

Abstract

An 11-step, 8-pot synthesis of the antimalarial drug tafenoquine succinate was achieved in 42% overall yield using commercially available starting materials. Compared to the previous manufacturing processes that utilize environmentally egregious organic solvents and toxic reagents, the current route features a far greener (as measured by Sheldon’s E Factors) and likely more economically attractive sequence, potentially expanding the availability of this important drug worldwide.
Keywords: malaria, antimalarial drug, tafenoquine, SNAr reaction, nitro reduction, reductive amination, sustainability
Short abstract
The potent, Food and Drug Administration-approved antimalarial drug tafenoquine has been made using a streamlined, efficient, and environmentally responsible route.
Introduction
Malaria is a life-threatening mosquito-borne parasitic disease responsible for an estimated 627,000 deaths globally in 2020.1 Many effective antimalarials have been developed (e.g., quinine, chloroquine, mefloquine, primaquine, and artemisinin-based combination therapies);2−4 however, their intensive use has led to the emergence of resistant Plasmodium strains as well as toxicological concerns. In response, a new antimalarial drug, tafenoquine (1), has been developed (Figure 1). Tafenoquine was recently approved by the US Food and Drug Administration as the first new single-dose treatment for Plasmodium vivax malaria in over 60 years.5,6 It is currently sold as the racemic succinate salt (2), under the names Krintafel (tablets of 150 mg) by GlaxoSmithKline (GSK)7 as well as Arakoda and Kodatef (tablets of 100 mg) by 60 Degrees Pharmaceuticals LLC.8 In comparison to previous generations of antimalarials, tafenoquine is considerably less toxic, has a longer plasma life (2–3 weeks),9 and is 10 times more potent.10−12 These combined features allow for single-dose treatment, compared to, e.g., the standard 14-day course of treatment associated with primaquine. Two synthetic routes to tafenoquine have previously been disclosed. The first involved 16 steps, ultimately affording the final drug in low overall yield (0.8%).13 An improved route reported by GSK involves 11 steps, leading to 2 in 14% overall yield.14−16 The key limitations of these routes include excess use of organic solvents along with toxic reagents (arsenic pentoxide)13 and low-yielding overall syntheses.14,15 Furthermore, recent increases in the stringency of environmental regulations are forcing pharmaceutical companies to pursue more sustainable processes.17−22 Therefore, there exists an urgent need for the development of both a green and economically attractive synthesis of tafenoquine.
Figure 1.

Structures of tafenoquine and its succinate salt.
In continuation of our group efforts to develop scalable routes to active pharmaceutical ingredients under cost-effective and environmentally friendly conditions,23−26 and in an ongoing collaboration with the Bill and Melinda Gates Foundation focused to date on pyronaridine (an antimalarial drug)24 and nirmatrelvir (the key ingredient in Pfizer’s Paxlovid for treatment of COVID-19),23 we now describe an environmentally responsible route to tafenoquine that simultaneously addresses these issues while maximizing both time and pot economies (Scheme 1).27,28 This has been accomplished by taking advantage of neat reactions following the Sheldon philosophy that “the best solvent is no solvent...”,29−36 and multistep, one-pot processes using environmentally preferred solvents.37,38
Scheme 1. Overall Sequence to Tafenoquine Succinate (2).
