

Abstract
Plectin, encoded by PLEC, is a cytoskeletal linker and intermediate filament protein expressed in many cell types. Plectin consists of three main domains that determine its functionality: the N-terminal domain, the Rod domain, and the C-terminal domain. Molecular defects of PLEC correlating with the functional aspects lead to a group of rare heritable disorders, plectinopathies. These multisystem disorders include an autosomal dominant form of epidermolysis bullosa simplex (EBS-Ogna), limb girdle muscular dystrophy (LGMD), aplasia cutis congenita (ACC), and an autosomal recessive form of epidermolysis bullosa simplex (EBS), which may associate with muscular dystrophy (EBS–MD), pyloric atresia (EBS–PA), and/or congenital myasthenic syndrome (EBS-MyS). In this study, genotyping of over 600 Iranian patients with epidermolysis bullosa by next generation sequencing identified 15 patients with disease-causing PLEC variants. This mutation update analyzes the clinical spectrum of PLEC in our cohort and in the literature and demonstrates the relationship between PLEC genotype and phenotypic manifestations. This study has integrated our seven novel PLEC variants and phenotypic findings with previously published data (totaling 116 variants) to provide the most complete overview of pathogenic PLEC variants and related disorders.
Keywords: Plectinopathy, Epidermolysis Bullosa, Plectin, Variant Spectrum
1. BACKGROUND
Cytoskeletal proteins, made up of actin, intermediate filaments (IF), or tubulin, serve as foundations for structural support in many tissues. However, additional proteins are responsible for the assembly and linkage of these proteins, such as the plakin family of proteins, commonly known as cytolinkers (Bouameur et al., 2014; Sonnenberg & Liem, 2007). The plakin family harbors a large variety of versatile proteins integral to the structural integrity of multiple tissues; one of the most functionally diverse cytolinkers is plectin (Bouameur et al., 2014; Sonnenberg & Liem, 2007). The plectin gene (PLEC) is located on chromosome 8q24 and consists of 32 exons (Figure 1c) (Steinbock & Wiche, 1999). It encodes a >500 kDa protein that is widely distributed in many tissues, with the highest level of expression in the skin, muscle, and brain (Sonnenberg & Liem, 2007).
Figure 1.

Genetic Complexity and Protein Structure of Plectin. (A) Structure of plectin and its domains. (B) Rodless plectin isofrom. (C) Gene schematic of PLEC and alternate exon 1. Note that the myasthenia gravis phenotype is a result of disruption of post-synaptic intermediate filament network to acetylcholine receptor via rapsin-plectin 1f complex. (D) Cutaneous basement membrane and plectin located within the hemidesmosomal complex. This figure was generated in Biorender.
The molecular multidomain structure of PLEC provides clues to the functional activities of plectin. It consists of a central 200-nm rod domain, encoded by exon 31, flanked by large N-terminal and C-terminal globular domains, which are encoded by exons 1-30 and exon 32, respectively (Figure 1a). The N-terminal globular domain has a differentially spliced first exon followed by an actin-binding domain (ABD) consisting of two calponin homology domains and a plakin domain consisting of nine spectrin repeats and a SH3 domain (Figure 1a) (Sonnenberg & Liem, 2007). The N-terminal has binding sites for: (a) IFs such as vimentin, (b) microfilaments such as actin, (c) hemidesmosomal proteins such as COL17A1 and integrin α6β4, and (d) neuromuscular acetylcholine receptor clustering protein rapsyn (Koster et al., 2004; Mihailovska et al., 2014; Sonnenberg & Liem, 2007; Steinbock & Wiche, 1999)The C-terminal domain consists of six plectin repeat molecules with a linker region in between each segment (Figure 1a) (Bouameur et al., 2014; Sonnenberg & Liem, 2007). Within this region, there are binding sites for IFs (desmin, vimentin, and cytokeratins), glial fibrillary protein, and integrin α6β4 (Figure 1a) (Potokar & Jorgacevski, 2021; Sonnenberg & Liem, 2007).
In the skin, plectin is a member of the hemidesmosomal and focal adhesion complexes, with the highest binding affinity of plectin for integrin α6β4; consequently, in the skin plectin preferentially localizes to hemidesmosomes (Figure 1d) (Borradori & Sonnenberg, 1999). In the skeletal muscle, plectin is localized to the sarcolemma and neuromuscular junction where it acts as an anchor for desmin IFs, forming intermyofibrillar and subsarcolemmal scaffolds with sarcoplasmic reticulum, mitochondria, Z-disks, and costameres (Sonnenberg & Liem, 2007). In the brain, plectin is found to localize with the glial fibrillary and tau proteins, to aid in the structural integrity of astrocytes and neurons (Potokar & Jorgacevski, 2021). In the neuromuscular junction, it bridges acetylcholine receptors (AChRs) and IFs via scaffolding protein rapsyn (Mihailovska et al., 2014).
Plectin has been identified in multiple tissues due to its isoform diversity. Currently, 12 alternative first exons (1, 1a, 1b, 1c, 1d, 1e, 1f, 1g, 1h, 1i, 1j, 1k) have been identified to produce 12 isoforms, eight of which are found in humans, that alternatively splice directly into a common exon 2 (Figure 1c) (Castanon et al., 2013). Additionally, alternative splicing of exons 2 and 3 which encode parts of the ABD, adds to the complexity of the gene by creating additional alternative exons, 2α and 3α, inserted between exons 2 and 3 as well as 3 and 4. Exon 2α is expressed in the brain, heart, and skeletal muscles and 3α in the brain (Figure 1c). Some plectin isoforms are expressed in specific tissues, for example, sarcomeres express four plectin isoforms (P1, P1b, P1d, and P1f), the epidermis expresses four isoforms (P1, P1a, P1c, P1f), and the neuromuscular junction preferentially expresses P1f (Wiche & Winter, 2011). In addition to these isoforms, there are two commonly expressed isoforms, whose deficiencies have been related to distinct forms of plectinopathies, full-length and a rodless plectin isoform (Figure 1a, 1b). The rodless isoform is found in the skin and the muscle and is expressed when exon 31 is alternatively spliced out (Figure 1b) (Natsuga, Nishie, Akiyama, et al., 2010). Each isoform adds to the complex heterogeneity of plectin’s pathology.
Plectin’s predominant roles are highlighted by the spectrum of rare human disorders associated with mutations in the PLEC gene, referred to as “plectinopathies”. PLEC variants cause clinically recognizable phenotypes that range from mild forms of skin fragility localized to the skin in autosomal dominant EBS-Ogna to severe manifestations in autosomal recessive epidermolysis bullosa simplex (EBS) and ACC (Kariminejad et al., 2019). EBS can also be associated with extracutaneous manifestations leading to syndromic forms of plectinopathy associated with muscular dystrophy (EBS-MD), myasthenic syndrome (MyS), EBS with pyloric atresia (EBS-PA), and cardiomyopathy, while limb-girdle muscular dystrophy (LGMD) presents on its own (Vahidnezhad, Youssefian, Saeidian, & Uitto, 2019).
The locations and types of PLEC variants have been related to different forms of plectinopathies. However, there has not been a comprehensive overview of all PLEC variants and their relationship to phenotypic presentation. For this purpose, we collected all PLEC variants published in the literature up to March 2022, and we also report seven novel pathogenic PLEC variants that have not been described previously. In this study we performed a systematic analysis on the seven novel variants and the 109 distinct PLEC variants, totaling 116 reported to cause with cause the heritable disorders.
2. VARIANTS IN PLEC
All PLEC variants were documented based on current Human Genome Variation Society mutation nomenclature guidelines based on GenBank accession numbers NM_201384.2, NM_201378.2, and NM_00445.4 and novel variants were submitted to the Leiden Open Variation Database (LOVD). In total, we collected 109 unique PLEC variants in 116 patients that have been reported in the Human Gene Mutation Database and in 67 peer-reviewed articles up to March 2022 (Table 1) (Figure 2a). We identified 15 patients with PLEC variants in over 600 Iranian EB patients and found five novel homozygous variants and two novel compound heterozygous variants in seven patients that, to our knowledge, have not been reported, collectively making 116 unique PLEC variants in 131 patients (Table 1 and Table 2) (Figure 2a). The pathogenicity of all variants was analyzed via ACMG classification and Sherloc and the impact of intronic variants were analyzed with Human Splicing Finder (HSF) System by Genomnis prediction software (https://hsf.genomnis.com). HSF software is able to identify splicing signals, predict the impact of mutations on these signals and provide a pathogenicity prediction for any intronic or exonic mutation that potentially affects splicing. For technical details of whole exome sequencing, RNA sequencing, homozygosity mapping, targeted 21 EB associated gene panel and interpretation of genomic sequence variants in EB see these references (Uitto et al., 2021; Vahidnezhad, Youssefian, Saeidian, Touati, et al., 2019; Vahidnezhad et al., 2017; Vahidnezhad, Youssefian, Saeidian, Zeinali, et al., 2019; Youssefian et al., 2021). The combined annotation-dependent depletion score (CADD) of novel variants in our cohort were analyzed with the mutation significance cutoff specific for PLEC (Figure 2b).
Table 1.
