

ABSTRACT
Bats have attracted global attention because of their zoonotic association with severe acute respiratory syndrome coronavirus (SARS-CoV) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Previous and ongoing studies have predominantly focused on bat-borne viruses; however, the prevalence or abundance of bat-borne pathogenic bacteria and their potential public health significance have largely been neglected. For the first time, this study used both metataxonomics (16S rRNA marker gene sequencing) and culturomics (traditional culture methods) to systematically evaluate the potential public health significance of bat fecal pathogenic bacteria. To this end, fecal samples were obtained from five bat species across different locations in China, and their microbiota composition was analyzed. The results revealed that the bat microbiome was most commonly dominated by Proteobacteria, while the strictly anaerobic phylum Bacteroidetes occupied 35.3% of the relative abundance in Rousettus spp. and 36.3% in Hipposideros spp., but less than 2.7% in the other three bat species (Taphozous spp., Rhinolophus spp., and Myotis spp.). We detected 480 species-level phylotypes (SLPs) with PacBio sequencing, including 89 known species, 330 potentially new species, and 61 potentially higher taxa. In addition, a total of 325 species were identified by culturomics, and these were classified into 242 named species and 83 potentially novel species. Of note, 32 of the 89 (36.0%) known species revealed by PacBio sequencing were found to be pathogenic bacteria, and 69 of the 242 (28.5%) known species isolated by culturomics were harmful to people, animals, or plants. Additionally, nearly 40 potential novel species which may be potential bacterial pathogens were identified.
IMPORTANCE Bats are one of the most diverse and widely distributed groups of mammals living in close proximity to humans. In recent years, bat-borne viruses and the viral zoonotic diseases associated with bats have been studied in great detail. However, the prevalence and abundance of pathogenic bacteria in bats have been largely ignored. This study used high-throughput sequencing techniques (metataxonomics) in combination with traditional culture methods (culturomics) to analyze the bacterial flora in bat feces from different species of bats in China, revealing that bats are natural hosts of pathogenic bacteria and carry many unknown bacteria. The results of this study can be used as guidance for future investigations of bacterial pathogens in bats.
KEYWORDS: bat, metataxonomics, culturomics, pathogenic species, fecal microbiota
INTRODUCTION
Bats (order Chiroptera) are the only flying mammals and have been identified as a natural reservoir of emerging and reemerging infectious pathogens. They are also referred to as mobile “virus banks” (1). Recently, bats have attracted attention globally because of their zoonotic association with severe acute respiratory syndrome coronavirus (SARS-CoV) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (2). Thus far, prior and current studies have primarily focused on viruses borne by bats, while the prevalence and abundance of pathogenic bacteria in bats and their associated potential public health significance have largely been neglected (3, 4).
Apart from viruses, research has shown that bat feces and intestines contain potential pathogens which can cause serious disease to their hosts or other animals. These potential pathogens include Campylobacter (5, 6), Clostridium (7), Salmonella (8–10), Shigella (11), and Bartonella (12) species. Other bat intestinal and fecal microbes have not shown obvious pathogenicity but may be considered opportunistic pathogens (4), underscoring the importance of exploring the bat microbiota in more detail.
Amplicon sequencing of the 16S rRNA gene (containing nine variable regions, V1 to V9) is the most commonly used method and has proven to be a powerful strategy for the taxonomic classification of bacterial communities (13). The Illumina MiSeq sequencing platform has been used to identify the microbiota at the genus level based on the V3-V4 hypervariable region of the 16S rRNA gene (14). Single-molecule real-time (SMRT) sequencing from Pacific Bioscience (PacBio) has been used to gain a higher taxonomic resolution and identify specific effects (species level) within certain bacterial groups by providing near full-length reads of the 16S rRNA gene (15). Previous studies have indicated that the phyla Firmicutes and Proteobacteria most commonly dominated the bat microbiome, while the phylum Bacteroidetes is relatively rare in the bat microbiome; this is very similar to the microbiome makeup of other flying animals, but distinct from the microbiome compositions of other terrestrial mammals (especially humans and mice) (16, 17). However, no studies have investigated bat fecal microbiota composition through a large-scale culture of bat feces. In addition, the current understanding of the bat fecal microbial community is mainly limited to taxonomic features at the genus level (18).
Herein, we used next-generation sequencing (NGS) for the short-length 16S rRNA gene (V3-V4 region), metataxonomics based on the complete length of the 16S rRNA gene (19), and culturomics (20) to analyze the fecal microbiota of bats collected from five different geographical locations in China to the species level. This study describes the general features of natural bat microbiota at the species level and evaluates the microbiota cytotoxicity and hemolysis in cells while simultaneously analyzing the metabolic features of 16 strains according to their genomes. Numerous pathogenic bacteria with the potential to cause infectious diseases in humans can be found in the fecal microbiota of bats (21).
RESULTS
Bat species metadata.
The five species of bats whose feces were sampled in this study come from different habitats (caves in mountains) in southern China, including Yunnan (N25°09′10″, E102°04′39″; N24°33′58″, E102°25′57″), Guangxi (N22°20′54″, E106°49′20″), Chongqing (N30°02′15″, E107°07′4″), and Hubei (N29°46′56″, E114°18′13″) (Fig. S1 in the supplemental material). The temperature of the caves was between 11.5°C and 32.3°C, with the relative humidity ranging from 27.8% to 89.8%. Additionally, the largest number of bats was found in the caves in the Chuxiong Yi Autonomous Prefecture of Yunnan Province, at an altitude of nearly 2,000 m. The BLAST results for the cytb gene showed that the bats belonged to the species Rousettus leschenaultii, Taphozous perforates, Hipposideros cervinus, Rhinolophus macrotis, and Myotis scotti, with the percentage of cytb gene similarity being between 95.09% and 97.24% (Fig. S1). Furthermore, phylogenetic information deduced from the cytb gene also showed that the five bat species were located in separate clusters (Fig. S2).
The fecal microbiota of bats revealed by metataxonomics and culturomics.
The results of the metataxonomics for full-length 16S rRNA gene analysis revealed that Proteobacteria dominated the fecal microbiota of all five bat species, accounting for 50.2%, 49.1%, 31.1%, 79.08%, and 62.0% relative abundance, respectively. Firmicutes, which was also detected in all fecal samples, accounted for approximately one-third or even one-half of the microbial community in Hipposideros spp. (B3), Myotis spp. (B5), and Taphozous spp. (B2) Interestingly, Bacteroidetes occupied 35.3% of Rousettus spp. (B1) and 36.3% of Hipposideros spp. (B3) and thus can be regarded as dominant. Nevertheless, its proportion in the other three species (B2, B4, and B5) was less than 2.7% (Fig. 1A). According to the Illumina sequencing results, Proteobacteria similarly accounted for the major proportion of microbiota in the bat feces, especially in B2 samples, with an abundance of more than 84.5% (Fig. 1B). More detailed community composition at the family level is shown in Fig. 1C and D. B2 and B5 samples shared a common feature, as they all contained a large proportion of sequences from Enterobacteriaceae and Streptococcaceae. Significantly, Flavobacteriaceae occupied 26.7% in B3 and Yersiniaceae occupied 28.5% in B4 (Rhinolophus spp.) samples according to the PacBio sequencing results (Fig. 1C). These two families have been confirmed to include many pathogenic bacteria, such as Flavobacterium meningosepticum (22), Yersinia pestis, Y. pseudotuberculosis, Y. enterocolitica (23) and so on.
FIG 1.

Profile of bat microbiota at the phylum and family levels. Phylum and family levels using the PacBio sequel (A, C) and Illumina HiSeq (B, D) platforms, respectively. B1, Rousettus spp.; B2, Taphozous spp.; B3, Hipposideros spp.; B4, Rhinolophus spp.; B5, Myotis spp.