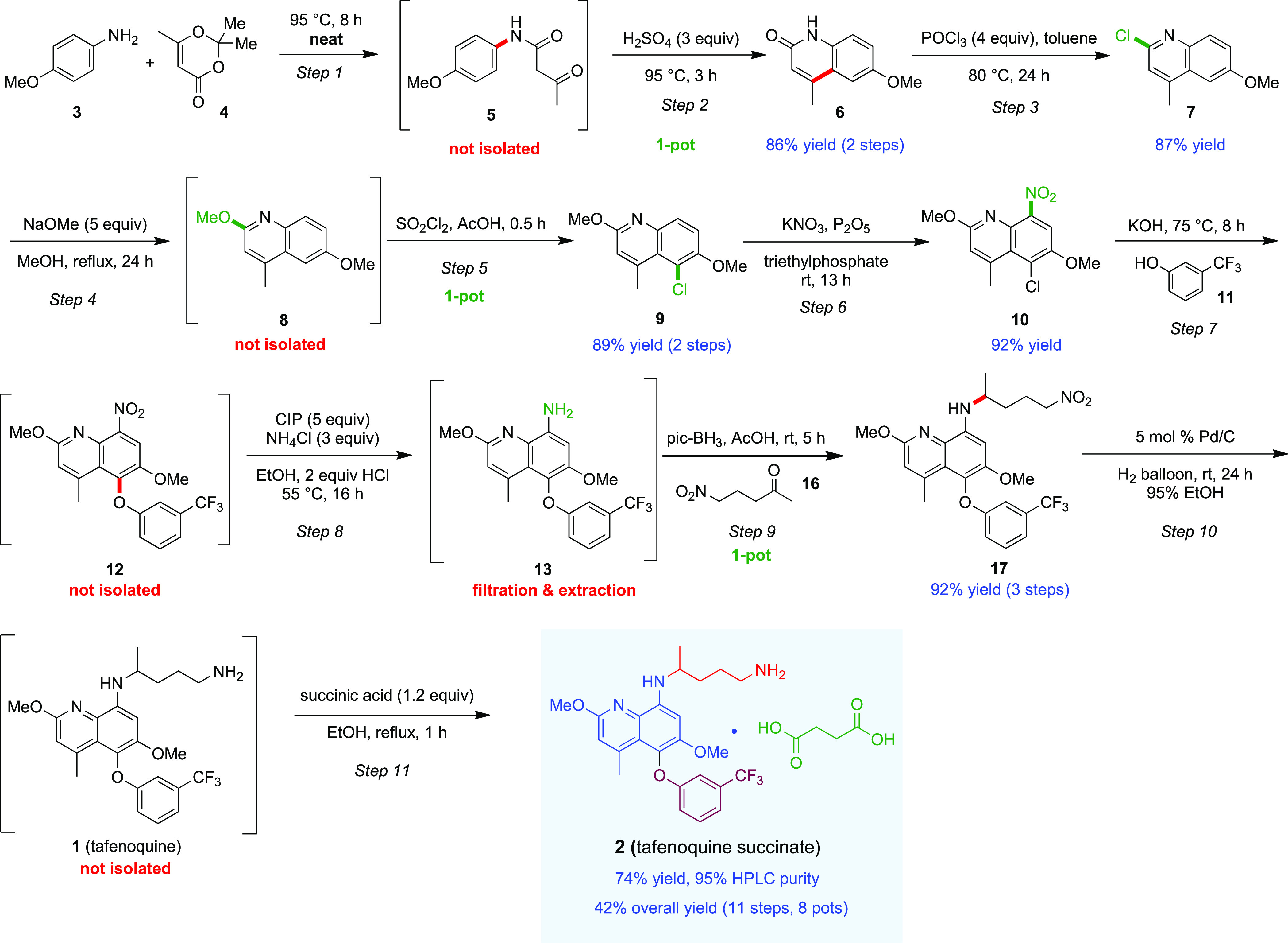
Results and Discussion
Two-Step, One-Pot Sequence En Route to Intermediate 6
Scheme 1 illustrates the synthesis of intermediate lactam 6. The first step, an amidation between 3 and methyl acetoacetate 4a (Table 1), was performed using an aqueous solution of 2 wt % TPGS-750-M/H2O39 at a global concentration of 0.5 m to form 5 in 75% yield, along with the undesired side product 5a (15% yield, entry 7) and unreacted starting material. In an effort to suppress side product formation and increase yield, the addition of an acid, such as BiBr3, HCl, and H2SO4, was screened. Unfortunately, none improved the reaction profile (entries 1–6). Scaling the best reaction conditions to 20 mmol led to only 41% isolated yield (following column chromatography) due to formation of byproduct 5a. This vinylogous carbamate likely forms as a result of a more rapid reaction of aniline 3 toward condensation with the keto group in methyl acetoacetate, rather than the desired reaction with the ester moiety.
Table 1. Optimization of Reaction Conditions to 5a.

To avoid this competing condensation, use of 2,2,6-trimethyl-4H-1,3-dioxin-4-one (4, TMD) was investigated, previously used by Clemens40,41 as an alternative coupling partner and shown to afford the same desired product. TMD is a stable equivalent of diketene which can be used to generate an acetylketene at higher temperatures (>82 °C)42 via a pseudo-retro-Diels-Alder reaction, eliminating acetone as the only byproduct. The resulting acetylketene intermediate can then be trapped by a nucleophile. The reaction between 3 and TMD (4) in 2 wt % TPGS-750-M/H2O at 85 °C over 12 h afforded the desired product in 87% isolated yield (Table 2, entry 1). Further increasing the reaction time to 24 h led to an improved yield of 95% (entry 2), whereas the reaction in the absence of this surfactant (i.e., “on water”) at reflux afforded product aniline 5 in a slightly lower yield (91%, entry 3). The latter result suggests that at higher temperatures the surfactant is not required to form the desired product 5. Running the reaction neat led to an 89% isolated yield (entry 5). Despite this slightly lower yield at this smaller scale, neat conditions were taken as optimal as they allowed for telescoping using 5 in the next step without its unwanted hydrolysis (vide infra). Given the apparent benefits of the micellar medium, further investigation into the role of other surfactants (e.g., PS-750-M)43 in this chemistry is of future interest in our group.
