

Abstract
In this paper, we report the structural analysis of dihydroorotase (DHOase) from the hyperthermophilic and barophilic archaeon Methanococcus jannaschii. DHOase catalyzes the reversible cyclization of N‐carbamoyl‐l‐aspartate to l‐dihydroorotate in the third step of de novo pyrimidine biosynthesis. DHOases form a very diverse family of enzymes and have been classified into types and subtypes with structural similarities and differences among them. This is the first archaeal DHOase studied by x‐ray diffraction. Its structure and comparison with known representatives of the other subtypes help define the structural features of the archaeal subtype. The M. jannaschii DHOase is found here to have traits from all subtypes. Contrary to expectations, it has a carboxylated lysine bridging the two Zn ions in the active site, and a long catalytic loop. It is a monomeric protein with a large β sandwich domain adjacent to the TIM barrel. Loop 5 is similar to bacterial type III and the C‐terminal extension is long.
Keywords: crystal structure, dihydroorotase, Methanococcus jannaschii, pyrimidine biosynthesis
1. INTRODUCTION
Dihydroorotases (DHOase) catalyze the third reaction of de novo pyrimidine biosynthesis, the reversible cyclization of N‐carbamoyl‐l‐aspartate (CA) to form l‐dihydroorotate (DHO). 1 , 2 They are Zn metalloenzymes and members of the amidohydrolase superfamily of proteins. 3 The core domain of all DHOases has the (βα)8‐barrel fold (TIM barrel). 3 Even though all DHOases share a common structural fold and catalyze the same reaction, they show high structural diversity from each other. 3 , 4 An early phylogenetic analysis classified DHOases into two major types. 4 Long (type I) are more ancient and larger with molecular weight ~45 kDa. Some function independently, others associate noncovalently with aspartate transcarbamoylase (ATCase), for example, Aquifex aeolicus DHOase (AaDHOase), and some are covalently linked to another enzyme of the pathway, for example, CAD in mammals which also has carbamoyl phosphate synthetase (CPSase) and ATCase activities. Short (type II) are more recent with molecular weight ~38 kDa. They are found in bacteria, fungi, and plants. Within a type, the sequence identity is ~40%. The sequence identity between the two types is <20%.
The first DHOase whose structure was determined was the enzyme from Escherichia coli (EcDHOase). 5 It is type II and a homodimer. Each subunit folds into the characteristic TIM barrel which is connected to an adjacent domain of two antiparallel β‐strands. The active site has two Zn ions coordinated by four invariant His residues and one invariant Asp. The two Zn ions are bridged by a carboxylated Lys residue (KCX) as well as a water molecule. In this structure, one subunit has CA bound and the other has DHO. This study allowed a structural description of the catalytic mechanism which was further biochemically verified. 6 It is a cyclization reaction where the substrate CA is stabilized by the active site Zn ions and a number of invariant active site residues and main chain atoms. The carboxylate of a conserved aspartate abstracts a proton from the amide nitrogen of CA allowing for a nucleophilic attack of the amide nitrogen on its carboxylate. Further structural work showed that a long flexible loop was also involved in catalysis, closing the active site as a lid when the substrate CA is bound. 7
The first structure of a type I DHOase was that of the eubacterium A. aeolicus. 8 This DHOase is active only in complex with ATCase. 9 The structure of the DHOase‐ATCase complex was a dodecamer consisting of two ATCase trimers and three DHOase dimers. 10 The DHOase active site contained only one Zn ion and the carboxylated lysine was replaced by an aspartate. The structure had a short flexible loop exposing the active site.
Since these first structures, several others have been determined. A more recent phylogenetic analysis was consistent with the earlier classification. 11 Long DHOases subdivide into archaeal, bacterial type I, bacterial type III, human CAD, and inactive CAD in fungi. Short DHOases correspond to bacterial type II, plant and active DHOases in fungi. Furthermore, the dihydroorotase structures in the Protein Data Bank were assigned into these subtypes. 12 These were 63 structures from 13 organisms giving 4 non‐redundant bacterial type I, 6 non‐redundant bacterial type II, 1 bacterial type III, 1 human DHOase, and 1 inactive from fungi. In this diverse family, each subtype has specific structural characteristics. More recently, the structure of an active fungal DHOase from Saccharomyces cerevisiae (ScDHOase) was determined. 13 At present, the only subtypes without representative structures are archaeal and plant DHOases.
In this study, we report the first crystal structure of an archaeal DHOase, that from the hyperthermophilic and barophilic Methanococcus jannaschii (MjDHOase). Its structural analysis adds to our understanding of the variability within the DHOase family of proteins. 11 , 14 Here, we focus on the similarities and differences between the archaeal DHOase and known representatives of all subtypes. Contrary to expectations from the phylogenetic analysis 11 and a recent physicochemical study, 14 we find that the two Zn ions in the active site are bridged with a carboxylated lysine, and the structure has a long flexible loop.