Comprehensive list of published PLEC cases
| Reference | # of Affected | cDNA Variant(s) | Protein Sequence | Variant Type | Impact Prediction | Exon/Intron | ACMG Classification | Sherloc | Genotype | Diagnosis |
|---|---|---|---|---|---|---|---|---|---|---|
| (Gostynska et al., 2017) | 1 | c.906 + 19_40del | p.Val303_Pro313ins11* | Indel | - | Intron 8 | Likely Pathogenic | 5P | Homozygous | EBS-MD, Cardiomyopathy |
| (Bauer et al., 2001) | 1 | c.873_875dup | p.Leu292dup | Indel | - | Exon 9 | VUS | 5P | compound heterozygous | EBS |
| c.4141C>T | p.Gln1381* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | ||||
| (Tu et al., 2020) | 1 | c.875T>C | p.Leu292Pro | Missense | - | Exon 9 | VUS | 4LP | compound heterozygous | EBS |
| c.2726G>A | p.Trp909* | Nonsense | - | Exon 22 | Likely Pathogenic | 5P | ||||
| (Tu et al., 2020) | 1 | c.875T>C | p.Leu292Pro | Missense | - | Exon 9 | VUS | 4LP | compound heterozygous | EBS |
| c.6874C>T | p.Arg2292* | Nonsense | - | Exon 31 | Pathogenic | 5P | ||||
| (Bolling et al., 2010) | 1 | c.887G>A | p.Arg296Gln | Missense | - | Exon 9 | Benign | 1B | compound heterozygous | EBS-MD, Cardiomyopathy |
| c.4759G>T | p.Glu1587* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | ||||
| (Uitto, 2004) | c.1449_1450ins36 | p.Ala483_Ile484ins | Indel | - | Exon 14 | VUS | 4LP | compound heterozygous | EBS-MD | |
| 1 | c.2596_2604del | p.Gln866_Ala868del | Indel | - | Exon 21 | Likely Pathogenic | 5P | |||
| (Khan et al., 2021) | 1 | c.1414C>G | p.Arg472Gly | Missense | - | Exon 14 | VUS | 4LP | Homozygous | EBS |
| (Alvarez et al., 2016) | 1 | c.2183_2185del | p.Phe728del | Indel | - | Exon 19 | VUS | 5P | Homozygous | EBS-MD |
| (Winter et al., 2016) | 1 | c.2183_2185del | p.Phe728del | Indel | - | Exon 19 | VUS | 5P | compound heterozygous | EBS-MD, Dilated Cardiomyopathy |
| c.3038_3039del | p.Lys1013Argfs*139 | Frameshift | - | Exon 24 | Likely Pathogenic | 5P | ||||
| (Natsuga et al., 2017) | 1 | c.2183_2185del | p.Phe728del | Indel | - | Exon 19 | VUS | 5P | compound heterozygous | EBS |
| c.9113dupT | p.Ser3039Glufs*55 | Frameshift | - | Exon 32 | Likely Pathogenic | 5P | ||||
| (Pulkkinen et al., 1996; Shimizu et al., 1999) | 1 | c.2596_2604del | p.Gln866_Ala868del | Indel | - | Exon 21 | Likely Pathogenic | 5P | Homozygous | EBS-MD |
| (Yiu et al., 2011) | 1 | c.2596_2604del | p.Gln866_Ala868del | Indel | - | Exon 21 | Likely Pathogenic | 5P | compound heterozygous | EBS-MD |
| c.4849C>T | p.Gln1617* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | ||||
| (Rouan et al., 2000) | 1 | c.2613-9_2624del | p.Leu872_Gln875del | Indel | - | Intron 22/Exon 23 | Likely Pathogenic | 5P | compound heterozygous | EBS-MD |
| c.4951del | p.Val1651Trpfs*65 | Frameshift | - | Exon 31 | Likely Pathogenic | 5P | ||||
| (Shimizu et al., 1999; Takizawa et al., 1999) | 1 | c.3076C>T | p.Gln1026* | Nonsense | - | Exon 24 | Likely Pathogenic | 5P | compound heterozygous | EBS-MD |
| c.5725C>T | p.Gln1909* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | ||||
| (Charlesworth et al., 2013) | 1 | c.3260+1G>T | N/A | Splicing | Broken WT Donor Site; Alteration of the WT Donor site most probably affecting splicing. | Intron 25 | Likely Pathogenic | 4P | compound heterozygous | EBS |
| c.6874C>T | p.Arg2292* | Nonsense | - | Exon 31 | Pathogenic | 5P | ||||
| (Charlesworth et al., 2013) | 1 | c.4045-4A>G | N/A | Splicing | No significant impact on splicing signals. | Intron 30 | Benign | 1B | compound heterozygous | EBS-MD |
| c.7723C>T | p.Gln2575* | Nonsense | - | Exon 32 | Likely Pathogenic | 5P | ||||
| (Charlesworth et al., 2013) | 1 | c.4135C>T | p.His1379Tyr | Nonsense | - | Exon 31 | VUS | 4LP | Homozygous | EBS-MD |
| (Dang et al., 1998) | 1 | c.4213_4225dup | p.Val1409Glyfs*40 | Frameshift | - | Exon 31 | Likely Pathogenic | 5P | compound heterozygous | EBS-MD |
| c.4284delC | p.Ser1429Argfs*93 | Frameshift | - | Exon 31 | Likely Pathogenic | 5P | ||||
| (Natsuga, Nishie, Akiyama, et al., 2010; Sawamura et al., 2007) | 1 | c.4267C>T | p.Gln1423* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS-MD |
| (Walker et al., 2017) | 1 | c.4468C>T | p.Arg1490* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS |
| (Natsuga, Nishie, Akiyama, et al., 2010) | 1 | c.4562_4586dup | p.Lys1531Glyfs*89 | Frameshift | - | Exon 31 | Likely Pathogenic | 5P | compound heterozygous | EBS |
| c.7039C>T | p.Gln2347* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | ||||
| (Pfendner et al., 2005) | 1 | c.4759G>T | p.Glu1587* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS-MD |
| (Winter et al., 2016) | 1 | c.4937_4955del | p.Leu1646Argfs*64 | Frameshift | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS-MD |
| (Mellerio et al., 1997) | 1 | c.4937_4955del | p.Leu1646Argfs*64 | Frameshift | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS |
| (McLean et al., 1996) | 1 | c.5024_5031del | p.Arg1675Glnfs*14 | Frameshift | - | Exon 31 | Pathogenic | 5P | Homozygous | EBS-MD |
| (Kunz et al., 2000) | 1 | c.5056C>T | p.Gln1686* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | compound heterozygous | EBS |
| c.6970C>T | p.Arg2324* | Nonsense | - | Exon 31 | Pathogenic | 5P | ||||
| (Yin et al., 2015) | 1 | c.4843C>T | p.Gln1615* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | compound heterozygous | EBS-MD |
| c.6874C>T | p.Arg2292* | Nonsense | - | Exon 31 | Pathogenic | 5P | ||||
| (Schara et al., 2004) | 1 | c.5176dup | p.Glu1726Glyfs*17 | Frameshift | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS-MD |
| (Koss-Harnes et al., 2004) | 2 | c.5329G>T | p.Glu1777* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS-MD |
| (Chavanas et al., 1996) | 1 | c.5647C>T | p.Gln1883* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS-MD |
| (Gache et al., 1996) | 2 | c.5647C>T | p.Gln1883* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS-MD |
| (Charlesworth et al., 2013) | 1 | c.5689C>T | p.Gln1897* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | NR | EBS-MD |
| (Pulkkinen et al., 1996) | 1 | c.5734delC | p.Leu1912Trpfs*6 | Frameshift | - | Exon 31 | Pathogenic | 5P | Homozygous | EBS-MD |
| (Shimizu et al., 1999) | 1 | c.5734delC | p.Leu1912Trpfs*6 | Frameshift | - | Exon 31 | Pathogenic | 5P | Homozygous | EBS-MD |
| (McMillan et al., 2007) | 1 | c.5734delC | p.Leu1912Trpfs*6 | Frameshift | - | Exon 31 | Pathogenic | 5P | Homozygous | EBS-MD |
| (Smith et al., 1996) | 1 | c.5768_5775dup | p.Glu1926Trpfs*8 | Frameshift | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS-MD |
| (McMillan et al., 2007; Mellerio et al., 1997) | 1 | c.5774_5775del | p.Glu1925Glyfs*60 | Frameshift | - | Exon 31 | Pathogenic | 5P | Homozygous | EBS |
| (Kyrova et al., 2016) | 1 | c.5821_5822del | p.Lys1941Glyfs*44 | Frameshift | - | Exon 31 | Likely Pathogenic | 5P | compound heterozygous | EBS-MD |
| c.9028_9044del | p.Val3010Cysfs*78 | Frameshift | - | Exon 32 | Likely Pathogenic | 5P | ||||
| (Rouan et al., 2000) | 1 | c.5932G>T | p.Glu1978* | Nonsense | - | Exon 31 | Pathogenic | 5P | compound heterozygous | EBS-MD |
| c.13297A>T | p.Lys4433* | Nonsense | - | Exon 32 | Likely Pathogenic | 5P | ||||
| (Chen et al., 2013) | 1 | c.6211C>T | p.Gln2071* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | compound heterozygous | EBS-MD |
| c.7708C>T | p.Gln2570* | Nonsense | - | Exon 32 | Likely Pathogenic | 5P | ||||
| (Natsuga, Nishie, Akiyama, et al., 2010) | 1 | c.6468_6501del | p.Leu2157Argfs*21 | Frameshift | - | Exon 31 | Likely Pathogenic | 5P | compound heterozygous | EBS-MD |
| c.12959dupG | p.Ile4321Hisfs*8 | Frameshift | - | Exon 32 | Likely Pathogenic | 5P | ||||
| (Charlesworth et al., 2013) | 1 | c.6601C>T | p.Gln2201* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | compound heterozygous | EBS-MD |
| c.10375C>T | p.Gln3459* | Nonsense | - | Exon 32 | Likely Pathogenic | 5P | ||||
| (Takahashi et al., 2005) | 1 | c.6874C>T | p.Arg2292* | Nonsense | - | Exon 31 | Pathogenic | 5P | Homozygous | EBS-MD |
| (Villa et al., 2015) | 1 | c.6970C>T | p.Arg2324* | Nonsense | - | Exon 31 | Pathogenic | 5P | Homozygous | EBS-MD, cardiomyopathy |
| (Argyropoulou et al., 2018) | 1 | c.7078G>T | p.Glu2360* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS-MD |
| (Shimizu et al., 1999; Takizawa et al., 1999) | 1 | c.7180C>T | p.Arg2394* | Nonsense | - | Exon 31 | Pathogenic | 5P | compound heterozygous | EBS-MD |
| c.12497_12500dup | p.Tyr4168Aspfs*41 | Frameshift | - | Exon 32 | Likely Pathogenic | 5P | ||||
| (Pfendner et al., 2005) | 1 | c.7180C>T | p.Arg2394* | Nonsense | - | Exon 31 | Pathogenic | 5P | Homozygous | EBS-MD |
| (Pfendner et al., 2005) | 1 | c.7312C>T | p.Arg2438* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS-MD |
| (McMillan et al., 2007; Smith et al., 1996) | 1 | c.7312C>T | p.Arg2438* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS-MD |
| (McMillan et al., 2007; Smith et al., 1996) | 5 | c.7312C>T | p.Arg2438* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS-MD |
| (Maccari et al., 2019) | 1 | c.7387C>T | p.Gln2463* | Nonsense | - | Exon 31 | Pathogenic | 5P | Homozygous | EBS |
| (Ahmad et al., 2018) | 1 | c.10498C>T | p.Arg3500Cys | Missense | - | Exon 32 | Likely Benign | 3VUS | Homozygous | EBS-MD |
| (Schroder et al., 2002; Winter et al., 2016) | 1 | c.13378_13393dup | p.Glu4465Glyfs*48 | Frameshift | - | Exon 32 | VUS | 5P | Homozygous | EBS-MD, Dilated Cardiomyopathy |
| (Forrest et al., 2010) | 1 | IVS11+2T>G | N/A | Splicing | - | Intron 11 | Likely Pathogenic | 4P | compound heterozygous | EBS-MD-Mys |
| c.10106_10109del | p.Val3369Alafs*11 | Frameshift | - | Exon 32 | Likely Pathogenic | 5P | ||||
| (Maselli et al., 2011) | 1 | c.1419_1420ins36 | p.Arg473_Val474ins12 | Indel | - | Exon 14 | VUS | 5P | Homozygous | EBS-Mys |
| (Argente-Escrig et al., 2021) | 1 | c.2458-2A>G | N/A | Splicing | Broken WT Acceptor Site; Alteration of the WT Acceptor site most probably affecting splicing. | Intron 21 | Likely Pathogenic | 4P | compound heterozygous | EBS-MD-Mys |
| c.11656delC | p.Arg3886Valfs*30 | Frameshift | - | Exon 32 | Likely Pathogenic | 5P | ||||
| (Gonzalez Garcia et al., 2019) | 3 | c.3005G>A | p.Arg1002His | Missense | - | Exon 24 | Likely Pathogenic | 4LP | compound heterozygous | EBS-MD-Mys |
| c.9598_9685del | p.Asp3202Valfs*21 | Frameshift | - | Exon 32 | VUS | 5P | ||||
| (Banwell et al., 1999; Selcen et al., 2011) | 1 | c.6088C>T | p.Gln2030* | Nonsense | - | Exon 31 | Pathogenic | 5P | compound heterozygous | EBS-MD-Mys |
| c.11962dupG | p.Glu3988Glyfs*69 | Frameshift | - | Exon 32 | Likely Pathogenic | 5P | ||||
| (Selcen et al., 2011) | 1 | c.6874C>T | p.Arg2292* | Nonsense | - | Exon 31 | Pathogenic | 5P | compound heterozygous | EBS-MD-Mys |
| c.11962dupG | p.Glu3988Glyfs*69 | Frameshift | - | Exon 32 | Likely Pathogenic | 5P | ||||
| (Pfendner & Uitto, 2005) | 1 | c.832C>T | p.Gln278* | Nonsense | - | Exon 9 | Pathogenic | 5P | Homozygous | EBS-PA, ACC |
