

Abstract
The intrinsic mechanisms sensing the imbalance of energy in cells are pivotal for cell survival under various environmental insults. AMP-activated protein kinase (AMPK) serves as a central guardian maintaining energy homeostasis by orchestrating diverse cellular processes, such as lipogenesis, glycolysis, TCA cycle, cell cycle progression and mitochondrial dynamics. Given that AMPK plays an essential role in the maintenance of energy balance and metabolism, managing AMPK activation is considered as a promising strategy for the treatment of metabolic disorders such as type 2 diabetes and obesity. Since AMPK has been attributed to aberrant activation of metabolic pathways, mitochondrial dynamics and functions, and epigenetic regulation, which are hallmarks of cancer, targeting AMPK may open up a new avenue for cancer therapies. Although AMPK is previously thought to be involved in tumor suppression, several recent studies have unraveled its tumor promoting activity. The double-edged sword characteristics for AMPK as a tumor suppressor or an oncogene are determined by distinct cellular contexts. In this review, we will summarize recent progress in dissecting the upstream regulators and downstream effectors for AMPK, discuss the distinct roles of AMPK in cancer regulation and finally offer potential strategies with AMPK targeting in cancer therapy.
INTRODUCTION
Energy balance is important for maintaining cell survival and body homeostasis. Imbalance of energy metabolism accompanied by obesity often results in the development of cancers, type 2 diabetes, cardiovascular disease, non-alcoholic fatty liver disease (NAFLD) and kidney disease. In order to cope with diverse stresses leading to energy imbalance, cells need to develop protective mechanisms for maintaining energy homeostasis and cell survival. AMP-activated protein kinase (AMPK) is a well-known energy sensor that is activated to regulate diverse signals and metabolic pathways in response to various stimuli including caloric restriction [1], exercise [2], aging [3–5] and obesity [6], thereby sustaining energy homeostasis and serving as the guardian of metabolism.
As key energy molecules, adenine nucleotide ATPs are constantly generated to meet cell energy demands through diverse metabolic pathways. As an energy sensor, AMPK senses the increased ratio of AMP/ATP or ADP/ATP and is activated by upstream kinases such as liver kinase B1 (LKB1) [7–9] and calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2 or CaMKKβ) [10–12]. As a consequence, AMPK turns on the catabolic processes such as glycolysis [13, 14], TCA cycle [15] and fatty acid oxidation [16, 17] to produce energy, but shuts off the anabolic metabolism including fatty acid synthesis, protein synthesis and steroid synthesis to limit energy usage by inactivating mTORC1 [18], thus maintaining the energy homeostasis.
Post-translational modification of AMPK has been demonstrated to play an important role in the stability of AMPK complexes and also directly impact on the AMPK enzymatic activity. Phosphorylation of AMPK on Threonine 172 (T172) residue dramatically increases AMPK activity [19, 20] to further phosphorylate its downstream substrates, thus governing various biological functions such as cellular metabolism, mitochondrial dynamics, Golgi assembly, gene transcription and cell cycle progression. ACC phosphorylation by AMPK leads to the inhibition of lipogenesis [16, 17]. MFF (mitochondrial fission factor) and ULK1 (unc-51 like autophagy activating kinase 1) phosphorylation by AMPK induces mitochondrial fission and mitophagy respectively [21–23]. Either inhibition of fatty acid synthesis or promotion of mitochondrial dynamics through AMPK activation eventually facilitates cellular adaptation to metabolic stress. Thereby, understanding how AMPK is activated as well as the downstream signals AMPK regulates will yield useful strategies to develop small molecules for targeting AMPK signals in the treatment of metabolic diseases or cancer.
For the past two decades, many small molecules have been developed for AMPK targeting. Metformin [24] and salicylate [25], widely used drugs for type 2 diabetes treatment, could indirectly activate AMPK, leading to the increase of glucose uptake. Additionally, several herbal medicines such as berberine [26–28] and resveratrol [27] have been shown to indirectly activate AMPK via the inhibition of mitochondrial respiration. Importantly, some compounds have been identified to activate AMPK by directly associating with its α, β or γ subunits, which in turn causes the allosteric conformational alteration of AMPK complex. Recently, sodium/glucose cotransporter 2 inhibitor (canagliflozin) [29, 30], PF-739 [31], PF-06409577 [32] and PAN-AMPK activator (MK-8722) [33] have emerged preclinical and clinical efficacy of reducing blood glucose similar to metformin and phenformin. Accumulated studies have shown that pharmacological activation of AMPK by several small molecules is useful for the treatment of metabolic disorders such as type 2 diabetes and obesity, although pharmacological targeting AMPK for cancer treatment still remains controversial in different cancer types.
In this review, we will describe the upstream regulation and downstream substrates of AMPK under physiological stresses. Moreover, we will elaborate the biological functions of AMPK in various cellular compartments including mitochondria, Golgi apparatus, ER (endoplasmic reticulum) and nucleus [34]. Finally, we will discuss the distinct roles of AMPK in cancer regulation and its targeting strategies by small molecules for cancer therapy in preclinical models.
AMPK COMPLEX
AMPK forms a heterotrimeric complex, which is composed of catalytic α subunits and regulatory β and γ subunits. There are multiple genes encoding each subunit, generating two α (α1, α2), two β (β1, β2) and three γ subunits (γ1, γ2, γ3). Thus, AMPK heterotrimers can form up to 12 αβγ combinations which display slightly different subcellular or tissue-specific distribution [35]. For instance, isoforms with catalytic α1 and regulatory β2 are predominantly expressed in human liver [36], while isoforms with γ2 subunit are highly expressed in human heart [36]. In rodent liver, α1 and α2 are expressed with similar levels, while β1 and γ1 are the major regulatory subunit isoforms [37]. Different from human heart, the isoforms with γ1 subunit are more predominant in rodent heart [36].
AMPKα subunits
AMPKα subunits consist of kinase domain (KD) containing N-lobe and C-lobe, autoinhibitory domain (AID), α-regulatory subunit-interacting motif (α-RIM) in linker peptides (α-linker), followed by the α-subunit carboxy-terminal domain (α-CTD) which binds to β subunits [38, 39]. The T172 residue within C-lobe is phosphorylated by upstream kinases such as LKB1 and CaMKK2, which largely accounts for the activation of AMPK complex [8, 10] (Fig. 1). Several small molecules such as 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) and PF-739 have been designed to target T172 phosphorylation of α-subunits, thereby manipulating AMPK activity [31, 40]. AID could compromise AMPK activity approximately tenfold, while removal of AID leads to constitutive activation of AMPK [39, 41]. Followed by AID, α-RIM (also known as α-hook) containing about 60 amino acids is essential for nucleotide-dependent regulation of AMPK [42, 43].
Fig.1. Functional domain of AMPK subunits.

Kinase domain (KD) of AMPKα subunits contains N-lobe and C-lobe and threonine 172 (T172) for phosphorylation by upstream kinases at C-lobe, followed by autoinhibitory domain (AID), aregulatory subunit-interacting motif (α-RIM) in linker peptides (α-linker) and α-subunit carboxy-terminal domain (α-CTD) which binds to b subunits. The functional domains of β-subunits include carbohydrate-binding module (β-CBM), binding to glycogen, and β-subunit C-terminal domain (β-CTD) which interacts with both α-subunits and the γ-subunits. Four tandem repeats of CBS motifs (CBS1, CBS2, CBS3 and CBS4) and two CBS repeats form a Batedomain in AMPKγ subunits are in charge of binding to adenine nucleotides (AMP, ADP or ATP) except for CBS2 motif.
AMPKβ subunits
AMPKβ subunits contain two functional domains including β-subunit carbohydrate-binding module (β-CBM) which associates with glycogen or starch in mammals and β-subunit C-terminal domain (β-CTD) which interacts with both the α-CTD and the γ-subunit (Fig. 1). AMPK complex phosphorylates glycogen synthase via the binding between β-CBM and glycogen, leading to the inactivation of glycogen synthase [44, 45]. Moreover, β-CBM also interacts with N-lobe of the kinase domain of α subunit through the surface opposite to its glycogen binding site [46, 47]. Importantly, the cleft between β-CBM and α subunit forms the binding site for ligands acting on AMPK, which is called allosteric drug and metabolite (ADaM) site. Once ligands bind to ADaM site, AMPK can be allosterically activated [46, 48].
AMPKγ subunits
ΑΜPΚγ subunits contain four tandem repeats named CBS1, CBS2, CBS3 and CBS4. Two CBS repeats assemble a Batedomain which binds to adenine nucleotides such as AMP, ADP and ATP (Fig. 1). In AMPKγ, the four CBS repeats in the γ-subunits form a flattened disk containing four potential adenine nucleotides-binding sites [49, 50]. In accordance with crystal structure of mammalian AMPK complexes, three AMP binding sites (sites 1, 2 and 3) identified within CBS1, CBS3 and CBS4 motifs respectively display conserved aspartate residue [51, 52]. In mammalian γ1 subunit, sites 1 and 2 represent the regulatory sites and are critically involved in the competition of AMP with ADP or ATP, whereas site 3 shows tight interaction with AMP, indicating that this non-exchangeable site is dispensable for AMP/ATP sensing [37]. In addition, ADP could also bind to two exchangeable adenine nucleotides binding sites on γ subunit, which in turn protects the enzyme from dephosphorylation without causing allosteric activation [38].
AMPK ACTIVATION
Adenine nucleotides dependent activation
AMPK is activated through both canonical and non-canonical regulation. AMPK, so called AMP-activated protein kinase, could be allosterically activated by 5’-AMP, a process referred as canonical adenine nucleotides dependent regulation [53, 54]. The association of AMP with AMPK leads to the enhancement of AMPKα T172 phosphorylation while inhibition of AMPKα T172 dephosphorylation, resulting in allosteric activation of AMPK complex. It has been demonstrated that LKB1 facilitates phosphorylation of AMPKα on T172 in response to elevated AMP/ATP ratio [47]. AMP has also been reported to promote AMPK T172 phosphorylation leading to allosteric activation of AMPK [55]. In addition, myristoylation of β subunit is required for adenine nucleotides-stimulated AMPKα T172 phosphorylation [56, 57]. Remarkably, AMPKγ subunits containing four CBS repeats whose structures have been solved with AMP, ADP and/or ATP bound at different sites [52, 58] play an important role in both T172 phosphorylation of AMPKα and allosteric activation of the whole complex [38, 52, 58, 59]. The R531G mutation in the AMPKγ2 subunit frequently observed in human heart disease abrogates allosteric activation and AMPKα T172 phosphorylation by AMP [60].
Activation via ADaM binding sites
Interestingly, the association of ligands with ADaM binding sites also leads to the activation of AMPK by protecting AMPKα T172 residue from dephosphorylation, in addition to nucleotide-binding [46, 48, 51]. Ligand binding at ADaM sites may potentiate the interaction between β-CBM and α-kinase domain, thereby abolishing dephosphorylation of AMPKα [51]. Furthermore, β1 subunit phosphorylation on Serine 108 (S108) residue also accounts for another important mechanism for AMPK activation by ADaM site ligand, A769662 [48]. Of importance, phosphorylation of S108 on β1 subunit enhances the binding affinity of ligands (A769662 and 991) at ADaM site [51]. Notably, the integrity of AMPK complex is indispensable for allosteric activation by ADaM site ligands. Crystal structure analysis reveals that outside of CBM consensus sequence of β subunit directly interacts with α-helix C region of the kinase domain (referred to as the C-interacting helix) of α subunit [51]. Mutation of Leucine 166 residue in the C-interacting helix dampens the association between α and β subunits, thus significantly compromising allosteric activation of AMPK by 991 [51]. All the evidence indicates that both the integrity of AMPK complex and β subunit phosphorylation are required for allosteric activation of AMPK by ADaM site ligands such as A769662 and 991. In addition, salicylate, a natural compound derived from willow bark, directly activates AMPK via binding to the ADaM site [25]. AMPK heterotrimers with various subunits combination display different binding affinity with ADaM site activators. For example, PF249 and PF06409577, the other two ADaM site activators, bind more tightly to β1-containing than to β2-containing AMPK complexes [32]. However, PF739 and MK-8722 both effectively bind to β1- or β2-containing AMPK complexes [31, 33]. Given that either nucleotide-binding to γ-CBS domain or ligand binding to ADaM sites can activate AMPK, it is conceivable that combination treatment targeting both nucleotide-binding and ADaM binding sites on AMPK complexes may lead to more robust and efficient activation of AMPK, representing the promising strategy to target AMPK for therapy [61–64].
Adenine nucleotides independent activation
AMPK activation can also be modulated in an adenine nucleotides independent manner, referred as non-canonical regulation. For decades, it has been speculated that decrease of AMP/ATP ratio serves as the main mechanism accounting for glucose deprivation induced AMPK activation. Thus, it is widely accepted that AMPK is activated through canonical adenine nucleotides dependent regulation under glucose restriction condition. However, the kinetic model suggests that only glucose removal is able to rapidly activate AMPK within 15 minutes without impact on the intracellular ADP/ATP or AMP/ATP ratio within the same time range, although complete restriction of carbon source could trigger more substantial and sustained activation of AMPK, correlated with elevated AMP/ATP ratio [65, 66]. These findings indicate that glucose deprivation alone activates AMPK in a fast but AMP/ADP-independent manner, while complete or prolonged removal of carbon source leads to hyperactivation of AMPK in a delayed and AMP/ADP-dependent manner. In line with this, cells with AMPKγ2 R531G mutation, insensitive to enhanced intracellular AMP or ADP level, still display rapid AMPK activation upon glucose deprivation [66], indicative of the other mechanism involved in low glucose driven AMPK activation. Recent study uncovers the glycolytic enzyme, aldolase, appears to play a non-canonical role for AMPK activation under glucose deprivation. Aldolase promotes the formation of lysosomal complexes containing the v-ATPase, Ragulator, AXIN, LKB1 and AMPK under glucose deprivation, leading to LKB1 mediated AMPKα T172 phosphorylation irrespective of the ADP/ATP or AMP/ATP [65]. In the glucose proficient condition, Fructose-1,6-biphosphate (FBP), a glycolytic intermediate generated from glycolysis, binds to aldolase and disrupts the association of AXIN/LKB1 with v-ATPase/Ragulator, resulting in defective AMPK activation [65]. Thus, aldolase serves as a new modulator for AMPK activation by sensing cellular FBP availability from glucose metabolism. Glucose starvation relieves the inhibitory effect of FBP on aldolase-mediated lysosome complexes formation, thereby facilitating AMPK activation in an adenine nucleotides independent manner [65].
Apart from glucose deprivation, intracellular Ca2+ ions can indirectly activate AMPK through CaMKK2 activation without alteration of adenine nucleotide levels. Other physiological stimuli including hypoxia, oxidative stress or growth factor stimulation could also activate AMPK without the change of AMP/ATP ratio [67]. Interestingly, AMPK activation can be also induced by DNA damage partially through ATM-LKB1 cascade [68–70]. However, other mechanisms through yet to be discovered are also involved in DNA damage induced AMPK activation [71]. Future studies aiming at identifying the additional mechanisms by which diverse stresses induce AMPK activation will be warranted to further unravel the complexity for AMPK activation.
