

Abstract
Background/Objective:
Moxifloxacin is a fluoroquinolone that is commonly used in adults, but not children. Certain clinical situations compel pediatric clinicians to use moxifloxacin, despite its potential for toxicity and limited pharmacokinetics (PK) data. Our objective was to further characterize the pharmacokinetics of moxifloxacin in children.
Methods:
We performed an opportunistic, open-label population PK study of moxifloxacin in children <18 years of age who received moxifloxacin as part of standard care. A set of structural PK models and residual error models were explored using non-linear mixed-effects modeling. Covariates with known biological relationships were investigated for their influence on PK parameters.
Results:
We obtained 43 moxifloxacin concentrations from 14 participants who received moxifloxacin intravenously (n=8) or orally (n=6). The dose of moxifloxacin was 10 mg/kg daily in participants ≤40 kg and 400 mg daily in participants >40kg. The population mean clearance and mean volume of distribution were 18.2 L/h and 167 L, respectively. The oral absorption was described by a first-order process. The estimated extent of oral bioavailability was highly variable (range 20–91%). Total body weight was identified as a covariate on clearance and volume of distribution, and substantially reduced the random unexplained inter-individual variability for both parameters. No participants experienced suspected serious adverse reactions related to moxifloxacin.
Conclusion:
These data add to the existing literature to support use of moxifloxacin in children in certain situations; however, further prospective studies on the safety and efficacy of moxifloxacin are needed.
Keywords: antibiotics, child, fluoroquinolone, pediatrics, pharmacodynamics, pharmacokinetics
1. Introduction
Moxifloxacin, a fourth-generation member of the fluoroquinolone class of antibacterial agents, is commonly prescribed in adults, but not children. Moxifloxacin’s bactericidal action is a result of inhibition of topoisomerases that are critical for bacterial DNA replication, transcription, repair, and recombination. Moxifloxacin has activity against Mycobacteria and both gram-negative and gram-positive organisms, with greater activity against gram-positive bacteria than earlier-developed agents in the fluoroquinolone class. Moxifloxacin is approved by the United States Food and Drug Administration (FDA) in adults for the treatment of acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, community-acquired pneumonia, uncomplicated and complicated skin and skin structure infections, and complicated intra-abdominal infections. Moxifloxacin is approximately 50% protein bound and widely distributed in the body, its tissue concentration often exceeding the plasma concentration. Moxifloxacin has volume of distribution in ranges from 1.7 to 2.7 L/kg, and apparent total body clearance and renal clearance are 12 L/hr and 2.6 L/hr, respectively, in healthy adult subjects. Oral moxifloxacin can be taken with or without food [1]. Moxifloxacin has a relatively long elimination half-life (about 12 hours reported in studies involving adults), so it can be administered once daily.
Despite its well-established use in adults, moxifloxacin is not approved by the U.S. FDA in children <18 years of age. Fluoroquinolones have been associated with arthropathy in juvenile animals [2]; due to concern for similar effects in humans, there have been limited studies of these drugs in children, and the majority of previous studies have not targeted moxifloxacin. In animal studies, chondrotoxicity associated with fluoroquinolones has been shown to be dependent on exposure [3]. To date, only 3 prospective studies and 1 case report have been published on moxifloxacin pharmacokinetics (PK) in pediatric populations [4–7].
Despite the limited PK data and potential for toxicity, pediatricians use moxifloxacin off-label in certain clinical situations. These situations occur primarily in children with infections caused by bacteria resistant to other antimicrobials or when other treatments are not available. Moxifloxacin’s broad spectrum of activity makes it versatile for use in infections at multiple sites, due to a variety of organisms. Successful moxifloxacin use has been reported in children with multi-drug resistant pulmonary tuberculosis [7, 8]. There are also case reports of moxifloxacin use in an infant with meningitis due to Mycoplasma hominis [6] and a 7-year-old boy with macrolide-resistant Mycoplasma pneumonia [9]. In each of these clinical situations, other treatment options were either limited or nonexistent. Our objective was to use opportunistic, minimal-risk methodologies and population PK analysis to further characterize the PK of moxifloxacin in children.
2. Methods
2.1. Study Design
We performed an opportunistic population PK study of moxifloxacin between March 2017 and March 2019. We aimed to enroll 30 children <18 years of age receiving moxifloxacin intravenously or enterally as part of standard of care as inpatients or outpatients at Duke University Medical Center. There were no exclusion criteria other than inability for the parent or legal guardian to provide consent.
2.2. Study Procedures
Information was collected prospectively from medical records including demographic data, clinical data (including medical history, moxifloxacin indication, concomitant medications of interest, concomitant food intake for participants receiving enteral moxifloxacin, standard of care laboratory values, clinical symptoms at start and end of therapy), results of all blood, urine, sputum, and other body fluid cultures within 7 days prior to start of moxifloxacin until end of moxifloxacin course, and moxifloxacin dosing information. Concomitant medications of interest were those identified from the moxifloxacin drug label to have potential interactions or adverse reactions with moxifloxacin and included: antacids containing aluminum or magnesium, sucralfate, iron, multivitamins containing iron or zinc, didanosine, warfarin, quinidine, ajmaline, procainamide, dispyramide, amiodarone, sotalol, ibutilide, dofetilide, dronedarone, or bretylium [1, 10]. Optimal sampling design analyses were conducted prior to the start of the study, based on the targeted enrollment of 30 subjects with at least 4 samples per subject, to identify the most informative sampling time windows to target when possible. WinPOPT software, a Windows interface for POPT (v3) was used. Optimal sampling was based on the fixed effects for CL, V1, and V2 with the aim to achieve a standard error of 30% or less. We targeted ideal sampling windows for blood collection (Table 1). Blood samples obtained were 0.2–0.5 mL in volume, and a maximum of 10 blood samples could be obtained per participant. When possible, blood samples were collected when labs were being obtained per standard of care; however, the child’s guardian was given the option to consent for additional blood sample collection. Samples were frozen at −70°C and shipped for analysis on dry ice.
Table 1.