| (Nakamura et al., 2005) | 1 | c.832C>T | p.Gln278* | Nonsense | - | Exon 9 | Pathogenic | 5P | compound heterozygous | EBS-PA, ACC |
| c.1263G>A | p.Ser421Ser | Synonymous/Splicing | Broken WT Donor Site; Alteration of the WT Donor site most probably affecting splicing. | Exon 12 | VUS | 4LP | ||||
| (Pfendner & Uitto, 2005) | 1 | c.1482_1485del | p.Gly495Trpfs*11 | Frameshift | - | Exon 14 | Likely Pathogenic | 5P | Homozygous | EBS-PA |
| (Charlesworth et al., 2003) | 1 | c.2599_2612del | p.Glu867Alafs*84 | Frameshift | - | Exon 21 | Pathogenic | 5P | Homozygous | EBS-PA, ACC |
| (Pfendner & Uitto, 2005) | 1 | c.2688_2707del | p.Trp896Cysfs*53 | Frameshift | - | Exon 22 | Likely Pathogenic | 5P | Homozygous | EBS-PA, ACC |
| (Walker et al., 2017) | 1 | c.2807dupT | p.Leu937Profs*19 | Frameshift | - | Exon 24 | Likely Pathogenic | 5P | compound heterozygous | EBS-PA |
| c.7018C>T | p.Gln2340* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | ||||
| (Charlesworth et al., 2013) | 1 | c.3261-2A>G | N/A | Splicing | Broken WT Acceptor Site; Alteration of the WT Acceptor site most probably affecting splicing. | Intron 26 | Likely Pathogenic | 4P | compound heterozygous | EBS-PA, ACC |
| c.3821_3822del | p.Gln1274Leufs*9 | Frameshift | - | Exon 28 | Likely Pathogenic | 5P | ||||
| (Nakamura et al., 2005) | 1 | c.3484C>T | p.Arg1162* | Nonsense | - | Exon 27 | Likely Pathogenic | 5P | Homozygous | EBS-PA, ACC |
| c.7531C>T | p.Gln2511* | Nonsense | - | Exon 27 | Pathogenic | 5P | Heterozygous | |||
| (Charlesworth et al., 2013) | 1 | c.4038_4039del | p.Glu1347Glyfs*5 | Frameshift | - | Exon 30 | Likely Pathogenic | 5P | compound heterozygous | EBS-PA |
| c.12418C>T | p.Arg4140* | Nonsense | - | Exon 32 | Pathogenic | 5P | ||||
| (Sawamura et al., 2007) | 1 | c.7315C>T | p.Gln2439* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | compound heterozygous | EBS-PA |
| c.7552C>T | p.Gln2518* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | ||||
| (Pfendner & Uitto, 2005) | 1 | c.9004C>T | p.Arg3002* | Nonsense | - | Exon 32 | Likely Pathogenic | 5P | Homozygous | EBS-PA |
| (Valari et al., 2019) | 1 | c.11831delA | p.Lys3944Argfs*10 | Frameshift | - | Exon 32 | Likely Pathogenic | 5P | compound heterozygous | EBS-PA-MD |
| c.12418C>T | p.Arg4140* | Nonsense | - | Exon 32 | Pathogenic | 5P | ||||
| (Natsuga, Nishie, Shinkuma, et al., 2010) | 1 | c.10903C>T | p.Gln3635* | Nonsense | - | Exon 32 | Likely Pathogenic | 5P | compound heterozygous | EBS-PA-MD |
| c.11372_11381del | p.Ile3791Argfs*90 | Frameshift | - | Exon 32 | Likely Pathogenic | 5P | ||||
| (Gundesli et al., 2010) | 3 | c.1_9del | N/A | Indel | Alteration of auxiliary sequences; Significant alteration of ESE / ESS motifs ratio (−2). New Donor splice site; Activation of a cryptic Donor site. Potential alteration of splicing | Exon 1 | VUS | 5P | Homozygous | LGMD |
| (Mroczek et al., 2020) | 4 | c.1_9del | N/A | Indel | Alteration of auxiliary sequences; Significant alteration of ESE / ESS motifs ratio (−2). New Donor splice site; Activation of a cryptic Donor site. Potential alteration of splicing | Exon 1 | VUS | 5P | Homozygous | LGMD-Mys |
| (Deev et al., 2017) | 1 | c.58G>T | p.Glu20* | Nonsense | - | Exon 1 | Likely Pathogenic | 5P | Homozygous | LGMD |
| (Fattahi et al., 2015) | 2 | c.2983C>T | p.Gln995* | Nonsense | - | Exon 24 | Likely Pathogenic | 5P | compound heterozygous | LGMD-Mys |
| c.11422G>A | p.Gly3808Ser | Missense | - | Exon 32 | VUS | 4LP | ||||
| (Zhong et al., 2017) | 1 | c.6037C>T | p.Arg2013Trp | Missense | - | Exon 31 | Likely Benign | 2B | compound heterozygous | LGMD |
| c.9982T>A | p.Phe3328Ile | Missense | - | Exon 32 | VUS | 3VUS | ||||
| (Vahidnezhad et al., 2017) | 1 | c.6874C>T | p.Arg2292* | Nonsense | - | Exon 31 | Pathogenic | 5P | compound heterozygous | EBS-MD |
| c.12611A>G | p.Asp4204Gly | Missense | - | Exon 32 | VUS | 3VUS | ||||
| (Walter et al., 2021) | 1 | c.8225C>G | p.Pro2742Arg | Missense | - | Exon 32 | VUS | 4LP | Homozygous | EBS-MD |
| c.7425+5C>G | N/A | Splicing | No significant impact on splicing signals. | Intron 31 | VUS | 3VUS | Homozygous | |||
| (Yu et al., 2021) | 1 | c.13C>T | p.Gln5* | Nonsense | - | Exon 1 | Likely Pathogenic | 4P | NR | EBS |
| c.1675C>T | p.Arg559* | Nonsense | - | Exon 14 | Likely Pathogenic | 5P | ||||
| (Koss-Harnes et al., 2002) | 9 | c.5917C>T | p.Arg1973Trp | Missense | - | Exon 31 | Likely Pathogenic | 4P | heterozygous | EBS-Ogna |
| (Kiritsi et al., 2013) | 4 | c.5917C>T | p.Arg1973Trp | Missense | - | Exon 31 | Likely Pathogenic | 4P | heterozygous | EBS-Ogna |
| (Bolling et al., 2014) | 2 | c.5917C>T | p.Arg1973Trp | Missense | - | Exon 31 | Likely Pathogenic | 4P | heterozygous | EBS-Ogna |
| (Bolling et al., 2014) | 1 | c.8587A>T | p.Thr2863Ser | Missense | - | Exon 32 | VUS | 4LP | heterozygous | EBS-Ogna |
| (Bolling et al., 2014) | 1 | c.10498C>T | p.Arg3500Cys | Missense | - | Exon 32 | Likely Benign | 3VUS | heterozygous | EBS-Ogna |
| (Mariath et al., 2019) | 1 | c.6616dupG | p.Asp2206Glyfs*45 | Frameshift | - | Exon 31 | Likely Pathogenic | 5P | NR | EBS |
| (Dai et al., 2015) | 1 | c.12746C>T | p.Ser4249Leu | Missense | - | Exon 32 | VUS | 3VUS | NR | MD |
| c.3176C>T | p.Ala1059Val | Missense | - | Exon 25 | VUS | 3VUS | ||||
| (Dai et al., 2015) | 1 | c.10660delA | p.Thr3554Profs*37 | Frameshift | - | Exon 32 | Likely Pathogenic | 5P | NR | MD |
| c.1738–3C>G | N/A | Splicing | New Donor splice site; Activation of a cryptic Donor site. Potential alteration of splicing. | Intron 14 | VUS | 4LP | ||||
| (Gostynska et al., 2015) | 1 | c.46C>T | p.Arg16* | Nonsense | - | Exon 1 | Pathogenic | 5P | Homozygous | EBS |
| (Martinez-Santamaria et al., 2022) | 1 | c.4180C>T | p.Gln1394* | Nonsense | - | Exon 31 | Likely Pathogenic | 5P | Homozygous | EBS-MD |
| (Kariminejad et al., 2019) | 1 | c.647_656delTGGAGAACCT | p.Leu216Argfs*14 | Frameshift | - | Exon 7 | Pathogenic | 5P | Homozygous | ACC |
Abbreviations: EBS, epidermolysis bullosa simplex; PLEC, Plectin; EBS–PA, epidermolysis bullosa simplex-pyloric atresia; EBS–MD, epidermolysis bullosa simplex-muscular dystrophy; LGMD, limb girdle muscular dystrophy; EBS-MyS, epidermolysis bullosa simplex-myasthenic syndrome; ACC, aplasia cutis congenita; P, Pathogenic; LP, Likely Pathogenic; VUS, variant of uncertain significance; y, year; d, day; m, male; f, female
PLEC variants were documented based on GenBank accession numbers NM_201384.2, NM_201378.2, and NM_00445.4.
Figure 2.

Distribution and assessment of pathogenicity of PLEC Variants. (a) Schematic representation of all 116 variants in PLEC. (b) The CADD scores of the unpublished variants. The CADD scores of variants are mapped on the y-axis in relation to the mutation significance cut-off (MSC) of 19.28 which is specific for PLEC. The X-axis depicts the allele frequency of the PLEC variants in the population database including aggregated gnomAD database. The CADD scores of novel variants: p.Thr3571Met (25.3), p.Trp531* (37), p.Ala2121Glnfs*68 (34), p.Gln414Arg (12.5), p.Arg1676Cys (23.8), p.Gln2425* (37), and c.1737+1G>A (23.7). This figure was generated in Biorender.
Table 2:
Unpublished and published variants in current cohort of plectinopathy patients
| Reference | Patient number, Ethnicity (age, gender) | Consanguinity | cDNA Variant | Protein Sequence | Variant Type | Exon/Intron | Impact Prediction | Prediction | gnomAD (Ho, He) | ACMG Classification | Sherloc | Genotype | Diagnosis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (Vahidnezhad et al., 2017) | P1, Shahin dej/west az (14y, M) | First Cousin | c.4504C>T | p.Gln1502* | Nonsense | Exon 31 | - | - | 0, 1 | P | 5P | Homozygous | EBS-MD-MyS |
| (McMillan et al., 2007; Pfendner et al., 2005; Vahidnezhad, Youssefian, Saeidian, Zeinali, et al., 2019) | P2, Marvdasht/shiraz (12y, M) | First Cousin | c.7312C>T | p.Arg2438* | Nonsense | Exon 31 | - | - | 0, 4 | P | 5P | Homozygous | EBS-MD-MyS |
| (Pfendner et al., 2005; Selcen et al., 2011; Vahidnezhad et al., 2017) | P3 Babol (6y, M) | None | c.6874C>T | p.Arg2292* | Nonsense | Exon 31 | - | - | 0, 5 | P | 5P | Compound Heterozygous | EBS-MD-MyS |
| (Vahidnezhad et al., 2017) | c.12611A>G | p.Asp4204Gly | Missense | Exon 32 | - | Damaging | 0, 1 | LP | 4LP | ||||
| (Youssefian et al., 2021) | P4, Tehran (22y, M) | None (Patients are siblings) |
c.5452C>T | p.Gln1818* | Nonsense | Exon 31 | - | - | 0, 0 | P | 5P | Homozygous | EBS-MD-MyS |
| (Youssefian et al., 2021) | P5, Tehran (30y, M) | c.5452C>T | p.Gln1818* | Nonsense | Exon 31 | - | - | 0, 0 | P | 5P | Homozygous | EBS-MD-MyS | |
| Unpublished | P6, Semnan/Damghan (15y, F) | First Cousin | c.10550C>T | p.Thr3571Met | Missense | Exon 32 | - | Damaging | 0, 11 | LP | 4LP | Homozygous | EBS-MD-MyS |
| (Takizawa et al., 1999) | P7, Mazandaran (5y, F) | First Cousin | c.5725C>T | p.Gln1909* | Nonsense | Exon 31 | - | - | 0, 0 | LP | 5P | Homozygous | EBS |
| Unpublished | P8, Tehran (45D, M) | First Cousin | c.1593G>A | p.Trp531* | Nonsense | Exon 14 | - | - | 0, 0 | P | 5P | Homozygous | EBS-PA |
| Unpublished | P9, Shiraz, (9y, F) | First Cousin | c.6361del | p.Ala2121Glnfs*68 | Frameshift | Exon 31 | - | - | 0, 0 | P | 5P | Homozygous | EBS-MD-MyS |
| (McMillan et al., 2007; Pfendner et al., 2005; Vahidnezhad, Youssefian, Saeidian, Zeinali, et al., 2019) | P10, Arak (7y, M) | First Cousin | c.7312C>T | p.Arg2438* | Nonsense | Exon 31 | - | - | 0, 4 | P | 5P | Homozygous | EBS |
| This study | P11, Tehran (17.5y, F) | First Cousin once-removed | c.1241A>G | p.Gln414Arg | Missense | Exon 12 | - | Damaging | 0, 0 | LP | 4P | Compound Heterozygous | EBS |
| This study | c.5026C>T | p.Arg1676Cys | Missense | Exon 31 | - | Damaging | 0, 70 | LP | 4LP | ||||
| This study | P12, Tehran (5.5y, M) | Second Cousin | c.7273C>T | p.Gln2425* | Nonsense | Exon 31 | - | - | 0, 0 | P | 5P | Homozygous | EBS |
| (Youssefian et al., 2021) | P13, Lorestan (8y, F) | First Cousin | c.5452C>T | p.Gln1818* | Nonsense | Exon 32 | - | - | 0, 0 | P | 5P | Homozygous | EBS-MyS |
| This study | P14, Urumia (Died at 9y, M) | First Cousin (Patients are twins) |
c.1737+1G>A | N/A | Splicing | Intron 14 | Broken WT Donor Site; Alteration of the WT Donor site most probably affecting splicing. | - | 0, 0 | LP | 4P | Homozygous | EBS, PKU |
| This study | P15, Urumia (Died at 9y, M) | c.1737+1G>A | N/A | Splicing | Intron 14 | Broken WT Donor Site; Alteration of the WT Donor site most probably affecting splicing. | - | 0, 0 | LP | 4P | Homozygous | EBS, PKU |
Abbreviations: EBS, epidermolysis bullosa simplex; PLEC, Plectin; EBS–PA, epidermolysis bullosa simplex-pyloric atresia; EBS–MD, epidermolysis bullosa simplex-muscular dystrophy; LGMD, limb girdle muscular dystrophy; EBS-MyS, epidermolysis bullosa simplex-myasthenic syndrome; PKU, phenylketonuria; Ho, homozygous; He, heterozygous; P, Pathogenic; LP, Likely Pathogenic; VUS, variant of uncertain significance; y, year; d, day; m, male; f, female
PLEC variants were documented based on GenBank accession numbers NM_201384.2.
