

Abstract
Primary mitochondrial diseases are a group of genetically and clinically heterogeneous disorders resulting from oxidative phosphorylation (OXPHOS) defects. COX11 encodes a copper chaperone that participates in the assembly of complex IV and has not been previously linked to human disease. In a previous study, we identified that COX11 knockdown decreased cellular adenosine triphosphate (ATP) derived from respiration, and that ATP levels could be restored with coenzyme Q10 (CoQ10) supplementation. This finding is surprising since COX11 has no known role in CoQ10 biosynthesis. Here, we report a novel gene‐disease association by identifying biallelic pathogenic variants in COX11 associated with infantile‐onset mitochondrial encephalopathies in two unrelated families using trio genome and exome sequencing. Functional studies showed that mutant COX11 fibroblasts had decreased ATP levels which could be rescued by CoQ10. These results not only suggest that COX11 variants cause defects in energy production but reveal a potential metabolic therapeutic strategy for patients with COX11 variants.
Keywords: coenzyme Q, COX11, mitochondrial disorders, OXPHOS
1. INTRODUCTION
Mitochondrial disorders impact more than one in 5000 live births, and often lead to the dysfunction of multiple organ systems and early death. The pathophysiology is believed to result from a primary or secondary disruption in the production of adenosine triphosphate (ATP) by mitochondria, which constitutes the major source of energy that the human body needs (Gorman et al., 2016) (Frazier et al., 2019). ATP is produced in the mitochondria through oxidative phosphorylation (OXPHOS) and involves five protein complexes. Mitochondrial respiratory chain complex IV (cytochrome c oxidase; COX; CIV) is composed of 14 subunits encoded by both mitochondrial DNA and nuclear DNA (nDNA) and catalyzes electron transfer from reduced cytochrome c to oxygen (O2), contributing to the electrochemical transmembrane gradient required to produce ATP (Gorman et al., 2016). Several proteins encoded by the nDNA are required for the proper assembly of CIV, with some previously linked to human mitochondrial disease. For instance, pathogenic variants in SURF1 are among the most frequent causes of Leigh syndrome (Lake et al., 2016). SURF1 participates in the assembly of CIV as part of the mitochondrial translation regulation assembly intermediate of cytochrome c oxidase complex (MITRAC) (Mick et al., 2012). COX11 (cytochrome c oxidase assembly factor 11) has not yet been linked to disease, however, COX11 also participates in COX assembly by mediating copper delivery into to the complex (Bourens & Barrientos, 2017). Moreover, knockdown studies using Clustered Regularly Interspaced Short Palindromic Repeats interference (CRISPRi) and a fluorescence resonance energy transfer (FRET)‐based ATP sensor showed that COX11 is required to maintain mitochondrial‐derived ATP levels. Supplementation with coenzyme Q10 (CoQ10) prevented this decrease in ATP due to COX11 deficiency, even though COX11 is not known to participate in CoQ10 synthesis (Mendelsohn et al., 2018). In this study, we describe for the first time two unrelated patients with biallelic COX11 variants leading to mitochondrial disease and show restoration of ATP levels in one of the patient's cell lines using CoQ10.
2. MATERIALS AND METHODS
2.1. Ethics and consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consents were obtained for the clinical genetic testing and the publication of the findings from all patients included in the study. This project was approved by the Human Research Ethics Committee of the Sydney Children's Hospitals Network (ID number 10/CHW/114).
2.2. Exome and genome sequencing
Trio genome sequencing of patient 1 (X1) was performed as previously described (Riley et al., 2020). In brief, samples were sequenced on an Illumina HiSeq X Ten sequencer (v3.0.29.0) to >x30 depth. Reads were aligned to the b37d5 reference genome using Burrows–Wheeler alignment (BWA MEM, v0.7.10) (Li & Durbin, 2009), sorted using Novosort (v.1.03.01), realigned and recalibrated using GATK (v.3.3). Variants were identified using GATK HaplotypeCaller (v.3.3) (DePristo et al., 2011) and annotated using Variant Effect Predictor (v79). Variant analysis was performed using Seave (Gayevskiy et al., 2019).
Trio exome sequencing of patient 2 (X2) was performed at the Department of Human Genetics, Bioscientia Healthcare GmbH. The fragmented genomic DNA was enriched using Roche NimbleGen capture technology (SeqCap MedExome Library), amplified and sequenced simultaneously using an Illumina NovaSeq. 6000 system. The target regions were sequenced with an average coverage of 124‐fold. For about 96% of the areas of interest, a 15‐fold coverage was obtained. Next ‐generation sequencing data were aligned to the hg19 genome assembly. An in‐house developed bioinformatics pipeline performed variant calling and annotation.
2.3. Blue native Polyacrylamide Gel Electrophoresis (BN‐PAGE) and immunoblotting
Blue native Polyacrylamide Gel Electrophoresis(BN‐PAGE) and immunoblot analysis used 30 μg of mitochondria isolated from fibroblasts and solubilized in 1% Digitonin or 1% Triton X‐100 as previously described (Formosa et al., 2020). Densitometry of BN‐PAGE data was performed using the ImageLab software (version 5.2.1) on the COX1 signal, and was taken as a ratio to Complex II succinate dehydrogenase complex subunit A. Total protein lysates were extracted from patient X1 fibroblasts for Western blot analysis using total Abcam OXPHOS human WB antibody cocktail (Abcam; ab11041) and Porin/VDAC1 (Abcam; ab14734) as previously published (Lake et al., 2017).