DOWNSTREAM TARGETS OF AMPK
As a kinase, AMPK regulates diverse biological processes primarily through phosphorylating its downstream targets. In essence, AMPK recognizes and phosphorylates its substrates through a consensus motif with hydrophobic residues at −5 and +4 positions of actual phosphorylation site [72]. The consensus motif has been identified including LRRVXSXXNL, MKKSXSXXDV, IXHRXSXXEI via quantitative phosphoproteomics datasets [72]. In addition, basic residues at −3 or −4 position and neutral polar residues (usually Serine) at −2 position are regarded as secondary AMPK consensus motif [73]. However, some AMPK targets such as H2B [74] with Arginine (R) at −5 position and PGC1α [75] with Asparagine (N) and Phenylalanine (F) at −5 position for two phosphorylation sites, respectively, suggesting the flexibility and complexity of AMPK consensus motif. Below are some key substrates of AMPK whose phosphorylation by AMPK critically regulates their activity and functions in diverse biological processes (Fig. 2).
Fig. 2. Physiological role modulated by downstream substrates of AMPK.
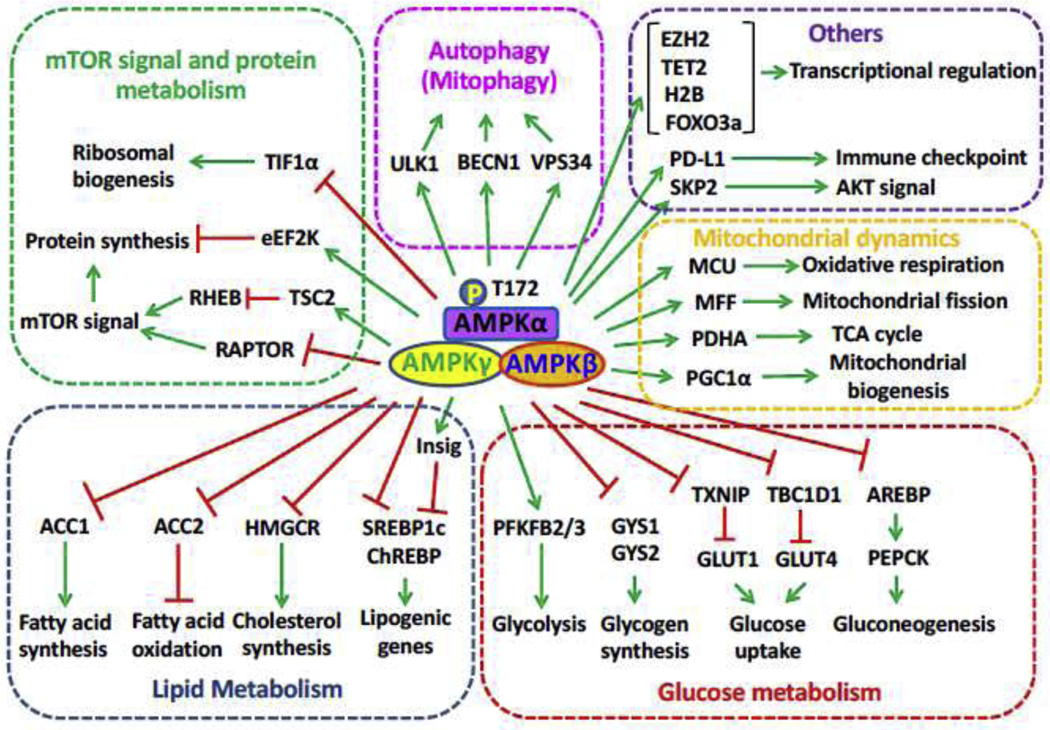
Protein synthesis, autophagy/mitophagy, lipid metabolism, glucose metabolism and transcriptional regulation are modulated by AMPK via phosphorylating its downstream substrates.
ACC
ACC (Acetyl-CoA carboxylase) is critically involved in fatty acid metabolism. Two isoforms of ACC (ACC1 and ACC2) have been identified, and both of which catalyze the carboxylation of Acetyl-CoA to malonyl-CoA. Unlike cytoplasmic ACC1, which plays an essential role in fatty acids synthesis, ACC2 is associated with mitochondria [76] and serves as a negative regulator for β-oxidation through allosteric inhibition of CPT-1 (carnitine palmitoyltransferase 1) critically involved in β-oxidation [77]. AMPK mediated phosphorylation of ACC has been unraveled for decades, and ACC1 Ser79 (S79) phosphorylation (S79 for mouse and rat, S80 for human) serves as a universal indicator for AMPK activation [78]. Although AMPK could trigger rat ACC1 phosphorylation on S79, S1200 and S1215 under in vitro condition, S79 phosphorylation is well characterized in vivo and is associated with ACC1 inactivation. Likewise, AMPK also induces S219 phosphorylation of ACC2 equivalent to S79 in ACC1, leading to impaired ACC2 activity and enhancing β-oxidation in mouse skeletal muscle [17]. Crystal structure study from yeast ACC reveals that AMPK mediated phosphorylation of ACC may dissociate the functional biotin-carboxylase domain dimer into inactive monomers, thereby repressing ACC catalytic activity [79]. It will be interesting to know whether the similar mechanism for ACC regulation can be also applied to mammalian cells.
ULK1
ULK1, together with FIP200 (focal adhesion kinase family interacting protein of 200 kDa), ATG13 (autophagy-related protein 13), and ATG101, serves as an important regulator for autophagy to maintain protein homeostasis, especially under nutrient starvation condition, by initiating the formation of autophagosome [80]. AMPK phosphorylates ULK1 on multiple sites from numerous studies including S467, S555, T574, S637 [23, 81], S317 and S777 [82]. Of note, AMPK mediated phosphorylation on ULK1 appears to be essential for ULK-mediated autophagy or mitophagy and cell survival upon stress [23, 82]. Other study reported that human ULK1 is hyper-phosphorylated on S638 and S758 (equivalent to S637 and S757 in mouse) under nutrient proficient condition, but dephosphorylation on S638 followed by S758 dephosphorylation occurs upon nutrient starvation. Importantly, phosphorylation of ULK on S638 and S758 is mediated by active mTOR under nutrient proficient condition, and dephosphorylation of ULK1 on S758 drives its dissociation with AMPK and activates ULK to elicit autophagy [81]. Interestingly, basal AMPK binds to ULK1 and limits its function by phosphorylating S638 under steady state, but activated AMPK promotes autophagy through phosphorylating ULK1 on more sites in response to starvation [81]. Thus, AMPK plays a dual role in mediating ULK1 phosphorylation towards autophagy regulation. Although AMPK-mediated ULK1 phosphorylation in governing autophagy has been unraveled for years, the exact phosphorylation sites and detailed mechanism underlying this process are still under debate.
MFF
Dynamic mitochondrial homeostasis between fusion and fission is essential for mitochondrial health and function [83, 84]. In general, mitochondrial fusion maximizes the energy production in response to mild nutrient shortage, while severe nutrient deprivation leads to mitochondrial fission and fragmentation followed by mitophagy [85]. MFF (mitochondrial fission factor), as a critical component of mitochondrial fission sensor, functions in the recruitment of DRP1 (dynamin-related protein 1) to the outer membrane of mitochondria to initiate fission process by inducing membrane constriction and scission [86]. AMPK elicits MFF phosphorylation on S155 and S172 upon energy stress [22]. Notably, compared with WT MFF, restoration of AMPK nonphosphorylatable SA2 (S155A, S172A) mutant MFF into Mff−/− MEFs attenuates the recruitment of DRP1 to mitochondrial outer membrane in response to AICAR or rotenone treatment. Interestingly, re-introduction of phosphomimetic SD2 (S155D, S172D) or SE2 (S155E, S172E) mutant MFF into Mff−/− MEFs leads to enhanced mitochondrial fragmentation even without extracellular stimuli, comparable to those cells with WT MFF restoration upon AICAR induction [22]. Thus, AMPK mediated phosphorylation of MFF on S155 and S172 critically drives mitochondrial fission process by facilitating the recruitment of DRP1.
TSC2 and RAPTOR
mTORC1 consisting of diverse proteins serves as the central regulator governing cell metabolism, survival, proliferation and growth, whose dysregulation results in various diseases including cancer and diabetes [18]. TSC2 (Tuberous sclerosis complex 2, also known as Tuberin) with GTPase activating protein (GAP) activity severs as a critical negative regulator of mTORC1 through inhibiting RHEB GTPase, which functions as a direct activator of mTORC1 [87]. TSC2 phosphorylation by mitogen-activated kinases including AKT, ERK, and RSK leads to its inactivation, in turn stimulating mTORC1 signaling [88]. In contrast, phosphorylation of TSC2 on T1227 and S1345 by AMPK in response to energy deprivation enhances the activity of TSC2 leading to impaired mTORC1 activity and protein translation [89, 90]. Thus, AMPK dependent TSC2 phosphorylation regulates protein homeostasis in response to various energy conditions [90].
Accumulated evidence reveals that TSC2 may not be the only substrate of AMPK for its regulation on mTORC1 signaling. First, although the negative correlation between mTORC1 and AMPK activation is observed throughout all eukaryotes, including C. elegans and S. cerevisiae, TSC2 ortholog has not been identified in either of those species [91], suggesting AMPK governs mTORC1 signaling through other mechanism in these species. Second, AMPK activation by AICAR or phenformin still profoundly compromises mTORC1 signaling in Tsc2 −/− MEFs or other TSC2 deficient cells [92, 93]. Using bioinformatic analysis based on AMPK consensus motif, mTOR binding protein RAPTOR [94] has been revealed as an ideal AMPK substrate. Further experiments demonstrated that AMPK phosphorylates RAPTOR on S722 and S792, and this phosphorylation is essential for energy stress-induced mTORC1 inhibition [93]. Mechanistically, RAPTOR phosphorylation by AMPK promotes the interaction of RAPTOR with 14–3-3 proteins, thus leading to inactivation of mTORC1, cell cycle arrest and cellular apoptosis upon metabolic stress.
Collectively, AMPK compromises mTORC1 activity through phosphorylating both TSC2 and RAPTOR, placing AMPK as a key energy sensor in coordinating cell growth and proliferation under distinct energy statuses.
H2B
Posttranslational modifications including phosphorylation on histones are critically involved in transcription regulation [95, 96]. The involvement of AMPK in transcriptomic reprogramming to elicit long term adaptation of cells to various stress conditions has been documented in multiple studies, although the mechanism underlying this regulation remains largely unknown [97]. The core histone H2B is phosphorylated by AMPK on S36 in response to metabolic stress [74]. Importantly, H2B S36 phosphorylation is abrogated in both Lkb1−/− and Ampk−/− MEFs upon energy stress or AMPK activators, suggesting LKB1 likely acts through AMPK activation to stimulate H2B S36 phosphorylation in response to diverse metabolic stresses. Further, S36 phosphorylation on H2B is required for its interaction with AMPK. ChIP analysis reveals that a positive correlation between AMPK and H2B S36 phosphorylation in the gene transcribed region is observed in response to glucose deprivation and UV treatment [74]. Interestingly, AMPK appears to be specifically recruited to the promoter region of p53 target genes such as p21 and cpt1c, reminiscent of the previous observation that AMPK induces a metabolic checkpoint in a p53 dependent manner [98]. Moreover, enhanced co-localization between AMPK and H2B S36 phosphorylation at p53 binding site and also along the transcription region is observed, suggesting AMPK, via H2B S36 phosphorylation, cooperates with p53 to modulate gene transcriptional elongation to elicit adaptive response, thus likely protecting cells from cell death upon stress conditions. Indeed, ablation of H2B S36 phosphorylation compromises RNA polymerase II association to transcribed region, leading to impaired transcription of AMPK target genes and reduced cell survival upon stresses [74]. Whether AMPK could similarly govern other stress-induced transcription factors such as FOXO3a [99] and PGC1α [100] through mediating H2B phosphorylation warrants further investigation.
SKP2
Ubiquitination, as an essential post-translational modification, plays an important role in diverse cellular and biological processes through maintaining protein homeostasis and activity [101]. SKP2, as a critical oncogenic F-Box E3 ligase [102], is involved in both protein degradation through mediating K48-linked ubiquitination [103] and non-proteolytic function by conjugating K63-linked ubiquitination [104]. Notably, activation of AMPK, via CaMKK2 upon EGF stimulation, could induce SKP2 phosphorylation on S256, thus promoting the interaction between SKP2 and Cullin1 to maintain the integrity of SKP2-SCF complex [67]. Moreover, AMPK mediated SKP2 phosphorylation on S256 is required for SKP2 E3 ligase activity to elicit K63-linked ubiquitination on AKT, leading to the activation of AKT and subsequent oncogenic processes [67]. Further, AMPK also protects cancer cells from hypoxia or glucose deprivation induced cell death partially through orchestrating SKP2 S256 phosphorylation and subsequent AKT activation [67]. Importantly, AMPK/SKP2 S256 phosphorylation/AKT cascade is activated in advanced breast cancer and its activation predicts poor survival outcome of breast cancer patients, indicating the targeting AMPK or AMPK mediated SKP2 phosphorylation is a novel strategy for breast cancer therapy. Given that SKP2 E3 ligase activity is indispensable for cellular response to other stress conditions such as DNA damage [105] and oxidative stress [106], it would be interesting to determine whether SKP2 phosphorylation on S256 by AMPK is also involved in these adaptive responses.
PDHA
Pyruvate dehydrogenase complex (PDHc) is a multi-enzyme complex and acts as a rate-limiting enzyme to maintain TCA cycle by catalyzing pyruvate to Acetyl-CoA [107]. The activity of PDHc largely depends on the phosphorylation status on its catalytic subunit PDHA. PDH kinase (PDHK) mediated PDHA phosphorylation on S293 serves as a negative signal to suppress the activation of PDHc [108]. Dephosphorylation of PDHA S293 site ensures the activity of PDHc to maintain TCA cycle through facilitating pyruvate metabolism, although the underlying mechanism remains elusive. Recently, AMPK is found to co-localize with PDHA in mitochondrial matrix and serve as a direct kinase to drive sequential PDHA phosphorylation on S314 and S295 sites [15]. Interestingly, PDHA S314 phosphorylation serve as a prime signal to compromise the interaction of PDHA with PDHK, resulting in the attenuated phosphorylation on S293. Due to the steric effect, dephosphorylation of S293 would provide space for AMPK to further induce PDHA phosphorylation on its neighboring S295 residue, which acts as an intrinsic catalytic site essential for PDHc activation and pyruvate metabolism [15]. Surprisingly, even with impaired S293 phosphorylation, PDHc with dephosphorylation on PDHA S295 site still largely compromise its ability to catalyze pyruvate to Acetyl-CoA [15], highlighting the essential role of AMPK mediated PDHA S295 phosphorylation in maintain PDHc activity [109]. Functionally, activation of AMPK-PDHc axis facilitates breast cancer lung metastasis through rewiring the metabolic pathway of cancer cells from glycolysis to TCA cycle, thus enabling disseminated cancer cells to cope with diverse stresses in the metastatic microenvironment [15]. Since dysregulation of TCA cycle is associated with multiple metabolic disorders including diabetes [110], obesity [111] and Alzheimer’s disease [112], AMPK mediated PDHA phosphorylation and PDHc activation may contribute to the development of these pathological disorders.
REGULATION OF AMPK EXPRESSION AND ACTIVITY
Dysregulation of AMPK activity is linked to aging process and various human diseases including metabolic syndrome, stroke, cardiovascular disease and cancer [113]. As the critical catalytic subunit, AMPKα expression and its kinase activity are tightly controlled through multiple layers of regulation including transcription, protein stability, and post-translational modifications under physiological condition (Fig. 3). We will briefly summarize how AMPK is regulated through transcription and post-translational modifications in this section.