Among the 116 total PLEC variants, the majority are loss-of-function (85%) with one synonymous variant at the exon-intron border (1%), eight splice site variants (7%), 8 insertions/deletions that were protein-truncating (7%), 34 frameshift (29%), and 48 nonsense (41%) variants (Figure 4a). Additionally, there were 17 missense variants (15%) (Figure 4a) (Table 1, Table 2).
Figure 4.
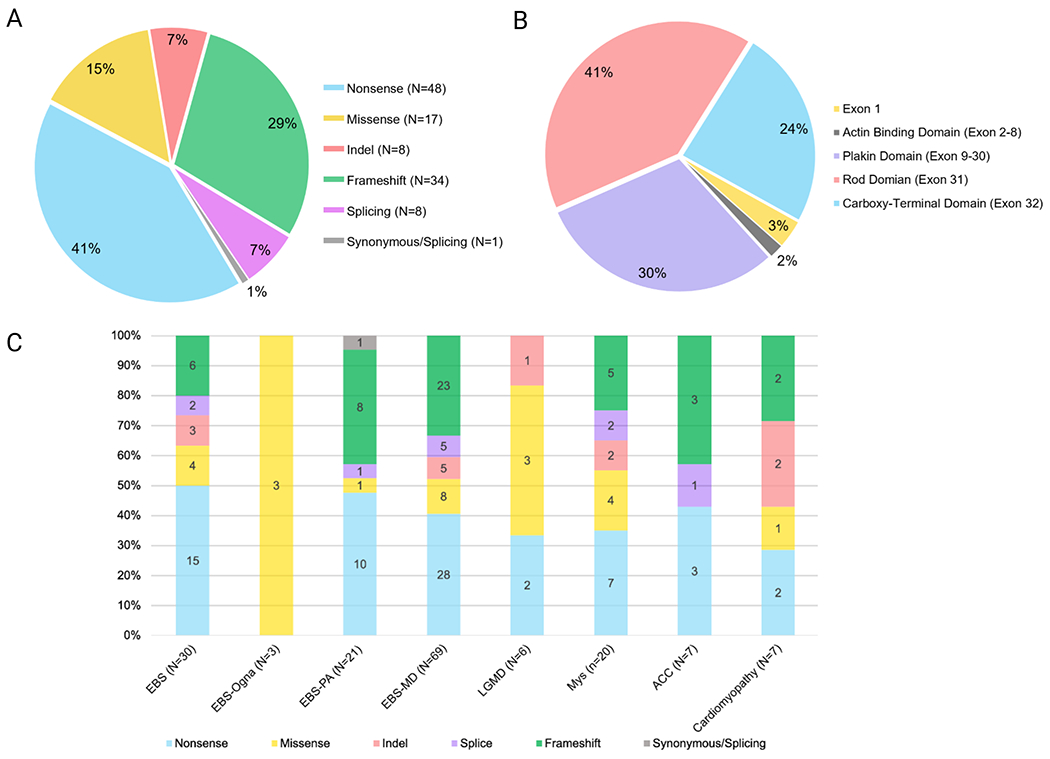
Prevalence of PLEC variants, types, and associated phenotypes. (A) Total number and percentage of nonsense, missense, frameshift, slice site, insertion, and deletion variants. (B) Total number and percentage of pathogenic variants by location in different protein domains. (C) The relative distribution of the type of PLEC variants by each plectinopathy subtype. Please note that EBS-Ogna is an autosomal dominant disorder and the presence of a missense variant causes the disease. All the other types of plectinopathies listed are autosomal recessive. This figure was generated in Biorender.
Multiple mutations in PLEC are found in the N-terminal alternative first exon of P1a and P1f isoforms and the plakin domain, rod domain, and C-terminal domain, however only two variants has been reported to affect the ABD encoded by exons 2-8 (Figure 4b) (Gostynska et al., 2017). One variant is an in-frame deletion, c. 906+19_40del*, p.Val303_P313ins11, found in intron 8 and noted to cause alternative splicing of exon 8 and produce a shortened transcript (Gostynska et al., 2017), and the other variant was a frameshift variant, c.647_656delTGGAGAACCT, found in exon 7 that caused extensive aplasia cutis congenita (ACC) (Kariminejad et al., 2019). Upon further analysis, the vast majority of the variants were located in the rod domain and C-terminal domain encoded by exon 31 and 32, respectively. In fact, 41% of all variants were located in exon 31 and 24% in exon 32 (Figure 4b).
The clinical manifestations of PLEC variants vary considerably. Plectinopathy is a spectrum of disorders with the same patients having multiple disorders, such as a patient with pyloric atresia manifesting muscular dystrophy later in life. In fact, 40.5% (66 patients) were diagnosed with muscular dystrophy, 14.1% (23 patients) of the patients were diagnosed with autosomal recessive EBS alone, 9.2% (15 patients) with autosomal dominant EBS-Ogna, 8.6% (14 patients) were diagnosed with pyloric atresia, 13.5% (22 patients) with MyS, 3.1% (five patients) with cardiac disease, 4.3% (seven patients) with ACC, and 6.7% (11 patients) with LGMD.
3. PLEC: BIOLOGICAL AND CLINICAL RELEVANCE
3.1. Epidermolysis bullosa simplex and aplasia cutis congenita
Epidermolysis bullosa (EB) is a group of heritable phenotypically diverse skin fragility disorders, characterized by trauma induced blisters, erosions, and wounds to the skin and mucous membranes. EB encompasses a vast phenotypic spectrum, varying from relatively mild lifelong cutaneous blistering to severe cutaneous and extracutaneous involvement with early mortality. EB is divided into four broad subtypes: EB simplex (EBS), junctional EB (JEB), dystrophic forms of EB (DEB), and kindler EB (KEB) (Fine et al., 2014; Has et al., 2020). To date, 16 genes involved in dermal-epidermal integrity and adhesion have been linked to the pathogenesis of different forms of EB. In EBS alone, seven genes have currently been identified as candidate genes: KRT5, KRT14, CD151, KLHL24, DST, EXPH5, and PLEC (Has et al., 2020; Khalesi et al., 2022; Vahidnezhad, Youssefian, Daneshpazhooh, et al., 2019; Vahidnezhad, Youssefian, Saeidian, Mahmoudi, et al., 2018; Vahidnezhad et al., 2016; Vahidnezhad, Youssefian, Saeidian, Touati, et al., 2018).
PLEC is believed to account for approximately 8% of all EBS cases (Bolling et al., 2014; McLean et al., 1996). The recessive subtype of EBS caused by PLEC variants is characterized by life-long, trauma-induced blistering of skin (Figure 3b, 3c, 3e, 3f) (Pfendner et al., 2005). However, there is significant phenotypic variability in this blistering subtype. One of the characteristic phenotypes seen in most plectin subtypes is the presence of hemorrhagic blisters and erosions (Figure 3a, 3d) (Table 3). However, in EBS-PA, skin fragility can range from trauma-induced erosions to extensive congenital ACC (Chung & Uitto, 2010; Pfendner & Uitto, 2005). ACC is a group of heterogeneous disorders characterized by absent skin at birth (Kariminejad et al., 2019). ACC can occur as part of a syndrome, as seen in EBS-PA, or as an isolated sign. Additionally, throughout all subtypes, some patients also present with nail dystrophy, palmoplantar keratoderma, tooth decay, edentulism, urethral strictures, respiratory infections, and mucosal erosions (Figure 3g–m) (Table 3).
Figure 3.

Phenotypic Manifestations of Plectinopathies. (A-G) Note the fragility of the skin with hemorrhagic blisters, erosions, and scarring. (H, I, K) Nail dystrophy associated with plectinopathies. (J) Plantar hyperkeratosis. (L, M) Dental abnormalities present in cases of plectinopathies. (N, O) Ptosis in patients with myasthenic syndrome. This figure was generated in Biorender.
Table 3:
Clinical Manifestations of Patients with PLEC mutations
| Ethnicity (Patient number, age, gender) | Consanguinity | cDNA Variant | Protein Sequence | Diagnosis | Clinical Manifestation |
|---|---|---|---|---|---|
| P1, Shahin dej/west az (14y, M) | First Cousin | c.4504C>T | p.Gln1502* | EBS-MD-MyS | Cutaneous: hemorrhagic blistering, alopecia Oropharyngeal: dental caries, no dysphonia Musculoskeletal: Proximal muscle weakness: 4 out of 5, ptosis, CPK 708 U/L, morning stiffness Respiratory: Respiratory involvement requiring hospitalization Cardiac: none Gastrointestinal: Intermittent abdominal pain Urologic: urinary burning |
| P2, Marvdasht/shiraz (12y, M) | First Cousin | c.7312C>T | p.Arg2438* | EBS-MD-MyS | Cutaneous: hemorrhagic blistering, nail dystrophy Oropharyngeal: dental caries, hoarseness Musculoskeletal: Proximal muscle weakness: 4 out of 5, decreased muscle bulk, ptosis, morning stiffness, CPK 238 U/L Cardiac: none Urologic: left cryptorchidism/surgery at age 10 |
| P3 Babol (6y, M) | None | c.6874C>T | p.Arg2292* | EBS-MD-MyS | Cutaneous: skin blistering, nail dystrophy, alopecia Oropharyngeal: hoarseness Musculoskeletal: Proximal muscle weakness: 4 out of 5, ptosis, CPK 343 U/L Cardiac: none Gastrointestinal: abdominal pain, constipation, GERD |
| c.12611A>G | p.Asp4204Gly | ||||
| P4, Tehran (22y, M) | None (Patients are siblings) |
c.5452C>T | p.Gln1818* | EBS-MD-MyS | Cutaneous: skin blistering, nail dystrophy Oropharyngeal: dental caries, dysphonia Musculoskeletal: Proximal muscle weakness: 4 out of 5, distal muscle weakness: 4 out of 5, ptosis, waddling gait Cardiac: none Respiratory: Respiratory involvement requiring hospitalization |
| P5, Tehran (30y, M) | c.5452C>T | p.Gln1818* | EBS-MD-MyS | Cutaneous: skin blistering, nail dystrophy Oropharyngeal: dental caries Musculoskeletal: Proximal muscle weakness: 4 out of 5, distal muscle weakness: 4 out of 5, ptosis, waddling gait Urologic: urethral stricture, hematuria, hospital admissions for urologic problems Cardiac: None |
|
| P6, Semnan/Damghan (15y, F) | First Cousin | c.10550C>T | p.Thr3571Met | EBS-MD-MyS | Cutaneous: skin blistering, nail dystrophy, alopecia Oropharyngeal: enamel hypoplasia, hoarseness, recurrent choking, required laryngotomy at age 7 due to ulcer in larynx Musculoskeletal: Proximal muscle weakness: 4 out of 5, distal muscle weakness: 4 out of 5, ptosis (left worse than right) Respiratory: admissions due to respiratory problems Cardiac: None |
| P7, Mazandaran (5y, F) | First Cousin | c.5725C>T | p.Gln1909* | EBS | Cutaneous: hemorrhagic blistering, alopecia, nail dystrophy Oropharyngeal: enamel hypoplasia, hoarseness Gastrointestinal: esophageal stenosis/intermittent oral G-tube feeding until age 2 years Cardiac: None Respiratory: recurrent pulmonary infection |
| P8, Tehran (45D, M) | First Cousin | c.1593G>A | p.Trp531* | EBS-PA | Cutaneous: hemorrhagic blistering Gastrointestinal: Pyloric Atresia, Abdominal distension |
| P9, Shiraz, (9y, F) | First Cousin | c.6361del | p.Ala2121Glnfs*68 | EBS-MD-MyS | Cutaneous: skin blistering Oropharyngeal: dental caries, hoarseness Musculoskeletal: Proximal muscle weakness: 4 out of 5, distal muscle weakness: 4 out of 5, ptosis, lower limbs spasticity, severe motor delay: sitting at age 2.5, speech delay |
| P10, Arak (7y, M) | First Cousin | c.7312C>T | p.Arg2438* | EBS | Cutaneous: skin blistering, nail dystrophy Oropharyngeal: hoarseness |
| P11, Tehran (17.5y, F) | First Cousin once-removed | c.1241A>G | p.Gln414Arg | EBS | Cutaneous: hemorrhagic blistering Oropharyngeal: hoarseness Respiratory: recurrent pulmonary infection |
| c.5026C>T | p.Arg1676Cys | ||||
| P12, Tehran (5.5y, M) | Second Cousin | c.7273C>T | p.Gln2425* | EBS | Cutaneous: hemorrhagic blistering, nail dystrophy |
| P13, Lorestan (8y, F) | First Cousin | c.5452C>T | p.Gln1818* | EBS-MyS | Cutaneous: skin blistering, nail dystrophy Urologic: urethral stricture |
| P14, Urumia (Died at 9y, M) | First Cousin (Patients are twins) |
c.1737+1G>A | N/A | EBS, PKU | Cutaneous: hemorrhagic blistering Childhood death due to missed diagnosis of PKU. |
| P15, Urumia (Died at 9y, M) | c.1737+1G>A | N/A | EBS, PKU | Cutaneous: hemorrhagic blistering Childhood death due to missed diagnosis of PKU. |
Abbreviations: EBS, epidermolysis bullosa simplex; PLEC, Plectin; EBS–PA, epidermolysis bullosa simplex-pyloric atresia; EBS–MD, epidermolysis bullosa simplex-muscular dystrophy; LGMD, limb girdle muscular dystrophy; EBS-MyS, epidermolysis bullosa simplex-myasthenic syndrome; PKU, phenylketonuria; y, year; d, day; m, male; f, female.
