

Abstract
Objectives:
Systemic sclerosis-associated interstitial lung Disease (SSc-ILD) is the leading cause of death in patients with systemic sclerosis (SSc) with unclear pathogenesis and limited treatment options. Evidence strongly supports an important role for profibrotic, SPP1-expressing macrophages in SSc-ILD. We sought to define the transcriptome and chromatin structural changes of SPP1 SSc-ILD macrophages, so as to better understand their role in promoting fibrosis and to identify transcription factors associated with open chromatin driving their altered phenotype.
Methods:
We performed single cell RNA-sequencing on 11 explanted SSc-ILD and healthy control lung samples, as well as single cell ATAC-sequencing on five lung samples to define altered chromatin accessibility of SPP1 macrophages. We predicted transcription factors regulating SPP1 macrophages using SCENIC and determined transcription factor binding sites associated with global alterations in SPP1 chromatin accessibility using Signac/Seurat.
Results:
We identified distinct macrophages subpopulations by scRNA-seq analysis in normal and SSc-ILD lungs and determined the occurring gene expression changes during the change of normal macrophages into SPP1 macrophages. Analysis of open chromatin validated SCENIC predictions, indicating that MITF, TFEB, ATF6, SREBF1, BHLHE40, KLF6 ETV5 and/or members of the AP-1 family of transcription factors regulate SPP1 macrophage differentiation.
Conclusions:
Our results shed light on the underlying changes in chromatin structure and transcription factor regulation of profibrotic SPP1 macrophages in SSc-ILD. Similar alterations in SPP1 macrophages may underpin fibrosis in other organs involved in SSc and point to novel targets for the treatment of SSc-ILD, targeting profibrotic macrophages.
INTRODUCTION
Systemic sclerosis (SSc) is a multisystem, autoimmune, fibrotic disease of unknown etiology with life-threatening fibrotic complications, such as SSc-associated interstitial lung disease (SSc-ILD) (1, 2). Despite advances in new SSc-ILD treatment options, such as nintedanib (3), tocilizumab (4, 5), and myeloablative autologous stem-cell transplantation (6), SSc-ILD remains difficult to treat. The consequences on survival and quality of life are important since approximately 50% of SSc patients develop SSc-ILD (2) and 33% of SSc patients die of SSc-ILD fibrotic complications (7).
Increasing evidence supports important roles for macrophages in SSc-ILD. Specifically, macrophage associated gene expression in lung biopsies correlates with progressive SSc-ILD, worsening lung fibrosis on high-resolution computed tomography (HRCT), and reduced performance on pulmonary function tests (PFTs) (8). SSc-ILD lungs show strongly upregulated expression of chemokine C-C motif ligand 18 (CCL18), which is expressed primarily by macrophages (9), and has been found to play a key role in pulmonary fibrotic disease by attracting immune cells and stimulating collagen overproduction (10, 11). Strikingly, elevated CCL18 in SSc sera are completely blocked by the anti-IL-6 receptor antagonist tocilizumab (4, 5), indicating that serum CCL18 is a biomarker for IL-6 activity in SSc. The distinct gene expression signature of SSc-ILD compared to healthy lungs also shows upregulation of macrophage marker genes, AIF1, CD163, MS4A4A, as well as secreted phosphoprotein I (SPP1) (8, 12), a marker of profibrotic macrophages in idiopathic pulmonary fibrosis (IPF) (13, 14). Lower lung lobes of IPF patients, where fibrosis is typically more advanced, demonstrated increased number of macrophages expressing SPP1 compared to upper IPF lung lobes and controls (14). Additionally, in murine bleomycin-induced lung fibrosis, intravenous administration of Ly6Chi inflammatory monocyte progenitor cells facilitates the progression of fibrosis through increase of alternatively activated lung macrophages, whereas macrophage depletion during fibrinogenesis is associated with less fibrosis (15).
In order to study the pathophysiology of SSc-ILD and define the role of profibrotic macrophages, we analyzed gene expression and altered chromatin structure of pulmonary macrophages from control and SSc-ILD lungs using single cell RNA sequencing (scRNA-seq) and single cell Assay for Transposase Accessible Chromatin sequencing (scATAC-seq). The conceptual basis for our work is that chromatin structure indicates the underlying transcriptional regulation. Chromatin structural changes or remodeling, where chromatin is open or accessible to transcription factors, is associated with regulated gene expression, affecting the transcriptome and consequently the cell phenotype (16). We sought to identify key transcription factors driving the altered transcriptome and differentiation of profibrotic lung macrophages, as well as to gain insight on signals, including cytokines, contributing to profibrotic macrophage differentiation.
METHODS
Single cell RNA-seq using 3’ v3 and 5’ v1 chemistries (10X Genomics) was performed on 11 explanted lung tissue samples, including both SSc-ILD and healthy control lungs (Table S1). Single Cell Reagents (10X Genomics, 3’ v3 or 5’ v1 chemistries) were used for the library preparation samples after digestion as described by Valenzi et al. (13). Library quantification and sequencing of the scRNA-seq cDNA libraries was carried out by the UPMC Genome Center, on an Illumina NextSeq-500 (14).
Healthy control and SSc-ILD lung samples were also analyzed for scATAC-seq (10X Genomics, Single Cell ATAC Reagent Kits, v1 Chemistry). Data were analyzed using the R packages Seurat v 4.0 (17) and Signac v 1.0.9004 (18), Loupe Browser 3.1.1 (10X Genomics) and Single-cell regulatory network inference and clustering (SCENIC) (19). Detailed methods are described in supplemental detailed methods file.
RESULTS
Profibrotic macrophages show discrete changes in transcriptomes
We analyzed lungs from six healthy control (HC) and five SSc-ILD patients by scRNA-seq (Table S1). We carried out bioinformatics analyses of samples in two groups because two different scRNA-seq chemistries were utilized, with different numbers of SSc-ILD versus healthy control lungs analyzed by each chemistry. Given the uneven number of sample types, batch correction between chemistries was problematic and might have obscured changes in macrophage transcriptomes associated with SSc-ILD. We first describe five lung samples including one healthy control and four SSc-ILD lungs, in which single cell cDNA libraries were prepared using 3’ v3 Chemistry (10X Genomics). After dimensional reduction and visualization by Uniform Manifold Approximation and Projection (UMAP) (20), we identified cell types in each cluster as we have described previously (13), using characteristic gene markers for each cell population (Figure 1A, Figures S1–S3). Myeloid cells were identified by markers: CD163, AIF1 and MARCO, and three macrophage subpopulations were identified by highly differentially expressed gene markers, similar to those seen in patients with idiopathic pulmonary fibrosis (13): SPP1 Mφ, Fatty Acid-Binding Protein 4 Mφ (FABP4, hereafter referred to as FABP4 Mφ) and Ficolin-1 Mφ (FCN1, hereafter referred to as FCN1 Mφ). The vast majority of the SPP1 Mφ cluster consisted of macrophages from the SSc-ILD patients, with few SPP1 Mφ from control lungs (Figures 1B, 1C). Notably, SPP1 was upregulated in the SSc macrophages compared to normal macrophages within the same cluster (Figures S1–S4). Dendritic cells (DCs) were rare and clustered together with the FCN1-expressing Mφ (14), (Figure 1, Figure S4).
Figure 1.
