

Abstract
The evolutionarily conserved Leucine Rich Repeat (LRR) protein domain is a unique structural motif found in many viral, bacterial, archaeal, and eukaryotic proteins. The LRR serves many roles including signaling domain and pathogen recognition receptor. In the human innate immune system, it serves an essential role by recognizing fragments of bacterial cell walls. Interestingly, the human fungal pathogen Candida albicans also uses an LRR domain containing protein, Cyrp1, to sense bacterial cell wall fragments. However, the dynamics of signaling and detection of bacterial peptidoglycan fragments by the LRR of Cyr1p remains poorly characterized. Here we develop optimal recombinant expression workflows and provide characterization of the entire region of the LRR as a peripheral membrane protein. Using a newly designed peptidoglycan enrichment bead assay, we demonstrate that this domain is able to bind bacterial peptidoglycan fragments under native conditions. The new membrane associated Cyr1p-LRR construct sets the stage for the development of antifungal agents via high throughput campaigns to inhibit cell wall-Cyr1p interactions.
Graphical Abstract

Introduction
The Leucine Rich Repeat (LRR) protein domain is a unique structural motif of 20–30 amino acids with a characteristic repetitive sequence rich in leucine residues1 2,containing the LxxLxLXXN/CxL3. LRR proteins have been identified in viruses, bacteria, archaea, and eukaryotes. The juxtaposition between the varies kingdoms that this domain is present in lies at the interface of innate immunity and protein-protein interactions4.
LRRs are especially important with regards to the molecular recognition and detection of pathogens and microbes in the human microbiome, which include bacteria, fungi, and viruses5. The ability to maintain human health is directly linked to a system of checks and balances that must occur between human and non-human cells in the microbiome6. The ability to sense and detect foreign entities makes the LRR domain a prime candidate to maintain proper symbiosis in the human host through innate immunity, and through molecular interactions between coexisting species of the microbiome7.
The importance of the LRR domain is readily observed in the polymorphic fungus Candida albicans (C. albicans), the most common fungal commensal in the human microbiome8. C. albicans can disseminate from its localized environment and invade almost every internal organ, producing invasive life-threatening infections, particularly if disseminated in the blood stream9. Whereas superficial skin infections pose no severe health complications, disseminated systemic infections known as Candida have a mortality rate exceeding 70%10.
The margin of commensalism and pathogenicity in C. albicans is a morphological transition from that of a budding (commensal) yeast to an extended hyphal form (pathogenic). The signal integrator for this morphological transition is the adenylyl cyclase, Cyr1p. The fungal adenylate cyclase Cyr1p, is a Class III adenylate Cyclase (AC) and catalyzes the formation of the universal secondary messenger cyclic adenosine-3’,5’-monophosphate (cAMP) from ATP11 (Figure 1)12. Though this protein remains poorly classified, previous studies have proven Cyr1p to be regulated by several strong hyphal inducing signals such as serum, carbon dioxide, and bacterial peptidoglycan13, 14. Xue, et al first noted that Cyr1p contains the evolutionarily conserved LRR domain that is also found in the human innate receptors, NOD1 and NOD214. Using biotin-streptavidin pull down assays, they demonstrated that the LRR domain of Cyr1p binds the synthetic bacterial peptidoglycan fragment muramyl dipeptide (MDP). The team has gone on to characterize this phenotype in the presence of peptidoglycan targeting antibiotics in mouse models15.
Figure 1:

A: The adenylate cyclase Cyr1p contains a Gα domain, a Ras associating domain, 14 Leucine Rich Repeats (LRR), a Protein Phosphatase 2 domain, a guanylate cyclase domain, and a catalytic binding domain. Our newly designed expression vector for Cyr1p LR LRR domain contains residues from 435 up to the PP2C domain. The construct contains 14 repeats of the LRR domain and is expressed in S. cerevisiae. Our previous construct did not include all of the Leucine Rich Repeat. B: Tertiary structure prediction of the 1690 amino acid adenylate cyclase by AlphaFold2. Model is depicted with respect to the LRR domain, red= Gα domain, Blue= Ras association domain, Teal+ Leucine Rich Repeat domain, Grey= Protein Phosphatase 2C domain, Orange= Adenylyl/ guanylyl cyclase domain. All uncharacterized, or disordered domains are depicted in green.
