

ABSTRACT
Our current understanding of plant viruses stems largely from those affecting economically important plants. Yet plant species in cultivation represent a small and biased subset of the plant kingdom. Here, we describe virus diversity and abundance in 1,079 transcriptomes from species across the breadth of the plant kingdom (Archaeplastida) by analyzing open-source data from the 1000 Plant Transcriptomes Initiative (1KP). We identified 104 potentially novel viruses, of which 40% were single-stranded positive-sense RNA viruses across eight orders, including members of the Hepelivirales, Tymovirales, Cryppavirales, Martellivirales, and Picornavirales. One-third of the newly described viruses were double-stranded RNA viruses from the orders Durnavirales and Ghabrivirales. The remaining were negative-sense RNA viruses from the Rhabdoviridae, Aspiviridae, Yueviridae, and Phenuiviridae and the newly proposed Viridisbunyaviridae. Our analysis considerably expands the known host range of 13 virus families to include lower plants (e.g., Benyviridae and Secoviridae) and 4 virus families to include alga hosts (e.g., Tymoviridae and Chrysoviridae). More broadly, however, a cophylogeny analysis revealed that the evolutionary history of these families is largely driven by cross-species transmission events. The discovery of the first 30-kDa movement protein in a nonvascular plant suggests that the acquisition of plant virus movement proteins occurred prior to the emergence of the plant vascular system. Together, these data highlight that numerous RNA virus families are associated with older evolutionary plant lineages than previously thought and that the apparent scarcity of RNA viruses found in lower plants likely reflects a lack of investigation rather than their absence.
IMPORTANCE Our knowledge of plant viruses is mainly limited to those infecting economically important host species. In particular, we know little about those viruses infecting basal plant lineages such as the ferns, lycophytes, bryophytes, and charophytes. To expand this understanding, we conducted a broad-scale viral survey of species across the breadth of the plant kingdom. We found that basal plants harbor a wide diversity of RNA viruses, including some that are sufficiently divergent to likely compose a new virus family. The basal plant virome revealed offers key insights into the evolutionary history of core plant virus gene modules and genome segments. More broadly, this work emphasizes that the scarcity of viruses found in these species to date most likely reflects the limited research in this area.
KEYWORDS: plant virus, virus discovery, alga virus, Benyviridae, Bunyavirales, Secoviridae, evolution
INTRODUCTION
Viruses are responsible for almost 50% of all emerging plant diseases (1). Historically, virus characterization has focused on pathogenic viruses that infect species of economic importance, with 69% of the current phytovirosphere—the total assemblage of plant viruses—identified in cultivated plant species even though they represent less than 0.17% of known plant diversity (2). Importantly, the advent of metagenomic sequencing technology enables the comprehensive screening of plant tissues for novel and known viruses (3). Despite this, virus diversity in the vast majority of plants remains unquantified (4).
Our ability to infer the origins and diversification of the phytovirosphere requires adequate sampling of the viruses across the plant kingdom. Several key plant groups are severely underrepresented or absent in previous studies, including charophytes, lower plants, gymnosperms and several angiosperm orders (4, 5). Improving knowledge across these groups will undoubtedly help uncover the evolutionary history of plant virus lineages. For instance, an analysis of the evolutionary history of viruses from rhodophytes and charophytes might reveal how key evolutionary transitions of plants—such as terrestrialization—have shaped the contemporary land plant virome (4). Similarly, through broad sampling across the plant kingdom, we can acquire a better understanding of the acquisition of viruses through cross-species transmission from plant-associated organisms such as invertebrates, fungi, or protists (4).
The majority (68%) of the currently documented genera of plant viruses have positive-sense single-stranded RNA (+ssRNA) genomes, and the majority of virus diversity is known only from angiosperms (6) (Fig. 1). Currently, 16 viruses belonging to 12 virus families have been found in gymnosperms (7–11). Outside of several viruses found in ferns, we know little of the diversity of viruses in the lycophytes, bryophytes, and charophytes, which together encompass ~27,000 species (12–15) (Fig. 1). A partial analysis of published transcriptome data detected homologs of the canonical RNA virus RNA-dependent RNA polymerase (RdRp) in algae, several lower plants, and gymnosperms (16). However, it is uncertain whether viruses that infect freshwater algae—which include the Zygnematophyceae ancestors of land plants—resemble those infecting angiosperms or those infecting the chlorophytes, which are dominated by double-stranded DNA (dsDNA) viruses, particularly from the Phycodnaviridae (17). To date, two +ssRNA viruses related to the benyvirids have been identified in freshwater algae (18, 19). Unlike the Chlorophyta, the Charophyta characteristically contain plasmodesmata and homologs of the key components of the land plant innate immune system, both of which have been speculated to explain the apparent absence of double-strand DNA (dsDNA) viruses in land plants (4, 20, 21). In particular, the plasmodesmata is thought to provide a physical barrier preventing the entry of medium to large virions which lack movement proteins (i.e., dsDNA viruses). An understanding of the viruses infecting the Charophyta and other lower plants is required to effectively test these ideas.
FIG 1.

(A) Phylogram of virus composition across the 1000 Plant Transcriptomes Initiative (1KP) samples. Plant-associated virus abundance was summarized for each plant species and normalized using a Box-Cox transformation. The height of each bar represents the percentage of virus reads detected in each plant species (after the removal of host reads). Plant clades are labeled and differentiated by shades of gray. The 1KP ASTRAL tree was used as the basis for this tree (29). Clade and abundance annotations were added using the Interactive Tree of Life (iTOL) Web-based tool (108). (B) The phytovirosphere across the Plantae and Phaeophyceae. A schematic tree of the evolution of major plant groups. Each bar represents the number of total viruses formally or likely associated with each host group and is colored by virus genome composition. The total number of viruses for each plant group plus those found in this study is also shown at the end of each bar. The Virus-Host (79) and NCBI virus databases (109) combined with literature searches were used to obtain virus counts. Lineage branches are not drawn to scale. To our knowledge, no viruses have been found in the glaucophytes. Plant and alga images were obtained from BioRender.com or drawn in Adobe Illustrator (https://www.adobe.com). *, Transcriptome scaffolds from libraries belonging to these host groups shared homology to virus RdRps and were partially analyzed but not assembled or deposited in GenBank (16).
Transcriptome mining has become an efficient method of virus discovery that leverages previous investment (22–28). To this end, we mined the transcriptome data generated by the One Thousand Plant Transcriptomes Initiative (1KP) using sequence homology searches of known plant viruses. The 1KP project provides a major untapped source of polyA-selected transcriptome data for virus discovery drawn from species across the breadth of the plants in a broad sense, including the Viridiplantae (green plants), Glaucophyta, and Rhodophyta (red algae) (29, 30). Our broad aim was to revise our understanding of the phytovirosphere using data across the plant kingdom as well as providing insights into the origins and diversification of plant viruses.
RESULTS
We characterized the viruses found in the transcriptomes of 960 plant species within the 1KP major release. The transcriptomes represented a broad taxonomic sampling across the Archaeplastida (green plants, glaucophytes, and rhodophytes). Sequencing libraries had a median of 25,187,714 paired reads (range, 10,156,464 to 46,650,336). A median of 82% of reads (range, 1% to 96%) in these libraries mapped to host genome scaffolds and were subsequently removed. De novo assembly of the sequencing reads resulted in a median of 36,015 contigs (range, 1,396 to 146,217) per library, with a total of 41,256,176 contigs generated (see Table S2 in the supplemental material).
Diversity and abundance of plant viruses.
In total, virus-like transcripts were found for 603 plant species; 69% of these were plant-associated, while numerous identified sequences shared high similarity to non-plant-associated viruses, including those known to infect fungal, invertebrate, and vertebrate hosts. Among the non-plant-associated virus transcripts, 34% were unclassified (10% of total virus-like transcripts) such that they were most closely related to a virus sequence with little to no taxonomic information (i.e., a virus sequence classified as only belonging to the Riboviria). If an RdRp-like region was detected in an unclassified virus-like transcript, we further assessed whether it could be plant-associated (see “Phylogenetic Analysis of Identified Viruses”). The remaining non-plant-associated virus transcripts were largely classified within the Orthomyxoviridae (vertebrate-associated) (25%), Rhabdoviridae (invertebrate-associated) (17%), Partitiviridae (fungus-associated) (10%), Mimiviridae (amoeboid-associated) (10%), and Adenoviridae (vertebrate-associated) (7%) and excluded from the remainder of this study. These sequences are discussed in more detail in the section “Presence of Contaminants in Sequencing Libraries.” Although some of these viruses could represent plant infection, we made the conservative decision to remove them from the analysis.
We detected transcripts closely associated with viruses containing single- and double-stranded DNA and RNA genomes. The majority of virus-like sequences belonged to families with +ssRNA genomes (61%) or reverse-transcribing dsDNA viruses (22%) (Fig. 1). The +ssRNA virus transcripts were predominately classified within the Betaflexiviridae (30%), Potyviridae (19%), Secoviridae (16%), and Alphaflexiviridae (10%) (Table S3). Negative-sense single-stranded RNA (–ssRNA) virus transcripts were classified within the Aspiviridae (0.04%), Rhabdoviridae (6%), and Tospoviridae (3%) (Phenuiviridae and Yueviridae transcripts were later detected in the unclassified virus-like transcripts) (Table S3). dsDNA virus transcripts with sequence similarities to the Phycodnaviridae were detected in the alga samples. These phycodna-like virus transcripts frequently encoded the chitinase and DNA ligase genes that are homologous to those in distantly related host organisms, including fungi and bacteria. As it is difficult to determine whether these transcripts represent Phycodnaviridae sequences or contamination, we excluded all phycodnavirus-related sequences. All remaining dsDNA viruses were exclusively reverse-transcribing viruses from the Caulimoviridae. We failed to detect any sequences that shared homology with several plant virus families, including Reoviridae, Nanoviridae, and Fimoviridae (although see Discussion for caveats).
There was a large range of viral abundance in each library (5 × 10−6% to 31% of reads after host-associated reads were removed). Viruses with +ssRNA genomes accounted for the vast majority (99.8%) of virus abundance (Fig. 1, Table S3). As expected, virus discovery was concentrated in the flowering plants (angiosperms). For instance, plant virus-like sequences were frequently discovered in the core eudicots and monocots (i.e., 73% of libraries in which plant virus transcripts were found). The detection rate of plant viruses was highest in the most basal angiosperms (57%) and monocots (50%). No significant difference in virus abundance was observed between sequencing platforms (Genome Analyzer II and Illumina HiSeq 2000; P = 0.327).
Presence of contaminants in sequencing libraries.
The bacterial, fungal, and insect species that live in or on plant tissues are commonly sampled within plant sequencing libraries (30), although contamination from other plants is also a possibility during sample preparation or sequencing. To quantify the extent of library contamination, we used the KMA and CCMetagen tools (Fig. 2). Among the libraries analyzed (n = 95), bacteria were consistently detected, representing a median of 1.5% of total abundance (range, 0.01% to 33%). A median of 2% of library abundance was associated with fungal sequences (range 0% to 53%). Arthropods and chordates were also commonly detected across libraries (found in 87 and 89 libraries, respectively) but at lower abundance (median, 0.15%; range, 0% to 11.4%). The presence of chordate-associated reads is likely attributed to various routes of sample contamination (e.g., feces) or sample processing and sequencing.
FIG 2.

Taxonomic assignments of reads in select 1000 Plant Transcriptomes Initiative (1KP) libraries. Each Krona graph illustrates the relative abundance of taxa in a metatranscriptome at various taxonomic levels. For clarity, a maximum depth of five taxonomic levels was chosen for each graph. The library Sequence Read Archive accession number, host species, and corresponding virus of interest are annotated above each graph. Segments are highlighted based upon the species taxonomic grouping (plants = green, chromista = blue, unclassified = orange, bacteria = red, metazoa = pink, fungi = purple, rhodophytes = light blue, other = yellow). Here, “plants” encompasses the Viridiplantae. Reads without any match in the nucleotide database are not shown.
The detection of four vertebrate-associated viruses across several libraries provided further evidence of library contamination. Sequences belonging to these viruses—influenza A virus (16 libraries), human mastadenovirus C (30 libraries), human immunodeficiency virus (15 libraries), and parainfluenza virus 5 (3 libraries)—were present at low abundance and showed little genetic variation between libraries. Notably, chordate-associated reads were only present in 66% of libraries in which these viruses were found. The failure to consistently detect potential hosts for these viruses suggests contamination during sequencing. The four vertebrate-associated viruses were largely absent in libraries in which novel plant-associated viruses were discovered, except for the Larix speciosa, Brachiomonas submarina, Climacium dendroides, Silene latifolia, and Oxera neriifolia transcriptomes.
In addition, the 1KP compared all assembled sequences to a reference set of nuclear 18S rRNA sequences from the SILVA small subunit rRNA database using BLASTn (30, 31). Where a sample had several alignments to any other plant sequences outside the expected source family, the sample was described as having “worrisome contamination” (30). This applied to 11 plant libraries in which novel viruses were identified. Below, we discuss library contaminates from the viewpoint of virus-host associations.
Phylogenetic analysis of identified viruses.
To infer phylogenetic relationships between identified viruses, order- and family-level phylogenetic trees were estimated using the highly conserved viral region that comprises the RdRp. In total, we assembled 104 RdRp contigs that likely represent novel virus species, 41 of which were considered unclassified or non-plant-associated due to their similarities to virus groups known to infect nonplant hosts (Table S4). Further analysis of these contigs revealed that they are likely plant-associated.
Positive-sense single-stranded RNA [(+)ssRNA] viruses.
(i) Hepelivirales. (a) Benyviridae. We identified three beny-like sequences that to our knowledge represent the first benyvirid found in lower plants. The first sequence, tentatively named Fern benyvirus (FeBV), was found in both the bird’s-nest fern (Asplenium nidus) and tomato fern (Lonchitis hirsuta). Together with Wheat stripe mosaic virus, FeBV represents a well-supported clade separate from the remaining plant benyviruses (Fig. 3).
