

Abstract
Dysfunction of the aquaporin-4 (AQP4)-dependent glymphatic waste clearance pathway has recently been implicated in the pathogenesis of several neurodegenerative diseases. However, it is difficult to unravel the causative relationship between glymphatic dysfunction, AQP4 depolarization, protein aggregation, and inflammation in neurodegeneration using animal models alone. There is currently a clear, unmet need for in vitro models of the brain’s waterscape, and the first steps towards a bona fide “glymphatics-on-a-chip” are taken in the present study. It is demonstrated that chronic exposure to lipopolysaccharide (LPS), amyloid-β(1-42) oligomers, and an AQP4 inhibitor impairs the drainage of fluid and amyloid-β(1-40) tracer in a gliovascular unit (GVU)-on-a-chip model containing human astrocytes and brain microvascular endothelial cells. The LPS-induced drainage impairment is partially retained following cell lysis, indicating that neuroinflammation induces parallel changes in cell dependent and matrisome-dependent fluid transport pathways in GVU-on-a-chip. AQP4 depolarization is also observed following LPS treatment, which demonstrates that AQP4 polarization status can be dynamically manipulated in vitro and suggests that LPS-induced drainage impairments on-chip may be driven by changes in AQP4-dependent fluid dynamics. These studies introduce a biomimetic model of glymphatics and reveal AQP4 polarization dynamics and fluid transport in the healthy and inflamed brain.
Keywords: glymphatics-on-a-chip, astrocytes, aquaporin-4, neuroinflammation, fluid transport
Graphical Abstract
AQP4 is a water channel expressed in a polarized manner by astrocytic endfeet lining the blood-brain-barrier. AQP4 depolarization was recently implicated in the progression of neurodegeneration; however, it has been difficult to study this phenomenon using mouse models alone. Here, we develop a new organ-on-a-chip model of the brain gliovascular unit in which neuroinflammatory cues induce glymphatic drainage dysfunction and the polarization status of AQP4 can be dynamically manipulated by neuroinflammatory cues.
1. Introduction:
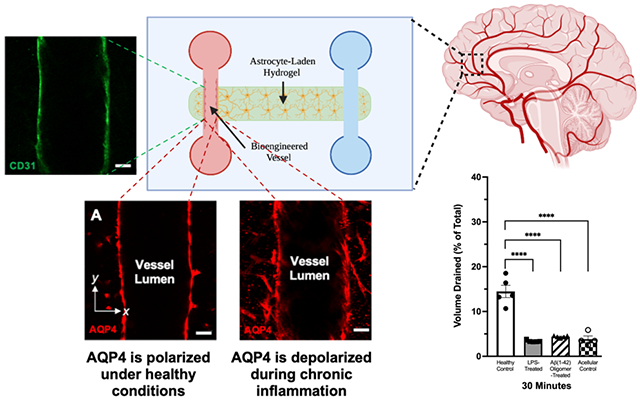
The gliovascular unit (GVU) is a network of astrocytes, endothelial cells, mural cells, and supportive connective tissues that forms a semipermeable barrier between the blood, cerebrospinal fluid (CSF), and interstitial fluid (ISF) compartments.[1] Reinforcing the CSF-ISF barrier is a layer of astrocytic endfeet—thin perivascular processes that ensheathe blood vessels throughout the central nervous system (CNS). Lacking traditional lymphatic vessels, brain parenchymal tissue is thought to rely on the exchange of CSF and ISF across astrocytic endfeet to remove harmful proteins and metabolic wastes from the cerebral cortex.[2] According to the glial-lymphatic (glymphatic) hypothesis, CSF accumulating in perivascular spaces (PVSs) around cortical arteries infiltrates brain tissue by passing through aquaporin-4 (AQP4) channels expressed on astrocytic endfeet (Figure 1). After entering the brain, CSF mixes with ISF and convects to PVSs surrounding cortical veins, generating a hydrostatic pressure gradient that helps clear wastes like amyloid-β (Aβ) peptides.[2–3] In healthy individuals, perivascular astrocytes only express AQP4 along their endfeet. This polarized pattern of AQP4 expression is thought to optimize the efficiency of CSF-ISF exchange.[2]
Figure 1. The gliovascular unit (GVU).

The gliovascular unit (GVU) forms a semipermeable barrier between the cerebrospinal, interstitial, and vascular fluid compartments. According to the glymphatic hypothesis, cerebrospinal fluid (CSF) is produced by the choroid plexus epithelium and accumulates in the subarachnoid space on the surface of the brain. According to the glymphatic hypothesis, CSF then penetrates the brain tissue by flowing along low-resistance peri-arterial spaces and through AQP4 channels expressed on astrocytic endfeet. It then mixes with interstitial fluid (ISF) in the brain parenchyma and drains waste products—such as Amyloid-β peptides—to the peri-venous space. From there, wastes are removed from the central nervous system through meningeal lymphatic vessels and subarachnoid granulations. Abbreviations: CSF, cerebrospinal fluid; ISF, interstitial fluid.
Recent studies have implicated dysfunction of AQP4-dependent fluid dynamics within the GVU in the pathogenesis of Alzheimer’s disease (AD),[4] traumatic brain injury (TBI),[5] amyotrophic lateral sclerosis (ALS),[6] and other neurodegenerative illnesses.[6–7] It is thought that depolarization of AQP4 from the endfeet of perivascular astrocytes reduces the efficiency with which wastes are cleared from the central nervous system in patients with these diseases, facilitating the formation of insoluble aggregates—such as Aβ plaques.[8] This hypothesis is supported by findings that AQP4 depolarizes from perivascular endfeet in murine models of AD, TBI,[8a, 9] ALS,[6, 10] and old age.[8f] Additionally, a positive correlation was recently observed between the degree of AQP4 depolarization, Aβ plaque density, and the severity of clinical symptoms at the time of death in AD patients[11]. Mestre et al. (2018) later demonstrated that inducing AQP4 depolarization through genetic deletion of the α-syntrophin adaptor protein reduced CSF influx into the murine cortex by 30-70%.[8c] However, the molecular mechanisms by which AQP4 depolarizes in the context of neurodegenerative disease remain unclear.
Given that AD, TBI, ALS, and ageing involve similar disturbances of the neuroimmune landscape, inflammation of perivascular astrocytes may represent a “final common pathway” by which AQP4 depolarization and glymphatic dysfunction occur.[8g, 12] It is well-known that the central nervous system of patients with TBI,[9c] ALS,[6] and advanced age[13] contain a greater proportion of immunoreactive astrocytes than those of healthy controls. This state of “reactive astrogliosis” is characterized by excessive proliferation, cytoskeletal remodeling, and AQP4 upregulation.[6, 9c, 14] Iliff et al. (2014) demonstrated that AQP4 depolarization and reactive astrogliosis reliably coincide in brain slices ipsilateral, but not contralateral to cerebral injury sites in mice with TBI.[8a] Peripheral immune challenges are also known to induce impairments in glymphatic clearance.[15] Similarly, AD is accompanied by significant, brain-wide inflammation and reactive astrogliosis.[2, 8e, 16] In fact, Aβ oligomers bind to toll-like receptor 4 (TLR4)—a cell-surface pattern recognition receptor—and trigger pro-inflammatory signaling cascades.[17] However, it is not yet known whether Aβ oligomer-induced reactive astrogliosis directly causes AQP4 depolarization. The temporal and functional relationship between reactive astrogliosis, AQP4 depolarization, glymphatic dysfunction, and protein aggregation in AD and other neurodegenerative illnesses thus remains controversial and unclear.[18]
However, it is difficult to unravel the causative relationships between these molecular biological and biophysical factors in the stepwise progression of neurodegenerative illness using conventional murine models alone. In vivo glymphatics experiments are subject to considerable variation depending upon the type of anesthetic used, the time of day, the mouse’s age, the location and speed of CSF tracer injection, and the identity of the tracer being studied[8c, 19]—creating real concerns about research reproducibility. Given the state-dependent nature of in vivo glymphatics, it has also been challenging to reliably interrogate the biophysical mechanisms by which AQP4 depolarization induces glymphatic dysfunction. It is also difficult and resource-intensive to generate the transgenic mouse lines required to isolate the discrete contributions of specific cell types and subcellular pathways to glymphatic drainage. Additionally, it is not yet possible to disentangle and compare the role of cell-dependent and extracellular matrix-dependent (which we refer to as “matrisome”-dependent) fluid transport pathways in the glymphatic system using animal models. Finally, the degree of AQP4 polarization is known to vary dramatically between humans and mice.[20] We therefore see a clear, unmet need for a humanized in vitro model of glymphatic physiology. Such a model could serve as an isolated system in which the biophysical and molecular biological factors that govern glymphatic waste clearance in healthy and diseased states can be sequentially manipulated and studied.
To develop a bona fide “glymphatics-on-a-chip” that can be applied in both basic and translational research contexts, current microfluidic models of the blood brain barrier will need to be improved in two ways. They must (1) recapitulate the complex, state-dependent patterns of fluid exchange within and between the human CSF, ISF, and blood compartments and (2) contain perivascular astrocytes that can dynamically modulate their polarization status to mimic healthy and diseased states. In this study, we take the first steps towards addressing these gaps in the literature by developing a “GVU-on-a-chip” containing human astrocytes seeded around a bioengineered blood vessel in which fluid drainage efficiency and AQP4 polarization status can be monitored and manipulated by chronic inflammation. Using a non-physiological drainage paradigm, we show that exposure to lipopolysaccharide (LPS), amyloid-β(1-42) oligomers, and the AQP4 inhibitor TGN-020[21] impairs the drainage of fluid and amyloid-β(1-40) tracer in GVU-on-a-chip. We also provide evidence that LPS-induced drainage impairments can be attributed to parallel changes in cell-dependent and matrisome-dependent fluid transport pathways. Finally, we demonstrate that LPS treatment is associated with AQP4 depolarization—suggesting a potential cell-dependent mechanism for the observed drainage impairment.
