

ABSTRACT
The reliability of sequence-based inference of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) transmission is not clear. Sequence data from infections among household members can define the expected genomic diversity of a virus along a defined transmission chain. SARS-CoV-2 cases were identified prospectively among 2,369 participants in 706 households. Specimens with a reverse transcription-PCR cycle threshold of ≤30 underwent whole-genome sequencing. Intrahost single-nucleotide variants (iSNV) were identified at a ≥5% frequency. Phylogenetic trees were used to evaluate the relationship of household and community sequences. There were 178 SARS-CoV-2 cases in 706 households. Among 147 specimens sequenced, 106 yielded a whole-genome consensus with coverage suitable for identifying iSNV. Twenty-six households had sequences from multiple cases within 14 days. Consensus sequences were indistinguishable among cases in 15 households, while 11 had ≥1 consensus sequence that differed by 1 to 2 mutations. Sequences from households and the community were often interspersed on phylogenetic trees. Identification of iSNV improved inference in 2 of 15 households with indistinguishable consensus sequences and in 6 of 11 with distinct ones. In multiple-infection households, whole-genome consensus sequences differed by 0 to 1 mutations. Identification of shared iSNV occasionally resolved linkage, but the low genomic diversity of SARS-CoV-2 limits the utility of “sequence-only” transmission inference.
IMPORTANCE We performed whole-genome sequencing of SARS-CoV-2 from prospectively identified cases in three longitudinal household cohorts. In a majority of multi-infection households, SARS-CoV-2 consensus sequences were indistinguishable, and they differed by 1 to 2 mutations in the rest. Importantly, even with modest genomic surveillance of the community (3 to 5% of cases sequenced), it was not uncommon to find community sequences interspersed with household sequences on phylogenetic trees. Identification of shared minority variants only occasionally resolved these ambiguities in transmission linkage. Overall, the low genomic diversity of SARS-CoV-2 limits the utility of “sequence-only” transmission inference. Our work highlights the need to carefully consider both epidemiologic linkage and sequence data to define transmission chains in households, hospitals, and other transmission settings.
KEYWORDS: SARS-CoV-2, genomic epidemiology, transmission, household
INTRODUCTION
RNA viruses evolve rapidly and accumulate mutations as outbreaks grow (1). As a result, the evolutionary relationships among sequenced cases hold important information about the processes that drive epidemics (2). For example, sequence data can help define transmission chains and outbreaks (3–5), the timing and location of viral introductions into communities (6–8), and larger patterns of spread (9–12). Over the course of the 2019 coronavirus disease (COVID-19) pandemic, SARS-CoV-2 sequences have been used to infer transmission linkage in hospitals and other congregate settings (13–18). Inferring these linkages with high confidence is necessary for subsequent studies of the biology of transmission and effectiveness of mitigation strategies.
To infer transmission, one can ask whether the sequences within a group of close contacts, such as a household, are more similar than sequences in the broader community. This approach depends on both the granularity of the sequence data and the amount of genomic diversity in the underlying community or metapopulation. The relatedness of viral sequences identified from potential transmission chains versus community virologic surveillance has been compared using phylogenetic trees of whole-genome consensus sequences or clustering of transmission-associated sequences (2). In the setting of insufficient community sampling and/or low genomic diversity, consensus trees can miss true linkages and identify false ones. Greater coverage sequencing can improve resolution by identifying intrahost single-nucleotide variants (iSNV) in host-derived viral populations that have yet to achieve consensus levels, or >50% within-host frequency, along a transmission chain (19, 20). While these approaches have proven useful for influenza virus and other viruses, the reliability of sequence-based inference of SARS-CoV-2 transmission is less clear. For example, we and others have found that participants without known epidemiologic linkage can share indistinguishable consensus sequences and even minority (<50%) iSNV (21–23).
Households are ideal settings for studies of the biology and epidemiology of viral transmission. Documentation of close contact and concurrent symptoms or test positivity provide strong epidemiologic evidence of within-household transmission. Sequence data from infected participants can therefore define the expected genomic diversity of a virus along a transmission chain and inform sequence-based studies in other transmission settings, where epidemiologic linkage may be uncertain. Here, we use whole-genome sequencing of SARS-CoV-2 populations from participants in two prospective household studies of COVID-19 that were conducted at three sites. To assess the utility of SARS-CoV-2 sequence data as a tool for inferring transmission, we used phylogenetic analysis of sequences from households with at least two SARS-CoV-2 infection cases to assess the clustering of within-household sequences relative to contemporaneous community sequences. We used iSNV to further resolve transmission linkages in selected households.
RESULTS
The C-HEaRT (Utah and New York City) and COCOVID (Puerto Rico) studies performed active surveillance for SARS-CoV-2 infection and COVID-like illness (CLI) in 706 households with 2,369 participants (Table 1). During September 2020 through August 2021, the cumulative incidence of SARS-CoV-2 infection was 11% (96/842 participants in 41/190 [22%] households under surveillance) at the Utah site and 7% (33/499 participants in 13/135 [10%] households) at the New York City site; during June 2020 through September 2021, cumulative incidence was 5% at the Puerto Rico site (49/1,028 infections detected in 28/381 households).
TABLE 1.
Distribution of SARS-CoV-2 test-positive cases across cohorts and households
| Characteristic | New York City | Utah | Puerto Rico |
|---|---|---|---|
| Households (n) | 135 | 190 | 381 |
| Participants (n) | 499 | 842 | 1,028 |
| Median household size (range) | 4 (2–9) | 4 (2–10) | 2 (1–6) |
| Unique SARS-CoV-2-positive casesa | 33 | 96 | 49 |
| Households with 1 case | 3 | 17 | 3 |
| Households with 2 casesb | 5 | 7 | 5 |
| Households with 3 casesb | 3 | 5 | 8 |
| Households with 4 casesb | 0 | 4 | 6 |
| Households with 5 casesb | 1 | 4 | 5 |
| Households with 6 casesb | 1 | 0 | 1 |
| Households with 7 casesb | 0 | 1 | 0 |
| Cases with sequence data/cases sequenced (n/n) | 28/29 | 52/86 | 26/32 |
Total number of cases over the study period.
Includes only households with cases testing positive within 14 days of each other.
Of the 191 participants with SARS-CoV-2 infections in these households, 147 (77%) in 70 households had samples with a threshold cycle (CT) value of <30 that were processed for whole-genome sequencing, of whom, 106 (72%) had samples that were successfully sequenced to sufficient breadth and depth of coverage (see Materials and Methods). Of the 706 households at the three sites, 56 included ≥2 participants who were test positive within a 14-day period, suggestive of within-household transmission (Table 1). Twenty-six households had high-quality sequence data on ≥2 of these contemporaneous infections. The SARS-CoV-2 clades and lineages identified (Table 2) were the same among participants of the same household and reflected viruses circulating in the corresponding time periods in the United States (www.outbreak.info) (Table 2).
TABLE 2.