2. MATERIALS AND METHODS
2.1. Protein preparation and crystallization
The purification was carried out as previously described from Rosetta‐gami 2 (DE3) cells harboring the pET‐21a plasmid with the M. jannaschii pyrC gene. 14 The purification involved a 35% ammonium sulfate precipitation step, a heat step at 85°C for 15 min and chromatography using an SP cation exchange column and a phenyl sepharose column. Initial crystallization conditions were obtained under oil by the High Throughput Crystallization Screening Center of the Hauptman Woodward Institute that samples 1536 different chemical conditions. The hits were modified for hanging drop and optimized by varying the precipitant concentration and buffer pH. For the crystals used for this study, the protein had concentration 6.7 mg/ml and the protein buffer was 50 mM Tris Cl, pH 7.5, 2 mM BME, 0.05 mM Zn acetate. 5‐Fluoroorotate was then added from a 10 mM aqueous solution so that the protein: 5‐fluoroorotate molar ratio would be 1:20. The reservoirs contained 35% PEG400, 0.1 M Na Hepes pH 7.5, 0.1 M Zn acetate. The drops consisted of 2 μl protein solution and 1 μl reservoir solution.
2.2. Data collection and processing
The crystal was prismatic and was mounted in a 0.8 mm quartz capillary by conventional means. 15 X‐ray data were measured at the ALS beamline 4.2.2 at a temperature ~298 K by irradiating four different parts of the crystal in a sweep of 180° corresponding to 360 frames using 0.5°/frame. The crystal to detector distance was 200 mm and the wavelength used was 1.0 Å. Images were processed with XDS. 16 Structure amplitudes were obtained by scaling and merging intensities using Aimless. 17 Processing Statistics are shown in Table 1. The crystals are trigonal P32 2 1 with cell constants a = b = 111.27 Å and c = 101.23 Å.
TABLE 1.
Data collection and final refinement statistics
| Data collection statistics | |
|---|---|
| X‐ray source | ALS 4.2.2 |
| Wavelength (Å) | 1.000 |
| Total oscillation angle (°) | 180 |
| Resolution (Å) | 48.18–1.90 (1.94–1.90) |
| Space group | P32 2 1 |
| Unit cell constants (Å) | a = b = 111.27, c = 101.23 |
| No. of observations | 625 862 (36 672) |
| Unique reflections | 57 394 (3683) |
| Multiplicity | 10.9 (10.0) |
| <I/σ(I)> | 9.7 (0.7) |
| R merge a (within I+/I−) | 0.227 (3.042) |
| (all I+ and I−) | 0.239 (3.188) |
| CC1/2 | 0.997 (0.310) |
| Completeness (%) | 100.0 (100.0) |
| Anomalous multiplicity | 5.4 (4.9) |
| Anomalous completeness (%) | 100.0 (100.0) |
| Final refinement statistics | |
| Resolution range (Å) | 48.18–1.90 |
| Reflections used in refinement | 57 234 |
| Reflections used for R‐free | 2703 |
| R work | 0.178 |
| R free | 0.215 |
| Wilson B‐factor (Å2) | 27.8 |
| Number of atoms/average B‐factors (Å2) | 3619/33.1 |
| Protein | 3389/32.6 |
| Ions (Zn) | 11/33.1 |
| Waters | 219/40.3 |
| Protein residues | 423 |
| RMSD bond length (Å) | 0.008 |
| RMSD bond angles (°) | 0.94 |
| Ramachandran plot | |
| Favored (%) | 95.93 |
| Outliers (%) | 0.96 |
| Rotamer outliers (%) | 0.26 |
| Clashscore | 2.32 |
Note: Values in parentheses are for the outer shell.
The merging R factor is defined as R merge = ∑ hkl ∑ i |I i (hkl) − < I(hkl) > | / ∑ hkl ∑ i I i (hkl).
2.3. Structure solution and refinement
Structure solution and refinement were carried out using PHENIX. 18 The structure was determined by molecular replacement with PHASER. The search model was chain A of the Bacillus anthracis DHOase 19 (BaDHOase, PDB: 3MPG), which has only 30% sequence identity with MjDHOase. All residues that were dissimilar in the two proteins were replaced with Ala in the search model. Iterative density modification, model building and refinement with AUTOBUILD starting from the molecular replacement solution gave a partial model. The remaining of the structure was manually built with Coot 20 and the refinement was carried out with phenix.refine. As the refinement progressed, 219 waters were added to the model with phenix.refine. Also, 11 Zn ions were found from electron density difference maps and were included in the refinement with variable occupancy factors. We did not find electron density that could be interpreted as 5‐fluoroorotate. Lys137 was found to be carboxylated. As the data showed significant anomalous signal for resolutions lower than 3.22 Å, the Zn sites were also directly determined from the anomalous data to 4 Å resolution with HySS in PHENIX and were at the same positions as those from the difference maps except for Zn508 and Zn509 for which only one Zn site was found by HySS in between the two. The final discrepancy indices are R = 0.178 and R free = 0.215. The quality of the structure was analyzed with MolProbity 21 as implemented in Phenix. The metal‐binding sites were validated with the CheckMyMetal (CMM) server. 22 , 23
3. RESULTS AND DISCUSSION
3.1. Overall structure and structure quality
The model presented in this paper consists of 3619 non‐hydrogen protein atoms in 423 amino acids, 11 Zn ions, and 219 water molecules. A summary of the model refinement statistics is given in Table 1. The Ramachandran plot showed that 95.93% of the residues lie in the most favored regions and only four residues are outliers with φ and ψ angles of 172.7° and 117.8° for Phe148; 24.7° and 69.6° for His227; −157.9° and −0.7° for Gly324; and −173.5° and 26.7° for Lys422. The electron density for Phe148, His227, Gly324 is well defined but poorly defined for Lys422. A representative section of the 2mFo‐DFc map is shown in Figure 1A. The final map, contoured at 1.2σ shows continuous electron density for all main and side chain atoms with a few exceptions. There is a gap in the electron density between residues Lys10 and Asp11, there is no electron density for the side chain of a few surface groups, mostly Lys and Glu, and the last two residues are poorly defined.