2. Results and Discussion:
2.1. Device Design and Optimization
GVU-on-a-chip consists of two parallel microchannels of diameter 250 μm fully embedded in a 3D hydrogel matrix (Figure 2A). In the left-sided channel, HBMECs are seeded to reconstitute the endogenous GVU (Figure 2B). Each microchannel feeds into a large fluid reservoir of diameter 6 mm and approximate height 6-7 mm on either end, and the entire structure is housed in a leucite polydimethylsiloxane (PDMS) enclosure of length 2.7 cm and width 2.0 cm (Figure 2C). Within the central hydrogel, brain-derived human astrocytes are seeded through hydrogel-casting ports. Our device is similar to that used by prior researchers to study lymphatic vessels, pancreatic ductal adenocarcinoma, and breast cancer in vitro.[22]
Figure 2. GVU-on-a-chip Design.
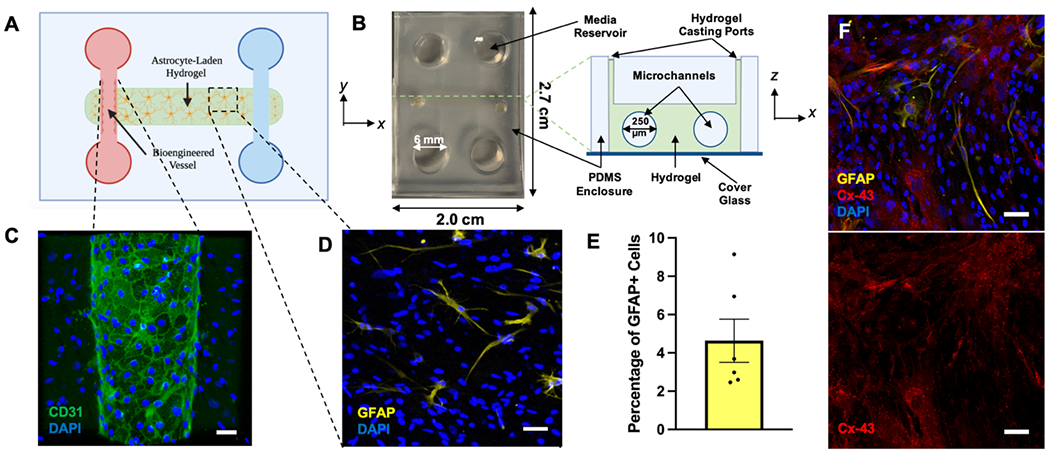
(A) A two-dimensional schematic diagram of our GVU-on-a-chip model. This device consists of two parallel microchannels fully embedded in a 3D, astrocyte-laden hydrogel. Human Brain Microvascular Endothelial Cells (HBMECs) seeded in the left microchannel interact with one another to form an endothelial sheet reinforced by tight junctions. (B) Left: A photograph of GVU-on-a-chip in the xy-plane. Right: A labeled, cross-sectional schematic of GVU-on-a-chip in the xz-plane. (C) A representative image of the bioengineered microvessel in GVU-on-a-chip stained for CD31 and DAPI. Scale bar is 50 μm. (D) A representative image of the astrocyte-laden hydrogel stained for GFAP and DAPI. This image was acquired from a chip with 2 x 106 cells mL−1. Scale bar is 50 μm. (E) The proportion of GFAP+ cells in six replicates of GVU-on-a-chip. Bars represent mean ± SEM. (F) Parenchymal astrocytes seeded in GVU-on-a-chip and stained for Cx-43, GFAP, and DAPI. Scale bars are 50 μm. Schematics were created with BioRender.com. Abbreviations: GVU, gliovascular unit; PDMS, polydimethylsiloxane; GFAP, glial fibrillary acidic protein; Cx-43, Connexin-43.
To optimize the cell culture conditions in GVU-on-a-chip, we evaluated the morphology and protein expression profile of astrocytes seeded at several densities in hydrogels of varying composition. In vivo, astrocytes exhibit vast morphological heterogeneity depending upon their location in the central nervous system as well as the physiological state of the individual.[23] However, a key attribute of healthy, functional astrocytes is a stellate morphology in which elaborate, tree-like processes extend radially from the cell body.[24] We found that astrocytes seeded at cell densities between 2 x 106 and 5 x 106 cells mL−1 in a 4:1:1 mixture of collagen I: Matrigel: hyaluronic acid (HA) begin to adopt a stellate morphology one day after seeding (Figure S1). The final concentrations of collagen I, Matrigel, and HA in this mixture were 2.35 mg mL−1, 0.6 mg mL−1, and 0.6 mg mL−1, respectively. This confirms prior observations that the inclusion of HA and Matrigel in collagen-based 3D hydrogels promotes astrocyte branching and elongation.[24b]
Figure 2D shows a representative maximum intensity z-projection of astrocytes seeded in our optimized hydrogel at a density of 2 x 106 cells mL−1 and stained for GFAP and DAPI. Only a small proportion of the astrocytes cultured under these conditions (each indicated by a DAPI+ nucleus) stained positive for GFAP. Using representative z-projections collected from six chips, we manually quantified the total proportion of GFAP+ astrocytes in GVU-on-a-chip. As shown in Figure 2E, less than 10% of astrocytes seeded in our hydrogel expressed GFAP. In fact, only 2-9% of astrocytes in most chips expressed GFAP at detectable levels. As discussed earlier, GFAP is a marker of reactive astrogliosis and is only expressed by 5-20% of astrocytes in the healthy brain.[25] Therefore, the data presented in Figures 2D–E confirms that astrocytes seeded in our optimized hydrogel do not adopt reactive or inflammatory phenotypes. We also demonstrate that parenchymal astrocytes cultured in GVU-on-a-chip interact with one another to form a robust network linked by connexin-43 (Cx-43) gap junctions (Figure 2F). Cx-43 channels are known to link the processes of adjacent astrocytes in vivo, creating a functional syncytium through which ions, nutrients, and signaling molecules can diffuse.[26] In this way, Cx-43 metabolically couples astrocytic networks to support cell growth,[27] migration,[28] and neuroglial communication.[29]
The extracellular matrix (ECM) found in the human brain is comprised primarily of laminin,[30] fibronectin,[30b] type IV collagen,[30b, 31] hyaluronan,[24b, 32] heparan sulfate proteoglycans,[33] chondroitin sulfate proteoglycans,[30b] and tenascin R scaffolds.[30b, 34] The hydrogel used to prepare GVU-on-a-chip contains all but two of the major ECM molecules found in vivo. While the two missing ECM components (chondroitin sulfate proteoglycan and tenascin R) do play an important role in astrocyte biology,[35] there is no evidence that they play a role in the glymphatic system. Additionally, to our knowledge, no currently established microfluidic models of the blood-brain barrier incorporate chondroitin sulfate proteoglycan and tenascin R in 3D culture with astrocytes.[24a, 36] That being said, Syková et al. (2005) did observe that the cortical interstitium of tenascin R-deficient mice is reduced in size.[37] Thus, the influence of specific ECM molecules on the brain’s waterscape must be more thoroughly investigated, and we predict that on-chip models of the brain’s waterscape will furnish the ideal platform on which to perform such studies in the future. Beyond ECM components, the Matrigel we used in our model contains several soluble factors known to critically regulate astrocyte function in vivo—including transforming growth factor β (TGF-β), and epidermal growth factor (EGF).[24b, 38]
2.2. Characterization of Astrocytic Endfeet in GVU-on-a-chip
We then examined the interactions between astrocytes and HBMECs in GVU-on-a-chip (Figure 3). Perivascular astrocytes wrap themselves around cortical microvessels in vivo, meaning that fluid transport in the brain is inherently dependent upon the gene expression profile and fluid-conducting properties of astrocytic endfeet. Human astrocytes seeded in GVU-on-a-chip at a density of 5 × 106 cells mL−1 spontaneously migrate to the bioengineered blood vessel and extend distinct endfeet processes within two days of HBMEC seeding (Figure 3A–C). We also sought to investigate the polarization status of AQP4 in our on-chip model. In vivo, astrocytes only express AQP4 along endfeet that line CSF compartments (i.e., the subarachnoid space and PVSs around blood vessels) in the brain and spinal cord.[39] As shown in Figure 3D and 3E, AQP4 is selectively expressed around the CD31+ vessel in the xy-plane of GVU-on-a-chip. When comparing Figure 3E with the corresponding brightfield image of the same chip shown in Figure 3D, the AQP4 peak corresponds with an astrocytic endfoot—mimicking the in vivo architecture. The histogram in Figure 3F plots these data quantitatively: AQP4 signal peaks in the region immediately adjacent to the blood vessel and quickly returns to negligible levels. Selective localization of AQP4 to endfeet around the CD31+ vessel is also observed in the xz-plane (Figure 3G). Conversely, astrocytes seeded in GVU-on-a-chip without HBMECs do not exhibit AQP-4 polarization around the chip microchannel (Figure 3H). Rather, astrocytes cultured under these conditions express AQP4 along the entire surface of their membrane. Taken together, these data confirm prior findings that BMEC-endfoot interactions are required to establish polarized AQP4 expression in vitro.[40]
Figure 3. GVU-on-a-chip Contains AQP4-Expressing Astrocytic Endfeet.