Households with two or more incident SARS-CoV-2 infections within a 14-day period
| Household | No. of specimens sequenced | Date of first specimen (mo/day/yr) | Days between first and last specimen | Nextclade cladea | PANGO lineageb | Mean consensus diffc |
|---|---|---|---|---|---|---|
| PR1 | 3 | 9/2/20 | 6 | 20C | B.1.426 | 0 |
| UT1 | 3 | 10/14/20 | 1 | 20G | B.1.2 | 0 |
| PR2 | 4 | 10/24/20 | 6 | 20C | B.1.588 | 0.5 |
| UT2 | 4 | 11/17/20 | 7 | 20B | B.1.1 | 0.5 |
| UT3 | 6 | 11/24/20 | 8 | 20G | B.1.2 | 1.4 |
| UT4 | 4 | 11/30/20 | 7 | 20B | B.1.1 | 1.5 |
| UT5 | 2 | 12/2/20 | 1 | 20A | B.1.400 | 1 |
| UT6 | 3 | 12/3/20 | 7 | 20G | B.1.2 | 0.67 |
| PR3 | 4 | 12/3/20 | 8 | 20B | B.1.1.486 | 1.17 |
| UT7 | 3 | 12/15/20 | 13 | 20A | B.1.596 | 0 |
| UT8 | 2 | 12/28/20 | 4 | 21C (Epsilon) | B.1.429 | 0 |
| UT9 | 2 | 1/12/21 | 2 | 20A | B.1.400 | 0 |
| NY1 | 3 | 1/29/21 | 6 | 21F (iota) | B.1.526 | 0.67 |
| PR4 | 2 | 2/8/21 | 0 | 20A | B.1.240 | 0 |
| NY2 | 3 | 2/9/21 | 0 | 21F (Iota) | B.1.526 | 0 |
| NY3 | 3 | 2/11/21 | 7 | 20C | B.1.582 | 0 |
| NY4 | 2 | 2/21/21 | 12 | 21F (iota) | B.1.526 | 0 |
| NY5 | 2 | 2/23/21 | 0 | 21F (Iota) | B.1.526 | 0 |
| NY6 | 6 | 2/24/21 | 15 | 20C | B.1.637 | 1.13 |
| UT10 | 2 | 3/1/21 | 14 | 21C (Epsilon) | B.1.427 | 2 |
| NY7 | 3 | 3/3/21 | 13 | 20C | B.1.637 | 0 |
| NY8 | 2 | 3/22/21 | 13 | 21F (Iota) | B.1.526 | 0 |
| PR5 | 2 | 3/23/21 | 7 | 20B | R.1 | 0 |
| PR6 | 3 | 4/23/21 | 0 | 20I (Alpha, V1) | Q.4 | 0 |
| UT11 | 3 | 7/26/21 | 15 | 21J (Delta) | AY.44 | 0 |
| UT12 | 2 | 8/4/21 | 0 | 21J (Delta) | AY.44 | 1 |
Defined using nextclade (https://clades.nextstrain.org).
Defined using pango (https://cov-lineages.org/resources/pangolin.html).
Total number of pairwise unambiguous consensus differences between sequences, divided by total number of sequences in a household.
We first used phylogenetic analysis of whole-genome sequences to infer transmission linkage within these 26 households. We used UShER (24) to obtain local sequences for each household and to place household sequences simultaneously on a phylogenetic tree. Over 7.8 million whole-genome SARS-CoV-2 sequences were available at the time of this analysis. Because most of these contextual sequences were from GISAID, we estimated the level of sampling at each study site over time by dividing the number of GISAID sequences by the number of reported cases (10). Sampling of locally circulating viruses was low in 2020 (<2% cases sequenced) and increased at all three sites beginning in early 2021 (>5% cases sequenced) (Fig. 1). In 2022, Utah was generally better sampled (~10 to 30%) than New York or Puerto Rico (5 to 15%).
FIG 1.
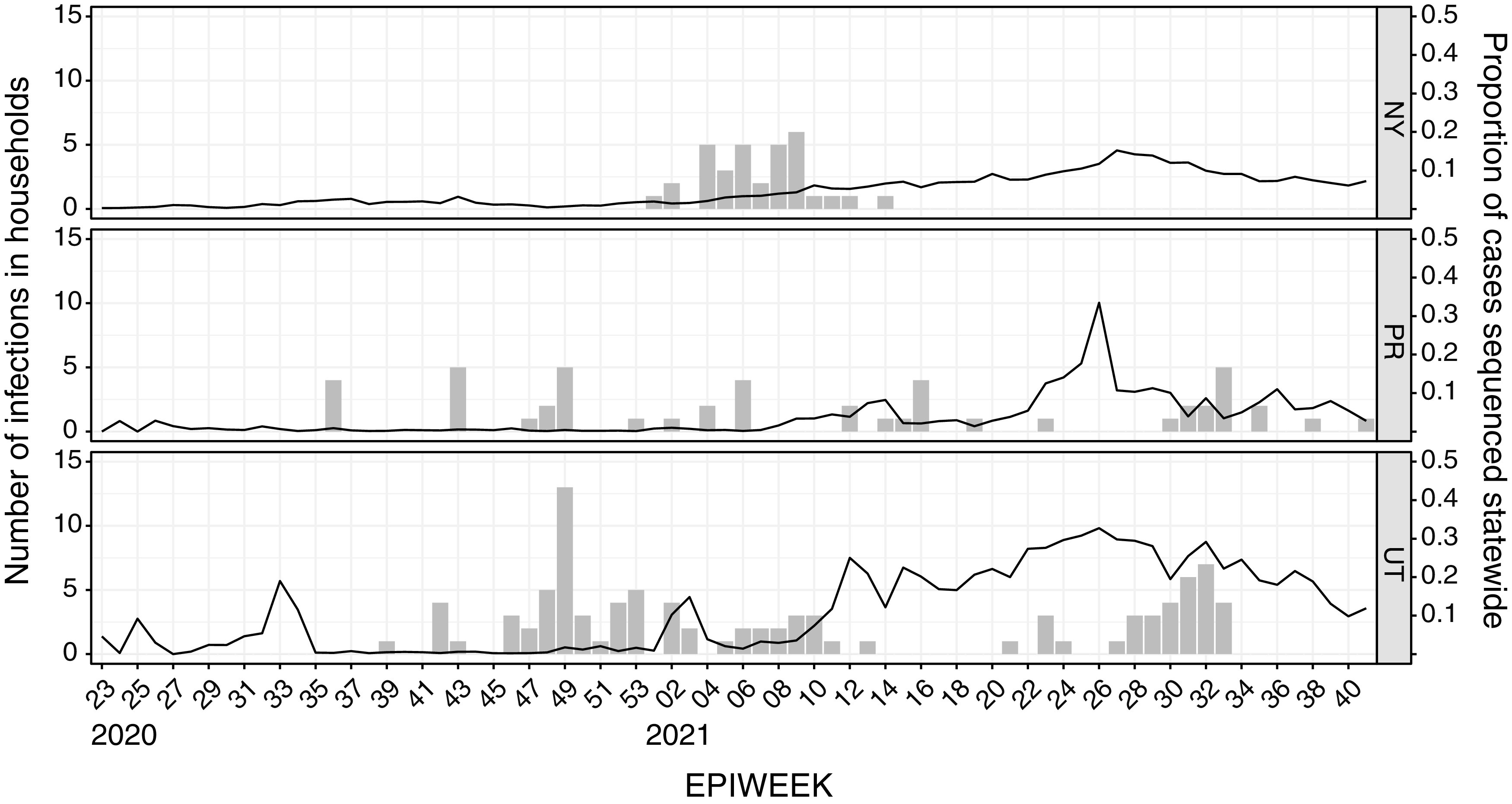
Cases and sampling density. Columns show the number of SARS-CoV-2 infections (left y axis) in households from New York (NY, top), Puerto Rico (PR, middle), and Utah (UT, bottom) cohorts by epiweek (x axis). The sampling density (line) for community genomes in each state or territory (right y axis) was estimated as the proportion of cases with sequences available on GISAID.
In 15 of the 26 households that we studied, the consensus sequences of all cases were indistinguishable and grouped together on their respective trees (representative trees are shown in Fig. 2, with additional trees in Fig. S1 to S4 in the supplemental material). Given the epidemiologic linkage in the same household, these can be considered sequence-confirmed transmission events. However, we also found several trees in which these monophyletic groupings also included indistinguishable, contemporaneous sequences from non-household members within the community of the same locality (see the trees in Fig. 2). In two of the New York households, there were many such sequences during a B.1.526 (Iota) variant wave (Fig. S1 and 2).