Table 2. Optimization of Modified Reaction Conditions En Route to 5a.

Reaction conditions: 1 mmol 1, 0.5 M.
Isolated yield.
Subsequent Knorr quinoline synthesis44−47 was employed in acidic media to convert the β-ketoanilide 5, without isolation, to the desired 2-hydroxyquinoline 6. Initially, the reaction was conducted in aqueous surfactant solution using three equiv. of conc. H2SO4, leading to quantitative formation of the undesired byproduct p-anisidine 3 (Table 3, entry 3). This outcome results from competitive hydrolysis of the amide bond in educt 5, thereby indicating that an aqueous medium was incompatible with this transformation.
Table 3. Optimization of Modified Reaction Conditions En Route to 6a.

Under neat conditions, however, the use of three equiv. of conc. H2SO4 (0.3 M) at 95 °C afforded the desired product in 88% yield, along with traces (<5%) of demethylated side product 6a (entry 5 and Figure 2). With the optimized stepwise synthesis of 6 in hand, a two-step, one-pot operation could then be devised to minimize handling (e.g., purification of initial product 6). As expected, the sequence involving initial amidation to afford 5, followed, in the same pot, by Knorr cyclization, smoothly afforded 6 in 86% isolated yield (see Scheme 1). It is interesting to note that the use of H3PO4 affords 6 in 89% isolated yield without any trace of byproduct 6a or 3 (Table 3, entry 1). However, scaling this reaction to 5 mmol led to the formation of only trace amounts of product, with the rest being unreacted starting material. Ultimately, due to the significantly lower cost of H2SO4 on a large scale compared to H3PO4, the use of this acid was not investigated further.
Figure 2.

Impurities observed at various stages of the route to tafenoquine.
Conversion of Intermediate 6 to 7
Deoxychlorination of intermediate 6 to form 2-chloroquinoline 7 was achieved using POCl3 (Scheme 1). Although water would not be tolerated in this step, toluene served nicely and could be recovered following isolation of product 7 (87% yield), thereby minimizing generation of organic waste. It was anticipated that generation of HCl during the reaction would lead to demethylation of the methoxy group, and thus triethylamine was initially included in the reaction mixture.48 However, omitting the base led to the same reaction outcome. Product 7 was purified by silica gel column chromatography to remove a brown impurity before proceeding to the next step. Alternatively, this material can be purified via recrystallization from EtOH.49
Two-Step, One-Pot SNAr/Chlorination Sequence En Route to Intermediate 9
The SNAr reaction between 7 and excess anhydrous sodium methoxide (5 equiv) in methanol under refluxing conditions led to product 8 in nearly quantitative yield (Scheme 1; specifically, see SI, section 3.5, Table S2, entry 5). Reducing the loading of sodium methoxide, e.g., from five to three equivalents led to a significant drop in conversion (to 53% yield; see SI, Section 3.5, Table S2). Chlorination at C-5 of quinoline 8 was effected with sulfuryl chloride in acetic acid at 60 °C for 30 min to afford the desired product 9 in 94% yield (see SI, Section 3.6, Table S3, entry 5). It should be noted that longer reaction times led to the formation of impurity 9a (Figure 2). This impurity was produced via intermediate I (as shown in Figure 2), formed as a result of demethylation of the methoxy group in 8 under acidic conditions, followed by chlorination of intermediate I.
Modification of the workup associated with the initial SNAr reaction allowed for direct conversion of 7 to 9 in a two-step, one-pot fashion. Thus, following the optimized SNAr protocol (vide supra), excess NaOMe was quenched using four equivalents of AcOH, after which the reaction was concentrated to dryness to remove all traces of MeOH that might interfere with the subsequent chlorination step. Acetic acid (as a solvent) was then added, and chlorination was carried out as described above to afford 9 in 89% yield over both steps (in one-pot).