FIG 3.

(A) Phylogenetic relationships of the beny-like viruses identified in this study. ML phylogenetic tree based on the RNA-1 replicase protein shows the topological position of virus-like sequences discovered in this study (black circles) in the context of their closest relatives. Branches are highlighted to represent host clade (land plants = green, lower plants = orange, invertebrates = red, vertebrates = pink, algae = blue, fungi = purple, environmental = yellow, chromista = light blue, rhodophytes = dark green). Here, “land plants” encompasses both angiosperms and gymnosperms, while “lower plants” includes the bryophytes, lycophytes, and ferns. All branches are scaled to the number of amino acid substitutions per site, and trees were midpoint rooted for clarity only. An asterisk indicates node support of >70% bootstrap support. Tip labels are bolded when the genome structure is shown on the right. (B) Genomic organization of the beny-like virus sequences identified in this study and representative species used in the phylogeny. Beet soilborne mosaic virus RNA 3and 4 are not pictured here. The data underlying this figure and definitions of acronyms used are presented in Table S5.
The triple gene block (TGB) is a hallmark gene module of the Benyviridae, among several other virus families in the class Alsuviricetes (32). In both fern libraries, proteins resembling the TGB were assembled (Fig. 3). The TGB proteins shared ~34% amino acid identity with the TGB protein of other benyvirids. To our knowledge, this is the first TGB protein found outside flowering plants. Phylogenetic analysis placed the TGB1 protein of FeBV basal to the Benyviridae (Fig. S1).
Two additional beny-like viruses, named here Leucodon julaceus associated beny-like virus (LjBV) and Wallace’s spikemoss associated beny-like virus (WasBV), were assembled. LjBV and WasBV cluster with unclassified algae, invertebrates, fungi, and soil-derived viruses forming a group basal to all plant benyvirids and potentially constitute a novel virus group (Fig. 3). LjBV contains a second open reading frame (ORF) with no detectable homology to known sequences (Fig. 3).
Due to the phylogenetic placement of LjBV and WasBV close to viruses infecting distant hosts (e.g., invertebrates and fungi), we investigated the potential of contamination from other eukaryotes as the source of these viruses. Of note, the Wallace’s spikemoss metatranscriptome contained reads that matched various fungus orders (7% of all reads) as well as those matching the plant-parasitic oomycete Albugo laibachii (7%), which makes inferring virus-host relationships challenging (Fig. 2). Reads belonging to various fungus species accounted for 10% of the bird’s-nest fern transcriptome and 12% of the tomato fern transcriptome (Fig. 2). Despite the presence of fungus-associated reads, the phylogenetic position of FeBV suggests that FeBV is likely plant-associated (Fig. 3). No concerning contaminants were detected in the Leucodon julaceus transcriptome.
(ii) Tymovirales. (a) Betaflexiviridae. We identified 18 virus sequences that fell in the order Tymovirales. Four virus transcripts were associated with the Betaflexiviridae. The first, Sea beet betaflexivirus (SbBV), clusters with Agapanthus virus A, an unclassified betaflexivirus (Fig. 4). The remaining sequences, denoted Iranian poppy betaflexivirus (IpBV), Linum macraei betaflexivirus (LimBV), and Lycopod associated betaflexivirus (LyBtV), resemble capilloviruses. Notably, LyBtV may extend the known host range of the Betaflexiviridae from angiosperms to lower plants. All sequences phylogenetically cluster with known capilloviruses and potentially represent novel virus species (Fig. 4). The Phylloglossum drummondii library in which LyBtV was assembled had contamination from lycopod and dicot species (Fig. 2). As the majority of plant-associated reads were assigned to lycophytes (50%), LyBtV has been tentatively assigned to this group.
FIG 4.

(Left) Phylogenetic relationships of the viruses within the order Tymovirales. ML phylogenetic tree based on the replication protein shows the topological position of virus-like sequences discovered in this study (black circles) in the context of their closest relatives. See Fig. 3 for the color scheme. All branches are scaled to the number of amino acid substitutions per site, and trees were midpoint rooted for clarity only. An asterisk indicates node support of >70% bootstrap support. Tip labels are bolded when the genome structure is shown on the right. (Right) Genomic organization of the virus sequences identified in this study and representative species used in the phylogeny.
(b) Tymoviridae. We identified 12 virus-like sequences that clustered within the Tymoviridae and related viruses. Ishige okamurae associated tymo-like virus (IoTV) was detected in the brown alga Ishige okamurae and likely represents the first virus in the order Tymovirales from brown algae. IoTV, along with 10 sequences assembled from species in the Anthocerotophyta (hornworts), Marchantiophyta (liverworts), and other bryophytes grouped with tymo-like viruses from fungus and environmental samples (Fig. 4). It is uncertain whether the true hosts of the novel tymo-like viruses discovered here are plants. Fungal contaminants were detected across these libraries but varied in abundance (range, 1% to 21%; mean, 6%). Despite their clustering with mycotymoviruses, Broom forkmoss associated tymo-like virus (BrfoTV) and Tree climacium moss associated tymo-like virus (TcmTV) were assembled from libraries with ~1% fungal reads, highlighting the inherent difficulties in host-virus assignment. Importantly, <1% of reads in the Ishige okamurae transcriptome belonged to species of fungi (Fig. 2).
We assembled two tymo-like virus sequences, denoted Oxera neriifolia tymo-like virus (OnTV) and Bloodroot marafivirus (BloMV). BloMV and OnTV grouped with the unclassified Glehnia littoralis marafivirus (Fig. 4). Marafiviruses and tymoviruses are commonly distinguished from each other based upon a highly conserved 16-nucleotide (nt) sequence known as the “tymobox” (GAGUCUGAAUUGCUUC) in tymoviruses and the “marafibox” [CA(G/A)GGUGAAUUGCUUC] in marafiviruses (33, 34). While these two novel viruses cluster together, they differ in terms of genome structure and motifs. A marafibox-like sequence is present in BloMV (CAACGCGAAUUGCUUU) (nt 5606 to 5621), albeit differing by several residues. This, combined with the BloMV genome likely consisting of a single large ORF, supports the assignment of BloMV as a Marafivirus. OnTV, like members of the Tymovirus genera, contains both a second ORF—likely encoding a coat protein (CP)—and a tymobox (nt 1493 to 1508) (Fig. 4). Phylogenetic analysis of the coat protein sequence places OnTV and BloMV in a clade with macula- and marafi-like viruses (Fig. S1).
(c) Deltaflexiviridae. We assembled two sequences that share similarities to members of the mycotymovirus family Deltaflexiviridae. The first sequence was detected in the liverwort Calypogeia fissa, tentatively named Calypogeia fissa associated deltaflexivirus (CafADV), and appeared distantly related to delta- and gammaflexiviruses. A second related partial sequence, named here Pinguicula agnata virus (PaV), shared 32% amino acid identity with the mycoflexivirus Botrytis virus F. In a phylogenetic analysis with members of the Tymovirales, CafADV and PaAGV are placed with the deltaflexivirids (Fig. 4).
It is unclear whether the source of these virus sequences is from plants or contamination from other eukaryotes. Indeed, the C. fissa library contained numerous contaminants, including algae, fungi, and bacteria, representing 1%, 15%, and 33% of total reads, respectively (Fig. 2). Interestingly, no fungus-associated reads were found in the Pinguicula agnata library, suggesting a potential plant origin (Fig. 2).
(iii) Picornavirales. (a) Secoviridae. We identified four sequences that shared similarities to members of the Secoviridae, denoted Common water moss secovirus (CwmSV), Salix dasyclados secovirus (SadSV), Tomato fern secovirus (TfSV), and Shostring fern secovirus (SfSV). CwmSV, TfSV and SfSV cluster within the nepoviruses and likely represent the first seco-like virus detected in the bryophytes and ferns (Fig. 5).
FIG 5.
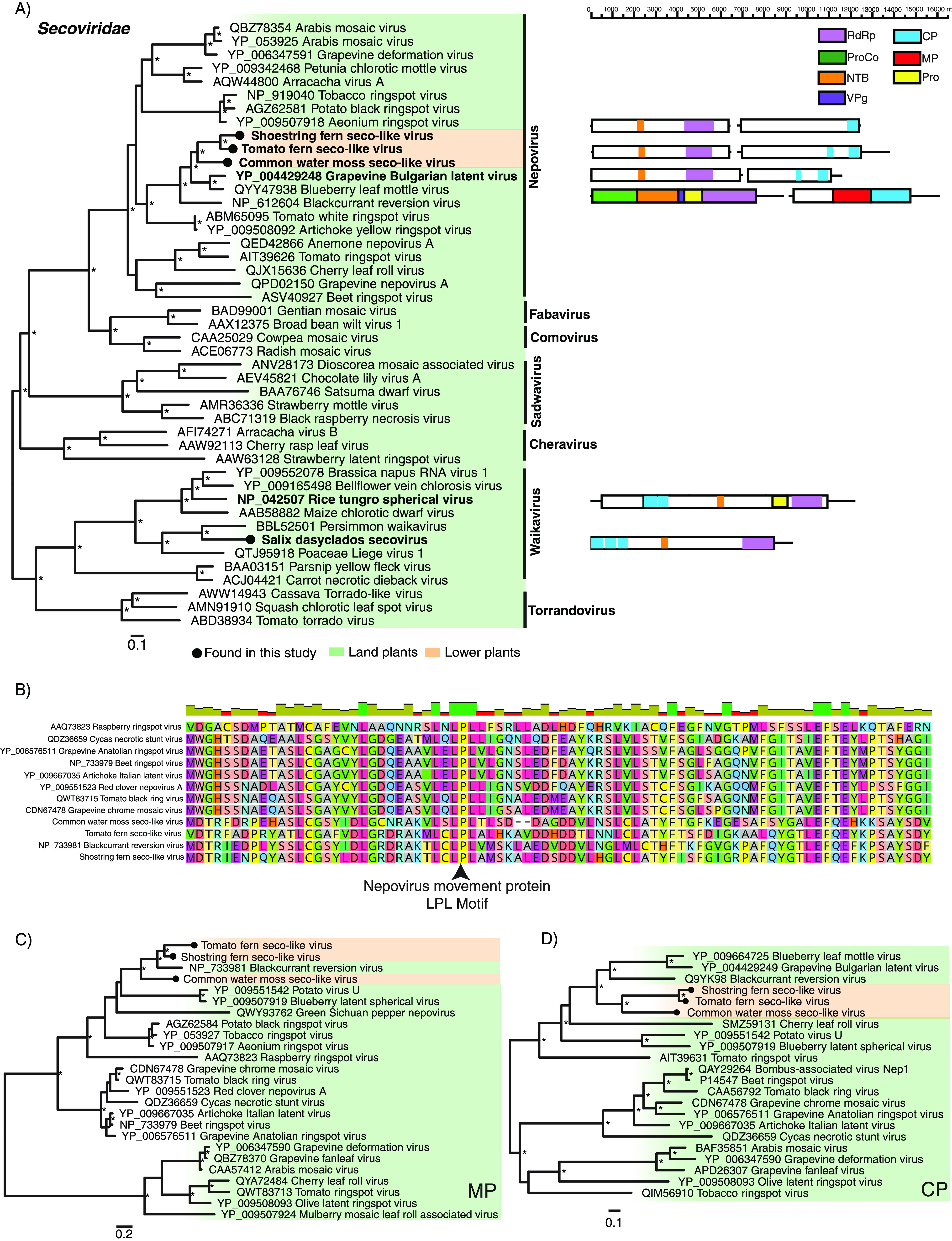
(A) (Left) Phylogenetic relationships of the viruses identified within the virus family Secoviridae. ML phylogenetic trees based on the Pro-pol region show the topological position of virus-like sequences discovered in this study (black circles) in the context of their closest relatives. (Right) Genomic organization of the seco-like sequences identified in this study and representative species used in the phylogeny. (B) Multiple amino acid sequence alignment of the 30K movement protein “LPL” motifs, which are highly conserved throughout the nepoviruses. (C) Phylogenetic relationships of the Nepovirus 30K movement proteins. (D) Phylogenetic relationships of the Nepovirus coat proteins. For all trees, branches are scaled to the number of amino acid substitutions per site, and trees were midpoint rooted for clarity only. An asterisk indicates node support of >70% bootstrap support. Tip labels are bolded when the genome structure is shown on the right. See Fig. 3 for the color scheme. Viruses discovered in this study are signified using a black circle on the tree tip.
A putative RNA2 ORF was assembled for the three nepovirus-like sequences, each containing a complete CP (Fig. 5). The CPs fall within the nepovirus subgroup C (Fig. 5D). While a movement protein (MP) domain was not formally detected, we predict that the region upstream of the CP contains a putative movement-like protein. For CwmSV, this region (amino acid positions 312 to 883) displayed sequence homology to the MP of Blackcurrant reversion virus (E value, 5.42e-86; amino acid identity, 46%). Both TfSV and SfSV displayed similar levels of sequence identity in this region. We detected the LPL motif which is commonly found in nepovirus MPs in all three viruses (Fig. 5B). Phylogenetic analysis of the putative MPs placed these viruses with Blackcurrant reversion virus in the genus Nepovirus (Fig. 5C).
We found little evidence that these viruses reflect contamination by land plants or other eukaryotes. The Fontinalis antipyretica transcriptome was composed of reads closely related to a feather moss belonging to the order Hypnales in which F. antipyretica is also found. Furthermore, a large proportion of reads were assigned to an uncultured eukaryote 18S rRNA gene (54%) (HG421124.1) identical to the F. antipyretica 18S rRNA (AF023714.1), among other bryophyte 18S rRNA genes in a BLASTn search (E value, 2e-102; nucleotide identity, 100%) (Fig. 2). Fungi represented 12% of reads in the Lonchitis hirsute transcriptome. Despite this, it is unlikely that TfSV is fungus-associated, as no fungal contamination was detected in the Vittaria lineata transcriptome, in which the closely related SfSV sequence (amino acid identity: 78%) was assembled (Fig. 2).
