

Abstract
Purpose:
HIF2α is a key driver of kidney cancer. Using a belzutifan analogue (PT2399), we previously showed in tumorgrafts (TGs) that ~50% of clear cell renal cell carcinomas (ccRCCs) are HIF2α dependent. However, prolonged treatment induced resistance mutations, which we also identified in humans. Here, we evaluated a tumor-directed, systemically-delivered, siRNA drug (siHIF2) active against wild-type and resistant mutant HIF2α.
Experimental Design:
Using our credentialed TG platform, we performed pharmacokinetic and pharmacodynamic analyses evaluating uptake, HIF2α silencing, target gene inactivation and anti-tumor activity. Orthogonal RNA-seq studies of siHIF2 and PT2399 were pursued to define the HIF2 transcriptome. Analyses were extended to a TG line generated from a study biopsy of a siHIF2 phase I clinical trial (NCT04169711) participant and the corresponding patient, an extensively pretreated individual with rapidly progressive ccRCC and paraneoplastic polycythemia likely evidencing a HIF2 dependency.
Results:
siHIF2 was taken up by ccRCC TGs, effectively depleted HIF2α, deactivated orthogonally-defined effector pathways (including Myc and novel E2F pathways), downregulated cell cycle genes and inhibited tumor growth. Effects on the study subject TG mimicked those in the patient, where HIF2α was silenced in tumor biopsies, circulating erythropoietin was downregulated, polycythemia was suppressed, and a partial response was induced.
Conclusions:
To our knowledge, this is the first example of functional inactivation of an oncoprotein and tumor suppression with a systemic, tumor-directed, RNA-silencing drug. These studies provide a proof-of-principle of HIF2α inhibition by RNA-targeting drugs in ccRCC and establish a paradigm for tumor-directed RNA-based therapeutics in cancer.
Keywords: A1HIF2, A2HIF2, ARO-HIF2, Belzutifan, E2F2, E2F8, PBK, PDX, PT2385, PT2977, RGD-DPC, siRNA
Introduction
Clear cell renal cell carcinoma (ccRCC), the most common type of kidney cancer, is characterized by inactivation of the von Hippel-Lindau (VHL) tumor suppressor gene [1]. Inactivated either through mutation (80%) or epigenetically (10%), VHL loss is regarded as the signature event of ccRCC [2–5]. The VHL protein normally functions to target hypoxia-inducible factor α (HIFα, both HIF1α and HIF2α) for degradation. When VHL is inactivated, HIFα subunits accumulate, bind their HIF1β partner, translocate to the nucleus and activate gene expression [6]. Among HIFα subunits, HIF2α is specifically regarded as the major oncogenic driver of ccRCC [7–11]. HIF2 (HIF2α/HIF1β) regulates a plethora of genes promoting cell survival, stemness, proliferation, and angiogenesis [10, 12–14]. Particularly prominent among the targets is vascular endothelial growth factor (VEGF), a secreted ligand that binds VEGF receptor 2 (VEGFR2) in endothelial cells promoting angiogenesis [10, 15–18]. The VEGF/VEGFR2 axis is of such importance that it is the target of 8 FDA-approved drugs for ccRCC to date [19]. However, as a more proximal and broader effector, HIF2 would be a more attractive candidate for drug targeting [20].
Historically regarded as undruggable [21, 22], a vulnerability was discovered in the HIF2a structure leading to the identification of small molecules that bound HIF2a, dissociating it from its obligatory partner HIF1b and inhibiting its function [23–25]. These chemicals were licensed to Peloton Therapeutics, Inc. who developed several analog drugs generally referred to as PT drugs (PT2385, PT2399, or the recently FDA-approved PT2977, also called belzutifan). In our previous study, we evaluated PT2399 for the treatment of ccRCC [26]. We showed that PT2399 inhibited tumor growth in approximately 50% of ccRCC tumorgrafts (TGs). However, when sensitive TG-bearing mice were subjected to prolonged treatment, resistance developed, and we identified an acquired gatekeeper HIF2α mutation (G323E) [26]. In vitro, HIF2α (G323E) was sufficient to prevent PT2399 from dissociating HIF2 complexes [26]. Subsequently, we identified the same mutation in patients treated with PT2385 in a phase I clinical trial [27]. A second resistance mutation was also discovered in HIF1β at the interface with HIF2α [26]. Resistance mutations are a well-known phenomenon with targeted therapies [28–30] and complementary therapeutic approaches are therefore needed.
There has been substantial progress with RNA-based and RNA-targeting therapeutics. mRNA-based vaccines have been developed for COVID-19 and the Food and Drug Administration (FDA) has approved several RNA-targeting drugs for metabolic and other diseases [31, 32]. However, to date, no RNA-targeting drug has been approved for cancer therapy. Recently, Arrowhead Pharmaceuticals reported the development of a HIF2α targeting siRNA drug [33]. Consisting of a specific HIF2α-targeting double stranded (ds)RNA, a dynamic polyconjugate (DPC), and a tumor-directed ligand (an RGD peptide mimetic that interacts with integrins αvβ3 and αvβ5 expressed in ccRCC [34, 35]), their first generation siHIF2 drug (referred herein as A1HIF2) was taken up by ccRCC cell lines and suppressed tumor growth [33].
Expanding upon these results, we show that A1HIF2 silences HIF2α, suppressing HIF2 target genes, and inhibits the growth of multiple patient-derived HIF2-dependent TG lines from the same platform that previously credentialed PT drugs [26]. Using orthogonal approaches integrating siHIF2 and PT2399, we characterize the HIF2 effector transcriptome in unprecedented detail. We show that A1HIF2 has activity against both wild-type as well as resistant mutant HIF2α. We present studies of a second-generation inhibitor and clinical candidate (A2HIF2), which shares the same HIF2α targeting sequence, but of significantly smaller size. We provide proof-of-principle of activity in a patient from whom a TG line was generated who participated in the phase I clinical trial [36], where A2HIF2 suppressed not only HIF2α protein in the tumor but also HIF2-dependent paraneoplastic polycythemia and induced a partial response. To our knowledge, this is the first example of functional inactivation of an oncoprotein and tumor suppression with a systemic tumor-directed RNA-silencing drug.
Materials and Methods
Nomenclature
XP refers to established tumorgraft lines [37, 38]. V and A represent respectively vehicle and A1/A2HIF2 treated mice. P refers to PT drug treated mice. An .x and .y were added to distinguish two vehicle treated samples from PT2399 and A1HIF2 trials that otherwise had the same name (XP374V1).
Tumorgrafts
Studies were conducted in accordance with the U.S. Common Rule and informed written consent was obtained from each subject. Investigations were performed after approval by an institutional review board and in accordance with an assurance filed with and approved by the U.S. Department of Health and Human Services. Patient derived TGs were generated and maintained as previously described [38, 39] . Briefly, tumor fragments were implanted orthotopically into 4- to 6-week-old male or female non-obese diabetic severe combined immunodeficient (NOD-SCID) mice according to a UT Southwestern Institutional Animal Care and Use Committee (IACUC) approved protocol. Mice were monitored weekly for tumor growth by palpation and by MRI as indicated.
A1HIF2 and A2HIF2 targeting sequence
The sequence of A1HIF2 is: Sense strand (5’ to 3’): ACGUAACGAUUUCAUGAAAT; Antisense strand (5’ to 3’): TUUCAUGAAAUCGUUACGUTT
The sequence of A2HIF2 is: Sense strand (5’ to 3’): CAACGUAACGAUUUCAUGAAA; Antisense strand (5’ to 3’): UUUCAUGAAAUCGUUACGUUG
Drug uptake experiments
NOD/SCID mice were implanted orthotopically with tumor fragments. Once tumors reached ~100–300 mm3, two mice per TG line (n=4 total) were administered RGD-DPC-Cy3 and one mouse per line (n=2 total) was administered DPC-Cy3 via intravenous injection. Tumors and normal kidneys were excised 4 hours after injection and frozen tissue sections were prepared. Tissue sections were counterstained with anti-Na+/K+-ATPase α1 (CST, 23565) for membrane (green) and DAPI for cell nuclei (blue). Cover glass mounted slides were analyzed using a LSM710 confocal microscope.
Drug trials
Typically, 6 NOD/SCID mice were implanted orthotopically with tissues from the TG lines. Once tumors reached ~6 mm in diameter, mice were allocated to vehicle or A1HIF2. A1HIF2 was administered at 10 mg/kg in D5W via intravenous injection every two weeks. Where applicable, A2HIF2 was administered at 20 mg/kg in D5W via intravenous injection once a week. D5W was used as a control. Tumor growth was evaluated by magnetic resonance imaging (MRI). Tumor volume was measured by multiplying tumor length, width, and depth to reduce bias. Toxicity was evaluated by monitoring mouse weights once weekly. Mice were euthanized according to IACUC guidelines at the end of drug trials, when they became ill or tumor reached 20 mm in diameter. Tumor tissues, plasma and serum were collected, processed, and stored as previously described [38, 39].
Immunohistochemistry
Immunohistochemistry (IHC) was performed using a Dako Autostainer Link 48. HIF2α IHC procedures and interpretation was standardized in our laboratory based on expression profiles in well-characterized cell lines and human ccRCC tissues with known expression by western blot analyses as described previously [26]. Tissues were stained with HIF2α antibodies (1:1800, clone A3, Bethyl Laboratories or 1:75, sc-46691, Santa Cruz) using citrate buffer antigen retrieval (K800521, Dako). Appropriate positive and negative controls were used with each run of immunostaining. The percentage of tumor cells staining in the entire section examined was recorded by a trained pathologist. Only nuclear reactivity was regarded as a positive expression.
Real-time PCR
Total RNA was isolated using TRIzol according to the manufacturer’s instructions (15596026, Invitrogen). 2 μg of total RNA for each sample was reverse transcribed using iScript Reverse Transcription Supermix (170–8841, Bio-Rad). RT–PCR was performed on an ABI 7500 Real-Time PCR machine using iTaq Universal SYBR Green SMX (1725124, Bio-Rad). Primers available upon request.
Western blot
For western blot analyses, HIF2α antibody (A700–003, Bethyl) was diluted at 1:1,000 in 5% BSA in Tris-Buffered Saline (TBS) and incubated overnight at 4 °C. Tubulin antibody (T5168, Sigma) was diluted at 1:5,000. Anti-rabbit and anti-mouse secondary antibodies (31460, 31430, Pierce) were used to detect primary antibodies. ECL substrate (1705061, Bio-rad) was used for signal development.
ELISA
ELISA was performed using human VEGF ELISA kit (DVE00, R&D Systems) as previously described [26].