FIG 2.
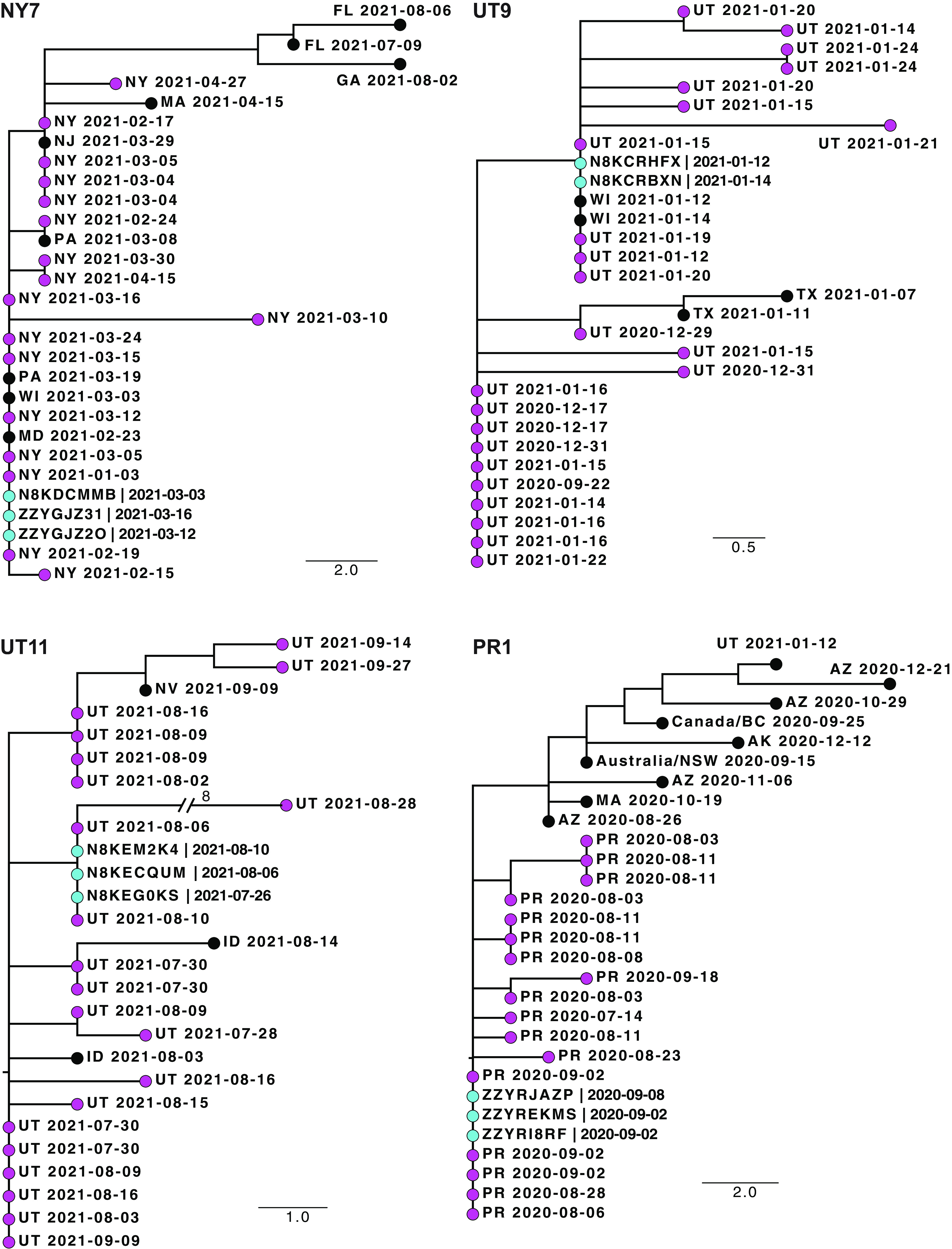
Phylogenetic trees of sequences from households where all participants had indistinguishable consensus sequences. Shown are four representative trees. Trees from 11 other households are shown in Fig. S1 to 4 in the supplemental material. Each tree is labeled with the household identifier (NY, New York; UT, Utah; PR, Puerto Rico). The tips of household sequences are colored cyan and those from nonhousehold participants in the same community in the same state or territory (2-letter abbreviation) are colored magenta. All other tips are colored black. The collection date for each specimen is indicated. Genetic distance is represented by the bar and corresponds to one mutation.
Phylogenetic trees of sequences from NY1 household where all participants had indistinguishable consensus sequences. The tips of household sequences are colored cyan, and those from the same state or territory (2-letter abbreviation) are colored magenta. All other tips are colored black. The collection date for each sample is indicated. Genetic distance is represented by the bar and corresponds to one mutation. Download FIG S1, EPS file, 2.2 MB (2.2MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
To better estimate the probability that members of the household would have viral sequences identical to those circulating in the community, we chose three households in which within-household consensus sequences were identical (NY7, UT11, and PR5). We downloaded sequences from GISAID from 2 weeks before to 2 weeks after the earliest symptom onset date within the specified households in each state and calculated the average number of mutational differences between household and community sequences and the number of identical sequences in the community. We found an average difference of 33 and 5/10,035 identical sequences for NY7 (epiweek 9), an average difference of 19 and 1/4,958 identical sequences for UT11 (epiweek 30), and an average difference of 45 and 0/602 identical sequences for PR5 (epiweek 12) (Fig. 1). Therefore, given sufficient sampling, it was not difficult to find indistinguishable viral sequences from individuals in the same region and time who presumably lacked a documented epidemiologic linkage.
In 11 households, the consensus sequences of the virus from one or more household members differed at 1 to 2 positions over the ~30-kb genome. This is not uncommon in transmission chains, particularly ones that are longer or in which the samples are collected 7 to 14 days apart (see Table 2 for time span). In nearly all cases, the trees from these households demonstrated linkage and/or an ancestor or descendant relationship for the viral sequences (representative trees are shown in Fig. 3, with additional trees in Fig. S5). In some cases, the household lineages were phylogenetically distinct from contemporaneous local sequences (e.g., UT2, UT4). These tree structures supported transmission linkage, but low sampling of community cases made it hard to rule out missed linkages between members of the household and the larger community. Indeed, there were some households in which there were sequences from the larger community included among the same branches of the within-household sequences (e.g., UT10, PR3). As above, the low genomic diversity of SARS-CoV-2 and modest sampling of community cases made it difficult to define a threshold to effectively rule in or rule out transmission.
FIG 3.