FIGURE 1.
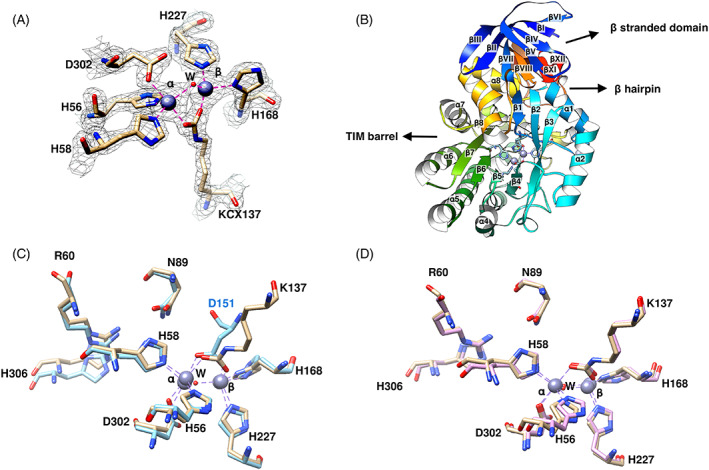
(A) The Zn‐binding site of MjDHOase superimposed on a 2mFo‐DFc electron density map contoured at 1.7σ. Zinc ions are presented as gray spheres, water molecules (W) are represented as red spheres, and coordination bonds are shown as magenta dashed lines. This figure was drawn with UCSF Chimera. The volume viewer tool was used for the electron density map. (B) Schematic ribbons diagram of MjDHOase. The protein is shown with rainbow colors from blue at the N‐terminus to red at the C‐terminus. The TIM barrel, its adjacent β stranded domain and the β hairpin are indicated with arrows. Most of the β hairpin (in orange color) is concealed in the back of the drawing as well as strands βIX and βX. The two active‐site Zn ions and the residues that coordinate them are also included in this figure. (C) Superposition of the active site of MjDHOase (wheat carbon atoms) and BaDHOase (cyan carbon atoms, PDB: 3MPG, chain A). (D) Superposition of the active site of MjDHOase (wheat carbon atoms) and EcDHOase (pink carbon atoms, PDB: 1XGE, chain A). The active site is highly conserved, and it is composed of the Zn‐binding residues H56, H58, H168, H227, D302, KCX137, and a water W molecule. The water in the active site of BaDHOase was not determined. In addition, the substrate binding residues N89, R60, H306 are shown but the main chain of residues 275 and 320 that also interact with the substrate are not. KCX137 in MjDHOase corresponds to KCX102 of EcDHOase as seen in (D) and its carboxylate group overlaps the carboxylate of D151 in BaDHOase as seen in (C). Coordinate bonds are drawn in purple dashed lines in (C) and (D)
The DHOase structure (Figure 1B) consists of a (βα)8 barrel (TIM barrel), residues 52–337, which is characteristic of the amidohydrolase superfamily, 3 connected to an adjacent β sandwich domain formed by residues 1–51 from the N‐terminal extension and residues 338–423 from the C‐terminal extension. The outer β sheet of the extra domain consists of the four strands βI, βIV, βV, and βVI from the N‐terminal extension and the inner consists of six strands, three from the N‐terminal extension βII, βIII, and βVII and three from the C‐terminal extension βVIII, βXI, and βXII. The C‐terminal extension also contributes a β hairpin between βIX and βX, that protrudes on one side of the TIM barrel toward the top of it. The labeling of the helices and strands in this paper follows Grande‐Garcia et al. 11 for ease of comparison.
The biological unit of MjDHOase is a monomer. The complexation significance score in PISA 24 for the interfaces between symmetry‐related molecules in the crystal ranges between 0.000 and 0.001, indicating that these interfaces are not significant for complex formation and are the result of crystal packing. A separate evaluation of these interfaces with the EPPIC server 25 also indicated that the biological unit of DHOase is a monomer. This is consistent with earlier SEC‐LS results in solution. 14
The active site (Figure 1A) is in a pocket at the carboxy end of the TIM barrel (Figure 1B), and contains two Zn ions, as expected from ICP‐MS studies. 14 Znα has trigonal bipyramidal coordination and Znβ has tetrahedral. Znα is coordinated by His56 and His58 and Znβ by His227 and His168. The carboxylate group of KCX137 coordinates both Zn ions. Asp302 also coordinates Znα. In addition, a water molecule (HOH692) coordinates the two Zn ions.