RNA-seq analyses
For A1HIF2 RNA-seq, 11 vehicle- and 11 A1HIF2-treated tumor RNA samples from XP165, XP283, XP289, and XP374, underwent RNA-seq at the New York Genome Center. For PT2399 RNAseq, 12 vehicle- and 12 PT2399-treated tumor RNA samples from XP144, XP164, XP373, XP374, and XP453 were accessed from Sequence Read Archive SRP073253. Downstream bioinformatic analyses were performed as follows. Briefly, quality control for raw data was accomplished using fastp to filter low-quality reads and trim adaptors [40]. Passed reads were aligned to both mouse and human reference sequences with bbmap [41]. Mouse reads were filtered out and human reads were re-mapped to the NCBI hg38 using Hisat2 [41]. PCR duplicates were further removed by picard (https://broadinstitute.github.io/picard/). Quantification of genes was performed using HTSeq [42]. Differential gene expression analysis was measured using DEseq2 [43]. A false discovery rate (FDR) cutoff of 0.05 was applied to identify statistically significant genes between comparison groups. GO enrichment analysis was performed using clusterProfiler in R [44]. Gene Set Enrichment Analysis (GSEA) was performed using GSEA software (https://www.gsea-msigdb.org/gsea/index.jsp) [40].
ChIP-seq analyses
ChIP-seq data was retrieved from Gene Expression Omnibus (Accession codes: GSE120887). Normalized bigwig files of called peaks were loaded into the Integrative Genomics Viewer (IGV). Peaks around the regulatory region of a gene locus were used to determine whether the gene may be a direct target of HIF2α.
Statistical analyses
Significance of relative mRNA expression values between A1HIF2 and vehicle groups was determined using Student t-tests. Significance of changes in tumor volume or weight was determined using a Mann–Whitney U test. Overlap of gene signatures was evaluated using a hypergeometric distribution. Data were analyzed and plotted using GraphPad Prism.
Data and materials availability statement
The data and materials, including TG lines, generated in this study.
Results
siHIF2 targeting of ccRCC tumorgrafts
We evaluated a first generation siHIF2 (A1HIF2) developed by Arrowhead Pharmaceuticals [33] and investigated its efficacy using the same TG platform previously credentialing PT drugs [26]. We evaluated four different patient-derived ccRCC TGs that were known (or suspected) to be HIF2α dependent and expressed the target integrins αvβ3 and αvβ5 (αvβ3/β5) (XP374, XP165, XP283 and XP289) [26, 45].
A summary of patient characteristics and treatment prior to TG line generation is shown in Table 1. XP374 was derived from a 48-year-old man who presented with a 14 cm ccRCC with sarcomatoid dedifferentiation that invaded beyond the kidney. XP165 was generated from an abdominal wall metastasis of a 42-year-old man with ccRCC, who had been previously treated with systemic therapy including an anti-VEGF antibody (bevacizumab). XP283 was derived from a high-grade locally invasive primary ccRCC from a 77-year-old woman who went on to develop metastatic disease. XP289 was derived from a 62-year-old female who presented with metastatic ccRCC and underwent a radical nephrectomy of a 6.5 cm locally advanced tumor.
TABLE 1.
Characteristics of patients corresponding to each tumorgraft line.
| XPID | Race | Age | Sex | Source | Histology | Sarcomatoid | Rhabdoid | Nucleolar Grade | Stage | Prior Therapy |
|---|---|---|---|---|---|---|---|---|---|---|
| XP165 | White | 42 | Male | Abdominal mass | ccRCC | NA | NA | 3 | - | HD-IL2, Interferon, Bevacizumab |
| XP283 | White | 77 | Female | Kidney | ccRCC | Absent | NA | 3 | T3aN0M0 | Treatment Naïve |
| XP289 | White | 62 | Female | Kidney | ccRCC | Absent | NA | 3 | T3aN0M1 | Treatment Naïve |
| XP374 | White | 48 | Male | Kidney | ccRCC | Present | Present | 4 | T4N0M0 | Treatment Naïve |
| XP1487 * | White | 70 | Female | Lymph node | ccRCC | Absent | Absent | 3 | - | Sunitinib, Nivolumab, Axitinib, ARO-HIF2 |
TG line from tumor biopsy of phase I trial participant. NA, not available.
To evaluate A1HIF2 tumor targeting, we determined tumor uptake in two different TG lines in the presence and absence of the RGD integrin-targeting ligand. While an RGD-conjugated Cy3-labeled dynamic polyconjugate (RGD-DPC-Cy3) was taken up by the XP283 TG, this was not the case for DPC-Cy3 lacking the RGD peptide mimetic (Sup Fig. 1A). Similar results were observed in a second TG line, XP289 (Sup Fig. 1B). Of note, RGD-DPC-Cy3 uptake was specific for the tumor with little uptake in normal kidney cells. Thus, the RGD ligand is necessary and sufficient for ccRCC tumor targeting.
Tumor growth inhibition by siHIF2
Next, we evaluated the impact of A1HIF2 on the growth of orthotopically implanted TGs. Cohorts of TG-bearing mice were established and subjected to magnetic resonance imaging (MRI). Once tumors reached ~100–300 mm3, mice were allocated to either A1HIF2 (10 mg/kg intravenously [IV] every two weeks) or vehicle. Tumor growth was monitored longitudinally using MRI and tumors were collected at the end of the study. To assess HIF2α downregulation, immunohistochemistry (IHC) analyses were performed. When sufficient tissue was available, the impact of A1HIF2 on HIF2α protein was also measured by western blot.
A1HIF2 inhibited tumor growth by >75% in XP374 (p = 0.0049, Fig. 1A, B and Sup Fig. 2A). Similar results were observed by evaluating tumor volumes and weights at the end of the study (Fig. 1C, D). IHC showed significant suppression of HIF2α by A1HIF2 with <5% of the tumor cells staining positively (Fig. 2A). HIF2α suppression was confirmed by western blot (Fig. 2B). Unlike studies we previously performed with other FDA-approved kidney cancer drugs, such as sunitinib [26, 38], A1HIF2 did not induce weight loss (Sup Fig. 2B).
Fig. 1. Tumor growth inhibition by siHIF2 in ccRCC TGs.

A. Representative T2-weighted MRI images at baseline and after administration of A1HIF2 with corresponding tumor volume quantitation. B. Tumor volume by MRI (n=3 per arm) at baseline and at the end of treatment. C. Actual tumor volume measurements at the end of treatment. D. Tumor weight at the end of treatment. *, p < 0.05; **, p<0.01; ****, p < 0.0001. Error bars represent standard deviation.
Fig. 2. HIF2α silencing and VEGF downregulation by siHIF2.

A. Representative immunohistochemistry images illustrating HIF2α protein depletion by A1HIF2 in TGs. B. Western blot analyses of HIF2α in A1HIF2 treated TG-bearing mice. C. Human VEGF ELISA of circulating tumor-produced VEGF in A1HIF2 or vehicle treated mice. *, p < 0.05. Error bars represent standard deviation.
For XP165, an MRI on day 21 of A1HIF2 treatment, showed significant tumor growth inhibition (p = 0.0094, Fig. 1A, B and Sup Fig. 2A). Physical analyses revealed a ~60% reduction in tumor size compared to vehicle-treated mice and a similar reduction in tumor weight (Fig. 1C, D). IHC showed depletion of HIF2α by A1HIF2 with <5% of tumor cells staining positively (Fig. 2A), and results were confirmed by western blot (Fig. 2B). There was no evidence of toxicity (Sup Fig. 2B).
For XP283, MRI analyses after 3 doses of A1HIF2 showed >90% reduction in tumor growth (p = 0.017, Fig. 1A, B and Sup Fig. 2A), and this was confirmed at the end of the trial (Fig. 1C, D). A1HIF2 suppressed HIF2α expression by IHC (Fig. 2A), but due to the small tumor size, western blot analyses could not be performed on trial tumors. However, significant depletion of HIF2α was achieved by A1HIF2 in this TG line as well (Fig. 2B).
In XP289, A1HIF2 inhibited tumor growth (p=0.0049) decreasing tumor size and weight by > 60% (Fig. 1 and Sup Fig. 2A). A1HIF2 suppressed HIF2α expression by IHC (Fig. 2A) and suppression was also observed by western blot (Fig. 2B).
To further evaluate the impact of HIF2α suppression, we measured VEGF, which plays a key role in ccRCC tumorigenesis. We leveraged the species difference between the tumor and host using an ELISA assay specific for human VEGF (hVEGF) and measured the effect of A1HIF2 on circulating VEGF levels. A1HIF2 markedly suppressed tumor VEGF production (Fig. 2C). While VEGF may also be regulated by HIF1α [15, 46], as expected, quantitative RT-PCR showed suppression of HIF2α (EPAS1) by A1HIF2 but not HIF1α (HIF1A) (Sup Fig. 3). In addition, no significant change was observed in mRNA levels of HIF1α canonical target genes (LDHA, PGK1) (Sup Fig. 3). Overall, these data show that A1HIF2 specifically depletes HIF2α in human ccRCC transplants in mice, where it inhibits VEGF production and tumor growth with no appreciable toxicity.
Orthogonal analyses illuminate HIF2 program in ccRCC
Previous studies of HIF2 in ccRCC tumorigenesis have been largely conducted using RCC tumor cell lines and how these cell lines reflect the role of HIF2 in RCC tumorigenesis is unknown. We evaluated the impact of A1HIF2 on gene expression by RNA-seq. We performed RNA-seq on 22 TGs from mice implanted with the 4 TG lines (XP165, XP283, XP289 and XP374) treated with either vehicle or A1HIF2. We identified 294 genes that were downregulated at least 2-fold (logFC < −1) at an FDRq < 0.05.
To isolate the effect of HIF2, we performed orthogonal studies leveraging the same ccRCC TG platform and the HIF2 inhibitor, PT2399 [26]. We reasoned that by examining the convergence between these two very different HIF2 inhibitors, the HIF2 program may be defined with unprecedented accuracy. Furthermore, because TGs (i) recapitulate the biology of human ccRCC (as shown for example by their co-clustering with the corresponding patient tumors in unsupervised gene expression analyses [38, 45]), and (ii) enable direct studies of tumor cells without contamination from the stroma (which is murine), how HIF2 supports ccRCC transformation could be precisely dissected.
We integrated data from A1HIF2 with PT2399 RNA-seq studies involving 12 PT2399-treated and 12 controls from XP144, XP164, XP373, XP374, and XP453 [26]. Focusing again on genes downregulated at least 2-fold at an FDRq < 0.05, we identified 419 genes downregulated by PT2399 in TGs. Speaking to the specificity of the two drugs, unsupervised gene ontology (GO) analyses of the individual gene lists showed that among the top 10 categories, 8 biological process, 7 cellular components, and 6 molecular functions were shared between A1HIF2 and PT2399 (Sup Fig. 4A). KEGG analysis further confirmed these results (Sup Fig. 4B).
Remarkably, 50% of the genes downregulated by A1HIF2 (147 out of 294) overlapped with the list of PT2399 downregulated genes (Fig. 3A and Fig. 3B). The probability of such convergence at random was quite low (p < 0.0001). The significance is further highlighted by considering that (i) the studies with A1HIF2 and PT2399 were performed on TG lines that only partially overlapped; (ii) both drugs are administered to mice systemically (one intravenously [A1HIF2] and the other one by gavage [PT2399]); (iii) a single timepoint was analyzed; and (iv) the studies were performed several years apart.
Fig. 3. Orthogonal analyses of siHIF2 and PT2399 in TGs define the HIF2 transcriptome in ccRCC.