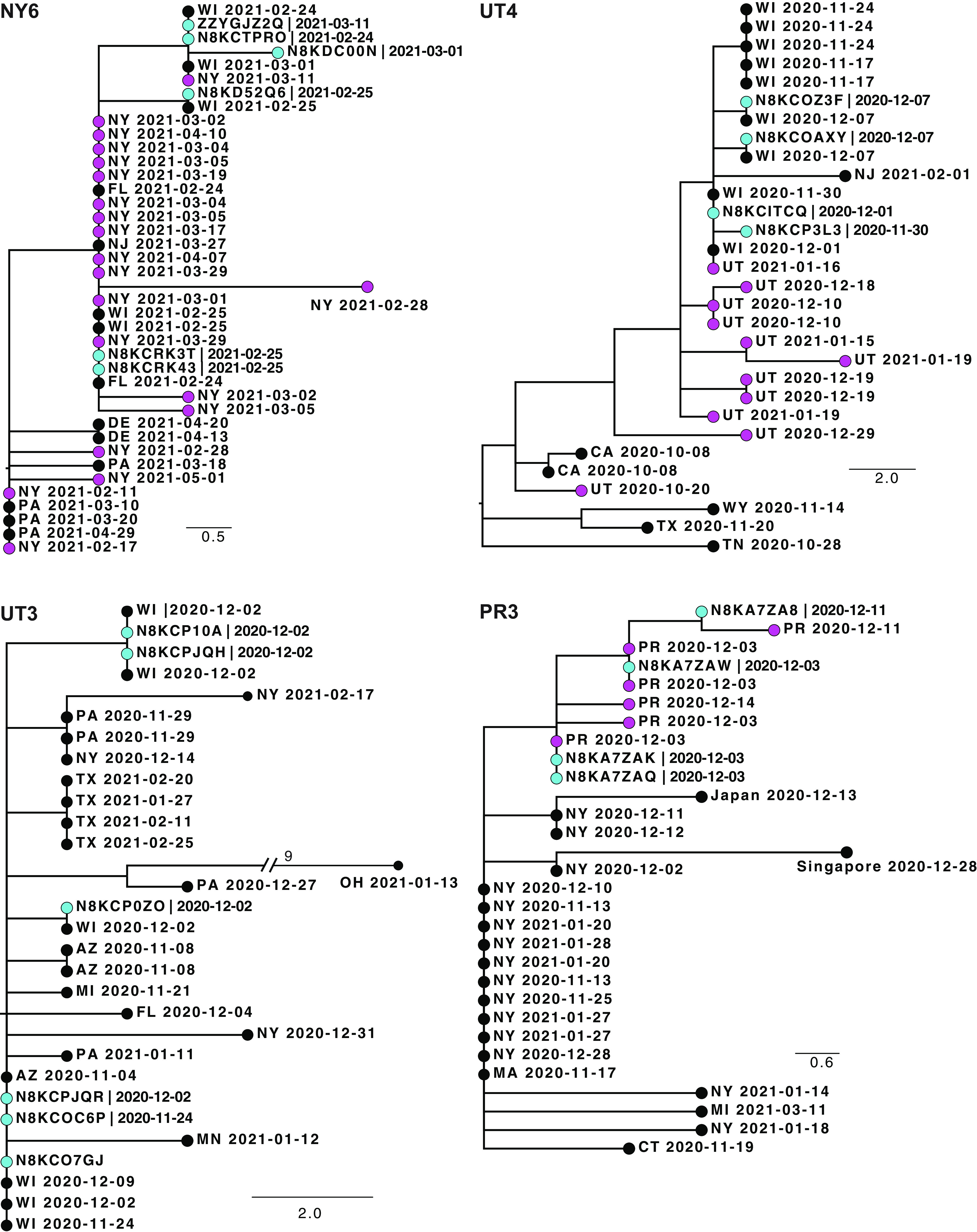
Phylogenetic trees of sequences from households where participants had distinct consensus sequences. Shown are four representative trees. Trees from 7 other households are shown in Fig. S5. Each tree is labeled with the household identifier (NY, New York; UT, Utah; PR, Puerto Rico). The tips of household sequences are colored cyan and those from the same state or territory (2-letter abbreviation) are colored magenta. All other tips are colored black. The collection date for each sample is indicated. Genetic distance is represented by the bar and corresponds to one mutation.
Phylogenetic trees of sequences from NY2 household where all participants had indistinguishable consensus sequences. The tips of household sequences are colored cyan and those from the same state or territory (2-letter abbreviation) are colored magenta. All other tips are colored black. The collection date for each sample is indicated. Genetic distance is represented by the bar and corresponds to one mutation. Download FIG S2, EPS file, 2.6 MB (2.6MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Phylogenetic trees of sequences from the five households in Utah (UT1 and UT7) and Puerto Rico (PR4, PR5, and PR6) where all participants had indistinguishable consensus sequences. The tips of household sequences are colored cyan and those from the same state or territory (2-letter abbreviation) are colored magenta. All other tips are colored black. The collection date for each sample is indicated. Genetic distance is represented by the bar and corresponds to one mutation. Download FIG S3, EPS file, 2.1 MB (2.1MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Phylogenetic trees of sequences from New York households (NY3, NY4, NY5, NY8) where all participants had indistinguishable consensus sequences. The tips of household sequences are colored cyan and those from the same state or territory (2-letter abbreviation) are colored magenta. All other tips are colored black. The collection date for each sample is indicated. Genetic distance is represented by the bar and corresponds to one mutation. Download FIG S4, EPS file, 2.0 MB (2MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Phylogenetic trees of sequences from the seven households where participants had distinct consensus sequences. Each tree is labeled with the household identifier (NY, New York; UT, Utah; PR, Puerto Rico). The tips of household sequences are colored cyan and those from the same state or territory (2-letter abbreviation) are colored magenta. All other tips are colored black. The collection date for each sample is indicated. Genetic distance is represented by the bar and corresponds to one mutation. Download FIG S5, EPS file, 2.5 MB (2.5MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
We next determined whether transmission inference could be improved by identifying iSNV that were shared among members of a household. These would manifest as polymorphic sites where the alternative allele, or mutation, was present but not fixed in the transmission chain. While there was just one household where two participants shared a minority iSNV (PR5, Fig. 4), several had iSNV in at least one individual at a consensus level (i.e., frequency of >0.5) but that had not yet achieved fixation (i.e., frequency if >0.95). In the 15 households with indistinguishable consensus sequences, each of the two participants in households NY5 and PR5 shared an iSNV that was consensus level but not fixed. In three (UT12, UT5, UT3) of the 11 households with distinct consensus sequences (Table 2), the consensus differences were due to one or more participants having a nonreference iSNV that achieved consensus level but not fixation. Household UT4 had two participants with consensus-level iSNV (Fig. 4). In household PR3, there was one site where one out of four members had a consensus-level iSNV and another member had this as a fixed mutation.
FIG 4.

Shared single-nucleotide polymorphisms within households. Each panel shows 1 of the 8 households in which members shared a polymorphic site. The frequency (5 to 95%) of the indicated mutation (relative to the Wuhan/Hu-1 reference) is shown on the y axis, and the individual and sequence identifier is shown on the x axis. Shared variants that did and did not lead to a consensus-level difference between household members are shown as circles and diamonds, respectively. Mutations that were fixed (>95% frequency) are shown as squares.
DISCUSSION
We evaluated the utility of SARS-CoV-2 sequence data in transmission inference using data from two studies of household cohorts at three sites. In the household setting, where at least two incident infections occurring within 14 days of one another are strongly suggestive of transmission, we found that sequencing generally confirmed transmission linkage. The whole-genome consensus sequences of participants within a household were nearly always indistinguishable or differed by one mutation. In some cases, these links were further supported by the identification of iSNV shared among members of the household. Of the 26 households evaluated, there was just one (UT10) in which the high average number of consensus differences (two) and absence of shared iSNV called linkage into doubt. Importantly, we frequently found multiple sequences from the community that were indistinguishable from those within the household with even modest sampling (<5%) over the course of the pandemic. This highlighted the limits of “sequence-only” inference of transmission in hospitals or other congregate settings where epidemiologic linkage is less certain.