Fig. 3. Regulation of AMPK by transcription, protein stability and post-translational modifications.

AID, autoinhibitory domain; CTD, C terminus domain; NTD, N terminus domain; CBM, carbohydrate-binding module; CBS, cystathionineb-synthase repeats; RIM, regulatory-subunit-interacting motif; ST-loop, serine/threonine-enriched loop
Regulation of AMPK expression
Transcription regulation and post-transcriptional regulation
ΔNp63α, the predominant p63 isoform expressed in epithelial cells, has been identified as a crucial transcription factor for AMPKα1 transcription. Four putative p63 binding element (CNNGNNNNNNCNNG) [114] on AMPKα1 promoter have been identified. Notably, ectopic expression of ΔNp63α, but not the DNA-binding defective mutant ΔNp63αC306R, significantly elevates AMPKα1 mRNA expression, suggesting ΔNp63α likely serves as a direct transcription factor to transactivate AMPKα1 gene expression [115].
Sestrins are stress-induced proteins conserved throughout the species. Enhanced sestrin proteins account for the acquired metabolic benefits of exercise [116]. Interestingly, sestrins could compromise mTORC1 signaling by activating AMPK [117]. Further, sestrins are found to not only physically associate with AMPK, but also modulate the transcription of AMPK subunits including AMPKα1 and AMPKβ1 [118], although the underlying mechanism and transcription factors involved remain unclear.
MicroRNAs (miRs), as single-strand non-coding RNAs, typically compromise the translation of target mRNAs through recognizing the complementary sequences of 3ÙTR regions, thus governing gene expression [119]. MiR-27b is shown to target AMPKα2 and leads to its downregulation. Moreover, pharmacological ablation of MiR-27b in mice improves the neurogenesis and neurological function upon permanent middle cerebral artery occlusion in an AMPK-dependent manner [120]. Through in silico study, AMPKα1 3` UTR region contains multiple recognition sites of miR-19b, miR-130, miR-101, and miR-19a [121], indicating that AMPKα1 expression may be also regulated by these miRs.
Regulation of AMPK protein stability
Ubiquitin-Proteasome System (UPS) is essential for the protein homeostasis through tightly regulating protein degradation [122]. The role of UPS in governing AMPK function in response to energy and metabolic stresses has started to emerge.
Makorin ring finger protein 1 (MKRN1), as an E3 ligase, is linked to tumorigenesis and adipocyte differentiation by mediating ubiquitination and proteasomal degradation of its target proteins such as p53 and PPARγ [123, 124]. Recently, MKRN1 is identified as a direct E3 ligase for AMPKα subunit (AMPKα1 and AMPKα2) and mediates proteasome-dependent degradation of AMPKα subunit [125]. Interestingly, MKRN1 depletion leads to the accumulation of AMPKα proteins, without impacts on AMPKβ and AMPKγ protein level, suggesting the specific role of MKRN1 in the regulation of AMPKα subunit. Notably, Mkrn1 null mice display chronic AMPK activation in the liver as well as white and brown adipose tissues, indicative of enhanced AMPK signal upon Mkrn1 deficiency in vivo. Moreover, activated AMPK in Mkrn1 null mice prevent high fat diet induced metabolic syndromes including insulin resistance, obesity and non-alcoholic fatty liver disease [125], which could be reversed by the genetic ablation of AMPK. As MKRN deficiency alleviates obesity and its related comorbidities through AMPK activation, targeting MKRN to boost AMPK signal represents a potential strategy to treat metabolic syndromes.
MAGE-A3 and MAGE-A6 are expressed mostly in testis [126], but aberrantly activated during malignant transformation to drive robust anchorage-independent growth and cancer metastasis [127, 128]. Interestingly, MAGE-A3/6 is found to specifically associate with TRIM28 E3 ubiquitin ligase and targets ubiquitination and proteolysis of p53 [129, 130], accounting for the oncogenic property of MAGE-A3/6. Using systematic protein arrays containing more than 9,000 recombinant proteins for screening additional substrates, AMPKα1 is identified as the most consistent and robust target of MAGE-A3/6-TRIM28 [131]. Depletion of MAGE-A3/6 or TRIM28 results in the elevation of AMPKα1 protein level without affecting its mRNA level. Conversely, ectopic expression of MAGE-A3 in MAGE-A3/6 deficient cells promotes proteasome-dependent AMPKα1 degradation [131]. As such, restriction of AMPKα1 by MAGEA3/6-TRIM28 is associated with mTOR activation, leading to impaired autophagy. Notably, enhanced anchorage-independent growth upon MAGE-A6 overexpression is compromised by the treatment of AMPK agonists such as AICAR and metformin, suggestive of a potential strategy for targeting MAGE-A3/6-positive tumors by inducing AMPK activation. Collectively, AMPKα1 is a bona fide substrate of MAGE-A3/6-TRIM28 E3 ligase complex whose inactivation accounts for oncogenic activity regulated by MAGE-A3/6.
GID (glucose induced degradation deficient) ubiquitin ligase complex has been shown to be another E3 ligase targeting phosphorylated AMPKα for ubiquitination and proteasomal degradation, thus preventing sustained activation of AMPK upon prolonged starvation [132]. Remarkably, depletion of GID complex components in C. elegans. drives AMPK hyperactivation and profoundly enhances the lifespan partially due to mTOR inactivation [132].
UBE2O (Ubiquitin-conjugating enzyme E2O), unlike the traditional E2 ubiquitin-conjugation enzyme, acts as an E2/E3 hybrid ubiquitin-protein ligase that contains both E2 and E3 ligase activities [133]. AMPKα2 appears to be a direct substrate of UBE2O, as UBE2O could directly induce K48-linked ubiquitination of AMPKα2 on Lys (K) 470, which is conserved among various species, in the presence of only the E1 enzyme, thus leading to degradation of AMPKα2 [134]. However, this K470 residue is replaced by Arginine (R475) in AMPKα1, likely accounting for the selective ubiquitination of AMPKα2 by UBE2O. As such, the protein expression level of AMPKα2, but not AMPKα1, is robustly elevated in Ube2o−/− mouse tissues, further confirming the specificity of AMPKα2 degradation by UBE2O. In MMTV-PyVT and TRAMP transgenic mouse models, UBE2O deficiency attenuates breast cancer and prostate cancer progression and metastasis, which could be rescued upon Ampkα2 ablation [134], underscoring the oncogenic role of UBE2O in cancer regulation through promoting AMPKα2 degradation.
Additionally, WWP1 (WW domain-containing E3 ubiquitin protein ligase 1) could also associates with and downregulates AMPKα2 in response to high energy status [135]. However, it remains to be determined whether WWP1 could directly target AMPKα2 for ubiquitination and degradation. Moreover, Cidea interacts with AMPK complex in the endoplasmic reticulum through specific association with AMPKβ subunit, leading to its ubiquitination and degradation [136]. Enhanced expression and enzyme activity of AMPK observed in Cidea−/− mice critically overcome diet induced obesity by facilitating energy expenditure [136].
Collectively, these studies underscore the importance of UPS in the regulation of various subunits of AMPK complex, thereby affecting enzyme activity and biological functions of AMPK.
Post-translational modifications of AMPK
Phosphorylation
Phosphorylation on T172 residue of AMPK α subunit (T185 in AMPKα1, T172 in AMPKα2) bolsters the activity of AMPK complex up to 100 fold [19]. LKB1 is the well characterized upstream kinase mediating AMPKα T172 phosphorylation in response to elevated AMP/ATP ratio under metabolic stress [8]. LKB1 inactivation through disruption of LKB1-STRAD-MO25 complex abolishes AMPK activation [137, 138]. CaMKK2 is another well defined kinase for AMPK activation by phosphorylating AMPK at T172 [12, 67]. It appears that TAK1 (TGF-β-activated kinase 1) may be the third kinase for AMPKα T172 phosphorylation, since TAK1 along with TAB1 (TAK1-binding proteins) could also stimulate AMPKα T172 phosphorylation both in vitro and in vivo [139]. However, the notion that TAK1 could act as a direct kinase is still under debate [140].
A recent study indicates that activation of LKB1/AMPK axis upon energy stress elevates the expression of the long non-coding RNA NBR2 (neighbor of BRCA1 gene 2), which then binds to the kinase domain of AMPKα subunit and boosts its T172 phosphorylation and activation irrespective of LKB1 status [141]. This feed-forward loop is considered necessary for the maintenance of AMPK activity in response to long-term energy crisis [142], but the detailed mechanism by which NBR2 governs AMPK activation remains elusive.
AMPKα T172 dephosphorylation leads to AMPK inactivation. This dephosphorylation process is mediated by at least three phosphatases including protein phosphatase 2A (PP2A) [143], protein phosphatase 2C (PP2C) [144] and Mg2+-/Mn2+-dependent protein phosphatase 1E (PPM1E) [145]. Of note, the binding of AMPKγ to AMP during energy stress relieves the inhibitory effect of these phosphatases on AMPKα T172 phosphorylation, leading to AMPK activation [146].
Additionally, phosphorylation on other residues, especially in ST-loop of AMPKα, by other kinases serves as a negative signal for AMPK activation [147]. PKA (cyclic-AMP-dependent protein kinase) and AKT mediated phosphorylation of S485 in AMPKα1 (S491 in AMPKα2) limits AMPK activation through antagonizing T172 phosphorylation during gluconeogenesis or insulin stimulation [148, 149]. Similarly, S6K-mediated phosphorylation on S491 of AMPKα2 upon leptin treatment impairs AMPK activity in hypothalamus [150]. Moreover, PKA triggers AMPKα1 phosphorylation on multiple residues including S173, S485, S497 in adipocyte context, and S173 phosphorylation impedes LKB1 mediated phosphorylation on T172 likely due to the potential steric hindrance, leading to AMPK inactivation and promoting lipolysis [151]. Several other kinases could also drive AMPKα phosphorylation include GSK3β (T479 in AMPKα1) [152], PKD1 (S491 in AMPKα2) [153] and PKC (S485 in AMPKα1) [154], indicative of the comprehensive regulation of AMPK activity by multiple phosphorylation events in response to various cellular contexts.
Ubiquitination and SUMOylation
In addition to promoting classic proteolysis, ubiquitination also plays an important role in the regulation of the activity of its substrates through non-K48 linked ubiquitination [102]. Genetic ablation of UCHL3 deubiquitinating enzyme in mice specifically potentiates AMPK activity in skeletal muscle without impact on AMPK expression level, indicative of the casual linkage between ubiquitination modification and AMPK activation [155]. Further study shows that ubiquitination on AMPKα abrogates its association with LKB1, and removal of this ubiquitination by USP10 (Ubiquitin Specific Peptidase 10) is required for LKB1 mediated phosphorylation and activation of AMPK during metabolic stress [156]. Interestingly, USP10 phosphorylation on S76 residue by activated AMPK acts as a positive feed-forward loop to further amplify AMPK activity upon energy crisis [156]. Although four lysine residues on AMPKα (K71, K285, K396, K485 in AMPKα1 and K60, K379, K391, K470 in AMPKα2) are identified as potential sites for USP10-mediated deubiquitination [156], the E3 ligase(s) triggering ubiquitination on these sites remains to be determined.
SUMOylation, analogous to ubiquitination, involves conjugating small ubiquitin-like modifiers SUMOs to its targets and enhances the interaction with other proteins through SUMO-interacting motif (SIM) [157]. In yeast, Snf1, the ortholog of mammalian AMPKα, is SUMOylated on K549 residue likely by the Mms21 SUMO (E3) ligase in glucose-proficient condition [158]. SUMOylation of Snf1 leads to its inactivation through both stabilizing auto-inhibitory conformation and facilitating ubiquitination mediated degradation. Similarly, in mammalian cells, PIAS4 SUMO E3 ligase induces AMPKα1 SUMOylation on K118 and compromises AMPK activity without alteration of either AMPKα protein levels or T172 phosphorylation [159], suggesting PIAS4-mediated SUMOylation affects AMPK activity through unknown mechanism. Surprisingly, PIAS4 modulates AMPK activity specifically towards mTORC1 signal, but not other canonical substrates of AMPK such as ACC [159]. Conversely, PIASy specifically conjugates SUMO2 to AMPKβ2, but not AMPKβ1, and potentiates AMPK activity [160]. Thus, SUMOylation appears to affect AMPK activity, although the underlying mechanism need to be further studies.
Oxidation and S-glutathionylation
AMPK plays a fundamental role in protecting cells against ROS (reactive oxygen species) [15]. While ROS itself could regulate AMPK activity, the underlying mechanism is still controversial. Two mechanisms are proposed for AMPK regulation by ROS. On one hand, exposure of H2O2 boosts AMPK activation through either LKB1 or CaMKK2 dependent on various cellular contexts [8, 12]. Interestingly, both exogenous treatment of H2O2 and endogenous H2O2 generated by glucose oxidase stimulate S-glutathionylation of AMPKα subunit on Cysteine (C) 299 and C304 residues to promote AMPK activation [161]. It is likely that these modifications compromise the interaction between auto-inhibitory domain and α-helix C region of AMPKα, thus endowing AMPK activation by upstream kinases.
On the other hand, ROS may also impair LKB1-AMPK axis in certain cellular context. In cardiomyocytes, both glucose deprivation and H2O2 treatment induce the oxidation of AMPKα on C130 and C174 residues, leading to the aggregation of AMPKα and abolishing its phosphorylation and activation by upstream kinase such as LKB1 [162]. Of note, Trx1 (Thioredoxin1), which is an essential enzyme that mediates the cleavage of disulfide bonds in proteins [163], acts as an important cofactor of AMPK activation by inhibiting oxidation of AMPKα upon metabolic stress. Thus, three different pools of AMPK have been proposed: oxidized (inactive), reduced (activatable), and reduced and phosphorylated (active), and exactly which type of AMPK pools is formed could largely depends on ROS level, Trx1 activity, energy status and AMPK upstream kinase. Interestingly, activated AMPK could further upregulate Trx expression to drive a feed-forward mechanism to amply AMPK activity for maintaining the redox homeostasis. As a result, down-regulation of TRX1 upon high-fat diet consumption is associated with elevated AMPK oxidation and inactivation, leading to enhanced cardiomyocyte death and heart failure during myocardial ischemia [163], highlighting the physiological relevance of TRX1 in regulating AMPK activation.
Collectively, these studies suggest that ROS or oxidative stress has a complicated role in regulating AMPK activity, but exactly which role it plays may be influenced by various cellular contexts, different dosage and/or nature of oxidative stresses as well as diverse nutrition and energy conditions.
THE BIOLOGICAL ROLE OF AMPK IN DIFFERENT ORGANELLES
Mitochondrial functions regulated by AMPK
AMPK not only serves as a guardian for cellular energy balance, but also maintains mitochondrial homeostasis by orchestrating mitochondrial biogenesis. AMPK stimulates mitochondrial biogenesis for ATP generation that fulfill the energy demand of cells in response to metabolic stress. This is achieved by AMK-mediated transcriptional regulation of numerous mitochondrial proteins. AMPK directly phosphorylates PGC1α (peroxisome-proliferator-activated receptor gamma coactivator 1α) on T177 and S538, leading to promoting PGC1α transcription activation to induce the expression of TFAM (Mitochondrial transcription factor A) [75]. TFAM then boosts mitochondrial biogenesis by promoting mtDNA replication and transcriptional regulation of mitochondrial biogenesis [75]. AMPK also phosphorylates chromatin remodeling proteins including methyltransferase 1 (DNMT1) and histone acetyltransferase 1 (HAT1) to promote mitochondrial biogenesis [164].