A. Venn diagram showing shared downregulated genes (FDR < 0.05 and LogFC < −1) by A1HIF2 and PT2399 in TGs (11 vehicle- and 11 A1HIF2-treated tumors from XP165, XP283, XP289, and XP374; and 12 vehicle- and 12 PT2399-treated tumors from XP144, XP164, XP373, XP374, and XP453). B. Heatmap of the overlapping 147 significantly downregulated genes. C. Unsupervised GSEA of overlapping 147 genes showing convergence on MYC, E2F, and G2M targets.
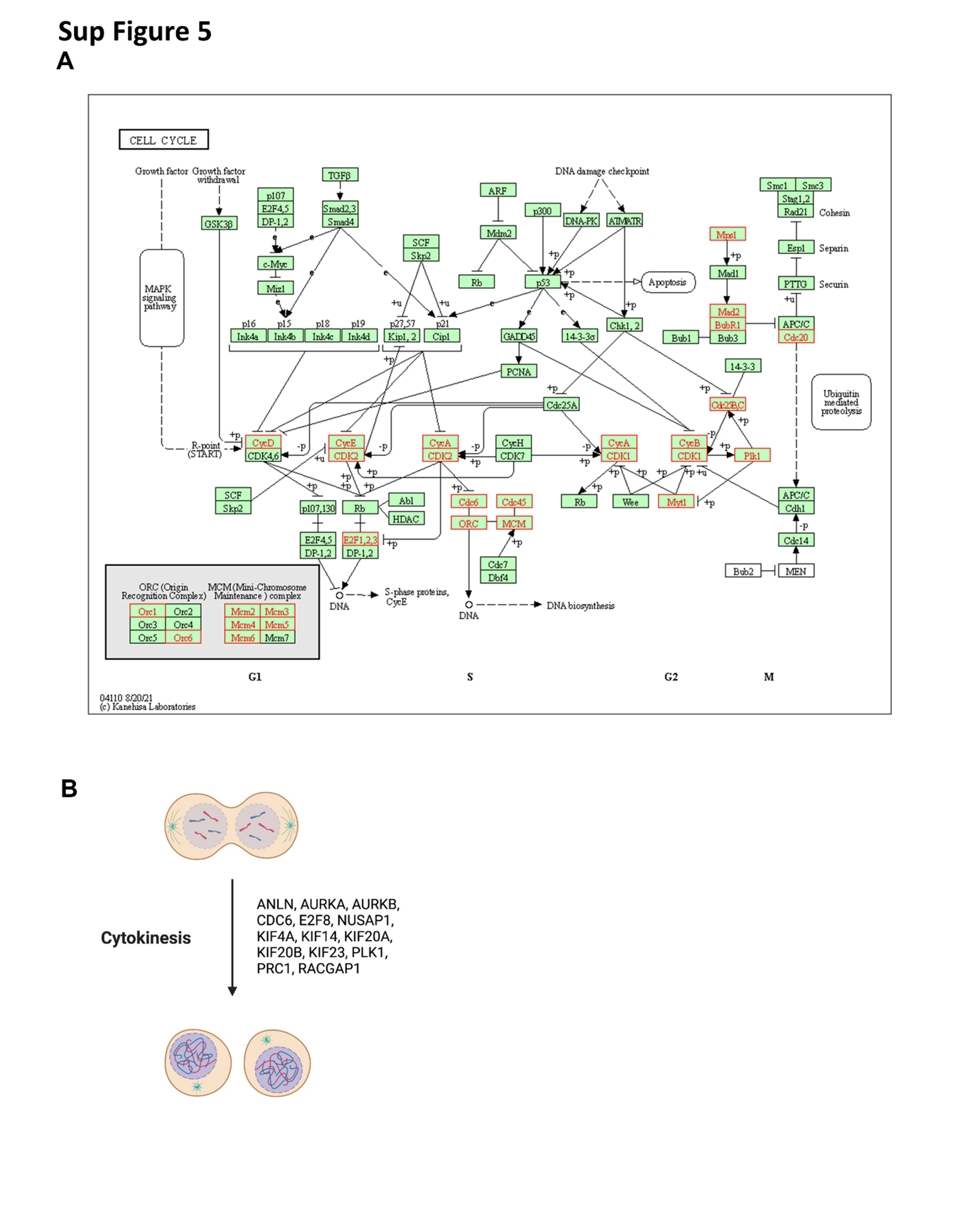
An investigation of the 147 gene list showed extensive cell cycle genes (Sup Fig. 5A). They included genes encoding cyclins (Cyclin D, Cyclin E, Cyclin A); cyclin-dependent and other cell cycle kinases such as CDK2, CDK1 and PLK1; origin recognition complex (ORC) proteins (ORC1, ORC6); mini-chromosome maintenance (MCM) complex proteins (MCM2, MCM3, MCM4, MCM5 and MCM6); ORC/MCM regulatory proteins (CDC6 and CDC45); anaphase promoting complex (APC) regulators (CDC20, BUBR1, MAD2 and MPS1) as well as proteins implicated in cytokinesis including ANLN, AURKA, KIF4A, KIF20A, KIF14, NUSAP1, KIF23, KIF20B. RACGAP1, AURKB and PRC1 (Sup Fig. 5B).
Notably, the 147 gene list included an independently defined and previously reported 11-gene network that was found to be upregulated across 32 cancer types [47]. Every member of the list was present (PBK, BIRC5, CCNB1, CDC20, CDK1, DLGAP5, MAD2L1, MELK, PLK1, TOP2A, and TTK). Assuming 25,000 genes in the human genome, the total number of 11-gene combinations is ~6×1040, and the probability that a particular such group be found among 147 genes is p= 1.98×10-25. Interestingly, all 11 genes contain HIF-binding sites (hypoxia-response elements) in the promoter region (−1000 to 100 from the transcription start site [TSS]). These data suggest that this previously defined pan-cancer network is regulated by HIF2.
Gene Set Enrichment Analysis (GSEA) of overlapping genes showed a convergence on Myc and E2F programs (Fig. 3C and Sup Fig. 4C). While Myc has been extensively implicated as an effector downstream of HIF2 [11, 48], our data implicate E2F in ccRCC development. Both E2F2 and E2F8 were downregulated by A1HIF2 and PT2399. In addition, from among the 147 commonly downregulated genes, 44 are putative E2F target genes (Sup Table 1).
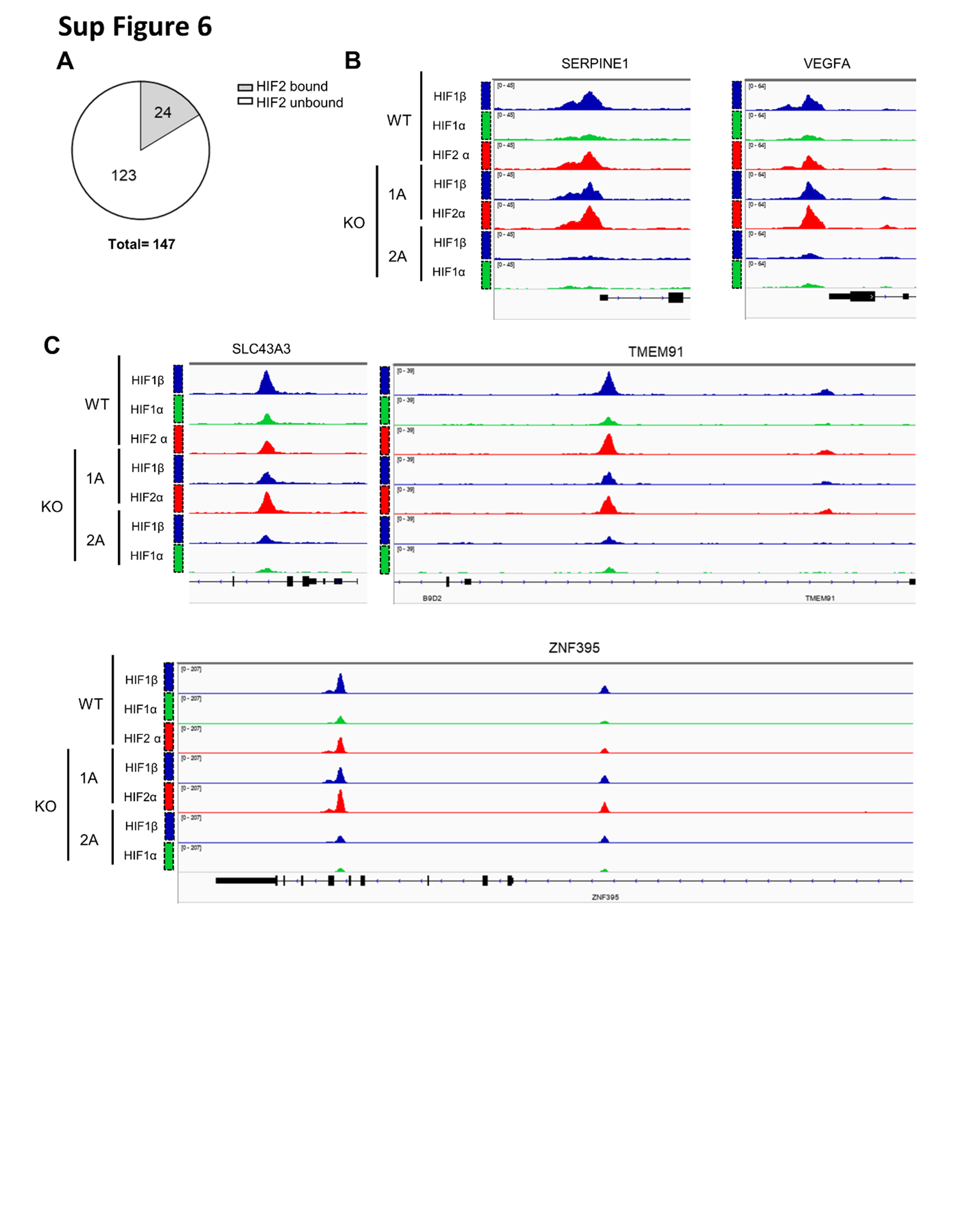
To further characterize the 147 gene list, we integrated our results with elegant HIF2α chromatin immunoprecipitation and sequencing (ChIP-Seq) experiments in human kidney proximal tubule (HKC8) cells [49–51]. In these experiments, HKC8 cells were edited to disrupt HIF2α, HIF1α or HIF1β and subjected to ChIP for all 3 proteins (GSE120887) (Sup Fig. 6A and Sup Table 2). To identify putative direct HIF2α target genes, we focused on genes: (i) with predominant HIF2α (compared to HIF1α) binding (TSS ± 5 Kb); (ii) unaffected by HIF1α disruption; and (iii) dampened by HIF2α knockout (Sup Fig. 6B, C). We found 24 HIF2 target genes, including canonical targets such as SERPINE1 and VEGFA (Sup Fig. 6B, C and Sup Table 2). However, many genes not previously implicated as putative direct HIF2 target genes in ccRCC were discovered, including cell cycle genes, such as ASPM, AURKA, BLM, CDCA2, CDCA3, CHAF1A, FEN1, GTSE1, KIF15, KNL1, MAD2L1, MCM2, NUF2 and PCLAF.