Strengths of the study include our reliance on samples from active surveillance of longitudinal cohorts and our use of quality-controlled, deep sequencing. With weekly sampling of all participants from a household, we were able to identify asymptomatic or mildly symptomatic cases and avoid some of the bias of case-ascertainment studies, in which cases are recruited based on a test-positive index. Together with our use of contemporaneous community specimens collected from participants not in the households but from the same site, our data provide a valuable benchmark for the expected SARS-CoV-2 diversity in households relative to that in the community. The cohorts are also drawn from diverse geographic areas with varied household sizes and composition (21, 25). Our assessment of viral diversity is strengthened by our criteria for identifying consensus and minority iSNV (22). The low observed diversity in this study, in part, reflects the stringent thresholds applied to the sequence data. This conservative approach reduces sequencing errors, which can be systematic and lead to incorrect ascertainment of shared iSNV among unrelated participants (21–23, 26).
This study had several notable limitations. First, we were relatively stringent in our criteria for identifying iSNV and therefore may have underascertained shared diversity in the rare (<5%) variant fraction within households. Second, given the limited number of households with sequenced cases, we were unable to formulate a statistically robust approach to sequence-based inference with clear cutoffs and associated positive and negative predictive values. Case-ascertained cohorts or contact tracing studies offer a more efficient way to capture and sequence many putative transmission pairs and will be useful as a setting in which to further develop this approach. Third, while we believe that our data provided an important framework for interpreting sequence data in studies of SARS-CoV-2 transmission, data from households may not translate completely to hospitals and other congregate living settings, which may differ in case density, contact frequency, and force of infection. Fourth, we assumed that household cases testing positive for SARS-CoV-2 within 14 days of one another were linked by transmission. If these cases represented distinct introductions into the household, we could overestimate expected within-household diversity. Fifth, it is possible, but in our opinion unlikely, that some of the community cases in our analysis actually had an epidemiologic linkage to participants in these households.
Despite the limitations identified in this study, integration of sequence and epidemiologic data can be a powerful approach to studies of SARS-CoV-2 transmission. In settings where there is strong epidemiologic linkage among cases (e.g., known exposure or clear temporal and spatial association), indistinguishable consensus sequences with or without shared iSNV should be confirmatory. In these situations, single mutation differences among consensus sequences in a cluster are not uncommon; mutations can fix along a transmission chain, particularly longer ones over a greater timespan. However, if epidemiologic linkage is less certain, sequence identity can only confirm transmission if the metapopulation is highly sampled and genetically diverse. For example, early in the pandemic when circulating SARS-CoV-2 diversity was low, many inpatients and employees in hospitals were found to share indistinguishable consensus sequences, and even iSNV, without any apparent epidemiologic linkage (22, 27). This contrasts with other studies of hospital outbreaks where the combination of contact tracing and sequence data confirmed suspected transmission chains and identified new ones. We expect that future studies of transmission in households, hospitals, and other congregate settings will benefit from Bayesian methods, which can integrate epidemiologic and sequence data for improved inference (28).
MATERIALS AND METHODS
Cohorts.
The Coronavirus Household Evaluation and Respiratory Testing (C-HEaRT) study enrolled households in Utah (Salt Lake, Weber, Davis, Box Elder, Cache, Tooele, Wasatch, Summit, Utah, and Iron Counties) and New York City (29) during August 2020 through February 2021 and followed them with surveillance for SARS-CoV-2 infection during September 2020 through August 2021. The Communities Organized for the Prevention of Arboviruses (COPA) was expanded to include investigation of the epidemiology of COVID-19, creating the COCOVID study, and recruited households in Ponce, Puerto Rico. For C-HEaRT, household eligibility criteria included the following: ≥1 child aged 0 to 17 years, ≥75% of household members met individual level eligibility (all members if a 2- or 3-person household), one adult member was willing to complete monthly questionnaires, and adult members could communicate in English or Spanish. Individual eligibility criteria included the following: anticipated residence in the household for ≥3 consecutive months and willingness to complete study surveys, weekly symptom assessments, and self-collect respiratory specimens. For COCOVID, household members were eligible if they were aged ≥1 year, slept in the house ≥4 nights per week, had no definite plans to move in the next year, and were willing and able to comply with study requirements.
Ethics statement.
For both the C-HEaRT and COCOVID studies, written informed consent (paper or electronic) was obtained from adults (aged >18 years in C-HEaRT and >20 years in COCOVID). Parents or legal guardians of minor children provided written informed consent on behalf of their children; older children (aged 12 to 17 years in C-HEaRT and 7 to 20 years in COCOVID) also provided assent to study participation. The C-HEaRT study protocol was reviewed and approved by the University of Utah Institutional Review Board (IRB) as the single IRB for all collaborators. The COCOVID study protocol was reviewed and approved by the Ponce Medical School Foundation IRB.
Sample collection and testing.
Participants were asked to self-collect (or for a parent or guardian to collect for children) midturbinate nasal swabs every week, regardless of illness symptoms, and place the swabs in viral transport media. Participants were also contacted by text message or email every week to ascertain if they had COVID-19-like illness (CLI) or any other illness symptoms; they were asked to self-collect an additional midturbinate flocked nasal swab once with onset of CLI symptoms. CLI was defined as 1 or more of the following: fever or feverishness, cough, shortness of breath, sore throat, diarrhea, muscle aches, chills, or change in taste or smell. Respiratory specimens were shipped overnight to a central lab and tested using either the Quidel Lyra SARS-CoV-2 assay or the ThermoFisher Combo kit platform. The assays were approved under emergency use authorization for the diagnosis of SARS-CoV-2 infection prior to use in this study. Test-positive infections in the same household that were first detected by reverse transcription-PCR (RT-PCR) within 14 days of each other (including those detected on the same date) were considered epidemiologically linked and likely to have resulted from within-household transmission.
SARS-CoV-2 genomic sequencing.
SARS-CoV-2 genomic sequencing was attempted on all specimens, with an RT-PCR cycle threshold (CT) of ≤30 on either the nucleocapsid protein 1 or 2 target. SARS-CoV-2 genomes were sequenced as described previously (10). Briefly, RNA was extracted from midturbinate nasal swab specimens with the MagMax MVPII viral nucleic acid isolation kit on a Kingfisher Flex apparatus (ThermoFisher) and reverse transcribed with Lunascript (NEB). We amplified SARS-CoV-2 cDNA in two pools using the ARTIC Network v3 primers and protocol. Amplicon pools were combined in equal volumes for a given sample and purified with magnetic beads. Barcoded sequencing libraries were prepared using the NEBNext ARTIC SARS-CoV-2 library prep kit with magnetic bead size selection. Individual barcoded sample libraries were pooled (up to 96) and sequenced on an Illumina MiSeq (v2 chemistry; 2 × 250 cycles).
Reads were mapped to the Wuhan/Hu-1/2019 reference genome (GenBank MN908947.3) with BWA-MEM (30). We used iVar 1.2.1 (31) to trim ARTIC amplification primer sequences and to determine consensus sequences using bases with >50% frequency and placing a designated unknown base N at positions covered by fewer than 10 reads. Genomes with 29,000 or more unambiguous bases (>97% completeness) were used in downstream analysis. We identified iSNV with iVar using the following parameters: sample with a minimum consensus genome length of 29,000 bases; sample with an average genome sequencing coverage depth of greater than 200 reads per position; iSNV frequency of 5 to 95%; read depth of 400 at iSNV sites with a Phred score of >30; iVar P value of <0.00001. We masked sites commonly affected by sequencing errors in both consensus sequences and iSNV calls (32).
Phylogenetic analysis.
Consensus sequences for each household were placed on the global SARS-CoV-2 phylogenetic tree using UShER (24). The tree versions used were from the week of 14 February 2022 and included over 7.8 million genome sequences from GISAID, GenBank, COG-UK, and CNCB. The level of genomic sampling of the state or territory of each study site (Fig. 1) was estimated with subsampler (10) using case data and GISAID submission data. Subtrees were initially constructed with 30 samples and then reconstructed with additional samples as needed to visualize all genomes from a household in a single subtree (e.g., when samples existed within large clusters of indistinguishable samples). The JSON files for each master tree and subtree are available in Data Set S1 in the supplemental material and can be visualized in the auspice viewer at https://auspice.us/. Trees were annotated and edited in FigTree using the subtree.nwk files generated by UShER.