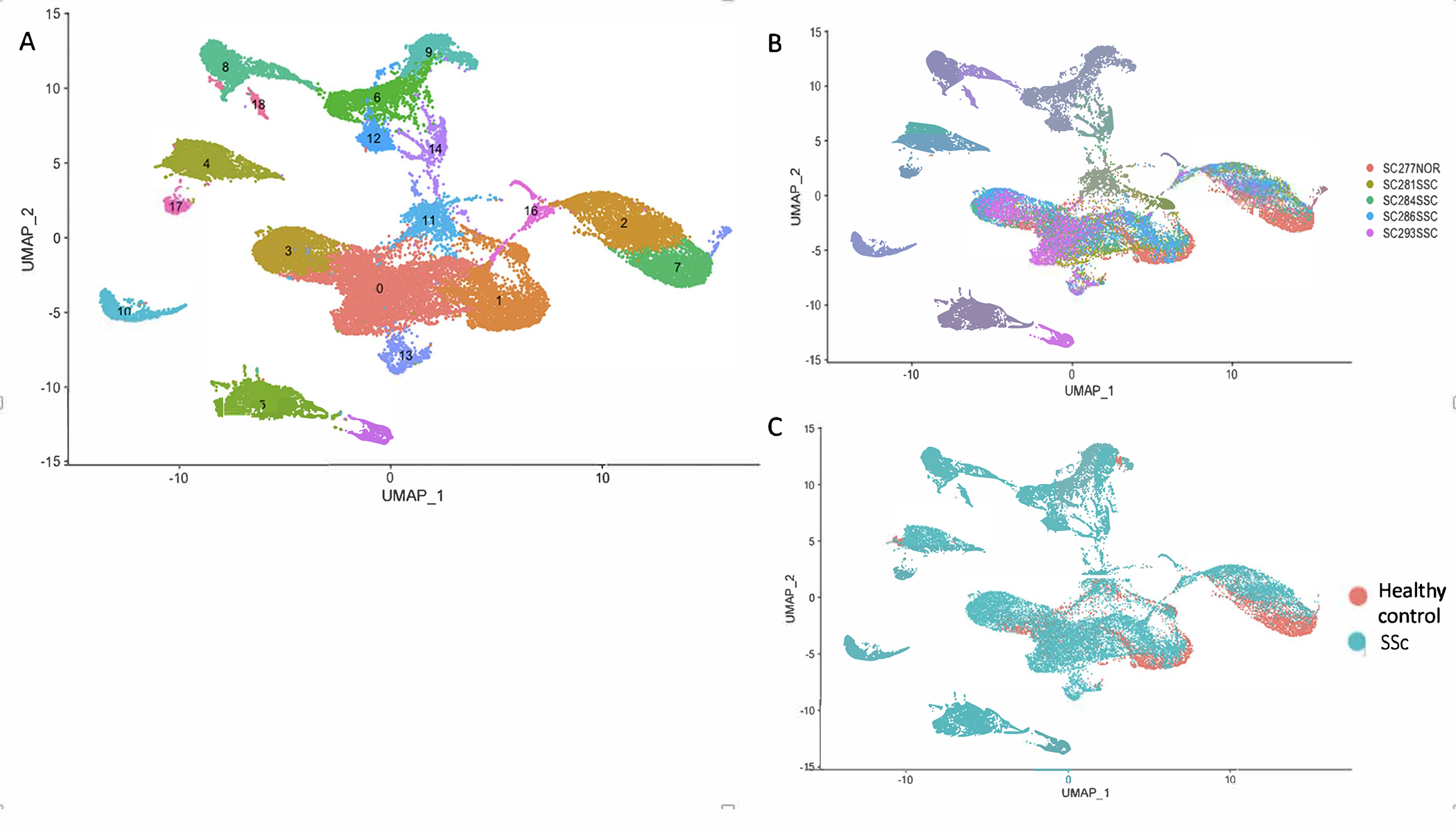
Single-cell RNA-sequencing analysis of 5 lung samples in 3’ v3 chemistry. (A) Visualization of clustering by UMAP per cell type: 0-SPP1 macrophages, 1-FCN1 macrophages and Dendritic cells, 2-T cells, 3-FABP4 macrophages, 4-Endothelial cells, 5-Fibroblasts, 6-Goblet cells and Alveolar Type 1 cells, 7-NK cells, 8-Ciliated cells and Goblet cells, 9-Alveolar Type 2 cells, 10-Mast cells, 11-Low quality cells, 12-Basal cells, 13-Proliferating cells, 14-Goblet cells, 15-Pericytes and Smooth muscle cells, 16-B cells, 17-Lymphatic endothelial cells, 18- Ciliated cells. Macrophage subpopulations, dendritic cells and proliferating cells are located along the center of the UMAPs. (B) Visualization of clustering by UMAP per individual identity. (C) Visualization of clustering by UMAP per health status. SPP1 macrophages cluster 0 is formed primarily of macrophages from the SSc-ILD patients.
Comparing SPP1 Mφs to the FABP4 and FCN1 Mφs revealed multiple differentiatially expressed genes (Table S1), including SPP1, Legumain (LGMN), seen co-regulated in our previous analyses (14), and Phospholipase A2 Group VII (PLA2G7), a target of transcriptional regulation discussed below (Figure S4). Differentially expressed genes associated with each macrophage subpopulation compared to the other two subpopulations, were determined and queried for enriched gene ontology pathways. Upregulated genes in SPP1 Mφ compared to FABP4 Mφ and FCN1 Mφs revealed enrichment of genes implicated in lipid metabolism and myeloid cell activation (Figures S5, S6, Table S2). Lipid metabolism plays a key role in macrophage activation by fatty acid synthesis to be utilized as precursors for inflammatory mediators synthesis with significant effect on the course of many metabolic diseases (21, 22). Upregulated differentially expressed genes in FABP4 Mφ compared to SPP1 Mφ and FCN1 Mφs demonstrated enrichment for negative regulation of macrophage differentiation, regulation of cholesterol storage and myeloid leukocyte activation pathways (23–25), (Table S3).
Lung myeloid cell subpopulations
We reclustered the myeloid cells from these samples, including macrophages, dendritic cells and proliferating Mφ guided by Clustree (Figure S7). This subclustering segregated most cells from the SSc and healthy samples into different clusters (Figure 2). The largest cluster, SSc-ILD SPP1 Mφ, selectively expressed MER proto-oncogene, Tyrosine Kinase (MERTK) and LGMN; SSc FABP4 Mφ selectively expressed Inhibin Subunit Beta A (INHBA); while HC and SSc FCN1 Mφ selectively expressed Interleukin 1 Beta (IL1B) as well as Sialic Acid Binding Ig Like Lectin 10 (SIGLEC10) gene markers (Figure 2). These and other marker genes defined these macrophage subpopulations (Figure S4, Table S4).
Figure 2.
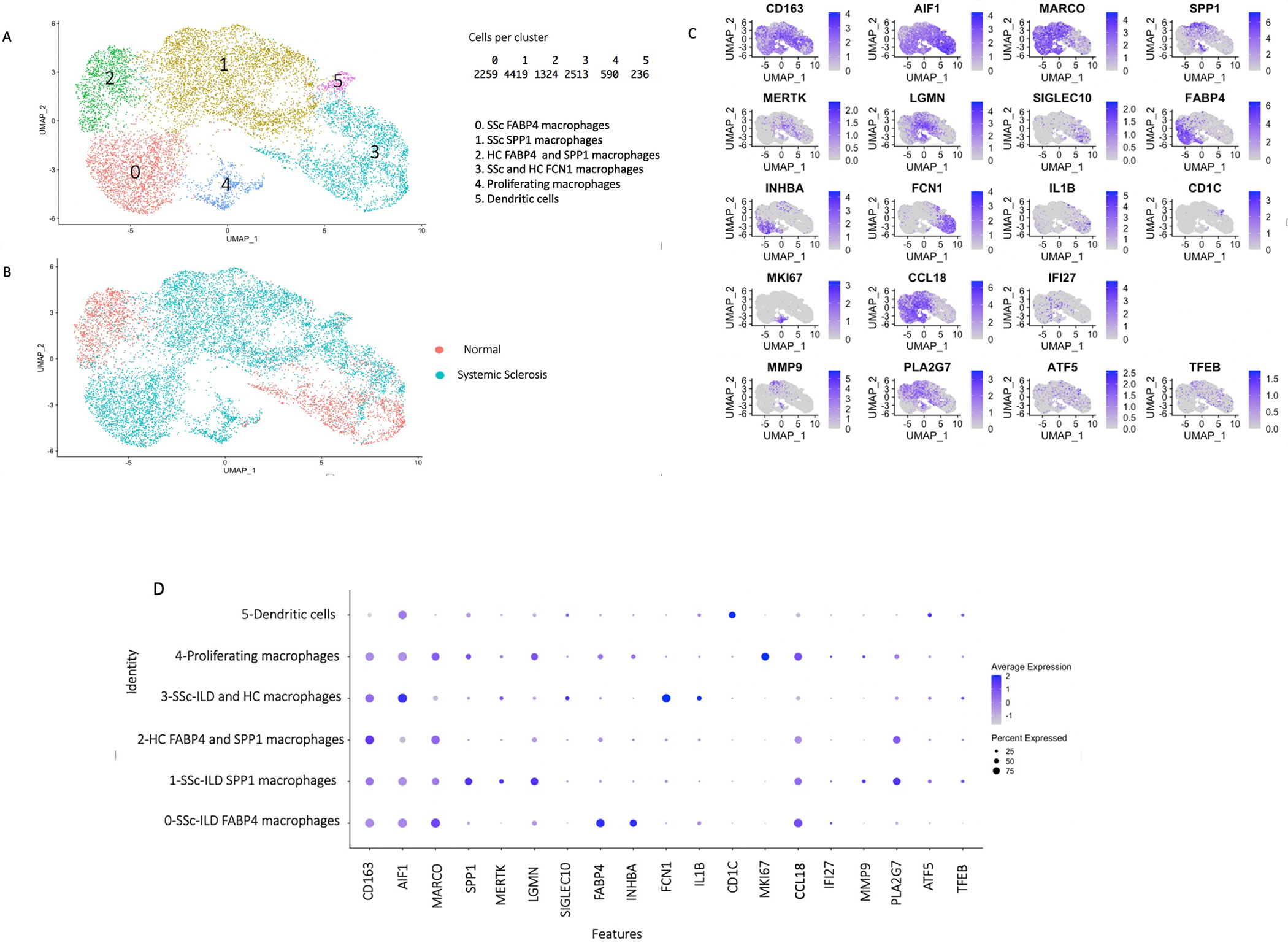
Single-cell RNA-sequencing analysis of macrophage subpopulations, dendritic cells, proliferating Mφ from 5 lung samples in 3’ v3 chemistry, 1 healthy control and 4 SSc-ILD lung samples. (A) Visualization of clustering by UMAP per cell type. (B) Visualization of clustering by UMAP per health status. (C) Visualization of gene expression by FeaturePlot of macrophage subpopulations gene markers, dendritic cells, proliferating Mφ and other genes expression during analysis of only macrophages, dendritic cells, proliferating Mφ. (D) Visualization of gene expression by DotPlot of macrophages, dendritic cells, proliferating Mφ from 1 healthy control and 4 SSc-ILD lung samples in scRNA-seq 3’ v3 Chemistry. DotPlot shows upregulation of SPP1, MERTK, LGMN genes in SSc-ILD SPP1 Mφ and proliferating Mφ. MMP9 and PLA2G7 genes markers are upregulated in SSc-ILD SPP1 Mφ compared to all other macrophages.