Nitration of Intermediate 9 to Afford 10
Various reagents were investigated for the nitration of 9 to arrive at nitroarene 10 (see SI, Section 3.7, Table S4). It was eventually found that in situ generation of N2O5 via dehydration of KNO3 (2 equiv) with P2O5 (4 equiv) in triethylphosphate (as a solvent) was optimal,14 leading to 10 in 92% isolated yield. The product was isolated simply by neutralizing the reaction mixture with aqueous NaHCO3 and collecting the resulting precipitate via filtration. Largely due to issues of solubility, very low yields (<10%) were obtained when other solvents (e.g., MeOH, CH3CN, sulfolane, 2-MeTHF, and DMSO) were used instead of triethylphosphate. Attempts to employ conventional nitrating conditions involving, e.g., HNO3 in H2SO4 led to rapid demethylation of the methoxy groups leading to impurity 10a (Figure 2), as did the use of nitronium tetrafluoroborate (NO2BF4).
Three-Step, One-Pot Sequence En Route to Intermediate 17
The SNAr reaction between nitroarene intermediate 10 and 3-(trifluoromethyl)phenol 11 to afford intermediate biaryl ether 12 was screened in both an aqueous surfactant medium and organic solvents. No conversion to the desired product was observed under aqueous conditions, and of the organic solvents tested, DMSO led to the highest yield 76% (see SI Section 3.8, Table S5, entry 4). Unfortunately, incomplete conversion under these conditions led to isolation problems, as 12 could not be easily separated from the starting material 10 via either column chromatography or recrystallization owing to close Rf values and poor solubility in several solvents. To obtain full consumption of starting material and facilitate product purification, use of neat conditions proved to be ideal (using 2 equiv 11 and equimolar base at 75 °C for 8 h), leading to 12 in 92% isolated yield (see SI, Section 3.8, Table S5, entry 9). The resulting product was of sufficiently high quality such that no further purification was needed at this stage. Nitro group reduction of intermediate 12 (without its isolation) to give aniline 13 (Scheme 1) was performed using two different protocols: (1) conventional Pd/C under H2 pressure, or (2) using carbonyl iron powder (CIP) that, as shown previously,50 smoothly reduces nitro groups in water. Under aqueous surfactant conditions, both Pd/C and CIP gave moderate yields of the desired product. These yields could be improved to 94% and 97%, respectively, by switching the medium to 95% EtOH (see SI, section 3.9, Table S6, entries 2, 4). The addition of one equivalent conc. HCl was necessary to achieve these results. Interestingly, a mixture of both types of media, such as 2 wt % TPGS-750-M/H2O and EtOH, led to only trace amounts of product formation.
An initial attempt at reductive amination between aniline 13 and ketone 14 (Table 4) was made by using the previously established protocol applied to the synthesis of Takeda’s drug TAK-954.22 These conditions called for an aqueous solution containing 2 wt % TPGS-750-M/H2O, together with MeOH (10 v/v %) as a cosolvent, and α-picoline borane as a hydride source (1.5 equiv). Under these conditions, the desired product was not formed; rather, only the starting material was fully recovered (Table 4, entry 2). Switching the cosolvent from MeOH to acetic acid led to a 54% yield (Table 4, entry 3). This suggested that AcOH was essential, presumably shifting the equilibrium toward desired product formation. Replacing the aqueous surfactant mixture by AcOH led to full conversion at rt to targeted product 15 in 91% isolated yield (after purification by column chromatography; entry 4). Although this reductive amination sequence is quite efficient, the approach suffers from inherent disadvantages, specifically with regard to the deprotection of the phthalimide group requiring hydrazinolysis, as well as separation of the phthalhydrazide byproduct that must be treated as waste. Hence, an alternative coupling partner 16 (i.e., 5-nitro-2-pentanone; Scheme 1) was selected for this final sequence, readily prepared via Michael addition of nitromethane to methyl vinyl ketone in the presence of catalytic amounts of sodium hydroxide.51 The reductive amination conditions initially optimized for the synthesis of intermediate 15 could be applied to the reaction between aniline 13 and ketone 16 to afford 17 in 90% yield (Table 4, entry 8) without further optimization. Once optimized conditions associated with each step had been determined, a three-step, one-pot synthesis was developed starting with 10 and ultimately affording 17 in 92% overall isolated yield, following recrystallization from ethanol. No column chromatography was needed at any stage for purification.
Table 4. Optimization of Reductive Amination Conditions to Arrive at 15a.