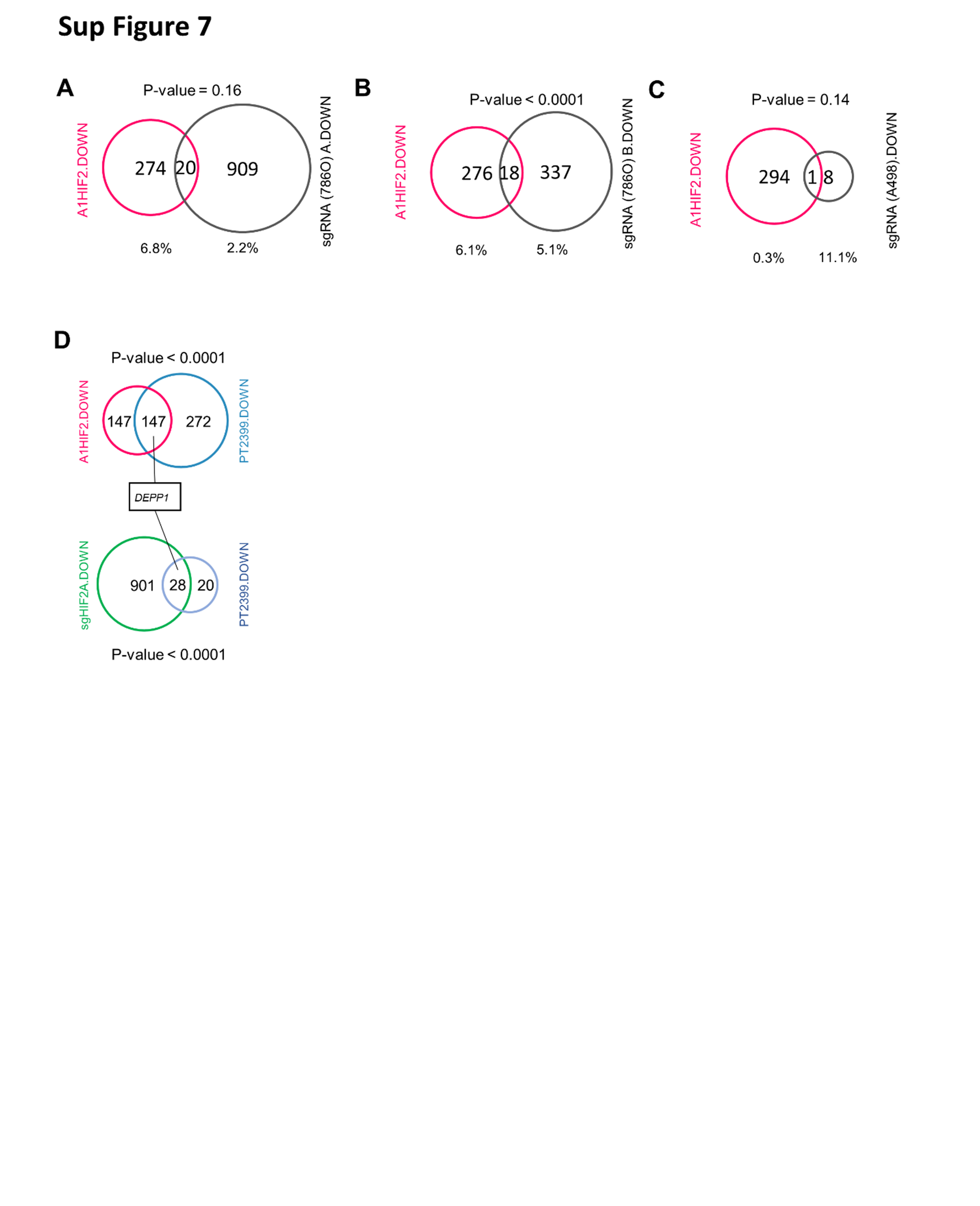
To assess the value of studying HIF2 function directly in TGs, we contextualized our findings with previous studies in ccRCC cell lines. We compared our RNA-seq results using the A1HIF2 siRNA to 3 previously published studies in which HIF2α was depleted in cell lines using sgRNA or siRNA. The first dataset (GSE72959), which involves microarray analysis of 786O ccRCC cells after CRISPR-based HIF2α gene (EPAS1) editing [52] identified 929 downregulated genes (Sup Fig. 7A). Only 20 genes overlapped with the 294 A1HIF2 downregulated genes (Sup Fig. 7A). A second similar dataset where EPAS1 was edited in 786O cells and output was evaluated by RNA-seq (GSE149005) [53] identified 355 downregulated genes, but only 18 genes overlapped (Sup Fig. 7B). Finally, microarray analysis of A498 cells treated with a HIF2α siRNA (GSE16622) [13] identified 9 downregulated genes and only one that overlapped (Sup Fig. 7C). Thus, a very small percentage of genes deregulated by HIF2α targeting in cell lines were validated in TGs. Furthermore, fewer than 10% of genes regulated by HIF2 in ccRCC TGs were identified in experiments using cell lines.
One cell line dataset involved orthogonal analyses similar to ours. In the GSE72959 dataset, 786O cells were treated with either a HIF2α sgRNA or PT2399. 929 genes were downregulated by the EPAS1 sgRNA, 48 by PT2399, and 28 overlapped (Sup Fig. 7D). Thus, whereas 50% of the genes downregulated by A1HIF2 in TGs were also downregulated by PT2399, only 3% of the genes downregulated by EPAS1 sgRNA were also downregulated by PT2399 in cell lines. When comparing the 28 genes to the 147 overlapping genes in TGs, only one gene overlapped, DEPP1. DEPP1 (also known as DEPP or C10orf10) is a hypoxia-responsive gene [54] implicated in autophagy and is likely a target of HIF2 in ccRCC [55].
HIF2α silencing by siHIF2 clinical candidate (A2HIF2)
Next, we evaluated a second-generation siHIF2, which preserves the RGD targeting moiety and the RNAi trigger sequence (see methods), but is otherwise significantly smaller and is the clinical candidate, ARO-HIF2 (referred herein as A2HIF2) (Fig. 4A). XP165 TG-bearing mice were treated with A1HIF2 (10 mg/kg in D5W via intravenous injection every two weeks), A2HIF2 (20 mg/kg in D5W via intravenous injection once a week) or vehicle. Western blot of tumor samples showed suppression of HIF2α by both A1HIF2 and A2HIF2 (Fig. 4B). However, despite A2HIF2 administration in molar excess compared to A1HIF2, HIF2α appeared to be downregulated to a lesser extent (Fig. 4B). Similar results were observed in other TG lines.
Fig. 4. First (A1HIF2) and second generation siHIF2 (A2HIF2).

A. Schematic illustration of A1HIF2 and A2HIF2, which share the same RNAi trigger. B. Comparative analyses of HIF2α depletion by A1HIF2 and A2HIF2. C. Depletion of ectopically expressed HIF2α with acquired resistance mutation (G323E) in HEK293T by siHIF2 (A1HIF2). D. Clinical trial schema of A2HIF2 (NCT04169711).
Resistant mutant HIF2α is inhibited by siHIF2
Our previous studies showed that TGs undergoing prolonged treatment with PT2399 acquired resistance and we identified a gatekeeper HIF2α mutation (G323E) [26]. The same mutation was subsequently found in patients who developed acquired resistance to the related drug (PT2385) in the phase I clinical trial [27]. This mutation is outside the HIF2α siRNA targeting sequence and would not be expected to affect siHIF2 activity. Indeed, A1HIF2 effectively depleted resistant-mutant HIF2α (Fig. 4C).
siHIF2 activity in TG from clinical trial participant
A2HIF2 advanced to a phase I clinical trial (Study of ARO-HIF2 in patients with advanced clear cell renal cell carcinoma; NCT04169711), which opened at UT Southwestern Medical Center (UTSW) in 2020. A2HIF2 was evaluated in three sequential dose escalation cohorts (Fig. 4D) of patients with advanced ccRCC previously treated with conventional anti-angiogenic therapy and immune checkpoint inhibitors. The study included pretreatment and on-treatment (after two doses) tumor biopsies for pharmacodynamic analyses.
Biopsy samples from UTSW patients participating in the study were implanted orthotopically in NOD/SCID mice using previously published protocols [38, 39]. A post-treatment biopsy specimen from a supraclavicular lymph node of a participant (106–00C) successfully engrafted and 69 days after implantation, the tumor was passaged into a second cohort of mice, where it grew again. Histological analyses showed that the TG line (XP1487) retained the features of the parent tumor (Fig. 5A).
Fig. 5. HIF2α inhibition and tumor suppression by siHIF2 in TG from clinical trial participant (106–00C).

A. Schematic illustrating orthotopic TG line generation from ultrasound-guided biopsy (yellow arrows) of a supraclavicular lymph node (white arrowheads) from 106–00C (day 16) along with representative H&E images of the patient core biopsy and corresponding TG (XP1487). B. MRI images of orthotopic tumor-bearing TGs treated with A1HIF2 (or vehicle). C. Tumor volume and tumor weight analyses. D. Representative HIF2α immunohistochemistry images from A1HIF2-treated or control TGs. E. Western blot analysis of HIF2α protein in TGs treated with A1HIF2 or vehicle. F. Integrated analyses of two A1HIF2 trials in TGs from 106–00C (see also Sup Fig. 8). *, p < 0.05. Error bars represent standard deviation.