PLEC variants were documented based on GenBank accession numbers NM_201384.2.
Altogether, 29 variants identified in 23 autosomal recessive EBS index cases were assessed (Table 1 and Table 2) (Figure 2a, 4c) (Bauer et al., 2001; Charlesworth et al., 2013; Gostynska et al., 2015; Khan et al., 2021; Kunz et al., 2000; Maccari et al., 2019; Mariath et al., 2019; McMillan et al., 2007; Mellerio et al., 1997; Natsuga, Nishie, Akiyama, et al., 2010; Natsuga et al., 2017; Tu et al., 2020; Walker et al., 2017; Yu et al., 2021). The variants associated with EBS are found throughout the entire gene including 15 nonsense mutations, four missense, three insertions/deletions, two splice variants, and five frameshift variants (Figure 4c). Of the 23 cases of EBS, 16 patients had at least one variant in exon 31 or 32 and they all resulted in PTCs. With evaluation of the variant type and the location in the rod domain, it has been predicted that patients with variants in this domain are likely to develop MD. In fact, several patients have been diagnosed with EBS-MD, without MD signs and symptoms and/or diagnostic testing, due to the location of the mutation. While it is common to have rod domain and c-terminal involvement, there are isoform specific mutations that can be predicted to lead to EBS alone. In fact, one patient had a homozygous nonsense variant (c.46C>T, p.Arg16*) specific to P1a isoform, and has developed, by the age of 1-year-of-age, skin fragility only (Figure 2a) (Gostynska et al., 2015). This isoform is primarily expressed in keratinocytes of the epidermal basal cell layer, and it was found that P1a transcription level was undetectable.
Altogether, we found seven variants in seven patients that was associated with either an isolated or syndromic ACC (Table 1). An isolated case of ACC was identified in one patient from a consanguineous family who harbored the pathogenic PLEC variant, c.647_656delTGGAGAACCT (Table 1) (Figure 2a) (Kariminejad et al., 2019). This variant was found in exon 7 in the ABD and is therefore noted to affect all isoforms of plectin. Interestingly, three other patients in this family also suffered from isolated ACC, however no mutation analysis was done to confirm the genetic cause. Additionally, all four patients from this family also had underdeveloped facies and facial dysmorphism. Further analysis of the literature also found three studies where patients with EBS-PA and ACC, also had underdeveloped ears, nose, or limbs (Charlesworth et al., 2013; Charlesworth et al., 2003; Pfendner & Uitto, 2005). ACC is typically found in syndromic cases, as seen in EBS-PA. In fact, analysis of this cohort found that six out of the 13 patients with EBS-PA also suffered from ACC. These patients harbored PTC nonsense and frameshift variants in domains that are noted to affect all isoforms of plectin and one patient harbored splice site variant in intron 26 that is predicted to affect the acceptor site of all plectin isoforms (Table 1) (Figure 2a and 4c).
3.2. Epidermolysis bullosa simplex-Ogna
In contrast to the severe recessive subtype, the autosomal dominant EBS-Ogna has a relatively mild presentation. This dominant form was first described in a large Norwegian family in the 1970s in the town of Ogna, however, the sequence variant in PLEC causing this EBS-Ogna was not described until some three decades later in a German family and members of a Norwegian family residing in Ogna. They found a recurrent heterozygous missense variant (c.5917C>T, p.Arg1973Trp) in exon 31 which encodes the rod domain (Koss-Harnes et al., 2002) (Figure 2a); the patients exhibit small, mechanically induced hemorrhagic blisters. Furthermore, clinically distinctive EBS-Ogna patients do not develop MD, cardiomyopathy, or MyS. Comparable findings were reported in an EBS-Ogna mouse model, where c.5917C>T, p.Arg1973Trp was induced in knock-in mice that had cutaneous microlesions and fragility (Walko et al., 2011). They also found that the plectin 1a isoform was selectively degraded in comparison to plectin 1c isoform, which is also found in keratinocytes. Of note, only P1a is recruited to hemidesmosomes, while P1c is found in suprabasal cell layers and at lateral and inter-hemidesmosome basal cell membranes, indicating EBS-Ogna to be a consequence of hemidesmosomal dysfunction (Walko et al., 2011). Additionally, they found that plectin’s rod domain formed dimers that added to plectin stability, which was reduced in the knock-in mice. Therefore, it was proposed that EBS-Ogna phenotype could also be attributed to decreased plectin dimer stabilization. Interestingly, in addition to the recurrent rod domain variant, two additional heterozygous missense variants that were dominantly inherited, c.8587A>T, p.Thr2863Ser, and c.10498C>T, p.Arg3500Cys, were found in exon 32 (Table 1) (Figure 4c) (Bolling et al., 2014; Kiritsi et al., 2013). Therefore, rod-domain stability may not contribute to EBS-Ogna, however, further functional studies of how these variants specifically affect plectin is necessary. Interestingly, this heterozygous variant c.10498C>T, p.Arg3500Cys, found in this Ogna patient, was also reported as a homozygous variant in an EBS-MD patient.
3.3. EBS-pyloric atresia
EBS-PA is one of the most severe consequences of mutations in the PLEC gene. The patients often present with polyhydramnios during pregnancy, severe blistering and ACC, pyloric or duodenal atresia, and early postnatal demise. Currently, there are 20 distinct variants in 14 cases of PLEC associated PA (Table 1, Table 2, Table 3) (Figure 2a and 4c) (Charlesworth et al., 2013; Charlesworth et al., 2003; Nakamura et al., 2005; Natsuga, Nishie, Shinkuma, et al., 2010; Pfendner & Uitto, 2005; Sawamura et al., 2007; Valari et al., 2019; Walker et al., 2017). All the patients, except for three patients, died in the neonatal period or the first year of life (Charlesworth et al., 2013; Valari et al., 2019; Walker et al., 2017).
All patients with PLEC variants and EBS-PA had at least one variant that resulted in a PTC (Table 1 and Table 2). Interestingly, of the 14 cases, 12 had variants outside of the rod domain. Of the two patients with variants in the rod domain, one patient had a PTC mutation within exon 24 and the other patient had both mutations in exon 31 (Table 1) (Figure 2a). With mutations outside of the rod domain, all plectin isoforms are potentially affected. In fact, one study found the expression of two distinct isoforms, and found that protein truncating mutations led to two different subtypes of plectinopathies, EBS-PA and EBS-MD (Natsuga, Nishie, Akiyama, et al., 2010). It was found that patients with EBS-PA had deficiency of both full-length and rodless plectin isoforms, which correlates with the severity and early demise seen in this cohort (Figure 1a, 1b) (Natsuga, Nishie, Akiyama, et al., 2010). However, EBS-MD patients had conserved rodless expression but no full-length plectin expression. Due to the loss of both isoforms, it was postulated that if an EBS-PA patient lived for a longer period, then it is likely that he/she may develop muscular dystrophy due to residual plectin expression.
Two studies have found patients who developed MD after PA. In one study, a patient lived until 3 months of age and began showing clinical signs of muscular dystrophy (Natsuga, Nishie, Shinkuma, et al., 2010). This patient had compound heterozygous variants (c.10903C>T, p.Gln3635*; c.11372_11381del, p.Ile3791Argfs*90) in exon 32 where binding of IF, such as desmin, occurs, explaining to the relatively early onset MD (Table 1). Similar to the previous patient, another study also denoted a patient who at the time of the study, had MD at the age of 2.5 years. This patient has compound heterozygous PTC mutations, c.11831delA, p.Lys3944Argfs*10, and c.12418C>T, p.Arg4140*, in exon 32 (Valari et al., 2019) (Table 1). Two additional patients have also survived past the 1st year of life; however, they have not developed MD. One patient was 13-years of age with compound heterozygous mutations in exon 32 and 30, and the other patient was 6-years of age and had no MD symptoms but had mildly elevated creatine kinase levels (Charlesworth et al., 2013; Walker et al., 2017). This 6-year-old patient had PTC mutations in exons 24 and 31 (Walker et al., 2017).
3.4. The skin-muscle connection: Epidermolysis bullosa simplex-muscular dystrophy (EBS-MD)
One of the most common PLEC related extracutaneous manifestations is muscular dystrophy. In EBS-MD, the patients typically present with skin blistering at birth, however, the onset of muscular symptoms onset is variable. MD defining symptoms may develop in infancy, however, most cases present later in life, with some cases reported to occur in the third and fourth decade of life (Chavanas et al., 1996; Pulkkinen et al., 1996). Most patients note the presentation of muscular weakness in their second or third decade of life and the muscular manifestations are slowly progressive. Patients typically present with stepwise loss of motor function: in the early stages of muscular dystrophy, the patients tend to present with upper and lower extremities muscle weakness and fatigue, while infants tend to present with delayed motor developmental milestones (Table 3). In the later stages, the patients lose their ability to ambulate and frequently experience respiratory insufficiency, which is the primary cause of early demise. The diagnosis of MD is made based on a spectrum of clinical findings including clinical symptoms, positive EMG results, elevated CPK, and muscle biopsy (Table 3). Muscle biopsies show atrophic angulated muscle fibers, widened spaces between the plasma membrane and muscle sarcomere, disordered desmin aggregation, and accumulation of sarcolemmal and intermyofibrillar nuclear aggregates which represent end-stage muscle fibers with residual nuclei (Alvarez et al., 2016; Chiaverini et al., 2010).
In total, 69 PLEC variants were found in 66 patients diagnosed with EBS-MD (Table 1 and Table 2) (Figure 4c). Among them, c.4937_4955del, was shared with one patient who only manifested EBS (Mellerio et al., 1997; Winter et al., 2016) (Table 1). The variants are present throughout the different domains of the gene (Figure 2a). Of the 69 variants, 32 were located in exon 31 and 22 were located in exon 32 and of the 66 patients, only six patients had pathogenic variants outside of exon 31 or 32 (Table 1 and Table 2).
Consequently, it has been suggested that patients with variants in exon 31 are likely to develop MD, but, since there are cases with PLEC variants in exon 31 and these patients have not developed MD, this prediction is not absolute. However, it is postulated that these patients may develop MD later in life, as the oldest reported patient with EBS alone was 31-years of age and there has been a case of EBS with late onset MD that presented in the patient’s 40’s (Tu et al., 2020; Uitto, 2004). In fact, further analysis of plectin expression found that EBS-MD patients had decreased to absent full-length plectin but found residual expression of the rodless isoform (Natsuga, Nishie, Akiyama, et al., 2010). Initially, it was postulated that presentation of rodless plectin was the primary cause of MD, however, rodless plectin knock-in mice were generated and it was found that the rod domain was not necessary for tissue integrity (Ketema et al., 2015). In fact, they found that mice with rodless plectin did not develop any skin or muscular pathology and rodless plectin was able to compensate for full-length plectin (Ketema et al., 2015). However, it is the low expression of the rodless plectin, rather than absence of the rod domain that contributes to the presentation of EBS-MD (Ketema et al., 2015). Further, as we investigated the location of the mutations in EBS-MD patients we found that a large majority of patients had variants in exon 32. Exon 32 of PLEC encodes the IF binding site used by desmin for sarcolemma structural integrity significant for muscular function.
3.5. Limb-girdle muscular dystrophy
Limb-girdle muscular dystrophy (LGMD) is a heterogeneous group of disorders characterized by weakness and atrophy of proximal muscles in the arms and legs. There are over 40 types of LGMD that are subcategorized by the causative gene and inheritance pattern. Variants in PLEC are known to cause autosomal recessive LGMD type 17 (LGMDR17, formerly LGMD2Q). Similar to EBS-MD patients, they present with classic axial muscular weakness in early childhood and progressive physical decline resulting in wheelchair/bed-bound individuals by late 20’s to early 30’s, however, no cutaneous involvement is reported (Deev et al., 2017; Fattahi et al., 2015; Gundesli et al., 2010; Mroczek et al., 2020).