Furthermore, metataxonomics analysis with almost full-length 16S rRNA gene sequencing generated by the PacBio system allowed precise identification of the microbiome composition up to the species level, especially for unknown taxa (19). After quality filtering and chimera removal, the USEARCH pipeline was used to obtain a total of 12,600 (accession no. PRJNA781098, Table S2) nearly full-length 16S rRNA gene sequences, which were clustered into 1,337 OTUs at 98.7% identity (the boundary for distinguishing between newly discovered and previously known bacterial species) (24). Next, 480 operational phylogenetic units (OPUs; 135 ± 48.08 per sample, Table S2) were detected based on visual inspection of the final de novo phylogenetic tree that was generated using the LTP128 (25) or SILVAREF 128 NR databases (26) in order to reconstruct the bat fecal microbiota community, which was ultimately achieved by selecting the most frequent representative sequences within each OTU. The fecal microbiota of the five bat species was identified as 89 known species, 330 potentially novel species, and 61 potentially higher taxa (Fig. 2). The 89 known species were affiliated with 65 genera, 39 families, 21 orders, 8 classes, and 4 phyla, and accounted for 35.2% (4,435 sequences) of the total sequences (Fig. 2A). Taxonomically, 81.5% (391/480) of the members of the bat fecal microbiota comprised unknown bacteria species (Fig. 2B and C). The 330 potentially new species were affiliated with 200 genera, 97 families, 50 orders, 25 classes, and 12 phyla, and accounted for 51.9% (6,544 sequences) of the total sequences (Fig. 2B). The remaining 61 potentially higher taxa included 1 SLP at the genus level, 47 SLPs at the family level, 36 SLPs at the order level, and 43 SLPs at the class or phylum level, and accounted for 12.9% (1,621 sequences) of the total sequences (Fig. 2C).
FIG 2.
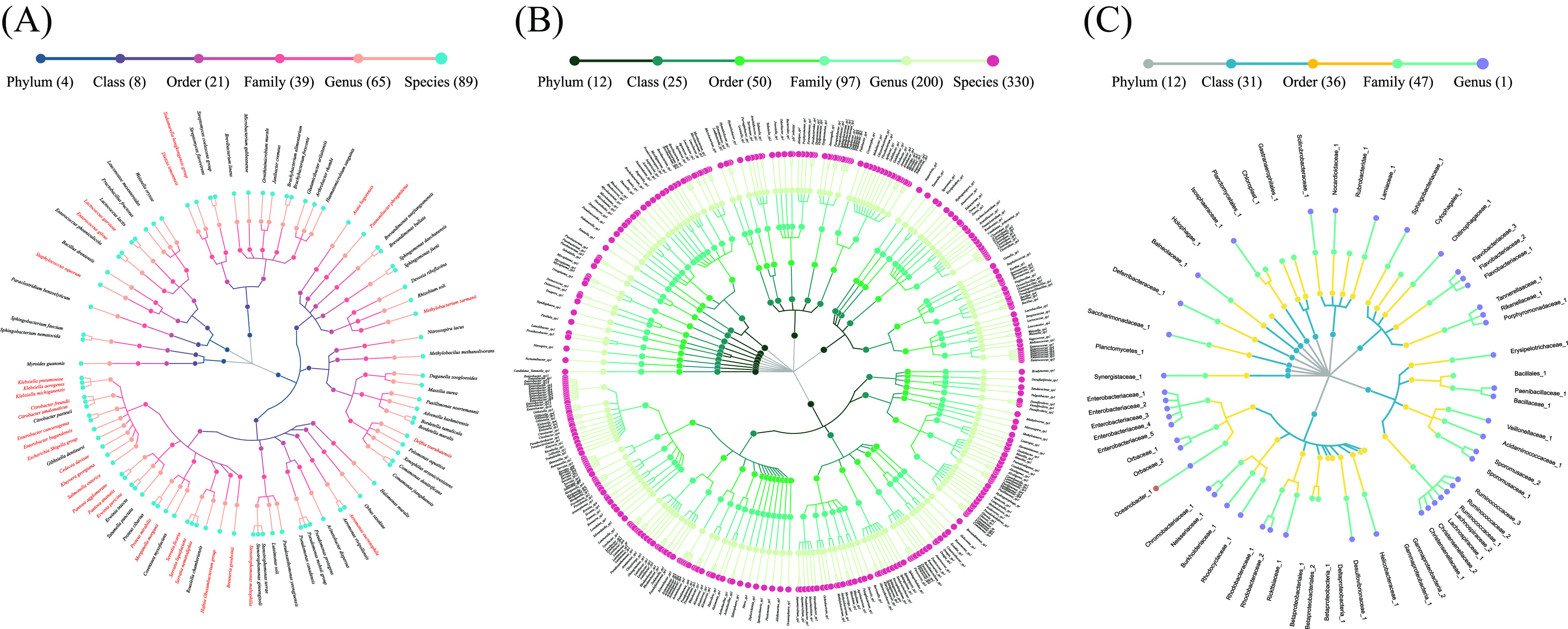
Taxonomic profiles of 480 species-level phylotypes (SLPs) in the gut microbial community of bats. A taxonomic tree of (A) 89 known species (bacteria associated with human, animal, or plant diseases appear in red letters) (27), (B) 330 potentially new species, and (C) 61 potentially higher taxa. Each dot represents an SLP. Descending hierarchical levels are expressed from the inner to the outer rings. The total numbers of SPLs at different hierarchical levels are shown in parentheses.
With the culturomics strategy (Table S1), a total of 1,820 strains were isolated from the fecal samples of the five bat species, and most of the isolates were classified as belonging to the phyla Firmicutes, Actinobacteria, Proteobacteria, and Bacteroidetes (Fig. 3A). On average, approximately 98 species ( 97.80 ± 31.93) were isolated from each sample, of which 5 (Enterococcus faecalis, E. gallinarum, Klebsiella grimontii, Lactococcus lactis, and L. garvieae) were found in all samples (Fig. 3B). A total of 325 species were identified, and these were classified as 242 known and 83 potentially novel species. In addition, 69 species were identified as those which can potentially cause disease in humans, animals, or plants; of these, 46.4% (27) belonged to Firmicutes, 31.9% (22) belonged to Proteobacteria, and only 18.8% (13) and 2.9% (2) belonged to Actinobacteria and Bacteroidetes, respectively (Fig. 3C). At the species level, 89 and 242 known species were identified by metataxonomics and culturomics, respectively, and a total of 22 species were detected by metataxonomics and culturomics methods (Fig. S3).
FIG 3.

Bacterial diversity in bat feces through culturomics. (A) Number of strains at the phylum level. (B) Number of strains at the species level (325, including 242 known species and 83 putative new species). The number in the Venn diagram represents the species in common between different samples. (C) Cladogram depicting the taxonomic classification of all 325 species. The cladogram is color-coded according to phyla and the outside circle of the graph represents the source of the isolated samples, as shown in the boxes. Red stars indicate bacteria associated with human, animal, or plant diseases:32 Firmicutes, 22 Proteobacteria, 13 Actinobacteria, and 2 Bacteroidetes. Potentially novel species which were described in the present study appear in red (yellow background indicates novel bacterial species which have been validly published).
The major microbiota in bat feces.
Of the 480 OPUs detected by metataxonomics, only 4 were shared by all five bat species, including three known species (Enterobacter cancerogenus, Lactococcus lactis, and Citrobacter freundii) and one potentially novel species (Enterococcus sp5). A total of 41 OPUs were detected in three-fifths (60% positive rate) of the bat species, including 15 known species, 22 potentially new species, 3 potentially higher taxa, and 1 unclassified group (Fig. S4A). Therefore, 41 of the 480 (8.5%) OPUs were designated high-frequency microbiota, and these were shared by 60% (3/5) of bat species. Of the top 41 most abundant OPUs (to be consistent with the number of species with a 60% positive rate), only 8 (19.5%) were known species, including Advenella kashmirensis, C. freundii, E. cancerogenus, Gibbsiella dentisursi, Klebsiella aerogenes, K. pneumoniae, L. lactis, and Morganella morganii. Of these, E. cancerogenus and L. lactis were the most prevalent and most abundant species, indicating their potential ecological and biological significance. The four most abundant species (Lactococcus sp1, E. cancerogenus, K. pneumoniae, and L. lactis) accounted for 30.2% of the total sequences obtained (Fig. S4B).
The multivariate correlation analysis between the top 41 highest-frequency bacteria and the top 41 highest-abundance bacteria across with the metadata showed that most of the bacteria had no statistically significant correlation with the temperature, relative humidity, or altitude of the caves (P > 0.05). Interestingly, most of the potentially novel bacteria were positively correlated with feeding habits or the number of bats in the cave (P < 0.05) (Fig. S5).