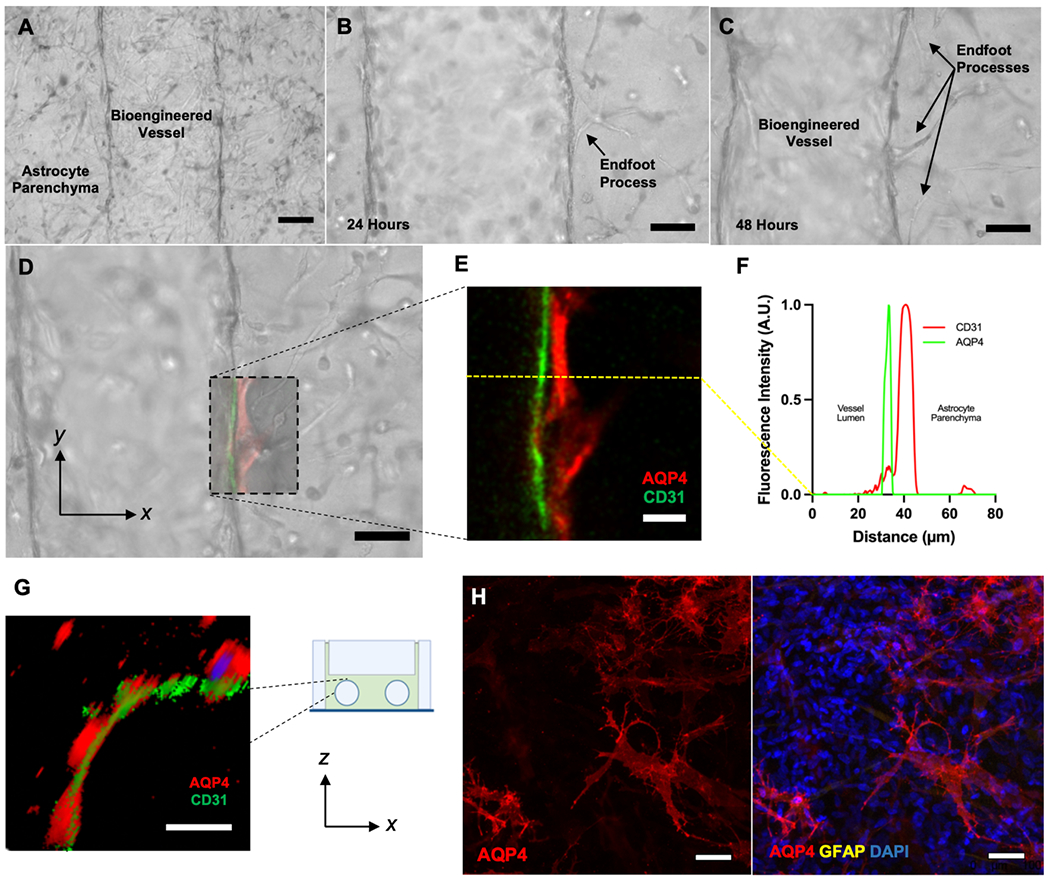
(A-D) Live brightfield micrographs of perivascular astrocytes interacting with the bioengineered cortical microvessel in GVU-on-a-chip. Images (A), (C), and (D) were taken 48 hours following HBMEC seeding. Image (B) was taken 24 hours after HBMEC seeding. Scale bars are 100 μm (A), 50 μm (B), 50 μm (C), and 50 μm (D). (E) A confocal (xy-plane) micrograph of a portion of the vessel-endfoot interface in (D) stained for AQP4 (red) and CD31 (green). Scale bar is 20 μm. (F) A histogram of the mean fluorescence intensity, measured in Arbitrary Fluorescence Units (A.U.), across the yellow line in (E). (G) An xz-plane cross-section of GVU-on-a-chip stained for AQP4 (red) and CD31 (green) digitally rendered from a confocal micrograph. Scale bar is 50 μm. (H) Images of astrocytes seeded in GVU-on-a-chip without HBMECs and stained for AQP4 (red), GFAP (yellow), and DAPI (blue). Without HBMECs, perivascular astrocytes do not exhibit polarized AQP-4 expression. Scale bars are 50 μm. Abbreviations: AQP4, aquaporin-4.
2.3. Functional Drainage Properties of Healthy and Inflamed GVU-on-a-chip Replicates
2.3.1. Neuroinflammation impairs the clearance of Aβ(1-40) in GVU-on-a-chip
Given the proposed role of neuroinflammation in glymphatic dysfunction,[8, 12] we assessed the drainage properties of GVU-on-a-chip in healthy and inflammatory contexts. We could not recapitulate glymphatic drainage as it occurs in vivo because the three fluid compartments of the GVU—the blood-filled vascular lumen, CSF-filled PVS, and ISF-filled brain parenchyma—are not segregated in our model. Rather, we performed the non-physiological drainage experiments schematically outlined in Figure S2A to rapidly screen the fluid volume and Aβ(1-40) tracer mass drained by GVU-on-a-chip replicates. We chose to model neuroinflammation by chronically exposing our chips to lipopolysaccharide (LPS) and Aβ(1-42) oligomers—both of which have been shown to induce TLR4-dependent reactive astrogliosis[17a]—according to the treatment schedule shown in Figure S2B. We found that 30 and 60 minutes after initiating interstitial flow, healthy control chips drained a significantly higher fluid volume (Figure 4A) and tracer mass (Figure 4B) than LPS-treated (p <0.0001 for volume and mass at both time points) and oligomer-treated (p <0.0001 for volume and mass at both time points) chips. These findings mirror prior in vivo studies demonstrating that peripheral LPS challenge impairs CSF-ISF exchange[15b] and Aβ clearance from the brain.[15a] Although we do not directly recapitulate glymphatic biology in our model, these data convey that chronic neuroinflammation generally disrupts the GVU barrier in vitro, leading to widespread shifts in fluid and solute transport between the blood, CSF, and ISF fluid compartments.
Figure 4. Characterizing the fluid-conducting properties of GVU-on-a-chip in healthy and inflamed states.
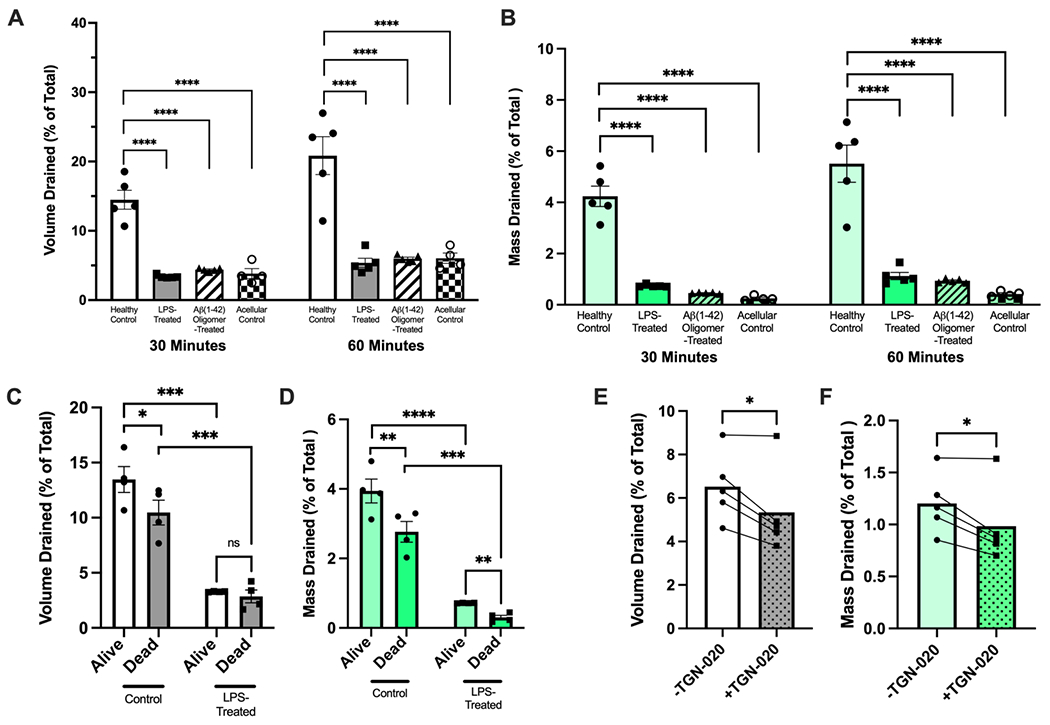
(A and B) The percent of total media volume drained (A) and percent of total Aβ (1-40) tracer mass drained (B) by control (n = 5 individual replicates), LPS-treated (n = 5 individual replicates), Aβ (1-42) oligomer-treated (n = 5 individual replicates) and acellular control (n = 5 individual replicates) chips 30 and 60 minutes after initiation of interstitial flow. Groups were compared at 30 and 60 minutes using a one-way ANOVA test with Dunnett’s multiple comparisons test. (C and D) The percent of total media volume drained (C) and percent of total Aβ (1-40) tracer mass drained (D) by control and LPS-treated chips before (“alive”) and after (“dead”) osmotic lysis (n = 4 paired “alive” and “dead” chip replicates in control and LPS-treated groups). Alive and dead chips pairs within treatment groups (control and LPS-treated) were compared using a paired t-test, while alive and dead chips between treatment groups (ie. alive control vs. alive LPS-treated, etc.) were compared using unpaired t-tests. (E and F) The percent of total media volume drained (E) and percent of total Aβ (1-40) tracer mass drained (F) by chips before and after treatment with TGN-020, an AQP4 inhibitor (n = 5 paired chip replicates). Chip replicated were compared before and after TGN-020 treatment using paired t-tests. All error bars represent mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
2.3.2. Healthy control chips drain fluid and tracer more efficiently than acellular controls
In the drainage experiments shown in Figure 4A and 4B, healthy chips were also observed to drain nearly three to four times as much fluid and tracer as acellular controls (p < 0.0001 for volume and mass) after 30 and 60 minutes. These findings suggest one of three possibilities: (1) the astrocytes in GVU-on-a-chip remodel the hydrogel to expand low-resistance extracellular fluid transport pathways, (2) there is a low-resistance, cell-dependent (potentially AQP4-dependent) pathway in healthy control chips that allows fluid to bypass the hydrogel, or (3) a combination of both. Much evidence has been gathered suggesting that glymphatic clearance in the mouse and human brain depends upon cell-dependent fluid transport pathways involving AQP4,[8g, 12] supporting explanation (1). It has also been well-established that astrocytes play a critical role in remodeling the matrisome in vivo.[41] However, limitations of current animal models have prevented researchers from investigating the role of matrisome-dependent fluid transport pathways in glymphatic function—and dysfunction—across the lifespan. This underscores the utility of in vitro models like GVU-on-a-chip, in which the function of intracellular and extracellular drainage pathways can be isolated and compared across a range of healthy and diseased states. Therefore, we next sought to identify the relative contributions of (1) matrisome-dependent and (2) cell-dependent fluid transport pathways to the inflammation-induced drainage impairment observed in GVU-on-a-chip (Figure 4A and 4B).