Mitochondrial dynamic including mitochondrial fission and fusion plays an important role in the maintenance of mitochondrial homeostasis in response to environmental insults. Upon energy stress, AMPK localizes to mitochondria and phosphorylates mitochondrial fission factor MFF on S146 that recruits dynamin-related Protein 1 (DRP1) to outer membrane of mitochondria, leading to mitochondrial fission [21, 22, 165]. AMPK also phosphorylates armadillo repeat-containing protein 10 (ARMC10) to orchestrate mitochondrial fission [166].
Mitophagy is a conserved biological process that degrades excessive, aged or damaged mitochondria to maintain the balance of mitochondrial mass and healthy mitochondria upon energy crisis. Acute energy depletion results in activation of AMPK, which phosphorylates ULK1 to induce mitophagy [23, 82, 167]. AMPK also phosphorylates several core autophagy/mitophagy components such as PAQR3 on T32 [168], ATG9 on S761 [169], Beclin1 on S91 and S94 [170], VPS34-associated protein RACK1 on T50 [171] and VPS34 on T133 and S135 [172] to regulate autophagy/mitophagy upon metabolic stress.
In addition, AMPK could phosphorylate mitochondrial MCU (Ca2+ uniporter) to elicit a rapid mitochondrial Ca2+ entry upon energy stress, which in turn boosts mitochondrial respiration to restore energy homeostasis [173]. AMPK also localizes in mitochondrial matrix to phosphorylate PDHA, catalytic subunit of pyruvate dehydrogenase complex (PDHc), on S295 and S314, leading to PDHc activation and TCA cycle entry [15]. Collectively, AMPK localized in different sub-fraction of mitochondria from outer membrane, inner membrane to matrix to regulate mitochondrial functions in response to metabolic stress or nutrition deprivation.
AMPK signaling at Golgi, ER, lysosome and nucleus
Interestingly, AMPK also regulates Golgi fragmentation in addition to mitochondrial fission/fusion. Upon energy stress, AMPK phosphorylates GBF1 on T1337, which is required for disassembly of Golgi apparatus and subsequent mitosis entry [174, 175]. Mechanistically, phosphorylation of GBF1, a guanine nucleotide exchange factor (GEF) for ADP-ribosylation factor (ARF)-GTPases, by AMPK induces GBF1 dissociation from Golgi membrane, thus leading to impaired ARF1 recruitment to Golgi and Golgi fragmentation [174]. Moreover, AMPKα2, activated and phosphorylated by CaMKK2 at G2/M phase, localizes in Golgi to facilitate mitotic Golgi fragmentation and G2/M transition [176].
Lysosomes generated from the endosomes containing hydrolytic acid from Golgi apparatus provide the platform for AMPK activation via an AMP/ADP-independent mechanism upon acute glucose deprivation [65]. The assembly of active lysosome complexes containing v-ATPase, Ragulator, AXIN, LKB1 and AMPK is essential for AMPK activation, and such process requires aldolase [65]. However, how AMPK is recruited to lysosome for its activation is not well understood. It would be interesting to identify the regulators that recruit and/or maintain AMPK in lysosomes.
Additionally, the ER protein, STIM2, has been identified as a new partner of AMPK. Through sensing the enhancement of intracellular calcium level, STIM2 facilitates the association of AMPK with CaMMK2, thus promoting the activation of AMPK [177]. Interestingly, as a scaffold, STIM2 specifically modulates calcium induced AMPK activation through CaMMK2, but fails to impact on LKB1 mediated AMPK activation in response to energy stress [177]. This evidence provides the novel insight into how AMPK signal is orchestrated on ER.
Nuclear localization of AMPK is indispensable for its role in transcription regulation. Both AMPKα1 and AMPKα2 contain nuclear localization signal (NLS) in N-terminal of catalytic domain and nuclear export signal (NES) in C-terminal regulatory domain, ensuring its shuttling between nucleus and cytoplasm [178–180]. AMPK translocates into nucleus and turns on PPARα transcription, contributing to leptin-induced fatty acid oxidation during myogenic differentiation [179]. Moreover, AMPK nuclear translocation is required for the transcription of PGC-1α during myogenesis [181]. Interestingly, nuclear AMPK orchestrates circadian clock by phosphorylating and destabilizing CRY1 (circadian component cryptochrome circadian regulator 1) [182].
Collectively, subcellular localization of AMPK in Golgi, lysosome, ER and nucleus regulates mitosis, AMPK activation and gene transcription.
PHYSIOLOGICAL FUNCTIONS OF AMPK SUBUNITS IN VIVO
Several evidence reveals the physiological functions of AMPK subunits including glucose metabolism, lipid metabolism, insulin sensitivity, glycogen metabolism and mitochondrial biogenesis using genetically engineered mouse models (GEMMs).
Ampkα2−/− but not Ampkα1−/− mice display resistance to hypoglycemic effects by AICAR, suggesting that AMPKα2 complexes mainly contribute to AMPK activation upon AICAR stimulation [183, 184]. Ampkα2 deficient mice also cause mild insulin resistance and impaired glucose tolerance correlated with defect of insulin secretion, whereas Ampkα1 deficient mice exhibit no obvious metabolic phenotypes [183, 184]. Interestingly, Ampkα2 deficient mice show rapid and severe ischemia contracture in heart, indicating that loss of Ampkα2 deteriorates heart functions during ischemia [185, 186].
Liver-specific deletion of Ampkα2 in mice results in mild hepatic hyperglycemia and maintains blood glucose in the physiological range under fasting [187]. Additionally, simultaneous deletion of Ampkα1 and Ampkα2 in mouse liver reduces mitochondrial biogenesis via downregulation of PGC-1α, cytochrome c oxidase I (COX I), COX IV and cytochrome c genes [188].
Muscle-specific deletion of Ampkβ1 in mice do not show detectable phenotypes, but muscle-specific deletion of Ampkβ2 in mice modestly reduces mitochondrial content and contraction-stimulated glucose uptake [189]. Notably, simultaneous deletion of Ampkβ1 and Ampkβ2 in mouse muscle dramatically reduces mitochondrial content and contraction-stimulated glucose uptake without affecting normal insulin sensitivity [189]. Moreover, both muscle-specific deletion of Ampkγ3 mice and transgenic mice overexpressing a kinase dead Ampkα2 mutant (Ampkα2-KD) do not change glycogen resynthesis after exercise [190, 191], but muscle glycogen content in skeletal muscle in transgenic mice overexpressing Ampkγ2 R225Q mutant is increased upon high exercise training [192]. Collectively, tissue-specific deletion of Ampkα2 or Ampkβ2 mice show modest phenotypes due to the functional compensation by AMPKα1 or AMPKβ2 subunit. Hence, deletion of Ampkα1α2 or Ampkβ1β2 in mice show significant phenotypes including hyperglycemia and decreased glucose uptake and mitochondrial biogenesis [189].
AMPK activation in adipocytes leads to inhibition of lipogenesis and an increase in fatty acid oxidation [193]. Consistently, Ampkα2−/− mice exhibit increased body weight and adipose tissue mass upon high fat diet treatment, indicating that loss of AMPKα2 subunit may contribute to the development of obesity [194]. In muscle cells, AMPK activation by AICAR stimulates glucose uptake and glycogen storage [195]. This observation is consistent with impaired glucose tolerance in mice with muscle-specific deletion of Ampkα2 [183, 184] and impaired glycogen storage in mice with muscle-specific deletion of Ampkγ3 [190].
THE ROLE OF AMPK IN CANCER
AMPK as a tumor suppressor
mTORC1 signaling, which governs protein synthesis, cell proliferation and growth, is directly inhibited by AMPK through phosphorylation of TSC2 and RAPTOR [89, 93]. Thus, AMPK is initially thought as a tumor suppressor because of its function in suppressing oncogenic mTORC1 signaling (Fig. 4). Subsequently, AMPK activation could also restrain prostate cancer cell growth partly by repressing de novo lipogenesis [196]. AMPK has been shown to cooperate with Hippo tumor suppressive signal to induce phosphorylation and inactivation of oncogenic YAP upon glucose deprivation [197]. Importantly, AMPK also phosphorylates and destabilizes GLI1, the core transcriptional factor controlling cell proliferation and differentiation involved in Hedgehog pathway, thus antagonizing its oncogenic activity in medulloblastoma [198]. Phosphorylation of BRAF on S729 by AMPK switches its binding partner from KSR1 scaffolding protein to 14–3-3 proteins, leading to the impairment of cell proliferation and cell cycle progression by compromising MEK-ERK cascades [199]. AMPK also induces cell cycle arrest by activating various tumor suppressors, such as p27, p53 and retinoblastoma protein (pRb) [98, 200, 201]. Consistently, LKB1, a well-known upstream kinase for AMPK activation, has been shown to exert tumor suppressive activity, whose loss or inactivation has been observed in diverse human cancers [202–204].
Fig. 4. Tumor suppressive and oncogenic role of AMPK regulated by various signaling cascades.

Phosphorylation of several downstream substrates by AMPK and ubiquitination or phosphorylation of AMPK by upstream enzymes contributes to inhibition or promotion of cancer progression.
Interestingly, AMPK also orchestrates epigenetic reprogramming to exert its tumor suppressive signal. AMPK has been reported to phosphorylate EZH2 on T311, which disrupts the interaction between EZH2 and SUZ12, leading to the impairment of PRC2-dependent methylation on histone H3 K27 [205]. Notably, AMPK-mediated phosphorylation of EZH2 suppresses PRC2 oncogenic function and correlates with better survival outcome in breast and ovarian cancer patients [205]. AMPK also impacts on DNA methylation through phosphorylating and activating tumor suppressor TET2 on S99 [206], which serves as a methyl-cytosine dioxygenase to convert methyl-cytosine to 5-hydroxymethylcytosine (5hmC), thus preventing dysregulation of 5hmc and neoplasms. Intriguingly, metformin-mediated AMPK activation triggers checkpoint blocker PD-L1 phosphorylation on S195, resulting in PD-L1 degradation via endoplasmic reticulum-associated protein degradation (ERAD) and promotes antitumor immunity [207].
Numerous studies using genetic mouse model has supported the role of AMPK as a tumor suppressor. First, Ampkβ1 depletion in prostate promotes earlier onset of prostate adenocarcinoma in conditional Pten knockout mice [196], indicative of the tumor suppressor function of AMPKβ1 in restricting prostate cancer development. Second, MAGE-A3/6-TRIM28, a cancer-specific ubiquitin ligase, has been demonstrated to drive AMPK ubiquitination and degradation, leading to promoting mTOR signaling and malignant transformation in lung, breast and colon tissues [131]. Third, UBE2O amplified in several human cancer targets AMPKα2 for ubiquitination and degradation, in turn promoting tumor initiation and progression [134]. Fourth, loss of Ampkα1 accelerates MYC-driven lymphomagenesis by inducing HIF-1α dependent aerobic glycolysis [208, 209] Fifth, in T cell acute lymphoblastic leukemia/lymphoma (T-ALL) mouse model induced by Pten deficiency in thymic T cell progenitor, Ampkα1 loss in T-ALL promotes leukemia/lymphoma development [210]. Finally, Ampkβ1 depletion promotes earlier onset of T-cell lymphoma upon p53 knockout [211]. Collectively, these studies provide the proof of principle evidence that AMPK could act as a tumor suppressor to restrict cancer development at least in certain contexts.
AMPK as an oncogene
Although above studies suggest the tumor suppressive role of AMPK in certain contexts, accumulated studies have uncovered the oncogenic role of AMPK in cancer promotion in other contexts (Fig. 4). Several mechanisms have been documented to account for the oncogenic activity of AMPK. First, AMPK maintains NADPH homeostasis to promote cancer cell survival and breast cancer progression by phosphorylating and inactivating ACC1/2 [212]. Second, AMPK is also involved in EGF (epidermal growth factor)/AKT signaling, which is deregulated during malignant transformation and cancer metastasis. AMPK promotes SKP2 phosphorylation on S256 in response to EGF stimulation, resulting in the ubiquitination and activation of AKT as well as subsequent oncogenic processes [67]. Remarkably, elevated AMPK T172 phosphorylation, SKP2 S256 phosphorylation and AKT S473 phosphorylation have been detected in advanced breast cancer and significantly correlates with poor disease-specific and metastasis-free survival, highlighting the clinical significance of AMPK-SKP2-AKT axis in breast cancer progression [67]. Third, phosphorylation of PDHA by AMPK critically maintains PDHc activation and TCA cycle, leading to promoting breast cancer metastasis. Importantly, activation of AMPK-PDHc cascade predicts poor metastasis-free survival in human breast cancer [15]. Fourth, inhibition of aerobic glycolysis and promotion of mitochondrial oxidative metabolism by AMPK mitigate metabolic stress and apoptosis in T-ALL, resulting in promoting T-ALL cell survival [213]. Finally, AMPK in the bone marrow endows the cellular resistance to oxidative stress and DNA damage upon metabolic stress [214].
Two studies using genetic mouse models further provide the evidence in support of oncogenic role of AMPK in cancer promotion. 1) In an MLL-AF9 induced AML models, Ampk loss depletes leukemia-initiating cells (LIC) and delays leukemogenesis [214]. 2) In an aggressive KrasG12Dp53−/− lung cancer model, Ampk ablation markedly impairs tumor burden, indicative of the requirement of AMPK activation for tumorigenesis [215].
Collectively, AMPK could elicit numerous oncogenic mechanisms to promote cancer progression and metastasis, offering potential therapeutic strategies to target AMPK pathways for cancer treatment.
Alterations of AMPK in human cancer
Copy number alterations of AMPK pathway genes through loss- or gain of function features have been identified in diverse human cancers. These genes were retrieved from the Kyoto encyclopedia of genes and genomes (KEGG) database. AMPKα2 is amplified in 17 cancer types including stomach and esophageal carcinoma (STES), stomach adenocarcinoma (STAD), sarcoma (SARC), head and neck squamous cell carcinoma (HNSC), esophageal carcinoma (ESCA), cervical squamous cell carcinoma, endocervical adenocarcinoma (CESC), pancreatic adenocarcinoma (PAAD), colon adenocarcinoma (COAD), kidney renal papillary cell carcinoma (KIRP), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), pan-kidney cohort (KIPAN), kidney renal clear cell carcinoma (KIRC), glioblastoma multiforme (GBM), bladder urothelial carcinoma (BLCA) and breast invasive carcinoma (BRCA) [216]. Of note, whole genome sequencing to examine human cancer somatic alterations reveals that AMPK inactivation was detected in lung adenocarcinoma due to LKB1 loss or inactivation mutations, correlated with higher mTOR activity [217]. It will be interesting to know whether there are somatic alterations in other AMPK subunit genes in human cancer.