Taking inspiration from Xu et al., we recombinantly expressed the LRR domain of Cyr1p using an E. coli expression system, conducted bioanalytical binding assays using a Surface Plasmon Resonance assay (SPR) and quantified a M binding event (Figure 1a)16. However, the E. coli expressed LRR was limited in further characterization experiments due to its instability and expression as aggregates in insoluble inclusion bodies. Purified protein did not remain soluble in solution once cleaved from the MBP fusion tag and readily precipitated upon dialysis or buffer exchange. To circumvent these limitations, the recombinant LRR was redesigned here utilizing a biophysical and bioinformatics approach. Biophysical data obtained from a Kyte-Doolittle hydropathy plots suggested several possible transmembrane/amphipathic helices (Figure S2, S3)17. In addition, C. albican’s adenylate cyclase is 43% homologous to Saccharomyces cerevisiae’s (S. cerevisiae) adenylate cyclase, which is a peripheral membrane protein18–20. These parameters led us to hypothesize that Cyr1p may also be a membrane associated protein.
Here, detailed characterization of a more stable recombinantly expressed LRR domain that spans the entire region of the predicted LRR using a new expression system with a smaller epitope tag is reported (Figure 1a). Inspiration for the selection of amino acid sequences to capture the entire span of the LRR domain was gathered from homology between the adenylate cyclases of 4 other Candida spp. (Figure S1). Using this information, a construct was synthesized that contains the amino acid residues 435–1004, with an N terminus His6 tag, and the C-terminus 1.4 kDa SpotTag, a 12-amino acid peptide tag recognized by a nanobody21,22. The newly designed recombinant Cyr1p-LRR domain was found to be more stable than the previously expressed E. coli construct, localized to cellular membranes and retained the ability to bind bacterial peptidoglycan fragments16.
Results
The 68.5 kDa LRR domain was expressed in an S. cerevisiae expression system containing both a His and SpotTag(Figure 1a) (Figure S4–5). Low protein expression levels propelled the investigation into biophysical parameters to better predict protein behavior. A Kyte-Doolittle hydropathy plot of the LRR domain suggests several possible membrane associated helices (Figure S2). In addition, secondary structure determination by the AlphaFold2 algorithm depicts localized hydrophobic segments on the surface of the LRR domain (Figure S3)23–24. Taken together, these data suggested the possibility that the LRR domain may localize to the cellular membrane.
To assess the cellular membrane as a possible site of protein localization, clarified total lysate, insoluble cellular debris, and fractionized cellular membranes were analyzed for Cyr1p-LRR levels. Following cellular lysis and membrane fractionization, the majority of Cyr1p-LRR was detected in the total lysate, and significantly decreased when membranes were fractionized from the total lysate by ultracentrifugation. Immunoblotting confirmed that Cyr1p-LRR abundance significantly decreased in the cytosolic fraction after ultracentrifugation, indicating that the majority of the protein was localized to the cellular membranes (Figure 2A).
Figure 2:

Membrane solubilization of the Cyr1p-LRR. Expected MW is 68.5 kD. A: LRR can be extracted from cellular membranes using high ionic strength alone. Solubilization of the LRR can be achieved with 0.5 M NaCl in the absence of detergent. B: Detergent screens with a salt concentration of 0.5 M demonstrate that the Fos-choline class of detergents is optimal. C: Fos-Choline 16 is optimal from the Fos-family. D Chemical Structure of Fos-Choline-16.
Confirmation that the LRR domain was preferentially distributed in the cellular membrane warranted characterization of the protein as either a transmembrane or a peripheral membrane protein. Protein solubilization was conducted from fractionized membranes in the presence of increasing salt concentrations. Immunoblotting confirmed the protein can be solubilized from the membrane effectively at 0.5 M NaCl. Densiometric analysis of immunoblots displayed that at a concentration of 1 M NaCl, Cyr1p-LRR solubility slightly decreased, possibly due to the salting out effect (Figure 2a).25. The solubilization of the protein in high salt concentrations without the presence of detergent is indicative of a peripheral membrane protein, as extraction of transmembrane proteins require complete disruption of the lipid bilayer through the use of organic solvents, or detergents26.