These results mostly agree with our previous work examining macrophages in IPF (14). Interferon Alpha Inducible Protein 27 (IFI27), an interferon regulated gene, and CCL18, a strong serum pharmacodynamic biomarker for tocilizumab (4, 5), were both more highly expressed in SSc SPP1- and FABP4-Mφ and the proliferating Mφ clusters compared to HC SPP1- and FABP4 Mφ (Figure 2). Although IFI27 was also expressed by other lung cell populations, CCL18 was almost exclusively expressed by lung macrophages. The proliferating Mφ cluster included both SPP1- and FABP4-Mφ, as this cluster showed a group of cells expressing SPP1 (Figure 2C) and other markers of SPP1 cells (MERTK and LGMN, Figure 2D), and another group of cells expressing FABP4 (Figure 2C) and other markers of FABP4 cells (INHBA, Figure 2D). These proliferating macrophages originated almost exclusively from the SSc-ILD patient lungs (Figure 2B), indicating that proliferating Mφ potentially differentiate into profibrotic SPP1 Mφ phenotype. The dendritic cells, also originated almost entirely from the SSc-ILD lungs, clustered into a discrete cluster expressing CD1C (Figure 2C), and other markers of cDC2 dendritic cells (not shown), (26).
Validation of macrophage subsets in SSc-ILD 5’ v1 Chemistry
We extended the results above by analyzing scRNA-seq data from an additional five HC and one SSc-ILD lungs, in which single cell cDNA libraries were prepared using 5’ v1 chemistry (10X Genomics). Myeloid subpopulations reflected those found in the analogous 3’ v3 subclustering (Figure 3, Figures S8–S11). Of note, most of the macrophages in the SPP1 Mφ cluster originated from the SSc-ILD patient, consistent with the 3’ v3 chemistry clustering. The proliferating Mφ population was relatively small as there was only one SSc-ILD lung and five HC lungs, however both SPP1 and FABP4 expression was noted (Figure S9). The proliferating cell cluster also included some control natural killer (NK) cells.
Figure 3.
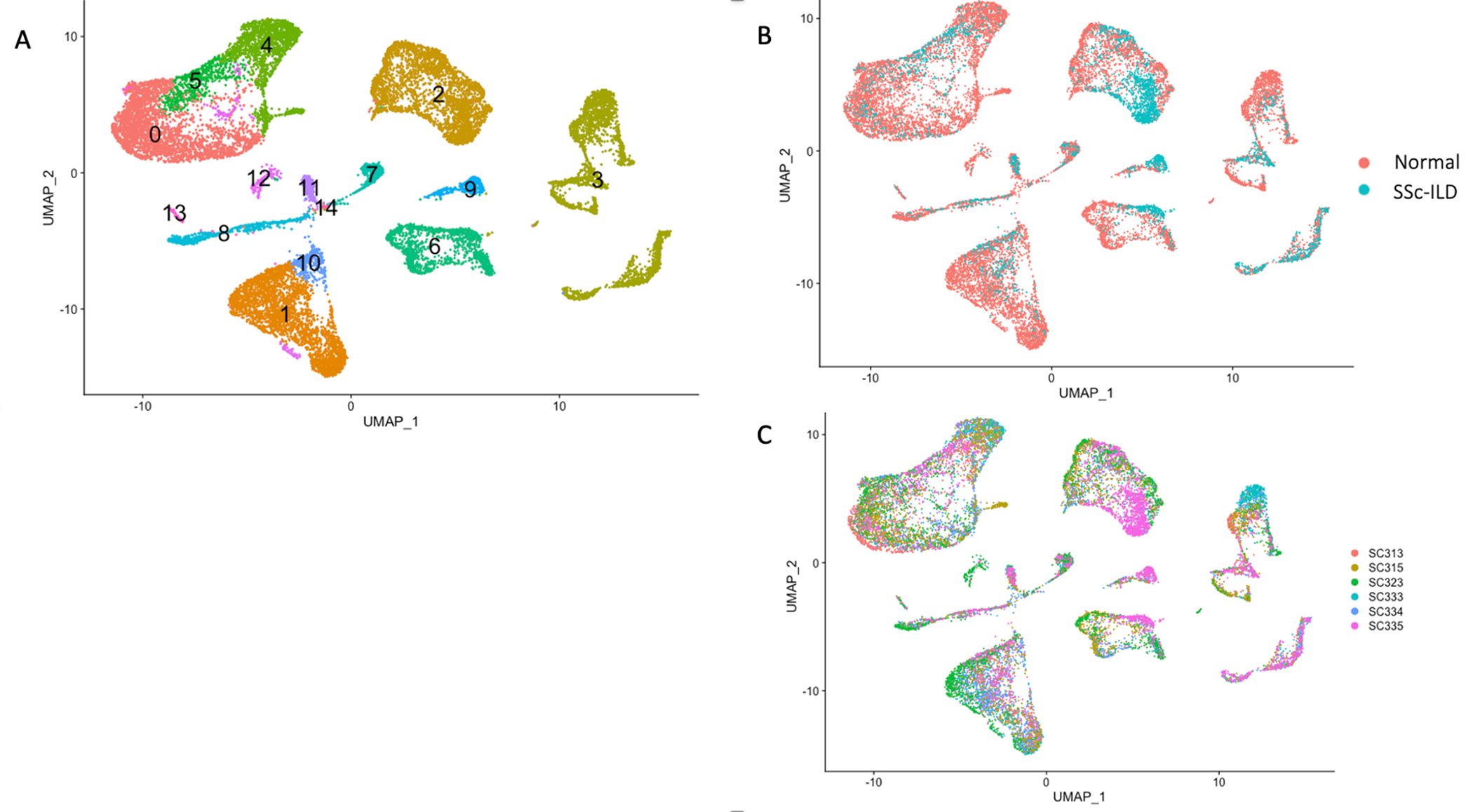
Single-cell RNA-sequencing analysis of 6 lung samples in 5’ v1 chemistry, including 1 SSc-ILD and 5 healthy control lungs. (A) Visualization of clustering by UMAP per cell type. Macrophage subpopulations, dendritic cells and proliferating cells are located along the left upper part of the UMAPs. Cell clusters were identified as follows: 0-FCN1 macrophages and Dendritic cells, 1-T cells and NK cells, 2-Endothelial cells, 3-Epithelial cells, 4-FABP4 macrophages, 5-SPP1 macrophages, 6-Fibroblasts, 7-Lymphatic endothelial cells, 8-B cells, 9-Pericytes and Smooth muscle cells, 10-Mitochondrial related genes, 11-Mast cells, 12-Proliferating cells, 13-Dendritic cells, 14-Hemoglobin related cells. (B) Visualization of clustering by UMAP per health status. SPP1 macrophages cluster 5 is formed primarily of macrophages from the SSc-ILD patient SC335. (C) Visualization of clustering by UMAP per individual identity.