Ideal Sampling Windows for: a) Intravenous; and b) Oral Administration of Moxifloxacin
| a) Intravenous administration | |||
|---|---|---|---|
| Dosing Interval (Hours) | |||
| Window** | 12 | 24 | 48 |
| Window #1 | 0a | 0a | 0a |
| Window #2 | 2–4 | 3–7 | 3–12 |
| Window #3 | 5–8 | 8–12 | 13–24 |
| Window #4 | 11–12 (Trough) | 23–24 (Trough) | 47–48 (Trough) |
| Window #5 (elimination) | 24–36 | 48–72 | 96–144 |
| b) Oral administration | |||
| Dosing Interval (Hours) | |||
| Window #1 | 0.5–2 | 0.5–2 | 0.5–2 |
| Window #2 | 2–4 | 3–7 | 3–12 |
| Window #3 | 5–8 | 8–12 | 13–24 |
| Window #4 | 11–12 (Trough) | 23–24 (Trough) | 47–48 (Trough) |
| Window #5 (elimination) | 24–36 | 48–72 | 96–144 |
Time 0 = end of infusion; sample collected after end of flush from location other than infusion line
2.3. Biologic Sample Analysis
Concentrations of moxifloxacin in plasma were determined using a validated liquid chromatography tandem mass spectrometry (LC-MS/MS) using moxifloxacin-d4 as the internal standard (Frontage Laboratories, Inc; Exton, PA). Calibration standards and quality control samples were prepared in drug-free human ethylenediaminetetraacetic acid (EDTA) plasma. The linear concentration range of moxifloxacin was from 0.01 mg/L to 5 mg/L. The lower limit of quantitation (LLOQ) was 0.01 mg/L. The intra-assay accuracies of the quality control samples (0.01, 0.03, 0.35, 3.8, and 76 mg/L) were within ± 15.0% (within ±20.0% for LLOQ) and the inter-run precision range values were no more than 15.0% (20.0% for LLOQ). Moxifloxacin stability analysis revealed that moxifloxacin was stable in human plasma for 12 months, and all samples were analyzed within 12 months of collection.
2.4. PK Analysis
The primary outcome of the study was the PK of moxifloxacin. The dosing, sampling, and demographic information were merged with the bioanalytical information to create a PK dataset. Data were analyzed using non-linear mixed effects modeling with NONMEM software (version 7.2, Icon Solutions, Ellicott City, MD). For determination of a base model, one-, two-, and three-compartment structural PK models were evaluated. Models with linear elimination and non-linear elimination were tested. Moxifloxacin input (administration) was modeled as a zero-order process with a fixed duration of infusion of 1 hour for intravenous doses. First-order, zero-order, and saturable processes were evaluated to describe the oral absorption of moxifloxacin. Bioavailability (F) of oral doses relative to IV dosing was included using a logit transformation to restrict F between 0 and 1. Due to this being a parallel study (i.e., IV and oral doses were not compared within subjects), estimation of F assumed that there was no systematic difference in CL between subjects receiving IV moxifloxacin and those receiving oral moxifloxacin. Exponential models were used to describe the inter-individual variability (IIV) for clearance, volume, and the bioavailability parameter (applied prior to logit transformation).
The residual unidentified variability was described by a combined additive and proportional error model. For models where the additive component of the combined residual error model could not be estimated, it was fixed to a small value. The half-life (t1/2) was calculated using the population estimates of clearance and volume of distribution.
2.5. Model Building Steps
Once the base model was identified, covariates were investigated for their influence on PK parameters. Due to the relatively small number of subjects, only potential covariates with known biological relationships to drug PK were considered. Covariates evaluated included total body weight, lean body weight [11], and creatinine clearance (CLcr). Lean body weight was calculated as follows:
- For females:
- For males:
Total body weight and lean body weight were evaluated as continuous covariates on clearance (CL) and volume of distribution (V) parameters. For some subjects in the study, body weights were recorded at multiple time points. Total body weight and lean body weight were evaluated as time-varying covariates, as this allowed better description of the observations in subjects that had changes in body weight during the course of the study. Allometric scaling using a coefficient of 0.75 for CL and 1.0 for V was applied [12–14]. The effect of CLcr on renal clearance was evaluated using 3 approaches: 1) Schwartz bedside equation for subjects <12 years old and Cockcroft-Gault equation for children 12 years and older, with CLcr normalized to 1.73 m2 for all subjects using body surface area; 2) the same approach as in 1), but with CLcr in units of mL/min (not normalized by body surface area); and 3) the univariate Schwartz equation for all subjects in units of mL/min [15, 16]. Maturation models were not formally evaluated as the maturation half-time for the clearance of common drugs has been reported at ~0 to 4 months after full-term gestation and maturation is expected to be complete by 2 years of age [17], and only 1 enrolled participant was <2 years of age.
2.6. Model Evaluation
Competing models were distinguished by their predictive performance assessed via the log-likelihood ratio test (−2LL) for nested models and the Akaike information criterion for non-nested models. The goodness of fit was evaluated via standard diagnostic plots, including plots of individual and population predictions versus observations, and conditional weighted residuals versus time. The predictive performance was evaluated by the normalized prediction distribution error (NPDE). Normal distribution of the NPDE values was evaluated using the Shapiro-Wilk test, a one-sample t-test was used to evaluate whether the mean was significantly different from 0, and the Wilcoxon signed-rank test was used to evaluate whether the median was significantly different from 0. The intention was to evaluate the predictive performance of candidate models by generating prediction-corrected visual predictive checks [18]. However, as a result of sparse sampling and a small number of subjects, insufficient data were available for either route of administration to be meaningfully evaluated by a prediction-corrected visual predictive checks. Instead, simulations (n=1,000 profiles) were conducted from the final model for a dose of 400 mg and body weight of 43 kg, and overlaid on the observed data that were normalized to a 400 mg dose and allometrically scaled to 43 kg (median weight in the study was 43.45 kg). In addition to the statistical model selection criteria, the plausibility of the parameter estimates with respect to known physiology and pathophysiology was considered.
2.7. Evaluation of the Applicability of a Published PK Model
We also evaluated whether a recently published population PK model [5, 19–22]] for moxifloxacin in pediatric patients could be applied to, and be predictive of, our study. The model developed by Willmann et al. was used in NONMEM to generate the post-hoc Bayesian estimates for the data in the current study. In addition, the same model was applied in simulations (n=1,000 profiles) for a dose of 400 mg and body weight of 43 kg, and overlaid on our observed data that were normalized to a 400 mg dose and allometrically scaled to 43 kg.
2.8. Monte Carlo simulations
Simulations including IIV were performed using the final covariate model, physician-selected intravenous moxifloxacin dosage regimens, and body weights of the study participants who received intravenous dosing. The protein binding of moxifloxacin has been reported to range between 47% and 55%. Therefore, an average protein binding of 50% was used for moxifloxacin in plasma [23]. Based on previous studies, the pharmacokinetic-pharmacodynamic (PK-PD) targets used were the area under the free concentration-time curve in a 24-hour interval (fAUC24h)/minimum inhibitory concentration (MIC) ≥34 for Streptococcus pneumoniae, and fAUC24h/MIC ≥75 for Gram-negative bacteria [24–26]. A range of MICs from 0.03125 to 4 mg/L was evaluated. The plasma concentration-time curves for 1,000 virtual patients were simulated at steady-state in the absence of residual error using NONMEM. The probability of target attainment (PTA) was derived by calculating the fraction of subjects who attained the PK-PD target at each MIC. The PK-PD breakpoint was defined as the highest MIC for which the PTA was at least 90%.