2.3.3. Characterizing the contributions of matrisome-dependent fluid pathways to inflammation-induced drainage impairments in GVU-on-a-chip
The optimization data shown in Figure S3 indicate that astrocytes seeded in GVU-on-a-chip readily interact with and remodel the cell-laden hydrogel. In fact, within five days of astrocyte seeding, the hydrogel is remodeled so much that it begins to detach from the PDMS enclosure. Therefore, it is possible that healthy and immunoreactive astrocytes exhibit different ECM-remodeling behaviors. If this were the case, then healthy and inflamed chips would contain hydrogels with different fluid-conducting properties, which could in part explain the drainage impairment observed in Figure 4A and 4B. To test this hypothesis, we performed drainage assays in healthy and inflamed chips before and after the cells in each chip were killed by osmotic lysis (which we refer to as “dead” chips”) (Figure S2C). While fluid in living chips can pass through both intact cells and extracellular matrix, fluid transport in dead chips is restricted to the extracellular space. Therefore, if inflamed chips continue to display a drainage impairment following cell death, we can infer that chronic inflammatory conditions perturb the fluid-conducting properties of the biomimetic hydrogel in GVU-on-a-chip. For these experiments, we used chronic LPS exposure as a general model of CNS inflammation.
We indeed observed that LPS-treated chips retained their drainage impairment following cell death (p < 0.001 for volume and mass), indicating that inflammation-related matrisome changes are at least partially responsible for the observations in Figures 4A and 4B. Our data suggest a fundamental difference in how astrocytes in the healthy and LPS-treated conditions interact with and remodel their ECM scaffolding. These findings build on a recent report that astrocytes activated by LPS-treated endothelial cells—to which astrocytes in our model are exposed—exhibit a robust shift in their ECM remodeling profile.[42] Johnson et al. (2022) also recently found that, of 44 protein modules (families of related proteins) studied, enrichment in the matrisome protein module (collection of ECM-associated proteins) was one of four modules most significantly correlated with AD neuropathology in both preclinical and clinical AD. Furthermore, this matrisome enrichment was conserved across brain regions and data sets.[43] Taken together with these findings, our data suggest that glymphatic dysfunction observed in neuroinflammatory and neurodegenerative diseases[8g, 12] may be mediated in part by shifts in astrocyte-dependent matrisome remodeling that favor ECM accumulation. In future studies, changes in the composition, porosity, and hydration status of the cell-laden hydrogel in healthy and inflamed GVU-on-a-chip replicates should be compared.
2.3.4. Characterizing the contributions of cell-dependent fluid pathways to inflammation-induced drainage impairments in GVU-on-a-chip
We also observed that, compared with living healthy control chips, dead healthy control chips exhibited reduced drainage of fluid and Aβ(1-40) tracer (p = 0.0126 for volume; p = 0.0053 for mass; Figure 4C). These data support the hypothesis that a cell-dependent fluid transport pathway parallel to the matrisome-dependent pathway mediates drainage in GVU-on-a-chip. By contrast, drainage in LPS-treated chips did not differ between live and dead chips, suggesting either dysfunction or absence of a cell-dependent pathway. We also demonstrate that treatment with the AQP4 inhibitor TGN-020[21] reduces the drainage of fluid (p = 0.0270) and tracer (p = 0.0270) in GVU-on-a-chip. Synthesizing these data, we hypothesized that dysfunction of AQP4-dependent fluid dynamics may contribute to the observed inflammation-induced drainage impairment. Given that AQP4 depolarization is a known cause of glymphatic dysfunction in several neurodegenerative and neuroinflammatory diseases,[4–8, 8c–g, 9–11] we next compared the polarization status of AQP4 in healthy in LPS-treated chips replicates.
2.4. Neuroinflammation Drives AQP4 Depolarization in GVU-on-a-chip
We qualitatively and quantitatively compared the polarization status of AQP4 in control and LPS-treated chips using confocal microscopy. Figure 4A shows xy-plane cross sections of control and LPS-treated chips stained for AQP4, CD31, and DAPI. While AQP4 expression in the control chip is limited to endfeet surrounding the bioengineered vessel, that in the LPS-treated chip is comparatively depolarized. The difference in polarization status can be appreciated quantitatively in Figures 5B–C. These two histograms display the normalized AQP4 and CD31 fluorescence intensity for the control (Figure 5B) and LPS-treated (Figure 5C) chips imaged in Figure 5A as a function of distance along the x-axis. Both graphs have intensity peaks on either side of the CD31+ blood vessel that correspond to AQP4 expressed by astrocytic endfeet. However, while the control chip’s plot quickly returns to near-baseline intensity levels within 20 μm of the endfoot peak (Figure 5B), that of LPS-treated chips remains relatively high—even 150 μm from the vessel (Figure 5C). We also observed that the mean division-xy-plane AQP4 polarization index (API) of control chips (n = 5)—a previously-established metric of AQP4 polarization status[44]—is more than double that of LPS-treated chips (Figure 5D).
Figure 5. Neuroinflammation drives AQP4 depolarization in vitro.
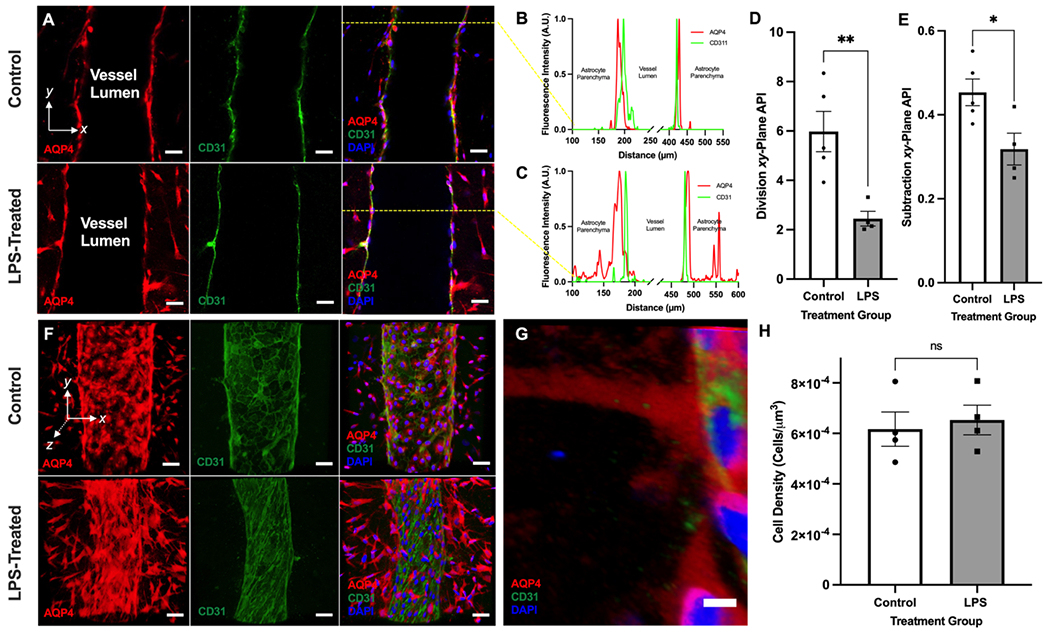
(A) Representative xy-plane images of control and LPS-treated chips stained for AQP4, CD31, and DAPI. Scale bars are 50 μm. (B) A histogram of the mean fluorescence intensity, measured in Arbitrary Fluorescence Units (A.U.), across the x-axis of the control chip imaged in the top panel sequence of (A). The AQP4 and CD31 signals are displayed in red and green, respectively. (C) A histogram of the mean fluorescence intensity, measured in Arbitrary Fluorescence Units (A.U.), across the x-axis of the LPS-treated chip shown in the bottom panel sequence of (A). The AQP4 and CD31 signals are displayed in red and green, respectively. (D) A summary of the division xy-plane AQP4 polarization index (API) of control (n = 5) and LPS-treated (n = 4) chips. (E) A summary of the subtraction xy-plane AQP4 polarization index (API) of control (n = 5) and LPS-treated (n = 4) chips. (F) Representative 3D reconstructions of control and LPS-treated chips stained for AQP4, CD31, and DAPI. Scale bars are 50 μm. (G) A close-up 3D reconstruction of an astrocytic endfoot process exhibiting AQP4 depolarization. Scale bar is 5 μm. (H) Quantification of the parenchymal astrocyte cell density in 3D reconstructions of control (n = 4) and LPS-treated (n = 4) chips shown in (F). All LPS-treated chips analyzed in this figure were treated with 1,000 ng mL−1 LPS. Bars on all graphs represent mean ± SEM. *p<0.05, **p<0.01 (two-tailed homoscedastic t-test). ‘ns’ means not significant. Abbreviations: AQP4, aquaporin-4; API, Aquaporin-4 Polarization Index; 3D, three-dimensional.
According to this API, chips that are “depolarized” (have a polarization index < 1) exhibit AQP4 enrichment in non-endfeet regions of astrocytic cell membranes without necessarily experiencing a decrease in AQP4 accumulation at the endfeet. This would be the case if perivascular astrocytes experienced both a global increase in AQP4 expression (which has been observed in both AD[11, 45] and TBI[46]) as well as a reduced capacity—for reasons of physical space or subcellular pathway dysfunction—to localize AQP4 to perivascular endfeet. We have also developed a secondary, subtraction-based xy-plane AQP4 polarization index (API) in which parenchymal AQP4 signal is subtracted from endfoot AQP4 signal. Chips in which AQP4 is enriched in the parenchyma and reduced at the endfeet are more likely to be distinguished from polarized control chips in which AQP4 is enriched in the parenchyma, but unchanged at the endfeet when using this subtraction-based index (compared to the division-based index). Therefore, according to this API, chip groups that are relatively “depolarized” (ie. having a significantly lower API than another chip group) are likely experiencing AQP4 enrichment in non-endfoot regions of astrocytic cell membranes along with a corresponding loss of AQP4 at the endfeet. LPS-treated chips also scored lower on this subtraction-based API (p = 0.0277; Figure 5E). Additionally, post-hoc analysis shows that there is no difference in the density of astrocytes in control and LPS-treated chips (p = 0.7012) (Figure 5H). This demonstrates that the increased parenchymal AQP4 signal observed in LPS-treated chips is not simply due to an increased density of AQP4-expressing astrocytes.