CCL18 (4, 5), IFI27, Matrix Metallopeptidase 9 (MMP9), and PLA2G7 (27) were upregulated in the SPP1 Mφ from the SSc-ILD lungs compared to SPP1 Mφ from HC lungs (Figure S9). Notably, IFI27 gene expression in the serum of SSc patients is associated with digital ulcers (28). MMP9 is also elevated in the serum of SSc patients and correlates with the Modified Rodnan Skin Score (MRSS) (29). In addition to SPP1, PLA2G7, discussed further below, differentiated SPP1 Mφ compared to FABP4 and FCN1 Mφ (Figures S12, S13).
Combined batch corrected datasets
In order to examine a larger dataset including both SSc-ILD and controls, we Harmony batch corrected and combined our two datasets (Figures S14–S20). Similar to our previous analyses, we identified the same cell types including myeloid cells after dimensional reduction and visualization by UMAP. Consistent with our previous results, the SPP1 Mφ cluster was formed mainly by Mφ from SSc-ILD patients. We subsequently reclustered the myeloid cells. As above, SPP1 macrophages, expressing also gene markers MERTK, LGMN, CCL18, MMP9, TFEB, PLA2G7, were almost exclusively from SSc-ILD samples. Thus, the results of this combined analysis are consistent with analyses of the uncombined datasets.
Pathway analysis of upstream regulators of SPP1 Mφ.
To addresss upstream drivers of macrophage differentiation in SSc-ILD, we analyzed genes differentially upregulated in SPP-1 Mφ using Ingenuity Pathway Analysis. This analysis supported several different mediators as potential upstream drivers of the SSc-ILD phenotype, including LPS and other TLR activators, IL-6 and TGF-β, all of which have been implicated in previous studies, as well as IL-33 and other cytokines (Table S5)
Skin microarray data suggest similar pathologic changes occur in other SSc organs
Our data examining the epigenetics of the profibrotic macrophages in SSc-ILD may also be relevant for skin fibrosis and other target organs in SSc. IL-6 inhibition with tocilizumab in the faSScinate clinical trial, while just missing a statistically significant p-value in both phase 2 and phase 3 trials, downregulated genes associated with M2 macrophages (30), and decreased expression of TGF-β regulated genes in explant fibroblast cultures (31). In order to better understand whether SPP1 macrophages contribute to fibrosis in other SSc organs, we examined gene expression in SSc skin. SPP1 and CCL18 expression was markedly increased in bulk SSc skin RNA. Previously described skin scRNA-seq data indicated that the expression of these genes comes primarily from myeloid cells in the skin (Figures S21–S23).
Prediction of transcription factors ATF5 and TFEB regulating SPP1 macrophage differentiation
Single-cell regulatory network inference and clustering (SCENIC) provides a bioinformatics method for reconstructing gene regulatory networks, as well as identifying cell states, providing insights into the transcription factors leading to cellular heterogeneity (19). SCENIC identifies regulons, a transcription factor and its associated downstream target genes, by examining coregulated gene expression in a single cell dataset and using RcisTarget to confirm transcription factor binding sites in downstream target genes. We used SCENIC to predict regulons and transcription factors regulating SPP1 Mφ differentiation, comparing the transcriptomes of SSc-ILD SPP1 Mφ to all FABP4 Mφ, all FCN1 Mφ and proliferating Mφ. These cells were then clustered by their regulon activity score, and labeled according to the cell population, patient individual identity and health status (Figure 4A). Notably, SPP1 Mφ formed the biggest cluster, composed almost entirely of cells from SSc-ILD lungs and included cells from each SSc-ILD sample. SPP1 Mφ were further examined by SCENIC for regulons predicited to selectively regulate the SSc SPP1 Mφ transcriptome (Figure 4B–D). Multiple regulons (Figure 4D) and their associated transcription factors (Figure 4C) were selectively upregulated in SPP1 Mφ compared to the other macrophage populations. Most of these regulons were also upregulated in the proliferating macrophage population, of which most of the cells expressed SPP1 and are thus likely contributing to the SSc SPP1 Mφ population. The regulons for Activating transcription factor 5 (ATF5) and Transcription factor EB (TFEB) both have SPP1 and MMP9 as target genes (Figures S24, S25). SPP1 and MMP9 are not only marker genes of the SPP1 Mφ population, but also have profibrotic activity (12, 14, 29). SPP1 gene expression is upregulated in SSc-ILD and IPF lungs compared to control lungs based on findings from our previous work as well as from other groups (12, 14). SPP1 is included in the differentiating genes between SSc-ILD and IPF lungs in comparison to control lungs (12). Increased serum MMP9 correlates with the degree of skin fibrosis in SSc patients through fibroblast activation and acceleration of fibrosis under the effect of proinflammatory cytokines such as tumor necrosis factor β (TGF-β) and interleukin 1β (IL-1β) (29). Thus SCENIC particularly implicates TFEB and ATF5 as key transciption factors regulating SPP1 Mφ cell differentiation, likely promoting the profibrotic activity of these cells.
Figure 4.
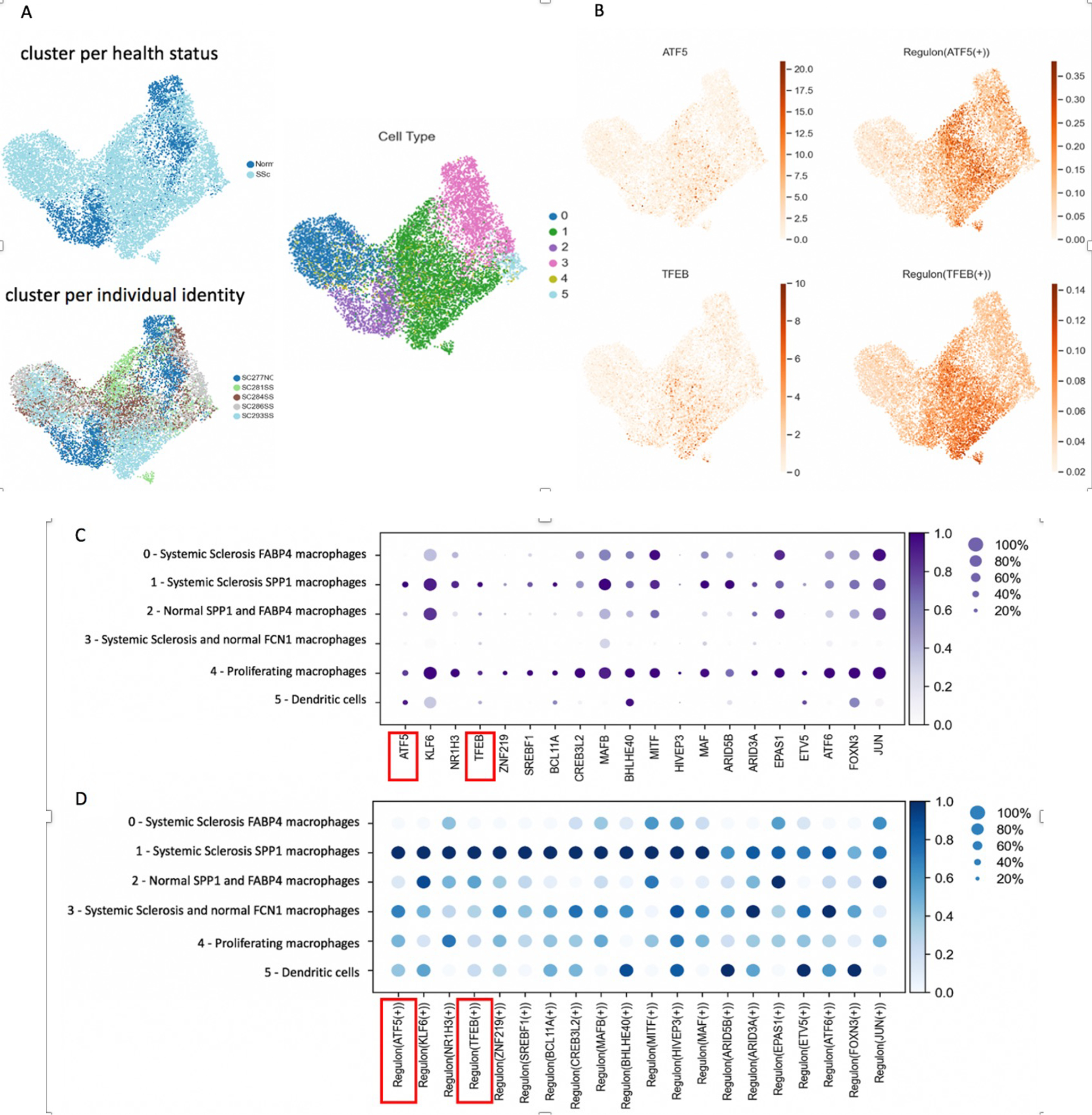
Regulons and Transcription Factors predicted to be important for SPP1 Mφ during analysis of macrophages, dendritic cells, proliferating Mφ from 5 lung samples in scRNA-seq 3’ v3 chemistry by SCENIC. (A) Cell clusters are identified as follows: 0- SSc-ILD FABP4 Mφ (blue), 1- SSc-ILD SPP1 Mφ (green), 2- HC SPP1 and FABP4 Mφ (purple), 3- SSc-ILD and HC FCN1 Mφ (pink), 4- Proliferating Mφ (yellow), 5- Dendritic cells (light blue). (B) SPP1 is a target gene for ATF5 and TFEB transcription factors and regulons. (C) Transcription factor DotPlot predicted to be important for SPP1 Mφ by SCENIC including transcription factors ATF5 and TFEB indicated in red box. (D) Regulon DotPlot predicted to be important for SPP1 Mφ by SCENIC including regulons ATF5 and TFEB indicated in red box.