Pathogenic bacteria revealed by metataxonomics and culturomics.
It must be mentioned that 32 of the 89 (36.0%) known species revealed by metataxonomics were pathogenic species known to cause infection, as evidenced by the published literature (Fig. 2A, Table S3). Moreover, of the 242 known species isolated by means of culturomics, 69 (28.5%) were pathogenic, as also evidenced by the published literatures (Fig. 3C, Table S4).
Among these, Proteobacteria accounted for the highest proportion of all isolates (46.8%, 22/47), followed by Firmicutes (37.6%, 32/85), whereas the lowest proportion of isolates belonged to the phylum Actinobacteria (14.0%, 13/93). Three pathogenic species—namely, E. cancerogenus, C. freundii, and Lactococcus garvieae—were isolated from all five bat species, and the first two species were the top 2 and top 20 most abundant OPUs among all five bat species (Fig. S4B).
Forty-one of the most prevalent OPUs were detected in more than 3 bat species, and 15 (36.6%) were known species. Of these 15 species, 11 (73.3%) were pathogenic, and these included Citrobacter amalonaticus (28), C. freundii, Enterobacter bugandensis, E. cancerogenus, Enterococcus gilvus, K. aerogenes, K. pneumoniae, Methylobacterium zatmanii, Morganella morganii (29), Pannonibacter phragmitetus, and Pantoea ananatis (Fig. S4A). Of the 41 most abundant OPUs detected in bats, 8 (19.5%) were named species, 5 of which (62.5%) were pathogenic bacterial species: C. freundii, E. cancerogenus, K. pneumoniae, M. morganii, and K. aerogenes (Fig. S4B). Interestingly, K. pneumoniae was detected in 4 of the 5 bat species and was among the top three most abundant species in bat samples tested in this study (30). C. freundii seems to be an important bat microbiome species and was found to be in the top 20 most abundant species (31). E. cancerogenus was both the most prevalent and the most abundant fecal bacterium across the five bat species.
Assessment of the pathogenic potential of the cultured potential new species.
Of the 83 potential new species, 46 (55.4%) were affiliated with 41 genera in which known pathogenic species have been reported (Table S3). According to the phylogenetic tree built in this study, based on the 16S rRNA gene obtained with metataxonomics and culturomics, 28 potentially new species (belonging to 19 genera) were in the same branch or group as known pathogenic bacteria (Fig. S6, blue color: metataxonomics). In addition, it is worth noting that 12 potentially new species belonging to 6 genera (Acinetobacter, Bartonella, Brevibacterium, Enterococcus, Hafnia, and Rhodococcus) were isolated by culture; phylogenetically, these species were most closely related to known pathogenic species (Fig. S6, green color: culturomics). In particular, Bartonella sp1. HY038 and B. sp2. HY328 are closely related to B. tamiae, which causes acute and chronic infectious diseases (32).
One strain was selected from each isolated species (n = 325) to test cytotoxicity using hemolytic activity and lactose dehydrogenase (LDH) cytotoxicity assays. The results revealed that 20 (6.2%) species (>20% hemolytic) were toxic to sheep blood, and 26 (8.0%) species (>20% cytotoxic) were toxic to BV2 cells (Fig. S7). The variations in hemolytic and cytotoxicity assays were stable without abnormal values (Fig. S8). There were 10 strains which had no destructive effect on BV2 cells but had greater than 20% hemolytic activity against sheep red blood cells (RBCs; Fig. S7A, double “##”). Twenty-two strains did not show significant hemolytic activity against sheep RBCs but were cytotoxic to BV2 cells (Fig. S7B, single “#”). Moreover, 8 species exhibited >20% toxicity toward sheep RBCs and BV2 cells (Fig. S7, single asterisk [*]). Significantly, there were five potential new strains (Enterococcus sp1. HY326, Enterococcus sp2. HY1045, Microvirga sp2. HY445, Pseudocitrobacter spp. HY512, and Rhodococcus spp. HY359) which may be potentially pathogenic bacterial by being >20% toxic to RBCs or BV2 cells. In particular, Proteus cibarius, Serratia fonticola, Serratia liquefaciens, and Staphylococcus aureus exhibited greater than 50% toxicity (Fig. S9).
Hemolytic and cytotoxic genes were extracted from 16 bacterial genomes based on the gene annotation and functional prediction, and these included offensive virulence factors and hemolysin genes (Table S5). The statistical results showed that hemolysis and cytotoxicity were positively correlated and that they were both proportional to adherence genes (P < 0.005). Additionally, the secretion system genes were also positively correlated with the cytotoxicity of strains (P < 0.005), but there was no statistically significant correlation with hemolysis. Of note, the number of toxin genes and hemolysin genes was not significantly different from the cytotoxicity and hemolytic results (Fig. S10). Moreover, the Clusters of Orthologous Groups (COG) database was used for the classification of the genes in the sequenced genomes (Fig. 4A). The results revealed that the highest numbers of genes contained in these genomes were associated with transcription (COG-K), translation (COG-J), and DNA replication and repair (COG-L) for information storage and processing. For cellular processes and signaling, the genes involved in cell wall/membrane/envelope biogenesis (COG-M) and signal transduction mechanisms (COG-T) were commonly abundant in the sequenced genomes. Based on our analysis of the genes associated with metabolism, we found that the genes in the bacteria in bat feces were involved in the preference of carbon sources composed of amino acids (COG-E) and in carbohydrate transport and metabolism (COG-G), which was aligned with the subsystem feature counts shown in Fig. 4B. We observed that the functions of inorganic ion transport and metabolism (COG-P), energy production and conversion (COG-C), and protein metabolism were also abundant in the bacterial genomes. However, the proportions of genes involved in secondary metabolism, potassium metabolism, and sulfur metabolism were relatively small. Notably, a large quantity of the genes in these bacterial genomes were poorly characterized, and their functions remain to be identified (COG-S) (Fig. 4).
FIG 4.

Distributions of the Clusters of Orthologous Groups (COGs) (A) and subsystem categories (B) in the 16 sequenced genomes isolated from the bat feces. The COGs and subsystems are bubble coded, and bubble size indicates the number of genes.
Isolation origins and oxygen tolerance of the bat fecal microbiota.
Detailed information on the known species detected by metataxonomics or isolated by culturomics was obtained from the LPSN (List of Prokaryotic names with Standing in Nomenclature, https://www.bacterio.net/species) and BacDive (https://bacdive.dsmz.de/). It was found that 71.9% and 71.5% of the aerobic bacteria detected by metataxonomics and culturomics, respectively, were originally from environmental sources, clinical specimens, food, insects, and so on. The facultatively anaerobic or anaerobic species could also be isolated from various sources (Fig. S11).
DISCUSSION
Compared to the number of studies on bat viruses that have drawn much research attention over the past few decades, knowledge about bat bacteria seems to be lagging behind. Because of the wide array of uncultivable microorganisms, studies relying only on traditional culture methods were obviously limited. Similarly, sequencing analysis alone without isolation and culture cannot provide a comprehensive understanding of strains (16). In addition, current understanding of the bat fecal microbial community is mainly limited to taxonomic features at the genus level or higher. For example, Vengust reported that five genera from the phylum Proteobacteria accounted for 50% of sequences (18). In another study, a heatmap constructed from data on 30 major genera showed that Lactococcus and Enterococcus were consistently detected in all samples and belonged to the top three abundances (16), which was similar to the findings of our study; however, we were able to obtain results at the species level (Lactococcus spp., L. lactis, and Enterococcus spp.). Furthermore, a total of 35 bacterial species belonging to different genera were isolated from the bat gut by the conventional plating method as described by Selvin (27), and a variety of pathogenic bacteria or novel species of bacteria could also be isolated from bat feces (33–36); however, it is rare to obtain pure strains from bat feces through large-scale isolation with different media, atmospheres, and temperatures (Table S1). Notably, this is the first study to analyze the bat fecal microbial community at the species level by combining metataxonomics with culturomics.