Though high ionic conditions are often sufficient to solubilize peripheral membrane proteins, once solubilized in high ionic conditions it is often necessary to remove high salts for further downstream biochemical assays, such as binding assays. Removal of proteins from high ionic environments can confer structural instability and lead to protein aggregation. As such, detergent screens were conducted with the aim of obtaining purified protein (Figure 2b–c, Figure S6)
Following isolation, membranes were resuspended in lysis buffer. The protein fraction was analyzed via immunoblotting against the SpotTag to ensure that protein loss did not occur due to precipitation upon introduction of detergent. Detergents were added to resuspended membranes at varying percentages (w/v) to ensure that the amount of detergent was orders of magnitude higher than critical micelle concentration (CMC) values Table 1. In lysis buffer, the best solubilizing detergent for the LRR domain was found to be the detergent Fos-C12 (Figure 2b).
Table 1:
Detergents used in this study.
| Detergent | Abbreviation | MW | *CMC mM |
|---|---|---|---|
| Octaethylene glycol monododecyl ether | C12E8 | 538.75 | 0.09 |
| n-Octyl-β-D-Glucoside | OG | 292.40 | 19 |
| n-Dodecylphosphocholine | Fos-C12 | 351.50 | 1.5 |
| n-Hexadecylphosphocholine | Fos-C16 | 407.50 | 0.013 |
| Dodecylpho-n-Methylethanolamine | Fos-MEA-12 | 323.00 | 0.43 |
| n-Lauroylsarcosine sodium salt | Sarkosyl | 293.40 | 14.4 |
| n-Dodecyl-B-D-Maltoside | DDM | 510.60 | 0.17 |
| Triton-x-100 | NA | 647.00 | 0.016 |
CMC values obtained from anatrace.com.
In the presence of 0.5 M NaCl, an increase in the solubilization of Cyr1p-LRR was observed from the detergent Fos-C12 and Fos-C16, which has a slightly longer lipid chain (Figure 2d). Protein solubilization failed in the presence of Fos-MEA 12, and other common detergents such as DDM and Sarkosyl. The longer lipid chain of Fos-C16 may aid in the additional solubilization of some membrane interacting lipids necessary for the stability and subsequent solubility of the LRR, as detergents with longer lipid chains tend to be more solubilizing than detergents with shorter chains27. Interestingly, the methyl group in Fos-MEA-12 inhibited the solubilization of the LRR and promoted protein precipitation, indicating some specificity for the charge of the amine head group.
Purification was successful in the presence of Fos-C16, as immunoblots were positive against both the N-terminal His6 Tag, and the C-terminal Spot-Tag, demonstrating that the entire LRR domain was successfully purified in detergent complexes (Figure S7). Upon purification, Cyr1p-LRR was dialyzed into buffer containing 0.01% Fos-C16, with no precipitation observed. Circular Dichroism (CD) spectroscopy of purified detergent solubilized protein determined that the LRR domain was properly folded, and agreed with our previous observations (Figure 3)16. Using the K2D2 program for estimation of protein secondary structure from CD spectra, the membrane purified construct was predicted to contain 28.8% alpha helixes and 16.41% β strands28, which is in visual agreement with the secondary structure model from AlphaFold2 (Figure 1B). CD spectroscopy was also used to analyze Cyr1p-LRR secondary structure variation under increasing temperatures. Though limited in the ability to produce high concentrations of purified protein in a monomeric state, data suggest that there is a change in secondary structure in the presence of high temperatures (Figure S8, S9).
Figure 3: Secondary Structure of Cyr1p-LRR.