Altered chromatin structure implicates transcription factors regulating pro-fibrotic SSc-ILD SPP1 macrophage differentiation
In order to better understand the transcription factors regulating differentiation of SSc-ILD SPP1 Mφ, using an orthogonal technology to SCENIC predictions, we analyzed lung samples by scATAC-seq in parallel with scRNA-seq (Table S1). ScATAC-seq examines the chromatin structure of nuclei from each cell by cleaving open chromatin with a Tn5 transposase, and then partitioning cells and ligating cell specific bar-codes to the DNA fragments. The resulting fragments indicate open chromatin structure on a cell-by-cell basis. We analyzed scATAC-seq by clustering nuclei for both SSc-ILD samples using two bioinformatics approaches: Loupe software (10X Genomics) and Signac (18), (Figures S26–S28). For Loupe, the cell types in each cluster were identified by examining the accessibility of promoters of gene markers for each cell type. Macrophages were identified as having open chromatin for the promoters of CD163, AIF1 and MARCO, and FABP4 and SPP1 Mφ populations identified by open chromatin in the respective 5’ promoters (Figure S27). In a complementary analysis we identified nuclei clusters by integrating with the paired scRNA-seq data to predicted cell types (Signac) (18). Signac software maps the cells by examining open chromatin in promoters of differentially expressed genes defined by scRNA-seq cell clusters (Figures S27, S28). We used both Loupe and Signac to determine enriched, accessible motifs for transcription factors, reflecting open chromatin across the genome, comparing SPP1 Mφ to FABP4 Mφ for each SSc-ILD sample (Table S1, Figures S25, S29). Both of these programs use the Jaspar database to identify transcription factor binding motifs (32).
Strikingly, many of the transcription factors binding motifs enriched in open chromatin of SPP1 Mφ were the same transcription factor associated regulons predicted by SCENIC to regulate the differentiation of profibrotic SPP1 Mφs: TFEB, Activating Transcription factor 6 (ATF6), Microphthalmia-associated transcription factor (MITF), Basic Helix-Loop-Helix Family member E40 (BHLHE40), Kruppel-Like Factor 6 (KLF6), ETS Variant Transcription Factor 5 (ETV5), Nuclear Receptor Subfamily 1 Group H Member 3 (NR1H3), CAMP Responsive Element Binding Protein 3 Like 2 (CREB3L2), MAF BZIP Transcription Factor B (MAFB), and AT-Rich Interaction Domain 3A (ARID3A) (Table 1 and Figure 4C). In addition, multiple members of the activating protein-1 (AP-1) transcription factor family, binding as homo and heterodimers, were identified across both software outputs. AP-1 proteins include the JUN, FOS, ATF and musculoaponeurotic fibrosarcoma (MAF) protein families (33). Specifically, the JUN family proteins c-JUN, JUNB, JUND, FOS family proteins c-FOS (FOS), FOSB, FRA1 (FOSL1), FRA2 (FOSL2), and MAF family proteins c-MAF (MAF), MAFA, MAFB, MAFF, MAFG and MAFK were enriched. Of these, the MAF regulon was also predicted by SCENIC to regulate SPP1 Mφ differentiation (Figure 4D).
Table 1.
Accessible motifs to transcription factors for SPP1 macrophages from single-cell ATAC sequencing analysis of SSc-ILD lung samples. Enriched, accessible motifs to transcription factors for SPP1 macrophages compared to FABP4 macrophages as predicted by Signac and SCENIC software for SSc-ILD samples SC336 and SC294, including, respectively: MITF, TFEB, ATF6, SREBF1, BHLHE40, KLF6 and ETV5; MITF, SREBF1, TFEB, BHLHE40 and ETV5 as indicated in red boxes. Also indicated are AP-1 transcription family members including FOS, FOSL1, JUND, JUNB, BATF3, FOSL2, JUN, MAF, NFE2.
| Motif Name | Fold enrichment | p value |
|---|---|---|
| SSc-ILD sample SC336 | ||
| FOS | 3.19010822 | 6.86E-145 |
| FOSL1::JUNB | 3.4413536 | 1.02E-143 |
| BATF3 | 2.9786597 | 2.52E-140 |
| FOSL2: :JUN | 3.32140474 | 5.48E-131 |
| JUN(var.2) | 3.04331528 | 1.07E-128 |
| FOSL1::JUND | 3.26643938 | 1.59E-127 |
| FOSL2: :JUNB | 3.23793531 | 9.57E-126 |
| MAF::NFE2 | 2.66199575 | 5.99E-56 |
| MITF | 1.82166811 | 1.01E-05 |
| TFEB | 1.73941338 | 3.75E-05 |
| ATF6 | 1.70764316 | 0.00072468 |
| SREBF1 | 1.43184691 | 0.0011076 |
| BHLHE40 | 1.09720706 | 0.36788806 |
| KLF6 | 0.90018323 | 0.88278759 |
| ETV5 | 0.71966307 | 0.99424075 |
| SSc-ILD sample SC294 | ||
| NFKB2 | 3.01107011 | 0.00589081 |
| RELA | 2.74533414 | 0.00589081 |
| NFKB1 | 2.70067517 | 0.00589081 |
| FOSB::JUNB | 2.65102267 | 0.00589081 |
| FOSL2::JUND | 2.63504611 | 0.00589081 |
| JDP2 | 2.61442107 | 0.00589081 |
| NFE2 | 2.52034217 | 0.00589081 |
| JUN(var.2) | 2.42862887 | 0.00589081 |
| MITF | 2.19428571 | 0.00589081 |
| SREBF1 | 1.58342582 | 0.00589081 |
| TFEB | 1.81588903 | 0.00589081 |
| BHLHE40 | 1.65048544 | 0.00589081 |
| ETV5 | 0.62227754 | 0.00589081 |
As examples of the epigenetic modifications associated with differentiation into SPP1 macrophages, we examined accessible chromatin of selected marker genes: SPP1, MMP9 and FABP4, comparing SPP1 Mφ and FABP4 Mφ in SSc-ILD (Figure 5, Figures S30–S32). The SPP1 gene showed more accessible chromatin for the SSc-ILD SPP1 Mφ compared to FABP4 Mφ in the promoter region immediately proximal to the transcriptional start site, as well as regions further upstream of the promoter and the 4th intronic region (Figure 5). The MMP9 gene also showed more accessible chromatin in SPP1 Mφ, but in this case increased accessiblity was not seen at the promoter but rather in regions around exon 6, exons 9–12 and introns (Figure 5). In contrast, the FABP4 gene showed strikingly more accessible chromatin in a region proximal to the transcriptional start site and in a broad second region 3’ from the gene in FABP4 Mφ compared to SPP1 Mφ (Figure 5).