To date, there have been six variants reported in 11 cases of LGMDR17 caused by PLEC variants (Table 1) (Figure 2a and 4c) (Deev et al., 2017; Fattahi et al., 2015; Gundesli et al., 2010; Mroczek et al., 2020; Zhong et al., 2017). Among them, there have been seven cases of a recurrent homozygous deletion (c.1_9del *, p.?) and three cases of homozygous nonsense variant (c.58G > T, p.Glu20*) in the first exon of the P1f isoform, which localizes to striated muscle and acts as a structural scaffold for muscle sarcolemma (Rezniczek et al., 2007). Additionally, they found that the 1f isoform expression was ~100-fold lower than in normal skeletal tissue. Interestingly, prediction software on how this variant would affect the transcript, predicted significant alterations in the exonic splicing enhancers and silencers and postulated the activation of a cryptic donor site that would lead to altered splicing (Table1).
LGMDR17 was initially associated with the P1f isoform, but additional cases identified patients with variants that would affect all isoforms, yet presented with LGMDR17 phenotypes. One patient harbored compound heterozygous missense variants, c.6037C>T, p.Arg2013Trp in exon 31 and c.9982T>A, p.Phe3328Ile in exon 32 (Zhong et al., 2017) (Table 1). This patient was a 7-year-old male who exhibited delayed independent walking at 2 years of age, and muscle biopsy confirmed cytoskeletal disorganization; the patient had no prominent skin manifestations. Similarly, another study also reported two additional patients in their 30’s with LGMDR17 phenotypes and compound heterozygous PLEC variants (c.2983C>T, p.Gln995*; c.11422G>A, p.Gly3808Ser) in exons 24 and 32, respectively (Table 1) (Fattahi et al., 2015). However, an additional variant was found in candidate gene titin (TTN) which is related to LGMD but PLEC was implicated due to the co-segregation analysis in the family. Interestingly, as we analyzed these variants, we found that both ACMG and Sherloc analysis would designate the PLEC variants c.2983C>T and c.11422G>A as a benign variant and a variant of unknown significance, respectively. Additionally, these patients had variants in domains that are likely to affect multiple tissue isoforms, however there was no reported EBS. Whether these patients may present with skin findings later in life is currently unknown and further studies on skin hemidesmosome involvement in these patients is needed.
3.6. Congenital myasthenic syndrome
Congenital myasthenic syndromes (CMS) or myasthenic syndrome (MyS) is an inherited disorder characterized by muscle weakness and fatigability, especially of ocular and cranial muscles (Engel et al., 2015). MyS patients are diagnosed based on clinical manifestations, decremental electromyography (EMG) response or abnormal single fiber EMG, and genetic testing (Engel et al., 2015). In MyS, the most common phenotypic presentations are ptosis, ophthalmoparesis, facial and bulbar weakness, and generalized muscle weakness in the neonatal period or early childhood (Figure 3n, 3o) (Table 3) (Engel et al., 2015). However, clinical manifestations may present in adolescence and late adulthood, and the pattern of muscle weakness may vary, making the diagnosis difficult. Currently, there are over 30 genes associated with impaired neuromuscular transmission in CMS, including PLEC (Engel et al., 2015). Plectin is located in the postsynaptic neuromuscular junction and is crucial in the formation and maintenance of acetylcholine receptors (AChR) clusters at synaptic sites (Mihailovska et al., 2014). Specifically, P1f interlinks desmin IF with dystrophin glycoprotein complexes which stabilize the synaptic junction for AChRs incorporation. Additionally, P1f binds rapsyn-AChR complexes to maintain the stability of AChR clusters (Mihailovska et al., 2014).
MyS secondary to PLEC mutations is an extremely rare presentation, which is oftentimes overshadowed by and merged into a consequence of MD. Currently, 20 variants have been associated with 22 cases of MyS in patients with PLEC mutations, however, an additional five reported patients had classic myasthenic signs that were diagnosed as muscular dystrophy (Figure 2a, 4c) (Table1 and Table 2) (Argente-Escrig et al., 2021; Banwell et al., 1999; Fattahi et al., 2015; Forrest et al., 2010; Gache et al., 1996; Gonzalez Garcia et al., 2019; Gostynska et al., 2017; Kyrova et al., 2016; Maselli et al., 2011; Selcen et al., 2011; Villa et al., 2015; Walker et al., 2017). In the 22 patients diagnosed with MyS, 20 patients also exhibited EBS-MD and harbored compound heterozygous and homozygous variants (Table 1, Table 2, Table 3). In one case, a 63-year-old Russian woman harbored a homozygous insertion, c.1419_1420ins36, p.Arg473_Val474ins12, in exon 14 of PLEC (Maselli et al., 2011). This patient had EBS that presented in infancy and MyS that did not present until 50-years-of-age and no signs of MD (Maselli et al., 2011). Interestingly, this patient also had a homozygous insertion, c.1293insG in CHRNE, which has protein expression in the neuromuscular junction (Maselli et al., 2011). Following this trend, in our cohort, we found one patient with a recurrent nonsense variant who had no clinical and laboratory signs of MD, however, this 8-year-old female manifested EBS-MyS (Table 2 and Table 3). Interestingly, apart from five cases, all MyS patients had at least one variant in exon 31 or 32. In four cases, the patients harbored the recurrent muscle and neuromuscular specific P1f isoform deletion, c.1_9del in exon 1, commonly causing LGMD (Mroczek et al., 2020) and the other patient was mentioned above (Maselli et al., 2011).
After diagnosing a patient with compound heterozygous variants, c.6955C > T, Arg2319*; c.12043dupG, p.Glu4015Glyfs*69 in exons 31 and 32, respectively, one study focused on the pathomechanisms of PLEC mutations causing MyS. They found that colocalization of plectin and AChR at the end-plates of neuromuscular junction was strong in normal patient samples, however, in affected patients the AChR expression was normal but plectin expression was essentially undetectable, leading to loss of the junctional folds (Selcen et al., 2011). Comparable to these findings, in a plectin knock-out mice that disrupted muscle specific P1f isoform, neuromuscular junctions were disorganized, and the mice showed phenotypes similar to that of EBS-MD-MyS, such as weakness and impaired balance (Mihailovska et al., 2014).
While a general genotype-phenotype correlation can be made about the type of mutations, isoforms, and locations affected in plectin-induced MyS, it is not fully understood why some patients with EBS-MD have MyS and some do not. In our cohort alone, we found 8 patients with MyS, therefore, to avoid underdiagnosis of MyS, it should always be considered and investigated in PLEC patients.
3.7. Plectin and the heart
Several studies have found a connection between aberrations in PLEC and systemic pathologies. For example, plectin is predominantly expressed in striated muscle explaining the classic characteristic of MD, however, plectin is also found in the intercalated disks of the myocardium. Indeed, there have been five reported mutation-confirmed cases of EBS-MD who harbored cardiac manifestations, including dilated cardiomyopathy with ventricular arrhythmias (Figure 2a) (Bolling et al., 2010; Winter et al., 2016), left ventricular hypertrophy (Schroder et al., 2002; Winter et al., 2016), left ventricular non-compaction hypertrophy (Villa et al., 2015), and cardiomyopathy (Bolling et al., 2010).
The PLEC mutations associated with the reported cardiac pathologies vary. Currently, there have been three homozygous variants and two compound heterozygous variants (Table 1) (Figure 4c). The homozygous variants include one deletion in intron 8, one duplication in exon 32, and a nonsense variant in exon 31. Interestingly, the variant in intron 8 was found to result in alternative splicing of exon 8 that expressed a shortened PLEC transcript in the muscle, myocardium, and skin (Gostynska et al., 2017). The compound heterozygous variants were a missense and nonsense variant in exon 9 and 31, respectively, and two deletions, one in exon 19 and the other in exon 31 (Bolling et al., 2010; Winter et al., 2016). Interestingly, all patients had at least one PTC variant and all patients had clinically severe EBS-MD and three patients had classic myasthenic symptoms. In addition to the cardiomyopathy, a genome-wide association study found a missense variant, p.Gly4098Ser in exon 32, in 14,255 atrial fibrillation patients in Iceland that associated with a 55% increased risk of atrial fibrillation and 64% increased risk of sick sinus syndrome, however, they did not find any association with cardiomyopathy (Thorolfsdottir et al., 2017; Vahidnezhad, Youssefian, Saeidian, & Uitto, 2019).
The cardiac manifestations expressed in these patients are an emerging consequence of PLEC variants. Therefore, future exploration into the cardiac manifestations of these patients is needed and should be investigated in all patients, due to the potentially fatal cardiac consequences in these patients and their families.
4. THERAPEUTIC APPROACH AND ADVANCEMENTS
Currently, plectinopathies are incurable. However, there are traditional and genetic approaches for symptomatic relief. In EBS, symptomatic approaches typically focus on protection via topical treatments, wound management, and avoidance of trauma. In cases of MyS, 13 patients have been treated for myasthenic symptoms, and all but two patients had significant symptomatic improvement (Argente-Escrig et al., 2021; Fattahi et al., 2015; Forrest et al., 2010; Gonzalez Garcia et al., 2019; Mroczek et al., 2020; Selcen et al., 2011). These patients received targeted treatments for MyS, such as pyridostigmine, 3,4-diaminopyridine, ephedrine, prednisone, and/or salbutamol. They variably experienced improvement in ptosis, climbing stairs, decreased fatigability, fewer chest infections, and improved strength, however, the decrease in repetitive nerve stimulation did not improve in two patients who experienced symptomatic improvement (Figure 3n, 3o) (Gonzalez Garcia et al., 2019).
Further exploration for curative options has led to the use of systemic gentamicin in approximately 40 percent of the patients (see Figure 4A). Gentamicin is an aminoglycoside, which beyond its antimicrobial actions can read through premature termination codons (PTCs) and has been tested in several genetic diseases. In fact, a recent study reported that systemic gentamicin in an EBS-MD patient led to an improvement in daily quality of life, strength, mobility, maximal inspiratory and expiratory pressures, and decreased mucosal blistering (Martinez-Santamaria et al., 2022). Additionally, they found that keratinocytes cultured with gentamicin had increased plectin expression. These findings point to the importance of further exploration of gentamicin as a treatment for plectinopathy, with the notion that it has appreciable results in patients with PTC mutations. Molecular genetics have revolutionized therapeutic approaches to genetic diseases and future approaches focus on targeted gene and cell-based treatment. Thus, interdisciplinary approaches to plectinopathies and other genetic diseases are necessary.
5. CONCLUSION AND FUTURE PROSPECTS
This study presents a comprehensive update of all PLEC cases to date and the associated clinical manifestations that will help investigators and clinicians. The clinical manifestations of individuals with plectinopathy vary regarding the onset, progression, and the type of manifestations a patient may develop. The cause of this variability is not fully understood and can only be partially explained by the location of mutations in different domains and the predicted affected isoforms. Therefore, a better understanding of clinical features of plectinopathies and associated molecular mechanisms are needed. Notably, patients with the same PLEC mutations might have a different phenotype, and consequently, other molecular, genetic, epigenetic, or environmental factors in the context of specific disease manifestations require further detailed studies.
Acknowledgments
Carol Kelly assisted in manuscript preparation.
Funding:
This study was supported by the Jefferson Institute of Molecular Medicine Institutional Funds and the NIH Grants R01 AR028450 and R01 AI143810.
Abbreviations:
- EB
epidermolysis bullosa
- EBS
epidermolysis bullosa simplex
- PLEC
Plectin
- EBS–PA
epidermolysis bullosa simplex-pyloric atresia
- EBS–MD
epidermolysis bullosa simplex-muscular dystrophy
- LGMD
limb girdle muscular dystrophy
- EBS-MyS
epidermolysis bullosa simplex-myasthenic syndrome
- JEB
junctional epidermolysis bullosa
- DEB
dystrophic epidermolysis bullosa
- KEB
kindler epidermolysis bullosa
- IF
intermediate filament
- ABD
actin-binding domain
- AChR
acetylcholine receptor
- LOVD
Leiden Open Variation Database
- CADD
combined annotation-dependent depletion score
- CMS
congenital myasthenic syndrome
- MyS
myasthenic syndrome
- EMG
electromyography
- PTC
premature termination codon
Footnotes
Web Resources
Mutalyzer: https://mutalyzer.nl
Leiden Open Variation Database: https://www.lovd.nl
Human Splice Finder System: https://hsf.genomnis.com/about
Pubmed: https://pubmed.ncbi.nlm.nih.gov
Ensembl: https://useast.ensembl.org/index.html
Mutation taster: https://www.mutationtaster.org
Conflict of Interests
The authors state no conflict of interest.