2.9. Safety and Effectiveness
Although the primary objective of our study was to evaluate the PK of moxifloxacin, we also collected clinical data related to safety and efficacy during the study period. We recorded any evidence of QTc prolongation by electrocardiogram (if obtained per usual clinical care), tendinopathy, and suspected serious adverse reactions related to moxifloxacin. Because of the expected underlying comorbidities of the enrolled participants, other adverse events and serious adverse events were not recorded. We also evaluated clearance of positive bacterial cultures in normally sterile body fluids.
3. Results
3.1. Participant Characteristics
A total of 14 children <18 years of age were enrolled in the study (Table S1). Enrollment was halted before the target of 30 participants were enrolled because of decreased standard-of-care moxifloxacin utilization following institution of hospital-wide antibiotic stewardship initiatives. Enrolled participants ranged between 1 and 16 years of age; most participants (10/14, 71%) were 12 years or older, 1 (7%) was 11 years of age, and 2 (14%) were 2 years of age, and 1 (7%) was 15 months of age. A total of 10/14 (71%) of participants were male; 6 (43%) were Caucasian, 4 (29%) were Black or African American, 1 (7%) was Asian, 1 (7%) was multi-racial, and 2 (14%) had unknown race. Three (21%) participants had Hispanic ethnicity. The most common indications for moxifloxacin were lower respiratory tract infection or pneumonia in 5 participants (36%), fever without respiratory tract infection or pneumonia in 3 (21%), and skin or soft tissue infection in 2 (14%); one participant had each of the following indications: culture-proven bacteremia, typhlitis, post-operative hardware-associated infection, and sinusitis. Most of the participants had substantial comorbidities, including 9/14 (64%) with cancer or hematologic disorders and 3/14 (21%) with severe genetic or metabolic disorders. Only 1 participant received a concomitant medication of interest (magnesium oxide); this participant had low bioavailability (0.20), and 7 of the 10 blood samples included in the analysis were collected before magnesium oxide was given.
Eight (57%) participants received moxifloxacin as a 1-hour intravenous infusion, 4 (29%) received moxifloxacin orally as a tablet, and 2 (14%) received moxifloxacin orally as a suspension. The prescribed dose was 10 mg/kg daily in participants ≤40 kg and 400 mg daily in participants >40kg. From the 14 participants, we collected 43 blood samples (median [25th–75th percentiles] number of samples 2.5 [1–4] per participant). All concentrations were quantifiable and none was below the limit of quantification of the assay.
3.2. Population PK Model Development and Evaluation and PTA analysis
Details of the structural models evaluated are presented in Table S2. While a number of strategies were employed to estimate two-compartment models, the parameters of a two-compartment could not be robustly and reproducible estimated. In addition, a one-compartment model with non-linear (saturable) elimination was evaluated, but this model did not converge and provided biologically implausible parameters. Therefore, a one-compartment disposition model with linear elimination was used as the structural model. This one-compartment model described the data adequately as evidenced by standard diagnostic plots of the final model (Figure 1). The distribution of the NPDE values (Figure S1) for the final model was normal (p=0.91) and the mean was not significantly different from 0 (p=0.26). In addition, the median was not significantly different from 0 (p=0.31).
Figure 1. Selected Diagnostic Plots.
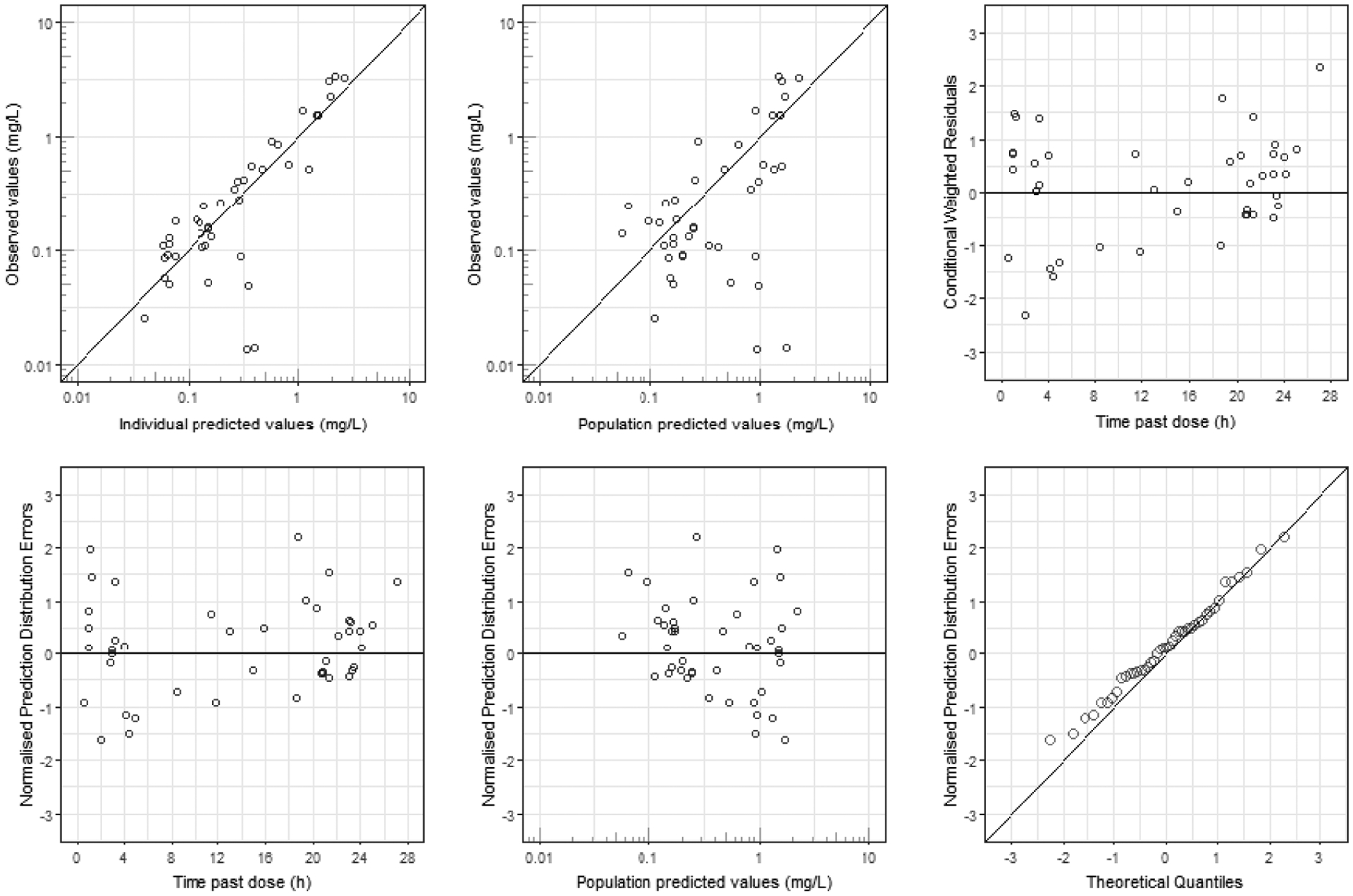
Selected diagnostic plots for the final pharmacokinetic model.