The carboxylation of Lys137 was unexpected from LC–MS studies. 14 According to these studies, Lys137 was not carboxylated. In addition, the carboxylation of Lys137 was surprising because of the expected similarity of archaeal DHOases with bacterial type I, 11 in which an Asp residue coordinates the two Zn ions in the active site. 19 The earlier homology model was built with an aspartate bridging the two Zn ions by analogy to BaDHOase. 14 Figure 1C,D shows the superposition of the active site of MjDHOase with B. anthracis and E. coli DHOase, respectively, and demonstrate a closer resemblance of the active site with EcDHOase (bacterial type II) rather than BaDHOase (bacterial type I). It may be noted in Figure 1C that the carboxylate group of Asp151 in BaDHOase overlaps the carboxylate group of KCX137 in MjDHOase.
The active site residues His56, His58, His168, His227, and Asp302, that coordinate the two Zn ions (Figure 1A), are invariant among DHOase structures. 5 , 11 , 12 , 13 , 14 KCX137 is invariant in bacterial type II, III, and human, 11 as well as active fungal 13 DHOases. The aspartate that coordinates the two Zn ions in bacterial type I, for example, BaDHOase, is invariant in this subtype. 11 Residues Arg60, Asn89, His306 in the active site, which are included in Figure 1C,D, interact with the substrate in the known active DHOase structures and are invariant. 5 , 11 , 26 , 27 The substrate also interacts with the main chain residues at 275 and 320, which are not shown.
It may be noted that, apart from the two active site Zn ions, the remaining nine are on the surface of the protein and are responsible for crystal packing.
3.2. Comparison with other dihydroorotases
The structure based multiple sequence alignment of MjDHOase with known representatives from the other active DHOase types and subtypes is given in Figure 2A. The structure based multiple sequence alignment was computed with the Match ➔ Align tool in UCSF Chimera 28 after the corresponding structures were superimposed with the matchmaker tool. An identity matrix giving a comparison of the sequence identity and root mean square deviation (RMSD) between corresponding Cα atoms for any two of these structures is given in Figure 2B. The RMSD's were computed for the 268 fully populated columns in the alignment (atoms with Cα…Cα distance <5 Å). The sequence identities and RMSD's of MjDHOase with these proteins are comparable with the corresponding values among them. The furthest relatives from the long DHOases are the bacterial type II (E. coli) and active fungal (S. cerevisiae) DHOases. These observations are consistent with archaeal DHOases being a separate subtype of long DHOases. 11 Even though the highest sequence identity of MjDHOase is with BaDHOase, the archaeal enzyme exhibits some traits similar to the other subtypes.
FIGURE 2.
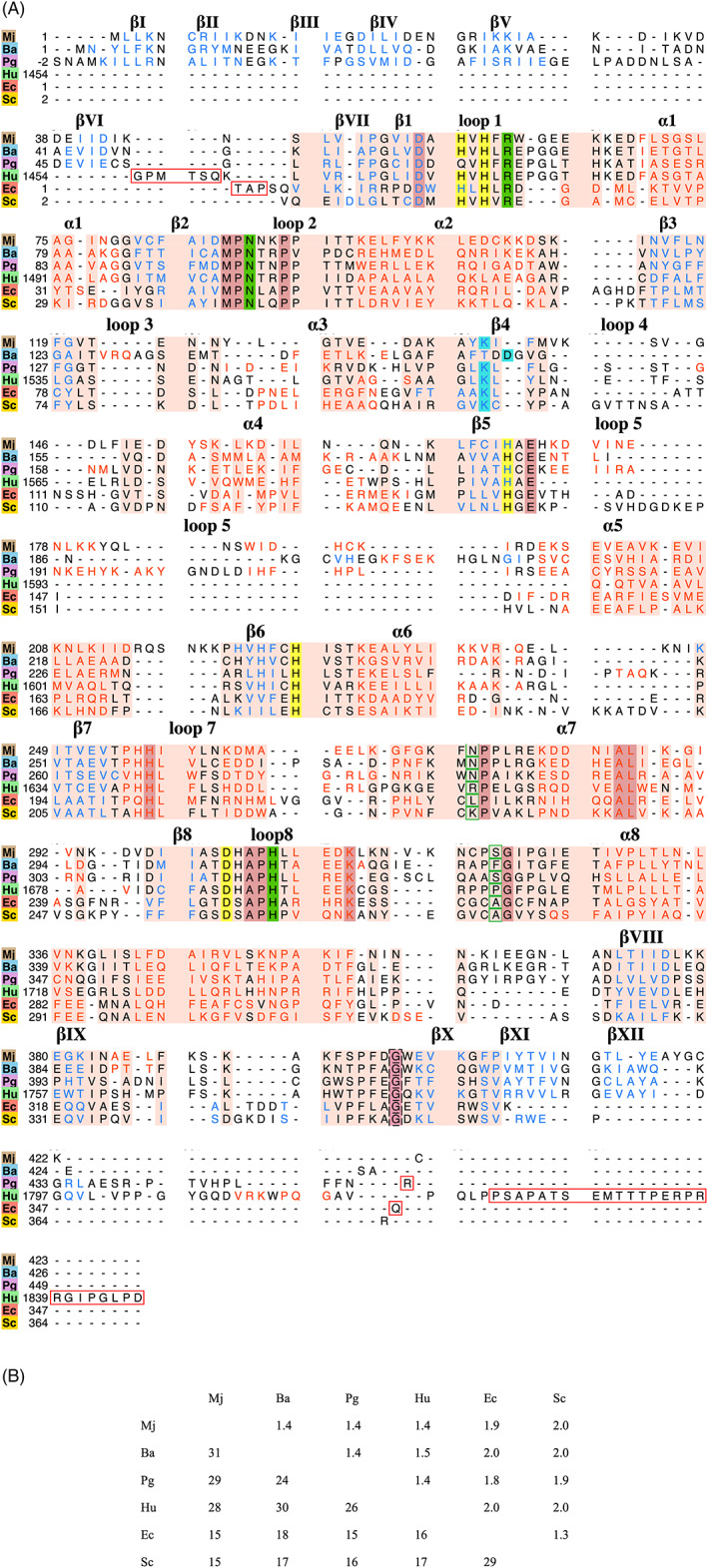
(A) Structure‐based multiple sequence alignment of MjDHOase with representatives of the different DHOase types: Ba (Bacillus anthracis, PDB: 3MPG chain A, bacterial type I), Pg (Porphyromonas gingivalis, PDB: 2GWN, bacterial type III), hu (PDB: 4C6C, human CAD), Ec (Escherichia coli, PDB: 1XGE chain A, bacterial type II), and Sc (Saccharomyces cerevisiae, PDB: 6L0J chain A, active fungal). The secondary structural elements are shown with blue lettering for β strands, red lettering for helices, and black lettering for loops. The secondary structural elements of the TIM barrel are labeled β1 to α8. The β strands in the adjacent domain are labeled as βI to βXII. Helices and β strands within loops are not labeled. Invariant residues involved in zinc binding are highlighted in bright yellow. Invariant residues involved in substrate binding are highlighted in dark green and invariant