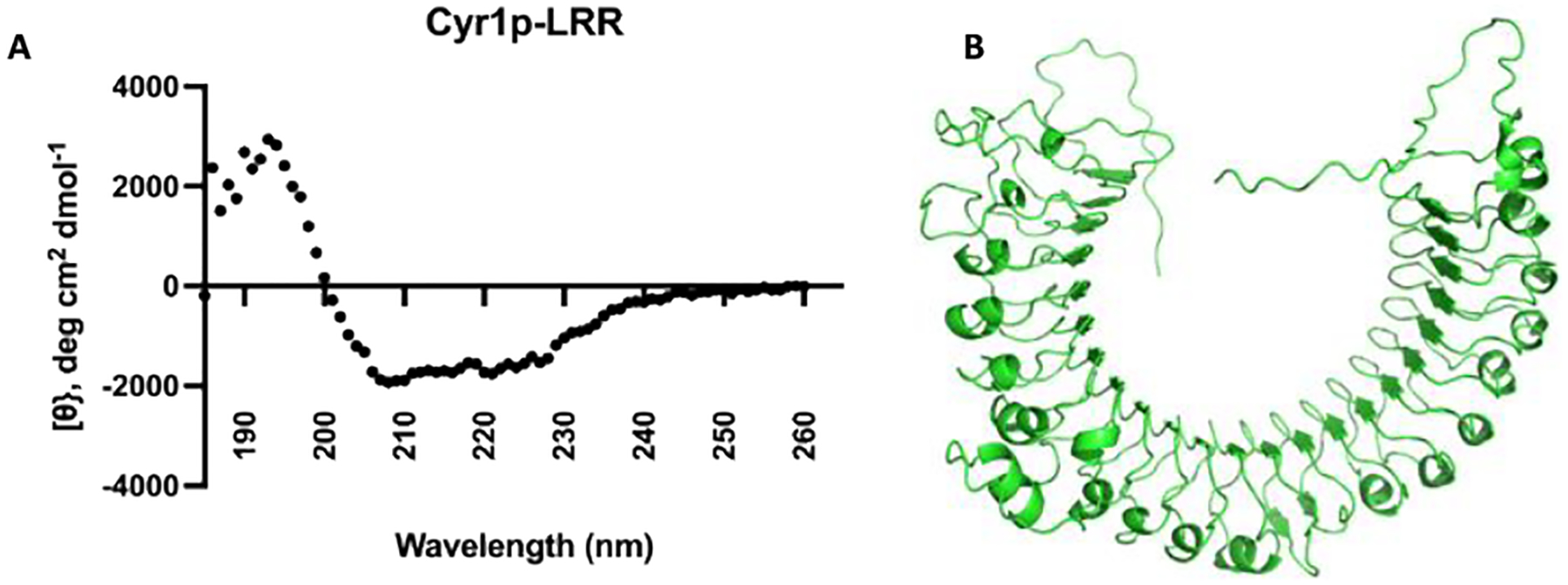
A: Circular Dichroism spectroscopy reveals that the LRR domain is well folded and consists of 28.8% alpha helix, and 16.41% β strands. B: AlphaFold2 secondary structure prediction agrees with the experimental observations from CD spectroscopy.
It has previously been reported that the LRR binds to bacterial peptidoglycan fragments (PG)16. The newly expressed Cyr1p-LRR’s functionality was assessed for the ability to bind PG fragments (Figure 4). Binding was observed for the peptidoglycan fragments relative to the ethanolamine control in cellular lysates (Figure S10–S12). However, binding preferences changed when membrane purified Cyr1p-LRR was assessed for binding. Specifically, limited background binding to ethanolamine was observed (Figure S13) and purified Cyr1p-LRR bound to MTP (Figure 4); these data are in accordance with the previously published SPR assay using E. coli LRR. These binding preferences could shift depending on the presence of accessory proteins, as it was observed that Cyr1p-LRR in total lysate did not display the same binding preferences as purified protein (Figure S10–S13). This is supported by recent work by Hang et al., that shows the binding of NOD2 for its peptidoglycan ligand can be enhanced in the presence of the accessory protein Arf629.
Figure 4:

The membrane associated Cyr1p-LRR can bind synthetic peptidoglycan fragments. a) Schematic of the magnetic bead functionalization. Terminal carboxylic acids are activated with NHS and subsequently coupled to amine functionalized MDP derivative. b) Structures of 2-NH2 Muramyl dipeptide (MDP), Muramyl tripeptide (MTP), 2-NH2 muramic acid (MA) coupled to the magnetic beads through amine functionality. c) Western Blot analysis and quantification of Cyr1p-LRR affinity purification relative to the unfunctionalized beads (ethanolamine control) (full gels and additional biological replicates/analysis are available in the supplemental information); error bars represent SD between replicates.