2.4. Quantitative polymerase chain reaction (PCR) and complementary DNA sequencing
Total RNA was isolated from blood of patient X2 using the RNeasy kit (Qiagen). Complementary DNA (cDNA) was amplified with SuperScript III First‐Strand synthesis kit (Thermo Fisher Scientific) using manufacturer's instructions. Quantitative PCR (qPCR) was conducted using TaqMan Fast Advanced Master Mix (Thermofisher) and TaqMan probes for COX11 (hs01680112) and normalized to HPRT1 (Hs03929096_g1).
Sequencing of COX11 cDNA (NM_004375.4) was conducted using primers situated in exon 1 and exon 3 (5′AGAGGGTTATGGGAGGGCTCTG3′, 5′AAACGCCAGTGCAGTCTCTC3′).
2.5. Real‐time ATP assay
A FRET‐based ATP assay was conducted as previously described (Mendelsohn et al., 2018). In brief, patient and control fibroblasts were immortalized using a lenti‐hTERT‐Neo Virus following the manufacturer's protocol. A FRET‐based ATP sensor consisting of Clover and mApple surrounding an ATP‐sensitive domain was transduced into fibroblasts, which were treated with either 50 μM CoQ10 (ubiquinone # C9538; Sigma) or dimethylformamide‐vehicle (DMF) control for 5 days. The fibroblasts were then subjected to metabolic perturbations: restricted metabolism to respiration‐only (phosphate buffered saline, [PBS], with 2% Fetal bovine serum, FBS, 10 mM pyruvate and 10 mM 2‐deoxy‐d‐glucose), ATP depletion (PBS with 2% FBS, 5 μM oligomycin and 10 mM 2‐deoxy‐d‐glucose), or unrestricted basal metabolism (PBS with 2% FBS, 5 mM pyruvate and 10 mM glucose) for 6 h before measuring their ATP level by fluorescence‐activated cell sorting on a FACS Aria II. FRET was detected by excitation from the 488‐nm laser, and emission was detected via a 610/20 or 615/30‐nm filter, on a linear scale.
2.6. Platereader luciferase assay for ATP
Patient and control fibroblasts were immortalized using a lenti‐hTERT‐Neo Virus. Fibroblasts were plated into 96‐well plates, 4000 cells per well, subjected to the above metabolic perturbations for 8 h. Following metabolic perturbation, ATP was measured using using CellTiter Glo 2.0 cell viability assay before measuring ATP on a Spectramax M4 platereader. Luminescence was normalized to Hoescht nuclear stain intensity.
2.7. MitoSOX‐based flow cytometric assay
Control and Patient X1 fibroblasts were cultured with 50 μM CoQ10 (ubiquinone; catalog # C9538; Sigma) or DMF vehicle control for 5 days before staining with 2.5 μM MitoSOX (Thermo Fisher). for 15 min at 37°C. Between 4000 and 15,000 cells were sorted per condition. Cells were then incubated in respiration‐only metabolic substrate for 5 h before analysis on a FACS Aria II. Unstained cells were used to set gates, and cells incubated with 2 μM rotenone, a complex I inhibitor, were used as positive controls. MitoSOX was detected by excitation from the 488‐nm laser, and emission was detected via a 610/20 or 615/30‐nm filter.
3. RESULTS
Patient X1 (X1) was the first child of consanguineous parents. She presented with congenital sensorineural deafness in the newborn period. She subsequently presented at 6 weeks of age with hypotonia, poor feeding and two clinical events of loss of responsiveness suspected to be seizures. Initial magnetic resonance imaging (MRI) was normal, and phenobarbitone was started. At 3 months of age, she experienced episodes of intermittent respiratory distress and clinical clonic seizures progressed, associated with developmental stasis, visual impairment and poor feeding, necessitating a 2 month admission. Brain MRI showed new lesions in the cerebellar peduncles, cerebrospinal fluid lactate was elevated 2.7 mmol/L (N: <2 mmol/L), and plasma lactate were intermittently elevated to 5 mmol/L. Phenobarbitone, levetiracetam and clobazam appeared to initially stabilize her, although she developed a dyskinetic movement disorder, and a repeat brain MRI at 7 months revealed progressive brain atrophy, dentate nucleus lesions, and diffuse white matter disease (Supporting Information: Figure S1). She required nasogastric feeding and was treated with three antiepileptic drugs. She was also treated with CoQ10 (in the form of ubiquinone) starting at 25 mg twice a day (weight 6 kg) from 5 months of age. However, her neurologic symptoms worsened, and her clinical course was complicated by several respiratory infections with respiratory distress, and the patient sadly died at 9 months of age.