positions that interact with the substrate via their main chain (but not invariant in sequence) are shown within green border. Other residues that show 100% identity in all sequences are highlighted in purple. The carboxylated lysine which is invariant in archaeal, bacterial type II and III, human and active fungal is highlighted in cyan color. The invariant aspartate in bacterial type I is also highlighted in cyan color. Regions depicting missing structure are enclosed within red border. The salmon background corresponds to the fully populated columns. This figure was drawn with UCSF Chimera. (B) An identity matrix giving the sequence identity and RMSD between corresponding Cα's in the 265 fully populated columns between any two of the structures in (A). % sequence identity is below the diagonal on the bottom left and RMSD (Å) is above the diagonal on the top right. The diagonal (100% for sequence identity and 0.0 Å for RMSD) is not included
3.2.1. Adjacent domain
A major difference between the two DHOase types is the molecular weight which correlates with extra residues at the N‐ and C‐termini in long compared to short DHOases. The extra amino acids in long bacterial DHOases typically create a large β sandwich domain adjacent to the TIM barrel. The structures of the adjacent domains were reviewed. 11 , 12 The domain adjacent to the TIM barrel in MjDHOase is large, containing an outer β sheet of four strands from the N‐terminal extension and an inner β sheet of six strands, three from the N‐terminal extension, and three from the C‐terminal extension (Figure 1B). The architecture of the domain is mostly similar to bacterial type I (BaDHOase). The domain in bacterial type III (Porphyromonas gingivalis DHOase, PgDHOase) is larger with four strands in the outer β sheet and seven in the inner due to a longer C‐terminal extension. In contrast to the long bacterial DHOases, the human DHOase has a smaller N‐terminal and a longer C‐terminal extension and this results in an adjacent domain with only a five‐stranded antiparallel β sheet. The longer C‐terminal extension is responsible for protein solubility. 29 Short E. coli and S. cerevisiae DHOases have only two and three strands, respectively, in the antiparallel β sheet of the adjacent domain. 11 , 13 The role of the large adjacent domain in long DHOases is not known. 11
3.2.2. Active site
The two Zn ions in the active site of MjDHOase are bridged with a carboxylated lysine (KCX137) (Figure 1A–D). This is similar to bacterial types II 5 , 7 , 12 and III, human, 11 and active fungal 13 DHOases, in which this carboxylated lysine is invariant. In contrast, bacterial type I use an aspartate, that is invariant in this subtype, to bridge the two Zn ions. 19 There is some variability in the number of Zn ions among DHOases. Some bacterial type I have only one Zn in the active site, for example, AaDHOase, and this aspartate forms a coordinate bond with it. 8 , 10 , 30 The human DHOase has three Zn ions, with the third Zn ion also involved in catalysis. 11 Furthermore, in contrast to all other DHOase structures, there is not an α3 helix in MjDHOase, and the latter has only a loop with fewer residues between β3 and β4. These two attributes bring KCX137 at the right position to coordinate the two Zn ions. In the aligned sequences in Figure 2A, the invariant Asp151 of BaDHOase neighbors the carboxylated lysine in MjDHOase, in bacterial types II and III, in active fungal, and in human DHOases. Also, as shown in Figure 1C, the carboxylate group of KCX137 overlaps the carboxylate group of Asp151 of BaDHOase. MjDHOase has two histidine residues coordinating the α Zn ion like the other DHOases, except for bacterial type III in which one of the histidines is replaced by glutamine.