The final model included allometric scaling of clearance and volume parameters by total body weight [11]. Allometric scaling of CL and V by total body weight resulted in a decrease of the IIV of CL from 41% to 18%, a decrease of the IIV of V from 51% to 33%, and a significant improvement in the objective function by 7.1 without adding any degrees of freedom (Table 2). Allometric scaling by lean body weight [11] did not improve the model performance compared to allometric scaling by total body weight. The objective function value of the model incorporating lean body weight as a time-varying covariate on CL and V was within one unit of that for the final model with allometric scaling by total body weight as a time-varying covariate on CL and V.
Table 2.
Selected Covariate Models Evaluated
| Covariate model | Covariance CL, V | −2LL | Δ−2LL | Comment |
|---|---|---|---|---|
| None | Yes | −77.167 | Base model | |
| WT on V | No | −69.784 | +7.383 | Did not converge |
| WT on V | Yes | −73.998 | +3.169 | Did not converge, covariance term not estimable |
| WT on CL | No | −81.188 | −4.021 | |
| WT on CL | Yes | −82.204 | −5.037 | Did not converge |
| LBW on CL and V | No | −83.288 | −6.121 | |
| WT on CL and V | No | −84.218 | −7.051 | Final model |
| WT on CL and V | Yes | −84.469 | −7.302 | Covariance term implausible |
| CLcr on CLr 1 | No | −87.277 | −10.11 | IIV CL not estimable (estimated at 1%, biologically implausible), covariance step aborted |
| CLcr on CLr 3 | No | −87.803 | −10.64 | IIV CL not estimable, covariance step aborted |
| CLcr on CLr 2 | No | −90.256 | −13.09 | Did not converge, IIV CL not estimable |
Covariate models including WT or LBW had fixed allometric exponents; therefore, no degrees of freedom were added.
CL, clearance; CLcr, creatinine clearance; CLr, renal clearance; IIV, inter-individual variability; LBW, lean body weight; TBW, total body weight; V, volume of distribution; −2LL, objective function value; Δ−2LL, change in objective function value
Models that included CLcr as a covariate on renal clearance and estimated a separate non-renal clearance did not converge successfully, did not allow estimation of IIV on clearance, or both. Therefore, the analyses did not support the inclusion of CLcr over or in addition to total body weight. In addition, plotting the individual estimates of moxifloxacin clearance from the base model versus CLcr (calculated by any of the three approaches) did not reveal an apparent correlation between moxifloxacin clearance and CLcr. While the subject with the lowest CLcr displayed the lowest moxifloxacin clearance, this subject also had the lowest body weight.
The parameter estimates, standard errors (SE%) and shrinkage values for the final model are presented in Table 3. Based on the average body weight in the study population of 43.7 kg, the population estimate for CL was 18.2 L/h, and the population estimate for V was 167 L. The individual estimates of F were 0.20 and 0.71 for the two subjects who received moxifloxacin as a suspension and ranged from 0.29 to 0.91 for the four subjects who received moxifloxacin as a tablet. Therefore, low or high absolute bioavailability was not linked to the oral formulation that was administered, and the type of formulation was not evaluated as a covariate on F. The correlation between CL and V was estimated at a physiologically implausible (negative) value following the inclusion of total body weight as a covariate on CL and V. This suggests that the correlation was not estimable, which may partly be due to the effect of total body weight explaining (most of) the covariance between CL and V that was observed with the base model. Therefore, the final model did not include a correlation between CL and V (Table 3).
Table 3.
Parameter Estimates of the Final Model with Allometric Scaling of Clearance and Volume of Distribution by Total Body Weight, Based on the Average Body Weight in the Study Population (43.7 kg)
| Parameter | Unit | Population Estimate or Median | Standard Error of the Population Estimate | IIV or Range | Eta Shrinkage |
|---|---|---|---|---|---|
| CL | L/h | 18.2 | 35% | 18% | 49% |
| V | L | 167 | 38% | 33% | 37% |
| F | - | 0.76 | - | 0.20 – 0.91 | 54% |
| ka | h−1 | 2.21 | 272% | - | - |
| t1/2 | h | 6.36 | - | - | - |
| CVCP | - | 0.49 | 81% | - | - |
| SDCP | mg/L | 0.055 (fixed) | - | - | - |
CL, clearance; CVCP, proportional residual error; F, absolute bioavailability of the oral dose (expressed as fraction of the administered dose), the median and range of the individual estimates are presented due to the wide distribution and use of a logit transformation to constrain F between 0 and 1 for each individual subject; IIV, inter-individual variability; ka, first-order absorption rate constant for oral dosing as a tablet or suspension; SDCP, additive residual error; t1/2, half-life; V, volume of distribution
The concentrations observed following oral dosing were highly variable. The current study collected 9 samples within 12 hours time past oral dose, and all except one of these concentrations were below 0.6 mg/L when normalized to a dose of 400 mg and allometrically scaled to 43 kg body weight. The concentrations did not correlate with the absence or presence of food in any participants.
The median (range) area under the concentration-time curve over 24 hours at steady-state (AUCss,24) was 16.0 (13.1–19.1) mg·h/L following intravenous dosing and 17.8 (4.69–25.5) mg·h/L following oral administration. The maximum concentration (Cmax) was 2.02 (1.47–2.87) mg/L following intravenous dosing and 1.92 (0.37–2.11) mg·h/L following oral administration. Simulations from the final model revealed that the 10th percentile (P10), median (P50), and 90th percentile (P90) of the model predictions agreed well with the normalized allometrically-scaled observed data (Figure 2).
Figure 2. Simulations for Intravenous and Oral Moxifloxacin Dose.