(iv) Lenarviricota. (a) Mitoviridae. We identified five virus sequences that cluster within the Mitoviridae—denoted Chinese swamp cypress mitovirus (CscMV), Asian bayberry mitovirus (AsbaMV), False cloak ferns mitovirus (FcfMV), Delta maidenhair fern mitovirus (DmfMV), and Lycopod associated mitovirus (LycoMV). The fern (FcfMV and DmfMV)- and lycophyte (LycoMV)-associated sequences cluster with the fern Azolla filiculoides mitovirus 1 form a sister group to the plant mitoviruses and nonretroviral endogenous RNA viral elements (NERVEs) (Fig. 6) (15). The gymnosperm-associated sequences form a sister group of all the plant-associated mitoviruses and NERVEs. LycoMV extends the known host range of plant mitoviruses from ferns to lycophytes. Another mitovirus sequence was detected in the chlorophyte Bolbocoleon piliferum, denoted Bolbocoleon piliferum mito-like virus (BopiMV). BopiMV falls basal to the mitoviruses, distinct from various unclassified mito-like viruses, including the chlorophyte associated Mito-like picolinusvirus (QOW97241) (Fig. 6). All novel sequences show strong conservation of the motifs characteristic of mitovirus RdRps (Fig. S2) (35).
FIG 6.

(Left) Phylogenetic relationships of the viruses within the virus families Narnaviridae and Mitoviridae. ML phylogenetic trees based on the replication protein show the topological position of virus-like sequences discovered in this study (black circles) in the context of their closest relatives. See Fig. 3 for the color scheme. Blue stars signify mitovirus sequences identified in reference 15. Red stars signify nonretroviral endogenous RNA viral elements (NERVEs). All branches are scaled to the number of amino acid substitutions per site, and trees were midpoint rooted for clarity only. An asterisk indicates node support of >70% bootstrap support. Tip labels are bolded when the genome structure is shown on the right. (Right) Genomic organization of the virus sequences identified in this study and representative species used in the phylogeny.
There is little evidence to suggest that these sequences are derived from a nonplant organism. While the FcfMV and DmfMV libraries were contaminated with fungi, (12% and 15% of reads, respectively) fungus-associated reads were absent in the libraries of all other mitoviruses. As the codon UGA encodes tryptophan (Trp) in fungal mitochondria, this codon assignment is also present in fungal mitoviruses (36–38). In contrast, the UGA codon in plant mitochondria is a stop codon and hence absent from plant mitovirus sequences except as a stop codon (15). The absence of internal UGA codons in these sequences is further evidence that these sequences are plant-derived (15, 39). In addition, we found no evidence through searches of the 1KP genome scaffolds and the whole-genome sequencing (WGS) shotgun database that these sequences are mitochondrial or nuclear NERVEs. Furthermore, CscMV, AsbaMV, and LycoMV contain complete RdRps, and their untranscribed regions (UTRs) share similarities in length and identity with plant mitoviruses.
(b) Narnaviridae. A partial narna-like virus sequence was identified in the rhodophyte Heterosiphonia pulchra denoted Heterosiphonia pulchra narna-like virus (HspuNV). HspuNV clusters with unclassified trypanosomatid associated viruses. While ~5% of reads in this library were associated with fungi the phylogenetic position of this virus suggests that it is not derived from fungi (Fig. 2 and 6).
(v) Tolivirales. (a) Tombusviridae. An alphacarmo-like virus tentatively named Ihi tombusvirus (IhiTV) was identified in an ihi (Portulaca molokiniensis) sample. IhiTV is phylogenetically positioned within the alphacarmoviruses (Fig. S3).
(vi) Patatavirales. (a) Potyviridae. We identified three virus-like sequences that clustered with plant viruses in the family Potyviridae—Traubia modesta potyvirus (TramPV), Common milkweed potyvirus (ComPV), and Salt wort potyvirus (SawPV). TramPV and ComPV shared 87% amino acid identity and may therefore represent a single virus species. The potyvirus-like sequences discovered all group with known potyviruses in a phylogenetic analysis of the Nib gene (Fig. S3).
(vii) Martellivirales. (a) Endornaviridae. Six alphaendorna-like virus sequences were detected in the five green algae species and one lycophyte. The green alga- and lycophyte-associated alphaendorna-like viruses, termed Bolbocoleon piliferum endorna-like virus (BopiEV), Volvox aureus endorna-like virus (VoauEV), Carteria obtusa associated endorna-like virus (CaobEV), Brachiomonas submarina associated endorna-like virus (BrsuEV), Staurastrum sebaldi endornavirus (SsEV), and Krauss’ spikemoss associated endorna-like virus (KrspEV), fall across the alphaendornavirus phylogeny and predominately cluster with alga- and fungus-associated viruses (Fig. 7). There was little evidence of algal (nonhost) or fungal contamination in the Staurastrum sebaldi, Bolbocoleon piliferum, and Volvox aureus transcriptomes with <1% of all reads associated with these groups (Fig. 2). Non-green algal contaminants were present in the Carteria obtusa (28%), Brachiomonas submarina (7%), and Selaginella kraussiana (4%) transcriptomes, where fungi also appeared as a notable contaminant representing 11% of all reads (Fig. 2). To our knowledge, these sequences represent the first endornavirus associated with charophytes, chlorophytes, and lycophytes, although further work is needed to confirm the virus-host associations.
FIG 7.
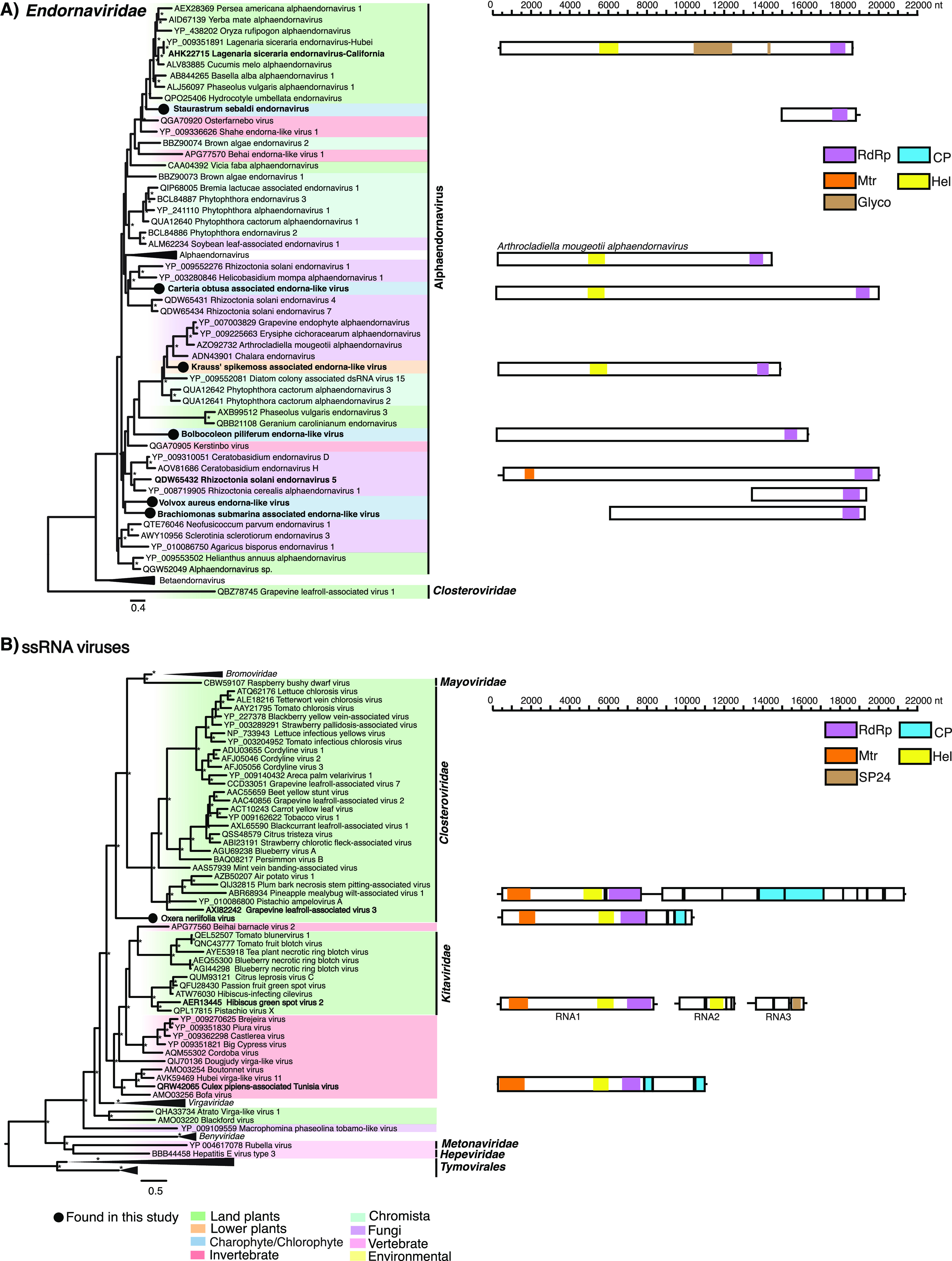
(Left) Phylogenetic relationships of the (A) endorna-like and (B) unclassified (+)ssRNA viruses identified in this study. ML phylogenetic trees based on the replication protein show the topological position of virus-like sequences discovered in this study in the context of those obtained previously. (Right) Genomic organization of the (A) endorna-like and (B) unclassified ssRNA virus sequence identified in this study and representative species used in the phylogeny. For all trees, branches are scaled to the number of amino acid substitutions per site, and trees were midpoint rooted for clarity only. An asterisk indicates node support of >70% bootstrap support. Tip labels are bolded when the genome structure is shown on the right. See Fig. 3 for the color scheme. Viruses discovered in this study are signified using a black circle on the tree tip.
(viii) Unclassified. We identified a virus-like sequence in an Oxera neriifolia library, termed Oxera neriifolia associated virus (OnV). The sequence, 10,214 nt in length, contained four ORFs. The first ORF (7,536 nt) was composed of a viral methyltransferase, helicase, and RNA polymerase, while the third ORF (513 nt) most closely resembled a CP. ORF 1 and ORF 3 shared the greatest sequence similarity with Culex pipiens associated Tunisia virus (32% amino acid identity). The second and fourth ORF had no homology to known viruses. The genome organization of OnV is distinct from that of the other related plant virus families (Fig. 7). OnV forms a distinct and well-supported outgroup to the Closterviridae, Bromoviridae, and Mayoviridae families. As such, OnV may potentially constitute a new virus family (Fig. 7). We found little evidence that OnV was detected due to contamination by other eukaryotes (Fig. 2).
Negative-sense single-stranded RNA [(–)ssRNA] viruses.
(i) Bunyavirales. (a) Phenuiviridae. A phenui-like virus sequence termed Brown algae phenui-like virus (BralPV) was recovered from a Sargassum thunbergii transcriptome. The partial L segment clusters with the unclassified plant and fungi viruses (Fig. 8A). No additional phenui-like virus segments were recovered. No concerning contaminants were detected in the S. thunbergii transcriptome (Fig. 2).
FIG 8.