Figure 5.
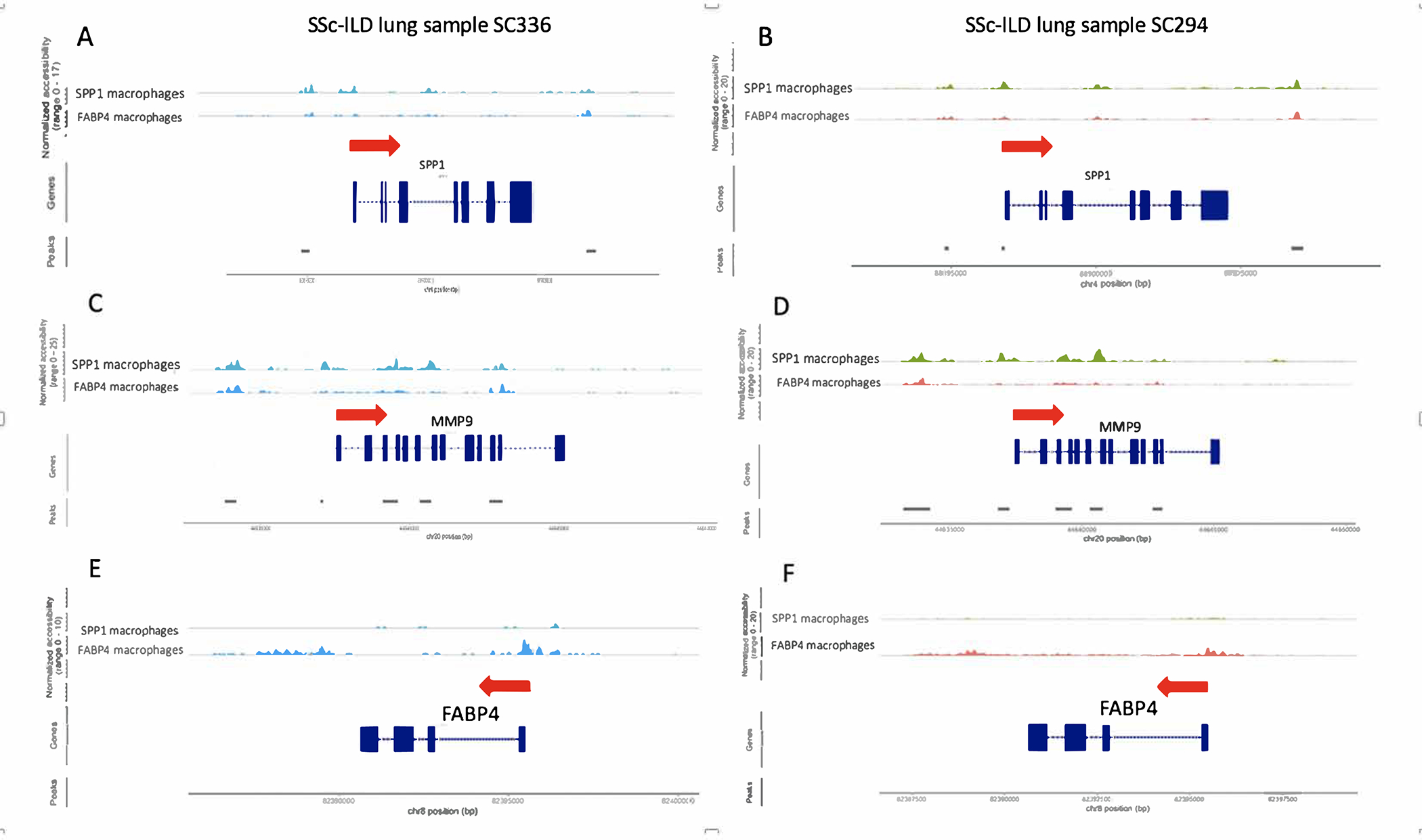
ScATAC-sequencing analysis of 2 SSc-ILD lung samples by Signac showing chromatin pattern changes for SPP1 macrophages compared to FABP4 macrophages. The red arrow indicates the direction of transcription. Exons are shown in blocks and introns flank exons. (A, B) SPP1 gene showed more accessible chromatin for the SSc-ILD SPP1 Mφ compared to FABP4 Mφ in the region proximal to the transcriptional start site for a SSc-ILD lung, as well as regions further 5’ of the promoter and in intron 4. (C, D) MMP9 gene also showed more accessible chromatin in SPP1 Mφ, but in this case increased accessibility was not seen around the promoter but rather in regions around exon 6, exons 9–12 and introns. (E, F) FABP4 gene showed strikingly more accessible chromatin in a region proximal to the transcriptional start site and in a broad second region 3’ from the gene in FABP4 Mφ compared to SPP1 Mφ.
Although we also examined scATAC-seq from several control lungs, technical variability and the inability to clearly distinguish in the scATAC-seq assay between macrophage populations precluded direct comparison of SSc-ILD and control macrophages (Figures S33–S37).
Increased SPP1/MERTK Mφ and TFEB expression in SSc-ILD.
Finally, in order to show increased SPP1 Mφ in SSc-ILD, we identified these cells by staining the co-expressed surface marker MERTK (Figure S38). In addition, we found that a subpoplation of these cells strongly co-expressed TFEB (Figures S39–S40). Available antibodies did not allow us to detect ATF5.
DISCUSSION
The mechanisms driving fibrosis in SSc-ILD are currently uncertain, however the association of macrophage gene expression, particularly of SPP1, with progressive fibrosis makes these cells likely mediators of profibrotic signals to fibroblasts (8). This study confirms the highly increased numbers of a subpopulation of macrophages, characterized by upregulated expression of SPP1 and MMP9, as well as a broader transcriptome in SSc-ILD, in agreement with previous studies both of our group and others (9–16). Analysis of the differentially expressed genes between SPP1 Mφ compared to all other Mφ subpopulations linked SPP1 Mφ to lipid metabolism pathways. Lipid metabolism plays an important role in macrophage function (21). Several transcription factors, including Peroxisome proliferator-activated family of receptors (PPARs) and CCAAT enhancer binding proteins (SREBPs), have been implicated in lipid metabolism in macrophages (21). Further investigation of the effect of lipid metabolism on the development of Mφ phenotype would be important, in view of drugs targeting metabolic pathways (21–25).
SPP1 and MMP9 genes were selectively upregulated by SSc SPP1 Mφ. Osteopontin (OPN), the product of SPP1, is increased in the serum of patients with systemic sclerosis (34, 35) and has been implicated in idiopathic pulmonary fibrosis (36), as well as renal and bone marrow fibrosis (37, 38), suggesting that SPP1 Mφ may have a more general role in promoting fibrosis. MMP9 gene expression is also elevated in idiopathic pulmonary fibrosis (39). Additionally, MMP9 activity is increased in bronchoalveolar lavage fluid of rapidly progressive IPF and in SSc-ILD (40). The expression of MMP-9 in SSc-ILD lungs uniquely by SPP1 Mφ and proliferating SPP1 Mφ suggests that increased levels in serum and BAL in SSc-ILD patients may reflect the activity and/or degree of lung infiltration by these cells, and thus might serve as a biomarker for their activity (35).
We used two approaches to definine likely transcription factors controlling the differentiation of SPP1 Mφ: SCENIC analysis of the SPP1 Mφ transcriptome and scATAC-seq analysis of open chromatin in SPP1 Mφ. SCENIC analyzes gene regulatory networks, i.e., it looks at genes co-regulated with transcriptions factors of each cell. Several approaches exist for identifying target genes directly regulated by transcription factors, (41), including: 1) direct TF-DNA binding assays; 2) computational predictions of transcription factor–target interactions, i.e., position weight matrices (PWMs); and 3) gene regulatory networks that assume transcription factor expression levels regulate targeted genes. SCENIC uses approach 3) in its initial screening, with the resulting gene regulatory network then filtered with CisTarget, a database incorporating results used in methods 1 and 2 (42). The concordance between the predictions from SCENIC with our scATAC-seq data are mutually reinforcing, but in particular strongly reinforce the value of SCENIC in analysing scRNA-seq transcriptomes.