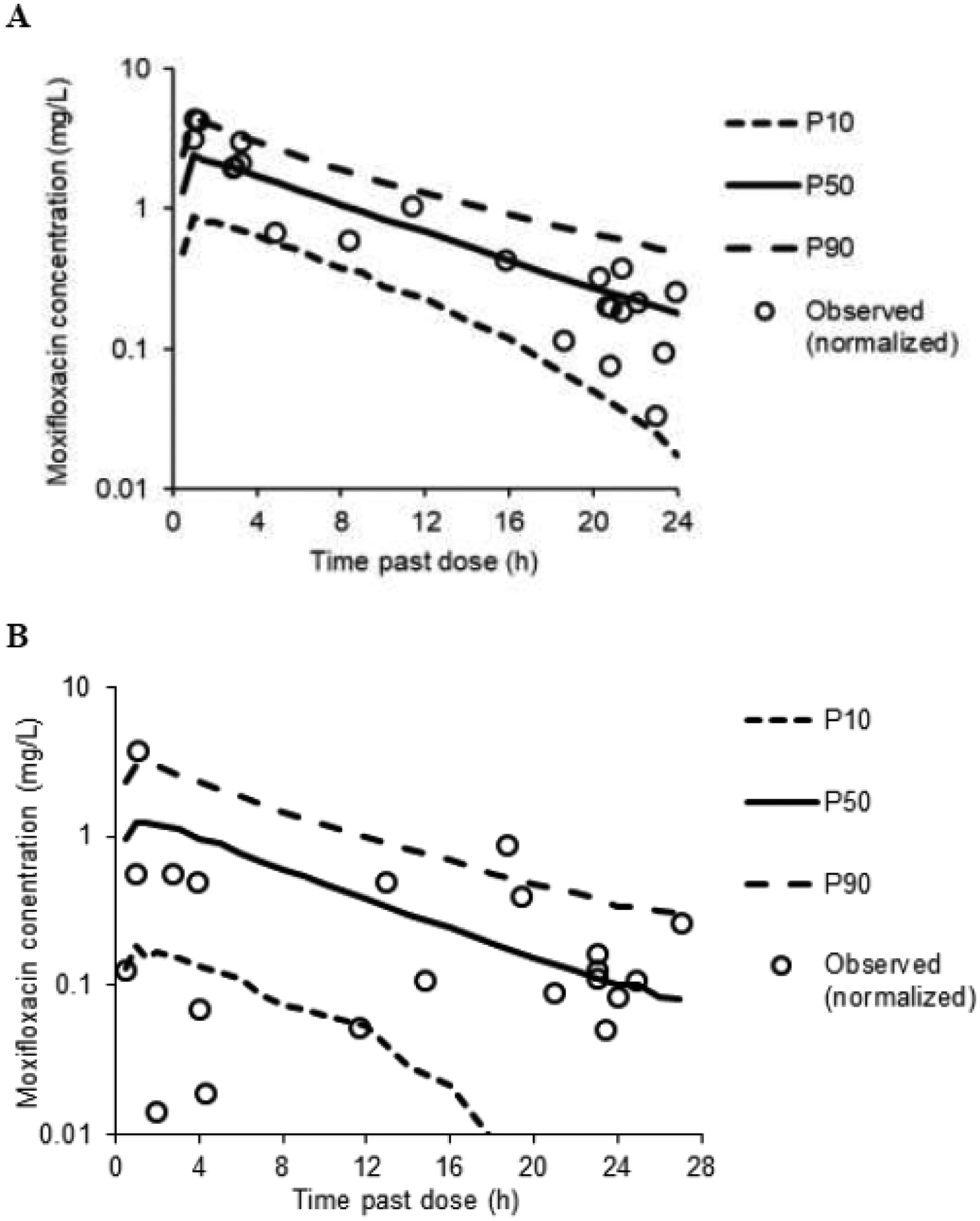
Simulations from the final model for an: A) intravenous; and B) oral dose of 400 mg and total body weight of 43 kg. Observed data shown are normalized (i.e., to a 400 mg dose and allometrically scaled to 43 kg).
P10, 10th percentile of the model predictions; P50, median percentile of the model predictions; P90, 90th percentile of the model predictions
Applying the recently published population PK model for moxifloxacin in pediatric patients to generate the post-hoc Bayesian estimates resulted in systematic overprediction of the moxifloxacin concentrations observed in the current study. Simulations from the same model also substantially overpredicted the moxifloxacin concentrations that were observed in our study (Figure S2), indicating that the populations were pharmacokinetically different.
The PK-PD breakpoints for the intravenous dosage regimens administered in the study were 0.125 mg/L at the target for S. pneumoniae and 0.0625 mg/L at the target for Gram-negative bacteria (Figure 3).
Figure 3. Probabilities of Target Attainment.
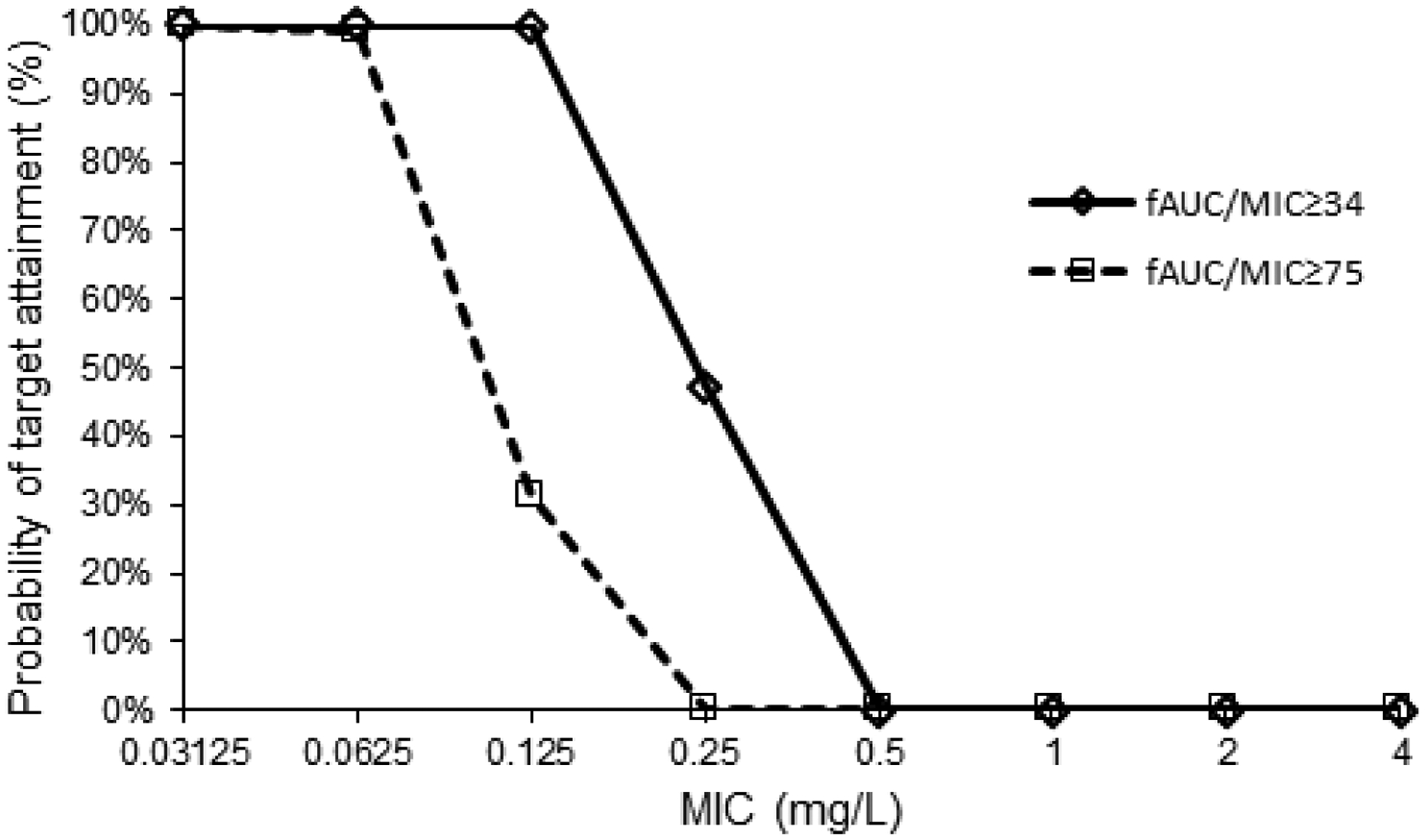
Based on Monte Carlo simulations of the intravenous dosage regimens administered in the study. PK-PD targets used: fAUC/MIC ≥34 for S. pneumoniae and fAUC/MIC ≥75 for Gram-negative bacteria.