3.2.3. Flexible loop
Loop 4 is flexible, and studies have shown that it is part of the catalytic cycle. 7 , 11 , 13 It shows high variability in size, sequence, and structure among DHOases. The loop in MjDHOase is long and similar in length to bacterial type III and human DHOases. This loop is longer in short DHOases, both bacterial type II and active fungal. Studies of bacterial type II and human DHOases with bound ligands show that in the presence of the substrate in the biosynthetic direction, that is, CA, 5 , 7 , 11 or its analogs, 12 this loop undergoes a conformational change to a closed conformation enclosing the active site and stabilizing the substrate in it. In the apo form 11 , 12 , 31 or in the presence of the substrate in the degradative direction, that is, DHO, 7 , 11 or its analogs, 11 , 31 this loop is in an open conformation exposing the active site. This loop is in the open conformation in the apo form of bacterial type III (PgDHOase). In the current structure, the active site is exposed, which is similar to what is found in the apo form of these DHOases. In contrast, bacterial type I DHOases have a shorter loop 4 leaving the active site open irrespective of whether CA is bound or not. 19 , 26 Surprisingly, the fungal DHOase from S. cerevisiae has been found only in the closed form in the presence of analogs of both its natural substrates, 13 , 32 , 33 interacting with them, but no structure of the apo form or in the presence of the natural substrates is known. The interaction of the catalytic loop with the substrate CA in bacterial type II 7 and its analog, malate, in active fungal 13 involves the two Thr residues of the loop. Human DHOase has one Thr residue in the flexible loop, which also interacts with the substrate CA. 11 By inference, M. jannaschii and P. gingivalis DHOases are expected to adopt the closed conformation upon binding of the substrate CA and interact with it, MjDHOase using Ser143 and PgDHOase using one or both of Ser155 and Thr156. Loop 4 in type I cannot seal the active site from the solvent, but the Asp that replaces KCX in them is followed by a conserved Gly which could interact with CA. It was also speculated that this may not be needed for bacterial type I DHOases as the active site of AaDHOase faces an electropositive reaction chamber in the complex with ATCase sealing it from the exterior, and all type I DHOases may associate with ATCase in a similar manner. 11
3.2.4. Loop 5
Loop 5 in MjDHOase is structurally similar to PgDHOase (bacterial type III) except that the loop in the latter is longer by five additional residues at the end of the first helix of the loop. Both are different from all other DHOases. Loop 5 is structurally similar among the known bacterial type I DHOases. 11 It is involved in interactions with ATCase in AaDHOase 7 and possibly in BaDHOase 34 and, because of the similarity of this loop among bacterial type I DHOases, it was suggested that they all are posed to interact with ATCase in a similar manner. 11 This idea is also supported by the short loop 4 in bacterial type I DHOases. Such an interaction is unlikely for MjDHOase as it has been shown that the catalytic trimers of ATCase in M. jannaschii associate with three regulatory dimers to form a type B ATCase. 35 There is no loop 5 in human DHOase. In human CAD, DHOase and ATCase are covalently linked and it is considered that the covalent link is necessary for the assembly of the hexameric DHOase‐ATCase structure of CAD. 36 The non‐covalent interaction between the DHOase and ATCase in CAD is considered to be loose if any. Bacterial type II and active fungal DHOases have a shorter loop 5 compared to long DHOases and it is involved in dimerization in both. 11 , 13 It is considered that the differences in loop 5, loop 4, and the dimerization interface among DHOases reflect their adaptation to interact or not with ATCase. 11
3.2.5. Oligomeric structure
Most known DHOases are either monomers or dimers. MjDHOase is a monomer in both crystal and solution. 14 This also appears to be the case for bacterial type III (PgDHOase). Both bacterial type I and type II DHOases form dimers, at least in the crystal. The mode of dimerization seems to be different in the two types, bacterial type I through lateral association and bacterial type II through the top of the barrel. 11 Even though the human DHOase uses the same lateral interface as AaDHOase for dimerization, the relative orientations of the two subunits of the dimer are different. 11 However, the active fungal DHOase from S. cerevisiae forms a tetramer in the crystalline state while it forms dimers and mostly tetramers in solution. 13 The mode of dimerization is similar to bacterial type II but most interacting residues are different. It was suggested that ScDHOase may form a tetramer for stability, even though it is not known whether it needs to function as a tetramer in association with S. cerevisiae Ura2. 13 The different oligomeric states of DHOases may be related to evolutionary diversity. 13
4. CONCLUSIONS
MjDHOase is the first archaeal DHOase studied by x‐ray diffraction. Even though the phylogenetic analysis of DHOases 11 suggested that archaeal DHOases are most closely related to bacterial type I, the current work showed that they also share features with the other DHOase types and subtypes. In particular, the two Zn ions in the active site are bridged with KCX and they have a long catalytic loop. These traits are similar to bacterial types II and III, human, and active fungal DHOases, but are different from bacterial type I that use an Asp to bridge the two Zn ions and have a short catalytic loop. Furthermore, MjDHOase is a monomer in contrast to bacterial type I and II DHOases that form dimers, at least in the crystal, and similar to type III PgDHOase. Loop 5 is mostly similar to type III but differs from the other DHOases. Finally, the extra β sandwich domain and the C‐terminal extension are similar to bacterial type I DHOases.