Our scATAC-seq data show characteristic chromatin pattern changes for SPP1 Mφ with increased accessibility of SPP1 and MMP9, and decreased accessibility of FABP4 reflecting the altered binding of transcription factors and enhancers comparing SPP1 Mφ to FABP4 Mφ. These observations provided confidence in our scATAC-seq data. However, altered open chromatin peaks are too broad to implicate specific transcription factors on the basis of individual genes, and thus Signac bioinformatics, examining transcription factor binding sites in open chromatin across the genome provided a more robust method for detecting the likely role of specific transcription factors regulating the phenotype of SPP1 Mφ. This identified numerous transcription factors, including ATF5, TFEB, BCL11A, ETV5, JUN, and others, requiring further experimental validation for their specific roles in regulating SPP1 Mφ differentiation.
Several of the transcription factors identified to participate in SPP1 Mφ differentiation have been implicated in profibrotic phenotypes in other settings. TFEB potentially plays role in silicosis, an occupational irreversible fibrotic lung disease, through disruption of lysosomal autophagy in alveolar macrophages (43). In particular, pulmonary fibrosis in silicosis is promoted with phagocytosis of crystalline silica (CS) particles by alveolar macrophages resulting in apoptosis and inflammation. There is increasing evidence of TFEB interplay with TGF-β (44). TGF-β inhibition associated with blockade of BRAF inhibition and TFEB phosphorylation contributes to malignant cells responsiveness to chemotherapy in melanoma (45). Thus, the effect of TFEB on profibrotic SPP1 Mφ differentiation requires further study. ATF6 and its role in macrophage endoplasmic reticulum stress has also been implicated in the pathophysiology of lung fibrosis (46). Similarly, Sterol Regulatory Element Binding Transcription Factor 1 (SREBF1) was implicated in progression of murine pulmonary fibrosis (47), and BHLHE40 has been identified as an important homeostatic regulator of macrophages in lungs through pulmonary surfactant turnover (48).
Members of the AP-1 transcription factor family, predicted in our analysis to play an important role in the profibrotic phenotype of SPP1 Mφ, have been investigated in the pathophysiology of many fibrotic diseases. As the binding sites for these family members are similar, our data do not clearly discriminate which family member is most important in SPP1 Mφ differentitation. Overexpression of JUN, a prototype AP-1 family member, is associated with severe multi-organ fibrosis in murine models (49). Fos-related antigen-2 transcription factor (Fra-2 or FOSL2) overexpression has been associated with murine SSc-like lung fibrosis (50). Myofibroblasts are activated in vitro by macrophages in a Fra-2-dependent manner, consequently promoting fibrosis. In addition, both myeloid cell inactivation of Fra-2 and Fra-2/AP-1 inhibitors attenuate pulmonary fibrosis in murine bleomycin models (50). Thus, our data strongly support these murine studies, indicating a critical role for AP-1 transcription factors in the development of profibrotic SPP1 macrophages in human SSc-ILD. Defining the specific AP-1 family member(s) most important in regulating SSc-ILD SPP1 Mφ will be key for potential targeting of Fra-2 or other AP-1 family members in SSc-ILD.
Proliferating cells in our dataset were largely composed of proliferating SPP1 and FABP4 Mφ from SSc-ILD lungs, consistent with our previous work showing proliferating macrophages in idiopathic pulmonary fibrosis (14). While the stimulus for proliferation of these cells is uncertain, regeneration of tissue resident Mφ through M-CSF and Granulocyte-Macrophage colony-stimulating factor (GM-CSF) is known to occur in repopulation of inflamed tissues following resolution of inflammation (51). IPF scRNA-seq also indicates increased CSF1 expression in lung mast cells and upregulation of LGMN and other genes regulated by IL-4 (14, 52). CSF1 is upregulated by SSc-ILD SPP1 Mφ and mast cells, as well as by control pericytes, smooth muscle cells and fibroblasts. These findings support proliferating macrophages as the primary source for profibrotic SPP1, suggesting that antiproliferative agents or drugs blocking macrophage cytokines might have efficacy in treating SSc-ILD.
There is increasing evidence linking chemokine CCL18 with pulmonary inflammation, supporting its potential future use as early marker of SSc-ILD (9, 30, 35, 53). Our data reflected upregulation of CCL18 in SPP1 Mφ from the SSc-ILD lungs compared to HC. Elevated CCL18 in the serum and supernatants of cultured bronchoalveolar lavage (BAL) of SSc patients is associated with more advanced ILD by pulmonary function testing (53). CCL18 concentration in the serum of SSc patients was strikingly decreased following treatment with tocilizumab and CCL18 gene expression in skin biopsies of SSc patients was downregulated following tocilizumab treatment, along with other macrophages markers (30). In light of the recent FDA approval of tocilizumab for decreasing progression of SSc-ILD, CCL18 has emerged as a marker of IL-6 in SSc-ILD. By extention, the upregulation of CCL18 in SSc-ILD SPP1 Mφ compared to HC seen on our results supports the role of SPP1 Mφ as drivers of fibrosis in SSc-ILD. Possibly, CCL18 and SPP1 expression characterize macrophages with common functional background and profibrotic potential (35).
The limitations of this study include preclusion of direct comparison of SSc-ILD and control macrophages with scATAC-seq due to technical variability and bioinformatics challenges to clearly distinguish macrophage subpopulations after scATAC-seq in normal lungs. Additionally, since the lung explants are obtained at the time of lung transplant indicating advanced stage of SSc-ILD, they may reflect later stages of SSc-ILD. It is possible that earlier stages of SSc-ILD might have different mechanisms driving disease. Furthermore, it is noted that sample size is small, scRNA-seq analysis groups 3’ and 5’ are skewed respectively towards SSc-ILD vs HC lungs and that single-cell analyses, including data integration, is parameterizable.
In summary, our transcriptomal analysis demonstrates the transcriptome of profibrotic SPP1 Mφ in SSc-ILD and uncovers critical transcription factors in the profibrotic phenotype through changes in chromatin accessibility. These findings support further investigation of key transcription factors, particularly of ATF5 and TFEB, that activate SPP1 Mφ signature genes.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to acknowledge the University of Pittsburgh Medical Center Lung Transplantation team for lungs procurement, the Center for Organ Research and Education (CORE), and the organ donors and their families for the generous lung tissue donation used in the study.
Research reported in this study was supported by the National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases under award number 2P50AR060780 (RL) and the National Heart, Lung, and Blood Institute under award numbers R01HL123766 (RL) and 5T32AI89443–10 (AP). The context is solely responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The University of Pittsburgh holds a Physician-Scientist Institutional Award from the Burroughs Wellcome Fund (AP).
Footnotes
Conflicts of interest R.L. served as a consultant for Bristol Myers Squibb, Formation, Sanofi, Biocon, Boehringer-Mannheim, Boehringer-Ingleheim, Merck and Genentech/Roche, and holds or recently had research grants from Corbus, Formation, Elpidera, Regeneron, Pfizer and Kiniksa.