Patient Consent
All subjects and parents of minors gave written informed consent to participate in this study and publish their images.
Data Availability Statement
All data associated with this study are presented in the paper. All novel variants have been submitted to the LOVD database (https://www.lovd.nl)
REFERENCES
- Ahmad F, Shah K, Umair M, Jan A, Irfanullah, Khan S, Muhammad D, Basit S, Wakil SM, Ramzan K, & Ahmad W (2018). Novel autosomal recessive LAMA3 and PLEC variants underlie junctional epidermolysis bullosa generalized intermediate and epidermolysis bullosa simplex with muscular dystrophy in two consanguineous families. Clin Exp Dermatol, 43(6), 752–755. 10.1111/ced.13610 [DOI] [PubMed] [Google Scholar]
- Alvarez VC, Penttila ST, Salutto VL, Udd B, & Mazia CG (2016). Epidermolysis bullosa simplex with muscular dystrophy associated with PLEC deletion mutation. Neurol Genet, 2(6), e109. 10.1212/NXG.0000000000000109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argente-Escrig H, Schultheis D, Kamm L, Schowalter M, Thiel C, Turk M, Clemen CS, Muelas N, Castanon MJ, Wiche G, Herrmann H, Vilchez JJ, & Schroder R (2021). Plectin-related scapuloperoneal myopathy with treatment-responsive myasthenic syndrome. Neuropathol Appl Neurobiol, 47(2), 352–356. 10.1111/nan.12652 [DOI] [PubMed] [Google Scholar]
- Argyropoulou Z, Liu L, Ozoemena L, Branco CC, Senra R, Reis-Rego A, & Mota-Vieira L (2018). A novel PLEC nonsense homozygous mutation (c.7159G > T; p.Glu2387*) causes epidermolysis bullosa simplex with muscular dystrophy and diffuse alopecia: a case report. BMC Dermatol, 18(1), 1. 10.1186/s12895-018-0069-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banwell BL, Russel J, Fukudome T, Shen XM, Stilling G, & Engel AG (1999). Myopathy, myasthenic syndrome, and epidermolysis bullosa simplex due to plectin deficiency. J Neuropathol Exp Neurol, 58(8), 832–846. 10.1097/00005072-199908000-00006 [DOI] [PubMed] [Google Scholar]
- Bauer JW, Rouan F, Kofler B, Rezniczek GA, Kornacker I, Muss W, Hametner R, Klausegger A, Huber A, Pohla-Gubo G, Wiche G, Uitto J, & Hintner H (2001). A compound heterozygous one amino-acid insertion/nonsense mutation in the plectin gene causes epidermolysis bullosa simplex with plectin deficiency. Am J Pathol, 158(2), 617–625. 10.1016/S0002-9440(10)64003-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolling MC, Jongbloed JDH, Boven LG, Diercks GFH, Smith FJD, Irwin McLean WH, & Jonkman MF (2014). Plectin mutations underlie epidermolysis bullosa simplex in 8% of patients. J Invest Dermatol, 134(1), 273–276. 10.1038/jid.2013.277 [DOI] [PubMed] [Google Scholar]
- Bolling MC, Pas HH, de Visser M, Aronica E, Pfendner EG, van den Berg MP, Diercks GF, Suurmeijer AJ, & Jonkman MF (2010). PLEC1 mutations underlie adult-onset dilated cardiomyopathy in epidermolysis bullosa simplex with muscular dystrophy. J Invest Dermatol, 130(4), 1178–1181. 10.1038/jid.2009.390 [DOI] [PubMed] [Google Scholar]
- Borradori L, & Sonnenberg A (1999). Structure and function of hemidesmosomes: more than simple adhesion complexes. J Invest Dermatol, 112(4), 411–418. 10.1046/j.1523-1747.1999.00546.x [DOI] [PubMed] [Google Scholar]
- Bouameur JE, Favre B, & Borradori L (2014). Plakins, a versatile family of cytolinkers: roles in skin integrity and in human diseases. J Invest Dermatol, 134(4), 885–894. 10.1038/jid.2013.498 [DOI] [PubMed] [Google Scholar]
- Castanon MJ, Walko G, Winter L, & Wiche G (2013). Plectin-intermediate filament partnership in skin, skeletal muscle, and peripheral nerve. Histochem Cell Biol, 140(1), 33–53. 10.1007/s00418-013-1102-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth A, Chiaverini C, Chevrant-Breton J, DelRio M, Diociaiuti A, Dupuis RP, El Hachem M, Le Fiblec B, Sankari-Ho AM, Valhquist A, Wierzbicka E, Lacour JP, & Meneguzzi G (2013). Epidermolysis bullosa simplex with PLEC mutations: new phenotypes and new mutations. Br J Dermatol, 168(4), 808–814. 10.1111/bjd.12202 [DOI] [PubMed] [Google Scholar]
- Charlesworth A, Gagnoux-Palacios L, Bonduelle M, Ortonne JP, De Raeve L, & Meneguzzi G (2003). Identification of a lethal form of epidermolysis bullosa simplex associated with a homozygous genetic mutation in plectin. J Invest Dermatol, 121(6), 1344–1348. 10.1111/j.1523-1747.2003.12639.x [DOI] [PubMed] [Google Scholar]
- Chavanas S, Pulkkinen L, Gache Y, Smith FJ, McLean WH, Uitto J, Ortonne JP, & Meneguzzi G (1996). A homozygous nonsense mutation in the PLEC1 gene in patients with epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest, 98(10), 2196–2200. 10.1172/JCI119028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Lin ZM, Wang HJ, Zhang J, Yin JH, & Yang Y (2013). New mutations in the PLEC gene in a Chinese patient with epidermolysis bullosa simplex with muscular dystrophy. Clin Exp Dermatol, 38(7), 792–794. 10.1111/ced.12116 [DOI] [PubMed] [Google Scholar]
- Chiaverini C, Charlesworth A, Meneguzzi G, Lacour JP, & Ortonne JP (2010). Epidermolysis bullosa simplex with muscular dystrophy. Dermatol Clin, 28(2), 245–255, viii. 10.1016/j.det.2010.01.001 [DOI] [PubMed] [Google Scholar]
- Chung HJ, & Uitto J (2010). Epidermolysis bullosa with pyloric atresia. Dermatol Clin, 28(1), 43–54. 10.1016/j.det.2009.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Wei X, Zhao Y, Ren H, Lan Z, Yang Y, Chen L, & Cui L (2015). A comprehensive genetic diagnosis of Chinese muscular dystrophy and congenital myopathy patients by targeted next-generation sequencing. Neuromuscul Disord, 25(8), 617–624. 10.1016/j.nmd.2015.03.002 [DOI] [PubMed] [Google Scholar]
- Dang M, Pulkkinen L, Smith FJ, McLean WH, & Uitto J (1998). Novel compound heterozygous mutations in the plectin gene in epidermolysis bullosa with muscular dystrophy and the use of protein truncation test for detection of premature termination codon mutations. Lab Invest, 78(2), 195–204. https://www.ncbi.nlm.nih.gov/pubmed/9484717 [PubMed] [Google Scholar]
- Deev RV, Bardakov SN, Mavlikeev MO, Yakovlev IA, Umakhanova ZR, Akhmedova PG, Magomedova RM, Chekmaryeva IA, Dalgatov GD, & Isaev AA (2017). Glu20Ter Variant in PLEC 1f Isoform Causes Limb-Girdle Muscle Dystrophy with Lung Injury. Front Neurol, 8, 367. 10.3389/fneur.2017.00367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel AG, Shen XM, Selcen D, & Sine SM (2015). Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol, 14(4), 420–434. 10.1016/S1474-4422(14)70201-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattahi Z, Kahrizi K, Nafissi S, Fadaee M, Abedini SS, Kariminejad A, Akbari MR, & Najmabadi H (2015). Report of a patient with limb-girdle muscular dystrophy, ptosis and ophthalmoparesis caused by plectinopathy. Arch Iran Med, 18(1), 60–64. https://doi.org/0151801/AIM.0014 [PubMed] [Google Scholar]
- Fine JD, Bruckner-Tuderman L, Eady RA, Bauer EA, Bauer JW, Has C, Heagerty A, Hintner H, Hovnanian A, Jonkman MF, Leigh I, Marinkovich MP, Martinez AE, McGrath JA, Mellerio JE, Moss C, Murrell DF, Shimizu H, Uitto J, Woodley D, & Zambruno G (2014). Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol, 70(6), 1103–1126. 10.1016/j.jaad.2014.01.903 [DOI] [PubMed] [Google Scholar]
- Forrest K, Mellerio JE, Robb S, Dopping-Hepenstal PJ, McGrath JA, Liu L, Buk SJ, Al-Sarraj S, Wraige E, & Jungbluth H (2010). Congenital muscular dystrophy, myasthenic symptoms and epidermolysis bullosa simplex (EBS) associated with mutations in the PLEC1 gene encoding plectin. Neuromuscul Disord, 20(11), 709–711. 10.1016/j.nmd.2010.06.003 [DOI] [PubMed] [Google Scholar]
- Gache Y, Chavanas S, Lacour JP, Wiche G, Owaribe K, Meneguzzi G, & Ortonne JP (1996). Defective expression of plectin/HD1 in epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest, 97(10), 2289–2298. 10.1172/JCI118671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez Garcia A, Tutmaher MS, Upadhyayula SR, Sanchez Russo R, & Verma S (2019). Novel PLEC gene variants causing congenital myasthenic syndrome. Muscle Nerve, 60(6), E40–E43. 10.1002/mus.26703 [DOI] [PubMed] [Google Scholar]
- Gostynska KB, Lemmink H, Bremer J, Pas HH, Nijenhuis M, van den Akker PC, Sinke RJ, Jonkman MF, & Pasmooij AMG (2017). A PLEC Isoform Identified in Skin, Muscle, and Heart. J Invest Dermatol, 137(2), 518–522. 10.1016/j.jid.2016.09.032 [DOI] [PubMed] [Google Scholar]
- Gostynska KB, Nijenhuis M, Lemmink H, Pas HH, Pasmooij AM, Lang KK, Castanon MJ, Wiche G, & Jonkman MF (2015). Mutation in exon 1a of PLEC, leading to disruption of plectin isoform 1a, causes autosomal-recessive skin-only epidermolysis bullosa simplex. Hum Mol Genet, 24(11), 3155–3162. 10.1093/hmg/ddv066 [DOI] [PubMed] [Google Scholar]
- Gundesli H, Talim B, Korkusuz P, Balci-Hayta B, Cirak S, Akarsu NA, Topaloglu H, & Dincer P (2010). Mutation in exon 1f of PLEC, leading to disruption of plectin isoform 1f, causes autosomal-recessive limb-girdle muscular dystrophy. Am J Hum Genet, 87(6), 834–841. 10.1016/j.ajhg.2010.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Has C, Bauer JW, Bodemer C, Bolling MC, Bruckner-Tuderman L, Diem A, Fine JD, Heagerty A, Hovnanian A, Marinkovich MP, Martinez AE, McGrath JA, Moss C, Murrell DF, Palisson F, Schwieger-Briel A, Sprecher E, Tamai K, Uitto J, Woodley DT, Zambruno G, & Mellerio JE (2020). Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol, 183(4), 614–627. 10.1111/bjd.18921 [DOI] [PubMed] [Google Scholar]
- Kariminejad A, Vahidnezhad H, Ghaderi-Sohi S, Ghannadan AR, Youssefian L, Parsimehr E, Faraji Zonooz M, Kariminejad MH, Uitto J, Najmabadi H, & Hennekam RC (2019). Widespread aplasia cutis congenita in sibs with PLEC1 and ITGB4 variants. Am J Med Genet A, 179(8), 1547–1555. 10.1002/ajmg.a.61260 [DOI] [PubMed] [Google Scholar]
- Ketema M, Secades P, Kreft M, Nahidiazar L, Janssen H, Jalink K, de Pereda JM, & Sonnenberg A (2015). The rod domain is not essential for the function of plectin in maintaining tissue integrity. Mol Biol Cell, 26(13), 2402–2417. 10.1091/mbc.E15-01-0043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalesi R, Harvey N, Garshasbi M, Kalamati E, Youssefian L, Vahidnezhad H, & Uitto J (2022). Pathogenic DST sequence variants result in either epidermolysis bullosa simplex (EBS) or hereditary sensory and autonomic neuropathy type 6 (HSAN-VI). Exp Dermatol. 10.1111/exd.14562 [DOI] [PubMed] [Google Scholar]