fAUC/MIC, area under the free concentration-time curve in a 24-hour interval/minimum inhibitory concentration; PK-PD, pharmacokinetic-pharmacodynamic
3.3. Safety and Effectiveness
No suspected adverse reactions related to moxifloxacin, QTc prolongation, or tendinopathy were observed in any of the participants. Only 3 participants had positive cultures for bacterial organisms during the study period. One participant had positive sputum cultures for rare organisms that were not speciated, which did not represent clinically important infection. The 2 other participants had clinically important infections. The first of these 2 participants was prescribed moxifloxacin for Leuconostoc lactis bacteremia and had 2 positive blood cultures on the 2 days preceding start of therapy. While susceptibility of this organism to moxifloxacin or other fluoroquinolones was not reported, the participant did clear the bacteremia with negative cultures while on moxifloxacin therapy. The individual estimate for AUCss,24 was 16.1 mg*h/L. The second participant was prescribed moxifloxacin for outpatient treatment of an orbital abscess following inpatient treatment with intravenous antibiotics. Initial culture of the abscess prior to treatment revealed Streptococcus intermedius and rare growth of Staphylococcus species with intermediate susceptibility to moxifloxacin. After completing 3 days of moxifloxacin treatment, the participant returned with worsening symptoms and reaccumulation of the abscess, requiring additional surgery and replacing moxifloxacin with other antibiotics. The individual estimate for AUCss,24 was 25.5 mg*h/L.
4. Discussion
We combined an opportunistic methodology with population PK analysis to characterize the PK of moxifloxacin in 14 children with substantial comorbidities. When we initially planned this study, PK data for moxifloxacin in children was almost nonexistent, and despite this, moxifloxacin was often used according to clinician preference for broad-spectrum empirical coverage at Duke, particularly in vulnerable, immunosuppressed pediatric populations (such as children undergoing bone marrow transplant). Over the course of the study, standard-of-care use of moxifloxacin decreased at our hospital, primarily because of the institution of antimicrobial stewardship policies that limited the use of broad-spectrum therapies without infectious disease team consultation. These policies originated due to knowledge of increasing fluoroquinolone resistance worldwide [27], suggesting that judicious use is needed. As a result, we did not reach our target enrollment of 30 children in this opportunistic study; however, the population PK analysis methodology allowed us to develop a model to estimate population clearance and volume of distribution.
In a recent population PK study of 186 children aged 3 months to <18 years who received IV and oral moxifloxacin, the authors developed a linear 3-compartment model [5]. The population mean CL was 0.45 L/h/kg0.75 (34.6% IIV), which translates to a CL of 7.65 L/h for a 43.7 kg subject and is lower than the population mean CL of 18.2 L/h in the current study. The sum of the population mean volumes of distribution of the central and peripheral compartments was 2.25 L/kg, translating to 98.3 L for a 43.7 kg subject, which is also somewhat lower than the V of 167 L in the current study. The population mean for absolute bioavailability was 86.6% (IIV was not estimated and fixed to 0), which is similar to the mean bioavailability of 76% in the current study. The population mean absorption rate constant was 0.537 h−1 compared to 2.21 h−1 in the current study. Nevertheless, data during the absorption phase in the current study were sparse and highly variable even within subjects. The previous study included allometric scaling by total body weight on the disposition parameters a priori, and no further covariates (age, serum creatinine, estimated glomerular filtration rate, study phase I or III, sex) were identified on top of that, which agrees with the population PK model from the current study. Given the substantial differences in parameter estimates between our study and the previous study, it was not unexpected that application of the previous model to our data resulted in systematic overprediction of the observed concentrations. The very different populations evaluated, including our inclusion of multiple participants with concurrent medical conditions including neoplasms, could partially explain the low performance of the previous model in our population.
Following oral dosing, the majority of the concentrations observed in the current study were substantially lower than those reported previously by a study involving 23 children with drug-resistant tuberculosis (including 6 HIV-infected) receiving oral moxifloxacin [7]. The average apparent clearance (CL/F) calculated by non-compartmental analysis was 10.53 L/h (range 7.23–14.14 L/h) in that study. This would translate to a CL/F of 14.4 L/h when allometrically scaled to the median body weight of 43.7 kg in the current study, and would be slightly lower than the estimate of CL in the current study assuming bioavailability in the previous study was high. Of note, the large uncertainty in the absorption rate constant was related to the extremely variable data with small sample size. In addition, we did not record details on dairy intake, whether tablets were ingested whole or crushed, or the receipt of other medications that could have impacted absorption. Therefore, our results following oral dosing should be interpreted with caution.
In a recent PK study in 31 children receiving a single IV dose of moxifloxacin, the participants were divided into three cohorts: ≥6 to ≤14 years (cohort 1, n=12), ≥2 to <6 years (cohort 2, n=12), and >3 months to <2 years (cohort 3, n=7) [4]. The elimination half-life was 7.89 hours for subjects in cohort 1 that received a dose of 5 mg/kg (n=7) and 6.16 hours for subjects in cohort 1 that received a 6 mg/kg dose (n=5). This is similar to the elimination half-life of 6.36 hours in the current study.