The difference in polarization status between control and LPS-treated chips can also be appreciated qualitatively in the 3D projections shown in Figure 5E. In control chips, AQP4 expression is limited to 1) perivascular endfeet and 2) the region of the cell body directly surrounding the nucleus (likely indicating protein expression in the endoplasmic reticulum). By comparison, AQP4 can be observed along the entire membrane of perivascular astrocytes in LPS-treated chips. In fact, the AQP4 signal allows us to visualize the extensive processes that link an astrocyte’s cell body with its endfeet. This is evident in the close-up 3D rendering of an endfoot experiencing AQP4 depolarization in Figure 5G, offering a stark contrast to the polarized endfood shown in Figure 3E. To our knowledge, this is the first evidence that chronic pro-inflammatory stimuli can be used to manipulate AQP4 polarization status in vitro. Interestingly, Manouchehrian et al. (2021) recently observed that AQP4 polarization status was unaffected three hours after peripheral LPS injection in mice.[15b] Taken together with our in vitro findings, this suggests that chronic exposure to inflammatory stimuli—as in AD, TBI, and ALS—rather than acute exposure is necessary to depolarize AQP4. In the future, we plan to use mouse models in synergy with GVU-on-a-chip to better understand the temporal relationship between LPS-induced neuroinflammation and AQP4 depolarization in vivo and in vitro.
Taken together with the drainage data shown in Figure 4, we hypothesize that LPS-induced AQP4 depolarization (Figure 5) contributed to the drainage impairment observed in inflamed GVU-on-a-chip replicates (Figure 4). As described earlier, both AQP4 depolarization[8c] and AQP4 knockout[2] have been shown to cause reduced fluid infiltration into the murine cortex. Additionally, evidence of AQP4 depolarization and reduced glymphatic drainage have been jointly observed in AD,[11, 47] TBI,[8a, 9] ALS,[6, 10] and old age.[8f] Interestingly though, Manouchehrian et al. (2021) recently demonstrated that peripheral LPS injections can cause glymphatic drainage impairments without inducing AQP4 depolarization. This suggests that inflammation-related glymphatic dysfunction is likely caused by multiple changes in the biology and fluid-conducting properties of perivascular astrocytes, supporting mural cells, and vascular endothelial cells. In addition to AQP4 depolarization, chronic neuroinflammation could induce swelling in astrocytic endfeet,[48] alter the tight junctions between adjacent HBMECs,[49] and alter the expression of transport proteins (such as lipoprotein receptor-related protein 1, an Aβ transport protein) in HBMECs.[15a] We also observed striking changes in the morphology of HBMECs in control and LPS-treated chips—while HBMECs in control chips retain oblong, oval-like shapes, those exposed to LPS appear thin and elongate. In summary, we observed that inflammation-induced drainage impairments are mediated by dysfunction of both matrisome-dependent and cell-dependent pathways. Furthermore, we identified AQP4 depolarization as a candidate cell-dependent pathway—establishing analogies between in vivo glymphatics and GVU-on-a-chip. These findings contribute to a growing body of literature suggesting that neuroinflammation may be a final common pathway by which GVU dysfunction occurs in neurodegenerative disease.[8g]
2.5. Future Directions
In this paper, we demonstrate that inflammatory cues impair the drainage of fluid and tracer in GVU-on-a-chip through matrisome and cell-dependent pathways. In future investigations, GVU-on-a-chip can be used to study the changes in these two pathways caused by inflammation in further detail. Specifically, the composition and porosity of the biomimetic hydrogel in healthy control and LPS-treated chips should be compared. GVU-on-a-chip can also be used to study and compare the molecular mechanisms by which healthy and immunoreactive astrocytes remodel their ECM microenvironment. We also provide evidence in this study that AQP4 polarization status can be dynamically manipulated in vitro by LPS. In the short-term, it would be important to assess whether chronic treatment with Aβ (1-42) oligomers—which we observed to impair drainage in GVU-on-a-chip—also induces AQP4 depolarization. In the long term, the next step towards developing a physiologically relevant glymphatics-on-a-chip model would be to recapitulate the complex patterns of fluid exchange that occur within and between the blood, CSF, and ISF compartments. Additionally, we believe it would be critical to further develop our current GVU-on-a-chip model by adding Aβ tracer-expressing neurons to the hydrogel-bulk and seeding pericytes—supportive mural cells—around our engineered blood vessel.
Unbound by the constraints of in vivo work, we believe a bona fide microfluidic model of the glymphatic system would help elucidate the molecular mechanisms responsible for AQP4 delocalization from astrocytic endfeet in neurodegenerative disease. Additionally, a “glymphatics-on-a-chip” could be used for high-throughput screening of drug candidates seeking to repolarize AQP4 and restore glymphatic drainage in patients with neurodegenerative disease. A glymphatics-on-a-chip model would also provide an isolated system in which the precise biophysical and molecular biological factors responsible for glymphatic waste clearance can be elucidated and applied clinically. Since Iliff et al.’s seminal 2012 paper, ongoing basic science research has generated several important controversies in the field.[2] Specifically, the directionality of waste clearance,[50] the relative contributions of convection and diffusion,[51] and the role of arterial pulsations and PVS geometry,[52] in the glymphatic system have been the subject of contentious debate. It also remains unclear how AQP4 depolarization impairs the drainage of metabolic wastes from the brain—does it reduce the infiltration of CSF into the parenchyma, alter the fluid dynamics involved CSF-ISF exchange, or some combination of both? Microfluidic technologies afford researchers unprecedented insights into the biophysics that govern fluid exchange across biological barriers. We envision on-chip models of the brain’s waterscape being used in synergy with traditional murine models to answer outstanding questions in the field, and this paper represents the first step towards that objective.
3. Conclusion
In the present study, we establish and validate a novel microfluidic model of the GVU comprised of human astrocytes and a bioengineered blood vessel. Through structural and functional analyses, we confirm that astrocytes in GVU-on-a-chip extend AQP4-expressing astrocytic endfeet and that fluid transport in our model is AQP4-dependent. We also demonstrate that chronic TLR4 agonism by LPS and disease modeling with Aβ(1-42) oligomers impairs the drainage of fluid and tracer through matrisome-dependent and cell-dependent pathways. Additionally, we found that LPS treatment depolarizes AQP4 in GVU-on-a-chip, suggesting a potential cell-dependent mechanism by which chronic inflammation impairs drainage in our model. These findings contribute to a growing body of literature demonstrating the central role of inflammation in dysfunction of the GVU. This model represents the first step towards a bona fide “glymphatics-on-a-chip” platform on which researchers can 1) study the contributions of glymphatic dysfunction to neurodegenerative disease, 2) develop and screen personalized therapeutics, and 3) investigate the biophysics underlying glymphatic drainage.
4. Experimental Section
Cell Culture:
All cell lines were propagated in Nunclon™ Delta surface treated T175 flasks (ThermoFisher Scientific, USA) and stored in a standard tissue culture incubator set to 37°C, 95% humidity, and 5% CO2. Human astrocytes purchased from ScienCell Research Laboratories (USA) were cultured in Astrocyte Medium (ScienCell) containing 98% basal medium, 2% fetal bovine serum (FBS), 1% astrocyte growth supplement (AGS), and 1% penicillin/streptomycin solution. Human Brain Microvascular Endothelial Cells (HBMECs) were purchased from Cell Systems (USA) and cultured in complete classic medium with 10% serum (Cell Systems). All HMBECs were maintained in T175 flasks that were pre-treated with attachment factor (Cell Systems), as instructed by the supplier. Human Astrocytes and HBMECs were used for experiments at passage numbers 4-10 and 4-7, respectively.
Device Fabrication:
The silicon master molds used for device fabrication were designed using the photolithography technique outlined in Kwak et al[22c, 22d]. To fabricate GVU-on-a-chip, a 10:1 weight ratio mixture of PDMS and cross-linker curing agent (SYLGARD™ 184 Silicone Elastomer Kit, The Dow Chemical Company, MI, USA) were degassed, cast into the silicon master mold, and baked for twelve hours at 80 °C. The solidified PDMS was then removed from the master mold and cut to create a device of length 2.7 cm, width 2 cm, and height 0.5 cm. Standard biopsy punches (Fisher Scientific) of diameter 2 mm and 6 mm were used to punch two hydrogel-casting ports and four media reservoirs, respectively, into each chip. The patterned face of the chips was then exposed to a plasma reacted ion etch (RIE) surface treatment (PE-25 Plasma Cleaner, Plasma Etch Inc., NV, USA), bonded to a 22 mm (length) x 40 mm (width) x 0.13-0.17 mm (height) cover glass and cured at 80 °C for at least ten minutes. To make the channels and the hydrogel bulk reservoir hydrophilic, chips were then treated with 0.01% (w/v) poly-L-lysine (Sigma) diluted in deionized water (DW) for one hour, washed thrice with PBS, and treated again for 30 minutes with 1% glutaraldehyde (Electron Microscopy Sciences) diluted in DW. The chips were then immersed in a DW bath overnight.
Casting of Astrocyte-Laden Hydrogel:
Casting needles of thickness 0.25 mm (0.25 x 50 mm, Hwato) were soaked in 70% ethanol overnight and blocked for one hour with 1% bovine standard albumin (BSA) (VWR) diluted in phosphate buffered saline (PBS). Pressurized air was used to thoroughly dry the collagen reservoir of each chip in preparation for hydrogel casting. BSA-coated needles were then threaded through each microchannel, and the chips were sterilized under an ultraviolet light for 30 minutes. The liquid hydrogel base was prepared by mixing Medium 199 10x (10% of total volume) (Gibco, USA) with 1N NaOH (2.5% volume of collagen type I to be added) (Electron Microscopy Science, USA). Collagen type I (Corning, USA), reduced growth factor, growth-factor reduced, LDEV-free Matrigel Matrix (Corning, USA), and Glycosil Hylauronic Acid (Advanced BioMatrix, USA) were then added to the mixture in the appropriate ratio (varied between experiments). Additionally, extralink-Lite PEDGA cross-linker was added in a 1:4 ratio with the volume of Glycosil. All materials were kept on ice under a sterilized tissue culture hood during hydrogel preparation.