Proteobacteria was the dominant phylum in the microbiomes of the five bat species (Fig. 1A and Fig. 3C). The presence of Proteobacteria is well recognized as a possible microbial signature of pathogenicity (37). However, strictly anaerobic bacteria from phylum Bacteroidetes were almost absent in the bat species. This phenomenon was similar to that reported in birds (Ovipara) but different from that seen in other mammals (16, 17). One reason for this may be that the intestinal lengths of bats and birds are shorter than those in mice and other mammals to facilitate more efficient powered flight by reducing intestinal content retention times and decreasing mass (38–40). Additionally, shorter intestines also do not provide a suitable anaerobic environment for anaerobic bacteria such as those from the phylum Bacteroidetes (17). In addition, the most likely reason is that the feces were exposed to the air of the caves for some time, resulting in the death of some strictly anaerobic bacteria, reducing the probability of detection and culture. Interestingly, Proteobacteria has also been reported as the most abundant phylum in the airborne microbiome (41), suggesting that there is a positive correlation between the intestinal flora of flying animals and the air to which flighted animals are more constantly exposed as a source of oxygen.
It should be mentioned that the air microbiome in the cave might have the potential to contaminate the fecal microbiota of the bats living in the same cave. However, we mixed all samples in a given cave into one pooled sample. The potential contamination effect of the air microbiome should have been significantly diminished for the cave-specific sample and would have no effect on the samples from other caves. Indeed, the high frequency and abundance of bacteria in bats were independent of temperature, relative humidity, and altitude according to the statistical analysis results (Fig. S5).
Moreover, it is well known that intestinal bacterial diversity is influenced by various factors, including host diet, age, phylogeny, and gut type (42–44). In particular, host diet emerges as a pivotal determinant of gut microbiota community structure and function (45). Based on diet, bats can be divided into frugivorous, nectarivorous, insectivorous, and sanguivorous (46). In our study, five fecal samples were obtained from bats belonging to the genera Rousettus, Taphozous, Hipposideros, Rhinolophus, and Myotis, which can be mainly divided into fruit bats (Rousettus spp.) and insectivorous bats (Taphozous spp., Hipposideros spp., Rhinolophus spp., and Myotis spp.). Metataxonomics revealed that only three known microbial species were shared by all five bat species. Two of these were pathogenic and can cause brain abscesses associated with conditions such as meningitis, osteomyelitis, and pneumonia (47–49). These species were E. cancerogenus and C. freundii, which have been detected or isolated from Dendrolimu kikuchii and pollen (50, 51), Hyalessa maculaticollis, Galleria mellonella (52, 53), and leafhoppers, respectively. The materials in which the two pathogenic species have been detected or isolated can be used as a food source for bats. Interestingly, some potential novel species may be mostly related to feeding habits or bat numbers (Fig. S5), which can explain why we could isolate so many novel bacteria from the feces of Rousettus spp. (Fig. 3C) and how the pathogenic bacteria in bat feces might be obtained from the food they ingest.
Significantly, a novel species of the genus Lactococcus was the most abundant in bats, and L. lactis was a common to all five bat species. L. lactis not only contributes to digestion but also generates a proton motive force during citrate metabolism (54). In addition, L. lactis can regulate intestinal immunity and help suppress pathogen infection (55). It is commonly known that L. lactis is widespread in the food industry and is generally recognized as safe (56). However, it is unexpected that L. lactis may also be an opportunistic bacterium for causing septicemia, cerebellar abscess, peritonitis, or endocarditis (57). Furthermore, L. garvieae can also cause sepsis, endocarditis, etc., and is well known as a potential zoonotic bacterium. This bacterium was detected (positive rate: 40%, Table S2) and cultured (100%, Fig. 3) in our bat fecal samples (58, 59), suggesting that we should increase discussion of Lactococcus spp. as potential pathogens, especially when they are detected in and cultured from wild animals, such as bats.
Some previous studies have shown that the type of hemolysis experiment performed in this study is related to four hemolysin genes (tlyA, tlyB, tlyC, and hlyA) and four putative hemolysin genes (hemolysin, hemolysin activation protein, hemolysin III, and hemolysin channel protein) found in bacteria (60, 61). However, there was no statistically significant correlation in this study between the hemolysin gene and hemolysis results (P > 0.05). Although some isolates caused hemolysis, hemolysin genes were not detected in the isolates, indicating that the hemolytic phenotype may be regulated by different genes and will require more research to be identified.
It is generally believed that the LDH cytotoxicity experiment is related to offensive virulence factors (such as adherence genes, toxin genes, and secretion system genes) in bacteria (62). Consistently, the numbers of adherence genes and secretion system genes were positively correlated with the cytotoxicity of the strains (P < 0.005). Unexpectedly, there was no correlation between the number of toxin genes and the cytotoxic results, which may be due to the lack of expression of relevant genes and warrants further research and elucidation. Significantly, the adherence genes were both proportional to the hemolysis and cytotoxicity results. Studies have shown that the adhesion ability of bacteria plays a critical early role in the pathogenesis of infectious diseases (63). These results indicate that the number of adhesion genes can be used as an area of focus for pathogenicity evaluation.
In summary, our findings indicated that bats carry numerous bacteria with pathogenic importance and that these include a variety of known and unknown pathogenic bacteria. Pathogen shedding by bats would result in the dissemination of those pathogens through feces since bats are widely distributed and prefer to inhabit eaves, woodlands, caves, and other places and therefore can easily come into contact with humans (64). Further investigations are warranted to identify the microbiota of bats in greater detail and their potential implications for human health.
MATERIALS AND METHODS
Fecal sample collection and identification of bat species.
Five genera of bats—Rousettus, Taphozous, Hipposideros, Rhinolophus, and Myotis—were sampled from 2011 to 2013 in the southern part of China (across four provinces and five regions) (Fig. S1). None of the bats showed any apparent clinical signs of illness according to their normal behavior and smooth fur appearance, and no dead bats were found in the caves (16). Collection, storage, and transportation of bat fecal samples and isolation of bacterial species were carried out as described in previous studies (65–67). In brief, fresh fecal samples were collected from bats using an aseptic plastic cloth which was placed under known natural bat roosting sites (usually in caves) during the afternoon. The next morning, the fecal samples were transferred from the plastic cloth into sterile tubes, and immediately placed into a constant temperature box with ice bags. The samples were transported to the laboratory and stored at −80°C until use. Fecal samples obtained from the same bat species were mixed together in the laboratory to minimize variations within the same species (16). Bat species were identified by amplifying the cytochrome b (cytb) gene with the primers L14724ag-F and H15915ag-R (16, 68) and then blasting the sequence within the National Center of Biotechnology Information (NCBI), and the species were determined by finding the species in the database with the highest homology to the sequence in the sample. To further verify the relationships of bat species, we generated a phylogenetic tree based on cytb genes using MEGA X (https://www.megasoftware.net/) with the neighbor-joining (NJ) algorithms (69). The robustness of the tree was evaluated based on 1,000 bootstrap replicates (70).
DNA extraction from bat fecal samples.
Genomic DNA was extracted from the mixed bat fecal samples using the QIAamp Fast DNA Stool minikit (Qiagen, cat. no. 51604) according to the manufacturer’s instructions, and a bead-beating (zirconia/silica beads) and violent mechanical oscillation step was introduced to facilitate bacteria lysis and improve yield. Once DNA extraction was complete, the purity of the extracts was tested using NanoDrop (Thermo Fisher Scientific, USA).
The V3-V4 region of 16S rRNA gene sequencing and data analysis.
Briefly, the variable V3-V4 region of the bacterial 16S rRNA genes was amplified and sequenced on an Illumina MiSeq instrument using paired-end 2 × 300 reads with barcode-indexed universal primers 341F (5′-CCT ACG GGN GGC WGC AG-3′) and 805R (5′-GAC TAC HVG GGT ATC TAA TCC-3′) at Tianjin Biochip Corporation, China (71, 72). The cycling conditions for amplifying V3-V4 were 94°C and 5 min for initial denaturation; then 25 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s; and finally 7 min of final elongation at 72°C. Real-Time Analysis (RTA) software was used to perform image analysis and base calling and to assign base-by-base quality scores (19). The forward and reverse reads were merged using FLASH (73). After joining the paired-end reads, the quality control phase retained sequences with a mean sequence quality score of >20 using FASTX-Toolkit v0.0.14 (http://hannonlab.cshl.edu/fastx_toolkit/). Potential chimeric sequences were screened out using VSEARCH v1.11.1. (74). Next, quality-checked sequence reads were clustered into operational taxonomic units (OTUs) at 98.7% sequence identity beforehand using USEARCH v9.2.64 (24). Finally, all OTUs were classified at the lowest possible taxonomic rank using QIIME based on the Greengenes reference database (https://greengenes.secondgenome.com) with the default parameters (75).