Targeting AMPK signaling by small molecules in cancers
Therapeutic targeting AMPK has been widely applied in the treatment of multiple metabolic disorders including diabetes. Since AMPK has been shown to display tumor suppressive roles in certain contexts, several small molecules that trigger AMPK activation has been tested in preclinical models for cancer targeting. For example, the clinical proven diabetic medication, canagliflozin activates AMPK and inhibits cell proliferation, survival and tumorigenicity of prostate and lung cancer cells [30]. Salicylate allosterically activates AMPK and compromises clonogenic survival in prostate and lung cancer cells. Notably, combined treatment of salicylate and metformin induces synergistically inhibition of cell survival through restricting de novo lipogenesis [218]. MT 63–78, a small molecule of AMPK activator, suppresses lipogenesis, mTORC1 pathway and tumorigenicity potential of prostate cancer cells [219]. Also, Phenformin, a metabolic drug activating AMPK, restricts tumor progression and enhances mouse overall survival in KrasG12DLkb1−/−, but not KrasG12Dp53−/− mice, suggesting that Phenformin can be used for a metabolism-based therapy in selectively targeting LKB1-deficeint NSCLC [220]. Of note, activation of AMPK by metformin has been shown to enhance antitumor immunity in combination with CTLA4 blockade to enhance the efficacy of immunotherapy [207].
In addition to target AMPK itself, a specific ACC inhibitor, ND-654, or maintenance of ACC phosphorylation by AMPK suppresses hepatic lipogenesis, inflammation and hepatocellular carcinoma [221]. Hence, AMPK activation results in the inhibition of cell proliferation in part due to the disruption of de novo fatty acid synthesis which fuels cell cycle progression [222].
Given that AMPK activation also protects keratinocytes from hyperproliferation via phosphorylation and inactivation of BRAF, pharmacological treatment using AMPK agonists such as Phenformin represents a promising strategy to overcome BRAF inhibitor-induced cutaneous squamous cell carcinoma (cSCC) [199]. Moreover, oral administration of phenformin, but not metformin, slows T-ALL progression in an AMPK dependent manner [210]. Taken together, multiple AMPK agonists display anti-tumor properties in preclinical models, although it remains to be determined whether AMPK activation indeed contributes to tumor suppression from these agents.
Because AMPK also displays tumor promoting activity in certain tumor contexts such as breast cancer and lung cancer, therapeutic strategies against AMPK can be also considered. Although compound C has been known to be as an AMPK inhibitor, it is highly toxic and displays numerous off-target effects [223–225], prohibiting it from being used for in vivo study. Thus, there is a great need to develop specific small molecule inhibitors of AMPK and then verify their efficacy in cancer targeting, ultimately offering novel and effective strategies to target those cancer driven by AMPK activation.
In summary, both AMPK agonists and inhibitors can be potentially used alone or in combination with other therapeutic strategies for cancer prevention or treatment under different tumor contexts that impacts the functional role of AMPK in acting as tumor suppression or tumor promotion.
CONCLUSION AND PERSPECTIVE
AMPK activation is regulated by diverse physiological and pathological conditions such as metabolic stress, caloric restriction, exercise, ageing and obesity. It regulates cell cycle progression, metabolism and mitochondria biology, and epigenetic reprogramming to maintain energy homeostasis and cell survival in response to diverse stresses. AMPK activity and stability can be regulated by diverse post-translational modifications such as phosphorylation and ubiquitination. Through extensive studies in the past decades, AMPK has been shown to act as a double edge sword that elicits either tumor suppressive activity or tumor promoting role, which may be influenced by distinct cellular contexts, offering potential strategies to target cancer by manipulating AMPK activation and inactivation. Several outstanding questions remain to be addressed. First, it will be important in the future to dissect the mechanisms by which distinct cellular contexts impact the role of AMPK in cancer regulation. Second, since AMPK activation could promote breast cancer progression and metastasis, it will be important to develop specific AMPK inhibitors for targeting those cancers driven by AMPK activation. Third, since AMPK is activated and localized in diverse organelles including lysosome and mitochondria with unknown mechanisms, it will be important to dissect the mechanisms by which AMPK localization to distinct subcellular compartments are regulated. Finally, AMPK activity is regulated by SUMOylation through unknown mechanisms, thereby dissecting the mechanisms underlying this regulation will be critical to advance our current understanding of AMPK regulation. Understanding these key questions will not only provide new insights into AMPK signaling regulation, but also offer new paradigms and strategies for cancer targeting as well as other diseases associated with AMPK deregulation.
Acknowledgements
We are grateful for the members in Lin’s laboratory for the inputs and suggestions throughout the course of this study. We deeply apologize to those scientists whose studies were not cited in this review due to the space limitation. This work was supported by start-up funds from Wake Forest School of Medicine, Anderson Discover Endowed Professorship award, NIH grants (R01CA182424 and 1R01CA193813) and Breast Cancer Pilot Award from Wake Forest Cancer Center supported by the NCI Cancer Center Support Grant (P30CA012197).
Footnotes
Declaration of Competing Interest
There are no conflicts of interest declared by the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Witters LA, Gao G, Kemp BE, Quistorff B, Hepatic 5’-AMP-activated protein kinase: zonal distribution and relationship to acetyl-CoA carboxylase activity in varying nutritional states, Arch Biochem Biophys 308(2) (1994) 413–9. [DOI] [PubMed] [Google Scholar]
- [2].Winder WW, Hardie DG, Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise, Am J Physiol 270(2 Pt 1) (1996) E299–304. [DOI] [PubMed] [Google Scholar]
- [3].Li M, Verdijk LB, Sakamoto K, Ely B, van Loon LJ, Musi N, Reduced AMPK-ACC and mTOR signaling in muscle from older men, and effect of resistance exercise, Mech Ageing Dev 133(11–12) (2012) 655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, Befroy D, Pypaert M, Hardie DG, Young LH, Shulman GI, Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis, Cell Metab 5(2) (2007) 151–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Qiang W, Weiqiang K, Qing Z, Pengju Z, Yi L, Aging impairs insulin-stimulated glucose uptake in rat skeletal muscle via suppressing AMPKalpha, Exp Mol Med 39(4) (2007) 535–43. [DOI] [PubMed] [Google Scholar]
- [6].Yavari A, Stocker CJ, Ghaffari S, Wargent ET, Steeples V, Czibik G, Pinter K, Bellahcene M, Woods A, Martinez de Morentin PB, Cansell C, Lam BY, Chuster A, Petkevicius K, Nguyen-Tu MS, Martinez-Sanchez A, Pullen TJ, Oliver PL, Stockenhuber A, C Nguyen M. Lazdam JF O’Dowd, Harikumar P, Toth M, Beall C, Kyriakou T, Parnis J, Sarma D, Katritsis G, Wortmann DD, Harper AR, Brown LA, Willows R, Gandra S, Poncio V, de Oliveira Figueiredo MJ, Qi NR, Peirson SN, McCrimmon RJ, Gereben B, Tretter L, Fekete C, Redwood C, Yeo GS, Heisler LK, Rutter GA, Smith MA, Withers DJ, Carling D, Sternick EB, Arch JR, Cawthorne MA, Watkins H, Ashrafian H, Chronic Activation of gamma2 AMPK Induces Obesity and Reduces beta Cell Function, Cell Metab 23(5) (2016) 821–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG, Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade, J Biol 2(4) (2003) 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC, The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress, Proc Natl Acad Sci U S A 101(10) (2004) 3329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D, LKB1 is the upstream kinase in the AMP-activated protein kinase cascade, Curr Biol 13(22) (2003) 2004-8. [DOI] [PubMed] [Google Scholar]
- [10].Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG, Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase, Cell Metab 2(1) (2005) 9–19. [DOI] [PubMed] [Google Scholar]
- [11].Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA, The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases, J Biol Chem 280(32) (2005) 29060–6. [DOI] [PubMed] [Google Scholar]
- [12].Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D, Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells, Cell Metab 2(1) (2005) 21–33. [DOI] [PubMed] [Google Scholar]
- [13].Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L, Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia, Curr Biol 10(20) (2000) 1247–55. [DOI] [PubMed] [Google Scholar]
- [14].Marsin AS, Bouzin C, Bertrand L, Hue L, The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase, J Biol Chem 277(34) (2002) 30778–83. [DOI] [PubMed] [Google Scholar]
- [15].Cai Z, Li CF, Han F, Liu C, Zhang A, Hsu CC, Peng D, Zhang X, Jin G, Rezaeian AH, Wang G, Zhang W, Pan BS, Wang CY, Wang YH, Wu SY, Yang SC, Hsu FC, D’Agostino RB Jr., Furdui CM, Kucera GL, Parks JS, Chilton FH, Huang CY, Tsai FJ, Pasche B, Watabe K, Lin HK, Phosphorylation of PDHA by AMPK Drives TCA Cycle to Promote Cancer Metastasis, Mol Cell 80(2) (2020) 263–278 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O’Neill HM, Ford RJ, Palanivel R, O’Brien M, Hardie DG, Macaulay SL, Schertzer JD, Dyck JR, van Denderen BJ, Kemp BE, Steinberg GR, Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin, Nat Med 19(12) (2013) 1649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].O’Neill HM, Lally JS, Galic S, Thomas M, Azizi PD, Fullerton MD, Smith BK, Pulinilkunnil T, Chen Z, Samaan MC, Jorgensen SB, Dyck JR, Holloway GP, Hawke TJ, van Denderen BJ, Kemp BE, Steinberg GR, AMPK phosphorylation of ACC2 is required for skeletal muscle fatty acid oxidation and insulin sensitivity in mice, Diabetologia 57(8) (2014) 1693–702. [DOI] [PubMed] [Google Scholar]
- [18].Laplante M, Sabatini DM, mTOR signaling at a glance, J Cell Sci 122(Pt 20) (2009) 3589–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Willows R, Sanders MJ, Xiao B, Patel BR, Martin SR, Read J, Wilson JR, Hubbard J, Gamblin SJ, Carling D, Phosphorylation of AMPK by upstream kinases is required for activity in mammalian cells, Biochem J 474(17) (2017) 3059–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG, Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase, J Biol Chem 271(44) (1996) 27879–87. [DOI] [PubMed] [Google Scholar]
- [21].Ducommun S, Deak M, Sumpton D, Ford RJ, Nunez Galindo A, Kussmann M, Viollet B, Steinberg GR, Foretz M, Dayon L, Morrice NA, Sakamoto K, Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate, Cell Signal 27(5) (2015) 978–88. [DOI] [PubMed] [Google Scholar]
- [22].Toyama EQ, Herzig S, Courchet J, Lewis TL Jr., Loson OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, Shaw RJ, Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress, Science 351(6270) (2016) 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ, Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy, Science 331(6016) (2011) 456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE, Role of AMP-activated protein kinase in mechanism of metformin action, J Clin Invest 108(8) (2001) 1167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ, Kemp BE, Sakamoto K, Steinberg GR, Hardie DG, The ancient drug salicylate directly activates AMP-activated protein kinase, Science 336(6083) (2012) 918–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ, Shen Y, Ye JM, Lee CH, Oh WK, Kim CT, Hohnen-Behrens C, Gosby A, Kraegen EW, James DE, Kim JB, Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states, Diabetes 55(8) (2006) 2256–64. [DOI] [PubMed] [Google Scholar]
- [27].Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B, Wierzbicki M, Verbeuren TJ, Cohen RA, Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice, Diabetes 55(8) (2006) 2180–91. [DOI] [PubMed] [Google Scholar]
- [28].Brusq JM, Ancellin N, Grondin P, Guillard R, Martin S, Saintillan Y, Issandou M, Inhibition of lipid synthesis through activation of AMP kinase: an additional mechanism for the hypolipidemic effects of berberine, J Lipid Res 47(6) (2006) 1281–8. [DOI] [PubMed] [Google Scholar]
- [29].Hawley SA, Ford RJ, Smith BK, Gowans GJ, Mancini SJ, Pitt RD, Day EA, Salt IP, Steinberg GR, Hardie DG, The Na+/Glucose Cotransporter Inhibitor Canagliflozin Activates AMPK by Inhibiting Mitochondrial Function and Increasing Cellular AMP Levels, Diabetes 65(9) (2016) 2784–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Villani LA, Smith BK, Marcinko K, Ford RJ, Broadfield LA, Green AE, Houde VP, Muti P, Tsakiridis T, Steinberg GR, The diabetes medication Canagliflozin reduces cancer cell proliferation by inhibiting mitochondrial complex-I supported respiration, Mol Metab 5(10) (2016) 1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cokorinos EC, Delmore J, Reyes AR, Albuquerque B, Kjobsted R, Jorgensen NO, Tran JL, Jatkar A, Cialdea K, Esquejo RM, Meissen J, Calabrese MF, Cordes J, Moccia R, Tess D, Salatto CT, Coskran TM, Opsahl AC, Flynn D, Blatnik M, Li W, Kindt E, Foretz M, Viollet B, Ward J, Kurumbail RG, Kalgutkar AS, Wojtaszewski JFP, Cameron KO, Miller RA, Activation of Skeletal Muscle AMPK Promotes Glucose Disposal and Glucose Lowering in Non-human Primates and Mice, Cell Metab 25(5) (2017) 1147–1159 e10. [DOI] [PubMed] [Google Scholar]
- [32].Salatto CT, Miller RA, Cameron KO, Cokorinos E, Reyes A, Ward J, Calabrese MF, Kurumbail RG, Rajamohan F, Kalgutkar AS, Tess DA, Shavnya A, Genung NE, Edmonds DJ, Jatkar A, Maciejewski BS, Amaro M, Gandhok H, Monetti M, Cialdea K, Bollinger E, Kreeger JM, Coskran TM, Opsahl AC, Boucher GG, Birnbaum MJ, DaSilva-Jardine P, Rolph T, Selective Activation of AMPK beta1-Containing Isoforms Improves Kidney Function in a Rat Model of Diabetic Nephropathy, J Pharmacol Exp Ther 361(2) (2017) 303311. [DOI] [PubMed] [Google Scholar]
- [33].Myers RW, Guan HP, Ehrhart J, Petrov A, Prahalada S, Tozzo E, Yang X, Kurtz MM, Trujillo M, Gonzalez Trotter D, Feng D, Xu S, Eiermann G, Holahan MA, Rubins D, Conarello S, Niu X, Souza SC, Miller C, Liu J, Lu K, Feng W, Li Y, Painter RE, Milligan JA, He H, Liu F, Ogawa A, Wisniewski D, Rohm RJ, Wang L, Bunzel M, Qian Y, Zhu W, Wang H, Bennet B, LaFranco Scheuch L, Fernandez GE, Li C, Klimas M, Zhou G, van Heek M, Biftu T, Weber A, Kelley DE, Thornberry N, Erion MD, Kemp DM, Sebhat IK, Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy, Science 357(6350) (2017) 507–511. [DOI] [PubMed] [Google Scholar]