Human astrocytes (P4-10) were washed twice with PBS, detached using TrypLE Express (Gibco, USA), neutralized with complete culture media, and counted. The cell suspension was then centrifuged and resuspended in a 100 μL aliquot of the hydrogel mixture at the appropriate cell density (varied between experiments). After resuspending the cell pellet in the hydrogel aliquot, the homogenate was returned to the hydrogel stock solution and thoroughly mixed. At this stage, any treatments being introduced into the chip (eg. vehicle (0.2% DMSO in complete astrocyte media), lipopolysaccharide, or Aβ(1-42) oligomers) were added to the hydrogel mixture. The stock (50 μL) was then loaded into each chip through the hydrogel-casting ports using gel-loading pipette tips. Following hydrogel casting, all chips were placed in a standard tissue culture incubator for one hour to promote hydrogel polymerization. Fresh astrocyte media was then added to the media reservoirs as well as the tops of the hydrogel-casting ports, and the chips were placed in an incubator overnight.
Production of Bioengineered Cortical Microvessels:
24 hours after astrocyte-seeding, the casting needles were removed and each microchannel was sealed using high vacuum grease (Dow Corning, USA). The chips were then placed in petri dishes lined with DW-soaked Kimwipes and stored in a tissue culture incubator for six hours. Gel-loading tips were then used to remove the excess collagen blocking the hydrogel-casting port. HBMECs (P4-7) were then washed twice with PBS, detached with TrypLE Express (Gibco, USA) and resuspended at a density of 1,000,000-2,000,000 cells mL−1. Cell suspension was added to each media reservoir and the chips were inverted and placed in the tissue culture incubator for ten minutes. After confirming that cells were adhering to the top of the vessel, the chips were flipped right-side-up and allowed to remain in the incubator for ten additional minutes. Each media reservoir was then washed twice with PBS to remove excess HBMECs that had not adhered to the microchannels, and the chips were placed in the tissue culture incubator overnight. The formation of a continuous endothelium was confirmed using brightfield microscopy 24 hours after cell seeding. Each day that followed, the media reservoirs were replaced with a 1:1 mixture of fresh astrocyte and complete classic media. 0.2% DMSO was added to the culture media of all chips in all treatment groups each day.
Preparation of Lipopolysaccharide-Treated Chips:
Chips being treated with lipopolysaccharide (LPS) (ThermoFisher Scientific, USA) were fabricated using slightly modified hydrogel-casting protocols. To mimic chronic neuroinflammation, astrocytes were pre-treated with LPS before being seeded on the chip. LPS was delivered into the cell culture media seven and three days before astrocyte seeding. LPS was also mixed into the liquid hydrogel before casting on the day of astrocyte seeding and added to the cell culture media one and two days after astrocyte seeding. All LPS treatments were delivered at a dosage of 1,000 ng mL−1 (vehicle 0.2% DMSO in media). This dose has been shown to induce TLR4-mediated reactive astrogliosis in human astrocytes.[17a]
Preparation of Aβ(1-42) Oligomer-Treated Chips:
Amyloid-β (Aβ) oligomers were prepared following the protocol of Stine et al (2011). In short, the recombinant human Aβ (1-42) peptide film (rPeptide, GA, USA) was suspended in DMSO (Sigma Aldrich, MO, USA) to create a 5mM solution, vortexed for 30 seconds, and then sonicated for 10 minutes in a bath sonicator. To this room temperature solution, cold phenol-free F-12 cell media (Gibco, ThermoFisher Scientific, MA, USA) was added to create a sterile 100μM solution of Aβ, vortexed for 15 seconds, and then incubated at static conditions in a 2°C environment for at least 24 hours.
Chips being treated with Aβ(1-42) oligomers (ThermoFisher Scientific, USA) were fabricated using slightly modified hydrogel-casting protocols. To mimic chronic neuroinflammation, astrocytes were pre-treated with Aβ(1-42) oligomers before being seeded on the chip. Aβ(1-42) oligomers was delivered into the cell culture media seven and three days before astrocyte seeding. Aβ(1-42) oligomers was also mixed into the liquid hydrogel before casting on the day of astrocyte seeding and added to the cell culture media one and two days after astrocyte seeding. All Aβ(1-42) oligomers treatments were delivered at a dosage of 1 μM (vehicle 0.2% DMSO in media).
Drainage Quantification Assays:
Functional drainage experiments were performed by filling the left-sided microchannel on GVU-on-a-chip with tracer-filled media and measuring the media volume, tracer concentration, and tracer mass that drained into the right-handed microchannel. Lyophilized Beta-Amyloid (1-40) TM-Fluor 488-Labelled (Anaspec) tracer was initially dissolved in 1% (v/v) ammonium hydroxide to a concentration of 2.5 mg mL−1. This stock solution was diluted further in phosphate buffered saline (PBS) to a final concentration of 0.1 mg mL−1 (21.334 μM), aliquoted, and stored at −20 °C until the drainage experiment was run. One day after seeding HMBECs, all media reservoirs were emptied and the hydrogel-casting ports on each chip were sealed with a 3D-printed PDMS plug. On the day of the drainage experiment, an aliquot of Aβ(1-40) was thawed and diluted in astrocyte media to a final concentration of 3 μg mL−1.
Diluted Aβ(1-40) was added to both media reservoirs attached to the left-sided vessel (150 μL/reservoir). The tracer was then allowed to convect through the vessel and across the astrocyte-laden hydrogel. Media was then collected from the reservoirs attached to the right-sided channel after 30 minutes or 60 minutes, and the total volume that drained in each chip was measured using a micropipette. Additionally, the fluorescence intensity of the drainage samples collected from each chip was quantified using a spectrofluorometer, and the absolute concentration of Aβ(1-40) tracer that drained was calculated using a standard curve. These volume and concentration data were then used to calculate the total mass of Aβ(1-40) that drained over the course of the experiment. All drainage quantification assays were performed on GVU-on-a-chip models containing 5 x 106 astrocytes mL−1 two days after HBMEC seeding. The cell density and timing of all drainage experiments were informed by the optimization data shown in Figure S3. A comprehensive timeline of the drainage experiments performed on control and LPS-treated chips is shown in Figure S2B. Additionally, a schematic of the drainage experiment is shown in Figure S2A.
Comparison of Drainage in Live vs. Dead Chips:
“Live” GVU-on-a-chip replicates were prepared according to the healthy chip manufacture procedure described above. Two days after HBMEC seeding, the drainage properties of live chips were studied using the drainage quantification assay described above. The cells in live chips were then exposed to distilled water (DW) for 12 hours to produce “dead” chips via osmotic lysis. Another drainage assay was then performed at this time, and drainage data collected from live and dead chips was compared using paired analyses (see Statistical Analysis). Data collected from any chips exhibiting delamination at the hydrogel-PDMS interface following DW treatment were excluded from the study. Following collection of drainage data, live and dead chip replicates were stained with 0.04% trypan blue (Gibco, ThermoFisher Scientific, USA) to confirm cell viability status. HBMECs and astrocytes in dead chips that have undergone osmotic lysis should accumulate the trypan blue stain, while alive cells in live chips should not.
TGN-020 Inhibition Experiment:
AQP4 inhibition studies were performed using paired chips groups to minimize batch effects. The AQP4 inhibitor 2-(nicotinamide)-1,3,4-thiadiazole (TGN-020) was diluted in DMSO at an initial concentration of 50 mg mL−1. This solution was then diluted in media to a working concentration of 200 μg mL−1 and stored at 4°C. A group of healthy chips prepared using the chip manufacture protocol described above (including continuous exposure to 0.4% DMSO vehicle) were then subjected to two 30-minute drainage quantification assay. This same batch of chips was then treated with the 200 μg mL−1 stock solution of TGN-020 for five hours, after which another two 30-minute drainage quantification assays were performed. Each individual chip was treated as a biological replicate. Each of the two 30-minute drainage experiments performed in the pre/post-TGN-020 treatment groups were treated as technical replicates and averaged. The two technical replicates were performed to control for the stochastic and age (meaning length of time from astrocyte seeding)-dependent variability in each individual chip’s drainage between successive trials. Drainage data collected from pre-TGN-020 and post-TGN-020 chips was compared using paired analyses (see Statistical Analysis).
Immunohistochemistry and Confocal Microscopy:
After being studied in functional drainage experiments, all chips were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) diluted in endothelial basal medium-2. The chips were placed on a rocker at room temperature for 40 minutes during fixation, washed once with PBS, then immersed in a PBS bath at 4 °C overnight. A PBST (0.3% Triton X in PBS) solution was then added to each media reservoir for 45-60 minutes to permeabilize cells in the hydrogel. Permeabilized chips were blocked with 3% BSA diluted in PBS and stored at 4 °C overnight. They were next incubated with primary antibodies targeting GFAP (Abcam, 1:100, ab53554,), AQP4 (ThermoFisher Scientific, 1:150, PA5-53234), and CD31 (Dako, 1:100, GA610) diluted in a blocking buffer (3% BSA in PBS) at 4 °C overnight. All devices were then washed in a 4 °C PBS bath overnight. Secondary antibodies were diluted (ThermoFisher Scientific, 1:500) in 3% BSA and added to the media reservoirs. All devices were incubated with secondary antibodies overnight at 4 °C shielded from light. Finally, fully stained chips were washed for 24 hours in a light-protected PBS bath at 4 °C to remove any off-target background signal. All images were then acquired using a Lecia TCS SP8 confocal laser scanning microscope (Lecia Microsystems Inc., USA).