More data on other archaeal DHOases are important to generalize the similarities and differences between archaeal DHOases and the other subtypes.
AUTHOR CONTRIBUTIONS
Jacqueline Vitali designed the project and the experiments, carried out several of them, solved the crystal structure, carried out the interpretation of crystallographic results, and wrote the manuscript. Jay C. Nix measured and reduced the diffraction data. Undergraduate student Haley E. Newman carried out purification and crystallization experiments and obtained the first good crystals. Michael J. Colaneri helped in the interpretation of crystallographic results and the writing of the manuscript.
CONFLICT OF INTERESTS
The authors declare no potential conflict of interest.
ACKNOWLEDGMENTS
This work was supported in part by grant GM071512 (Jacqueline Vitali) from the National Institutes of Health, two Undergraduate Student Research Awards (USRA) from Cleveland State University (Jacqueline Vitali) and an Undergraduate Research Award (URA) from Cleveland State University (Haley E. Newman). The computations were supported in part by an allocation of computing time from the Ohio Supercomputer Center. Most figures were drawn with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.
Vitali J, Nix JC, Newman HE, Colaneri MJ. Crystal structure of Methanococcus jannaschii dihydroorotase. Proteins. 2023;91(1):91‐98. doi: 10.1002/prot.26412
Funding information National Institutes of Health, Grant/Award Number: GM071512; Cleveland State University
DATA AVAILABILITY STATEMENT
Coordinates and structure factors have been deposited with the Protein Data bank and will be released upon publication; PDB code: 7UOF.
REFERENCES
- 1. Cooper C, Wilson DW. Biosynthesis of pyrimidines. Fed Proc. 1954;13:194. [Google Scholar]
- 2. Lieberman I, Kornberg A. Enzymatic synthesis and breakdown of a pyrimidine, orotic acid. I. Dihydroorotic acid, ureidosuccinic acid, and 5‐carboxymethylhydantoin. J Biol Chem. 1954;207:911‐924. [PubMed] [Google Scholar]
- 3. Holm L, Sander C. An evolutionary treasure: unification of a broad set of amidohydrolases related to urease. Proteins. 1997;28:72‐82. [PubMed] [Google Scholar]
- 4. Fields C, Brichta D, Shepherdson M, Farinha M, O'Donovan G. Phylogenetic analysis and classification of dihydroorotases: a complex history for a complex enzyme. Paths Pyrimidines. 1999;7:49‐63. [Google Scholar]
- 5. Thoden JB, Phillips GN Jr, Neal TM, Raushel FM, Holden HM. Molecular structure of dihydroorotase: a paradigm for catalysis through the use of a binuclear metal center. Biochemistry. 2001;40:6989‐6997. [DOI] [PubMed] [Google Scholar]
- 6. Porter TN, Li Y, Raushel FM. Mechanism of the dihydroorotase reaction. Biochemistry. 2004;43:16285‐16292. [DOI] [PubMed] [Google Scholar]
- 7. Lee M, Chan CW, Mitchell Guss J, Christopherson RI, Maher MJ. Dihydroorotase from Escherichia coli: loop movement and cooperativity between subunits. J Mol Biol. 2005;348:523‐533. [DOI] [PubMed] [Google Scholar]
- 8. Martin PD, Purcarea C, Zhang P, et al. The crystal structure of a novel, latent dihydroorotase from Aquifex aeolicus at 1.7A resolution. J Mol Biol. 2005;348:535‐547. [DOI] [PubMed] [Google Scholar]
- 9. Ahuja A, Purcarea C, Ebert R, Sadecki S, Guy HI, Evans DR. Aquifex aeolicus dihydroorotase: association with aspartate transcarbamoylase switches on catalytic activity. J Biol Chem. 2004;279:53136‐53144. [DOI] [PubMed] [Google Scholar]
- 10. Zhang P, Martin PD, Purcarea C, et al. Dihydroorotase from the hyperthermophile Aquifex aeolicus is activated by stoichiometric association with aspartate transcarbamoylase and forms a one‐pot reactor for pyrimidine biosynthesis. Biochemistry. 2009;48:766‐778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grande‐García A, Lallous N, Díaz‐Tejada C, Ramón‐Maiques S. Structure, functional characterization, and evolution of the dihydroorotase domain of human CAD. Structure. 2014;22:185‐198. [DOI] [PubMed] [Google Scholar]
- 12. Lipowska J, Miks CD, Kwon K, et al. Pyrimidine biosynthesis in pathogens – structures and analysis of dihydroorotases from Yersinia pestis and Vibrio cholerae. Int J Biol Macromol. 2019;136:1176‐1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guan HH, Huang YH, Lin ES, Chen CJ, Huang CY. Structural analysis of Saccharomyces cerevisiae dihydroorotase reveals molecular insights into the tetramerization mechanism. Molecules. 2021;26:7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vitali J, Singh AK, Colaneri MJ. Characterization of the dihydroorotase from Methanococcus jannaschii . Protein J. 2017;36:361‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McPherson A. Preparation and Analysis of Protein Crystals. Krieger Publishing Company; 1989. [Google Scholar]