Two-Step, Tandem Sequence En Route to Tafenoquine Succinate (2)
Tafenoquine (1) was obtained by reduction in 95% EtOH of the nitro group-containing biaryl ether intermediate 17 using 5 mol % Pd/C under H2 pressure at rt for 24 h. Upon completion, the reaction mixture was filtered through a short plug of Celite to remove Pd/C. Tafenoquine succinate (2) was then formed by addition of succinic acid in EtOH. The precipitated amine succinate salt 2 was collected via filtration and obtained in 74% yield and 95% purity (by HPLC) over two steps (42% overall yield, 11 steps in 8 pots). Additional purification can be accomplished via recrystallization from EtOH, as described by GSK.15
Alternative Gabriel Amine Synthesis Approach to Tafenoquine Succinate (2)
Although hydrazinolysis of phthalimide intermediate 15 in hot EtOH was found to be somewhat lower yielding and less attractive (vide supra and Scheme 2), it could be used to arrive at tafenoquine (1) in 73% yield following aqueous workup. This crude material was then converted to its succinic acid salt (2) in MeCN/MeOH. Collection of the precipitated product via filtration afforded 2 in 66% yield and >99% purity by HPLC (26% overall yield, 11 steps in 8 pots; ca. 42% using the nitro reduction route, vide supra).
Scheme 2. Alternative Route to Tafenoquine Succinate 2 from Phthalimide 15.

E Factor Determination
To compare the environmental footprint associated with our optimized route to 17 versus that by GSK,14,15 a complete E factor (cEF) was determined following the Roschangar procedure,52 as one measure of “greenness”, calculated as the ratio of the mass of waste generated to the mass of product. This was evaluated for the three-step tandem sequence leading to compound 17, starting with 5-chloro-2,6-dimethoxy-4-methyl-8-nitroquinoline 10 (Scheme 1).
The results are indicative of a 3-fold decrease in waste creation, leading to a very low value of E = 17 (including aqueous waste streams (see SI, section 6) exemplifying the environmental friendliness of the described sequence. By contrast, the calculated cEF of E = 69 for the GSK sequence was characteristic of many environmentally egregious pharmaceutical processes that tend to have associated E factors between 25 and 100.53 Direct comparisons between the major reaction parameters between the GSK route14,15 and the current, far greener synthesis of tafenoquine are summarized in Table 5.
Table 5. Comparisons between GSK and This Route to Tafenoquine Succinate (2).
| reaction parameter | GSK14,15 | this work |
|---|---|---|
| amide bond formation (step 1) | solvent: xylene | solvent: none |
| reaction temperature: reflux | reaction temperature: 95 °C | |
| reagent: ethylacetoacetate, triethanolamine | reagent: TMD | |
| deoxychlorination (step 3) | POCl3 as a solvent | recoverable toluene minimal amount of POCl3 |
| SNAr reaction (step 7) | DMSO, reaction temperature: 100 °C product contains black tar impurity, requires activated carbon treatment and precipitation by toluene/hexane | neat, reaction temperature: 75 °C, no post purification |
| E factor for 10 to 17 | 69 | 17 |
| nitro reduction (step 8) | Pd/C | carbonyl iron powder (CIP) |
| overall yield | 14% | 42% |
Conclusions
In summary, an alternative synthetic route to the antimalarial drug tafenoquine relative to those currently known has been provided resulting in a more efficient and far greener process. This combination may significantly reduce both the cost and environmental footprint associated with this especially effective drug, potentially increasing its availability to those in need throughout the world.
Acknowledgments
Insight and guidance provided by both BMGF consultants John Dillon and Trevor Laird throughout this project are very much appreciated. Initial studies by Dr. Balaram Takale are warmly acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssuschemeng.2c05628.
Experimental procedures; analytical data for all new compounds; and NMR spectra of the products (PDF)
Financial support provided by the Bill & Melinda Gates Foundation (BMGF; INV-005858) is gratefully acknowledged with thanks. A pre-doctoral award from the National Science Foundation Graduate Research Fellowship Program is also warmly acknowledged (Grant No. 1650114 to J.R.A.K.).
The authors declare no competing financial interest.