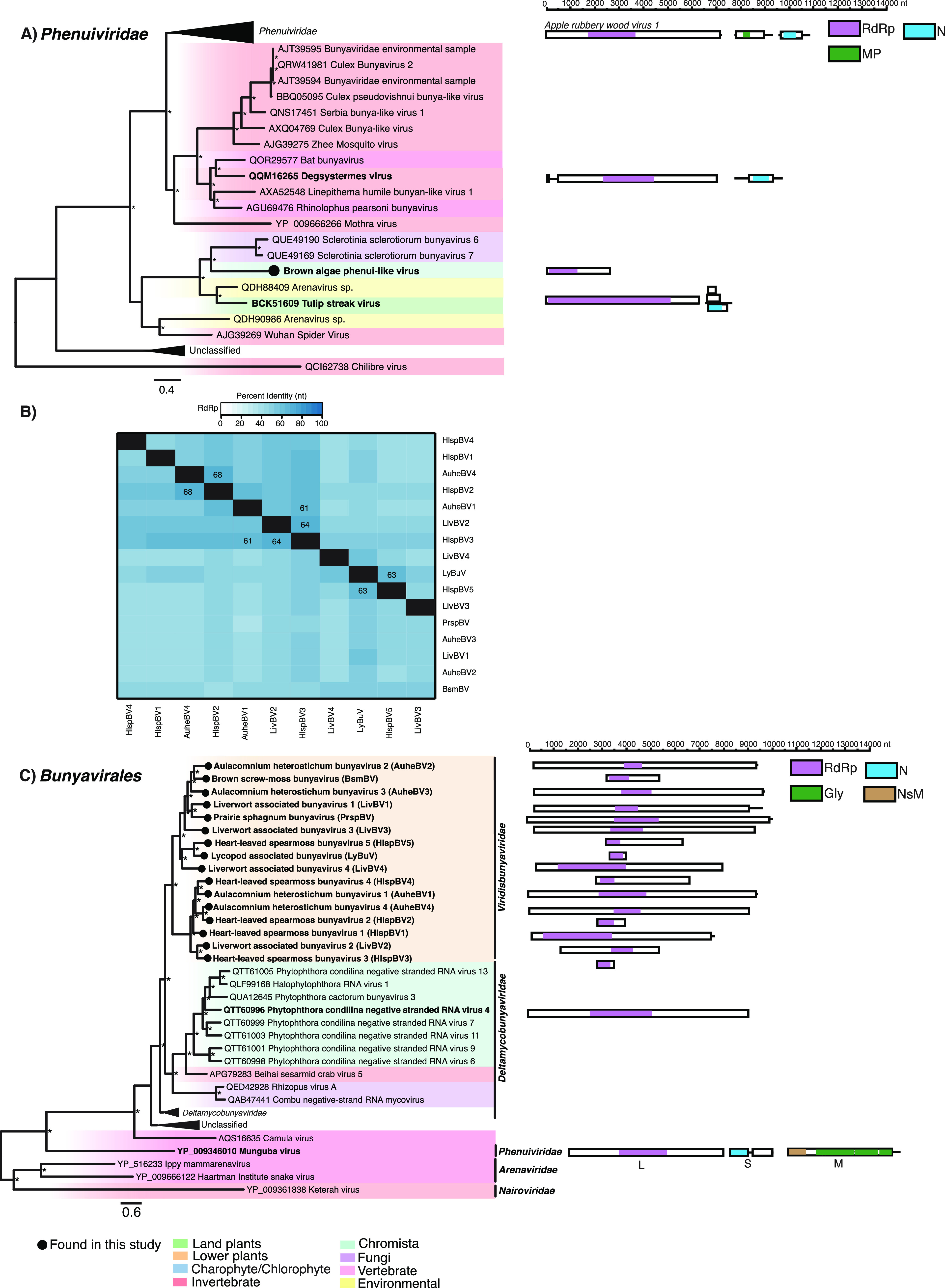
Phylogenetic relationships of the viruses (A) (Left) A phylogeny depicting a novel clade of viruses related to the Deltamycobunyaviridae in the context of the Bunyavirales. (Right) Genomic organization of the virus sequences identified in this study and representative species used in the phylogeny. (B) Percent identity matrix of the novel bunya-like viruses. Identity scores are calculated from an alignment of the RdRp protein-coding sequence. For clarity, the 100% identity along the diagonal has been removed. Where sequence identity is ≥60%, the value is shown. (C) (Left) A phylogeny depicting the phenui-like virus identified in this study in the context of the Phenuiviridae. (Right) Genomic organization of the virus sequences identified in this study and representative species used in the phylogeny. For all trees, branches are scaled to the number of amino acid substitutions per site, and trees were midpoint rooted for clarity only. An asterisk indicates node support of >70% bootstrap support. Tip labels are bolded when the genome structure is shown on the right. See Fig. 3 for the color scheme. Viruses discovered in this study are signified using a black circle on the tree tip.
(b) Viridisbunyaviridae. We identified 16 bunya-like virus sequences from 8 bryophyte libraries. Three libraries contained multiple distinct putative complete and partial viruses. The overall pairwise nucleotide identity was <70% between each sequence (Fig. 8B). As such, we consider each a different bunya-like virus. These sequences group together to form a novel clade of unclassified bunya-like viruses distantly related to oomycete, fungus, and invertebrate viruses (Fig. 8C). Bunyaviruses typically comprise three segments (L, M, and S), although only the L segment was recovered for these sequences. These sequences represent the first plant-associated viruses that cluster near the unofficially named Deltamycobunyaviridae (40) (Fig. 8C). As the complete coding sequences of the viruses discovered share <30% amino acid identity to the nearest relatives in the Deltamycobunyaviridae, they may constitute a new virus family. We tentatively name this virus family the Viridisbunyaviridae, (viridis meaning green, while bunya is derived from the virus order Bunyavirales, in which this clade falls within). There was no evidence that these sequences originated from nonplant contaminants. Host assignment was unclear for Lycopod associated bunyavirus (LyBuV) and Liverwort associated bunyavirus 1:4, as reads belonging to several lycophyte and liverwort species, respectively, were found in the source transcriptomes (Fig. 2).
(ii) Mononegavirales. (a) Rhabdoviridae. We identified seven sequences that clustered with plant viruses in the family Rhabdoviridae, denoted Canadian violet rhabdovirus 1 (CvRV1), Canadian violet rhabdovirus 2 (CvRV2), Common ivy rhabdovirus (CoiRV), Indian pipe rhabdovirus (InpRV), Tree fern varicosa-like virus (TfVV), Monoclea gottschei varicosa-like virus (MgVV), and Bug moss associated rhabdo-like virus (BmRV). Notably, TfVV and MgVV expand the host range of the rhabdoviruses from angiosperms and gymnosperm to ferns and liverworts. RNA2 segments were recovered for both viruses; TfVV RNA2 contained five genes, while MgVV contained four (Fig. 9A). Two partial segments sharing similarities to the nucleocapsid (N) of Black grass varicosavirus-like virus (YP_009130620.1) were found in the Indian pipe library and share 50% amino acid identity. All sequences likely represent novel species within known plant-infecting genera (Fig. 9A).
FIG 9.

(Left) Phylogenetic relationships of the viruses within the families (A) Rhabdoviridae, (B) Aspiviridae, and (C) Yue- and Qinviridae. (Right) Genomic organization of the virus sequences identified in this study and representative species used in the phylogeny. For all trees, branches are scaled to the number of amino acid substitutions per site, and trees were midpoint rooted for clarity only. An asterisk indicates node support of >70% bootstrap support. Tip labels are bolded when the genome structure is shown on the right. See Fig. 3 for the color scheme. Viruses discovered in this study are signified using a black circle on the tree tip. For trees is panels B and C, the RdRp motif C trimer of each sequence is shown in parentheses at the end of the tip label.
BmRV is a partial sequence (693 nt) most closely related to the unclassified Hubei rhabdo-like virus 2 (44% amino acid identity). Further evidence is needed to confirm BmRV as the first bryophyte rhabdovirus, but the relatively low proportion of contaminates in this library (3% algae and 3% fungi) suggests that this virus is plant-associated (Fig. 2). While 53% of reads in the MgVV library were fungi-associated, the phylogenetic position of MgVV suggests it is derived from plants (Fig. 2 and 9A).
(iii) Serpentovirales. (a) Aspiviridae. We identified an aspi-like sequence termed Nees’ Pellia aspi-like virus (NpAV). A complete RNA1 segment (6,989 nt) was assembled, although no other segments were recovered (Fig. 9B). NpAV most closely resembles Rhizoctonia solani negative-stranded virus 3 (amino acid identity, 22%) and falls basal to all the unclassified aspi-like viruses, including those found in fungi, invertebrates, and oomycetes. NpAV is the first aspi-like virus identified in plants outside of the angiosperms and may constitute a novel virus group (Fig. 9B). Notably, unlike the other aspiviruses that possess an SDD sequence in motif C of the RdRp—a known signature for segmented negative-stranded RNA viruses—NpAV has a GDD sequence (Fig. 9B).
(iv) Goujianvirales. (a) Yueviridae. A yue-like virus sequence termed Meadow spikemoss associated yue-like virus (MsYV) was found in the lycophyte Selaginella apoda and most closely resembles alga-associated Bremia lactucae associated yuevirus-like virus 1 (amino acid identity, 26%). Phylogenetic analysis supports the assignment of MsYV as the first plant yuevirus (Fig. 9C).
A second partial yue-like virus sequence was detected in a Pityrogramma trifoliata library and termed Goldenrod fern associated yue-like virus (GfYV). GfYV falls with a group of oomycete-associated viruses. Consistent with the qin-like viruses, GfYV has an IDD (Ile-Asp-Asp) sequence motif instead of the common GDD (Gly-Asp-Asp) in the catalytic core of its RdRp, while MsYV contains SDD (Ser-Asp-Asp) in the same manner as many yue-like viruses (Fig. 9C). The libraries from which GfYV and MsYV were assembled are contaminated with fungal reads (5% and 21%, respectively), and as such, host assignment is made with caution (Fig. 2). Reads belonging to oomycetes were not found in either library.
Double-stranded RNA (dsRNA) viruses.
(i) Durnavirales. (a) Amalgaviridae. We detected five sequences that cluster with amalga-like viruses. Lycopod associated amalgavirus (LycoAV) is a partial RdRp containing a sequence that falls basal to the Amalgaviridae and represents the first amalga-like virus in the lycophytes (Fig. 10). Three amalga-like sequences were discovered in green algae and rhodophyte transcriptomes and cluster with Diatom colony associated dsRNA virus 2 (Fig. 10). As noted in the case of Bryopsis mitochondria-associated dsRNA virus and several green alga-associated viruses (41), when translated into amino acids using the protozoan mitochondrial code, two overlapping ORFs are present: the first encoding a hypothetical protein, and the second, a replicase through a –1 ribosomal frameshift (42). For two of the amalga-like sequences identified in this study—Nucleotaenium eifelense virus (NueiV) and Rhodella violacea virus (RhviV)—a similar structure was observed, but we were unable to identify any ribosomal frameshift motifs in either sequence (Fig. 10). Further work is needed to confirm if these sequences should be translated through the mitochondrial genetic code.
FIG 10.

(Left) Phylogenetic relationships of the viruses within the order Durnavirales. ML phylogenetic trees based on the replication protein show the topological position of the virus-like sequences discovered in this study (black circles) in the context of their closest relatives. See Fig. 3 for the color scheme. Green stars are used to signify sequences that have been translated using the protozoan mitochondrial genetic code. Red stars are used to signify sequences for which a dsRNA3 coat protein-like segment has been described. All branches are scaled to the number of amino acid substitutions per site, and trees were midpoint rooted for clarity only. An asterisk indicates node support of >70% bootstrap support. Tip labels are bolded when the genome structure is shown on the right. (Right) Genomic organization of the virus sequences identified in this study and representative species used in the phylogeny.
A contig containing what appears to be a complete coding sequence (3,259 nt) and RdRp motifs was assembled in the Woodsia scopulina transcriptome and tentatively named Rocky Mountain woodsia associated virus (RmwPV). The predicted RdRp region (nt 918 to 1,921) of RmvPV shares similarity to both partiti-like viruses (e.g., Ustilaginoidea virens nonsegmented virus 2, 26% amino acid [aa] identity) and the unclassified Phytophthora infestans RNA virus 1 (42% aa identity), which likely constitutes a novel virus family (43). The resemblance RmwPV shares with two seemingly distantly related virus groups suggests its position within the Durnavirales should be treated with caution (Fig. 10).
The transcriptome in which RmwPV was discovered is contaminated with fungal reads (10%) (Fig. 2). If RmwPV was derived from fungal contaminates, this could potentially explain the phylogenetic placement of RmwPV (Fig. 10). The Lycopodiella appressa transcriptome in which LycoAV was discovered is contaminated by reads belonging to species across various land plant groups. Reads belonging to land plants composed 35% of plant-associated reads, while lycopod-associated reads composed 65% (Fig. 2).
(b) Partitiviridae. We detected 14 sequences that share a resemblance with members of the Partitiviridae. For each of these sequences, complete dsRNA1 and dsRNA2 segments were recovered. Ten sequences were found in nonflowering plants that cluster within the deltapartitiviruses. A clade within the deltapartitiviruses is known to encode a third segment comprising a divergent dsRNA2 full-length capsid protein with unknown function (Fig. 10). We identified dsRNA3 segments in related conifer-associated sequences but not in those found in bryophyte and lycophyte libraries (Fig. 10). Phylogenies estimated on the coding sequences of dsRNA2 and dsRNA3 reveal essentially the same grouping, which is largely consistent with the host phylogeny (Fig. S4). We extend the known host range of the deltapartitiviruses to include bryophytes and lycophytes. The remaining sequences were found in eudicots and cluster with known plant partitiviruses (Fig. 10). The white campion was judged to have contamination from ginseng and chickweed (30). However, the relatively low proportion of the library these contaminates compose (<1%) suggests that it is unlikely these species are the host of White campion partitivirus (WcPV) (Fig. 2). There is no evidence that the other partiti-like sequences discovered are derived from contaminates.
(ii) Ghabrivirales. (a) Chrysoviridae. We identified two partial sequences that share resemblance with members of the alphachrysoviruses, denoted Mesotaenium kramstae alphachyrso-like virus (MkACV) and Tree fringewort alphachyrso-like virus (TwACV) (Fig. 11). A complete RNA2 segment was recovered for MkACV, which shared similarity with the p98 of various chrysoviruses (Fig. 11). The MkACV RNA2 segment did not contain the PGDGXCXXHX motif commonly found in this protein (44). To our knowledge, these sequences represent the first chryrsoviruses in the Marchantiophyta and Chlorophyta. While reads belonging to fungi were found in the libraries MkACV and TwACV were assembled from, the phylogenetic positioning of the viruses suggest that they are plant-derived (Fig. 2 and Fig. 11).
FIG 11.

(Left) Phylogenetic relationships of the viruses within the order Ghabrivirales. (A) A phylogeny of the Chrysoviridae; (B) an order level phylogeny. ML phylogenetic trees based on the replication protein show the topological position of the virus-like sequences discovered in this study (black circles) in the context of their closest relatives. See Fig. 3 for the color scheme. All branches are scaled to the number of amino acid substitutions per site, and trees were midpoint rooted for clarity only. An asterisk indicates node support of >70% bootstrap support. Virus taxonomic names are labeled on the right. (Right) Genomic organization of the virus sequences identified in this study and representative species used in the phylogeny.
(b) Totiviridae. A total of 13 sequences sharing similarities to toti-like viruses were discovered in eight red and green alga transcriptomes. All sequences share less than 50% amino acid identity across their coding sequence, and as such, we consider each a putative toti-like virus. Among these sequences, four cluster with Delisea pulchra totivirus IndA (AMB17469.1) to form a rhodophyte-associated clade basal to the totiviruses (Fig. 11). Gracilaria vermiculophylla toti-like virus (GrveTV) along with Red algae totivirus 1 (BBZ90082) form a sister group to the protozoan-infecting leishmaniaviruses (Fig. 11). The remaining sequences are phylogenetically positioned across the tree of toti-like viruses, commonly occupying basal positions (Fig. 11).