Metataxonomic analysis of fecal microbiota at the species level.
Full-length 16S rRNA high-throughput sequencing was performed on a PacBio RS II platform at Tianjin Biochip Corporation (76). Near full-length 16S rRNA genes were amplified using 10 μL of total DNA as the template and the universal primer set 27F (5′-AGA GTT TGA TCC TGG CTC AG-3′) and 1492R (5′-GNT ACC TTG TTA CGA CTT-3′) with 16-nt barcodes tagged at the 5′ end (100 μL of 2× PCR buffer, 8 μL of forward primer [10 μM], 8 μL of reverse primer [10 μM], 2 μL of KODFX, and 72 μL of H2O). PCR was performed using KODFX DNA polymerase (TOYOBO), and the amplification parameters were as follows: initial denaturation for 2 min at 94°C; 25 cycles of 10 s at 98°C, 30 s at 52°C, and 1 min 30 s at 68°C; and finally a final elongation step of 10 min at 68°C. The primary sequences generated were processed through SMRT Portal (v2.3.0) provided by Pacific Biosciences (www.pacb.com/devnet/). To ensure that the barcoded reads were appropriately assigned to their original samples, a minimum barcode score of 22 was chosen to achieve an accuracy of 99.5%. Data containing ambiguous bases were deleted, primer sequences and adapters from the filtered reads were removed, and sequences outside nucleotide positions 10 to 1,490 were trimmed. The 16S cyclic consensus sequence was analyzed using standard tools in Mot package 2, UCHIME, and Arb (77, 78). Operational phylogenetic unit analyses were performed as previously described (76).
Culturomics analysis.
Bacteria were isolated as described previously (67, 79). In brief, bat fecal samples collected from the same place were mixed and ground. Approximately 1 g of the ground fecal matter was placed in a sterile 1.5-mL EP centrifuge tube and mixed thoroughly with 1 mL of 0.85% normal saline. Then, 150 μL of this solution was transferred with a pipette and spread onto culture plates, including brain heart infusion (BHI) agar, BHI supplemented with 5.0% sheep blood agar, tryptone soy agar (TSA), R2A agar, and thiosulfate citrate bile salt sucrose agar. The uniformly coated medium was placed in an incubator for 7 days. Culture conditions were the result of the free combination of two temperatures (28°C and 37°C) and 3 gas conditions (aerobic, 5.0% CO2, and anaerobic [80.0% N2, 10% CO2, and 10% H2]; Table S1). Next, emerging colonies with different morphologies were selected, purified, and stored at −80°C in BHI broth supplemented with glycerol (20%, vol/vol) for further study.
The genomes were sequenced on an Illumina HiSeq 2000 platform with the respective extracted DNA of the bacterial pellet of the pure strains using the Wizard Genomic DNA purification kit (Promega), according to the manufacturer’s instructions, and assembled by SOAPdenovo (80). Gene calling and annotation were performed using the RAST (Rapid Annotation using Subsystem Technology) server (https://rast.nmpdr.org/) and the SEED viewer framework (81).
Phylogenetic analyses.
The genomic DNA of each isolate was extracted and purified using a Wizard Genomic DNA purification kit (Promega). Pure culture strains were sequenced on the Illumina HiSeq 2000 platform and assembled using Velvet to obtain their respective draft genome sequences (82). The almost-complete 16S rRNA genes of strains were amplified and sequenced using the universal bacterial primers 27F and 1492R (67). The parameters for amplification were as follows: initial denaturation at 94°C for 5 min; followed by 30 cycles at 94°C for 30 s and 55°C for 30 s, and an extension step of 72°C for 90 s; with a final extension at 72°C for 10 min. The amplified sequences of our isolates were determined and compared with their corresponding sequences in the EzBioCloud database (https://www.ezbiocloud.net/identify) and GenBank by basic local alignment search tool (BLAST) search (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to locate taxonomic positions. Phylogenetic trees were constructed using the NJ method (69) using MEGA X software (www.megasoftware.net) with a bootstrap analysis of 1,000 replications (70), and Kimura’s two-parameter method (83) was used to reconstruct NJ trees.
Pathogen detection and virulence potential analysis.
A database containing 16S rRNA genes of well-identified bacterial pathogens was constructed from existing pathogen databases, including the National Microbial Pathogen Database Resource and Risk Group Database from the American Biological Safety Association (https://my.absa.org/Riskgroups). 16S rRNA genes in bacterial pathogens were retrieved from the Ribosomal Database Project (http://rdp.cme.msu.edu/index.jsp). Local BLAST was performed using our sequencing data (including the 16S rRNA gene sequences of the isolated strains) with the database and pathogen database as the query. To ensure maximum reliability, only results with sequence identity higher than 99% were recorded and the corresponding reports were searched in the PubMed database. For potential new bacterial species (16S rRNA gene sequence similarity <98.7%), phylogenetic trees were used to locate their taxonomic positions using the constructed NJ algorithms with the corresponding 16S rRNA gene sequences in the same pathogenic genus or family.
The virulence potential of isolated strains was analyzed by hemolytic analysis and cytotoxicity to cell lines. Three parallel controls were set for each experiment, and the experiment was repeated three times. The variation in assay results was statistically analyzed by the box plots, and the correlation between experimental results and genomic genes was shown with a correlation heatmap (https://www.chiplot.online/). For the bacterial hemolysis test, 8 mL of sterile defibrillated sheep blood was divided into two tubes and centrifuged at 1,000 × g for 10 min. Next, the supernatant was carefully pipetted out and discarded while taking care to avoid picking up red blood cells. Then, 1 mL of red blood cells was mixed with 49 mL of sterile phosphate-buffered saline to prepare 2% sheep red blood cells, and 150 μL of this mixture was then used to obtain 2-fold dilutions of the culture supernatant of experimental strains and the positive strain (Streptococcus suis). The microplate was incubated at 37°C (ambient air plus 5.0% CO2) for 2 h and centrifuged at 800 × g for 10 min. Then, 150 μL of the supernatant was transferred to a new plate and read at 540 nm with a microplate reader (BioTek Instruments, Elx808) (84).
The LDH cytotoxicity assay is a common method for determining cytotoxicity based on measuring the activity of cytoplasmic enzymes released by damaged cells. LDH is rapidly released into the cell culture supernatant when the plasma membrane is damaged, a key feature of cells undergoing apoptosis, necrosis, and other forms of cellular damage (85). Cytotoxicity was assessed using a Cytotox 96 Non-Radioactive Cytotoxicity assay kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. Briefly, BV2 cells were purchased from the American Type Culture Collection (https://webstore.ansi.org/sdo/ATCC). BV2 cells (1 × 106 cells/mL) were cultured for 2 days at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum. Target strains were incubated with BV2 cells for 12 h at 37°C. LDH released from lysed target cells was measured in a microplate reader at 490 nm. To investigate the interaction between target strains and BV2 cells, cells were incubated with DMEM supplemented with 5% fetal bovine serum at 37°C for 12 h before the assay.
The hemolysis and cytotoxicity were calculated using the following formulas: hemolysis (%) = {[OD540 (optical density at 540 nm) sample − OD540 buffer]/[OD540 max − OD540 buffer]} × 100 and cytotoxicity (%) = (OD490 sample − OD490 negative control)/(OD490 max − OD490 buffer) × 100, calculated based on the average of replicates (86). Approximately 20% was regarded as the minimal hemolysis value compared to the positive control for hemolytic activity of FX-compounds (87), and because lactate dehydrogenase release over 20% has been used to evaluate the chemotherapy effect of advanced non-small cell lung cancer patients with platinum-based chemotherapy (88), 20% was listed as the cutoff for evaluating hemolysis and cytotoxicity in this study.