Supplementary Material
References
- https://www
- Camille T.; Dassonville-Klimpt A.; Gosselet F.; Sonnet P. Antimalarial Drug Discovery: From Quinine to the Most Recent Promising Clinical Drug Candidates. Curr. Med. Chem. 2022, 29, 3326–3365. 10.2174/0929867328666210803152419. [DOI] [PubMed] [Google Scholar]
- Vijayan K.; Wei L.; Glennon E. K. K.; Mattocks C.; Bourgeois N.; Staker B.; Kaushansky A. Host-targeted Interventions as an Exciting Opportunity to Combat Malaria. Chem. Rev. 2021, 121, 10452–10468. 10.1021/acs.chemrev.1c00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K. Y.; Derbyshire E. R. Tafenoquine: A Step toward Malaria Elimination. Biochemistry 2020, 59, 911–920. 10.1021/acs.biochem.9b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters W. The evolution of tafenoquine-antimalarial for a new millennium?. J. R. Soc. Med. 1999, 92, 345–352. 10.1177/014107689909200705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hounkpatin A. B.; Kreidenweiss A.; Held J. Clinical utility of tafenoquine in the prevention of relapse of Plasmodium vivax malaria: a review on the mode of action and emerging trial data. Infect. Drug Resist. 2019, 12, 553–570. 10.2147/IDR.S151031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210795s000lbl.pdf
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210607lbl.pdf
- Brueckner R. P.; Lasseter K. C.; Lin E. T.; Schuster B. G. First-time-in-humans safety and pharmacokinetics of WR 238605, a new antimalarial. Am. J. Trop. Med. Hyg. 1998, 58, 645–649. 10.4269/ajtmh.1998.58.645. [DOI] [PubMed] [Google Scholar]
- Puri S. K.; Dutta G. P. Blood schizontocidal activity of WR 238605 (Tafenoquine) against Plasmodium cynomolgi and Plasmodium fragile infections in rhesus monkeys. Acta Trop. 2003, 86, 35–40. 10.1016/S0001-706X(02)00289-9. [DOI] [PubMed] [Google Scholar]
- Practical Chemotherapy of Malaria; World Health Organization Technical Report Series no. 805; World Health Organization: Geneva, 1990; p 128. [PubMed] [Google Scholar]
- Davidson D. E.; Ager A. L.; Brown J. L.; Chapple F. E.; Whitmire R. E.; Rossan R. N. New tissue schizontocidal antimalarial drugs. Bull. W. H. O. 1981, 59, 463–479. [PMC free article] [PubMed] [Google Scholar]
- LaMontagne M. P.; Blumbergs P.; Smith D. C. Antimalarials. 16. Synthesis of 2-Substituted Analogues of 8-[(4-Amino-l-methylbutyl)amino]-6-methoxy-4-methyl-5-[3-(trifluoromethyl)phenoxyjquinoline] as Candidate Antimalarials. J. Med. Chem. 1989, 32, 1728–1732. 10.1021/jm00128a010. [DOI] [PubMed] [Google Scholar]
- Ugwuegbulam C. O.; Foy J. E.. Process for the Preparation of Anti-malarial Drugs. WO1997013753A1.
- Bell D.; Davies J. B.; Kincey P. M.. Process for the preparation of quinoline derivatives. WO03/093239A2.
- For a detailed scheme of GSK route to synthesis of tafenoquine, see:; Zhu W.; Wang J.; Wang S.; Gu Z.; Aceña J. L.; Izawa K.; Liu H.; Soloshonok V. A Recent advances in the trifluoromethylation methodology and new CF3-containing drugs. J. Fluorine Chem. 2014, 167, 37–54. 10.1016/j.jfluchem.2014.06.026. [DOI] [Google Scholar]
- Summary of the Pollution Prevention Act Available online: http://www.epa.gov/laws-regulations/summary-pollution-prevention-act (accessed on Oct 19, 2022).