To determine pairwise distances in community samples, we downloaded sequences from GISAID from 2 weeks before to 2 weeks after the earliest symptom onset date within the specified household for each state. Community sequences and household sequences were aligned to the Wuhan/Hu-1/2019 reference, and sequences with >5% ambiguous sites were removed. We used the R package snp-dists to calculate the number of sites that differed between the household sequence and each set of community sequences.
Data availability.
Primary sequence data and analysis code for the generation of consensus sequences and phylogenetic analysis are available at https://github.com/lauringlab/SARS-CoV-2_Household_diversity. The GISAID identifiers for community sequences can be found by accessing the .json files for each household in https://github.com/lauringlab/SARS-CoV-2_Household_diversity/tree/main/Data/Household_Trees, uploading the .json files to auspice (https://auspice.us/), and visualizing the tips on the trees. Laboratories responsible for submissions are acknowledged in Table S1 in the supplemental material. The consensus genomes that we generated for this study are publicly available at https://github.com/lauringlab/SARS-CoV-2_Household_diversity.
Submitting laboratories for GISAID sequences used in this study (GISAID identifiers can be found in the unedited trees in the Data Set S1). Download Table S1, CSV file, 0.00 MB (3.8KB, csv) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
ACKNOWLEDGMENTS
We thank the participants in the C-HEaRT and COCOVID cohorts and all GISAID submitting laboratories. We acknowledge Anderson Britto for developing and suggesting the subsampler tool used to generate Fig. 1.
We do not have a commercial or other association that might pose a conflict of interest. This work was supported by the Centers for Disease Control and Prevention through a contract to Abt Associates Inc.
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
This work has not been presented previously at a meeting.
Contributor Information
Adam S. Lauring, Email: alauring@med.umich.edu.
Nicole M. Bouvier, Mount Sinai School of Medicine
REFERENCES
- 1.Lauring AS. 2020. Within-host viral diversity: a window into viral evolution. Annu Rev Virol 7:63–81. doi: 10.1146/annurev-virology-010320-061642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kao RR, Haydon DT, Lycett SJ, Murcia PR. 2014. Supersize me: how whole-genome sequencing and big data are transforming epidemiology. Trends Microbiol 22:282–291. doi: 10.1016/j.tim.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lemieux JE, Siddle KJ, Shaw BM, Loreth C, Schaffner SF, Gladden-Young A, Adams G, Fink T, Tomkins-Tinch CH, Krasilnikova LA, DeRuff KC, Rudy M, Bauer MR, Lagerborg KA, Normandin E, Chapman SB, Reilly SK, Anahtar MN, Lin AE, Carter A, Myhrvold C, Kemball ME, Chaluvadi S, Cusick C, Flowers K, Neumann A, Cerrato F, Farhat M, Slater D, Harris JB, Branda JA, Hooper D, Gaeta JM, Baggett TP, O’Connell J, Gnirke A, Lieberman TD, Philippakis A, Burns M, Brown CM, Luban J, Ryan ET, Turbett SE, LaRocque RC, Hanage WP, Gallagher GR, Madoff LC, Smole S, Pierce VM, Rosenberg E, et al. 2021. Phylogenetic analysis of SARS-CoV-2 in Boston highlights the impact of superspreading events. Science 371:eabe3261. doi: 10.1126/science.abe3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siddle KJ, Krasilnikova LA, Moreno GK, Schaffner SF, Vostok J, Fitzgerald NA, Lemieux JE, Barkas N, Loreth C, Specht I, Tomkins-Tinch CH, Paull JS, Schaeffer B, Taylor BP, Loftness B, Johnson H, Schubert PL, Shephard HM, Doucette M, Fink T, Lang AS, Baez S, Beauchamp J, Hennigan S, Buzby E, Ash S, Brown J, Clancy S, Cofsky S, Gagne L, Hall J, Harrington R, Gionet GL, DeRuff KC, Vodzak ME, Adams GC, Dobbins ST, Slack SD, Reilly SK, Anderson LM, Cipicchio MC, DeFelice MT, Grimsby JL, Anderson SE, Blumenstiel BS, Meldrim JC, Rooke HM, Vicente G, Smith NL, Messer KS, et al. 2022. Transmission from vaccinated individuals in a large SARS-CoV-2 Delta variant outbreak. Cell 185:485–492.e10. doi: 10.1016/j.cell.2021.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeller M, Gangavarapu K, Anderson C, Smither AR, Vanchiere JA, Rose R, Snyder DJ, Dudas G, Watts A, Matteson NL, Robles-Sikisaka R, Marshall M, Feehan AK, Sabino-Santos G, Bell-Kareem AR, Hughes LD, Alkuzweny M, Snarski P, Garcia-Diaz J, Scott RS, Melnik LI, Klitting R, McGraw M, Belda-Ferre P, DeHoff P, Sathe S, Marotz C, Grubaugh ND, Nolan DJ, Drouin AC, Genemaras KJ, Chao K, Topol S, Spencer E, Nicholson L, Aigner S, Yeo GW, Farnaes L, Hobbs CA, Laurent LC, Knight R, Hodcroft EB, Khan K, Fusco DN, Cooper VS, Lemey P, Gardner L, Lamers SL, Kamil JP, Garry RF, et al. 2021. Emergence of an early SARS-CoV-2 epidemic in the United States. Cell 184:4939–4952.e15. doi: 10.1016/j.cell.2021.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Worobey M, Pekar J, Larsen BB, Nelson MI, Hill V, Joy JB, Rambaut A, Suchard MA, Wertheim JO, Lemey P. 2020. The emergence of SARS-CoV-2 in Europe and North America. Science 370:564–570. doi: 10.1126/science.abc8169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du Plessis L, McCrone JT, Zarebski AE, Hill V, Ruis C, Gutierrez B, Raghwani J, Ashworth J, Colquhoun R, Connor TR, Faria NR, Jackson B, Loman NJ, O'Toole Á, Nicholls SM, Parag KV, Scher E, Vasylyeva TI, Volz EM, Watts A, Bogoch II, Khan K, Aanensen DM, Kraemer MUG, Rambaut A, Pybus OG, COVID-19 Genomics UK (COG-UK) Consortium . 2021. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science 371:708–712. doi: 10.1126/science.abf2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Candido DS, Claro IM, de Jesus JG, Souza WM, Moreira FRR, Dellicour S, Mellan TA, Du Plessis L, Pereira RHM, Sales FCS, Manuli ER, Thézé J, Almeida L, Menezes MT, Voloch CM, Fumagalli MJ, Coletti TM, da Silva CAM, Ramundo MS, Amorim MR, Hoeltgebaum HH, Mishra S, Gill MS, Carvalho LM, Buss LF, Prete CA, Ashworth J, Nakaya HI, Peixoto PS, Brady OJ, Nicholls SM, Tanuri A, Rossi ÁD, Braga CKV, Gerber AL, de C Guimarães AP, Gaburo N, Alencar CS, Ferreira ACS, Lima CX, Levi JE, Granato C, Ferreira GM, Francisco RS, Granja F, Garcia MT, Moretti ML, Perroud MW, Castiñeiras TMPP, Lazari CS, et al., for the Brazil-UK Centre for Arbovirus Discovery, Diagnosis, Genomics and Epidemiology (CADDE) Genomic Network. 2020. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science 369:1255–1260. doi: 10.1126/science.abd2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viana R, Moyo S, Amoako DG, Tegally H, Scheepers C, Althaus CL, Anyaneji UJ, Bester PA, Boni MF, Chand M, Choga WT, Colquhoun R, Davids M, Deforche K, Doolabh D, Du Plessis L, Engelbrecht S, Everatt J, Giandhari J, Giovanetti M, Hardie D, Hill V, Hsiao N-Y, Iranzadeh A, Ismail A, Joseph C, Joseph R, Koopile L, Kosakovsky Pond SL, Kraemer MUG, Kuate-Lere L, Laguda-Akingba O, Lesetedi-Mafoko O, Lessells RJ, Lockman S, Lucaci AG, Maharaj A, Mahlangu B, Maponga T, Mahlakwane K, Makatini Z, Marais G, Maruapula D, Masupu K, Matshaba M, Mayaphi S, Mbhele N, Mbulawa MB, Mendes A, Mlisana K, et al. 2022. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 603:679–686. doi: 10.1038/s41586-022-04411-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alpert T, Brito AF, Lasek-Nesselquist E, Rothman J, Valesano AL, MacKay MJ, Petrone ME, Breban MI, Watkins AE, Vogels CBF, Kalinich CC, Dellicour S, Russell A, Kelly JP, Shudt M, Plitnick J, Schneider E, Fitzsimmons WJ, Khullar G, Metti J, Dudley JT, Nash M, Beaubier N, Wang J, Liu C, Hui P, Muyombwe A, Downing R, Razeq J, Bart SM, Grills A, Morrison SM, Murphy S, Neal C, Laszlo E, Rennert H, Cushing M, Westblade L, Velu P, Craney A, Cong L, Peaper DR, Landry ML, Cook PW, Fauver JR, Mason CE, Lauring AS, St George K, MacCannell DR, Grubaugh ND, et al. 2021. Early introductions and transmission of SARS-CoV-2 variant B.1.1.7 in the United States. Cell 184:2595–2604.e13. doi: 10.1016/j.cell.2021.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valesano AL, Fitzsimmons WJ, Blair CN, Woods RJ, Gilbert J, Rudnik D, Mortenson L, Friedrich TC, O’Connor DH, MacCannedd DR, Petrie JG, Martin ET, Lauring AS. 2021. SARS-CoV-2 genomic surveillance reveals little spread from a large university campus to the surrounding community. Open Forum Infect Dis 8:ofab518. doi: 10.1093/ofid/ofab518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aggarwal D, Warne B, Jahun AS, Hamilton WL, Fieldman T, Du Plessis L, Hill V, Blane B, Watkins E, Wright E, Hall G, Ludden C, Myers R, Hosmillo M, Chaudhry Y, Pinckert ML, Georgana I, Izuagbe R, Leek D, Nsonwu O, Hughes GJ, Packer S, Page AJ, Metaxaki M, Fuller S, Weale G, Holgate J, Brown CA, Howes R, McFarlane D, Dougan G, Pybus OG, Angelis DD, Maxwell PH, Peacock SJ, Weekes MP, Illingworth C, Harrison EM, Matheson NJ, Goodfellow IG, COVID-19 Genomics UK (COG-UK) Consortium . 2022. Genomic epidemiology of SARS-CoV-2 in a UK university identifies dynamics of transmission. Nat Commun 13:751. doi: 10.1038/s41467-021-27942-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lucey M, Macori G, Mullane N, Sutton-Fitzpatrick U, Gonzalez G, Coughlan S, Purcell A, Fenelon L, Fanning S, Schaffer K. 2021. Whole-genome sequencing to track severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) transmission in nosocomial outbreaks. Clin Infect Dis 72:e727–e735. doi: 10.1093/cid/ciaa1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francis RV, Billam H, Clarke M, Yates C, Tsoleridis T, Berry L, Mahida N, Irving WL, Moore C, Holmes N, Ball JK, Loose M, McClure CP, COVID-19 Genomics UK (COG-UK) Consortium . 2022. The impact of real-time whole-genome sequencing in controlling healthcare-associated SARS-CoV-2 outbreaks. J Infect Dis 225:10–18. doi: 10.1093/infdis/jiab483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meredith LW, Hamilton WL, Warne B, Houldcroft CJ, Hosmillo M, Jahun AS, Curran MD, Parmar S, Caller LG, Caddy SL, Khokhar FA, Yakovleva A, Hall G, Feltwell T, Forrest S, Sridhar S, Weekes MP, Baker S, Brown N, Moore E, Popay A, Roddick I, Reacher M, Gouliouris T, Peacock SJ, Dougan G, Török ME, Goodfellow I. 2020. Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: a prospective genomic surveillance study. Lancet Infect Dis 20:1263–1272. doi: 10.1016/S1473-3099(20)30562-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamilton WL, Fieldman T, Jahun A, Warne B, Illingworth CJR, Jackson C, Blane B, Moore E, Weekes MP, Peacock SJ, De Angelis D, Goodfellow I, Gouliouris T, Török ME, Cambridge COVID-19 Group . 2021. Applying prospective genomic surveillance to support investigation of hospital-onset COVID-19. Lancet Infect Dis 21:916–917. doi: 10.1016/S1473-3099(21)00251-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arons MM, Hatfield KM, Reddy SC, Kimball A, James A, Jacobs JR, Taylor J, Spicer K, Bardossy AC, Oakley LP, Tanwar S, Dyal JW, Harney J, Chisty Z, Bell JM, Methner M, Paul P, Carlson CM, McLaughlin HP, Thornburg N, Tong S, Tamin A, Tao Y, Uehara A, Harcourt J, Clark S, Brostrom-Smith C, Page LC, Kay M, Lewis J, Montgomery P, Stone ND, Clark TA, Honein MA, Duchin JS, Jernigan JA, Public Health–Seattle and King County and CDC COVID-19 Investigation Team . 2020. Presymptomatic SARS-CoV-2 infections and transmission in a skilled nursing facility. N Engl J Med 382:2081–2090. doi: 10.1056/NEJMoa2008457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Addetia A, Crawford KHD, Dingens A, Zhu H, Roychoudhury P, Huang M-L, Jerome KR, Bloom JD, Greninger AL. 2020. Neutralizing antibodies correlate with protection from SARS-CoV-2 in humans during a fishery vessel outbreak with a high attack rate. J Clin Microbiol 58:e02107-20. doi: 10.1128/JCM.02107-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCrone JT, Woods RJ, Martin ET, Malosh RE, Monto AS, Lauring AS. 2018. Stochastic processes constrain the within and between host evolution of influenza virus. Elife 7:e35962. doi: 10.7554/eLife.35962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Worby CJ, Lipsitch M, Hanage WP. 2017. Shared genomic variants: identification of transmission routes using pathogen deep-sequence data. Am J Epidemiol 186:1209–1216. doi: 10.1093/aje/kwx182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braun KM, Moreno GK, Wagner C, Accola MA, Rehrauer WM, Baker DA, Koelle K, O'Connor DH, Bedford T, Friedrich TC, Moncla LH. 2021. Acute SARS-CoV-2 infections harbor limited within-host diversity and transmit via tight transmission bottlenecks. PLoS Pathog 17:e1009849. doi: 10.1371/journal.ppat.1009849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valesano AL, Rumfelt KE, Dimcheff DE, Blair CN, Fitzsimmons WJ, Petrie JG, Martin ET, Lauring AS. 2021. Temporal dynamics of SARS-CoV-2 mutation accumulation within and across infected hosts. PLoS Pathog 17:e1009499. doi: 10.1371/journal.ppat.1009499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tonkin-Hill G, Martincorena I, Amato R, Lawson AR, Gerstung M, Johnston I, Jackson DK, Park N, Lensing SV, Quail MA, Gonçalves S, Ariani C, Spencer Chapman M, Hamilton WL, Meredith LW, Hall G, Jahun AS, Chaudhry Y, Hosmillo M, Pinckert ML, Georgana I, Yakovleva A, Caller LG, Caddy SL, Feltwell T, Khokhar FA, Houldcroft CJ, Curran MD, Parmar S, Alderton A, Nelson R, Harrison EM, Sillitoe J, Bentley SD, Barrett JC, Torok ME, Goodfellow IG, Langford C, Kwiatkowski D, COVID-19 Genomics UK (COG-UK) Consortium . 2021. Patterns of within-host genetic diversity in SARS-CoV-2. Elife 10:e66857. doi: 10.7554/eLife.66857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turakhia Y, Thornlow B, Hinrichs AS, De Maio N, Gozashti L, Lanfear R, Haussler D, Corbett-Detig R. 2021. Ultrafast sample placement on existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nat Genet 53:809–816. doi: 10.1038/s41588-021-00862-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walter KS, Kim E, Verma R, Altamirano J, Leary S, Carrington YJ, Jagannathan P, Singh U, Holubar M, Subramanian A, Khosla C, Maldonado Y, Andrews JR. 2022. Shared within-host SARS-CoV-2 variation in households. medRxiv. 