- [34].Chauhan AS, Zhuang L, Gan B, Spatial control of AMPK signaling at subcellular compartments, Crit Rev Biochem Mol Biol 55(1) (2020) 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ross FA, MacKintosh C, Hardie DG, AMP-activated protein kinase: a cellular energy sensor that comes in 12 flavours, FEBS J 283(16) (2016) 2987–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wu J, Puppala D, Feng X, Monetti M, Lapworth AL, Geoghegan KF, Chemoproteomic analysis of intertissue and interspecies isoform diversity of AMP-activated protein kinase (AMPK), J Biol Chem 288(50) (2013) 35904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Woods A, Salt I, Scott J, Hardie DG, Carling D, The alpha1 and alpha2 isoforms of the AMP-activated protein kinase have similar activities in rat liver but exhibit differences in substrate specificity in vitro, FEBS Lett 397(2–3) (1996) 347–51. [DOI] [PubMed] [Google Scholar]
- [38].Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ, Structure of mammalian AMPK and its regulation by ADP, Nature 472(7342) (2011) 230–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Crute BE, Seefeld K, Gamble J, Kemp BE, Witters LA, Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase, J Biol Chem 273(52) (1998) 35347–54. [DOI] [PubMed] [Google Scholar]
- [40].Corton JM, Gillespie JG, Hawley SA, Hardie DG, 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells?, Eur J Biochem 229(2) (1995) 558–65. [DOI] [PubMed] [Google Scholar]
- [41].Pang T, Xiong B, Li JY, Qiu BY, Jin GZ, Shen JK, Li J, Conserved alpha-helix acts as autoinhibitory sequence in AMP-activated protein kinase alpha subunits, J Biol Chem 282(1) (2007) 495–506. [DOI] [PubMed] [Google Scholar]
- [42].Chen L, Xin FJ, Wang J, Hu J, Zhang YY, Wan S, Cao LS, Lu C, Li P, Yan SF, Neumann D, Schlattner U, Xia B, Wang ZX, Wu JW, Conserved regulatory elements in AMPK, Nature 498(7453) (2013) E8–10. [DOI] [PubMed] [Google Scholar]
- [43].Xin FJ, Wang J, Zhao RQ, Wang ZX, Wu JW, Coordinated regulation of AMPK activity by multiple elements in the alpha-subunit, Cell Res 23(10) (2013) 1237–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bultot L, Guigas B, Von Wilamowitz-Moellendorff A, Maisin L, Vertommen D, Hussain N, Beullens M, Guinovart JJ, Foretz M, Viollet B, Sakamoto K, Hue L, Rider MH, AMP-activated protein kinase phosphorylates and inactivates liver glycogen synthase, Biochem J 443(1) (2012) 193–203. [DOI] [PubMed] [Google Scholar]
- [45].Jorgensen SB, Nielsen JN, Birk JB, Olsen GS, Viollet B, Andreelli F, Schjerling P, Vaulont S, Hardie DG, Hansen BF, Richter EA, Wojtaszewski JF, The alpha2–5’AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading, Diabetes 53(12) (2004) 3074–81. [DOI] [PubMed] [Google Scholar]
- [46].Goransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, Viollet B, Hardie DG, Sakamoto K, Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase, J Biol Chem 282(45) (2007) 32549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ross FA, Jensen TE, Hardie DG, Differential regulation by AMP and ADP of AMPK complexes containing different gamma subunit isoforms, Biochem J 473(2) (2016) 189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D, Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family, J Biol Chem 282(45) (2007) 32539–48. [DOI] [PubMed] [Google Scholar]
- [49].Chen L, Wang J, Zhang YY, Yan SF, Neumann D, Schlattner U, Wang ZX, Wu JW, AMP-activated protein kinase undergoes nucleotide-dependent conformational changes, Nat Struct Mol Biol 19(7) (2012) 716–8. [DOI] [PubMed] [Google Scholar]
- [50].Li X, Wang L, Zhou XE, Ke J, de Waal PW, Gu X, Tan MH, Wang D, Wu D, Xu HE, Melcher K, Structural basis of AMPK regulation by adenine nucleotides and glycogen, Cell Res 25(1) (2015) 50–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Xiao B, Sanders MJ, Carmena D, Bright NJ, Haire LF, Underwood E, Patel BR, Heath RB, Walker PA, Hallen S, Giordanetto F, Martin SR, Carling D, Gamblin SJ, Structural basis of AMPK regulation by small molecule activators, Nat Commun 4 (2013) 3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, Martin SR, Carling D, Gamblin SJ, Structural basis for AMP binding to mammalian AMP-activated protein kinase, Nature 449(7161) (2007) 496–500. [DOI] [PubMed] [Google Scholar]
- [53].Sim AT, Hardie DG, The low activity of acetyl-CoA carboxylase in basal and glucagon-stimulated hepatocytes is due to phosphorylation by the AMP-activated protein kinase and not cyclic AMP-dependent protein kinase, FEBS Lett 233(2) (1988) 294–8. [DOI] [PubMed] [Google Scholar]
- [54].Yeh LA, Lee KH, Kim KH, Regulation of rat liver acetyl-CoA carboxylase. Regulation of phosphorylation and inactivation of acetyl-CoA carboxylase by the adenylate energy charge, J Biol Chem 255(6) (1980) 2308–14. [PubMed] [Google Scholar]
- [55].Gowans GJ, Hawley SA, Ross FA, Hardie DG, AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation, Cell Metab 18(4) (2013) 556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Oakhill JS, Chen ZP, Scott JW, Steel R, Castelli LA, Ling N, Macaulay SL, Kemp BE, beta-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK), Proc Natl Acad Sci U S A 107(45) (2010) 19237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, Kemp BE, AMPK is a direct adenylate charge-regulated protein kinase, Science 332(6036) (2011) 1433–5. [DOI] [PubMed] [Google Scholar]
- [58].Jin X, Townley R, Shapiro L, Structural insight into AMPK regulation: ADP comes into play, Structure 15(10) (2007) 1285–95. [DOI] [PubMed] [Google Scholar]
- [59].Townley R, Shapiro L, Crystal structures of the adenylate sensor from fission yeast AMP-activated protein kinase, Science 315(5819) (2007) 1726–9. [DOI] [PubMed] [Google Scholar]
- [60].Salt IP, Hardie DG, AMP-Activated Protein Kinase: An Ubiquitous Signaling Pathway With Key Roles in the Cardiovascular System, Circ Res 120(11) (2017) 1825-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Bultot L, Jensen TE, Lai YC, Madsen AL, Collodet C, Kviklyte S, Deak M, Yavari A, Foretz M, Ghaffari S, Bellahcene M, Ashrafian H, Rider MH, Richter EA, Sakamoto K, Benzimidazole derivative small-molecule 991 enhances AMPK activity and glucose uptake induced by AICAR or contraction in skeletal muscle, Am J Physiol Endocrinol Metab 311(4) (2016) E706–E719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Timmermans AD, Balteau M, Gelinas R, Renguet E, Ginion A, de Meester C, Sakamoto K, Balligand JL, Bontemps F, Vanoverschelde JL, Horman S, Beauloye C, Bertrand L, A-769662 potentiates the effect of other AMP-activated protein kinase activators on cardiac glucose uptake, Am J Physiol Heart Circ Physiol 306(12) (2014) H1619–30. [DOI] [PubMed] [Google Scholar]
- [63].Ducommun S, Ford RJ, Bultot L, Deak M, Bertrand L, Kemp BE, Steinberg GR, Sakamoto K, Enhanced activation of cellular AMPK by dual-small molecule treatment: AICAR and A769662, Am J Physiol Endocrinol Metab 306(6) (2014) E688–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Boyle KE, Patinkin ZW, Shapiro ALB, Bader C, Vanderlinden L, Kechris K, Janssen RC, Ford RJ, Smith BK, Steinberg GR, Davidson EJ, Yang IV, Dabelea D, Friedman JE, Maternal obesity alters fatty acid oxidation, AMPK activity, and associated DNA methylation in mesenchymal stem cells from human infants, Mol Metab 6(11) (2017) 1503–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhang CS, Hawley SA, Zong Y, Li M, Wang Z, Gray A, Ma T, Cui J, Feng JW, Zhu M, Wu YQ, Li TY, Ye Z, Lin SY, Yin H, Piao HL, Hardie DG, Lin SC, Fructose-1,6bisphosphate and aldolase mediate glucose sensing by AMPK, Nature 548(7665) (2017) 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM, Hardie DG, Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation, Cell Metab 11(6) (2010) 554–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Han F, Li CF, Cai Z, Zhang X, Jin G, Zhang WN, Xu C, Wang CY, Morrow J, Zhang S, Xu D, Wang G, Lin HK, The critical role of AMPK in driving Akt activation under stress, tumorigenesis and drug resistance, Nat Commun 9(1) (2018) 4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Luo L, Huang W, Tao R, Hu N, Xiao ZX, Luo Z, ATM and LKB1 dependent activation of AMPK sensitizes cancer cells to etoposide-induced apoptosis, Cancer Lett 328(1) (2013) 114–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Fu X, Wan S, Lyu YL, Liu LF, Qi H, Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation, PLoS One 3(4) (2008) e2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Sanli T, Rashid A, Liu C, Harding S, Bristow RG, Cutz JC, Singh G, Wright J, Tsakiridis T, Ionizing radiation activates AMP-activated kinase (AMPK): a target for radiosensitization of human cancer cells, Int J Radiat Oncol Biol Phys 78(1) (2010) 221–9. [DOI] [PubMed] [Google Scholar]
- [71].Vara-Ciruelos D, Dandapani M, Gray A, Egbani EO, Evans AM, Hardie DG, Genotoxic Damage Activates the AMPK-alpha1 Isoform in the Nucleus via Ca(2+)/CaMKK2 Signaling to Enhance Tumor Cell Survival, Mol Cancer Res 16(2) (2018) 345–357. [DOI] [PubMed] [Google Scholar]
- [72].Schaffer BE, Levin RS, Hertz NT, Maures TJ, Schoof ML, Hollstein PE, Benayoun BA, Banko MR, Shaw RJ, Shokat KM, Brunet A, Identification of AMPK Phosphorylation Sites Reveals a Network of Proteins Involved in Cell Invasion and Facilitates Large-Scale Substrate Prediction, Cell Metab 22(5) (2015) 907–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Hardie DG, Schaffer BE, Brunet A, AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs, Trends Cell Biol 26(3) (2016) 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, Berger SL, Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation, Science 329(5996) (2010) 1201–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Jager S, Handschin C, St-Pierre J, Spiegelman BM, AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha, Proc Natl Acad Sci U S A 104(29) (2007) 12017–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Abu-Elheiga L, Brinkley WR, Zhong L, Chirala SS, Woldegiorgis G, Wakil SJ, The subcellular localization of acetyl-CoA carboxylase 2, Proc Natl Acad Sci U S A 97(4) (2000) 1444–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Munday MR, Regulation of mammalian acetyl-CoA carboxylase, Biochem Soc Trans 30(Pt 6) (2002) 1059–64. [DOI] [PubMed] [Google Scholar]
- [78].Witters LA, Nordlund AC, Marshall L, Regulation of intracellular acetyl-CoA carboxylase by ATP depletors mimics the action of the 5’-AMP-activated protein kinase, Biochem Biophys Res Commun 181(3) (1991) 1486–92. [DOI] [PubMed] [Google Scholar]
- [79].Wei J, Tong L, Crystal structure of the 500-kDa yeast acetyl-CoA carboxylase holoenzyme dimer, Nature 526(7575) (2015) 723–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zachari M, Ganley IG, The mammalian ULK1 complex and autophagy initiation, Essays Biochem 61(6) (2017) 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Shang L, Chen S, Du F, Li S, Zhao L, Wang X, Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK, Proc Natl Acad Sci U S A 108(12) (2011) 4788–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kim J, Kundu M, Viollet B, Guan KL, AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1, Nat Cell Biol 13(2) (2011) 132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Youle RJ, van der Bliek AM, Mitochondrial fission, fusion, and stress, Science 337(6098) (2012) 1062–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Westermann B, Mitochondrial fusion and fission in cell life and death, Nat Rev Mol Cell Biol 11(12) (2010) 872–84. [DOI] [PubMed] [Google Scholar]
- [85].Zhang CS, Lin SC, AMPK Promotes Autophagy by Facilitating Mitochondrial Fission, Cell Metab 23(3) (2016) 399–401. [DOI] [PubMed] [Google Scholar]
- [86].Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K, Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells, J Cell Biol 191(6) (2010) 1141–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Inoki K, Li Y, Xu T, Guan KL, Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling, Genes Dev 17(15) (2003) 1829-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Sabatini DM, mTOR and cancer: insights into a complex relationship, Nat Rev Cancer 6(9) (2006) 729–34. [DOI] [PubMed] [Google Scholar]
- [89].Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC, The LKB1 tumor suppressor negatively regulates mTOR signaling, Cancer Cell 6(1) (2004) 91–9. [DOI] [PubMed] [Google Scholar]
- [90].Inoki K, Zhu T, Guan KL, TSC2 mediates cellular energy response to control cell growth and survival, Cell 115(5) (2003) 577–90. [DOI] [PubMed] [Google Scholar]
- [91].Martin TD, Chen XW, Kaplan RE, Saltiel AR, Walker CL, Reiner DJ, Der CJ, Ral and Rheb GTPase activating proteins integrate mTOR and GTPase signaling in aging, autophagy, and tumor cell invasion, Mol Cell 53(2) (2014) 209–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N, Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity, J Biol Chem 280(37) (2005) 32081–9. [DOI] [PubMed] [Google Scholar]
- [93].Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ, AMPK phosphorylation of raptor mediates a metabolic checkpoint, Mol Cell 30(2) (2008) 214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM, mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery, Cell 110(2) (2002) 163–75. [DOI] [PubMed] [Google Scholar]
- [95].Berger SL, The complex language of chromatin regulation during transcription, Nature 447(7143) (2007) 407–12. [DOI] [PubMed] [Google Scholar]
- [96].Ivaldi MS, Karam CS, Corces VG, Phosphorylation of histone H3 at Ser10 facilitates RNA polymerase II release from promoter-proximal pausing in Drosophila, Genes Dev 21(21) (2007) 2818–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].McGee SL, Hargreaves M, AMPK and transcriptional regulation, Front Biosci 13 (2008) 3022–33. [DOI] [PubMed] [Google Scholar]
- [98].Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB, AMP-activated protein kinase induces a p53-dependent metabolic checkpoint, Mol Cell 18(3) (2005) 283–93. [DOI] [PubMed] [Google Scholar]
- [99].Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A, The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor, J Biol Chem 282(41) (2007) 30107–19. [DOI] [PubMed] [Google Scholar]