- 16. Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr. 2013;69:1204‐1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adams PD, Afonine PV, Bunkóczi G, et al. PHENIX: a comprehensive python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mehboob S, Mulhearn DC, Truong K, Johnson ME, Santarsiero BD. Structure of dihydroorotase from Bacillus anthracis at 2.6 Å resolution. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:1432‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Emsley P, Cowtan K. Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126‐2132. [DOI] [PubMed] [Google Scholar]
- 21. Davis IW, Leaver‐Fay A, Chen VB, et al. MolProbity: all‐atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35(Web Server issue):W375‐W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zheng H, Chordia MD, Cooper DR, et al. Validation of metal‐binding sites in macromolecular structures with the CheckMyMetal web server. Nat Protoc. 2014;9:156‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zheng H, Cooper DR, Porebski PJ, Shabalin IG, Handing KB, Minor W. CheckMyMetal: a macromolecular metal‐binding validation tool. Acta Crystallogr D Struct Biol. 2017;73(Pt 3):223‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774‐797. [DOI] [PubMed] [Google Scholar]
- 25. Bliven S, Lafita A, Parker A, Capitani G, Duarte JM. Automated evaluation of quaternary structures from protein crystals. PLoS Comput Biol. 2018;14:e1006104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rice AJ, Lei H, Santarsiero BD, Lee H, Johnson ME. Ca‐asp bound X‐ray structure and inhibition of Bacillus anthracis dihydroorotase (DHOase). Bioorg Med Chem. 2016;24:4536‐4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Edwards BF, Fernando R, Martin PD, et al. The mononuclear metal center of type‐I dihydroorotase from Aquifex aeolicus. BMC Biochem. 2013;14:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605‐1612. [DOI] [PubMed] [Google Scholar]
- 29. Lallous N, Grande‐García A, Molina R, Ramón‐Maiques S. Expression, purification, crystallization and preliminary x‐ray diffraction analysis of the dihydroorotase domain of human CAD. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012;68:1341‐1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prangé T, Girard E, Fourme R, et al. Pressure‐induced activation of latent dihydroorotase from Aquifex aeolicus as revealed by high pressure protein crystallography. FEBS J. 2019;286:1204‐1213. [DOI] [PubMed] [Google Scholar]
- 31. Lee M, Chan CW, Graham SC, Christopherson RI, Guss JM, Maher MJ. Structures of ligand‐free and inhibitor complexes of dihydroorotase from Escherichia coli: implications for loop movement in inhibitor design. J Mol Biol. 2007;370:812‐825. [DOI] [PubMed] [Google Scholar]
- 32. Guan HH, Huang YH, Lin ES, Chen CJ, Huang CY. Structural basis for the interaction modes of dihydroorotase with the anticancer drugs 5‐fluorouracil and 5‐aminouracil. Biochem Biophys Res Commun. 2021;551:33‐37. [DOI] [PubMed] [Google Scholar]
- 33. Guan HH, Huang YH, Lin ES, Chen CJ, Huang CY. Complexed crystal structure of Saccharomyces cerevisiae dihydroorotase with inhibitor 5‐fluoroorotate reveals a new binding mode. Bioinorg Chem Appl. 2021;2021:2572844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kankanala R. Characterizing the Oligomeric Structure and Catalytic Activity of the Dihydroorotase and Aspartate Transcarbamoylase from the Bacterium, Bacillus anthracis. Master's theses and doctoral dissertations. Paper 339; 2011. http://commons.emich. edu/theses/339
- 35. Hack ES, Vorobyova T, Sakash JB, et al. Characterization of the aspartate transcarbamoylase from Methanococcus jannaschii . J Biol Chem. 2000;275:15820‐15827. [DOI] [PubMed] [Google Scholar]
- 36. Moreno‐Morcillo M, Grande‐García A, Ruiz‐Ramos A, Del Caño‐Ochoa F, Boskovic J, Ramón‐Maiques S. Structural insight into the core of CAD, the multifunctional protein leading de novo pyrimidine biosynthesis. Structure. 2017;25:912‐923. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Coordinates and structure factors have been deposited with the Protein Data bank and will be released upon publication; PDB code: 7UOF.