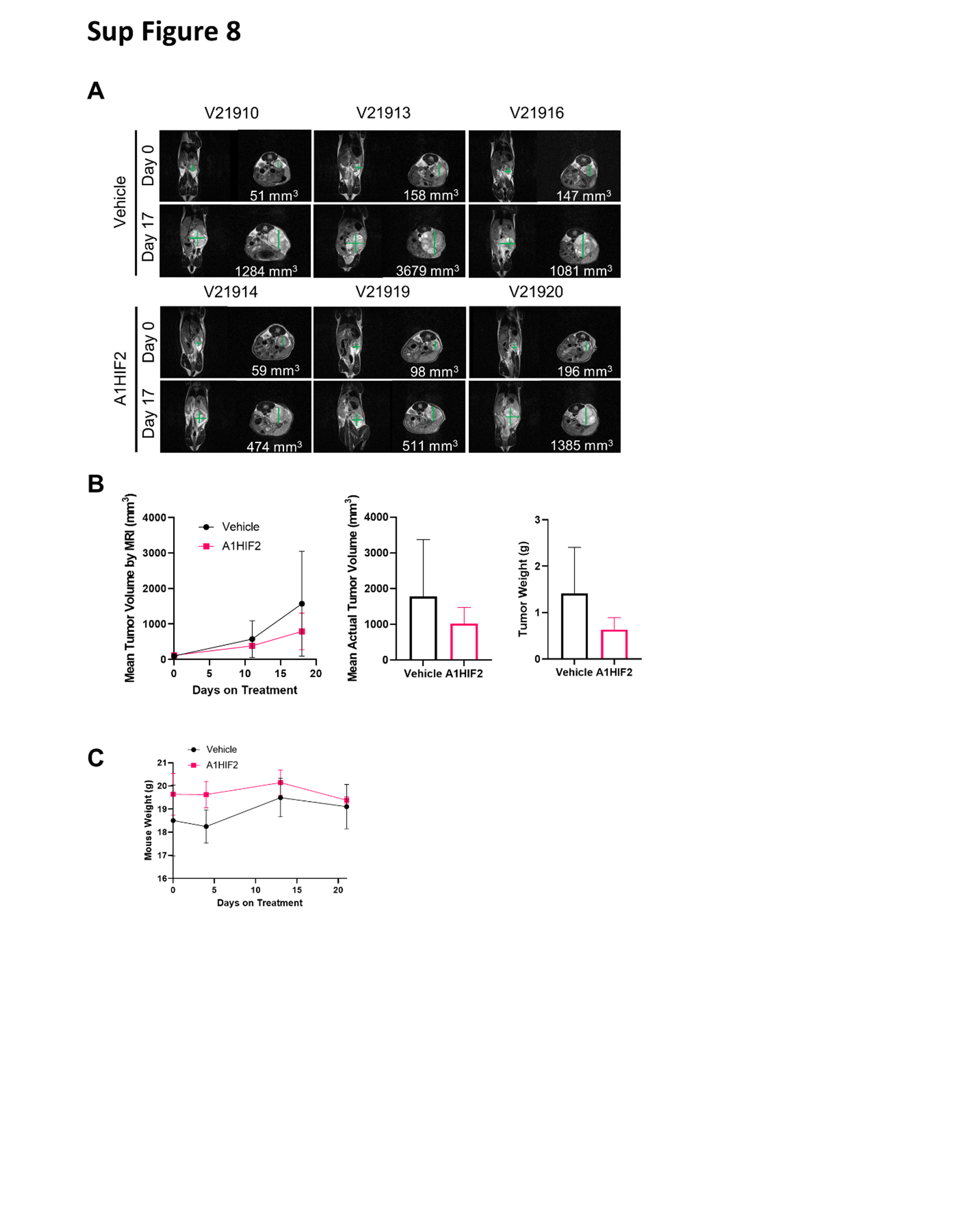
XP1487 TG samples were expanded for siHIF2 studies. We observed a >5-fold reduction in tumor size with A1HIF2 (Fig. 5B, C). HIF2α depletion was noted by IHC and confirmed by western blot analyses (Fig. 5D, E). Similar results were obtained with a second trial (Sup Fig. 8A, B) and a statistically significant difference in tumor growth was observed in combined analyses (Fig. 5F). As expected, A1HIF2 was well tolerated and the mice did not lose weight (Sup Fig. 8C).
HIF2α depletion, suppression of paraneoplastic polycythemia and tumor growth inhibition in clinical trial participant
Patient 106–00C, a 70-year-old female, was diagnosed with metastatic ccRCC in 2017. She had been treated with sunitinib (dose-reduced due to toxicity) for 7 months and had received nivolumab for 2 months, when she developed hyperprogression with new onset of brain, bone and scalp metastases. She received stereotactic radiation to brain and bone metastases and was switched to axitinib, on which she had rapid improvement. Her disease slowly progressed, however, necessitating a gradual increase in axitinib, on which she remained for 2 years. She developed paraneoplastic polycythemia, a syndrome resulting from tumor secretion of erythropoietin (Epo), which induces supranormal red blood cell counts (RBC) [56, 57]. Paraneoplastic polycythemia is most often observed with ccRCC, where it is likely driven by HIF2. To reduce RBC counts, phlebotomies were needed as often as every 2 weeks. In January 2021, the patient enrolled in cohort 2 (525 mg weekly) of the ARO-HIF2 phase I trial (NCT04169711). Her disease was particularly aggressive with clinical progression during the 2-week washout period mandated by the study necessitating repeat scans, which showed >30% increase by RECIST v1.1 criteria (Fig. 6A, B). She received two doses of weekly A2HIF2 and a biopsy on day 16 showed downregulation of HIF2α (compared to a pretreatment biopsy) (Fig. 6C). Unfortunately, she bled from an intestinal metastasis, which precipitated an acute coronary syndrome and she came off study. Despite stopping siHIF2 therapy, she had a deep partial response (>60%) (Fig. 6A, B). This was accompanied by a substantial decrease in her Epo, which normalized (Fig. 6B). During this time, she enjoyed an outstanding and unprecedented quality of life since her diagnosis. However, Epo levels began to rise over time and scans showed progression after 4 months. She was started on tivozanib, which reduced her Epo and controlled her disease, at the cost, however, of fatigue, diarrhea, loss of appetite and hypertension, which are common side effects of tyrosine kinase inhibitors (Fig. 6B).
Fig. 6. HIF2α downregulation, Epo suppression and tumor growth inhibition by siHIF2 in clinical trial participant (106–00C).

A. Axial (top) and coronal (bottom) CT images of subcarinal lymph node target lesion showing rapid progression during 2-week washout period and deep prolonged response after two doses of A2HIF2. While the first CT scan was performed without iodinated contrast, marked decrease in lymph node enhancement between baseline (second) CT and subsequent CTs illustrates profound antiangiogenic effect. B. Treatment timeline, tumor change (LD, longest diameter) and erythropoietin (EPO) levels prior to and following A2HIF2 administration (red lines). C. H&E and HIF2α immunohistochemistry of lymph node biopsy at baseline and 16 days after treatment onset with percent of tumor cells positively staining for HIF2α.
Discussion
Here, we show that systemic delivery of a tumor-directed HIF2α siRNA drug resulted in target inactivation and anti-tumor activity in ccRCC. siHIF2 effectively depleted HIF2α protein in human ccRCC tumors in mice inhibiting target gene expression and VEGF production, and this was associated with substantial tumor growth inhibition. Proof-of-principle of activity is also presented in a patient that participated in the phase I trial.
siHIF2 inhibited not only wild-type HIF2α, but also drug-resistant HIF2α. Previously, we reported that belzutifan analogues are associated with resistance mutations, including a gatekeeper mutation, which we first identified in preclinical models, and subsequently reported in patients [26, 27]. Resistance mutations are commonplace for targeted therapeutics. With the recent FDA approval of belzutifan, resistance mutations are likely to be more widely identified. Resistance mutations occur not only in HIF2α, but also in the HIF1β partner protein that is dislodged by belzutifan [27]. HIF1β mutations stabilize the HIF2α/HIF1β complex [58]. These mutations illustrate a fundamental dependency on HIF2 in ccRCC, and regardless of whether HIF2α or HIF1β are mutated, HIF2 should be inactivated by siHIF2.
We present proof-of-principle of activity in a patient (106–00C) that participated in the phase I clinical trial of A2HIF2 [36]. The patient enrolled in the second planned dose-escalation cohort. Her disease was characterized by sensitivity to anti-angiogenic therapy and resistance to immune checkpoint inhibitors and was particularly aggressive with objective progression in the two weeks washout period prior to A2HIF2. She had paraneoplastic polycythemia, which likely indicates a HIF2-driven ccRCC. She experienced a deep partial response, which was accompanied by suppression of Epo, normalization of her hemoglobin, and discontinuation of phlebotomies. IHC studies of pre- and on-treatment tumor biopsies showed HIF2α depletion by siHIF2. That tumor control was due to siHIF2 is supported by the following. First, there was substantial tumor growth preceding siHIF2 but the trend reversed following A2HIF2 administration. Second, tumor regression was accompanied by suppression of Epo. Third, there was tumor growth over time, which was accompanied by an Epo increase, and both responded to tivozanib, a potent FDA-approved ccRCC drug. Finally, her tumor was similarly responsive to siHIF2 in a TG line derived from a biopsy sample. Taken together, these data provide compelling evidence for siHIF2 activity against ccRCC.
In the phase I trial, A2HIF2 was administered weekly. However, in patient 106–00C the disease remained controlled and Epo suppressed for 4 months after just two A2HIF2 infusions. The prolonged activity is consistent with other systemically administered FDA approved siRNA therapeutics in non-oncologic conditions, which can be administered every 3 to 6 months [59]. While we cannot exclude idiosyncrasies about the particular patient, this case study illustrates the complexity of establishing administration schedules for RNA silencing therapeutics for oncologic indications. In this particular patient, erythropoietin could be used as a pharmacodynamic marker, but paraneoplastic polycythemia is uncommon, and in the majority of patients where tumors do not secrete Epo, Epo could not be used as a biomarker of HIF2α depletion in tumor cells. This emphasizes the need for approaches to evaluate target depletion by siRNA therapeutics. In the case of HIF2α, this could be accomplished by a HIF2α probe, and one has been generated by substituting a fluor atom in PT2385 for 18F[60, 61]. Preclinical dual TG models revealed selective uptake in HIF2α expressing ccRCCs by positron emission tomography (PET), and a clinical trial is ongoing (NCT04989959). Regardless, if generalizable, the data in 106–00C suggest that the A2HIF2 dosing interval could be significantly prolonged.
How many ccRCC patients may benefit from siHIF2 is unclear. In principle, all HIF2-dependent tumors should be responsive to siHIF2. Among these, a minority may have paraneoplastic polycythemia. However, tumors that are sensitive to belzutifan are likely HIF2-dependent. Furthermore, our discovery of HIF2 resistance mutations[27, 60] suggest that HIF2 represents a core dependency. This is unsurprising as ccRCC is thought to be initiated by VHL loss and HIF2 is possibly the major effector downstream. Thus, much like EGFR, where successive generations of inhibitors have been developed targeting resistant mutant EGFR, disabling mutant HIF2 which could be accomplished with siHIF2, should provide additional tumor control. However, the extent to which HIF2-dependent tumors will be responsive to siHIF2 will depend on siHIF2 uptake, which depends in turn on integrin expression [36].
We speculate that, as for targeted therapies, resistance will also arise to siHIF2 drugs. Resistance may involve mutations in the siRNA target sequence. However, RNA-targeting drugs could be re-designed to target other sequences in HIF2α. In addition, multiple sequences could be targeted simultaneously thereby reducing the chances of such resistance mutations. Nevertheless, other mechanisms of resistance may emerge leading for example to reduced drug uptake (i.e. downregulating integrin levels).
Given their specificity, siRNA-based therapeutics have the potential to be particularly well tolerated. Currently FDA-approved drugs targeting angiogenesis downstream of HIF2 cause a plethora of adverse effects including hypertension, diarrhea, thyroid dysfunction, and loss of appetite [19]. Many patients lose weight, which is also seen in mice [38]. Belzutifan and its analogues are significantly better tolerated, which is likely related to their inhibiting VEGF signaling just in the tumor (in other sites, VEGF production is uncoupled from HIF2) [26]. However, belzutifan and its analogues cause anemia and hypoxia [27, 62–65], which reflect two physiological processes that engage HIF2, red blood cell production [66] and respiration [67]. Anemia is frequently observed with chronic belzutifan administration and results from suppressing physiological Epo production [66]. However, hypoxia develops infrequently and appears to be associated with particular circumstances such as body habitus or insults such as anesthesia. By directing siRNA to tumor cells, siRNA drugs have the potential to be particularly well tolerated. Nevertheless, toxicities may be observed, as in the case of A2HIF2, that may pertain other aspects of the drug [36].