A spectrophotometric assay of OXPHOS enzyme activity was performed as previously described (Frazier et al., 2020). In muscle, all complexes were normal to elevated relative to protein, relative to complex II and relative to citrate synthase (CS). In the liver, complexes I, II and CS were elevated, so when the activities of complexes III and IV were expressed relative to complex II or CS they were borderline low (Table 1). Enzyme activities of Complex I and IV in fibroblasts were determined using dipstick activity assays (Abcam #ab109720; #ab109876), and showed a mild decrease in CIV (decreased by 40% relative to control A), but no difference in complex I activity (data not shown).
Table 1.
Spectrophotometric analysis of oxidative phosphorylation (OXPHOS) enzyme activities in muscle and liver from X1
| Muscle | Liver | |||||
|---|---|---|---|---|---|---|
| Enzyme |
Residual Activity (%) |
CS ratio (%) |
CII ratio (%) |
Residual Activity (%) |
CS ratio (%) |
CII ratio (%) |
| I |
221 (45−173) |
182 (70−149) |
141 (66−147) |
768 (82−118) |
218 (77−124) |
327 (92−121) |
| II |
160 (58−138) |
127 (80−109) |
‐ |
236 (88−119) |
67 (80−126) |
‐ |
| III |
106 (44−175) |
81 (30−171) |
65 (31‐161) |
74 (68‐135) |
21 (61−136) |
31 (76–125) |
| IV |
97 (51−139) |
78 (77−116) |
61 (83‐126) |
70 (64−123) |
20 (68−130) |
29 (65–120) |
| CS |
125 (66−138) |
346 (94−110) |
||||
Note: Activities of OXPHOS enzyme complexes I, II, III, IV and citrate synthase (CS) are expressed as % relative to protein (residual activity), citrate synthase (CS ratio) and CII (CII ratio) of control samples.
Trio GS identified a homozygous missense variant in COX11, NM_004375.4:c.730G > C NP_004366.1:p.(Ala244Pro), with both parents being heterozygous (Supporting Information: Figure S3A). Sanger sequencing in the patient's unaffected siblings showed they were both heterozygous for the variant. The alanine at position 244 has high conservation and is situated within the cytochrome c oxidase assembly functional domain. In silico software predictions are conflicting (Supporting Information: Table S1) and the variant has not been observed in the gnomAD population database (v3.1.2 accessed October 10, 2021) (Karczewski et al., 2020).
An unrelated patient (Patient X2) is the first child of consanguineous parents. Antenatal scans revealed severe symmetrical intrauterine growth restriction with absent end‐diastolic flow. The mother received prenatal dexamethasone, and the delivery was performed via cesarean section due to fetal distress at 34 + 3rd gestational weeks. Her APGAR scores were 7, 8, 8 at 1, 5 and 10 min, respectively. Her weight, length, and head circumference at birth were 970 g (−6.8 Z‐score), 36 cm (−7.0 Z‐score), and 26.5 cm (−6.2 Z‐score), respectively. She had feeding intolerance with abdominal distention suggestive of stage one necrotizing enterocolitis. The initial investigations showed hypoglycemia and increased serum lactate levels ranging from 3 to 6 mmol/L (N: 0.5‐2.2 mmol/L) without metabolic acidosis. Other laboratory investigations were unremarkable, including tandem mass spectrometry (MS) and urine gas chromatography‐MS. The upper gastrointestinal tract barium study showed dysmotility and moderate gastroesophageal reflux disease.
Electroencephalogram showed generalized epileptiform discharges at 7 months of age, controlled by Levetiracetam. Echocardiography showed an isolated aortopulmonary collateral artery with an insignificant hemodynamic effect. The auditory brainstem response testing showed normal bilateral hearing at a 2−4 kHz frequency range. At 1 year of age, the brain MRI showed bilateral basal ganglia hyperintensity, mild brain atrophy, and magnetic resonance spectroscopy showed elevated lactate levels in basal ganglia (Supporting Information: Figure S2).
On her last examination at the age of 3 years, a significant delay in acquiring age‐appropriate milestones and inattentiveness to surrounding visual and auditory stimuli and occasional focal seizures were noted. She was unable to roll or sit and could not speak any words. She lacked any attempt for vocal communication or a palmar grasp. She had some visual contact, followed her parents, and smiled occasionally. The physical examination revealed microcephaly (43.5 cm; −3.07 Z‐score), increased peripheral tone, and brisk deep tendon reflexes. Her weight was 7.67 kg (−6.89 Z‐score), her length was 76.5 cm (supine; 4.69 Z‐score), her body mass index was 13.1 (−2.8. Z‐score). Serum lactate level remained elevated with a value of 6.2 mmol/L (N: 0.5 to 2.2 mmol/L).
Trio exome sequencing identified a homozygous variant in COX11 NM_004375.4:c.35_36delinsG NP_004366.1:p.(Val12Glyfs*21), with both parents being heterozygous (Supporting Information: Figure S3B). The subsequent segregation analysis showed that the unaffected younger sister is also heterozygous for the NM_004375.4:c.35_36delinsG NP_004366.1:p.(Val12Glyfs*21) variant. This variant has not been observed in the gnomAD population database (v2.1.1 accessed December 10, 2021) (Karczewski et al., 2020).
