacuo to give crude intermediate 3days. Intermediate 3d (0.150 g, 0.255 mmol) was dissolved in CH2Cl2 (2.5 mL) and cooled to 0°C in an ice bath. This was treated with hydrogen chloride (0.765 mL, 0.765 mmol, 1 M in Et2O, Sigma-Aldrich, cat # 294837) by dropwise addition. Once the addition was complete, the reaction was stirred at room temperature for 1 h. The reaction was then concentrated in vacuo. The resulting yellowish foam was dissolved in anhydrous DMF (1.0 mL) and treated with cyanoguanidine (0.096 g, 1.47 mmol, Sigma-Aldrich, cat #D76609). The reaction was then heated to 160°C for 2 h. The reaction was concentrated in vacuo. The material was purified by silica column chromatography using amine-treated silica (1-30% v/v MeOH/DCM) to give 0.114 g (67%) of the title compound. 1H NMR (500 MHz, CD3OD) δ 7.61–7.47 (6H, m), 7.21–7.08 (6H, m), 3.83 (9H, s), 3.26–3.19 (2H, m), 3.12–3.05 (3H, m), 3.02–2.91 (6H, m), 1.60–1.50 (2H, m),1.47–1.35 (4H, m), 1.30–1.11 (10H, m). HRMS (+ESI) m/z: [M−Br]+ calculated for C33H47N3O5 592.3411; Found 592.3409.
Statistical analyses
All statistical analyses were performed using GraphPad Prism (La Jolla, CA). Unpaired analyses were calculated using Student’s t test to identify pairwise differences between experimental and control groups. Values provided represent mean ± SD. Statistical significance was defined as p ≤ 0.05 and denoted with asterisks as indicated in the figure legends.
Acknowledgments
This work is supported in part by grants from the National Cancer Institute, R01 CA226279 (MBD) and R01 CA229907 (PC-R), the National Institute of General Medical Sciences R35 GM128840 (BCS), the Advancing a Healthier Wisconsin Endowment, and continuing philanthropic support from the Bobbie Nick Voss Charitable Foundation. Seahorse-XFe96-based analyses were performed in the MCW Cancer Center Redox & Bioenergetics Shared Resource. The content is solely the responsibility of the author(s) and does not necessarily represent the official views of the NIH.
Author contributions
Conceptualization: MAE, RFK, BCS, and MBD; formal analysis: MAE, RFK, DM, and FP; funding acquisition: BCS and MBD, investigation: MAE, RFK, and DM; methodology: MAE, RFK, BCS, and MBD; project administration: MBDResources: BCS, IPK, PC-R, FP, and MBD; supervision: BCS and MBD; validation: MAE, RFK, DM, and FP; visualization: MAE, RFK, DJS, DM, and MBD; writing – original draft: MAE; writing – review and editing: MAE, RFK, DJS, BCS, and MBD.
Declaration of interests
MBD and FP are co-founders and have ownership and financial interests in Protein Foundry, LLC and Xlock Biosciences, LLC. Chemical modifications of mitochondria-targeted compounds, including those included in this report, are covered by a provisional US Patent, U.S. Application No. 63/285,374, to MBD, BCS, MAE, and RK. The remaining authors have no competing interests to disclose.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as living with a disability.
Published: December 22, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105670.
Contributor Information
Brian C. Smith, Email: brismith@mcw.edu.
Michael B. Dwinell, Email: mdwinell@mcw.edu.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Smith R.A.J., Porteous C.M., Gane A.M., Murphy M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA. 2003;100:5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frattaruolo L., Brindisi M., Curcio R., Marra F., Dolce V., Cappello A.R. Targeting the mitochondrial metabolic network: a promising strategy in cancer treatment. Int. J. Mol. Sci. 2020;21:6014. doi: 10.3390/ijms21176014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rovini A., Heslop K., Hunt E.G., Morris M.E., Fang D., Gooz M., Gerencser A.A., Maldonado E.N. Quantitative analysis of mitochondrial membrane potential heterogeneity in unsynchronized and synchronized cancer cells. FASEB J. 2021;35:e21148. doi: 10.21096/fj.202001693R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Momcilovic M., Jones A., Bailey S.T., Waldmann C.M., Li R., Lee J.T., Abdelhady G., Gomez A., Holloway T., Schmid E., et al. In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature. 2019;575:380–384. doi: 10.1038/s41586-41019-41715-41580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armstrong J.S. Mitochondrial medicine: pharmacological targeting of mitochondria in disease. Br. J. Pharmacol. 2007;151:1154–1165. doi: 10.1038/sj.bjp.0707288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zong W.X., Rabinowitz J.D., White E. Mitochondria and cancer. Mol. Cell. 2016;61:667–676. doi: 10.1016/j.molcel.2016.1002.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pustylnikov S., Costabile F., Beghi S., Facciabene A. Targeting mitochondria in cancer: current concepts and immunotherapy approaches. Transl. Res. 2018;202:35–51. doi: 10.1016/j.trsl.2018.1007.