Prasiola crispa is contaminated by reads from the fungus Candida albicans. Prasiola crispa toti-like virus (PrcrTV), clusters with unclassified protist, fungus, invertebrate, and alga viruses, including Elkhorn sea moss toti-like virus (EsmTV) (Fig. 2 and 11). The Kappaphycus alvarezii transcriptome in which EsmTV was found showed no evidence of contamination, suggesting that PrcrTV may also be derived from algae (Fig. 2). The Mazzaella japonica transcriptome in which Red algae toti-like virus 2:3 (RedTV2/3) was discovered was predominantly composed of reads associated with the rhodophyte genera Chondrus. As >99% of reads in this library belong to rhodophyte species, RedTV2 and RedTV3 have been assigned to this group. The Porphyridium purpureum transcriptome is highly contaminated by reads belonging to flowering plants and an unidentified cloning vector (M10197) (Fig. 2). The phylogenetic positioning of the viruses discovered from this transcriptome (Porphyridium purpureum toti-like virus 1 and 2) point toward it being derived from rhodophytes rather than flowering plants (Fig. 11).
Long-term virus-host evolutionary relationships.
To examine the frequency of cross-species transmission and codivergence among plant viruses, we estimated tanglegrams that depict pairs of rooted phylogenetic trees displaying the evolutionary relationship between a virus family and their hosts. This revealed cross-species transmission as the predominate macroevolutionary process predicted among all the RNA virus groups analyzed (median, 65%; range, 46% to 79%) (Fig. 12). Cross-species transmission was most frequent in the Betaflexiviridae (79%) and the subfamily Betarhabdovirinae (79%). However, virus-host codivergence (median, 23%; range, 14% to 29%) and to a lesser extent duplication (i.e., speciation) (median, 4.6%; range, 1.4% to 24%) and extinction events (median, 2.9%; range, 0% to 11%) were detected across plant virus families (Fig. 12). Codivergence was most frequently predicted in the Benyviridae and Tymoviridae, representing 29% and 26% of events, respectively. Importantly, however, the results of our cophylogenetic analysis are undoubtedly influenced by the sample of plant viruses and will likely change as the number of plant viruses identified increases.
FIG 12.
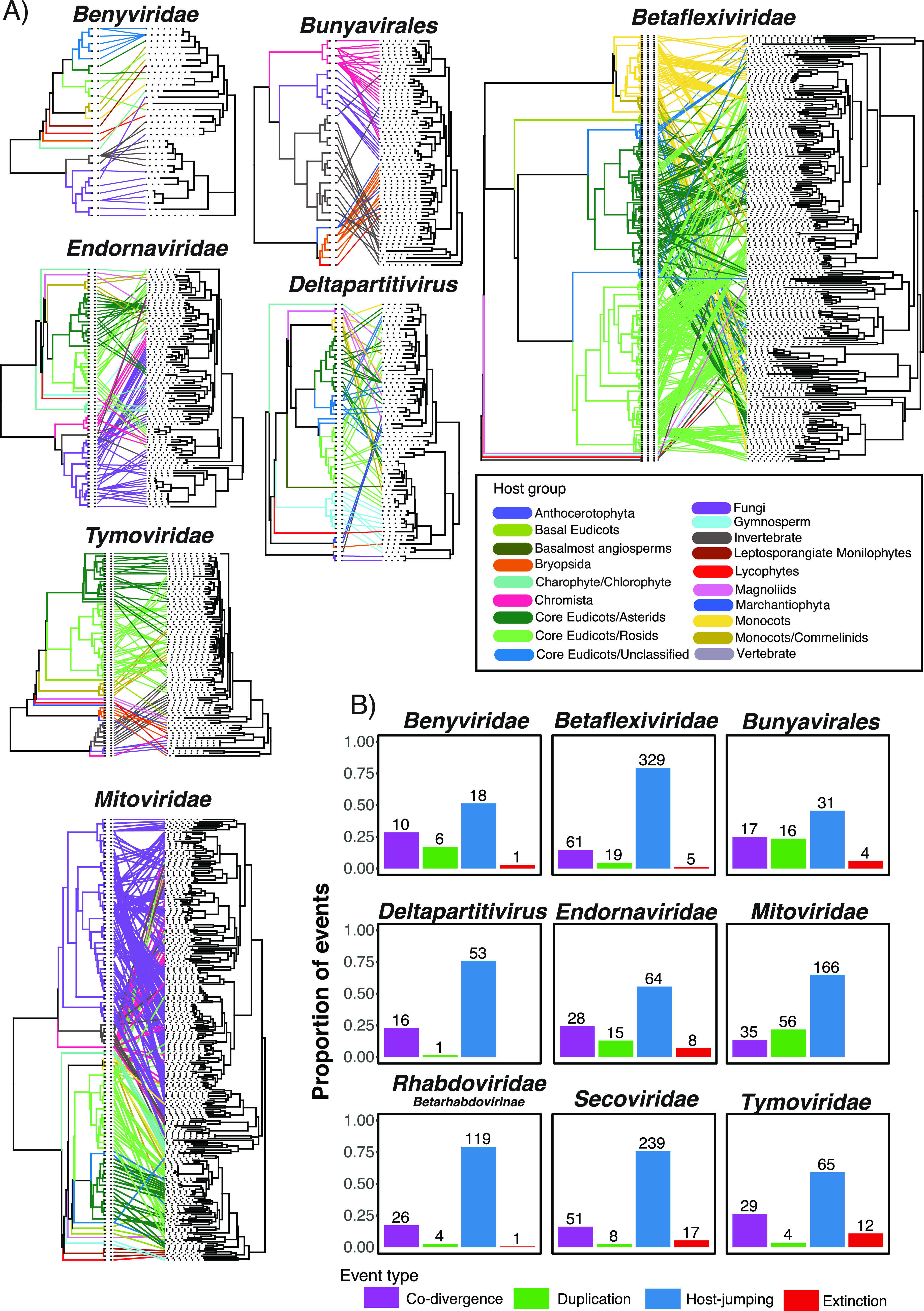
(A) Tanglegram of rooted phylogenetic trees for select virus groups and their hosts. Lines and branches are colored to represent the host clade. The cophylo function implemented in phytools (v0.7-80) was used to maximize the congruence between the host (left) and virus (right) phylogenies. Fig. S5 provides the names of the hosts and viruses along with additional tanglegrams for the Secoviridae and Rhabdoviridae. (B). Reconciliation analysis of select virus groups. Bar plots illustrate the range of the proportion of possible events and are colored by event type.
DISCUSSION
Our understanding of the interactions between plants and their viruses has been constrained by inadequate sampling across the enormous, extant diversity of plant species. Here, we provide a large-scale virus discovery project based on mining transcriptomes from across the entire breadth of the plant kingdom. In doing so, we have identified 104 potentially novel virus species, considerably expanding the known host range of 13 virus families to include lower plants. In addition, we expand the host range of four other virus families to include algae (including two viruses in brown algae that fall outside the Archaeplastida lineage). We also present the first evidence of a movement protein with a predicted molecular weight of ~30 kDa (here referred to as a “30K MP”) in a virus of nonvascular plants. Collectively, these results advance our understanding of RNA virus diversity across the Archaeplastida. It is important to note that the transcriptomes analyzed were prepared with enrichment of polyA+ RNA, limiting the detection of nonpolyadenylated viruses. Such biases do not detract from the findings of this study but do highlight the need for further virus discovery efforts in characterizing the phytovirosphere.
RNA viruses are widespread across lower plant lineages.
To date, viral surveys in basal plant lineages (namely, ferns, bryophytes, and algae) have revealed only the minimal occurrence of (+)RNA viruses (4, 16, 19, 41, 45, 46), supporting the idea that RNA viromes in angiosperms evolved as they diversified during the Cretaceous (47, 48). However, our results potentially challenge this paradigm, as we detected the first evidence of (+)ssRNA viruses in lower plants and algae, implying that these groups are associated with older lineages of plants. Several of these virus lineages are deep-branching and sit basal to angiosperm-infecting viruses (e.g., LycoAV and LyBuV) in phylogenetic trees. Other viruses discovered here occupy ambiguous positions between established plant virus families (e.g., OnV) or cluster in large numbers to form novel plant-associated clades (e.g., the Viridisbunyaviridae in the Bunyavirales). Benyviruses are typically transmitted by the root-infecting plasmodiophorids Polymyxa betae and Polymyxa graminis (49, 50). The phytomyxids (plasmodiophorids and phagomyxids) are parasites of plants, diatoms, oomycetes, and brown algae and have been shown to exhibit cross-kingdom host shifts (e.g., between angiosperms and oomycetes) (51). As such, the plasmodiophorids may be a vehicle for cross-species transmission between aquatic protists and land plants (4). FeBV, a beny-like virus identified in this study, formed a clade along with Wheat stripe mosaic virus distinct from members of the genus Benyvirus. Deciphering the evolutionary history and mode of transmission for the lower plant beny-like viruses will require further studies with particular emphasis on these taxa. Interestingly, no plasmodiophorid-associated reads were detected in any of the libraries from which we assembled a beny-like virus. LjBV and WasBV appear distantly related to the benyviruses. These viruses group with a suite of unclassified viruses assembled from a soil metatranscriptome study suggesting that, like the benyviruses, this larger group of unclassified viruses may involve soilborne parasites such as the plasmodiophorids (52). Our detection of tymovirid-like sequences in the lycophytes, bryophytes, and brown algae dramatically expands the known host range of the Tymovirales. Several of these viruses were similar to unclassified Riboviria species assembled from a recent survey of common wild oat soil rhizosphere and detritosphere (52) (Fig. 4). The metatranscriptome of the sequenced soil samples from the common wild oat study was largely composed of Viridiplantae, fungi, Amoebozoa, protists, nematodes, and other eukaryotes. Phylogenetic clusters to infer host associations of our viruses therefore remain challenging. Indeed, these viruses may result from contamination from other eukaryotes (e.g., fungi or invertebrates), although we found no consistent evidence among these viruses (Fig. 2). Assuming these viruses are plant-associated, their phylogenetic pattern suggests that they may have resulted from cross-kingdom transmission events that frequent the Alsuviricetes.
The partial deltaflexi-like virus we detected in P. agnata (PaADV) is particularly noteworthy. The deltaflexiviruses are only known to infect fungi, although no fungus-associated reads were found in the P. agnata metatranscriptome (Fig. 2). The mycovirus families Delta- and Gammaflexiviridae are thought to have been derived from the plant alpha- and betaflexivirids through cross-species transmission (4, 53). As such, PaAGV could potentially represent an intermediate between the plant and fungus flexiviruses or perhaps a more recent fungus to plant transmission. As only a fragment of the polymerase gene was assembled for this virus, future work should confirm the presence of PaAGV and its phylogenetic position.
The extension of the Mitoviridae to a lycophyte host.
Through the analysis of mitovirus-like, nonretroviral endogenous RNA viral elements (NERVEs), it was argued that the origin of plant mitovirus NERVEs was a single horizontal transfer from a fungal mitovirus before the origin of vascular plants in the early Silurian, ~400 million years ago (MYA) (54). Evidence of contemporary mitoviruses in flowering plants and a fern have challenged this view, suggesting that a lineage of plant rather than fungal mitoviruses are the immediate ancestors of plant mitovirus NERVEs (15). Indeed, plant-to-fungus transmission would eliminate code conflicts between fungus and plant mitochondrial genetic codes (39). Here, we demonstrate the existence of a lower plant-associated sister clade to the angiosperm mitoviruses and NERVEs. This clade includes a clubmoss-associated mitovirus, the most basal plant mitovirus sequence to date. This finding aligns with the estimation of the origin of plant mitovirus NERVEs occurring as early as the evolution of the clubmoss (53). The recent finding of mitoviruses in green algae—including BopiMV in this study—highlight the broad host range of mitoviruses (41, 46). The phylogenetic position of these viruses and the absence of NERVEs from these groups suggest that they are not the ancestors of land plant mitoviruses and NERVEs.
Establishment of a new virus family in the Bunyavirales: Viridisbunyaviridae.
We identified 16 bunya-like viruses assembled from 6 nonvascular plant libraries, including bryophyte and lycophyte species. These viruses form a novel clade within the Bunyavirales and represent the first viruses in this order to be associated with lower plants. This clade likely represents a novel virus family, which we have tentatively named the Viridisbunyaviridae. Several libraries contained up to five distinct viruses (each sharing <70% nucleotide identity). Virus coinfections are frequently observed in plants and have been reported in the closest relatives of these viruses, the Deltamycobunyaviridae (55, 56). As with previous studies, we were only able to recover the bunyavirus L segment (56, 57). Further studies are needed to recover the missing small and medium-sized segments and to confirm the presence of mixed infections in plants.
Discovery of the first 30 kDa movement protein in nonvascular plants.
Through the discovery of lower plant-associated viruses, we have gained insights into how the genome structure and composition of contemporary flowering plant viruses have evolved. The detection of secovirid-like sequences in bryophytes and ferns represents the first occurrence of plant secoviruses outside angiosperms and the first evidence of a 30K MP homolog in nonvascular plants. These proteins aid the cell-to-cell movement of viruses in plants. For example, the MP of Cucumber mosaic virus increases the size exclusion limit of plasmodesmata, allowing virus particles to pass through cell walls (58). To date, homologs of 30K MP have only been detected in plant viruses infecting angiosperms to the lycophytes (16, 59). Further work is needed to confirm the presence and function of 30K MPs in viruses infecting the bryophytes and other lower plants.
Detection of Deltapartitivirus dsRNA3 segments in gymnosperms but not in nonvascular plants.