- [100].Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC, The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin, Science 310(5754) (2005) 1642–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Swatek KN, Komander D, Ubiquitin modifications, Cell Res 26(4) (2016) 399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Cai Z, Moten A, Peng D, Hsu CC, Pan BS, Manne R, Li HY, Lin HK, The Skp2 Pathway: A Critical Target for Cancer Therapy, Semin Cancer Biol 67(Pt 2) (2020) 16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Xu D, Li CF, Zhang X, Gong Z, Chan CH, Lee SW, Jin G, Rezaeian AH, Han F, Wang J, Yang WL, Feng ZZ, Chen W, Wu CY, Wang YJ, Chow LP, Zhu XF, Zeng YX, Lin HK, Skp2-macroH2A1-CDK8 axis orchestrates G2/M transition and tumorigenesis, Nat Commun 6 (2015) 6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z, Huang HY, Tsai KK, Flores LG, Shao Y, Hazle JD, Yu D, Wei W, Sarbassov D, Hung MC, Nakayama KI, Lin HK, The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis, Cell 149(5) (2012) 1098–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Wu J, Zhang X, Zhang L, Wu CY, Rezaeian AH, Chan CH, Li JM, Wang J, Gao Y, Han F, Jeong YS, Yuan X, Khanna KK, Jin J, Zeng YX, Lin HK, Skp2 E3 ligase integrates ATM activation and homologous recombination repair by ubiquitinating NBS1, Mol Cell 46(3) (2012) 351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Wang JY, Liu GZ, Wilmott JS, La T, Feng YC, Yari H, Yan XG, Thorne RF, Scolyer RA, Zhang XD, Jin L, Skp2-Mediated Stabilization of MTH1 Promotes Survival of Melanoma Cells upon Oxidative Stress, Cancer Res 77(22) (2017) 6226–6239. [DOI] [PubMed] [Google Scholar]
- [107].Sun W, Liu Q, Leng J, Zheng Y, Li J, The role of Pyruvate Dehydrogenase Complex in cardiovascular diseases, Life Sci 121 (2015) 97–103. [DOI] [PubMed] [Google Scholar]
- [108].Patel MS, Nemeria NS, Furey W, Jordan F, The pyruvate dehydrogenase complexes: structure-based function and regulation, J Biol Chem 289(24) (2014) 16615–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Cai Z, Peng D, Lin HK, AMPK maintains TCA cycle through sequential phosphorylation of PDHA to promote tumor metastasis, Cell Stress 4(12) (2020) 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Gaster M, A primary reduced TCA flux governs substrate oxidation in T2D skeletal muscle, Curr Diabetes Rev 8(6) (2012) 458–79. [DOI] [PubMed] [Google Scholar]
- [111].Nagao H, Nishizawa H, Bamba T, Nakayama Y, Isozumi N, Nagamori S, Kanai Y, Tanaka Y, Kita S, Fukuda S, Funahashi T, Maeda N, Fukusaki E, Shimomura I, Increased Dynamics of Tricarboxylic Acid Cycle and Glutamate Synthesis in Obese Adipose Tissue: IN VIVO METABOLIC TURNOVER ANALYSIS, J Biol Chem 292(11) (2017) 4469–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Atamna H, Frey WH, 2nd, Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease, Mitochondrion 7(5) (2007) 297–310. [DOI] [PubMed] [Google Scholar]
- [113].Steinberg GR, Kemp BE, AMPK in Health and Disease, Physiol Rev 89(3) (2009) 1025–78. [DOI] [PubMed] [Google Scholar]
- [114].Yang A, Zhu Z, Kapranov P, McKeon F, Church GM, Gingeras TR, Struhl K, Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells, Mol Cell 24(4) (2006) 593–602. [DOI] [PubMed] [Google Scholar]
- [115].Yi Y, Chen D, Ao J, Zhang W, Yi J, Ren X, Fei J, Li F, Niu M, Chen H, Luo Y, Luo Z, Xiao ZJ, Transcriptional suppression of AMPKalpha1 promotes breast cancer metastasis upon oncogene activation, Proc Natl Acad Sci U S A 117(14) (2020) 8013–8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Kim M, Sujkowski A, Namkoong S, Gu B, Cobb T, Kim B, Kowalsky AH, Cho CS, Semple I, Ro SH, Davis C, Brooks SV, Karin M, Wessells RJ, Lee JH, Sestrins are evolutionarily conserved mediators of exercise benefits, Nat Commun 11(1) (2020) 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Budanov AV, Karin M, p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling, Cell 134(3) (2008) 451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Sanli T, Linher-Melville K, Tsakiridis T, Singh G, Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells, PLoS One 7(2) (2012) e32035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Bartel DP, MicroRNAs: target recognition and regulatory functions, Cell 136(2) (2009) 215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Wang Z, Yuan Y, Zhang Z, Ding K, Inhibition of miRNA-27b enhances neurogenesis via AMPK activation in a mouse ischemic stroke model, FEBS Open Bio 9(5) (2019) 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Prastowo S, Amin A, Sohel MH, Identifying Candidate MicroRNAs in MicroRNA-AMPK Gene Interaction Regulating Lipid Accumulation of Bovine Granulosa Cell Luteinization: An In Silico Study, in: Isnansetyo A, Nuringtyas TR (Eds.) Proceeding of the 1st International Conference on Tropical Agriculture, Springer International Publishing, Cham, 2017, pp. 431–438. [Google Scholar]
- [122].Wilkinson KD, Protein ubiquitination: a regulatory post-translational modification, Anticancer Drug Des 2(2) (1987) 211–29. [PubMed] [Google Scholar]
- [123].Lee EW, Lee MS, Camus S, Ghim J, Yang MR, Oh W, Ha NC, Lane DP, Song J, Differential regulation of p53 and p21 by MKRN1 E3 ligase controls cell cycle arrest and apoptosis, EMBO J 28(14) (2009) 2100–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Kim JH, Park KW, Lee EW, Jang WS, Seo J, Shin S, Hwang KA, Song J, Suppression of PPARgamma through MKRN1-mediated ubiquitination and degradation prevents adipocyte differentiation, Cell Death Differ 21(4) (2014) 594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Lee MS, Han HJ, Han SY, Kim IY, Chae S, Lee CS, Kim SE, Yoon SG, Park JW, Kim JH, Shin S, Jeong M, Ko A, Lee HY, Oh KJ, Lee YH, Bae KH, Koo SH, Kim JW, Seong JK, Hwang D, Song J, Loss of the E3 ubiquitin ligase MKRN1 represses diet-induced metabolic syndrome through AMPK activation, Nat Commun 9(1) (2018) 3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].De Plaen E, Arden K, Traversari C, Gaforio JJ, Szikora JP, De Smet C, Brasseur F, van der Bruggen P, Lethe B, Lurquin C, et al. , Structure, chromosomal localization, and expression of 12 genes of the MAGE family, Immunogenetics 40(5) (1994) 360–9. [DOI] [PubMed] [Google Scholar]
- [127].Jang SJ, Soria JC, Wang L, Hassan KA, Morice RC, Walsh GL, Hong WK, Mao L, Activation of melanoma antigen tumor antigens occurs early in lung carcinogenesis, Cancer Res 61(21) (2001) 7959–63. [PubMed] [Google Scholar]
- [128].Liu W, Cheng S, Asa SL, Ezzat S, The melanoma-associated antigen A3 mediates fibronectin-controlled cancer progression and metastasis, Cancer Res 68(19) (2008) 8104–12. [DOI] [PubMed] [Google Scholar]
- [129].Marcar L, Maclaine NJ, Hupp TR, Meek DW, Mage-A cancer/testis antigens inhibit p53 function by blocking its interaction with chromatin, Cancer Res 70(24) (2010) 10362–70. [DOI] [PubMed] [Google Scholar]
- [130].Doyle JM, Gao J, Wang J, Yang M, Potts PR, MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases, Mol Cell 39(6) (2010) 963–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Pineda CT, Ramanathan S, Fon Tacer K, Weon JL, Potts MB, Ou YH, White MA, Potts PR, Degradation of AMPK by a cancer-specific ubiquitin ligase, Cell 160(4) (2015) 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Liu H, Ding J, Kohnlein K, Urban N, Ori A, Villavicencio-Lorini P, Walentek P, Klotz LO, Hollemann T, Pfirrmann T, The GID ubiquitin ligase complex is a regulator of AMPK activity and organismal lifespan, Autophagy 16(9) (2020) 1618–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Berleth ES, Pickart CM, Mechanism of ubiquitin conjugating enzyme E2–230K: catalysis involving a thiol relay?, Biochemistry 35(5) (1996) 1664–71. [DOI] [PubMed] [Google Scholar]
- [134].Vila IK, Yao Y, Kim G, Xia W, Kim H, Kim SJ, Park MK, Hwang JP, Gonzalez-Billalabeitia E, Hung MC, Song SJ, Song MS, A UBE2O-AMPKalpha2 Axis that Promotes Tumor Initiation and Progression Offers Opportunities for Therapy, Cancer Cell 31(2) (2017) 208–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Lee JO, Lee SK, Kim N, Kim JH, You GY, Moon JW, Jie S, Kim SJ, Lee YW, Kang HJ, Lim Y, Park SH, Kim HS, E3 ubiquitin ligase, WWP1, interacts with AMPKalpha2 and down-regulates its expression in skeletal muscle C2C12 cells, J Biol Chem 288(7) (2013) 4673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Qi J, Gong J, Zhao T, Zhao J, Lam P, Ye J, Li JZ, Wu J, Zhou HM, Li P, Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue, EMBO J 27(11) (2008) 1537–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Godlewski J, Nowicki MO, Bronisz A, Nuovo G, Palatini J, De Lay M, Van Brocklyn J, Ostrowski MC, Chiocca EA, Lawler SE, MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells, Mol Cell 37(5) (2010) 620–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Lee SW, Li CF, Jin G, Cai Z, Han F, Chan CH, Yang WL, Li BK, Rezaeian AH, Li HY, Huang HY, Lin HK, Skp2-dependent ubiquitination and activation of LKB1 is essential for cancer cell survival under energy stress, Mol Cell 57(6) (2015) 1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Momcilovic M, Hong SP, Carlson M, Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro, J Biol Chem 281(35) (2006) 25336–43. [DOI] [PubMed] [Google Scholar]
- [140].Neumann D, Is TAK1 a Direct Upstream Kinase of AMPK?, Int J Mol Sci 19(8) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Liu X, Xiao ZD, Han L, Zhang J, Lee SW, Wang W, Lee H, Zhuang L, Chen J, Lin HK, Wang J, Liang H, Gan B, LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress, Nat Cell Biol 18(4) (2016) 431–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Liu X, Xiao ZD, Gan B, An lncRNA switch for AMPK activation, Cell Cycle 15(15) (2016) 1948-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Joseph BK, Liu HY, Francisco J, Pandya D, Donigan M, Gallo-Ebert C, Giordano C, Bata A, Nickels JT Jr., Inhibition of AMP Kinase by the Protein Phosphatase 2A Heterotrimer, PP2APpp2r2d, J Biol Chem 290(17) (2015) 10588–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Davies SP, Helps NR, Cohen PT, Hardie DG, 5’-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC, FEBS Lett 377(3) (1995) 421–5. [DOI] [PubMed] [Google Scholar]
- [145].Voss M, Paterson J, Kelsall IR, Martin-Granados C, Hastie CJ, Peggie MW, Cohen PT, Ppm1E is an in cellulo AMP-activated protein kinase phosphatase, Cell Signal 23(1) (2011) 114–24. [DOI] [PubMed] [Google Scholar]
- [146].Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D, Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade, Biochem J 403(1) (2007) 139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Hardie DG, AMPK--sensing energy while talking to other signaling pathways, Cell Metab 20(6) (2014) 939–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, Schlattner U, Wallimann T, Carling D, Hue L, Rider MH, Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491, J Biol Chem 281(9) (2006) 5335–40. [DOI] [PubMed] [Google Scholar]
- [149].Hurley RL, Barre LK, Wood SD, Anderson KA, Kemp BE, Means AR, Witters LA, Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP, J Biol Chem 281(48) (2006) 36662–72. [DOI] [PubMed] [Google Scholar]
- [150].Dagon Y, Hur E, Zheng B, Wellenstein K, Cantley LC, Kahn BB, p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake, Cell Metab 16(1) (2012) 104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, Viollet B, Makela TP, Wallimann T, Neumann D, Krek W, PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis, EMBO J 29(2) (2010) 469–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Suzuki T, Bridges D, Nakada D, Skiniotis G, Morrison SJ, Lin JD, Saltiel AR, Inoki K, Inhibition of AMPK catabolic action by GSK3, Mol Cell 50(3) (2013) 407–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Coughlan KA, Valentine RJ, Sudit BS, Allen K, Dagon Y, Kahn BB, Ruderman NB, Saha AK, PKD1 Inhibits AMPKalpha2 through Phosphorylation of Serine 491 and Impairs Insulin Signaling in Skeletal Muscle Cells, J Biol Chem 291(11) (2016) 5664–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Heathcote HR, Mancini SJ, Strembitska A, Jamal K, Reihill JA, Palmer TM, Gould GW, Salt IP, Protein kinase C phosphorylates AMP-activated protein kinase alpha1 Ser487, Biochem J 473(24) (2016) 4681–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Setsuie R, Suzuki M, Kabuta T, Fujita H, Miura S, Ichihara N, Yamada D, Wang YL, Ezaki O, Suzuki Y, Wada K, Ubiquitin C-terminal hydrolase-L3-knockout mice are resistant to diet-induced obesity and show increased activation of AMP-activated protein kinase in skeletal muscle, FASEB J 23(12) (2009) 4148–57. [DOI] [PubMed] [Google Scholar]
- [156].Deng M, Yang X, Qin B, Liu T, Zhang H, Guo W, Lee SB, Kim JJ, Yuan J, Pei H, Wang L, Lou Z, Deubiquitination and Activation of AMPK by USP10, Mol Cell 61(4) (2016) 614–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Geiss-Friedlander R, Melchior F, Concepts in sumoylation: a decade on, Nat Rev Mol Cell Biol 8(12) (2007) 947–56. [DOI] [PubMed] [Google Scholar]
- [158].Simpson-Lavy KJ, Johnston M, SUMOylation regulates the SNF1 protein kinase, Proc Natl Acad Sci U S A 110(43) (2013) 17432–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Yan Y, Ollila S, Wong IPL, Vallenius T, Palvimo JJ, Vaahtomeri K, Makela TP, SUMOylation of AMPKalpha1 by PIAS4 specifically regulates mTORC1 signalling, Nat Commun 6 (2015) 8979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Rubio T, Vernia S, Sanz P, Sumoylation of AMPKbeta2 subunit enhances AMP-activated protein kinase activity, Mol Biol Cell 24(11) (2013) 1801–11, S1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, Abraham E, Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase, J Biol Chem 285(43) (2010) 33154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Shao D, Oka S, Liu T, Zhai P, Ago T, Sciarretta S, Li H, Sadoshima J, A redoxdependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation, Cell Metab 19(2) (2014) 232–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Kallis GB, Holmgren A, Differential reactivity of the functional sulfhydryl groups of cysteine-32 and cysteine-35 present in the reduced form of thioredoxin from Escherichia coli, J Biol Chem 255(21) (1980) 10261–5. [PubMed] [Google Scholar]
- [164].Marin TL, Gongol B, Zhang F, Martin M, Johnson DA, Xiao H, Wang Y, Subramaniam S, Chien S, Shyy JY, AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1, Sci Signal 10(464) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Kalia R, Wang RY, Yusuf A, Thomas PV, Agard DA, Shaw JM, Frost A, Structural basis of mitochondrial receptor binding and constriction by DRP1, Nature 558(7710) (2018) 401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Chen Z, Lei C, Wang C, Li N, Srivastava M, Tang M, Zhang H, Choi JM, Jung SY, Qin J, Chen J, Global phosphoproteomic analysis reveals ARMC10 as an AMPK substrate that regulates mitochondrial dynamics, Nat Commun 10(1) (2019) 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Behrends C, Sowa ME, Gygi SP, Harper JW, Network organization of the human autophagy system, Nature 466(7302) (2010) 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Xu DQ, Wang Z, Wang CY, Zhang DY, Wan HD, Zhao ZL, Gu J, Zhang YX, Li ZG, Man KY, Pan Y, Wang ZF, Ke ZJ, Liu ZX, Liao LJ, Chen Y, PAQR3 controls autophagy by integrating AMPK signaling to enhance ATG14L-associated PI3K activity, EMBO J 35(5) (2016) 496–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Weerasekara VK, Panek DJ, Broadbent DG, Mortenson JB, Mathis AD, Logan GN, Prince JT, Thomson DM, Thompson JW, Andersen JL, Metabolic-stress-induced rearrangement of the 14–3-3zeta interactome promotes autophagy via a ULK1- and AMPK-regulated 14–3-3zeta interaction with phosphorylated Atg9, Mol Cell Biol 34(24) (2014) 4379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Zhang D, Wang W, Sun X, Xu D, Wang C, Zhang Q, Wang H, Luo W, Chen Y, Chen H, Liu Z, AMPK regulates autophagy by phosphorylating BECN1 at threonine 388, Autophagy 12(9) (2016) 1447–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Zhao Y, Wang Q, Qiu G, Zhou S, Jing Z, Wang J, Wang W, Cao J, Han K, Cheng Q, Shen B, Chen Y, Zhang WJ, Ma Y, Zhang J, RACK1 Promotes Autophagy by Enhancing the Atg14L-Beclin 1-Vps34-Vps15 Complex Formation upon Phosphorylation by AMPK, Cell Rep 13(7) (2015) 1407–1417. [DOI] [PubMed] [Google Scholar]