To analyze the effect of the NM_004375.4:c.730G > C NP_004366.1:p.(Ala244Pro) variant in patient X1 on CIV assembly, BN‐PAGE immunoblot analysis was performed on mitochondria isolated from fibroblasts from proband X1 and age‐matched controls, solubilized in digitonin (to preserve the supercomplex form) or Triton X100 detergent (to separate the individual OXPHOS complexes into holoenzymes) (Formosa et al., 2020). The results suggested a lower level of assembled monomeric CIV was more evident in the Triton X‐100 solubilized samples, consistent between COX2, COX4 (Figure 1a) and COX1 antibodies (Figure 1b), with normal levels of other OXPHOS subunits in isolated mitochondria compared to controls (Figure 1c). The modest reduction in CIV levels was consistent with the reduced levels of CIV from analysis of whole cell lysates by sodium dodecyl sulfate (SDS)‐PAGE western blot with COXII antibody (Figure 1d).
Figure 1.
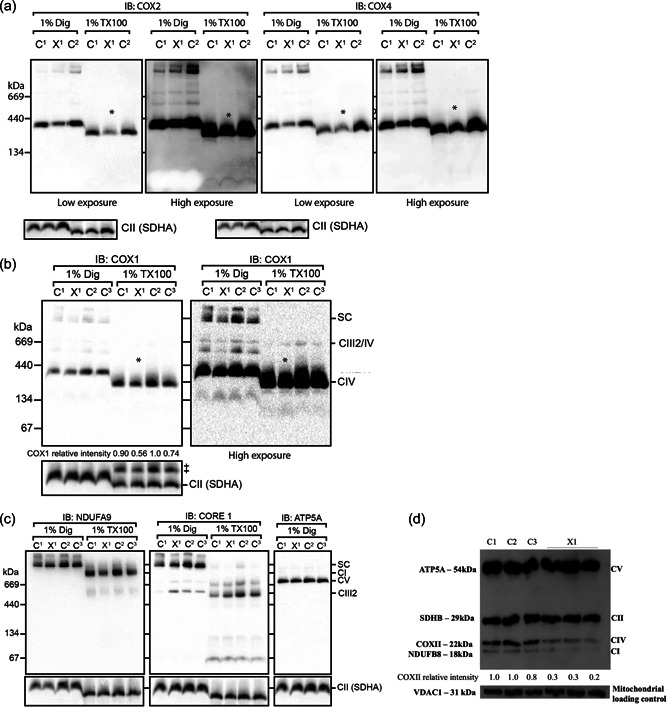
BN‐PAGE for complex IV and OXPHOS Western blot in COX11 fibroblasts. Mitochondria isolated from Patient X1 and control fibroblasts analyzed by BN‐PAGE immunoblotting for COX2, COX4, (a) and COX1 (b) suggest a decreased level of monomeric CIV more evident in mitochondria solubilized with Triton X‐100, which separates the individual OXPHOS complexes (marked by *); ‡indicates previous COX1 signal and complex II (SDHA) was used as a loading control. (c) The levels of other OXPHOS subunits in isolated mitochondria were similar to controls (CI‐NDUFA9, CIII‐CORE1, and CV‐ATP5A). (d) Representative SDS‐PAGE and western blot analysis in whole cell lysates with an antibody cocktail targeting individual OXPHOS complex subunits showed consistently lower levels of CIV in patient X1; porin (VDAC1) was used as a loading control. BN, Blue native; CIV, complex IV; OXPHOS, oxidative phosphorylation; BN‐ PAGE, Blue native polyacrylamide gel electrophoresis; SDHA, succinate dehydrogenase complex subunit A; SDS, sodium dodecyl sulfate.
To assess if the NM_004375.4:c.730G > C NP_004366.1:p.(Ala244Pro) homozygous variant identified in patient X1 could impact ATP levels, we assayed steady‐state ATP levels with different substrates and plus or minus CoQ10 supplementation using patient X1 fibroblasts.
Under basal conditions, where glycolysis and respiration are unrestricted, ATP levels were comparable between patient X1 and control cells derived from a healthy individual, in the presence or absence of 50 µM CoQ10. However, when glycolytic ATP generation was blocked with 2‐deoxy‐d‐glucose, the patient X1 (COX11) fibroblasts showed markedly decreased ATP levels compared with control cells (Figure 2). In the presence of 50 µM CoQ10, patient X1 (COX11) cells showed substantial restoration of ATP levels, while control cells showed no significant change (Figure 2).
Figure 2.
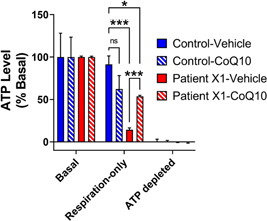
COX11 variation is associated with decreased respiration‐derived ATP levels, which can be restored with CoQ10. ATP levels were measured by flow cytometry after 6 h of metabolic substrate in patient X1 (COX11) and control fibroblasts expressing a genetically encoded FRET‐based ATP sensor. Basal ATP levels were similar between all conditions. Respiration‐only ATP levels were normalized within each cell line to basal and depleted conditions. Patient XI (COX11) and control fibroblast ATP levels remained high with unrestricted basal metabolism. When metabolism was restricted to respiration‐only by addition of 10 mM 2‐deoxy‐d‐glucose, ATP levels dropped in patient cells that were treated with the vehicle, while patient XI (COX11) cells maintained higher levels of ATP with CoQ10 treatment. Data show mean ± SEM; n= 2−8 replicates of at least 300 cells per condition, compiled from two independent experiments. ***p< 0.001, *<0.05 by two‐way analysis of variance with Tukey's multiple comparisons test. ATP adenosine triphosphate; FRET, fluorescence resonance energy transfer.