22275279. https://10.1101/2022.05.26.22275279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lythgoe KA, Hall M, Ferretti L, de Cesare M, MacIntyre-Cockett G, Trebes A, Andersson M, Otecko N, Wise EL, Moore N, Lynch J, Kidd S, Cortes N, Mori M, Williams R, Vernet G, Justice A, Green A, Nicholls SM, Ansari MA, Abeler-Dörner L, Moore CE, Peto TEA, Eyre DW, Shaw R, Simmonds P, Buck D, Todd JA, Connor TR, Ashraf S, da Silva Filipe A, Shepherd J, Thomson EC, Bonsall D, Fraser C, Golubchik T, on behalf of the Oxford Virus Sequencing Analysis Group (OVSG) . 2021. SARS-CoV-2 within-host diversity and transmission. Science 372eabg0821. doi: 10.1126/science.abg0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braun KM, Moreno GK, Buys A, Somsen ED, Bobholz M, Accola MA, Anderson L, Rehrauer WM, Baker DA, Safdar N, Lepak AJ, O'Connor DH, Friedrich TC. 2021. Viral sequencing to investigate sources of SARS-CoV-2 infection in US healthcare personnel. Clin Infect Dis 73:e1329–e1336. doi: 10.1093/cid/ciab281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindsey BB, Villabona-Arenas CJ, Campbell F, Keeley AJ, Parker MD, Shah DR, Parsons H, Zhang P, Kakkar N, Gallis M, Foulkes BH, Wolverson P, Louka SF, Christou S, State A, Johnson K, Raza M, Hsu S, Jombart T, Cori A, Evans CM, Partridge DG, Atkins KE, Hué S, de Silva TI, CMMID COVID-19 Working Group . 2022. Characterising within-hospital SARS-CoV-2 transmission events using epidemiological and viral genomic data across two pandemic waves. Nat Commun 13:671. doi: 10.1038/s41467-022-28291-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dawood FS, Porucznik CA, Veguilla V, Stanford JB, Duque J, Rolfes MA, Dixon A, Thind P, Hacker E, Castro MJE, Jeddy Z, Daugherty M, Altunkaynak K, Hunt DR, Kattel U, Meece J, Stockwell MS. 2022. Incidence rates, household infection risk, and clinical characteristics of SARS-CoV-2 infection among children and adults in Utah and New York City, New York. JAMA Pediatr 176:59–67. doi: 10.1001/jamapediatrics.2021.4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 13033997 [q-bio.GN]. https://arXiv.org/abs/1303.3997.
- 31.Grubaugh ND, Gangavarapu K, Quick J, Matteson NL, De Jesus JG, Main BJ, Tan AL, Paul LM, Brackney DE, Grewal S, Gurfield N, Van Rompay KKA, Isern S, Michael SF, Coffey LL, Loman NJ, Andersen KG. 2019. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol 20:8. doi: 10.1186/s13059-018-1618-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Maio N, Walker C, Borges R, Weilguny L, Slodkowicz G, Goldman N. 2020. Masking strategies for SARS-CoV-2 alignments. https://virological.org/t/masking-strategies-for-sars-cov-2-alignments/480/14. Accessed 10 March 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phylogenetic trees of sequences from NY1 household where all participants had indistinguishable consensus sequences. The tips of household sequences are colored cyan, and those from the same state or territory (2-letter abbreviation) are colored magenta. All other tips are colored black. The collection date for each sample is indicated. Genetic distance is represented by the bar and corresponds to one mutation. Download FIG S1, EPS file, 2.2 MB (2.2MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Phylogenetic trees of sequences from NY2 household where all participants had indistinguishable consensus sequences. The tips of household sequences are colored cyan and those from the same state or territory (2-letter abbreviation) are colored magenta. All other tips are colored black. The collection date for each sample is indicated. Genetic distance is represented by the bar and corresponds to one mutation. Download FIG S2, EPS file, 2.6 MB (2.6MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Phylogenetic trees of sequences from the five households in Utah (UT1 and UT7) and Puerto Rico (PR4, PR5, and PR6) where all participants had indistinguishable consensus sequences. The tips of household sequences are colored cyan and those from the same state or territory (2-letter abbreviation) are colored magenta. All other tips are colored black. The collection date for each sample is indicated. Genetic distance is represented by the bar and corresponds to one mutation. Download FIG S3, EPS file, 2.1 MB (2.1MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Phylogenetic trees of sequences from New York households (NY3, NY4, NY5, NY8) where all participants had indistinguishable consensus sequences. The tips of household sequences are colored cyan and those from the same state or territory (2-letter abbreviation) are colored magenta. All other tips are colored black. The collection date for each sample is indicated. Genetic distance is represented by the bar and corresponds to one mutation. Download FIG S4, EPS file, 2.0 MB (2MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Phylogenetic trees of sequences from the seven households where participants had distinct consensus sequences. Each tree is labeled with the household identifier (NY, New York; UT, Utah; PR, Puerto Rico). The tips of household sequences are colored cyan and those from the same state or territory (2-letter abbreviation) are colored magenta. All other tips are colored black. The collection date for each sample is indicated. Genetic distance is represented by the bar and corresponds to one mutation. Download FIG S5, EPS file, 2.5 MB (2.5MB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Submitting laboratories for GISAID sequences used in this study (GISAID identifiers can be found in the unedited trees in the Data Set S1). Download Table S1, CSV file, 0.00 MB (3.8KB, csv) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Data Availability Statement
Primary sequence data and analysis code for the generation of consensus sequences and phylogenetic analysis are available at https://github.com/lauringlab/SARS-CoV-2_Household_diversity. The GISAID identifiers for community sequences can be found by accessing the .json files for each household in https://github.com/lauringlab/SARS-CoV-2_Household_diversity/tree/main/Data/Household_Trees, uploading the .json files to auspice (https://auspice.us/), and visualizing the tips on the trees. Laboratories responsible for submissions are acknowledged in Table S1 in the supplemental material. The consensus genomes that we generated for this study are publicly available at https://github.com/lauringlab/SARS-CoV-2_Household_diversity.
Submitting laboratories for GISAID sequences used in this study (GISAID identifiers can be found in the unedited trees in the Data Set S1). Download Table S1, CSV file, 0.00 MB (3.8KB, csv) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.