- [172].Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL, Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy, Cell 152(1–2) (2013) 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Zhao H, Li T, Wang K, Zhao F, Chen J, Xu G, Zhao J, Li T, Chen L, Li L, Xia Q, Zhou T, Li HY, Li AL, Finkel T, Zhang XM, Pan X, AMPK-mediated activation of MCU stimulates mitochondrial Ca(2+) entry to promote mitotic progression, Nat Cell Biol 21(4) (2019) 476–486. [DOI] [PubMed] [Google Scholar]
- [174].Mao L, Li N, Guo Y, Xu X, Gao L, Xu Y, Zhou L, Liu W, AMPK phosphorylates GBF1 for mitotic Golgi disassembly, J Cell Sci 126(Pt 6) (2013) 1498–505. [DOI] [PubMed] [Google Scholar]
- [175].Miyamoto T, Oshiro N, Yoshino K, Nakashima A, Eguchi S, Takahashi M, Ono Y, Kikkawa U, Yonezawa K, AMP-activated protein kinase phosphorylates Golgi-specific brefeldin A resistance factor 1 at Thr1337 to induce disassembly of Golgi apparatus, J Biol Chem 283(7) (2008) 4430–8. [DOI] [PubMed] [Google Scholar]
- [176].Lee IJ, Lee CW, Lee JH, CaMKKbeta-AMPKalpha2 signaling contributes to mitotic Golgi fragmentation and the G2/M transition in mammalian cells, Cell Cycle 14(4) (2015) 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Chauhan AS, Liu X, Jing J, Lee H, Yadav RK, Liu J, Zhou Y, Gan B, STIM2 interacts with AMPK and regulates calcium-induced AMPK activation, FASEB J 33(2) (2019) 2957–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Kodiha M, Rassi JG, Brown CM, Stochaj U, Localization of AMP kinase is regulated by stress, cell density, and signaling through the MEK-->ERK1/2 pathway, Am J Physiol Cell Physiol 293(5) (2007) C1427–36. [DOI] [PubMed] [Google Scholar]
- [179].Suzuki A, Okamoto S, Lee S, Saito K, Shiuchi T, Minokoshi Y, Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor alpha gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the alpha2 form of AMP-activated protein kinase, Mol Cell Biol 27(12) (2007) 4317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Kazgan N, Williams T, Forsberg LJ, Brenman JE, Identification of a nuclear export signal in the catalytic subunit of AMP-activated protein kinase, Mol Biol Cell 21(19) (2010) 3433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Okamoto S, Asgar NF, Yokota S, Saito K, Minokoshi Y, Role of the alpha2 subunit of AMP-activated protein kinase and its nuclear localization in mitochondria and energy metabolism-related gene expressions in C2C12 cells, Metabolism 90 (2019) 52–68. [DOI] [PubMed] [Google Scholar]
- [182].Lamia KA, Sachdeva UM, DiTacchio L, Williams EC, Alvarez JG, Egan DF, Vasquez DS, Juguilon H, Panda S, Shaw RJ, Thompson CB, Evans RM, AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation, Science 326(5951) (2009) 437–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF, Knockout of the alpha2 but not alpha1 5’-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle, J Biol Chem 279(2) (2004) 1070–9. [DOI] [PubMed] [Google Scholar]
- [184].Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S, The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity, J Clin Invest 111(1) (2003) 91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Zarrinpashneh E, Carjaval K, Beauloye C, Ginion A, Mateo P, Pouleur AC, Horman S, Vaulont S, Hoerter J, Viollet B, Hue L, Vanoverschelde JL, Bertrand L, Role of the alpha2-isoform of AMP-activated protein kinase in the metabolic response of the heart to no-flow ischemia, Am J Physiol Heart Circ Physiol 291(6) (2006) H2875–83. [DOI] [PubMed] [Google Scholar]
- [186].Carvajal K, Zarrinpashneh E, Szarszoi O, Joubert F, Athea Y, Mateo P, Gillet B, Vaulont S, Viollet B, Bigard X, Bertrand L, Ventura-Clapier R, Hoerter JA, Dual cardiac contractile effects of the alpha2-AMPK deletion in low-flow ischemia and reperfusion, Am J Physiol Heart Circ Physiol 292(6) (2007) H3136–47. [DOI] [PubMed] [Google Scholar]
- [187].Andreelli F, Foretz M, Knauf C, Cani PD, Perrin C, Iglesias MA, Pillot B, Bado A, Tronche F, Mithieux G, Vaulont S, Burcelin R, Viollet B, Liver adenosine monophosphate-activated kinase-alpha2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin, Endocrinology 147(5) (2006) 2432–41. [DOI] [PubMed] [Google Scholar]
- [188].Guigas B, Taleux N, Foretz M, Detaille D, Andreelli F, Viollet B, Hue L, AMP-activated protein kinase-independent inhibition of hepatic mitochondrial oxidative phosphorylation by AICA riboside, Biochem J 404(3) (2007) 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jorgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA, Kemp BE, Richter EA, Steinberg GR, AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise, Proc Natl Acad Sci U S A 108(38) (2011) 16092–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Mu J, Brozinick JT Jr., Valladares O, Bucan M, Birnbaum MJ, A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle, Mol Cell 7(5) (2001) 1085–94. [DOI] [PubMed] [Google Scholar]
- [191].Barnes BR, Marklund S, Steiler TL, Walter M, Hjalm G, Amarger V, Mahlapuu M, Leng Y, Johansson C, Galuska D, Lindgren K, Abrink M, Stapleton D, Zierath JR, Andersson L, The 5’-AMP-activated protein kinase gamma3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle, J Biol Chem 279(37) (2004) 38441–7. [DOI] [PubMed] [Google Scholar]
- [192].Barnes BR, Glund S, Long YC, Hjalm G, Andersson L, Zierath JR, 5’-AMP-activated protein kinase regulates skeletal muscle glycogen content and ergogenics, FASEB J 19(7) (2005) 773–9. [DOI] [PubMed] [Google Scholar]
- [193].Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK, Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase, FEBS Lett 353(1) (1994) 33–6. [DOI] [PubMed] [Google Scholar]
- [194].Villena JA, Viollet B, Andreelli F, Kahn A, Vaulont S, Sul HS, Induced adiposity and adipocyte hypertrophy in mice lacking the AMP-activated protein kinase-alpha2 subunit, Diabetes 53(9) (2004) 2242–9. [DOI] [PubMed] [Google Scholar]
- [195].Hunter RW, Treebak JT, Wojtaszewski JF, Sakamoto K, Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle, Diabetes 60(3) (2011) 766–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Penfold L, Woods A, Muckett P, Nikitin AY, Kent TR, Zhang S, Graham R, Pollard A, Carling D, CAMKK2 Promotes Prostate Cancer Independently of AMPK via Increased Lipogenesis, Cancer Res 78(24) (2018) 6747–6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Wang W, Xiao ZD, Li X, Aziz KE, Gan B, Johnson RL, Chen J, AMPK modulates Hippo pathway activity to regulate energy homeostasis, Nat Cell Biol 17(4) (2015) 490–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Li YH, Luo J, Mosley YY, Hedrick VE, Paul LN, Chang J, Zhang G, Wang YK, Banko MR, Brunet A, Kuang S, Wu JL, Chang CJ, Scott MP, Yang JY, AMP-Activated Protein Kinase Directly Phosphorylates and Destabilizes Hedgehog Pathway Transcription Factor GLI1 in Medulloblastoma, Cell Rep 12(4) (2015) 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Shen CH, Yuan P, Perez-Lorenzo R, Zhang Y, Lee SX, Ou Y, Asara JM, Cantley LC, Zheng B, Phosphorylation of BRAF by AMPK impairs BRAF-KSR1 association and cell proliferation, Mol Cell 52(2) (2013) 161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB, The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis, Nat Cell Biol 9(2) (2007) 218–24. [DOI] [PubMed] [Google Scholar]
- [201].Dasgupta B, Milbrandt J, AMP-activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development, Dev Cell 16(2) (2009) 256–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR, LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1, EMBO J 23(4) (2004) 833–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, Liang MC, Cai D, Naumov GN, Bao L, Contreras CM, Li D, Chen L, Krishnamurthy J, Koivunen J, Chirieac LR, Padera RF, Bronson RT, Lindeman NI, Christiani DC, Lin X, Shapiro GI, Janne PA, Johnson BE, Meyerson M, Kwiatkowski DJ, Castrillon DH, Bardeesy N, Sharpless NE, Wong KK, LKB1 modulates lung cancer differentiation and metastasis, Nature 448(7155) (2007) 807–10. [DOI] [PubMed] [Google Scholar]
- [204].Sato N, Rosty C, Jansen M, Fukushima N, Ueki T, Yeo CJ, Cameron JL, Iacobuzio-Donahue CA, Hruban RH, Goggins M, STK11/LKB1 Peutz-Jeghers gene inactivation in intraductal papillary-mucinous neoplasms of the pancreas, Am J Pathol 159(6) (2001) 2017-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Wan L, Xu K, Wei Y, Zhang J, Han T, Fry C, Zhang Z, Wang YV, Huang L, Yuan M, Xia W, Chang WC, Huang WC, Liu CL, Chang YC, Liu J, Wu Y, Jin VX, Dai X, Guo J, Liu J, Jiang S, Li J, Asara JM, Brown M, Hung MC, Wei W, Phosphorylation of EZH2 by AMPK Suppresses PRC2 Methyltransferase Activity and Oncogenic Function, Mol Cell 69(2) (2018) 279–291 e5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [206].Wu D, Hu D, Chen H, Shi G, Fetahu IS, Wu F, Rabidou K, Fang R, Tan L, Xu S, Liu H, Argueta C, Zhang L, Mao F, Yan G, Chen J, Dong Z, Lv R, Xu Y, Wang M, Ye Y, Zhang S, Duquette D, Geng S, Yin C, Lian CG, Murphy GF, Adler GK, Garg R, Lynch L, Yang P, Li Y, Lan F, Fan J, Shi Y, Shi YG, Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer, Nature 559(7715) (2018) 637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, Li CW, Kim T, Chang SS, Lee HH, Hsu JL, Wang HL, Kuo CW, Chang WC, Hadad S, Purdie CA, McCoy AM, Cai S, Tu Y, Litton JK, Mittendorf EA, Moulder SL, Symmans WF, Thompson AM, Piwnica-Worms H, Chen CH, Khoo KH, Hung MC, Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1, Mol Cell 71(4) (2018) 606–620 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, Mamer OA, Avizonis D, DeBerardinis RJ, Siegel PM, Jones RG, AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo, Cell Metab 17(1) (2013) 113–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL, The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice, Nature 318(6046) (1985) 533–8. [DOI] [PubMed] [Google Scholar]
- [210].Vara-Ciruelos D, Dandapani M, Russell FM, Grzes KM, Atrih A, Foretz M, Viollet B, Lamont DJ, Cantrell DA, Hardie DG, Phenformin, But Not Metformin, Delays Development of T Cell Acute Lymphoblastic Leukemia/Lymphoma via Cell-Autonomous AMPK Activation, Cell Rep 27(3) (2019) 690–698 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Houde VP, Donzelli S, Sacconi A, Galic S, Hammill JA, Bramson JL, Foster RA, Tsakiridis T, Kemp BE, Grasso G, Blandino G, Muti P, Steinberg GR, AMPK beta1 reduces tumor progression and improves survival in p53 null mice, Mol Oncol 11(9) (2017) 1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Jeon SM, Chandel NS, Hay N, AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress, Nature 485(7400) (2012) 661–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ, Macintyre AN, Goraksha-Hicks P, de Cubas AA, Liu T, Warmoes MO, Abel ED, Yeoh AE, Gershon TR, Rathmell WK, Richards KL, Locasale JW, Rathmell JC, AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival, Cell Metab 23(4) (2016) 649–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Saito Y, Chapple RH, Lin A, Kitano A, Nakada D, AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow, Cell Stem Cell 17(5) (2015) 585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Eichner LJ, Brun SN, Herzig S, Young NP, Curtis SD, Shackelford DB, Shokhirev MN, Leblanc M, Vera LI, Hutchins A, Ross DS, Shaw RJ, Svensson RU, Genetic Analysis Reveals AMPK Is Required to Support Tumor Growth in Murine Kras-Dependent Lung Cancer Models, Cell Metab 29(2) (2019) 285–302 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Chang WH, Lai AG, An integrative pan-cancer investigation reveals common genetic and transcriptional alterations of AMPK pathway genes as important predictors of clinical outcomes across major cancer types, BMC Cancer 20(1) (2020) 773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Cancer N. Genome Atlas Research, Comprehensive molecular profiling of lung adenocarcinoma, Nature 511(7511) (2014) 543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].O’Brien AJ, Villani LA, Broadfield LA, Houde VP, Galic S, Blandino G, Kemp BE, Tsakiridis T, Muti P, Steinberg GR, Salicylate activates AMPK and synergizes with metformin to reduce the survival of prostate and lung cancer cells ex vivo through inhibition of de novo lipogenesis, Biochem J 469(2) (2015) 177–87. [DOI] [PubMed] [Google Scholar]
- [219].Zadra G, Photopoulos C, Tyekucheva S, Heidari P, Weng QP, Fedele G, Liu H, Scaglia N, Priolo C, Sicinska E, Mahmood U, Signoretti S, Birnberg N, Loda M, A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis, EMBO Mol Med 6(4) (2014) 519–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, Wei L, Fishbein MC, Czernin J, Mischel PS, Shaw RJ, LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin, Cancer Cell 23(2) (2013) 143–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Lally JSV, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, Erstad DJ, Fujiwara N, Leong V, Houde VP, Anagnostopoulos AE, Wang A, Broadfield LA, Ford RJ, Foster RA, Bates J, Sun H, Wang T, Liu H, Ray AS, Saha AK, Greenwood J, Bhat S, Harriman G, Miao W, Rocnik JL, Westlin WF, Muti P, Tsakiridis T, Harwood HJ Jr., Kapeller R, Hoshida Y, Tanabe KK, Steinberg GR, Fuchs BC, Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma, Cell Metab 29(1) (2019) 174–182 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Scaglia N, Tyekucheva S, Zadra G, Photopoulos C, Loda M, De novo fatty acid synthesis at the mitotic exit is required to complete cellular division, Cell Cycle 13(5) (2014) 859–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Boergermann JH, Kopf J, Yu PB, Knaus P, Dorsomorphin and LDN-193189 inhibit BMP-mediated Smad, p38 and Akt signalling in C2C12 cells, Int J Biochem Cell Biol 42(11) (2010) 1802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Vogt J, Traynor R, Sapkota GP, The specificities of small molecule inhibitors of the TGFss and BMP pathways, Cell Signal 23(11) (2011) 1831-42. [DOI] [PubMed] [Google Scholar]
- [225].Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P, The selectivity of protein kinase inhibitors: a further update, Biochem J 408(3) (2007) 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]