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng G., Zielonka J., Dranka B.P., McAllister D., Mackinnon A.C., Jr., Joseph J., Kalyanaraman B. Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 2012;72:2634–2644. doi: 10.1158/0008-5472.CAN-11-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng G., Zielonka J., Ouari O., Lopez M., McAllister D., Boyle K., Barrios C.S., Weber J.J., Johnson B.D., Hardy M., et al. Mitochondria-targeted analogues of metformin exhibit enhanced antiproliferative and radiosensitizing effects in pancreatic cancer cells. Cancer Res. 2016;76:3904–3915. doi: 10.1158/0008-5472.CAN-15-2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng G., Zielonka J., Hardy M., Ouari O., Chitambar C.R., Dwinell M.B., Kalyanaraman B. Synergistic inhibition of tumor cell proliferation by metformin and mito-metformin in the presence of iron chelators. Oncotarget. 2019;10:3518–3532. doi: 10.18632/oncotarget.26943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyle K.A., Van Wickle J., Hill R.B., Marchese A., Kalyanaraman B., Dwinell M.B. Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation. J. Biol. Chem. 2018;293:14891–14904. doi: 10.1074/jbc.RA117.001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zielonka J., Joseph J., Sikora A., Hardy M., Ouari O., Vasquez-Vivar J., Cheng G., Lopez M., Kalyanaraman B. Mitochondria-targeted triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem. Rev. 2017;117:10043–10120. doi: 10.1021/acs.chemrev.7b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalyanaraman B., Cheng G., Hardy M., Ouari O., Lopez M., Joseph J., Zielonka J., Dwinell M.B. A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. Redox Biol. 2018;14:316–327. doi: 10.1016/j.redox.2017.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy J.W., Cho Y., Sachpatzidis A., Fan C., Hodsdon M.E., Lolis E. Structural and functional basis of CXCL12 (stromal cell-derived factor-1 alpha) binding to heparin. J. Biol. Chem. 2007;282:10018–10027. doi: 10.1074/jbc.M608796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ross M.F., Prime T.A., Abakumova I., James A.M., Porteous C.M., Smith R.A.J., Murphy M.P. Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochem. J. 2008;411:633–645. doi: 10.1042/BJ20080063. [DOI] [PubMed] [Google Scholar]
- 16.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Murphy M.P. Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol. 1997;15:326–330. doi: 10.1016/S0167-7799(1097)01068-01068. [DOI] [PubMed] [Google Scholar]
- 18.Jauslin M.L., Meier T., Smith R.A.J., Murphy M.P. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003;17:1972–1974. doi: 10.1096/fj.1903-0240fje. [DOI] [PubMed] [Google Scholar]
- 19.Cheng G., Hardy M., Topchyan P., Zander R., Volberding P., Cui W., Kalyanaraman B. Potent inhibition of tumour cell proliferation and immunoregulatory function by mitochondria-targeted atovaquone. Sci. Rep. 2020;10:17872. doi: 10.11038/s41598-17020-74808-17870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.AbuEid M., McAllister D.M., McOlash L., Harwig M.C., Cheng G., Drouillard D., Boyle K.A., Hardy M., Zielonka J., Johnson B.D., et al. Synchronous effects of targeted mitochondrial complex I inhibitors on tumor and immune cells abrogate melanoma progression. iScience. 2021;24:102653. doi: 10.1016/j.isci.2021.102653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kulkarni C.A., Fink B.D., Gibbs B.E., Chheda P.R., Wu M., Sivitz W.I., Kerns R.J. A novel triphenylphosphonium carrier to target mitochondria without uncoupling oxidative phosphorylation. J. Med. Chem. 2021;64:662–676. doi: 10.1021/acs.jmedchem.1020c01671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meanwell N.A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 2018;61:5822–5880. doi: 10.1021/acs.jmedchem.5827b01788. [DOI] [PubMed] [Google Scholar]
- 23.Alvarez S. A cartography of the van der Waals territories. Dalton Trans. 2013;42:8617–8636. doi: 10.1039/c3dt50599e. [DOI] [PubMed] [Google Scholar]
- 24.Inoue M., Sumii Y., Shibata N. Contribution of organofluorine compounds to pharmaceuticals. ACS Omega. 2020;5:10633–10640. doi: 10.11021/acsomega.10630c00830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson D.F. Oxidative phosphorylation: regulation and role in cellular and tissue metabolism. J. Physiol. 2017;595:7023–7038. doi: 10.1113/JP273839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plitzko B., Loesgen S. Measurement of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) in culture cells for assessment of the energy metabolism. Bio. Protoc. 2018;8:e2850. doi: 10.21769/BioProtoc.22850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ehinger J.K., Piel S., Ford R., Karlsson M., Sjövall F., Frostner E.Å., Morota S., Taylor R.W., Turnbull D.M., Cornell C., et al. Cell-permeable succinate prodrugs bypass mitochondrial complex I deficiency. Nat. Commun. 