Transcription factor regulation of profibrotic macrophages in systemic sclerosis-Interstitital lung disease
References
- 1.Abraham DJ, Varga J. Scleroderma: from cell and molecular mechanisms to disease models. Trends Immunol. 2005;26(11):587–95. [DOI] [PubMed] [Google Scholar]
- 2.Steele R, Hudson M, Lo E, Baron M, Canadian Scleroderma Research G. Clinical decision rule to predict the presence of interstitial lung disease in systemic sclerosis. Arthritis Care Res (Hoboken). 2012;64(4):519–24. [DOI] [PubMed] [Google Scholar]
- 3.Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N Engl J Med. 2019;380(26):2518–28. [DOI] [PubMed] [Google Scholar]
- 4.Khanna D, Lin CJF, Furst DE, Goldin J, Kim G, Kuwana M, et al. Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med. 2020;8(10):963–74. [DOI] [PubMed] [Google Scholar]
- 5.Khanna D, Denton CP, Lin CJF, van Laar JM, Frech TM, Anderson ME, et al. Safety and efficacy of subcutaneous tocilizumab in systemic sclerosis: results from the open-label period of a phase II randomised controlled trial (faSScinate). Ann Rheum Dis. 2018;77(2):212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sullivan KM, Goldmuntz EA, Keyes-Elstein L, McSweeney PA, Pinckney A, Welch B, et al. Myeloablative Autologous Stem-Cell Transplantation for Severe Scleroderma. N Engl J Med. 2018;378(1):35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis. 2007;66(7):940–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christmann RB, Sampaio-Barros P, Stifano G, Borges CL, de Carvalho CR, Kairalla R, et al. Association of Interferon- and transforming growth factor beta-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis Rheumatol. 2014;66(3):714–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schutyser E, Richmond A, Van Damme J. Involvement of CC chemokine ligand 18 (CCL18) in normal and pathological processes. J Leukoc Biol. 2005;78(1):14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atamas SP, Luzina IG, Choi J, Tsymbalyuk N, Carbonetti NH, Singh IS, et al. Pulmonary and activation-regulated chemokine stimulates collagen production in lung fibroblasts. Am J Respir Cell Mol Biol. 2003;29(6):743–9. [DOI] [PubMed] [Google Scholar]
- 11.Prasse A, Pechkovsky DV, Toews GB, Jungraithmayr W, Kollert F, Goldmann T, et al. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med. 2006;173(7):781–92. [DOI] [PubMed] [Google Scholar]
- 12.Hsu E, Shi H, Jordan RM, Lyons-Weiler J, Pilewski JM, Feghali-Bostwick CA. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum. 2011;63(3):783–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valenzi E, Bulik M, Tabib T, Morse C, Sembrat J, Trejo Bittar H, et al. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann Rheum Dis. 2019;78(10):1379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J. 2019;54(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med. 2011;184(5):569–81. [DOI] [PubMed] [Google Scholar]
- 16.Voss TC, Hager GL. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet. 2014;15(2):69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stuart T, Srivastava A, Madad S, Lareau CA, Satija R. Single-cell chromatin state analysis with Signac. Nat Methods. 2021;18(11):1333–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 2018. [DOI] [PubMed] [Google Scholar]
- 21.Remmerie A, Scott CL. Macrophages and lipid metabolism. Cell Immunol. 2018;330:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Batista-Gonzalez A, Vidal R, Criollo A, Carreno LJ. New Insights on the Role of Lipid Metabolism in the Metabolic Reprogramming of Macrophages. Front Immunol. 2019;10:2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hume DA, Summers KM, Rehli M. Transcriptional Regulation and Macrophage Differentiation. Microbiol Spectr. 2016;4(3). [DOI] [PubMed] [Google Scholar]
- 24.Consortium F, Suzuki H, Forrest AR, van Nimwegen E, Daub CO, Balwierz PJ, et al. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat Genet. 2009;41(5):553–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cuchel M, Rader DJ. Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation. 2006;113(21):2548–55. [DOI] [PubMed] [Google Scholar]
- 26.Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. 2018;154(1):3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bauer Y, Tedrow J, de Bernard S, Birker-Robaczewska M, Gibson KF, Guardela BJ, et al. A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2015;52(2):217–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bos CL, van Baarsen LG, Timmer TC, Overbeek MJ, Basoski NM, Rustenburg F, et al. Molecular subtypes of systemic sclerosis in association with anti-centromere antibodies and digital ulcers. Genes Immun. 2009;10(3):210–8. [DOI] [PubMed] [Google Scholar]
- 29.Kim WU, Min SY, Cho ML, Hong KH, Shin YJ, Park SH, et al. Elevated matrix metalloproteinase-9 in patients with systemic sclerosis. Arthritis Res Ther. 2005;7(1):R71–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khanna D, Denton CP, Jahreis A, van Laar JM, Frech TM, Anderson ME, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet. 2016;387(10038):2630–40. [DOI] [PubMed] [Google Scholar]
- 31.Denton CP, Ong VH, Xu S, Chen-Harris H, Modrusan Z, Lafyatis R, et al. Therapeutic interleukin-6 blockade reverses transforming growth factor-beta pathway activation in dermal fibroblasts: insights from the faSScinate clinical trial in systemic sclerosis. Ann Rheum Dis. 2018;77(9):1362–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48(D1):D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gazon H, Barbeau B, Mesnard JM, Peloponese JM Jr. Hijacking of the AP-1 Signaling Pathway during Development of ATL. Front Microbiol. 2017;8:2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu M, Schneider DJ, Mayes MD, Assassi S, Arnett FC, Tan FK, et al. Osteopontin in systemic sclerosis and its role in dermal fibrosis. J Invest Dermatol. 2012;132(6):1605–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao X, Jia G, Guttman A, DePianto DJ, Morshead KB, Sun KH, et al. Osteopontin Links Myeloid Activation and Disease Progression in Systemic Sclerosis. Cell Rep Med. 2020;1(8):100140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pardo A, Gibson K, Cisneros J, Richards TJ, Yang Y, Becerril C, et al. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005;2(9):e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Merszei J, Wu J, Torres L, Hicks JM, Bartkowiak T, Tan F, et al. Osteopontin overproduction is associated with progression of glomerular fibrosis in a rat model of anti-glomerular basement membrane glomerulonephritis. Am J Nephrol. 2010;32(3):262–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruberti S, Bianchi E, Guglielmelli P, Rontauroli S, Barbieri G, Tavernari L, et al. Involvement of MAF/SPP1 axis in the development of bone marrow fibrosis in PMF patients. Leukemia. 2018;32(2):438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dancer RC, Wood AM, Thickett DR. Metalloproteinases in idiopathic pulmonary fibrosis. Eur Respir J. 2011;38(6):1461–7. [DOI] [PubMed] [Google Scholar]
- 40.Andersen GN, Nilsson K, Pourazar J, Hackett TL, Kazzam E, Blomberg A, et al. Bronchoalveolar matrix metalloproteinase 9 relates to restrictive lung function impairment in systemic sclerosis. Respir Med. 2007;101(10):2199–206. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Alonso L, Holland CH, Ibrahim MM, Turei D, Saez-Rodriguez J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019;29(8):1363–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herrmann C, Van de Sande B, Potier D, Aerts S. i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Res. 2012;40(15):e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He X, Chen S, Li C, Ban J, Wei Y, He Y, et al. Trehalose Alleviates Crystalline Silica-Induced Pulmonary Fibrosis via Activation of the TFEB-Mediated Autophagy-Lysosomal System in Alveolar Macrophages. Cells. 2020;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lafyatis R. Transforming growth factor beta--at the centre of systemic sclerosis. Nat Rev Rheumatol. 2014;10(12):706–19. [DOI] [PubMed] [Google Scholar]
- 45.Li S, Song Y, Quach C, Guo H, Jang GB, Maazi H, et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat Commun. 2019;10(1):1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burman A, Tanjore H, Blackwell TS. Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol. 2018;68–69:355–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shichino S, Ueha S, Hashimoto S, Otsuji M, Abe J, Tsukui T, et al. Transcriptome network analysis identifies protective role of the LXR/SREBP-1c axis in murine pulmonary fibrosis. JCI Insight. 2019;4(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rauschmeier R, Gustafsson C, Reinhardt A, N AG, Tortola L, Cansever D, et al. Bhlhe40 and Bhlhe41 transcription factors regulate alveolar macrophage self-renewal and identity. EMBO J. 2019;38(19):e101233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wernig G, Chen SY, Cui L, Van Neste C, Tsai JM, Kambham N, et al. Unifying mechanism for different fibrotic diseases. Proc Natl Acad Sci U S A. 2017;114(18):4757–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ucero AC, Bakiri L, Roediger B, Suzuki M, Jimenez M, Mandal P, et al. Fra-2-expressing macrophages promote lung fibrosis in mice. J Clin Invest. 2019;129(8):3293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14(10):986–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Birnhuber A, Biasin V, Schnoegl D, Marsh LM, Kwapiszewska G. Transcription factor Fra-2 and its emerging role in matrix deposition, proliferation and inflammation in chronic lung diseases. Cell Signal. 2019;64:109408. [DOI] [PubMed] [Google Scholar]
- 53.Prasse A, Pechkovsky DV, Toews GB, Schafer M, Eggeling S, Ludwig C, et al. CCL18 as an indicator of pulmonary fibrotic activity in idiopathic interstitial pneumonias and systemic sclerosis. Arthritis Rheum. 2007;56(5):1685–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.