Ethical approval.
The ethical practice was approved by Ethical Committee of the National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention (no. ICDC-2016004).
Data availability.
The data sets generated for this study can be accessed from GenBank under accession no. PRJNA781098. The GenBank/EMBL/DDBJ accession numbers for the 16S rRNA gene sequences of potential novel species (culturomics) can be found in Fig. S6. The cytochrome b (cytb) gene sequences of B1-B5 were deposited into the GenBank database under accession numbers OP076971 to OP076975.
ACKNOWLEDGMENTS
This work was supported by grants from National Key R&D Program of China (2019YFC1200500 and 2019YFC1200505) and Research Units of Discovery of Unknown Bacteria and Function (2018RU010).
J.X. conceived the study. Y.H., Y.S., Q.H., X.L., W.Z., and Z.S. performed the sampling and sequencing. Y.H., Y.S., J.P., S.L., D.J., and J.X. analyzed the data and drafted the manuscript. J.Y. and L.L. supervised the statistical analysis. All authors contributed to the article and approved the submitted version.
We declare that we have no conflicts of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Jianguo Xu, Email: xujianguo@icdc.cn.
Biao He, Changchun Veterinary Research Institute.
REFERENCES
- 1.Shi Z. 2010. Bat and virus. Protein Cell 1:109–114. doi: 10.1007/s13238-010-0029-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu B, Guo H, Zhou P, Shi ZL. 2021. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol 19:141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan CW, Yang X, Anderson DE, Wang LF. 2021. Bat virome research: the past, the present and the future. Curr Opin Virol 49:68–80. doi: 10.1016/j.coviro.2021.04.013. [DOI] [PubMed] [Google Scholar]
- 4.Mühldorfer K. 2013. Bats and bacterial pathogens: a review. Zoonoses Public Health 60:93–103. doi: 10.1111/j.1863-2378.2012.01536.x. [DOI] [PubMed] [Google Scholar]
- 5.Hazeleger WC, Jacobs-Reitsma WF, Lina PHC, de Boer AG, Bosch T, van Hoek A, Beumer RR. 2018. Wild, insectivorous bats might be carriers of Campylobacter spp. PLoS One 13:e0190647. doi: 10.1371/journal.pone.0190647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hatta Y, Omatsu T, Tsuchiaka S, Katayama Y, Taniguchi S, Masangkay JS, Puentespina R, Jr., Eres E, Cosico E, Une Y, Yoshikawa Y, Maeda K, Kyuwa S, Mizutani T. 2016. Detection of Campylobacter jejuni in rectal swab samples from Rousettus amplexicaudatus in the Philippines. J Vet Med Sci 78:1347–1350. doi: 10.1292/jvms.15-0621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hajkova P, Pikula J. 2007. Veterinary treatment of evening bats (Vespertilionidae) in the Czech Republic. Vet Rec 161:139–140. doi: 10.1136/vr.161.4.139. [DOI] [PubMed] [Google Scholar]
- 8.Reyes AWB, Rovira H, Baticados W, Masangkay JS, Ramirez T, Yoshikawa Y. 2011. Polymerase chain reaction assay and conventional isolation of Salmonella spp. from Philippine bats. Acta Scientiae Veterinariae 39. https://www.bvs-vet.org.br/vetindex/periodicos/acta-scientiae-veterinariae/39-(2011)-1/polymerase-chain-reaction-assay-and-conventional-isolation-of-salmonel/. [Google Scholar]
- 9.Moreno G, Lopes CA, Seabra E, Pavan C, Correa A. 1975. Bacteriological study of the intestinal flora of bats (Desmodus rotundus) (author’s transl). Arq Inst Biol (Sao Paulo) 42:229–232. [PubMed] [Google Scholar]
- 10.Adesiyun AA, Stewart-Johnson A, Thompson NN. 2009. Isolation of enteric pathogens from bats in Trinidad. J Wildl Dis 45:952–961. doi: 10.7589/0090-3558-45.4.952. [DOI] [PubMed] [Google Scholar]
- 11.Arata AA, Vaughn JB, Newell KW, Barth RA, Gracian M. 1968. Salmonella and Shigella infections in bats in selected areas of Colombia. Am J Trop Med Hyg 17:92–95. doi: 10.4269/ajtmh.1968.17.92. [DOI] [PubMed] [Google Scholar]
- 12.Nabeshima K, Sato S, Kabeya H, Kato C, Suzuki K, Maruyama S. 2020. Isolation and genetic properties of Bartonella in eastern bent-wing bats (Miniopterus fuliginosus) in Japan. Infect Genet Evol 83:104354. doi: 10.1016/j.meegid.2020.104354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuo Y, Komiya S, Yasumizu Y, Yasuoka Y, Mizushima K, Takagi T, Kryukov K, Fukuda A, Morimoto Y, Naito Y, Okada H, Bono H, Nakagawa S, Hirota K. 2021. Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinION nanopore sequencing confers species-level resolution. BMC Microbiol 21:35. doi: 10.1186/s12866-021-02094-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang N, Luo ZC, Zhang L, Zheng T, Fan P, Tao Y, Ouyang F. 2020. The association between gestational diabetes and microbiota in placenta and cord blood. Front Endocrinol (Lausanne) 11:550319. doi: 10.3389/fendo.2020.550319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brede M, Orton T, Pinior B, Roch FF, Dzieciol M, Zwirzitz B, Wagner M, Breves G, Wetzels SU. 2020. PacBio and Illumina MiSeq amplicon sequencing confirm full recovery of the bacterial community after subacute ruminal acidosis challenge in the RUSITEC system. Front Microbiol 11:1813. doi: 10.3389/fmicb.2020.01813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun DL, Gao YZ, Ge XY, Shi ZL, Zhou NY. 2020. Special features of bat microbiota differ from those of terrestrial mammals. Front Microbiol 11:1040. doi: 10.3389/fmicb.2020.01040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song SJ, Sanders JG, Delsuc F, Metcalf J, Amato K, Taylor MW, Mazel F, Lutz HL, Winker K, Graves GR, Humphrey G, Gilbert JA, Hackett SJ, White KP, Skeen HR, Kurtis SM, Withrow J, Braile T, Miller M, McCracken KG, Maley JM, Ezenwa VO, Williams A, Blanton JM, McKenzie VJ, Knight R. 2020. Comparative analyses of vertebrate gut microbiomes reveal convergence between birds and bats. mBio 11:e02901-19. doi: 10.1128/mBio.02901-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vengust M, Knapic T, Weese JS. 2018. The fecal bacterial microbiota of bats; Slovenia. PLoS One 13:e0196728. doi: 10.1371/journal.pone.0196728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meng X, Lu S, Yang J, Jin D, Wang X, Bai X, Wen Y, Wang Y, Niu L, Ye C, Rossello-Mora R, Xu J. 2017. Metataxonomics reveal vultures as a reservoir for Clostridium perfringens. Emerg Microbes Infect 6:e9. doi: 10.1038/emi.2016.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greub G. 2012. Culturomics: a new approach to study the human microbiome. Clin Microbiol Infect 18:1157–1159. doi: 10.1111/1469-0691.12032. [DOI] [PubMed] [Google Scholar]
- 21.Xu J. 2019. Reverse microbial etiology: a research field for predicting and preventing emerging infectious diseases caused by an unknown microorganism. J Biosaf Biosecur 1:19–21. doi: 10.1016/j.jobb.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujita J, Hata Y, Irino S. 1990. Respiratory infection caused by Flavobacterium meningosepticum. Lancet 335:544. doi: 10.1016/0140-6736(90)90780-9. [DOI] [PubMed] [Google Scholar]
- 23.Bruneteau M, Minka S. 2003. Lipopolysaccharides of bacterial pathogens from the genus Yersinia: a mini-review. Biochimie 85:145–152. doi: 10.1016/s0300-9084(03)00005-1. [DOI] [PubMed] [Google Scholar]