Our discovery of six tri-segmented Deltaparititivirus species provides insights into the evolution of the Deltapartitivirus dsRNA3 segment. dsRNA3 segments have been found in several alpha- and deltapartitiviruses infecting flowering plants (60–63). These segments typically encode a seemingly full-length capsid protein or in the case of the Alphapartitivirus Rosellinia necatrix partitivirus 2, a truncated version of the RdRp which may serve as an interfering RNA (64). There is some debate as to the source of dsRNA3 segments, particularly whether they are satellite viruses that co-opt the RdRp of the coinfecting helper viruses or that the additional segment is a result of coinfection of two different plant partitiviruses and the second RdRp-encoding segment is lost after the initial infection (65). For the first time, we found dsRNA3 segments in conifer-associated viruses but not in those found in lower plants, including bryophytes and lycophytes. The absence of dsRNA3 in nonvascular plants means that it is possible that this segment evolved after the divergence of vascular and nonvascular plants in the Silurian period (66). It is possible that dsRNA3 segments exist for the nonvascular plant-infecting deltapartitiviruses but was not detected due to the large degree of divergence between this segment and reference sequences (including those found in this study). However, the dsRNA1 and dsRNA2 segments of the putative lower plant deltapartitiviruses shared >50% aa identity with the tri-segmented deltapartitiviruses—well above the detection limit for tools such as DIAMOND BLASTx (67). dsRNA3 segments typically appear no more divergent than dsRNA2 segments, so it is unlikely that we would detect both the dsRNA1 and dsRNA2 segments without detecting dsRNA3. Further work is needed to confirm the presence of Deltapartitivirus dsRNA3 segments.
Discovery of an unsegmented varicosavirus-like virus in ferns and the Marchantiophyta.
Finally, the recently discovered gymnosperm varicose-like Pinus flexilis virus 1 in the family Rhabdoviridae contains an unsegmented genome organization that differs from the typical bi-segmented structure of the varicosaviruses (24, 68). We found the bi-segmented structure in varicosavirus-like viruses for the first time in ferns and species in the Marchantiophyta (TfVV and MgVV) which predate the gymnosperms.
Caveats.
The data generated under the 1KP were not explicitly created for virus discovery, such that there are important caveats associated with the methods and metatranscriptomic data mined for virus contigs. For instance, as axenic cultures are not a viable option in most instances, the 1KP samples are commonly contaminated by nucleic acids belonging to bacterial, fungal, and insect species. We addressed this by using a combination of host/virus abundance measurements and phylogenetic analyses to improve the accuracy of virus-host assignments. For most of the viruses described, phylogenetic placement within plant-infecting virus families strongly supports their association with plants. However, several of the viruses found in algae and lower plants were associated with lineages known to infect invertebrates and fungi or unclassified viruses recovered from environmental samples. The association between the viruses of lower plants and algae with those of fungi and invertebrate may reflect the absence of algal and lower plant viruses in reference sequence databases. Experimental confirmation is needed to formally assign the viruses discovered in this study to their hosts.
The average sequencing depth of the 1KP libraries was 1.99 Gb of sequence per sample (range, 1.3 to 3.0), lower than many other virus discovery studies (5, 69, 70). Sequencing depth has been shown to correlate with the ability to detect viruses present at low abundance (71, 72). Further, a large proportion of the virus transcripts detected were from viruses whose full-length genomic or subgenomic mRNAs were polyadenylated at the 3′ end (Table S4, Fig. 1). Although this was anticipated (i.e., the libraries generated by the 1KP initiative were prepared from polyA+ RNA), it limited the detection of nonpolyadenylated viruses and may have contributed to the lack of phycodnavirus sequences detected in algae (72).
To reduce the computational burden of assembly, we attempted to remove host-associated reads before contig assembly by mapping them to the host scaffolds provided by the 1KP initiative. While this step reduces the occurrence of false-positive virus detection, it also risks removing virus reads, particularly reverse-transcribing plant viruses (73). While we frequently detected transcripts associated with the reverse-transcribing family Caulimoviridae, no members of the Metaviridae or Pseudoviridae were detected.
MATERIALS AND METHODS
Transcriptome data generation.
The 1KP generated RNA sequencing libraries from 1,143 species across the breadth of the plant kingdom (29). A total of 30 chromista and rhodophyte species were also included. Due to the diversity of species examined, samples were obtained from multiple sources, including field collections, greenhouses, culture collections, and laboratory specimens (74). For the majority of species, young leaves or shoots were collected, although occasionally, a mix of vegetative and reproductive tissues was used. To avoid RNA degradation, RNA extraction was performed immediately after tissue collection or tissue was frozen in liquid nitrogen and stored at –80°C until extraction (74). Several extraction protocols were used, including CTAB and TRIzol (see reference 32 for complete details). All sequencing was conducted at BGI-Shenzhen, China, using a combination of in-house protocols and TruSeq chemistry (74). All libraries were prepared from polyA RNA. Paired-end sequencing was initially completed using Illumina GAII machines (11% of libraries) with an ~72-bp read length, but later, the HiSeq platform was used (89% of libraries) with a 90-bp read length (74).
Surveying for viruses in the 1KP.
Raw transcriptomes (n = 1,079, belonging to 960 plants species) from the 1KP major release were downloaded from the NCBI Sequence Read Archive (SRA) database (BioProject accession PRJEB21674) and converted to FASTQ format using the SRA Toolkit program fastq-dump in combination with the parallel-fastq-dump wrapper (https://github.com/rvalieris/parallel-fastq-dump) (75). A total of 100 transcriptomes within the BioProject were not publicly available (released August 22 2019) at the commencement of this study and thus were not analyzed. Transcriptomes from the 1KP pilot study (BioProject accession PRJEB4921) and secondary project (BioProject accession PRJEB8056) were similarly not analyzed. To reduce the downstream computing resources, raw sequences were mapped to their respective host genome scaffold using Bowtie 2 (76). Genome scaffolds were assembled as part of a previous study (29). Where genome scaffolds were not available (n = 2), reads were assembled de novo. Trinity RNA-seq (v2.1.1) was used to quality trim and assemble de novo the unaligned reads captured from mapping (77). The assembled contigs were then assigned to known virus families and annotated through similarity searches against the NCBI nucleotide database, the nonredundant protein database (nr), and a custom viral RdRp database using BLASTn and DIAMOND (BLASTx) (67, 78). To filter out weak BLAST sequence matches an E value cutoff of 1 × 10−10 was employed. To identify potential false positives, putative viral contigs were manually compared across the three BLAST searches (nucleotide, nr and RdRp) to ensure that matches to virus-associated sequences were consistent.
Virus filtering and abundance calculations.
For all analyses, we focused on virus families known to infect plants or algae. As our analyses rely on sequence-based similarity searches for virus detection, it is necessarily biased toward viruses that exhibit similarity to existing virus families. Together, the Virus-Host Database (79) and the International Committee on Taxonomy of Viruses (http://www.ictvonline.org/) were used to develop a list of plant virus families and genera to filter out virus-like contigs associated with vertebrate, invertebrate, or fungal hosts based upon their top BLASTx and BLASTn matches. Packages within the Tidyverse collection (v1.3.0) in RStudio were used to complete these tasks (80–82). Where the host was ambiguous (e.g., belonged to a family or genus known to infect both plant and fungal species), the contig was inspected manually.
The relative abundance of each transcript within the host transcriptome was calculated using RNA-Seq by Expectation-Maximization (v1.2.28) (83). To account for variation in the number of unaligned reads between libraries after mapping, contig abundance was standardized by the total number of unaligned paired reads. Contigs under 200 nucleotides in length were excluded from further analysis.
Genome extension and annotation.
Where a novel virus-like contig was discovered, we reassembled the complete library—without removing host reads—in an attempt to recover a complete virus genome. For all reassembled libraries, we recalculated abundance measurements to account for both host and nonhost reads (Table S4). We further reassembled all libraries belonging to nonflowering plants (n = 402). Reads were mapped onto virus-like contigs using BBMap, and heterogeneous coverage and potential misassemblies were manually resolved using Geneious (v11.0.9) (84, 85).
To determine whether a virus was novel, we followed the criteria as specified by the International Committee on Taxonomy of Viruses. Novel viruses were named using a combination of the host common name—if documented—and the associated virus taxonomic group (e.g., Interrupted club-moss deltapartitivirus). In cases where host assignment proved difficult, the suffix “associated” was added to the host name to signify this (e.g., Calypogeia fissa associated deltaflexivirus). Where the taxonomic position of a virus was ambiguous, the suffix “-like” was used (e.g., Goldenrod fern qin-like virus). Virus acronyms were created using a combination of the first and/or second letters of the host common name—if documented—and virus taxonomic group (e.g., Leucodon julaceus beny-like virus [LjBV]). Where multiple related viruses were found in the same host, we assigned each a number (e.g., Aulacomnium heterostichum bunyavirus 1 [AuheBV1]).
The percentage identity among virus sequences was calculated via multiple sequence alignments using Clustal Omega (v1.2.3) (86). The RdRp protein-coding domain was used for all sequence alignments. Percentage identity matrices were converted to heat map plots using a custom R script provided by reference (27).
To characterize functional domains, predicted protein sequences along with their closest viral relatives were subjected to a domain-based search using the Conserved Domains database (v3.18) (https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) and cross-referenced with the PFAM (v34.0) and Uniclust30 (v2018_08) databases available within the MMseqs2 webserver (87). To recover additional annotations, we used HHpred within the MPI Bioinformatics Toolkit webserver to query the PDB_mmCIF70 (v12_Oct), SCOPe70 (v2.08), UniProt-Swiss-Prot-viral70 (v3_Nov_2021), and TIGRFAMs (v15.0) databases (88). Virus genome diagrams were produced using the program littlegenomes (89). Where available, NCBI/GenBank CDS information was used to annotate reference virus sequences (90).
Detection of endogenous virus elements.
All genome scaffolds produced by the 1KP were used as a database which we queried using the protein translations of the viruses discovered in this study. Endogenous viral elements (i.e., EVEs) were detected using the TBLASTN algorithm (91). The search threshold was limited to 100 amino acids in length with an E value cutoff of 1 × 10−20. Where multiple hits across several plant scaffolds were observed, we manually examined the sequence. Suspected EVEs were queried against a subset whole-genome shotgun contig database which included green plants (taxid: 33090) and rhodophytes (taxid: 2763). In addition, the virus-like sequences discovered in this study were checked for host gene contamination using the contamination function implemented in CheckV (v0.8.1) (92). All EVEs were removed.
Assessing library contamination by eukaryotes, bacteria, and protozoa.
For libraries in which a novel virus was discovered, we investigated whether reads belonging to other eukaryotes were also present in the sequencing libraries. To achieve this, we obtained taxonomic identification for raw reads in each library—without the removal of host reads—by aligning them to the NCBI nucleotide database using the KMA aligner and the CCMetagen program (93, 94). Sequence abundance was calculated by counting the number of nucleotides matching the reference sequence with an additional correction for template length (the default parameter in KMA). Krona charts generated by CCMetagen were further edited in Adobe Illustrator (https://www.adobe.com) (95). Library contamination was also assessed by the 1KP and used to inform our host-virus assignments (30).
Phylogenetic analysis of plant viruses.
Phylogenetic trees of the plant-associated viruses discovered here were inferred using a maximum likelihood approach. We combined our translated virus contigs with known virus protein sequences from each respective virus family taken from NCBI/GenBank (90). Sequences were then aligned with the program Clustal Omega (v1.2.3) with default parameters (86). Sites of ambiguity were removed using trimAl (v1.2) with a gap threshold of 0.9 and a variable conserve value (96). To estimate phylogenetic trees, selection of the best-fit model of amino acid substitution was determined using the Akaike information criterion (AIC), the corrected AIC, and the Bayesian information criterion with the ModelFinder function (-m MFP) in IQ-TREE (97, 98). All phylogenetic trees were created using IQ-TREE with 1,000 bootstrap replicates. Phylogenetic trees were annotated with FigTree (v1.4.4) (99) and further edited in Adobe Illustrator (https://www.adobe.com).
To visualize the occurrence of cross-species transmission and virus-host codivergence across plant virus families, we reconciled the cophylogenetic relationship between viruses and their hosts. In each case a vascular plant host cladogram was constructed using trees from reference 100 and 101, using the R package V.PhyloMaker (v0.1.0) (102). As lower plants and nonplant species are not present in the V.PhyloMaker megatree, these hosts were added to the cladogram using the phyloT software, a phylogenetic tree generator based on NCBI taxonomy (http://phylot.biobyte.de/), as well as topologies available in the appropriate literature. Host information was obtained from the NCBI virus database (accessed 14 December 2021) and available literature (103) A tanglegram that graphically represents the correspondence between host and virus trees was created using the R packages phytools (v0.7-80) and ape (v5.5) (104, 105). Virus sequences from each family were obtained through a broad survey of all virus genomic data available on GenBank. The virus phylogenies used in the cophylogenies were constructed as detailed above. To quantify the relative frequencies of cross-species transmission versus virus-host codivergence, we reconciled the cophylogenetic relationship between viruses and their hosts using the Jane package (106). This employs a maximum parsimony approach to determine the best “map” of the virus phylogeny onto the host phylogeny. The cost of duplication, host-jump, and extinction event types were set to 1.0, while host-virus codivergence was set to zero, as it was considered the null event. The reconciliation proceeds by minimizing the total event cost. The number of generations and the population size were both set to 100. Jane was chosen over its successor eMPRess, as it allows for a virus to be associated with multiple host species and handles polytomies (107). For a multihost virus, we represented each association as a polytomy on the virus phylogeny.
Assigning plant host clades.
Each plant host was assigned to each clade in a previous study based upon their phylogenetic positioning and lineage information (29). To improve clarity when coloring the phylogenies (although not the tanglegrams), we reduced the number of clades from 25 to 10 (core eudicots, basal eudicots, monocots, basal-most angiosperms, gymnosperms, fern and fern allies, nonvascular, chlorophyte/charophyte, rhodophyte, and lastly, chromista) by combining those that were closely related or potentially overlapping to increase the number of species in each group (Table S1).