- https://echa.europa.eu/regulations/reach/understanding-reach
- Candeias N. R.; Branco L. S. C.; Gois P. M. P.; Afonso C. A. M.; Trindade A. F. More Sustainable Approaches for the Synthesis of N-Based Heterocycles. Chem. Rev. 2009, 109, 2703–2802. 10.1021/cr800462w. [DOI] [PubMed] [Google Scholar]
- Sheldon R. A. Green chemistry and resource efficiency: towards a green economy. Green Chem. 2016, 18, 3180–3183. 10.1039/C6GC90040B. [DOI] [Google Scholar]
- Becker J.; Manske C.; Randl S. Green chemistry and sustainability metrics in the pharmaceutical manufacturing sector. Curr. Opin. Green Sustainable Chem. 2022, 33, 100562 10.1016/j.cogsc.2021.100562. [DOI] [Google Scholar]
- Bailey J. D.; Helbling E.; Mankar A.; Stirling M.; Hicks F.; Leahy D. K. Beyond organic solvents: synthesis of a 5-HT4 receptor agonist in water. Green Chem. 2021, 23, 778–795. 10.1039/D0GC03316B. [DOI] [Google Scholar]
- Kincaid J. R. A.; Caravez J. C.; Iyer K. S.; Kavthe R. D.; Fleck N.; Aue D. H.; Lipshutz B. H. An Environmentally Responsible Synthesis of the SARS-CoV-2 Mpro Inhibitor Nirmatrelvir (PF-07321332), the Active Ingredient in Paxlovid. ChemRxiv 2022. This content is a preprint. Commun. Chem., 10.1038/242004-022-00758-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kincaid J. R. A.; Kavthe R. D.; Caravez J. C.; Takale B. S.; Thakore R. R.; Lipshutz B. H. Environmentally Responsible and Cost-Effective Synthesis of the Antimalarial Drug Pyronaridine. Org. Lett. 2022, 24, 3342–3346. 10.1021/acs.orglett.2c00944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.; Iyer K. S.; Lipshutz B. H. An environmentally responsible synthesis of the antitumor agent lapatinib (Tykerb). Green Chem. 2022, 24, 3640–3643. 10.1039/D2GC00598K. [DOI] [Google Scholar]
- Takale B. S.; Thakore R. R.; Kong F. Y.; Lipshutz B. H. An environmentally responsible 3-pot, 5-step synthesis of the antitumor agent sonidegib using ppm levels of Pd catalysis in water. Green Chem. 2019, 21, 6258–6262. 10.1039/C9GC03495A. [DOI] [Google Scholar]
- Hayashi Y. J. Time Economy in Total Synthesis. J. Org. Chem. 2021, 86, 1–23. 10.1021/acs.joc.0c01581. [DOI] [PubMed] [Google Scholar]
- Hayashi Y. Pot economy and one-pot synthesis. Chem. Sci. 2016, 7, 866–880. 10.1039/C5SC02913A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon R. A.; Arends I. W. C. E.; Hanefeld U.. Green Chemistry and Catalysis; Wiley-VCH: Weinheim, 2007; 448pp; ISBN 978-3-527-30715-9. [Google Scholar]
- Sarkar A.; Santra S.; Kundu S. K.; Hajra A.; Zyryanov G. V.; Chupakhin O. N.; Charushin V. N.; Majee A. A decade update on solvent and catalyst-free neat organic reactions: a step forward towards sustainability. Green Chem. 2016, 18, 4475–4525. 10.1039/C6GC01279E. [DOI] [Google Scholar]
- Tanaka K.; Toda F. Solvent-Free Organic Synthesis. Chem. Rev. 2000, 100, 1025–1074. 10.1021/cr940089p. [DOI] [PubMed] [Google Scholar]
- Garay A. L.; Pichon A.; James S. L. Solvent-free synthesis of metal complexes. Chem. Soc. Rev. 2007, 36, 846. 10.1039/b600363j. [DOI] [PubMed] [Google Scholar]
- Martins M. A. P.; Frizzo C. P.; Moreira D. N.; Buriol L.; Machado P. Solvent-Free Heterocyclic Synthesis. Chem. Rev. 2009, 109, 4140–4182. 10.1021/cr9001098. [DOI] [PubMed] [Google Scholar]
- Varma R. S. Solvent-free organic syntheses using supported reagents and microwave irradiation. Green Chem. 1999, 1, 43–55. 10.1039/a808223e. [DOI] [Google Scholar]
- Walsh P. J.; Li H.; de Parrodi C. A. A Green Chemistry Approach to Asymmetric Catalysis: Solvent-Free and Highly Concentrated Reactions. Chem. Rev. 2007, 107, 2503–2545. 10.1021/cr0509556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainath Z.; Pravinkumar P. A Review on Solvent-free Methods in Organic Synthesis. Curr. Org. Chem. 2019, 23, 2295–2318. 10.2174/1385272823666191016165532. [DOI] [Google Scholar]