These data suggest that the patient X1 (COX11) cells show a very similar functional defect to COX11 knock‐down cells in having inadequate ATP generation that can be rescued by CoQ10 (Mendelsohn et al., 2018).
To corroborate the results using a different method, ATP content was also measured using luciferase assay for ATP (Supporting Information: Figure S4), showing a similar decrease in respiration‐derived ATP levels in COX11 patient cells, which was rescued by CoQ10 supplementation.
RNA was extracted from total peripheral blood of patient X2 and reverse‐transcribed. qPCR was performed using TaqMan gene expression assays, showing no significant difference in COX11 transcript expression compared to controls (Supporting Information: Figure S5A). In addition, cDNA sequencing identified the homozygous NM_004375.4:c.35_36delinsG variant in the transcript (Supporting Information: Figure S5B). Together, these data suggest that the mutant transcript is not degraded by nonsense mediated decay and is therefore predicted to cause a truncated protein. Unfortunately, no tissue samples or cultured fibroblasts were available for further studies.
4. DISCUSSION
While COX11 is known to play an important role in the assembly of CIV by mediating copper delivery into the COX1 CuB center (Bourens & Barrientos, 2017), disease‐causing variants in COX11 have not previously been reported. We identify two such individuals with mitochondrial encephalopathies that overlap the Leigh Syndrome spectrum (Lake et al., 2016) and show that one of these variants causes a decrease in respiration‐derived ATP levels. CIV couples electron transfer to oxygen, resulting in the production of water and the pumping of protons across the mitochondrial inner membrane. Therefore, defects in CIV could lead to a decrease in mitochondrial membrane potential or protonmotive force that powers respiration‐derived ATP production. Cofactor therapies such as CoQ10 have been used to treat disorders in biosynthesis of CoQ10, and a synthetic analogue of CoQ10 (Idebenone) is licensed for the treatment in patients with Leber Hereditary Optic Neuropathy (Lyseng‐Williamson, 2016). However, there is very limited evidence on the effectiveness of CoQ10 for patients with other mitochondrial diseases, especially in the pediatric population (Neergheen et al., 2017). We observed that the decline in ATP levels in COX11 patient fibroblasts was ameliorated with CoQ10 supplementation.
Since COX11 has no known role in CoQ10 biosynthesis, it is unclear why CoQ10 supplementation would address functional deficits in cells with COX11 variants. We previously showed that CRISPRi knockdown of COX11 in human K562 cells does not significantly change total amounts of CoQ10, suggesting that COX11 loss of function may not create a deficit in CoQ10 (Mendelsohn et al., 2018). Here, we observe that patient X1 expressing a COX11 variant, has lower CIV levels in fibroblasts. Therefore, CoQ10 supplementation may restore ATP levels by restoring electron transport chain complex levels, for instance by promoting complex assembly, by preventing complex degradation, or by promoting mitochondrial biogenesis (Santos‐Ocana et al., 2002). Alternatively, CoQ10 supplementation could increase ATP levels by directly contributing electrons to complex III, however, our previously published results show that CoQ10 supplementation increased the fraction of oxidized versus reduced state (Mendelsohn et al., 2018). CoQ10 levels may also regulate enzyme activity within the electron transport chain, or levels of supercomplex, each of which are believed to regulate ATP production (Acin‐Perez et al., 2008; Salviati et al., 2005) (Lapuente‐Brun et al., 2013).
While we demonstrate that cells from a patient with COX11 variants have decreased respiration‐derived ATP, it is possible that disruptions of other non‐energetic functions of CIV or the electron transport chain also contribute to the disease pathophysiology. While there are no known sites of reactive oxygen species generation in CIV, it is possible that decreasing levels of CIV increase reactive oxygen species generation from other upstream sites in the electron transport chain (Quinlan et al., 2013). Furthermore, COX11 also acts as a copper chaperone, and facilitates the formation of copper centers in CIV. Knockdown or mutation in COX11 may result in disrupted copper mobilization. Free copper in the cytosol can contribute to the conversion of hydrogen peroxide to damaging hydroxyl radicals (Collin, 2019). Since CoQ10 can accept electrons and in some contexts has been considered an antioxidant, it is possible that CoQ10 supplementation may also address excessive ROS generation resulting from decreasing levels of CIV. However, mitochondrial superoxide was not significantly altered with CoQ10 treatment in patient X1 fibroblasts (Supporting Information: Figure S6).