This study provides unique insight into HIF2 effector pathways in ccRCC. We leverage a state-of-the-art experimental system characterized by: (i) two highly specific inhibitors acting through different mechanisms (PT2399 and siHIF2); (ii) TGs, which faithfully reproduce human ccRCC [38, 45]; and (iii) the ability to eliminate confounding effects from the stroma. The importance of the approach is highlighted by comparative studies to tumor cell lines. Whereas in TGs, 50% of the genes downregulated by siHIF2 were downregulated by PT2399, this was the case for only 3% of the genes in cell lines. Furthermore, fewer than 10% of HIF2-regulated genes in ccRCC TGs were previously identified in cell line studies.
Several interesting observations emerged from gene expression analyses, which defined the HIF2 transcriptome in ccRCC with unprecedented accuracy. First, our data suggest that E2F is an important effector downstream of HIF2. Interestingly, E2F was previously linked to HIF2 in a genetically-engineered mouse model [68]. While previous studies were performed in mice and did not characterize the process further, we found that HIF2 regulates the levels of E2F2, which promotes cell cycle entry [69, 70]. Our data suggest that HIF2 may be implicated in cell cycle regulation. One unresolved question in ccRCC is where mitogenic signaling comes from. Most tumors have activating mutations in growth factor receptors or downstream signal transduction proteins, but these are rarely mutated in ccRCC. Our data suggest that perhaps HIF2 itself is responsible for inducing cell proliferation and many cell cycle genes we discovered were previously implicated as putative direct HIF2 targets in normal kidney tubular cells. It is noteworthy that HIF2α was originally identified in endothelial cells where hypoxia, unlike in most other tissues, induces a proliferative response [18]. We also found that HIF2 regulates E2F8, which has been implicated in angiogenesis [71]. Previously, E2F8 was shown to form a transcriptional complex with HIF1 and induce VEGF expression [71, 72]. Given the high degree of similarity between HIF1 and HIF2, HIF2 may similarly interact with E2F8 to promote VEGF expression. Finally, we identified all 11 members of a previously reported multi-cancer gene network and given our finding of hypoxia-response elements in the promoters of all genes, they may be directly regulated by HIF2.
Another aspect of the gene expression analyses is the paucity of HIF2-independent effects. Previously, we showed that PT2399 had minimal effects on the transcriptome of ccRCC TGs devoid of HIF2α [26]. The low number of genes deregulated by siHIF2 suggests that despite engaging the RNAi machinery to generate the final active siRNA drug, siHIF2 is similarly highly specific.
To our knowledge, this is the first example of target depletion and anti-tumor activity using a tumor-directed, systemically administered siRNA. This provides a proof-of-concept for what may become a new generation of RNA silencing cancer therapeutics. The same approach could be deployed to target non-canonical VHL substrates including ZHX2 and SFMBT1 [73, 74] as well as VHL synthetic lethal partners such TBK1, CDK4 and GLS [75–77]. In addition, the same paradigm could find application in other tumor types.
Supplementary Material