AQP4 Polarization Analysis:
To obtain a robust index of AQP4 polarization in the xy-plane, Z-stacks were acquired from each control and LPS-treated chip. Three images in the xy-plane were then chosen at random from each Z-stack for further analysis. The images were imported into ImageJ[53], and 25 one-dimensional regions of interest were selected along the x-axis (see Figure 4 for dimensions). The fluorescence intensity data at every location was averaged between the 25 lines, normalized to the peak AQP4 intensity signal found in the dataset, and compiled into a histogram. The division xy-plane AQP4 polarization index (API) was then calculated as the ratio between the average peak endfeet fluorescence and the average parenchymal fluorescence using Equation (1). This division xy-plane polarization index is very similar to that calculated by Munk et al. (2019).[44] We have also developed a secondary, subtraction-based xy-plane AQP4 polarization index (API) in which parenchymal AQP4 signal is subtracted from endfoot AQP4 signal, as shown in Equation (2).
| (1) |
| (2) |
For both indices, the “endfeet” were considered to represent the horizontal distance between the two local minima on either side of the fluorescence peaks that flank each blood vessel. When the width of the peak exceeded 20 μm, the endfeet were usually defined as the horizontal distance between the local minima on the parenchymal side and the point 20 μm closer to the local minima on the vessel side. However, for each analysis, fluorescence intensity data were compared with their respective images to appropriately define the endfeet. Additionally, to account for variability in fluorescence intensity emitted from individual chips, the intensity of images acquired from all control and LPS-treated chips were adjusted to be similar using the LAS X Life Science Microscope Software (Lecia Microsystems Inc., USA).
Post-Hoc Cell Counting:
To calculate the astrocyte cell density in each chip, 3D projections were acquired and imported into ImageJ (NIH)[53]. A ratio was then obtained between the total number of cells in the astrocyte parenchyma (counted by eye) and the total parenchymal area in each projection.
Statistical Analysis:
The two-sample comparisons shown in Figures 4D, 4E, and 4H were performed using unpaired, two-tailed homoscedastic t-tests. Results were considered significant at an α-value of 0.05. The four-sample comparison shown in Figures 5A and 5B were performed at each indicated times point (30 and 60 minutes) using a one-way ANOVA test with Dunnett’s multiple comparisons test. The paired analyses shown in Figures 5B, 5C, 5D, 5E, and 5F were compared using unpaired t-tests. Error bars used in the study are SEM (Standard Error of the Mean). All statistical analyses were run using GraphPad Prism version 9.0.
Supplementary Material
Acknowledgements
P.A.S., A.R.H, and E.L. designed the experiments. P.A.S and A.R.H. performed the experiments. P.A.S wrote the manuscript. All co-authors edited the manuscript and have given approval to the final version. The authors would like to thank Dr. Julia Felice and Dr. Nozomi Nishimura for helpful discussions and guidance throughout the research process. This work was performed in part at the Cornell NanoScale Science & Technology Facility, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (Grant NNCI-2025233). The authors (P.A.S., A.R.H., E.L.) would also like to thank the Cornell University Start-up fund, Nancy and Peter Meinig Family Investigator fund, and NIH grants (AI166772; CA252162; HL165135). P.A.S. was supported by the Cornell University College of Human Ecology Alumni Association (HEAA) through the Alan D. Mathios Research and Service Grant program. A.R.H. was supported by the NSF Graduate Research Fellowships Program (NSF GRFP) and the Cornell Neurotech Mong Fellowship.
Footnotes
Supporting Information
Supporting information is available from the Wiley Online Library or from the authors.
Conflict of Interest
All authors declare no competing interests.
Contributor Information
Paul A. Soden, College of Human Ecology, Cornell University, Ithaca, NY 14853, USA; Present Address: Boston Children’s Hospital, Enders Building/11th floor, 320 Longwood Ave, Boston, MA 02115, USA
Aria R. Henderson, Nancy E. and Peter C. Meinig School of Biomedical Engineering, Cornell University, Ithaca, NY 14853, USA
Esak Lee, Nancy E. and Peter C. Meinig School of Biomedical Engineering, Cornell University, Ithaca, NY 14853, USA.
Data Availability Statement
All source data are available in the main text or the supporting information. Additional data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
- [1].Kugler EC, Greenwood J, MacDonald RB, Frontiers in Cell and Developmental Biology 2021, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M, Science Translational Medicine 2012, 4, 147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Bedussi B, Almasian M, de Vos J, VanBavel E, Bakker EN, J Cereb Blood Flow Metab 2018, 38, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hannocks MJ, Pizzo ME, Huppert J, Deshpande T, Abbott NJ, Thorne RG, Sorokin L, J Cereb Blood Flow Metab 2018, 38, 669. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Harrison IF, Siow B, Akilo AB, Evans PG, Ismail O, Ohene Y, Nahavandi P, Thomas DL, Lythgoe MF, Wells JA, Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, Deane R, Nedergaard M, J Neurosci 2013, 33, 18190. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lochhead JJ, Wolak DJ, Pizzo ME, Thorne RG, J Cereb Blood Flow Metab 2015, 35, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M, Science (New York, N.Y.) 2013, 342, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Yang L, Kress BT, Weber HJ, Thiyagarajan M, Wang B, Deane R, Benveniste H, Iliff JJ, Nedergaard M, Journal of Translational Medicine 2013, 11, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yang J, Lunde LK, Nuntagij P, Oguchi T, Camassa LMA, Nilsson LNG, Lannfelt L, Xu Y, Amiry-Moghaddam M, Ottersen OP, Torp R, Journal of Alzheimer’s Disease 2011, 27, 711. [DOI] [PubMed] [Google Scholar]
- [5].Liu H, Qiu G, Zhuo F, Yu W, Sun S, Li F, Yang M, Biomed Res Int 2015, 2015, 471631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Watanabe-Matsumoto S, Moriwaki Y, Okuda T, Ohara S, Yamanaka K, Abe Y, Yasui M, Misawa H, Neurosci Res 2018, 133, 48. [DOI] [PubMed] [Google Scholar]
- [7].Dai J, Lin W, Zheng M, Liu Q, He B, Luo C, Lu X, Pei Z, Su H, Yao X, Molecular medicine reports 2017, 16, 1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, Singh I, Deane R, Nedergaard M, The Journal of neuroscience : the official journal of the Society for Neuroscience 2014, 34, 16180. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jessen NA, Munk ASF, Lundgaard I, Nedergaard M, Neurochemical research 2015, 40, 2583. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mestre H, Hablitz LM, Xavier AL, Feng W, Zou W, Pu T, Monai H, Murlidharan G, Castellanos Rivera RM, Simon MJ, Pike MM, Plá V, Du T, Kress BT, Wang X, Plog BA, Thrane AS, Lundgaard I, Abe Y, Yasui M, Thomas JH, Xiao M, Hirase H, Asokan A, Iliff JJ, Nedergaard M, eLife 2018, 7, e40070. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Reeves BC, Karimy JK, Kundishora AJ, Mestre H, Cerci HM, Matouk C, Alper SL, Lundgaard I, Nedergaard M, Kahle KT, Trends Mol Med 2020, 26, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, Axel L, Rusinek H, Nicholson C, Zlokovic BV, Frangione B, Blennow K, Ménard J, Zetterberg H, Wisniewski T, de Leon MJ, Nat Rev Neurol 2015, 11, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, Xie L, Kang H, Xu Q, Liew JA, Plog BA, Ding F, Deane R, Nedergaard M, Annals of neurology 2014, 76, 845. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Mogensen FL, Delle C, Nedergaard M, Int J Mol Sci 2021, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Fukuda AM, Pop V, Spagnoli D, Ashwal S, Obenaus A, Badaut J, Neuroscience 2012, 222, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lu DC, Zador Z, Yao J, Fazlollahi F, Manley GT, J Neurotrauma 2011, DOI: 10.1089/neu.2011.2114 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ren Z, Iliff JJ, Yang L, Yang J, Chen X, Chen MJ, Giese RN, Wang B, Shi X, Nedergaard M, Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2013, 33, 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hirose M, Asano M, Watanabe-Matsumoto S, Yamanaka K, Abe Y, Yasui M, Tokuda E, Furukawa Y, Misawa H, Neuroscience Research 2020, DOI: 10.1016/j.neures.2020.10.006. [DOI] [PubMed] [Google Scholar]
- [11].Zeppenfeld DM, Simon M, Haswell JD, D’Abreo D, Murchison C, Quinn JF, Grafe MR, Woltjer RL, Kaye J, Iliff JJ, JAMA Neurology 2017, 74, 91. [DOI] [PubMed] [Google Scholar]
- [12].Price BR, Norris CM, Sompol P, Wilcock DM, J Neurochem 2018, 144, 644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sabbatini M, Barili P, Bronzetti E, Zaccheo D, Amenta F, Mechanisms of Ageing and Development 1999, 108, 165. [DOI] [PubMed] [Google Scholar]
- [14].a) Sofroniew MV, Vinters HV, Acta Neuropathologica 2010, 119, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sun D, Jakobs TC, The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry 2012, 18, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Erickson MA, Hartvigson PE, Morofuji Y, Owen JB, Butterfield DA, Banks WA, Journal of neuroinflammation 2012, 9, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Manouchehrian O, Ramos M, Bachiller S, Lundgaard I, Deierborg T, Journal of neuroinflammation 2021, 18, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Thrane AS, Rangroo Thrane V, Nedergaard M, Trends in neurosciences 2014, 37, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT, Alzheimer’s & dementia (New York, N. Y.) 2018, 4, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Hughes C, Choi ML, Yi J-H, Kim S-C, Drews A, George-Hyslop PS, Bryant C, Gandhi S, Cho K, Klenerman D, Communications Biology 2020, 3, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jones RS, Minogue AM, Connor TJ, Lynch MA, J Neuroimmune Pharmacol 2013, 8, 301. [DOI] [PubMed] [Google Scholar]