The ability of CoQ10 to restore ATP raises questions about its therapeutic potential for patients with COX11 variants. Although patient X1 declined despite CoQ10 supplementation later in her disease, our data suggest boosting CoQ10 may be a promising therapeutic strategy for COX11 deficiency. Therapeutic doses of CoQ10 supplementation in patients with CoQ10 deficiency range from 5 to 50 mg/kg/day (Desbats et al., 2015). Patient X1 received low dose CoQ10 (8 mg/kg/day), and it was administered late in the course of her disease. Therefore, we speculate that efficacy will require earlier intervention, higher doses and improved bioavailability in vulnerable tissues including the brain. CoQ10 is highly hydrophobic (Suarez‐Rivero et al., 2021), and higher doses may be needed for COX11 deficiency. Notably, the efficacy of CoQ10 therapy may also depend on whether toxicity ultimately results from insufficient energy, which also remains to be proven.
The modified mitochondrial disease criteria score suggests a probable mitochondrial disorder for patient X2, although functional studies could not be performed (Witters et al., 2018). When it is considered together with a biallelic truncating variant in COX11, it seems likely that the p.(Val12Glyfs*21) variant abolishes import of the protein into mitochondria as suggested by mitochondrial localization in silico prediction tools MitoFates (Fukasawa et al., 2015), and Tppred 3.0 (Savojardo et al., 2015) (Supporting Information: Table S2&S3).
While rare, variants affecting the mitochondrial targeting sequence have been described before in other genes causing mitochondrial disease such as MLYCD, PDHA1 and ISCA1, leading to defects such as reduced mitochondrial protein import or protein mistargeting (Takakubo et al., 1995; Torraco et al., 2018; Wightman et al., 2003). However, a cell line of patient X2 was not available and validation studies were not performed.
In summary, our study demonstrates that biallelic pathogenic variants in COX11 are a causative of mitochondrial encephalopathy. Further research including the interaction of CoQ10 supplementation in patients with COX11‐related mitochondrial disease could be of interest to elucidate potential treatment targets.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
The Chair in Genomic Medicine awarded to J. C. is generously supported by The Royal Children's Hospital Foundation. We are grateful to the Crane, Perkins and Miller families for their generous financial support. We thank the Kinghorn Center for Clinical Genomics for assistance with production and processing of genome sequencing data. L. E. F. acknowledges support from the Mito Foundation. M. J. C. acknowledges support from the Luminesce Alliance. We thank Dr. Michio Hirano for providing fibroblasts from healthy individuals for controls. K. N. and N. K. B. were supported by NIH R01AG065428 to K. N. N. K. B. was also supported by NIH award F32 AG063457‐01, and a Berkelhammer Award for Excellence in Neuroscience. This researchstudy was supported by a New South Wales Office of Health and Medical Research Council Sydney Genomics Collaborative grant (J. C.). We acknowledge funding from the National Health and Medical Research Council (NHMRC): Project Grants GNT1165217 (M. T. R.) and GNT1164479 (D. R. T.), Principal Research Fellowship GNT1155244 (D. R. T.), and Investigator Grant 2010149 to L. E. F. The research conducted at the Murdoch Children's Research Institute was supported by the Victorian Government's Operational Infrastructure Support Program. Open access publishing facilitated by The University of Melbourne, as part of the Wiley ‐ The University of Melbourne agreement via the Council of Australian University Librarians.
Rius, R. , Bennett, N. K. , Bhattacharya, K. , Riley, L. G. , Yüksel, Z. , Formosa, L. E. , Compton, A. G. , Dale, R. C. , Cowley, M. J. , Gayevskiy, V. , Tala, S. A. , Almehery, A. A. , Ryan, M. T. , Thorburn, D. R. , Nakamura, K. , & Christodoulou, J. (2022). Biallelic pathogenic variants in COX11 are associated with an infantile‐onset mitochondrial encephalopathy. Human Mutation, 43, 1970–1978. 10.1002/humu.24453
Rocio Rius and Neal K. Bennett Joint first authors.
Ken Nakamura and John Christodoulou Joint co‐last authors.