Data availability.
The raw 1000 Plant Transcriptomes Initiative sequence reads are available at BioProject PRJEB21674. All viral genomes and corresponding sequences assembled in this study have been deposited in the European Nucleotide Archive at EMBL-EBI under accession number PRJEB52092 (Table S4).
ACKNOWLEDGMENTS
We thank Richard Miller for computational support and Justine Charon for advice on alignments. This work would not have been possible without the data generously provided by the 1000 Plant Transcriptomes Initiative.
J.C.O.M. was supported by the William Macleay Microbiological Research Fund from the Linnean Society of New South Wales (NSW). R.V.G. was funded by a Discovery Early Career Researcher Award (DECRA) (DE170100208), and E.C.H. was funded by an ARC Australian Laureate Fellowship (FL170100022) from the Australian Research Council. J.L.G. was funded by a New Zealand Royal Society Rutherford Discovery Fellowship (RDF-20-UOO-007).
Footnotes
[This article was published on 31 May 2022 with accession numbers missing in the fourth table in the supplemental material file. The supplemental material was updated in the current version, posted on 5 December 2022.]
Supplemental material is available online only.
Contributor Information
Jemma L. Geoghegan, Email: jemma.geoghegan@otago.ac.nz.
Anne E. Simon, University of Maryland, College Park
REFERENCES
- 1.Anderson JT. 2016. Plant fitness in a rapidly changing world. New Phytol 210:81–87. 10.1111/nph.13693. [DOI] [PubMed] [Google Scholar]
- 2.Wren JD, Roossinck MJ, Nelson RS, Scheets K, Palmer MW, Melcher U. 2006. Plant virus biodiversity and ecology. PLoS Biol 4:e80. 10.1371/journal.pbio.0040080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roossinck MJ, Martin DP, Roumagnac P. 2015. Plant virus metagenomics: advances in virus discovery. Phytopathology 105:716–727. 10.1094/PHYTO-12-14-0356-RVW. [DOI] [PubMed] [Google Scholar]
- 4.Dolja VV, Krupovic M, Koonin EV. 2020. Deep roots and splendid boughs of the global plant virome. Annu Rev Phytopathol 58:23–53. 10.1146/annurev-phyto-030320-041346. [DOI] [PubMed] [Google Scholar]
- 5.Shates TM, Sun P, Malmstrom CM, Dominguez C, Mauck KE. 2018. Addressing research needs in the field of plant virus ecology by defining knowledge gaps and developing wild dicot study systems. Front Microbiol 9:3305. 10.3389/fmicb.2018.03305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dolja VV, Koonin EV. 2018. Metagenomics reshapes the concepts of RNA virus evolution by revealing extensive horizontal virus transfer. Virus Res 244:36–52. 10.1016/j.virusres.2017.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veliceasa D, Enünlü N, Kós PB, Köster S, Beuther E, Morgun B, Deshmukh SD, Lukács N. 2006. Searching for a new putative cryptic virus in Pinus sylvestris L. Virus Genes 32:177–186. 10.1007/s11262-005-6874-4. [DOI] [PubMed] [Google Scholar]
- 8.Sidharthan VK, Kalaivanan NS, Baranwal VK. 2021. Discovery of putative novel viruses in the transcriptomes of endangered plant species native to India and China. Gene 786:145626. 10.1016/j.gene.2021.145626. [DOI] [PubMed] [Google Scholar]
- 9.Han S, Karasev A, Ieki H, Iwanami T. 2002. Nucleotide sequence and taxonomy of Cycas necrotic stunt virus. Arch Virol 147:2207–2214. 10.1007/s00705-002-0876-5. [DOI] [PubMed] [Google Scholar]
- 10.Yang S, Shan T, Wang Y, Yang J, Chen X, Xiao Y, You Z, He Y, Zhao M, Lu J. 2020. Virome of riverside phytocommunity ecosystem of an ancient canal. Preprint (Version 1) available at Research Square. 10.21203/rs.3.rs-25620/v1. [DOI]
- 11.Nibert ML, Pyle JD, Firth AE. 2016. A +1 ribosomal frameshifting motif prevalent among plant amalgaviruses. Virology 498:201–208. 10.1016/j.virol.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dawes C. 2016. Macroalgae systematics, p 107–148. In Fleurence J, Levine I (ed), Seaweed in health and disease prevention. Academic Press, San Diego, CA. [Google Scholar]
- 13.Christenhusz MJ, Byng JW. 2016. The number of known plants species in the world and its annual increase. Phytotaxa 261:201–217. 10.11646/phytotaxa.261.3.1. [DOI] [Google Scholar]
- 14.Valverde RA, Sabanadzovic S. 2009. A novel plant virus with unique properties infecting Japanese holly fern. J Gen Virol 90:2542–2549. 10.1099/vir.0.012674-0. [DOI] [PubMed] [Google Scholar]
- 15.Nibert ML, Vong M, Fugate KK, Debat HJ. 2018. Evidence for contemporary plant mitoviruses. Virology 518:14–24. 10.1016/j.virol.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mushegian A, Shipunov A, Elena SF. 2016. Changes in the composition of the RNA virome mark evolutionary transitions in green plants. BMC Biol 14:68. 10.1186/s12915-016-0288-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Short SM, Staniewski MA, Chaban YV, Long AM, Wang DL. 2020. Diversity of viruses infecting eukaryotic algae. Curr Issues Mol Biol 39:29–61. 10.21775/cimb.039.029. [DOI] [PubMed] [Google Scholar]
- 18.Gibbs AJ, Torronen M, Mackenzie AM, Wood JT, Armstrong JS, Kondo H, Tamada T, Keese PL. 2011. The enigmatic genome of Chara australis virus. J Gen Virol 92:2679–2690. 10.1099/vir.0.033852-0. [DOI] [PubMed] [Google Scholar]
- 19.Vlok M, Gibbs AJ, Suttle CA. 2019. Metagenomes of a freshwater charavirus from British Columbia provide a window into ancient lineages of viruses. Viruses 11:299. 10.3390/v11030299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han G-Z. 2019. Origin and evolution of the plant immune system. New Phytol 222:70–83. 10.1111/nph.15596. [DOI] [PubMed] [Google Scholar]
- 21.Brunkard JO, Zambryski PC. 2017. Plasmodesmata enable multicellularity: new insights into their evolution, biogenesis, and functions in development and immunity. Curr Opin Plant Biol 35:76–83. 10.1016/j.pbi.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Greninger AL. 2018. A decade of RNA virus metagenomics is (not) enough. Virus Res 244:218–229. 10.1016/j.virusres.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller AK, Mifsud JCO, Costa VA, Grimwood RM, Kitson J, Baker C, Brosnahan CL, Pande A, Holmes EC, Gemmell NJ, Geoghegan JL. 2021. Slippery when wet: cross-species transmission of divergent coronaviruses in bony and jawless fish and the evolutionary history of the Coronaviridae. Virus Evol 7:veab050. 10.1093/ve/veab050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bejerman N, Dietzgen RG, Debat H. 2021. Illuminating the plant rhabdovirus landscape through metatranscriptomics data. Viruses 13:1304. 10.3390/v13071304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parry R, Wille M, Turnbull OMH, Geoghegan JL, Holmes EC. 2020. Divergent influenza-like viruses of amphibians and fish support an ancient evolutionary association. Viruses 12:1042. 10.3390/v12091042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grimwood RM, Holmes EC, Geoghegan JL. 2021. A novel rubi-like virus in the Pacific electric ray (Tetronarce californica) reveals the complex evolutionary history of the Matonaviridae. Viruses 13:585. 10.3390/v13040585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilbert KB, Holcomb EE, Allscheid RL, Carrington JC. 2019. Hiding in plain sight: new virus genomes discovered via a systematic analysis of fungal public transcriptomes. PLoS One 14:e0219207. 10.1371/journal.pone.0219207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lauber C, Seitz S, Mattei S, Suh A, Beck J, Herstein J, Börold J, Salzburger W, Kaderali L, Briggs JAG, Bartenschlager R. 2017. Deciphering the origin and evolution of hepatitis B viruses by means of a family of non-enveloped fish viruses. Cell Host Microbe 22:387–399.e6. 10.1016/j.chom.2017.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leebens-Mack JH, Barker MS, Carpenter EJ, Deyholos MK, Gitzendanner MA, Graham SW, Grosse I, Li Z, Melkonian M, Mirarab S, Porsch M, Quint M, Rensing SA, Soltis DE, Soltis PS, Stevenson DW, Ullrich KK, Wickett NJ, DeGironimo L, Edger PP, Jordon-Thaden IE, Joya S, Liu T, Melkonian B, Miles NW, Pokorny L, Quigley C, Thomas P, Villarreal JC, Augustin MM, Barrett MD, Baucom RS, Beerling DJ, Benstein RM, Biffin E, Brockington SF, Burge DO, Burris JN, Burris KP, Burtet-Sarramegna V, Caicedo AL, Cannon SB, Çebi Z, Chang Y, Chater C, Cheeseman JM, Chen T, Clarke ND, Clayton H, Covshoff S, et al. 2019. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574:679–685. 10.1038/s41586-019-1693-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carpenter EJ, Matasci N, Ayyampalayam S, Wu S, Sun J, Yu J, Jimenez Vieira FR, Bowler C, Dorrell RG, Gitzendanner MA, Li L, Du W, K Ullrich K, Wickett NJ, Barkmann TJ, Barker MS, Leebens-Mack JH, Wong GK-S. 2019. Access to RNA-sequencing data from 1,173 plant species: the 1000 Plant Transcriptomes Initiative (1KP). GigaScience 8:giz126. 10.1093/gigascience/giz126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morozov SY, Solovyev AG. 2003. Triple gene block: modular design of a multifunctional machine for plant virus movement. J Gen Virol 84:1351–1366. 10.1099/vir.0.18922-0. [DOI] [PubMed] [Google Scholar]
- 33.Hammond R, Ramirez P. 2001. Molecular characterization of the genome of Maize rayado fino virus, the type member of the genus Marafivirus. Virology 282:338–347. 10.1006/viro.2001.0859. [DOI] [PubMed] [Google Scholar]
- 34.Ding SW, Howe J, Keese P, Mackenzie A, Meek D, Osorio-Keese M, Skotnicki M, Srifah P, Torronen M, Gibbs A. 1990. The tymobox, a sequence shared by most tymoviruses: its use in molecular studies of tymoviruses. Nucleic Acids Res 18:1181–1187. 10.1093/nar/18.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie J, Ghabrial SA. 2012. Molecular characterization of two mitoviruses co-infecting a hypovirulent isolate of the plant pathogenic fungus Sclerotinia sclerotiorum. Virology 428:77–85. 10.1016/j.virol.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 36.Heinze C. 2012. A novel mycovirus from Clitocybe odora. Arch Virol 157:1831–1834. 10.1007/s00705-012-1373-0. [DOI] [PubMed] [Google Scholar]
- 37.Nibert ML. 2017. Mitovirus UGA(Trp) codon usage parallels that of host mitochondria. Virology 507:96–100. 10.1016/j.virol.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang T, Li W, Chen H, Yu H. 2015. Full genome sequence of a putative novel mitovirus isolated from Rhizoctonia cerealis. Arch Virol 160:1815–1818. 10.1007/s00705-015-2431-1. [DOI] [PubMed] [Google Scholar]
- 39.Shackelton LA, Holmes EC. 2008. The role of alternative genetic codes in viral evolution and emergence. J Theor Biol 254:128–134. 10.1016/j.jtbi.2008.05.024. [DOI] [PubMed] [Google Scholar]
- 40.Nerva L, Turina M, Zanzotto A, Gardiman M, Gaiotti F, Gambino G, Chitarra W. 2019. Isolation, molecular characterization and virome analysis of culturable wood fungal endophytes in esca symptomatic and asymptomatic grapevine plants. Environ Microbiol 21:2886–2904. 10.1111/1462-2920.14651. [DOI] [PubMed] [Google Scholar]
- 41.Charon J, Marcelino VR, Wetherbee R, Verbruggen H, Holmes EC. 2020. Metatranscriptomic identification of diverse and divergent RNA viruses in green and Chlorarachniophyte algae cultures. Viruses 12:1180. 10.3390/v12101180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koga R, Horiuchi H, Fukuhara T. 2003. Double-stranded RNA replicons associated with chloroplasts of a green alga, Bryopsis cinicola. Plant Mol Biol 51:991–999. 10.1023/A:1023003412859. [DOI] [PubMed] [Google Scholar]
- 43.Cai G, Myers K, Hillman BI, Fry WE. 2009. A novel virus of the late blight pathogen, Phytophthora infestans, with two RNA segments and a supergroup 1 RNA-dependent RNA polymerase. Virology 392:52–61. 10.1016/j.virol.2009.06.040. [DOI] [PubMed] [Google Scholar]
- 44.Covelli L, Coutts RH, Di Serio F, Citir A, Açıkgöz S, Hernandez C, Ragozzino A, Flores R. 2004. Cherry chlorotic rusty spot and Amasya cherry diseases are associated with a complex pattern of mycoviral-like double-stranded RNAs. I. Characterization of a new species in the genus Chrysovirus. J Gen Virol 85:3389–3397. 10.1099/vir.0.80181-0. [DOI] [PubMed] [Google Scholar]
- 45.Rousvoal S, Bouyer B, López‐Cristoffanini C, Boyen C, Collén J. 2016. Mutant swarms of a totivirus‐like entities are present in the red macroalga Chondrus crispus and have been partially transferred to the nuclear genome. J Phycol 52:493–504. 10.1111/jpy.12427. [DOI] [PubMed] [Google Scholar]
- 46.Charon J, Murray S, Holmes EC. 2021. Revealing RNA virus diversity and evolution in unicellular algae transcriptomes. Virus Evol 7:veab070. 10.1093/ve/veab070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kenrick P, Crane PR. 1997. The origin and early evolution of plants on land. Nature 389:33–39. 10.1038/37918. [DOI] [Google Scholar]