2016;7:12317. doi: 10.1038/ncomms12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piel S., Ehinger J.K., Chamkha I., Frostner E.Å., Sjövall F., Elmér E., Hansson M.J. Bioenergetic bypass using cell-permeable succinate, but not methylene blue, attenuates metformin-induced lactate production. Intensive Care Med. Exp. 2018;6:22. doi: 10.1186/s40635-40018-40186-40631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Callahan J., Kopecek J. Semitelechelic HPMA copolymers functionalized with triphenylphosphonium as drug carriers for membrane transduction and mitochondrial localization. Biomacromolecules. 2006;7:2347–2356. doi: 10.1021/bm060336m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelso G.F., Porteous C.M., Coulter C.V., Hughes G., Porteous W.K., Ledgerwood E.C., Smith R.A., Murphy M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 31.Scaduto R.C., Jr., Grotyohann L.W. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J. 1999;76:469–477. doi: 10.1016/S0006-3495(1099)77214-77210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernas T., Dobrucki J. Mitochondrial and nonmitochondrial reduction of MTT: interaction of MTT with TMRE, JC-1, and NAO mitochondrial fluorescent probes. Cytometry. 2002;47:236–242. doi: 10.1002/cyto.10080. [DOI] [PubMed] [Google Scholar]
- 33.Seyfried T.N., Arismendi-Morillo G., Mukherjee P., Chinopoulos C. On the origin of ATP synthesis in cancer. iScience. 2020;23:101761. doi: 10.101016/j.isci.102020.101761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herzig S., Shaw R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018;19:121–135. doi: 10.1038/nrm.2017.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thuronyi B.W., Privalsky T.M., Chang M.C.Y. Engineered fluorine metabolism and fluoropolymer production in living cells. Angew. Chem. Int. Ed. 2017;56:13637–13640. doi: 10.11002/anie.201706696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buskiewicz I.A., Montgomery T., Yasewicz E.C., Huber S.A., Murphy M.P., Hartley R.C., Kelly R., Crow M.K., Perl A., Budd R.C., Koenig A. Reactive oxygen species induce virus-independent MAVS oligomerization in systemic lupus erythematosus. Sci. Signal. 2016;9:ra115. doi: 10.1126/scisignal.aaf1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng G., Zielonka J., McAllister D., Hardy M., Ouari O., Joseph J., Dwinell M.B., Kalyanaraman B. Antiproliferative effects of mitochondria-targeted cationic antioxidants and analogs: role of mitochondrial bioenergetics and energy-sensing mechanism. Cancer Lett. 2015;365:96–106. doi: 10.1016/j.canlet.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng G., Hardy M., Zielonka J., Weh K., Zielonka M., Boyle K.A., Abu Eid M., McAllister D., Bennett B., Kresty L.A., et al. Mitochondria-targeted magnolol inhibits OXPHOS, proliferation, and tumor growth via modulation of energetics and autophagy in melanoma cells. Cancer Treat. Res. Commun. 2020;25:100210. doi: 10.1016/j.ctarc.2020.100210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng G., Hardy M., Topchyan P., Zander R., Volberding P., Cui W., Kalyanaraman B. Mitochondria-targeted hydroxyurea inhibits OXPHOS and induces antiproliferative and immunomodulatory effects. iScience. 2021;24:102673. doi: 10.101016/j.isci.102021.102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodríguez-Gascón A., Solinís M.Á., Isla A. The role of PK/PD analysis in the development and evaluation of antimicrobials. Pharmaceutics. 2021;13:833. doi: 10.3390/pharmaceutics13060833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCreight L.J., Bailey C.J., Pearson E.R. Metformin and the gastrointestinal tract. Diabetologia. 2016;59:426–435. doi: 10.1007/s00125-00015-03844-00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Graham G.G., Punt J., Arora M., Day R.O., Doogue M.P., Duong J.K., Furlong T.J., Greenfield J.R., Greenup L.C., Kirkpatrick C.M., et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011;50:81–98. doi: 10.2165/11534750-000000000-000000000. [DOI] [PubMed] [Google Scholar]
- 43.Smith R.A.J., Murphy M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- 44.Hwang R.F., Moore T., Arumugam T., Ramachandran V., Amos K.D., Rivera A., Ji B., Evans D.B., Logsdon C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herrera D., Peral D., Bayón J.C. Preparation of carboxylic arylphosphines by hydrolysis of the trifluoromethyl group. RSC Adv. 2022;12:7103–7114. doi: 10.1039/d7102ra00420h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fortun S., Schmitzer A.R. Synthesis and characterization of biguanide and biguanidium surfactants for efficient and recyclable application in the suzuki-miyaura reaction. ACS Omega. 2018;3:1889–1896. doi: 10.1021/acsomega.1887b01962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.