REFERENCES
- Acin‐Perez, R. , Fernandez‐Silva, P. , Peleato, M. L. , Perez‐Martos, A. , & Enriquez, J. A. (2008, November 21). Respiratory active mitochondrial supercomplexes. Molecular Cell, 32(4), 529–539. 10.1016/j.molcel.2008.10.021 [DOI] [PubMed] [Google Scholar]
- Bourens, M. & Barrientos, A. (2017, March). A CMC1‐knockout reveals translation‐independent control of human mitochondrial complex IV biogenesis. EMBO Reports, 18(3), 477–494. 10.15252/embr.201643103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin, F. (2019, May 15). Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. International Journal of Molecular Sciences, 20(10), 2407. 10.3390/ijms20102407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo, M. A. , Banks, E. , Poplin, R. , Garimella, K. V. , Maguire, J. R. , Hartl, C. , Philippakis, A. A. , del Angel, G. , Rivas, M. A. , Hanna, M. , McKenna, A. , Fennell, T. J. , Kernytsky, A. M. , Sivachenko, A. Y. , Cibulskis, K. , Gabriel, S. B. , Altshuler, D. , & Daly, M. J. (2011, May). A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nature Genetics, 43(5), 491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbats, M. A. , Lunardi, G. , Doimo, M. , Trevisson, E. , & Salviati, L. (2015, January). Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. Journal of Inherited Metabolic Disease, 38(1), 145–156. 10.1007/s10545-014-9749-9 [DOI] [PubMed] [Google Scholar]
- Formosa, L. E. , Muellner‐Wong, L. , Reljic, B. , Sharpe, A. J. , Jackson, T. D. , Beilharz, T. H. , Stojanovski, D. , Lazarou, M. , Stroud, D. A. , & Ryan, M. T. (2020, Apr 21). Dissecting the roles of mitochondrial complex I intermediate assembly complex factors in the biogenesis of complex I. Cell Reports, 31(3), 107541. 10.1016/j.celrep.2020.107541 [DOI] [PubMed] [Google Scholar]
- Frazier, A. E. , Thorburn, D. R. , & Compton, A. G. (2019, April 5). Mitochondrial energy generation disorders: genes, mechanisms, and clues to pathology. Journal of Biological Chemistry, 294(14), 5386–5395. 10.1074/jbc.R117.809194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier, A. E. , Vincent, A. E. , Turnbull, D. M. , Thorburn, D. R. , & Taylor, R. W. (2020). Assessment of mitochondrial respiratory chain enzymes in cells and tissues. Methods in Cell Biology, 155, 121–156. 10.1016/bs.mcb.2019.11.007 [DOI] [PubMed] [Google Scholar]
- Fukasawa, Y. , Tsuji, J. , Fu, S. C. , Tomii, K. , Horton, P. , & Imai, K. (2015, April). MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Molecular & Cellular Proteomics, 14(4), 1113–1126. 10.1074/mcp.M114.043083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayevskiy, V. , Roscioli, T. , Dinger, M. E. , & Cowley, M. J. (2019, Jan 1). Seave: a comprehensive web platform for storing and interrogating human genomic variation. Bioinformatics, 35(1), 122–125. 10.1093/bioinformatics/bty540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman, G. S. , Chinnery, P. F. , DiMauro, S. , Hirano, M. , Koga, Y. , McFarland, R. , Suomalainen, A. , Thorburn, D. R. , Zeviani, M. , & Turnbull, D. M. (2016, Oct 20). Mitochondrial diseases. Nature Reviews Disease Primers, 2, 16080. 10.1038/nrdp.2016.80 [DOI] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alfoldi, J. , Wang, Q. , Collins, R. L. , Laricchia, K. M. , Ganna, A. , Birnbaum, D. P. , Gauthier, L. D. , Brand, H. , Solomonson, M. , Watts, N. A. , Rhodes, D. , Singer‐Berk, M. , England, E. M. , Seaby, E. G. , Kosmicki, J. A. , … MacArthur, D. G. (2020, May). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake, N. J. , Compton, A. G. , Rahman, S. , & Thorburn, D. R. (2016, February). Leigh syndrome: One disorder, more than 75 monogenic causes. Annals of Neurology, 79(2), 190–203. 10.1002/ana.24551 [DOI] [PubMed] [Google Scholar]
- Lake, N. J. , Webb, B. D. , Stroud, D. A. , Richman, T. R. , Ruzzenente, B. , Compton, A. G. , Mountford, H. S. , Pulman, J. , Zangarelli, C. , Rio, M. , Bodaert, N. , Assouline, Z. , Sherpa, M. D. , Schadt, E. E. , Houten, S. M. , Byrnes, J. , McCormick, E. M. , Zolkipli‐Cunningham, Z. , Haude, K. , … Thorburn, D. R. (2017, August 3). Biallelic mutations in MRPS34 lead to instability of the small mitoribosomal subunit and leigh syndrome. American Journal of Human Genetics, 101(2), 239–254. 10.1016/j.ajhg.2017.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapuente‐Brun, E. , Moreno‐Loshuertos, R. , Acin‐Perez, R. , Latorre‐Pellicer, A. , Colas, C. , Balsa, E. , Perales‐Clemente, E. , Quiros, P. M. , Calvo, E. , Rodriguez‐Hernandez, M. A. , Navas, P. , Cruz, R. , Carracedo, A. , Lopez‐Otin, C. , Perez‐Martos, A. , Fernandez‐Silva, P. , Fernandez‐Vizarra, E. , & Enriquez, J. A. (2013, Junuary 28). Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science, 340(6140), 1567–1570. 10.1126/science.1230381 [DOI] [PubMed] [Google Scholar]