- 48.Friis EM, Pedersen KR, Crane PR. 2010. Diversity in obscurity: fossil flowers and the early history of angiosperms. Philos Trans R Soc Lond B Biol Sci 365:369–382. 10.1098/rstb.2009.0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Valente JB, Pereira FS, Stempkowski LA, Farias M, Kuhnem P, Lau D, Fajardo TVM, Nhani Junior A, Casa RT, Bogo A, da Silva FN. 2019. A novel putative member of the family Benyviridae is associated with soilborne wheat mosaic disease in Brazil. Plant Pathol 68:588–600. 10.1111/ppa.12970. [DOI] [Google Scholar]
- 50.Tamada T, Schmitt C, Saito M, Guilley H, Richards K, Jonard G. 1996. High resolution analysis of the readthrough domain of beet necrotic yellow vein virus readthrough protein: a KTER motif is important for efficient transmission of the virus by Polymyxa betae. J Gen Virol 77:1359–1367. 10.1099/0022-1317-77-7-1359. [DOI] [PubMed] [Google Scholar]
- 51.Neuhauser S, Kirchmair M, Bulman S, Bass D. 2014. Cross-kingdom host shifts of phytomyxid parasites. BMC Evol Biol 14:33. 10.1186/1471-2148-14-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Starr EP, Nuccio EE, Pett-Ridge J, Banfield JF, Firestone MK. 2019. Metatranscriptomic reconstruction reveals RNA viruses with the potential to shape carbon cycling in soil. Proc Natl Acad Sci USA 116:25900–25908. 10.1073/pnas.1908291116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghabrial SA, Caston JR, Jiang D, Nibert ML, Suzuki N. 2015. 50-plus years of fungal viruses. Virology 479–480:356–368. 10.1016/j.virol.2015.02.034. [DOI] [PubMed] [Google Scholar]
- 54.Bruenn JA, Warner BE, Yerramsetty P. 2015. Widespread mitovirus sequences in plant genomes. Peerj 3:e876. 10.7717/peerj.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moreno AB, López-Moya JJ. 2020. When viruses play team sports: mixed infections in plants. Phytopathology 110:29–48. 10.1094/PHYTO-07-19-0250-FI. [DOI] [PubMed] [Google Scholar]
- 56.Botella L, Jung T. 2021. Multiple viral infections detected in Phytophthora condilina by total and small RNA sequencing. Viruses 13:620. 10.3390/v13040620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Botella L, Janoušek J, Maia C, Jung MH, Raco M, Jung T. 2020. Marine oomycetes of the genus Halophytophthora harbor viruses related to bunyaviruses. Front Microbiol 11:1467. 10.3389/fmicb.2020.01467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Su S, Liu Z, Chen C, Zhang Y, Wang X, Zhu L, Miao L, Wang X-C, Yuan M. 2010. Cucumber mosaic virus movement protein severs actin filaments to increase the plasmodesmal size exclusion limit in tobacco. Plant Cell 22:1373–1387. 10.1105/tpc.108.064212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mushegian AR, Elena SF. 2015. Evolution of plant virus movement proteins from the 30K superfamily and of their homologs integrated in plant genomes. Virology 476:304–315. 10.1016/j.virol.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 60.Kumar S, Subbarao BL, Kumari R, Hallan V. 2017. Molecular characterization of a novel cryptic virus infecting pigeonpea plants. PLoS One 12:e0181829. 10.1371/journal.pone.0181829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sabanadzovic S, Ghanem-Sabanadzovic NA. 2008. Molecular characterization and detection of a tripartite cryptic virus from rose. J Plant Pathol 90:287–293. [Google Scholar]
- 62.Chen L, Chen J, Liu L, Yu X, Yu S, Fu T, Liu W. 2006. Complete nucleotide sequences and genome characterization of double-stranded RNA 1 and RNA 2 in the Raphanus sativus-root cv. Arch Virol 151:849–859. 10.1007/s00705-005-0685-8. [DOI] [PubMed] [Google Scholar]
- 63.Wu LP, Du YM, Xiao H, Peng L, Li R. 2020. Complete genomic sequence of tea-oil camellia deltapartitivirus 1, a novel virus from Camellia oleifera. Arch Virol 165:227–231. 10.1007/s00705-019-04429-0. [DOI] [PubMed] [Google Scholar]
- 64.Chiba S, Lin YH, Kondo H, Kanematsu S, Suzuki N. 2013. Effects of defective interfering RNA on symptom induction by, and replication of, a novel partitivirus from a phytopathogenic fungus, Rosellinia necatrix. J Virol 87:2330–2341. 10.1128/JVI.02835-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nibert ML, Ghabrial SA, Maiss E, Lesker T, Vainio EJ, Jiang D, Suzuki N. 2014. Taxonomic reorganization of family Partitiviridae and other recent progress in partitivirus research. Virus Res 188:128–141. 10.1016/j.virusres.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 66.Harrison CJ, Morris JL. 2018. The origin and early evolution of vascular plant shoots and leaves. Philos Trans R Soc Lond B Biol Sci 373:20160496. 10.1098/rstb.2016.0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60. 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 68.Walker PJ, Blasdell KR, Calisher CH, Dietzgen RG, Kondo H, Kurath G, Longdon B, Stone DM, Tesh RB, Tordo N, Vasilakis N, Whitfield AE, ICTV Report Consortium . 2018. ICTV virus taxonomy profile: Rhabdoviridae. J Gen Virol 99:447–448. 10.1099/jgv.0.001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi M, Lin X-D, Tian J-H, Chen L-J, Chen X, Li C-X, Qin X-C, Li J, Cao J-P, Eden J-S, Buchmann J, Wang W, Xu J, Holmes EC, Zhang Y-Z. 2016. Redefining the invertebrate RNA virosphere. Nature 540:539–543. 10.1038/nature20167. [DOI] [PubMed] [Google Scholar]
- 70.Hao X, Zhang W, Zhao F, Liu Y, Qian W, Wang Y, Wang L, Zeng J, Yang Y, Wang X. 2018. Discovery of plant viruses from tea plant (Camellia sinensis (L.) O. Kuntze) by metagenomic sequencing. Front Microbiol 9:2175. 10.3389/fmicb.2018.02175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maclot F, Candresse T, Filloux D, Malmstrom CM, Roumagnac P, van der Vlugt R, Massart S. 2020. Illuminating an ecological blackbox: using high throughput Sequencing to characterize the plant virome across scales. Front Microbiol 11:578064. 10.3389/fmicb.2020.578064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Visser M, Bester R, Burger JT, Maree HJ. 2016. Next-generation sequencing for virus detection: covering all the bases. Virol J 13:85. 10.1186/s12985-016-0539-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Llorens C, Muñoz-Pomer A, Bernad L, Botella H, Moya A. 2009. Network dynamics of eukaryotic LTR retroelements beyond phylogenetic trees. Biol Direct 4:41. 10.1186/1745-6150-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson MTJ, Carpenter EJ, Tian Z, Bruskiewich R, Burris JN, Carrigan CT, Chase MW, Clarke ND, Covshoff S, Depamphilis CW, Edger PP, Goh F, Graham S, Greiner S, Hibberd JM, Jordon-Thaden I, Kutchan TM, Leebens-Mack J, Melkonian M, Miles N, Myburg H, Patterson J, Pires JC, Ralph P, Rolf M, Sage RF, Soltis D, Soltis P, Stevenson D, Stewart CN, Surek B, Thomsen CJM, Villarreal JC, Wu X, Zhang Y, Deyholos MK, Wong GK-S. 2012. Evaluating methods for isolating total RNA and predicting the success of sequencing phylogenetically diverse plant transcriptomes. PLoS One 7:e50226. 10.1371/journal.pone.0050226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leinonen R, Sugawara H, Shumway M, Collaboration INSD, on behalf of the International Nucleotide Sequence Database Collaboration . 2011. The Sequence Read Archive. Nucleic Acids Res 39:D19–D21. 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, Leduc RD, Friedman N, Regev A. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512. 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 79.Mihara T, Nishimura Y, Shimizu Y, Nishiyama H, Yoshikawa G, Uehara H, Hingamp P, Goto S, Ogata H. 2016. Linking virus genomes with host taxonomy. Viruses 8:66. 10.3390/v8030066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.RStudio Team. 2020. RStudio: integrated development for R. RStudio, Inc., Boston, MA. [Google Scholar]
- 81.R Core Team. 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 82.Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, Grolemund G, Hayes A, Henry L, Hester J, Kuhn M, Pedersen T, Miller E, Bache S, Müller K, Ooms J, Robinson D, Seidel D, Spinu V, Takahashi K, Vaughan D, Wilke C, Woo K, Yutani H. 2019. Welcome to the Tidyverse. J Open Source Softw 4:1686. 10.21105/joss.01686. [DOI] [Google Scholar]
- 83.Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bushnell B. 2014. BBMap: a fast, accurate, splice-aware aligner. Lawrence Berkeley National Lab (LBNL), Berkeley, CA. [Google Scholar]
- 85.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mirdita M, Steinegger M, Söding J. 2019. MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics 35:2856–2858. 10.1093/bioinformatics/bty1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zimmermann L, Stephens A, Nam S-Z, Rau D, Kübler J, Lozajic M, Gabler F, Söding J, Lupas AN, Alva V. 2018. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J Mol Biol 430:2237–2243. 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 89.Lay CL. 2021. biolumber/littlegenomes: first release. 10.5281/ZENODO.5081375. [DOI]
- 90.Sayers EW, Cavanaugh M, Clark K, Pruitt KD, Schoch CL, Sherry ST, Karsch-Mizrachi I. 2021. GenBank. Nucleic Acids Res 49:D92–D96. 10.1093/nar/gkaa1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gertz EM, Yu Y-K, Agarwala R, Schäffer AA, Altschul SF. 2006. Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol 4:41–14. 10.1186/1741-7007-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nayfach S, Camargo AP, Schulz F, Eloe-Fadrosh E, Roux S, Kyrpides NC. 2021. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat Biotechnol 39:578–585. 10.1038/s41587-020-00774-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Marcelino VR, Clausen PTLC, Buchmann JP, Wille M, Iredell JR, Meyer W, Lund O, Sorrell TC, Holmes EC. 2020. CCMetagen: comprehensive and accurate identification of eukaryotes and prokaryotes in metagenomic data. Genome Biol 21:103. 10.1186/s13059-020-02014-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Clausen PT, Aarestrup FM, Lund O. 2018. Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinformatics 19:1–8. 10.1186/s12859-018-2336-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ondov BD, Bergman NH, Phillippy AM. 2011. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. 10.1186/1471-2105-12-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nguyen L-T, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kalyaanamoorthy S, Minh BQ, Wong TK, Von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589. 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rambaut A, Drummond A. 2012. FigTree: tree figure drawing tool, version 1.4.0. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh, Scotland. [Google Scholar]
- 100.Zanne AE, Tank DC, Cornwell WK, Eastman JM, Smith SA, FitzJohn RG, McGlinn DJ, O’Meara BC, Moles AT, Reich PB, Royer DL, Soltis DE, Stevens PF, Westoby M, Wright IJ, Aarssen L, Bertin RI, Calaminus A, Govaerts R, Hemmings F, Leishman MR, Oleksyn J, Soltis PS, Swenson NG, Warman L, Beaulieu JM. 2014. Three keys to the radiation of angiosperms into freezing environments. Nature 506:89–92. 10.1038/nature12872. [DOI] [PubMed] [Google Scholar]
- 101.Smith SA, Brown JW. 2018. Constructing a broadly inclusive seed plant phylogeny. Am J Bot 105:302–314. 10.1002/ajb2.1019. [DOI] [PubMed] [Google Scholar]
- 102.Jin Y, Qian H. 2019. V.PhyloMaker: an R package that can generate very large phylogenies for vascular plants. Ecography 42:1353–1359. 10.1111/ecog.04434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hatcher EL, Zhdanov SA, Bao Y, Blinkova O, Nawrocki EP, Ostapchuck Y, Schäffer AA, Brister JR. 2017. Virus Variation Resource: improved response to emergent viral outbreaks. Nucleic Acids Res 45:D482–D490. 10.1093/nar/gkw1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Paradis E, Schliep K. 2019. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35:526–528. 10.1093/bioinformatics/bty633. [DOI] [PubMed] [Google Scholar]
- 105.Revell LJ. 2012. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol 3:217–223. 10.1111/j.2041-210X.2011.00169.x. [DOI] [Google Scholar]
- 106.Conow C, Fielder D, Ovadia Y, Libeskind-Hadas R. 2010. Jane: a new tool for the cophylogeny reconstruction problem. Algorithms Mol Biol 5:16–10. 10.1186/1748-7188-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Santichaivekin S, Yang Q, Liu J, Mawhorter R, Jiang J, Wesley T, Wu Y-C, Libeskind-Hadas R. 2021. eMPRess: a systematic cophylogeny reconciliation tool. Bioinformatics 37:2481–2482. 10.1093/bioinformatics/btaa978. [DOI] [PubMed] [Google Scholar]
- 108.Letunic I, Bork P. 2019. Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47:W256–W259. 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brister JR, Ako-Adjei D, Bao Y, Blinkova O. 2015. NCBI viral genomes resource. Nucleic Acids Res 43:D571–7. 10.1093/nar/gku1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S5, Tables S1 to S5. Download jvi.00260-22-s0001.pdf, PDF file, 10.3 MB (10.3MB, pdf)
Data Availability Statement
The raw 1000 Plant Transcriptomes Initiative sequence reads are available at BioProject PRJEB21674. All viral genomes and corresponding sequences assembled in this study have been deposited in the European Nucleotide Archive at EMBL-EBI under accession number PRJEB52092 (Table S4).