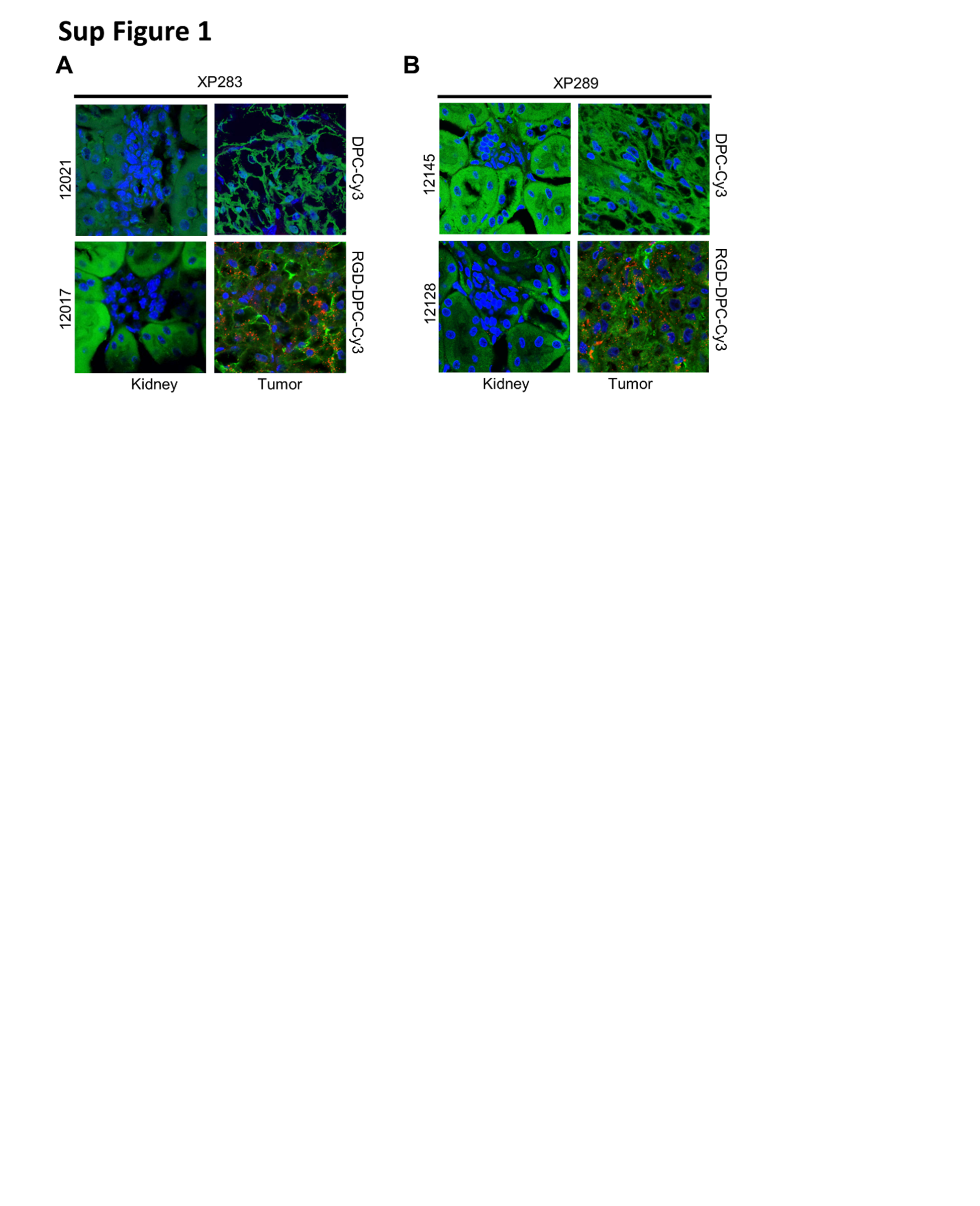{kind=link}
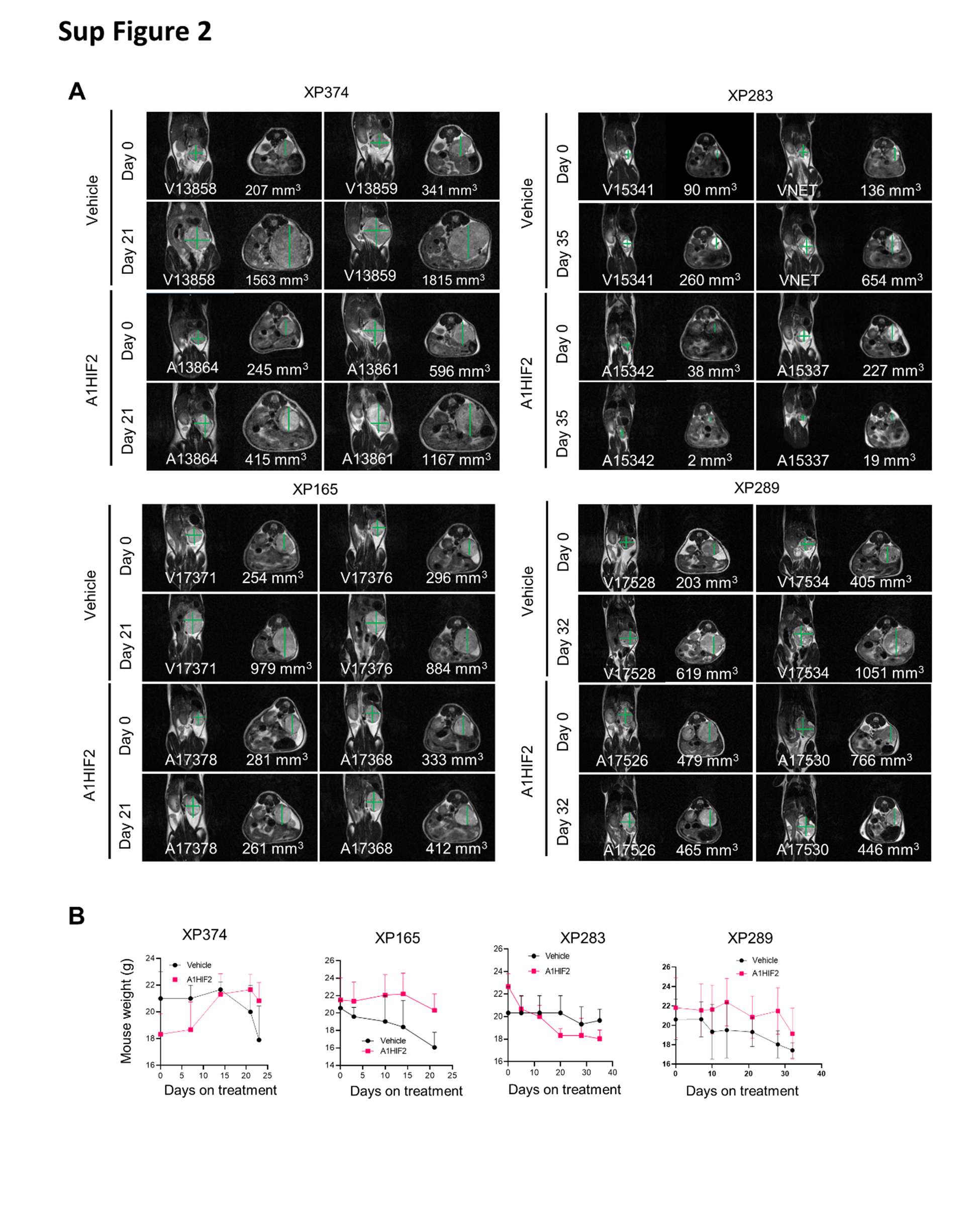{kind=link}
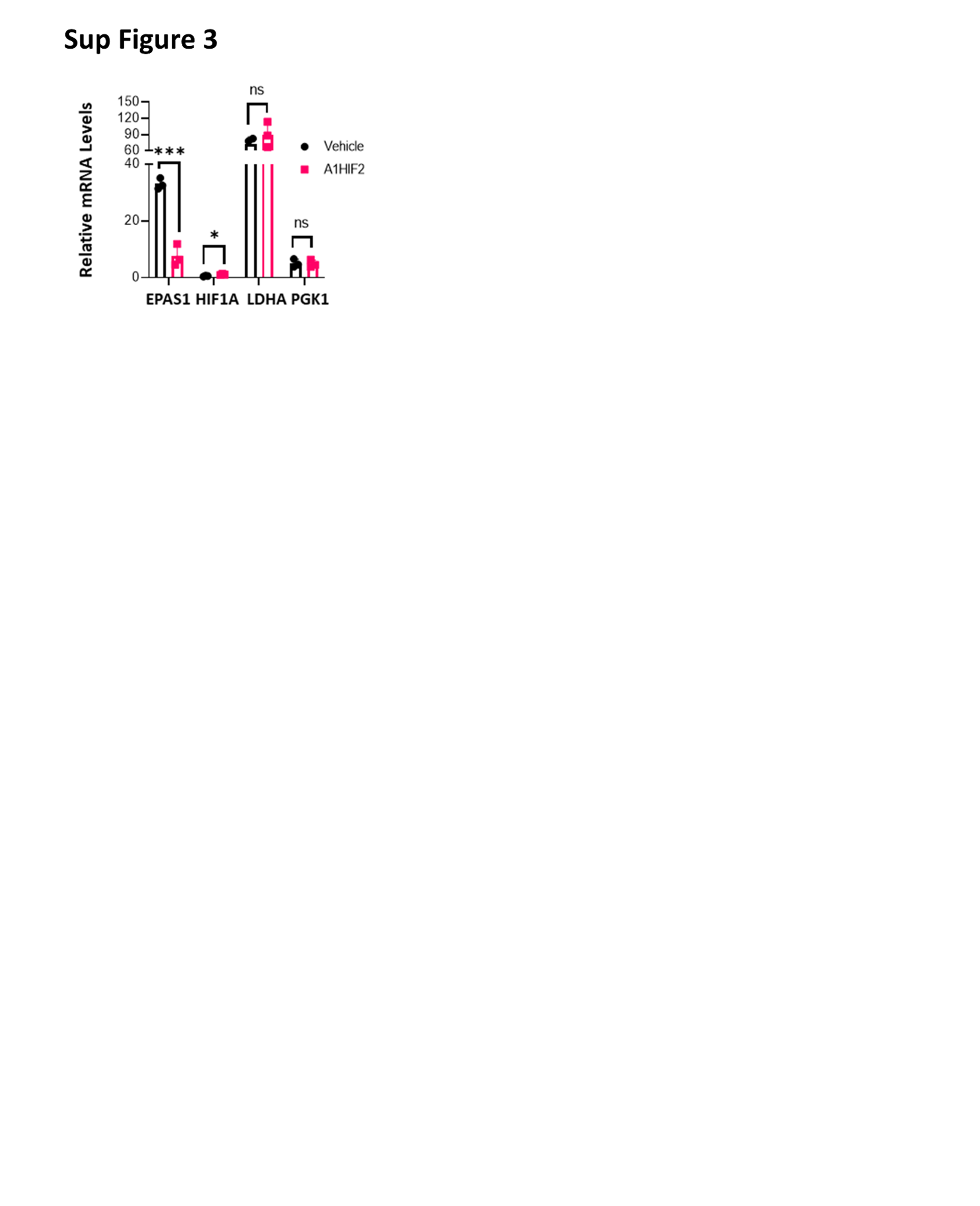{kind=link}
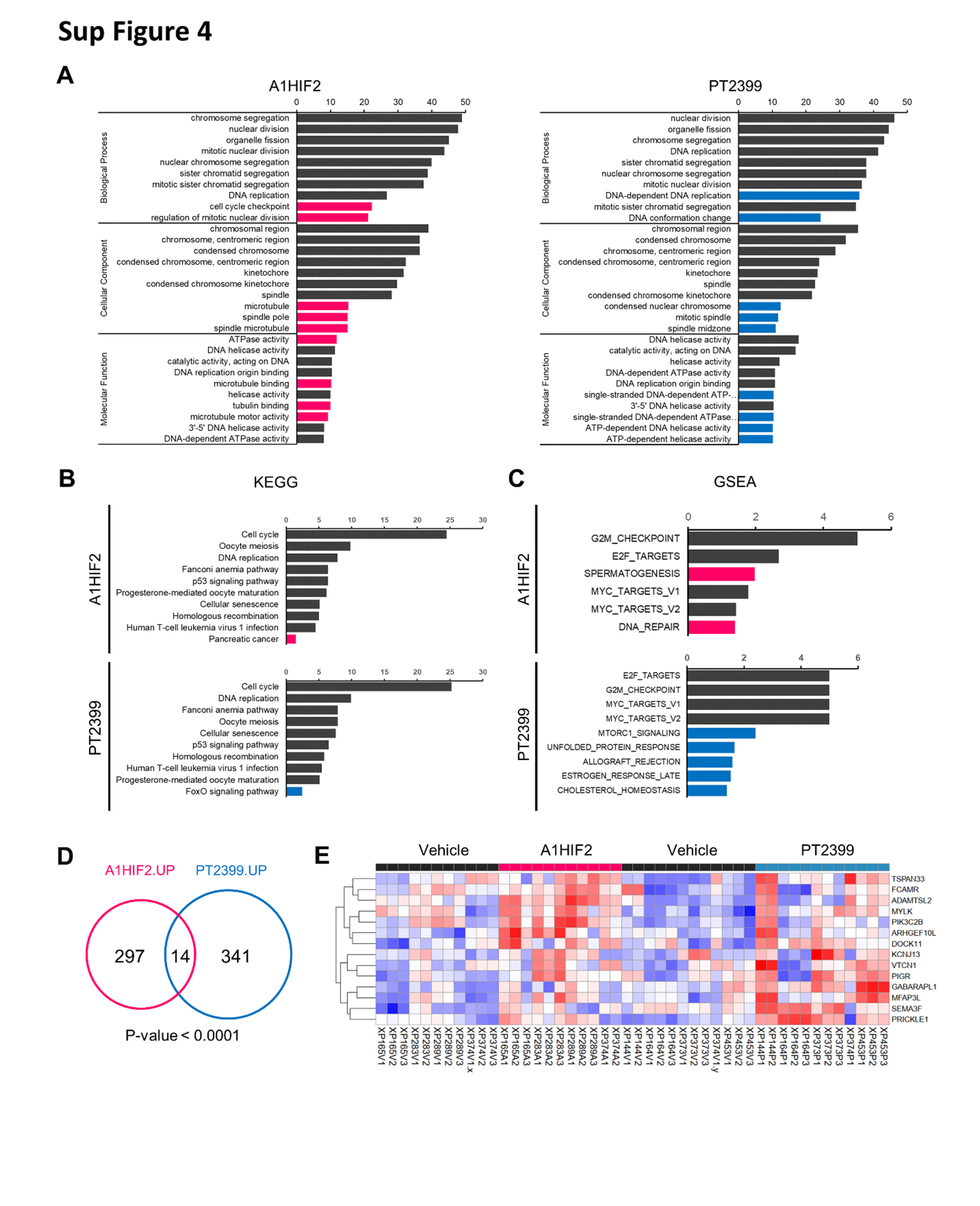{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Translational relevance.
In this report, we address the need for new therapeutic strategies in clear cell renal cell carcinoma (ccRCC), the most common type of kidney cancer. We evaluate an innovative, systemically delivered, tumor-directed HIF2α siRNA drug (siHIF2) targeting both wild-type and resistant mutant HIF2α and provide proof-of-principle of activity in mouse models and a patient. Further, using orthogonal RNA-seq studies of siHIF2 and a belzutifan-related inhibitor in TGs, we define the HIF2α transcriptome in ccRCC with unprecedented detail implicating not only traditional (i.e. Myc) but also novel pathways such as E2F2 and E2F8 as well as an 11-gene multi-cancer network (PBK, BIRC5, CCNB1, CDC20, CDK1, DLGAP5, MAD2L1, MELK, PLK1, TOP2A, and TTK) that was previously identified but is enigmatically controlled. These studies provide insight into HIF2α-mediated tumorigenesis and establish a paradigm for RNA-based therapies in cancer.
Acknowledgments
We acknowledge the patients who participated in this study and donated samples for research. This work was supported by funding from Arrowhead Pharmaceuticals as well as the UTSW SPORE grant (P50CA196516). YM, CS, VTT, AC, JM, DC, OO, TM, IP, PK and JB are supported by P50CA196516. FS is supported by DoD CDMRP Kidney Cancer Research Program (Award No. W81XWH2110631). Q. N. Do is supported by NICHD K25 award (K25HD104004). Imaging was facilitated by the Small Animal Imaging Research Program supported in part by U24CA126608 and Cancer Prevention and Research Institute of Texas (RP210099). We acknowledge the UTSW Tissue Resource, which is supported in part by the Harold C. Simmons Cancer Center through an NCI Cancer Center Support Grant, 1P30CA142543.
Footnotes
Competing Interests Statement
A. Christie reports grants from the NIH/NCI during the conduct of the study, as well as grants from CPRIT outside the submitted work. O. Fatunde reports other support from Arrowhead Pharmaceuticals during the conduct of the study. S.C. Wong reports other support from Arrowhead Pharmaceuticals, Inc during the conduct of the study, as well as other support from Arrowhead Pharmaceuticals, Inc outside the submitted work. I. Pedrosa reports personal fees from Merck outside the submitted work. P. Kapur reports personal fees from ClearNano Inc outside the submitted work. T. Schluep reports personal fees from Arrowhead Pharmaceuticals during the conduct of the study, as well as personal fees from Arrowhead Pharmaceuticals outside the submitted work. S.B. Kanner reports a patent for US20160348107 issued. J. Hamilton reports other support from Arrowhead Pharmaceuticals during the conduct of the study, as well as other support from Arrowhead Pharmaceuticals outside the submitted work. J. Brugarolas reports grants and personal fees from Arrowhead Pharmaceuticals during the conduct of the study, as well as personal fees from Eisai, Johnson and Johnson, Exelixis, and Calithera outside the submitted work; in addition, J. Brugarolas has patents for HIF2 biomarkers and mechanisms of resistance pending. No disclosures were reported by the other authors.