The data demonstrate that the LRR domain is able to bind bacterial peptidoglycan fragments with greater binding relative to an ethanolamine control (Figure S10–S13). While not a quantitative assay, this affinity-assay confirms the positive association of the LRR domain with peptidoglycan fragments, suggesting that the newly expressed LRR domain is functional. This non-quantitative experiment allows for rapid evaluation of the LRR domain with various PG ligands and is amenable to the limitations of working with a membrane protein. It provides an effective middle ground between a biotin-streptavidin pull down assay14 and an analytical SPR assay16. The former involves complex synthesis to access the bacterial cell wall fragments, while the later requires expensive instrumentation. We envision that this assay could be rapidly expanded with a host of bacterial PG fragments30,31,32 to quickly assess Cyr1p’s binding preferences both from cellular lysates and purified protein.
The LRR domain of the adenylate cyclase Cyr1p from the human commensal C. albicans is a membrane associated protein that is able to sense and detect bacterial peptidoglycan fragments. We have established a rigorous and reproducible workflow to recombinantly express and purify a stable Cyr1p-LRR domain from cellular membranes, and demonstrated interactions with bacterial peptidoglycan fragments. This difficult to characterize protein was expressed with an N-terminal His6 Tag and a small C-terminal SpotTag. To the best of our knowledge, this is the first time this protein has been expressed in a closely related host with such a small affinity tag. These findings permit a better understanding of the native properties governing the structure and function of Cyr1p’s LRR. We hypothesize that Cyr1p’s membrane association are important for protein stability and the ability to properly interact with bacterial PG fragments from its niche via membrane association. In future experiments, this construct will be used to identify accessory proteins and to study the inhibition of bacterial PG binding by small molecules, which will serve as therapeutic leads in the development of new anti-fungals.
Supplementary Material
Acknowledgments
We thank our colleagues at the University of Delaware: The the Perilla group for providing us with structural models for our construct through AlphaFold2 and Dr. Dennis Wykoff from Villanova University providing us with strain EY1202 and guidance with protein expression in yeast.
Funding Sources
This work was supported by the National Institute of Health (T32GM133395) through the Chemical Biology Interface Program and the National Science Foundation (NSF 1554967).
Abbreviations
- PG
peptidoglycan
- LRR
Leucine Rich Repeat
- Candida. albicans
C. albicans
- MA
Muramic Acid
- MTP
Muramyl tripeptide
- MDP
Muramyl Dipeptide
Footnotes
The authors declare no competing financial interests.
Supporting Information
Supplement methods such as materials, expression and purification of Cyr1p-LRR, biophysical data, CD spectroscopy, and PG affinity assays.
Uniprot ID: A0A1D8PR83 (A0A1D8PR83_CANAL)
References
- 1.Kobe B; Deisenhofer J, Trends in Biochemical Sciences 1994, 19 (10), 415–421. [DOI] [PubMed] [Google Scholar]
- 2.Ng A; Xavier RJ, Autophagy 2011, 7 (9), 1082–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kobe B; Kajava AV, Current Opinion in Structural Biology 2001, 11 (6), 725–732. [DOI] [PubMed] [Google Scholar]
- 4.Janeway CA Jr., Cold Spring Harb Symp Quant Biol 1989, 54 Pt 1, 1–13. [PubMed] [Google Scholar]