- Li, H. & Durbin, R. (2009, July 15). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyseng‐Williamson, K. A. (2016). May). idebenone: A review in leber's hereditary optic neuropathy. Drugs, 76(7), 805–813. 10.1007/s40265-016-0574-3 [DOI] [PubMed] [Google Scholar]
- Mendelsohn, B. A. , Bennett, N. K. , Darch, M. A. , Yu, K. , Nguyen, M. K. , Pucciarelli, D. , Nelson, M. , Horlbeck, M. A. , Gilbert, L. A. , Hyun, W. , Kampmann, M. , Nakamura, J. L. , & Nakamura, K. (2018, August). A high‐throughput screen of real‐time ATP levels in individual cells reveals mechanisms of energy failure. PLoS Biology, 16(8), e2004624. 10.1371/journal.pbio.2004624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mick, D. U. , Dennerlein, S. , Wiese, H. , Reinhold, R. , Pacheu‐Grau, D. , Lorenzi, I. , Sasarman, F. , Weraarpachai, W. , Shoubridge, E. A. , Warscheid, B. , & Rehling, P. (2012, December 21). MITRAC links mitochondrial protein translocation to respiratory‐chain assembly and translational regulation. Cell, 151(7), 1528–1541. 10.1016/j.cell.2012.11.053 [DOI] [PubMed] [Google Scholar]
- Neergheen, V. , Chalasani, A. , Wainwright, L. , Yubero, D. , Montero, R. , Artuch, R. , & Hargreaves, I. (2017). Coenzyme Q10 in the treatment of mitochondrial disease. Journal of Inborn Errors of Metabolism and Screening, 5, 1–8. 10.1177/2326409817707771 [DOI] [Google Scholar]
- Quinlan, C. L. , Perevoshchikova, I. V. , Hey‐Mogensen, M. , Orr, A. L. , & Brand, M. D. (2013). Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biology, 1, 304–312. 10.1016/j.redox.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley, L. G. , Cowley, M. J. , Gayevskiy, V. , Minoche, A. E. , Puttick, C. , Thorburn, D. R. , Rius, R. , Compton, A. G. , Menezes, M. J. , Bhattacharya, K. , Coman, D. , Ellaway, C. , Alexander, I. E. , Adams, L. , Kava, M. , Robinson, J. , Sue, C. M. , Balasubramaniam, S. , & Christodoulou, J. (2020, Jul). The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genetics in Medicine, 22(7), 1254–1261. 10.1038/s41436-020-0793-6 [DOI] [PubMed] [Google Scholar]
- Salviati, L. , Sacconi, S. , Murer, L. , Zacchello, G. , Franceschini, L. , Laverda, A. M. , Basso, G. , Quinzii, C. , Angelini, C. , Hirano, M. , Naini, A. B. , Navas, P. , DiMauro, S. , & Montini, G. (2005). Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: A CoQ10‐responsive condition. Neurology, 65(4), 606–608. 10.1212/01.wnl.0000172859.55579.a7 [DOI] [PubMed] [Google Scholar]
- Santos‐Ocana, C. , Do, T. Q. , Padilla, S. , Navas, P. , & Clarke, C. F. (2002, March 29). Uptake of exogenous coenzyme Q and transport to mitochondria is required for bc1 complex stability in yeast coq mutants. Journal of Biological Chemistry, 277(13), 10973–10981. 10.1074/jbc.M112222200 [DOI] [PubMed] [Google Scholar]
- Savojardo, C. , Martelli, P. L. , Fariselli, P. , & Casadio, R. (2015, October 15). TPpred3 detects and discriminates mitochondrial and chloroplastic targeting peptides in eukaryotic proteins. Bioinformatics, 31(20), 3269–3275. 10.1093/bioinformatics/btv367 [DOI] [PubMed] [Google Scholar]
- Suarez‐Rivero, J. M. , Pastor‐Maldonado, C. J. , Povea‐Cabello, S. , Alvarez‐Cordoba, M. , Villalon‐Garcia, I. , Munuera‐Cabeza, M. , Suarez‐Carrillo, A. , Talaveron‐Rey, M. , & Sanchez‐Alcazar, J. A. (2021, February 4). Coenzyme Q10 analogues: Benefits and challenges for therapeutics. Antioxidants (Basel), 10(2), 236. 10.3390/antiox10020236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakubo, F. , Cartwright, P. , Hoogenraad, N. , Thorburn, D. R. , Collins, F. , Lithgow, T. , & Dahl, H. H. (1995, October). An amino acid substitution in the pyruvate dehydrogenase E1 alpha gene, affecting mitochondrial import of the precursor protein. American Journal of Human Genetics, 57(4), 772–780. [PMC free article] [PubMed] [Google Scholar]
- Torraco, A. , Stehling, O. , Stumpfig, C. , Rosser, R. , De Rasmo, D. , Fiermonte, G. , Verrigni, D. , Rizza, T. , Vozza, A. , Di Nottia, M. , Diodato, D. , Martinelli, D. , Piemonte, F. , Dionisi‐Vici, C. , Bertini, E. , Lill, R. , & Carrozzo, R. (2018, August 1). ISCA1 mutation in a patient with infantile‐onset leukodystrophy causes defects in mitochondrial [4Fe‐4S] proteins. Human Molecular Genetics, 27(15), 2739–2754. 10.1093/hmg/ddy183 [DOI] [PubMed] [Google Scholar]
- Wightman, P. J. , Santer, R. , Ribes, A. , Dougherty, F. , McGill, N. , Thorburn, D. R. , & FitzPatrick, D. R. (2003, October). MLYCD mutation analysis: Evidence for protein mistargeting as a cause of MLYCD deficiency. Human Mutation, 22(4), 288–300. 10.1002/humu.10264 [DOI] [PubMed] [Google Scholar]
- Witters, P. , Saada, A. , Honzik, T. , Tesarova, M. , Kleinle, S. , Horvath, R. , Goldstein, A. , & Morava, E. (2018, April). Revisiting mitochondrial diagnostic criteria in the new era of genomics. Genetics in Medicine, 20(4), 444–451. 10.1038/gim.2017.125 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.