The median oral bioavailability in the current study was 76%, yet the two subjects for whom rich data were available had much lower estimated bioavailability of 20% and 29%. The remaining four subjects who contributed one or two data points each had estimated bioavailability of 71% to 91%. While one participant receiving magnesium oxide had low bioavailability (0.20), 7 of 10 PK samples were collected prior to magnesium oxide being started; consequently, administration of magnesium oxide did not appear to explain this participant’s low availability. Oral bioavailability of moxifloxacin was previously estimated in a population PK study involving 25 adult surgical intensive care unit patients with a population mean of 76% and a relatively small IIV of 20% [28]. The population mean oral bioavailability of moxifloxacin was 89% with an IIV of 30% in 18 adult patients with community-acquired pneumonia [26]. Therefore, while the median bioavailability was similar between the current study and adult studies in the literature, the IIV was higher in the current study.
While CL and V were estimated with good precision, shrinkage was 37% or greater for all parameters. The relatively high shrinkage values were a consequence of the smaller sample size, and as a result, the individual parameter estimates shrunk towards the population mean estimate. High shrinkage values are a limitation of our study and present a limitation for the use of a model in dosing simulations. In addition, while we reported individual parameter estimates for participants with positive bacterial cultures, it is important to interpret these individual exposure metrics with caution. Similarly, the results of the PTA analysis need to be interpreted with caution, due to the small sample size on which the model was based (eight participants received intravenous dosing), and consequently relatively high shrinkage values.
Our study had several additional limitations. Due to the decrease in standard-of-care use of moxifloxacin at our hospital, our total sample size was less than we had anticipated. As a result, while we had initially planned to examine the association between exposure and response to moxifloxacin with regard to effectiveness, we only enrolled 3 participants with positive cultures (2 with clinically important infection). Additionally, while we did not observe any moxifloxacin-related adverse effects, these results should be interpreted with caution. Participants did not have routine electrocardiograms pre- and post-initiation of moxifloxacin, and we only detected adverse events that were reported in the clinical record. Laboratory parameters were limited to those obtained by standard care and reported in the medical record. In addition, some adverse effects, such as tendinopathy, may occur longer after drug exposure, and our follow-up period ended after moxifloxacin was discontinued. Finally, because moxifloxacin was prescribed per standard care and we did not exclude any indications or diagnoses, many of our participants had substantial comorbidities and received numerous concomitant medications. Our small sample size did not permit us to include any additional covariates, such as drug-drug interactions not related to moxifloxacin, in the PK model.
5. Conclusions
By using an opportunistic design at a center with routine standard-of-care use of moxifloxacin, we were able to gather data to inform our PK analysis with minimal risk to participants. We estimated CL and V values that were comparable to previous PK studies in children following intravenous and oral dosing. Our data add to the growing body of literature to support the use of moxifloxacin in children in certain situations; however, further prospective studies on the safety and efficacy of moxifloxacin are needed.
Supplementary Material
Key Points.
We further characterized the pharmacokinetics of moxifloxacin in children by performing an opportunistic, open-label population pharmacokinetic (PK) study in those <18 years of age who received moxifloxacin as part of standard care.
We estimated CL and V values that were comparable to previous PK studies in children following intravenous and oral dosing.
No participants experienced suspected serious adverse reactions related to moxifloxacin.
Our data add to the growing body of literature to support the use of moxifloxacin in children in certain situations.
Funding:
This project was supported by the Division of Microbiology and Infectious Diseases (DMID), National Institute of Allergy and Infectious Diseases (NIAID) of NIH through the Vaccine and Treatment Evaluation Units (VTEU), and the US Department of Health and Human Service under contracts HHS (Duke University HHSN272201300017I, HHSN27200009, FY2016A1B1C1D1.0026; Emmes HHSN272201500002C).
Conflicts of interest/Competing interests:
EBW reports potential conflicts due to research support received from Pfizer and Moderna; EBW is a member of a scientific advisory board for Vaxcyte. RGG reports consulting services for Tellus Therapeutics. Other authors declare no conflicts or financial interest in any product or service mentioned in the manuscript, including grants, equipment, medications, employment, gifts, and honoraria. Carl Kirkpatrick, Cornelia Landersdorfer, Thomas Conrad, and Aya Nakamura had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. BO contributed to this manuscript while employed in the Division of Microbiology and Infectious Diseases, NIAID, NIH. This manuscript reflects the views of the author and should not be construed to represent FDA’s views or policies.
Footnotes
Ethics approval: The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national guidelines on human experimentation.
Consent to participate: The Duke University Institutional Review Board (Durham, NC) gave approval for the study to be conducted. All participants gave written informed consent and assent (if applicable) to participate in the study. Registration on clinicaltrials.gov was not required due to the observational nature of the study.
Availability of data and material:
Please contact the corresponding author for data requests.
References
- 1.DAILYMED. MOXIFLOXACIN HYDROCHLORIDE - moxifloxacin hydrochloride tablet (drug label), Mylan Pharmaceuticals Inc. Revised August 2015. DAILYMED web site. https://dailymed.nlm.nih.gov/dailymed/archives/fdaDrugInfo.cfm?archiveid=235974. Accessed May 7, 2021. [Google Scholar]
- 2.Burkhardt JE, Hill MA, Carlton WW, Kesterson JW. Histologic and histochemical changes in articular cartilages of immature beagle dogs dosed with difloxacin, a fluoroquinolone. Vet Pathol. 1990;27(3):162–70. doi: 10.1177/030098589002700303. [DOI] [PubMed] [Google Scholar]
- 3.Stahlmann R, Merker HJ, Hinz N, Chahoud I, Webb J, Heger W, et al. Ofloxacin in juvenile non-human primates and rats. Arthropathia and drug plasma concentrations. Arch Toxicol. 1990;64(3):193–204. doi: 10.1007/BF02010725. [DOI] [PubMed] [Google Scholar]