- 24.Edgar RC. 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 25.Yarza P, Ludwig W, Euzeby J, Amann R, Schleifer KH, Glockner FO, Rossello-Mora R. 2010. Update of the All-Species Living Tree Project based on 16S and 23S rRNA sequence analyses. Syst Appl Microbiol 33:291–299. doi: 10.1016/j.syapm.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selvin J, Lanong S, Syiem D, De Mandal S, Kayang H, Kumar NS, Kiran GS. 2019. Culture-dependent and metagenomic analysis of lesser horseshoe bats’ gut microbiome revealing unique bacterial diversity and signatures of potential human pathogens. Microb Pathog 137:103675. doi: 10.1016/j.micpath.2019.103675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warren JR, Farmer JJ, 3rd, Dewhirst FE, Birkhead K, Zembower T, Peterson LR, Sims L, Bhattacharya M. 2000. Outbreak of nosocomial infections due to extended-spectrum beta-lactamase-producing strains of enteric group 137, a new member of the family Enterobacteriaceae closely related to Citrobacter farmeri and Citrobacter amalonaticus. J Clin Microbiol 38:3946–3952. doi: 10.1128/JCM.38.11.3946-3952.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gautam V, Gupta V, Joshi RM, Sawhney G, Duhan S. 2003. Morganella morganii-associated arthritis in a diabetic patient. J Clin Microbiol 41:3451. doi: 10.1128/JCM.41.7.3451.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gharout-Sait A, Touati A, Ahmim M, Brasme L, Guillard T, Agsous A, de Champs C. 2019. Occurrence of carbapenemase-producing Klebsiella pneumoniae in bat guano. Microb Drug Resist 25:1057–1062. doi: 10.1089/mdr.2018.0471. [DOI] [PubMed] [Google Scholar]
- 31.Ewers C, Bethe A, Wieler LH, Guenther S, Stamm I, Kopp PA, Grobbel M. 2011. Companion animals: a relevant source of extended-spectrum beta-lactamase-producing fluoroquinolone-resistant Citrobacter freundii. Int J Antimicrob Agents 37:86–87. doi: 10.1016/j.ijantimicag.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 32.Kosoy M, Morway C, Sheff KW, Bai Y, Colborn J, Chalcraft L, Dowell SF, Peruski LF, Maloney SA, Baggett H, Sutthirattana S, Sidhirat A, Maruyama S, Kabeya H, Chomel BB, Kasten R, Popov V, Robinson J, Kruglov A, Petersen LR. 2008. Bartonella tamiae sp. nov., a newly recognized pathogen isolated from three human patients from Thailand. J Clin Microbiol 46:772–775. doi: 10.1128/JCM.02120-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vandzurova A, Backor P, Javorsky P, Pristas P. 2013. Staphylococcus nepalensis in the guano of bats (Mammalia). Vet Microbiol 164:116–121. doi: 10.1016/j.vetmic.2013.01.043. [DOI] [PubMed] [Google Scholar]
- 34.Qiu Y, Kajihara M, Nakao R, Mulenga E, Harima H, Hang’ombe BM, Eto Y, Changula K, Mwizabi D, Sawa H, Higashi H, Mweene A, Takada A, Simuunza M, Sugimoto C. 2020. Isolation of Candidatus Bartonella rousetti and other bat-associated Bartonellae from bats and their flies in Zambia. Pathogens 9:469. doi: 10.3390/pathogens9060469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Urushadze L, Bai Y, Osikowicz L, McKee C, Sidamonidze K, Putkaradze D, Imnadze P, Kandaurov A, Kuzmin I, Kosoy M. 2017. Prevalence, diversity, and host associations of Bartonella strains in bats from Georgia (Caucasus). PLoS Negl Trop Dis 11:e0005428. doi: 10.1371/journal.pntd.0005428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang Y, Yang J, Lu S, Lai XH, Jin D, Zhou J, Zhang S, Huang Q, Lv X, Zhu W, Pu J, Huang Y, Liu L, Xu J. 2022. Morphological and genomic characteristics of two novel halotolerant actinomycetes, Tomitella gaofuii sp. nov. and Tomitella fengzijianii sp. nov. isolated from bat faeces. Syst Appl Microbiol 45:126294. doi: 10.1016/j.syapm.2022.126294. [DOI] [PubMed] [Google Scholar]
- 37.Rizzatti G, Lopetuso LR, Gibiino G, Binda C, Gasbarrini A. 2017. Proteobacteria: a common factor in human diseases. Biomed Res Int 2017:9351507. doi: 10.1155/2017/9351507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klite PD. 1965. Intestinal bacterial flora and transit time of three neotropical bat species. J Bacteriol 90:375–379. doi: 10.1128/jb.90.2.375-379.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caviedes-Vidal E, McWhorter TJ, Lavin SR, Chediack JG, Tracy CR, Karasov WH. 2007. The digestive adaptation of flying vertebrates: high intestinal paracellular absorption compensates for smaller guts. Proc Natl Acad Sci USA 104:19132–19137. doi: 10.1073/pnas.0703159104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caviedes-Vidal E, Karasov WH, Chediack JG, Fasulo V, Cruz-Neto AP, Otani L. 2008. Paracellular absorption: a bat breaks the mammal paradigm. PLoS One 3:e1425. doi: 10.1371/journal.pone.0001425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caliz J, Triado-Margarit X, Camarero L, Casamayor EO. 2018. A long-term survey unveils strong seasonal patterns in the airborne microbiome coupled to general and regional atmospheric circulations. Proc Natl Acad Sci USA 115:12229–12234. doi: 10.1073/pnas.1812826115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI. 2008. Evolution of mammals and their gut microbes. Science 320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang C, Zhang M, Wang S, Han R, Cao Y, Hua W, Mao Y, Zhang X, Pang X, Wei C, Zhao G, Chen Y, Zhao L. 2010. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J 4:232–241. doi: 10.1038/ismej.2009.112. [DOI] [PubMed] [Google Scholar]
- 44.Enck P, Zimmermann K, Rusch K, Schwiertz A, Klosterhalfen S, Frick JS. 2009. The effects of maturation on the colonic microflora in infancy and childhood. Gastroenterol Res Pract 2009:752401. doi: 10.1155/2009/752401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zmora N, Suez J, Elinav E. 2019. You are what you eat: diet, health and the gut microbiota. Nat Rev Gastroenterol Hepatol 16:35–56. doi: 10.1038/s41575-018-0061-2. [DOI] [PubMed] [Google Scholar]
- 46.Li J, Li L, Jiang H, Yuan L, Zhang L, Ma JE, Zhang X, Cheng M, Chen J. 2018. Fecal bacteriome and mycobiome in bats with diverse diets in South China. Curr Microbiol 75:1352–1361. doi: 10.1007/s00284-018-1530-0. [DOI] [PubMed] [Google Scholar]
- 47.Plakkal N, Soraisham AS, Amin H. 2013. Citrobacter freundii brain abscess in a preterm infant: a case report and literature review. Pediatr Neonatol 54:137–140. doi: 10.1016/j.pedneo.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 48.Garazzino S, Aprato A, Maiello A, Masse A, Biasibetti A, De Rosa FG, Di Perri G. 2005. Osteomyelitis caused by Enterobacter cancerogenus infection following a traumatic injury: case report and review of the literature. J Clin Microbiol 43:1459–1461. doi: 10.1128/JCM.43.3.1459-1461.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Demir T, Baran G, Buyukguclu T, Sezgin FM, Kaymaz H. 2014. Pneumonia due to Enterobacter cancerogenus infection. Folia Microbiol (Praha) 59:527–530. doi: 10.1007/s12223-014-0330-6. [DOI] [PubMed] [Google Scholar]
- 50.Wang JH, Li B, Zhang WX, Xiong Z, Wang LF, Wang HL. 2013. Analysis of the diversity of intestinal aerobic bacteria from the 5th instar larvae of Dendrolimu kikuchii. Chinese J Appl Entomol 50:230–234. [Google Scholar]
- 51.Cheng K, Fang LX, Ge QW, Wang D, He B, Lu JQ, Zhong ZX, Wang XR, Yu Y, Lian XL, Liao XP, Sun J, Liu YH. 2021. Emergence of fosA3 and blaCTX-M-14 in multidrug-resistant Citrobacter freundii isolates from flowers and the retail environment in China. Front Microbiol 12:586504. doi: 10.3389/fmicb.2021.586504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y. 2015. Distribution of endosymbionts in cicadas (Hemiptera: Cicadidae). Northwest A&F University 4:49. https://cdmd.cnki.com.cn/Article/CDMD-10712-1015333896.htm. [Google Scholar]