References
- 1.Shen C. and Kaelin WG Jr., The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol, 2013. 23(1): p. 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Latif F, et al. , Identification of the von Hippel-Lindau disease tumor suppressor gene. Science, 1993. 260(5112): p. 1317–20. [DOI] [PubMed] [Google Scholar]
- 3.Gnarra JR, et al. , Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet, 1994. 7(1): p. 85–90. [DOI] [PubMed] [Google Scholar]
- 4.Herman JG, et al. , Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A, 1994. 91(21): p. 9700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nickerson ML, et al. , Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res, 2008. 14(15): p. 4726–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaelin WG Jr. and Ratcliffe PJ, Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell, 2008. 30(4): p. 393–402. [DOI] [PubMed] [Google Scholar]
- 7.Kondo K, et al. , Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol, 2003. 1(3): p. E83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zimmer M, et al. , Inhibition of hypoxia-inducible factor is sufficient for growth suppression of VHL−/− tumors. Mol Cancer Res, 2004. 2(2): p. 89–95. [PubMed] [Google Scholar]
- 9.Shen C, et al. , Genetic and functional studies implicate HIF1alpha as a 14q kidney cancer suppressor gene. Cancer Discov, 2011. 1(3): p. 222–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keith B, Johnson RS, and Simon MC, HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer, 2011. 12(1): p. 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordan JD, et al. , HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell, 2008. 14(6): p. 435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Covello KL, et al. , HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev, 2006. 20(5): p. 557–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertout JA, et al. , HIF2alpha inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc Natl Acad Sci U S A, 2009. 106(34): p. 14391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts AM, et al. , Suppression of hypoxia-inducible factor 2alpha restores p53 activity via Hdm2 and reverses chemoresistance of renal carcinoma cells. Cancer Res, 2009. 69(23): p. 9056–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ema M, et al. , A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A, 1997. 94(9): p. 4273–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Compernolle V, et al. , Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med, 2002. 8(7): p. 702–10. [DOI] [PubMed] [Google Scholar]
- 17.Turner KJ, et al. , Expression of hypoxia-inducible factors in human renal cancer: relationship to angiogenesis and to the von Hippel-Lindau gene mutation. Cancer Res, 2002. 62(10): p. 2957–61. [PubMed] [Google Scholar]
- 18.Tian H, McKnight SL, and Russell DW, Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev, 1997. 11(1): p. 72–82. [DOI] [PubMed] [Google Scholar]
- 19.Motzer RJ, et al. , NCCN Guidelines Insights: Kidney Cancer, Version 1.2021. J Natl Compr Canc Netw, 2020. 18(9): p. 1160–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brugarolas J, Renal-cell carcinoma--molecular pathways and therapies. N Engl J Med, 2007. 356(2): p. 185–7. [DOI] [PubMed] [Google Scholar]
- 21.Koehler AN, A complex task? Direct modulation of transcription factors with small molecules. Curr Opin Chem Biol, 2010. 14(3): p. 331–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lazo JS and Sharlow ER, Drugging Undruggable Molecular Cancer Targets. Annu Rev Pharmacol Toxicol, 2016. 56: p. 23–40. [DOI] [PubMed] [Google Scholar]
- 23.Key J, et al. , Principles of ligand binding within a completely buried cavity in HIF2alpha PAS-B. J Am Chem Soc, 2009. 131(48): p. 17647–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rogers JL, et al. , Development of inhibitors of the PAS-B domain of the HIF-2alpha transcription factor. J Med Chem, 2013. 56(4): p. 1739–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheuermann TH, et al. , Artificial ligand binding within the HIF2alpha PAS-B domain of the HIF2 transcription factor. Proc Natl Acad Sci U S A, 2009. 106(2): p. 450–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen W, et al. , Targeting renal cell carcinoma with a HIF-2 antagonist. Nature, 2016. 539(7627): p. 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Courtney KD, et al. , HIF-2 Complex Dissociation, Target Inhibition, and Acquired Resistance with PT2385, a First-in-Class HIF-2 Inhibitor, in Patients with Clear Cell Renal Cell Carcinoma. Clin Cancer Res, 2020. 26(4): p. 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fisher R, Pusztai L, and Swanton C, Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer, 2013. 108(3): p. 479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Housman G, et al. , Drug resistance in cancer: an overview. Cancers (Basel), 2014. 6(3): p. 1769–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dagogo-Jack I. and Shaw AT, Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol, 2018. 15(2): p. 81–94. [DOI] [PubMed] [Google Scholar]
- 31.Crooke ST, et al. , Antisense technology: an overview and prospectus. Nat Rev Drug Discov, 2021. 20(6): p. 427–453. [DOI] [PubMed] [Google Scholar]
- 32.Paunovska K, Loughrey D, and Dahlman JE, Drug delivery systems for RNA therapeutics. Nat Rev Genet, 2022. 23(5): p. 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong SC, et al. , HIF2alpha-Targeted RNAi Therapeutic Inhibits Clear Cell Renal Cell Carcinoma. Mol Cancer Ther, 2018. 17(1): p. 140–149. [DOI] [PubMed] [Google Scholar]
- 34.Vogetseder A, et al. , αv-Integrin isoform expression in primary human tumors and brain metastases. International Journal of Cancer, 2013. 133(10): p. 2362–2371. [DOI] [PubMed] [Google Scholar]
- 35.Wechsel HW, et al. , Renal cell carcinoma: immunohistological investigation of expression of the integrin alpha v beta 3. Anticancer research, 1999. 19(2C): p. 1529–1532. [PubMed] [Google Scholar]
- 36.Brugarolas J, et al. , Initial results from the phase 1 study of ARO-HIF2 to silence HIF2-alpha in patients with advanced ccRCC (AROHIF21001). Journal of Clinical Oncology, 2022. 40(6_suppl): p. 339–339. [Google Scholar]
- 37.Elias R, et al. , A renal cell carcinoma tumorgraft platform to advance precision medicine. Cell Reports, 2021. 37(8): p. 110055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sivanand S, et al. , A validated tumorgraft model reveals activity of dovitinib against renal cell carcinoma. Sci Transl Med, 2012. 4(137): p. 137ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pavía-Jiménez A, Tcheuyap VT, and Brugarolas J, Establishing a human renal cell carcinoma tumorgraft platform for preclinical drug testing. Nat Protoc, 2014. 9(8): p. 1848–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen S, et al. , fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 2018. 34(17): p. i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bushnell B, BBMap: A Fast, Accurate, Splice-Aware Aligner, in Conference: 9th Annual Genomics of Energy & Environment Meeting, Walnut Creek, CA, March 17–20, 2014. 2014: United States. p. Medium: ED. [Google Scholar]
- 42.Anders S, Pyl PT, and Huber W, HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics, 2015. 31(2): p. 166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Love MI, Huber W, and Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 2014. 15(12): p. 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu G, et al. , clusterProfiler: an R package for comparing biological themes among gene clusters. Omics, 2012. 16(5): p. 284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elias R, et al. , A renal cell carcinoma tumorgraft platform to advance precision medicine. Cell Rep, 2021. 37(8): p. 110055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu CJ, et al. , Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol, 2003. 23(24): p. 9361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, et al. , Omics- and Pharmacogenomic Evidence for the Prognostic, Regulatory, and Immune-Related Roles of PBK in a Pan-Cancer Cohort. Front Mol Biosci, 2021. 8: p. 785370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gordan JD, et al. , HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell, 2007. 11(4): p. 335–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smythies JA, et al. , Inherent DNA-binding specificities of the HIF-1alpha and HIF-2alpha transcription factors in chromatin. EMBO Rep, 2019. 20(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmid V, et al. , Co-incidence of RCC-susceptibility polymorphisms with HIF cis-acting sequences supports a pathway tuning model of cancer. Sci Rep, 2019. 9(1): p. 18768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lauer V, et al. , Hypoxia drives glucose transporter 3 expression through hypoxia-inducible transcription factor (HIF)-mediated induction of the long noncoding RNA NICI. J Biol Chem, 2020. 295(13): p. 4065–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cho H, et al. , On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature, 2016. 539(7627): p. 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hong K, et al. , USP37 promotes deubiquitination of HIF2alpha in kidney cancer. Proc Natl Acad Sci U S A, 2020. 117(23): p. 13023–13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klee K, et al. , The Expression of Decidual Protein Induced by Progesterone (DEPP) is Controlled by Three Distal Consensus Hypoxia Responsive Element (HRE) in Hypoxic Retinal Epithelial Cells. Genes (Basel), 2020. 11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stepp MW, et al. , The c10orf10 gene product is a new link between oxidative stress and autophagy. Biochim Biophys Acta, 2014. 1843(6): p. 1076–88. [DOI] [PubMed] [Google Scholar]
- 56.Morais C, et al. , Functional significance of erythropoietin in renal cell carcinoma. BMC Cancer, 2013. 13: p. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Palapattu GS, Kristo B, and Rajfer J, Paraneoplastic syndromes in urologic malignancy: the many faces of renal cell carcinoma. Rev Urol, 2002. 4(4): p. 163–70. [PMC free article] [PubMed] [Google Scholar]
- 58.Wu D, et al. , Bidirectional modulation of HIF-2 activity through chemical ligands. Nat Chem Biol, 2019. 15(4): p. 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ray KK, et al. , Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N Engl J Med, 2017. 376(15): p. 1430–1440. [DOI] [PubMed] [Google Scholar]
- 60.Chen W, et al. , Targeting renal cell carcinoma with a HIF-2 antagonist. Nature, 2016. 539(7627): p. 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Courtney KD, et al. , Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2α Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J Clin Oncol, 2018. 36(9): p. 867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choueiri TK, et al. , Inhibition of hypoxia-inducible factor-2alpha in renal cell carcinoma with belzutifan: a phase 1 trial and biomarker analysis. Nat Med, 2021. 27(5): p. 802–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Courtney KD, et al. , Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2alpha Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J Clin Oncol, 2018. 36(9): p. 867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Choueiri TK, et al. , Phase I/II study of the oral HIF-2 α inhibitor MK-6482 in patients with advanced clear cell renal cell carcinoma (RCC). Journal of Clinical Oncology, 2020. 38(6_suppl): p. 611–611. [Google Scholar]
- 65.Jonasch E, et al. , Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N Engl J Med, 2021. 385(22): p. 2036–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haase VH, Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev, 2013. 27(1): p. 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng X, et al. , Marked and rapid effects of pharmacological HIF-2alpha antagonism on hypoxic ventilatory control. J Clin Invest, 2020. 130(5): p. 2237–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoefflin R, et al. , HIF-1alpha and HIF-2alpha differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat Commun, 2020. 11(1): p. 4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beier R, et al. , Induction of cyclin E-cdk2 kinase activity, E2F-dependent transcription and cell growth by Myc are genetically separable events. Embo j, 2000. 19(21): p. 5813–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Joyce NC, et al. , Effect of overexpressing the transcription factor E2F2 on cell cycle progression in rabbit corneal endothelial cells. Invest Ophthalmol Vis Sci, 2004. 45(5): p. 1340–8. [DOI] [PubMed] [Google Scholar]
- 71.Weijts BG, et al. , E2F7 and E2F8 promote angiogenesis through transcriptional activation of VEGFA in cooperation with HIF1. Embo j, 2012. 31(19): p. 3871–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bakker WJ, et al. , HIF proteins connect the RB-E2F factors to angiogenesis. Transcription, 2013. 4(2): p. 62–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu X, et al. , Genome-wide Screening Identifies SFMBT1 as an Oncogenic Driver in Cancer with VHL Loss. Mol Cell, 2020. 77(6): p. 1294–1306 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang J, et al. , VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science, 2018. 361(6399): p. 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu L, et al. , TBK1 Is a Synthetic Lethal Target in Cancer with VHL Loss. Cancer Discov, 2020. 10(3): p. 460–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nicholson HE, et al. , HIF-independent synthetic lethality between CDK4/6 inhibition and VHL loss across species. Sci Signal, 2019. 12(601). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Okazaki A, et al. , Glutaminase and poly(ADP-ribose) polymerase inhibitors suppress pyrimidine synthesis and VHL-deficient renal cancers. J Clin Invest, 2017. 127(5): p. 1631–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.