- 4.Stass H, Lettieri J, Vanevski KM, Willmann S, James LP, Sullivan JE, et al. Pharmacokinetics, safety, and tolerability of single-dose intravenous moxifloxacin in pediatric patients: dose optimization in a phase 1 study. J Clin Pharmacol. 2019;59(5):654–67. doi: 10.1002/jcph.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willmann S, Frei M, Sutter G, Coboeken K, Wendl T, Eissing T, et al. Application of physiologically-based and population pharmacokinetic modeling for dose finding and confirmation during the pediatric development of moxifloxacin. CPT Pharmacometrics Syst Pharmacol. 2019;8(9):654–63. doi: 10.1002/psp4.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watt KM, Massaro MM, Smith B, Cohen-Wolkowiez M, Benjamin DK Jr., Laughon MM. Pharmacokinetics of moxifloxacin in an infant with Mycoplasma hominis meningitis. Pediatr Infect Dis J. 2012;31(2):197–9. doi: 10.1097/INF.0b013e31823980c3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thee S, Garcia-Prats AJ, Draper HR, McIlleron HM, Wiesner L, Castel S, et al. Pharmacokinetics and safety of moxifloxacin in children with multidrug-resistant tuberculosis. Clin Infect Dis. 2015;60(4):549–56. doi: 10.1093/cid/ciu868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garazzino S, Scolfaro C, Raffaldi I, Barbui AM, Luccoli L, Tovo P-A. Moxifloxacin for the treatment of pulmonary tuberculosis in children: a single center experience. Pediatr Pulmonol. 2014;49(4):372–6. doi: 10.1002/ppul.22755. [DOI] [PubMed] [Google Scholar]
- 9.Shen Y, Zhang J, Hu Y, Shen K. Combination therapy with immune-modulators and moxifloxacin on fulminant macrolide-resistant Mycoplasma pneumoniae infection: a case report. Pediatr Pulmonol. 2013;48(5):519–22. doi: 10.1002/ppul.22650. [DOI] [PubMed] [Google Scholar]
- 10.DAILYMED. MOXIFLOXACIN - moxifloxacin hydrochloride injection, solution. Revised June/2015, Fresenius Kabi USA, LLC. DAILYMED web site. https://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=a4e28b09-714b-46e7-b4e6-0163cad78fc5. Accessed May 7, 2021. [Google Scholar]
- 11.Al-Sallami HS, Goulding A, Grant A, Taylor R, Holford N, Duffull SB. Prediction of fat-free mass in children. Clin Pharmacokinet. 2015;54(11):1169–78. doi: 10.1007/s40262-015-0277-z. [DOI] [PubMed] [Google Scholar]
- 12.McCune JS, Bemer MJ, Barrett JS, Baker KS, Gamis AS, Holford NHG. Busulfan in infant to adult hematopoietic cell transplant recipients: a population pharmacokinetic model for initial and Bayesian dose personalization. Clin Cancer Res. 2014;20(3):754–63. doi: 10.1158/1078-0432.CCR-13-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ooi MH, Ngu SJ, Chor JK, Li J, Landersdorfer CB, Nation RL. Population pharmacokinetics of intravenous colistin in pediatric patients: implications for selection of dosage regimens. Clin Infect Dis. 2019;69(11):1962–8. doi: 10.1093/cid/ciz067. [DOI] [PubMed] [Google Scholar]
- 14.Landersdorfer CB, Findling RL, Frazier JA, Kafantaris V, Kirkpatrick CMJ. Lithium in paediatric patients with bipolar disorder: implications for selection of dosage regimens via population pharmacokinetics/pharmacodynamics. Clin Pharmacokinet. 2017;56(1):77–90. doi: 10.1007/s40262-016-0430-3. [DOI] [PubMed] [Google Scholar]
- 15.Llanos-Paez CC, Staatz C, Lawson R, Hennig S. Comparison of methods to estimate glomerular filtration rate in paediatric oncology patients. J Paediatr Child Health. 2018;54(2):141–7. doi: 10.1111/jpc.13752. [DOI] [PubMed] [Google Scholar]
- 16.Schwartz GJ, Schneider MF, Maier PS, Moxey-Mims M, Dharnidharka VR, Warady BA, et al. Improved equations estimating GFR in children with chronic kidney disease using an immunonephelometric determination of cystatin. C Kidney Int. 2012;82(4):445–53. doi: 10.1038/ki.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson BJ, Holford NH. Understanding dosing: children are small adults, neonates are immature children. Arch Dis Child. 2013;98(9):737–44. doi: 10.1136/archdischild-2013-303720. [DOI] [PubMed] [Google Scholar]
- 18.Jamsen KM, Patel K, Nieforth K, Kirkpatrick CMJ. A regression approach to visual predictive checks for population pharmacometric models. CPT Pharmacometrics Syst Pharmacol. 2018;7(10):678–86. doi: 10.1002/psp4.12319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andersson MI, Macgowan AP. Development of the quinolones. J Antimicrob Chemother. 2003;51 Suppl 1:1–11. doi: 10.1093/jac/dkg212. [DOI] [PubMed] [Google Scholar]
- 20.Siefert HM, Domdey-Bette A, Henniger K, Hucke F, Kohlsdorfer C, Stass HH. Pharmacokinetics of the 8-methoxyquinolone, moxifloxacin: a comparison in humans and other mammalian species. J Antimicrob Chemother. 1999;43 Suppl B:69–76. doi: 10.1093/jac/43.suppl_2.69. [DOI] [PubMed] [Google Scholar]
- 21.Stass H Dalhoff A, Kubitza D, Schühly U. Pharmacokinetics, safety, and tolerability of ascending single doses of moxifloxacin, a new 8-methoxy quinolone, administered to healthy subjects. Antimicrob Agents Chemother. 1998;42(8):2060–2065. doi: 10.1128/AAC.42.8.2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhanel GG, Ennis K, Vercaigne L, Walkty A, Gin AS, Embil J, et al. A critical review of the fluoroquinolones: focus on respiratory infections. Drugs. 2002;62(1):13–59. doi: 10.2165/00003495-200262010-00002. [DOI] [PubMed] [Google Scholar]
- 23.Landersdorfer CB, Kinzig M, Hennig FF, Bulitta JB, Holzgrabe U, Drusano GL, et al. Penetration of moxifloxacin into bone evaluated by Monte Carlo simulation. Antimicrob Agents Chemother. 2009;53(5):2074–2081. doi: 10.1128/AAC.01056-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ambrose PG, Bhavnani SM, Rubino CM, Louie A, Gumbo T, Forrest A, et al. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it’s not just for mice anymore. Clin Infect Dis. 2007;44(1):79–86. doi: 10.1086/510079. [DOI] [PubMed] [Google Scholar]
- 25.Ambrose PG, Bhavnani SM, Owens RC Jr. Clinical pharmacodynamics of quinolones. Infect Dis Clin North Am. 2003;17(3):529–543. doi: 10.1016/s0891-5520(03)00061-8. [DOI] [PubMed] [Google Scholar]
- 26.Öbrink-Hansen K, Hardlei TF, Brock B, Jensen-Fangel S, Thomsen MK, Petersen E, et al. Moxifloxacin pharmacokinetic profile and efficacy evaluation in empiric treatment of community-acquired pneumonia. Antimicrob Agents Chemother. 2015;59(4):2398–404. doi: 10.1128/AAC.04659-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Redgrave LS, Sutton SB, Webber MA, Piddock LJV. Fluoroquinolone resistance: mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 2014;22(8):438–445. doi: 10.1016/j.tim.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 28.Kees MG, Schaeftlein A, Haeberle HA, Kees F, Kloft C, Heininger A. Population pharmacokinetics and pharmacodynamic evaluation of intravenous and enteral moxifloxacin in surgical intensive care unit patients. J Antimicrob Chemother. 2013;68(6):1331–7. doi: 10.1093/jac/dkt040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Please contact the corresponding author for data requests.