- 5.Ursell LK; Metcalf JL; Parfrey LW; Knight R, Nutr Rev 2012, 70 Suppl 1 (Suppl 1), S38–S44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mohajeri MH; Brummer RJM; Rastall RA; Weersma RK; Harmsen HJM; Faas M; Eggersdorfer M, Eur J Nutr 2018, 57 (Suppl 1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crump GM; Zhou J; Mashayekh S; Grimes CL, Chemical Communications 2020, 56 (87), 13313–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mayer FL; Wilson D; Hube B, Virulence 2013, 4 (2), 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Tilburg Bernardes E; Pettersen VK; Gutierrez MW; Laforest-Lapointe I; Jendzjowsky NG; Cavin J-B; Vicentini FA; Keenan CM; Ramay HR; Samara J; MacNaughton WK; Wilson RJA; Kelly MM; McCoy KD; Sharkey KA; Arrieta M-C, Nature Communications 2020, 11 (1), 2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nature Microbiology 2017, 2 (8), 17120. [DOI] [PubMed] [Google Scholar]
- 11.Linder JU; Schultz JE, Cellular Signalling 2003, 15 (12), 1081–1089. [DOI] [PubMed] [Google Scholar]
- 12.Linder JU, Cellular and Molecular Life Sciences 2006, 63 (15), 1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klengel T; Liang W-J; Chaloupka J; Ruoff C; Schröppel K; Naglik JR; Eckert SE; Mogensen EG; Haynes K; Tuite MF; Levin LR; Buck J; Mühlschlegel FA, Curr Biol 2005, 15 (22), 2021–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu XL; Lee RT; Fang HM; Wang YM; Li R; Zou H; Zhu Y; Wang Y, Cell Host Microbe 2008, 4 (1), 28–39. [DOI] [PubMed] [Google Scholar]
- 15.Tan CT; Xu X; Qiao Y; Wang Y, Nat Commun 2021, 12 (1), 2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burch JM; Mashayekh S; Wykoff DD; Grimes CL, ACS Infectious Diseases 2018, 4 (1), 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kyte J; Doolittle RF, J Mol Biol 1982, 157 (1), 105–32. [DOI] [PubMed] [Google Scholar]
- 18.Mitts MR; Grant DB; Heideman W, Mol Cell Biol 1990, 10 (8), 3873–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Altschul SF; Madden TL; Schäffer AA; Zhang J; Zhang Z; Miller W; Lipman DJ, Nucleic Acids Res 1997, 25 (17), 3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altschul SF; Wootton JC; Gertz EM; Agarwala R; Morgulis A; Schäffer AA; Yu YK, Febs j 2005, 272 (20), 5101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Virant D; Traenkle B; Maier J; Kaiser PD; Bodenhöfer M; Schmees C; Vojnovic I; Pisak-Lukáts B; Endesfelder U; Rothbauer U, Nature Communications 2018, 9 (1), 930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braun MB; Traenkle B; Koch PA; Emele F; Weiss F; Poetz O; Stehle T; Rothbauer U, Scientific Reports 2016, 6 (1), 19211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skolnick J; Gao M; Zhou H; Singh S, Journal of Chemical Information and Modeling 2021, 61 (10), 4827–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; Bridgland A; Meyer C; Kohl SAA; Ballard AJ; Cowie A; Romera-Paredes B; Nikolov S; Jain R; Adler J; Back T; Petersen S; Reiman D; Clancy E; Zielinski M; Steinegger M; Pacholska M; Berghammer T; Bodenstein S; Silver D; Vinyals O; Senior AW; Kavukcuoglu K; Kohli P; Hassabis D, Nature 2021, 596 (7873), 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moringo NA; Bishop LDC; Shen H; Misiura A; Carrejo NC; Baiyasi R; Wang W; Ye F; Robinson JT; Landes CF, Proceedings of the National Academy of Sciences 2019, 116 (46), 22938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith SM, Methods Mol Biol 2011, 681, 485–96. [DOI] [PubMed] [Google Scholar]
- 27.le Maire M; Champeil P; Møller JV, Biochimica et Biophysica Acta (BBA) - Biomembranes 2000, 1508 (1), 86–111. [DOI] [PubMed] [Google Scholar]
- 28.Perez-Iratxeta C; Andrade-Navarro MA, BMC Structural Biology 2008, 8 (1), 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hespen CW; Zhao X; Hang HC, Chemical Communications 2022, 2598. [DOI] [PubMed] [Google Scholar]
- 30.Bersch KL; DeMeester KE; Zagani R; Chen S; Wodzanowski KA; Liu S; Mashayekh S; Reinecker H-C; Grimes CL, ACS Central Science 2021, 7 (4), 688–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.D’Ambrosio EA; Bersch KL; Lauro ML; Grimes CL, J Am Chem Soc 2020, 142 (25), 10926–10930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lazor KM; Zhou J; DeMeester KE; D’Ambrosio EA; Grimes CL, Chembiochem 2019, 20 (11), 1369–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.