- Byrne F. P.; Jin S.; Paggiola G.; Petchey T. H. M.; Clark J. H.; Farmer T. J.; Hunt A. J.; McElroy C. R.; Sherwood J. Tools and techniques for solvent selection: green solvent selection guides. Sustainable Chem. Processes 2016, 4, 7. 10.1186/s40508-016-0051-z. [DOI] [Google Scholar]
- Prat D.; Hayler J.; Wells A. A survey of solvent selection guides. Green Chem. 2014, 16, 4546–4551. 10.1039/C4GC01149J. [DOI] [Google Scholar]
- Lipshutz B. H.; Ghorai S.; Abela A. R.; Moser R.; Nishikata T.; Duplais C.; Krasovskiy A.; Gaston R. D.; Gadwood R. C. TPGS-750-M: A Second-Generation Amphiphile for Metal-Catalyzed Cross-Couplings in Water at Room Temperature. J. Org. Chem. 2011, 76, 4379–4391. 10.1021/jo101974u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens R. J.; Hyatt J. A. Acetoacetylation with 2,2,6-trimethyl-4H-1,3-dioxin-4-one: a convenient alternative to diketene. J. Org. Chem. 1985, 50, 2431–2435. 10.1021/jo00214a006. [DOI] [Google Scholar]
- Gama F. H. S.; de Souza R. O. M. A.; Garden S. J. An efficient green protocol for the preparation of acetoacetamides and application of the methodology to a one-pot synthesis of Biginelli dihydropyrimidines Expansion of dihydropyrimidine topological chemical space. RSC Adv. 2015, 5, 70915–70928. 10.1039/C5RA14355A. [DOI] [Google Scholar]
- Clemens R. J.; Witzeman J. S. Kinetic and spectroscopic studies on the thermal decomposition of 2,2,6-trimethyl-4H-1,3-dioxin-4-one Generation of acetylketene. J. Am. Chem. Soc. 1989, 111, 2186. 10.1021/ja00188a037. [DOI] [Google Scholar]
- Brals J.; Smith J. D.; Ibrahim F.; Gallou F.; Handa S. Micelle-Enabled Palladium Catalysis for Convenient sp2-sp3 Coupling of Nitroalkanes with Aryl Bromides in Water Under Mild Conditions. ACS Catal. 2017, 7, 7245–7250. 10.1021/acscatal.7b02663. [DOI] [Google Scholar]
- Knorr L. Synthesis of quinoline-derivatives. Liebigs Ann. Chem. 1884, 46, 72. [Google Scholar]
- Hauser C. R.; Reynolds G. Reactions of β-Keto Esters with Aromatic Amines. Syntheses of 2- and 4-Hydroxyquinoline Derivatives. J. Am. Chem. Soc. 1948, 70, 2402–2404. 10.1021/ja01187a025. [DOI] [Google Scholar]
- Staskun B. The Conversion of Benzoylacetanilides into 2- and 4-Hydroxyquinolines. J. Org. Chem. 1964, 29, 1153–1157. 10.1021/jo01028a038. [DOI] [Google Scholar]
- Solingapuram S. K. K.; Gilbert T. M.; Klumpp D. A. Knorr Cyclizations and Distonic Superelectrophiles. J. Org. Chem. 2007, 72, 9761–9764. 10.1021/jo7013092. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Zhang Z.; Wu A.; Yang X.; Zhu Y.; Zhao N. A Novel Process for Antimalarial Drug Pyronaridine Tetraphosphate. Org. Process Res. Dev. 2014, 18, 349–353. 10.1021/op400357f. [DOI] [Google Scholar]
- March L. C.; Romanchick W. A.; Bajaw G. S.; Joullie M. M. Antimalarials. 2. Dihydro-1,3-oxazinoquinolines and dihydro-1,3-pyridobenzoxazines. J. Med. Chem. 1973, 16, 337–342. 10.1021/jm00262a006. [DOI] [PubMed] [Google Scholar]
- Lee N. R.; Bikovtseva A. A.; Cortes-Clerget M.; Gallou F.; Lipshutz B. H. Carbonyl Iron Powder: A Reagent for Nitro Group Reductions under Aqueous Micellar Catalysis Conditions. Org. Lett. 2017, 19, 6518–6521. 10.1021/acs.orglett.7b03216. [DOI] [PubMed] [Google Scholar]
- Moussaoui Y.; Salem R. B. Michael Additions of Nitroalkanes to Conjugated Ketones, Carboxylic Esters and Nitriles in Water and Biphasic Conditions (Water-Dichloromethane). J. Soc. Chim. Tunis. 2009, 11, 37–43. [Google Scholar]
- Roschangar F.; Sheldon R. A.; Senanayake C. H. Overcoming barriers to green chemistry in the pharmaceutical industry - the Green Aspiration Level concept. Green Chem. 2015, 17, 752–768. 10.1039/C4GC01563K. [DOI] [Google Scholar]
- Sheldon R. A. The E factor 25 years on: the rise of green chemistry and sustainability. Green Chem. 2017, 19, 18–43. 10.1039/C6GC02157C. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.