- [18].Silva I, Silva J, Ferreira R, Trigo D, Neurological Research and Practice 2021, 3, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hablitz LM, Plá V, Giannetto M, Vinitsky HS, Stæger FF, Metcalfe T, Nguyen R, Benrais A, Nedergaard M, Nature Communications 2020, 11, 4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Eidsvaag VA, Enger R, Hansson HA, Eide PK, Nagelhus EA, Glia 2017, 65, 964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Sun C, Lin L, Yin L, Hao X, Tian J, Zhang X, Ren Y, Li C, Yang Y, Frontiers in Immunology 2022, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Igarashi H, Huber VJ, Tsujita M, Nakada T, Neurol Sci 2011, 32, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li J, Jia Z, Xu W, Guo W, Zhang M, Bi J, Cao Y, Fan Z, Li G, Life Sciences 2019, 222, 148. [DOI] [PubMed] [Google Scholar]; d) Igarashi H, Tsujita M, Suzuki Y, Kwee IL, Nakada T, NeuroReport 2013, 24, 324. [DOI] [PubMed] [Google Scholar]; e) Nakamura Y, Suzuki Y, Tsujita M, Huber VJ, Yamada K, Nakada T, ACS Chemical Neuroscience 2011, 2, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Rosu G-C, Catalin B, Balseanu TA, Laurentiu M, Claudiu M, Kumar-Singh S, Daniel P, Molecular Neurobiology 2020, 57, 4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Henderson AR, Ilan IS, Lee E, Microcirculation 2021, DOI: 10.1111/micc.12730e12730 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nguyen D-HT, Lee E, Alimperti S, Norgard RJ, Wong A, Lee JJ-K, Eyckmans J, Stanger BZ, Chen CS, Science advances 2019, 5, eaav6789. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kwak TJ, Lee E, Biofabrication 2020, DOI: 10.1088/1758-5090/abbd28 [DOI] [PubMed] [Google Scholar]; d) Kwak TJ, Lee E, Sci Rep 2020, 10, 20142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhou B, Zuo Y-X, Jiang R-T, CNS Neuroscience & Therapeutics 2019, 25, 665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Ahn SI, Sei YJ, Park H-J, Kim J, Ryu Y, Choi JJ, Sung H-J, MacDonald TJ, Levey AI, Kim Y, Nature Communications 2020, 11, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Placone AL, McGuiggan PM, Bergles DE, Guerrero-Cazares H, Quiñones-Hinojosa A, Searson PC, Biomaterials 2015, 42, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Balasubramanian S, Packard JA, Leach JB, Powell EM, Tissue engineering. Part A 2016, 22, 885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Kettenmann H, Verkhratsky A, Fortschr Neurol Psychiatr 2011, 79, 588. [DOI] [PubMed] [Google Scholar]; b) Bushong EA, Martone ME, Jones YZ, Ellisman MH, The Journal of Neuroscience 2002, 22, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yoon H, Walters G, Paulsen AR, Scarisbrick IA, PloS one 2017, 12, e0180697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Xing L, Yang T, Cui S, Chen G, Frontiers in molecular neuroscience 2019, 12, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liang Z, Wang X, Hao Y, Qiu L, Lou Y, Zhang Y, Ma D, Feng J, Frontiers in Neurology 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lagos-Cabré R, Burgos-Bravo F, Avalos AM, Leyton L, Frontiers in Pharmacology 2020, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Clasadonte J, Scemes E, Wang Z, Boison D, Haydon PG, Neuron 2017, 95, 1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].a) Gautam J, Zhang X, Yao Y, Scientific Reports 2016, 6, 36450. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lau LW, Cua R, Keough MB, Haylock-Jacobs S, Yong VW, Nature Reviews Neuroscience 2013, 14, 722. [DOI] [PubMed] [Google Scholar]; c) Yurchenco PD, Curr Top Membr 2015, 76, 1. [DOI] [PubMed] [Google Scholar]
- [31].Benarroch EE, Neurology 2015, 85, 1417. [DOI] [PubMed] [Google Scholar]
- [32].Konopka A, Zeug A, Skupien A, Kaza B, Mueller F, Chwedorowicz A, Ponimaskin E, Wilczynski GM, Dzwonek J, PloS one 2016, 11, e0155053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Thomsen MS, Birkelund S, Burkhart A, Stensballe A, Moos T, J Neurochem 2017, 140, 741. [DOI] [PubMed] [Google Scholar]
- [34].Morawski M, Dityatev A, Hartlage-Rübsamen M, Blosa M, Holzer M, Flach K, Pavlica S, Dityateva G, Grosche J, Brückner G, Schachner M, Philosophical Transactions of the Royal Society B: Biological Sciences 2014, 369, 20140046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].a) Okuda H, Tatsumi K, Morita S, Shibukawa Y, Korekane H, Horii-Hayashi N, Wada Y, Taniguchi N, Wanaka A, The Journal of biological chemistry 2014, 289, 2620. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pyka M, Wetzel C, Aguado A, Geissler M, Hatt H, Faissner A, European Journal of Neuroscience 2011, 33, 2187. [DOI] [PubMed] [Google Scholar]
- [36].a) Brown TD, Nowak M, Bayles AV, Prabhakarpandian B, Karande P, Lahann J, Helgeson ME, Mitragotri S, Bioengineering & translational medicine 2019, 4, e10126. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Campisi M, Shin Y, Osaki T, Hajal C, Chiono V, Kamm RD, Biomaterials 2018, 180, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Choi J-H, Santhosh M, Choi J-W, Micromachines 2019, 11, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Herland A, van der Meer AD, FitzGerald EA, Park T-E, Sleeboom JJF, Ingber DE, PloS one 2016, 11, e0150360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Syková E, Vorísek I, Mazel T, Antonova T, Schachner M, The European journal of neuroscience 2005, 22, 1873. [DOI] [PubMed] [Google Scholar]
- [38].a) Chan SJ, Niu W, Hayakawa K, Hamanaka G, Wang X, Cheah PS, Guo S, Yu Z, Arai K, Selim MH, Kurisawa M, Spector M, Lo EH, STEM CELLS Translational Medicine 2019, 8, 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Diniz L, Matias I, Siqueira M, Stipursky J, Gomes F, Molecular Neurobiology 2019, 56. [DOI] [PubMed] [Google Scholar]; c) Kornblum HI, Hussain R, Wiesen J, Miettinen P, Zurcher SD, Chow K, Derynck R, Werb Z, J Neurosci Res 1998, 53, 697. [DOI] [PubMed] [Google Scholar]
- [39].Haj-Yasein NN, Vindedal GF, Eilert-Olsen M, Gundersen GA, Skare Ø, Laake P, Klungland A, Thorén AE, Burkhardt JM, Ottersen OP, Nagelhus EA, Proc Natl Acad Sci U S A 2011, 108, 17815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Camassa LMA, Lunde LK, Hoddevik EH, Stensland M, Boldt HB, De Souza GA, Ottersen OP, Amiry-Moghaddam M, Glia 2015, 63, 2073. [DOI] [PubMed] [Google Scholar]
- [41].a) Yang T, Dai Y, Chen G, Cui S, Frontiers in Cellular Neuroscience 2020, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bradbury EJ, Burnside ER, Nat Commun 2019, 10, 3879. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jones EV, Bouvier DS, Neural Plast 2014, 2014, 321209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Taylor X, Cisternas P, Jury N, Martinez P, Huang X, You Y, Redding-Ochoa J, Vidal R, Zhang J, Troncoso J, Lasagna-Reeves CA, Communications Biology 2022, 5, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Johnson ECB, Carter EK, Dammer EB, Duong DM, Gerasimov ES, Liu Y, Liu J, Betarbet R, Ping L, Yin L, Serrano GE, Beach TG, Peng J, De Jager PL, Haroutunian V, Zhang B, Gaiteri C, Bennett DA, Gearing M, Wingo TS, Wingo AP, Lah JJ, Levey AI, Seyfried NT, Nature Neuroscience 2022, 25, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Munk AS, Wang W, Bèchet NB, Eltanahy AM, Cheng AX, Sigurdsson B, Benraiss A, Mäe MA, Kress BT, Kelley DH, Betsholtz C, Møllgård K, Meissner A, Nedergaard M, Lundgaard I, Cell Rep 2019, 26, 2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Moftakhar P, Lynch MD, Pomakian JL, Vinters HV, J Neuropathol Exp Neurol 2010, 69, 1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kapoor S, Kim SM, Farook JM, Mir S, Saha R, Sen N, J Neurosci 2013, 33, 17398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].a) Igarashi H, Suzuki Y, Kwee IL, Nakada T, Neurol Res 2014, 36, 1094. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nakada T, Igarashi H, Suzuki Y, Kwee I, Neurology 2014, 82, S58.001 [Google Scholar]; c) Suzuki Y, Nakamura Y, Yamada K, Igarashi H, Kasuga K, Yokoyama Y, Ikeuchi T, Nishizawa M, Kwee IL, Nakada T, PLoS One 2015, 10, e0123708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Xiang J, Tang Y, Li C, Su EJ, Lawrence DA, Keep RF, Acta Neurochir Suppl 2016, 121, 19. [DOI] [PubMed] [Google Scholar]
- [49].Erikson K, Tuominen H, Vakkala M, Liisanantti JH, Karttunen T, Syrjälä H, Ala-Kokko TI, Critical Care 2020, 24, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50] <j/>a).Albargothy NJ, Johnston DA, MacGregor-Sharp M, Weller RO, Verma A, Hawkes CA, Carare RO, Acta Neuropathol 2018, 136, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bakker ENTP, Naessens DMP, VanBavel E, Experimental physiology 2019, 104, 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH, Weller RO, Neuropathol Appl Neurobiol 2008, 34, 131. [DOI] [PubMed] [Google Scholar]; d) Hladky SB, Barrand MA, Fluids Barriers CNS 2014, 11, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Smith AJ, Verkman AS, FASEB J 2018, 32, 543. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Weller RO, Sharp MM, Christodoulides M, Carare RO, Møllgård K, Acta Neuropathol 2018, 135, 363. [DOI] [PubMed] [Google Scholar]
- [51].a) Faghih MM, Sharp MK, Fluids Barriers CNS 2018, 15, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Holter KE, Kehlet B, Devor A, Sejnowski TJ, Dale AM, Omholt SW, Ottersen OP, Nagelhus EA, Mardal KA, Pettersen KH, Proc Natl Acad Sci U S A 2017, 114, 9894. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jin B-J, Smith AJ, Verkman AS, Journal of General Physiology 2016, 148, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].a) Bedussi B AM., de Vos J, VanBavel E, & Bakker EN , Journal of Cerebral Blood Flow & Metabolism 2018, 38, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tithof J, Kelley DH, Mestre H, Nedergaard M, Thomas JH, Fluids Barriers CNS 2019, 16, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Schneider CA, Rasband WS, Eliceiri KW, Nat Methods 2012, 9, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All source data are available in the main text or the supporting information. Additional data that support the findings of this study are available from the corresponding authors upon reasonable request.