- 53.Chung JH, Jeong H, Ryu CM. 2018. Complete genome sequences of Enterobacter cancerogenus CR-Eb1 and Enterococcus sp. strain CR-Ec1, isolated from the larval gut of the greater wax moth, Galleria mellonella. Genome Announc 6:e00044-18. doi: 10.1128/genomeA.00044-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hugenholtz J, Perdon L, Abee T. 1993. Growth and energy generation by Lactococcus lactis subsp. lactis biovar diacetylactis during citrate metabolism. Appl Environ Microbiol 59:4216–4222. doi: 10.1128/aem.59.12.4216-4222.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu D, Xia Y, Ge L, Tan B, Chen S. 2021. Effects of Lactococcus lactis on the intestinal functions in weaning piglets. Front Nutr 8:713256. doi: 10.3389/fnut.2021.713256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Casalta E, Montel MC. 2008. Safety assessment of dairy microorganisms: the Lactococcus genus. Int J Food Microbiol 126:271–273. doi: 10.1016/j.ijfoodmicro.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 57.Mofredj A, Bahloul H, Chanut C. 2007. Lactococcus lactis: an opportunistic bacterium? Med Mal Infect 37:200–207. doi: 10.1016/j.medmal.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 58.Sahu KK, Sherif AA, Syed MP, Rajendran A, Mishra AK, Davaro R. 2019. A rare cause of sepsis: Lactococcus garvieae. QJM 112:447–448. doi: 10.1093/qjmed/hcz078. [DOI] [PubMed] [Google Scholar]
- 59.Lin YS, Kweh KH, Koh TH, Lau QC, Abdul Rahman NB. 2020. Genomic analysis of Lactococcus garvieae isolates. Pathology 52:700–707. doi: 10.1016/j.pathol.2020.06.009. [DOI] [PubMed] [Google Scholar]
- 60.Joerling J, Willems H, Ewers C, Herbst W. 2020. Differential expression of hemolysin genes in weakly and strongly hemolytic Brachyspira hyodysenteriae strains. BMC Vet Res 16:169. doi: 10.1186/s12917-020-02385-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huurne AA, Muir S, van Houten M, van der Zeijst BA, Gaastra W, Kusters JG. 1994. Characterization of three putative Serpulina hyodysenteriae hemolysins. Microb Pathog 16:269–282. doi: 10.1006/mpat.1994.1028. [DOI] [PubMed] [Google Scholar]
- 62.Cui J, Hu J, Du X, Yan C, Xue G, Li S, Cui Z, Huang H, Yuan J. 2019. Genomic analysis of putative virulence factors affecting cytotoxicity of Cronobacter. Front Microbiol 10:3104. doi: 10.3389/fmicb.2019.03104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adegbola RA. 1988. Bacterial adhesion and pathogenicity. Afr J Med Med Sci 17:63–69. [PubMed] [Google Scholar]
- 64.Medinas D, Ribeiro V, Marques JT, Silva B, Barbosa AM, Rebelo H, Mira A. 2019. Road effects on bat activity depend on surrounding habitat type. Sci Total Environ 660:340–347. doi: 10.1016/j.scitotenv.2019.01.032. [DOI] [PubMed] [Google Scholar]
- 65.Yang XL, Zhang YZ, Jiang RD, Guo H, Zhang W, Li B, Wang N, Wang L, Waruhiu C, Zhou JH, Li SY, Daszak P, Wang LF, Shi ZL. 2017. Genetically diverse filoviruses in Rousettus and Eonycteris spp. bats, China, 2009 and 2015. Emerg Infect Dis 23:482–486. doi: 10.3201/eid2303.161119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ge X, Li Y, Yang X, Zhang H, Zhou P, Zhang Y, Shi Z. 2012. Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J Virol 86:4620–4630. doi: 10.1128/JVI.06671-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang Y, Wang X, Yang J, Lu S, Lai XH, Jin D, Pu J, Huang Y, Ren Z, Zhu W, Liang H, Zhou P, Shi Z, Xu J. 2020. Apibacter raozihei sp. nov. isolated from bat feces of Hipposideros and Taphozous spp. Int J Syst Evol Microbiol 70:611–617. doi: 10.1099/ijsem.0.003801. [DOI] [PubMed] [Google Scholar]
- 68.Linacre A, Lee JC. 2016. Species determination: the role and use of the cytochrome b gene. Methods Mol Biol 1420:287–296. doi: 10.1007/978-1-4939-3597-0_20. [DOI] [PubMed] [Google Scholar]
- 69.Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 70.Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 71.Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, Ravel J. 2014. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2:6. doi: 10.1186/2049-2618-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glockner FO. 2013. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Magoc T, Salzberg SL. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rognes T, Flouri T, Nichols B, Quince C, Mahe F. 2016. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang J, Pu J, Lu S, Bai X, Wu Y, Jin D, Cheng Y, Zhang G, Zhu W, Luo X, Rossello-Mora R, Xu J. 2020. Species-level analysis of human gut microbiota with metataxonomics. Front Microbiol 11:2029. doi: 10.3389/fmicb.2020.02029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG. 2013. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10:57–59. doi: 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cheng Y, Bai Y, Huang Y, Yang J, Lu S, Jin D, Pu J, Zheng H, Li J, Huang Y, Wang S, Xu J. 2021. Agromyces laixinhei sp. nov. isolated from bat feces in China. J Microbiol 59:467–475. doi: 10.1007/s12275-021-0546-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, Tang J, Wu G, Zhang H, Shi Y, Liu Y, Yu C, Wang B, Lu Y, Han C, Cheung DW, Yiu SM, Peng S, Xiaoqian Z, Liu G, Liao X, Li Y, Yang H, Wang J, Lam TW, Wang J. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R. 2014. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42:D206–14. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 84.Pan X, Ge J, Li M, Wu B, Wang C, Wang J, Feng Y, Yin Z, Zheng F, Cheng G, Sun W, Ji H, Hu D, Shi P, Feng X, Hao X, Dong R, Hu F, Tang J. 2009. The orphan response regulator CovR: a globally negative modulator of virulence in Streptococcus suis serotype 2. J Bacteriol 191:2601–2612. doi: 10.1128/JB.01309-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kumar P, Nagarajan A, Uchil PD. 2018. Analysis of cell viability by the lactate dehydrogenase assay. Cold Spring Harb Protoc 2018:prot095497. doi: 10.1101/pdb.prot095497. [DOI] [PubMed] [Google Scholar]
- 86.Prabakaran M, Rajakannu S, Adhimoolam LK, Gupta M. 2021. In vitro degradation, haemolysis and cytotoxicity study of Mg-0.4Ce/ZnO2 nanocomposites. IET Nanobiotechnol 15:157–163. doi: 10.1049/nbt2.12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roolf C, Saleweski JN, Stein A, Richter A, Maletzki C, Sekora A, Escobar HM, Wu XF, Beller M, Junghanss C. 2019. Novel isoquinolinamine and isoindoloquinazolinone compounds exhibit antiproliferative activity in acute lymphoblastic leukemia cells. Biomol Ther (Seoul) 27:492–501. doi: 10.4062/biomolther.2018.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de Jong C, Deneer VHM, Kelder JC, Ruven H, Egberts TCG, Herder GJM. 2020. Association between serum biomarkers CEA and LDH and response in advanced non-small cell lung cancer patients treated with platinum-based chemotherapy. Thorac Cancer 11:1790–1800. doi: 10.1111/1759-7714.13449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1-S5, Fig. S1-S11. Download spectrum.01802-22-s0001.pdf, PDF file, 3.6 MB (3.6MB, pdf)
Data Availability Statement
The data sets generated for this study can be accessed from GenBank under accession no. PRJNA781098. The GenBank/EMBL/DDBJ accession numbers for the 16S rRNA gene sequences of potential novel species (culturomics) can be found in Fig. S6. The cytochrome b (cytb) gene sequences of B1-B5 were deposited into the GenBank database under accession numbers OP076971 to OP076975.