- Khan FF, Khan N, Rehman S, Ejaz A, Ali U, Erfan M, Ahmed ZM, & Naeem M (2021). Identification and Computational Analysis of Novel Pathogenic Variants in Pakistani Families with Diverse Epidermolysis Bullosa Phenotypes. Biomolecules, 11(5). 10.3390/biom11050620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiritsi D, Pigors M, Tantcheva-Poor I, Wessel C, Arin MJ, Kohlhase J, Bruckner-Tuderman L, & Has C (2013). Epidermolysis bullosa simplex ogna revisited. J Invest Dermatol, 133(1), 270–273. 10.1038/jid.2012.248 [DOI] [PubMed] [Google Scholar]
- Koss-Harnes D, Hoyheim B, Anton-Lamprecht I, Gjesti A, Jorgensen RS, Jahnsen FL, Olaisen B, Wiche G, & Gedde-Dahl T Jr. (2002). A site-specific plectin mutation causes dominant epidermolysis bullosa simplex Ogna: two identical de novo mutations. J Invest Dermatol, 118(1), 87–93. 10.1046/j.0022-202x.2001.01591.x [DOI] [PubMed] [Google Scholar]
- Koss-Harnes D, Hoyheim B, Jonkman MF, de Groot WP, de Weerdt CJ, Nikolic B, Wiche G, & Gedde-Dahl T Jr. (2004). Life-long course and molecular characterization of the original Dutch family with epidermolysis bullosa simplex with muscular dystrophy due to a homozygous novel plectin point mutation. Acta Derm Venereol, 84(2), 124–131. 10.1080/00015550310007094 [DOI] [PubMed] [Google Scholar]
- Koster J, van Wilpe S, Kuikman I, Litjens SH, & Sonnenberg A (2004). Role of binding of plectin to the integrin beta4 subunit in the assembly of hemidesmosomes. Mol Biol Cell, 15(3), 1211–1223. 10.1091/mbc.e03-09-0697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz M, Rouan F, Pulkkinen L, Hamm H, Jeschke R, Bruckner-Tuderman L, Brocker EB, Wiche G, Uitto J, & Zillikens D (2000). Mutation reports: epidermolysis bullosa simplex associated with severe mucous membrane involvement and novel mutations in the plectin gene. J Invest Dermatol, 114(2), 376–380. 10.1046/j.1523-1747.2000.00856.x [DOI] [PubMed] [Google Scholar]
- Kyrova J, Kopeckova L, Buckova H, Mrazova L, Vesely K, Hermanova M, Oslejskova H, & Fajkusova L (2016). Epidermolysis bullosa simplex with muscular dystrophy. Review of the literature and a case report. J Dermatol Case Rep, 10(3), 39–48. 10.3315/jdcr.2016.1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccari ME, Speckmann C, Heeg M, Reimer A, Casetti F, Has C, Ehl S, & Castro CN (2019). Profound immunodeficiency with severe skin disease explained by concomitant novel CARMIL2 and PLEC1 loss-of-function mutations. Clin Immunol, 208, 108228. 10.1016/j.clim.2019.06.004 [DOI] [PubMed] [Google Scholar]
- Mariath LM, Santin JT, Frantz JA, Doriqui MJR, Kiszewski AE, & Schuler-Faccini L (2019). An overview of the genetic basis of epidermolysis bullosa in Brazil: discovery of novel and recurrent disease-causing variants. Clin Genet, 96(3), 189–198. 10.1111/cge.13555 [DOI] [PubMed] [Google Scholar]
- Martinez-Santamaria L, Maseda R, de Arriba MDC, Membrilla JA, Siguenza AI, Mascias J, Garcia M, Quintana L, Esteban-Rodriguez I, Hernandez-Fernandez CP, Illera N, Duarte B, Guerrero-Aspizua S, Woodley DT, Del Rio M, de Lucas R, Larcher F, & Escamez MJ (2022). Evaluation of Systemic Gentamicin as Translational Readthrough Therapy for a Patient With Epidermolysis Bullosa Simplex With Muscular Dystrophy Owing to PLEC1 Pathogenic Nonsense Variants. JAMA Dermatol. 10.1001/jamadermatol.2022.0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maselli RA, Arredondo J, Cagney O, Mozaffar T, Skinner S, Yousif S, Davis RR, Gregg JP, Sivak M, Konia TH, Thomas K, & Wollmann RL (2011). Congenital myasthenic syndrome associated with epidermolysis bullosa caused by homozygous mutations in PLEC1 and CHRNE. Clin Genet, 80(5), 444–451. 10.1111/j.1399-0004.2010.01602.x [DOI] [PubMed] [Google Scholar]
- McLean WH, Pulkkinen L, Smith FJ, Rugg EL, Lane EB, Bullrich F, Burgeson RE, Amano S, Hudson DL, Owaribe K, McGrath JA, McMillan JR, Eady RA, Leigh IM, Christiano AM, & Uitto J (1996). Loss of plectin causes epidermolysis bullosa with muscular dystrophy: cDNA cloning and genomic organization. Genes Dev, 10(14), 1724–1735. 10.1101/gad.10.14.1724 [DOI] [PubMed] [Google Scholar]
- McMillan JR, Akiyama M, Rouan F, Mellerio JE, Lane EB, Leigh IM, Owaribe K, Wiche G, Fujii N, Uitto J, Eady RA, & Shimizu H (2007). Plectin defects in epidermolysis bullosa simplex with muscular dystrophy. Muscle Nerve, 35(1), 24–35. 10.1002/mus.20655 [DOI] [PubMed] [Google Scholar]
- Mellerio JE, Smith FJ, McMillan JR, McLean WH, McGrath JA, Morrison GA, Tierney P, Albert DM, Wiche G, Leigh IM, Geddes JF, Lane EB, Uitto J, & Eady RA (1997). Recessive epidermolysis bullosa simplex associated with plectin mutations: infantile respiratory complications in two unrelated cases. Br J Dermatol, 137(6), 898–906. https://www.ncbi.nlm.nih.gov/pubmed/9470905 [PubMed] [Google Scholar]
- Mihailovska E, Raith M, Valencia RG, Fischer I, Al Banchaabouchi M, Herbst R, & Wiche G (2014). Neuromuscular synapse integrity requires linkage of acetylcholine receptors to postsynaptic intermediate filament networks via rapsyn-plectin 1f complexes. Mol Biol Cell, 25(25), 4130–4149. 10.1091/mbc.E14-06-1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroczek M, Durmus H, Topf A, Parman Y, & Straub V (2020). Four Individuals with a Homozygous Mutation in Exon 1f of the PLEC Gene and Associated Myasthenic Features. Genes (Basel), 11(7). 10.3390/genes11070716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Sawamura D, Goto M, Nakamura H, McMillan JR, Park S, Kono S, Hasegawa S, Paku S, Nakamura T, Ogiso Y, & Shimizu H (2005). Epidermolysis bullosa simplex associated with pyloric atresia is a novel clinical subtype caused by mutations in the plectin gene (PLEC1). J Mol Diagn, 7(1), 28–35. 10.1016/S1525-1578(10)60005-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsuga K, Nishie W, Akiyama M, Nakamura H, Shinkuma S, McMillan JR, Nagasaki A, Has C, Ouchi T, Ishiko A, Hirako Y, Owaribe K, Sawamura D, Bruckner-Tuderman L, & Shimizu H (2010). Plectin expression patterns determine two distinct subtypes of epidermolysis bullosa simplex. Hum Mutat, 31(3), 308–316. 10.1002/humu.21189 [DOI] [PubMed] [Google Scholar]
- Natsuga K, Nishie W, Nishimura M, Shinkuma S, Watanabe M, Izumi K, Nakamura H, Hirako Y, & Shimizu H (2017). Loss of interaction between plectin and type XVII collagen results in epidermolysis bullosa simplex. Hum Mutat, 38(12), 1666–1670. 10.1002/humu.23344 [DOI] [PubMed] [Google Scholar]
- Natsuga K, Nishie W, Shinkuma S, Arita K, Nakamura H, Ohyama M, Osaka H, Kambara T, Hirako Y, & Shimizu H (2010). Plectin deficiency leads to both muscular dystrophy and pyloric atresia in epidermolysis bullosa simplex. Hum Mutat, 31(10), E1687–1698. 10.1002/humu.21330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfendner E, Rouan F, & Uitto J (2005). Progress in epidermolysis bullosa: the phenotypic spectrum of plectin mutations. Exp Dermatol, 14(4), 241–249. 10.1111/j.0906-6705.2005.00324.x [DOI] [PubMed] [Google Scholar]
- Pfendner E, & Uitto J (2005). Plectin gene mutations can cause epidermolysis bullosa with pyloric atresia. J Invest Dermatol, 124(1), 111–115. 10.1111/j.0022-202X.2004.23564.x [DOI] [PubMed] [Google Scholar]
- Potokar M, & Jorgacevski J (2021). Plectin in the Central Nervous System and a Putative Role in Brain Astrocytes. Cells, 10(9). 10.3390/cells10092353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulkkinen L, Smith FJ, Shimizu H, Murata S, Yaoita H, Hachisuka H, Nishikawa T, McLean WH, & Uitto J (1996). Homozygous deletion mutations in the plectin gene (PLEC1) in patients with epidermolysis bullosa simplex associated with late-onset muscular dystrophy. Hum Mol Genet, 5(10), 1539–1546. 10.1093/hmg/5.10.1539 [DOI] [PubMed] [Google Scholar]
- Rezniczek GA, Konieczny P, Nikolic B, Reipert S, Schneller D, Abrahamsberg C, Davies KE, Winder SJ, & Wiche G (2007). Plectin 1f scaffolding at the sarcolemma of dystrophic (mdx) muscle fibers through multiple interactions with beta-dystroglycan. J Cell Biol, 176(7), 965–977. 10.1083/jcb.200604179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouan F, Pulkkinen L, Meneguzzi G, Laforgia S, Hyde P, Kim DU, Richard G, & Uitto J (2000). Epidermolysis bullosa: novel and de novo premature termination codon and deletion mutations in the plectin gene predict late-onset muscular dystrophy. J Invest Dermatol, 114(2), 381–387. 10.1046/j.1523-1747.2000.00880.x [DOI] [PubMed] [Google Scholar]
- Sawamura D, Goto M, Sakai K, Nakamura H, McMillan JR, Akiyama M, Shirado O, Oyama N, Satoh M, Kaneko F, Takahashi T, Konno H, & Shimizu H (2007). Possible involvement of exon 31 alternative splicing in phenotype and severity of epidermolysis bullosa caused by mutations in PLEC1. J Invest Dermatol, 127(6), 1537–1540. 10.1038/sj.jid.5700707 [DOI] [PubMed] [Google Scholar]
- Schara U, Tucke J, Mortier W, Nusslein T, Rouan F, Pfendner E, Zillikens D, Bruckner-Tuderman L, Uitto J, Wiche G, & Schroder R (2004). Severe mucous membrane involvement in epidermolysis bullosa simplex with muscular dystrophy due to a novel plectin gene mutation. Eur J Pediatr, 163(4-5), 218–222. 10.1007/s00431-004-1410-4 [DOI] [PubMed] [Google Scholar]
- Schroder R, Kunz WS, Rouan F, Pfendner E, Tolksdorf K, Kappes-Horn K, Altenschmidt-Mehring M, Knoblich R, van der Ven PF, Reimann J, Furst DO, Blumcke I, Vielhaber S, Zillikens D, Eming S, Klockgether T, Uitto J, Wiche G, & Rolfs A (2002). Disorganization of the desmin cytoskeleton and mitochondrial dysfunction in plectin-related epidermolysis bullosa simplex with muscular dystrophy. J Neuropathol Exp Neurol, 61(6), 520–530. 10.1093/jnen/61.6.520 [DOI] [PubMed] [Google Scholar]
- Selcen D, Juel VC, Hobson-Webb LD, Smith EC, Stickler DE, Bite AV, Ohno K, & Engel AG (2011). Myasthenic syndrome caused by plectinopathy. Neurology, 76(4), 327–336. 10.1212/WNL.0b013e31820882bd [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Masunaga T, Kurihara Y, Owaribe K, Wiche G, Pulkkinen L, Uitto J, & Nishikawa T (1999). Expression of plectin and HD1 epitopes in patients with epidermolysis bullosa simplex associated with muscular dystrophy. Arch Dermatol Res, 291(10), 531–537. 10.1007/s004030050449 [DOI] [PubMed] [Google Scholar]
- Smith FJ, Eady RA, Leigh IM, McMillan JR, Rugg EL, Kelsell DP, Bryant SP, Spurr NK, Geddes JF, Kirtschig G, Milana G, de Bono AG, Owaribe K, Wiche G, Pulkkinen L, Uitto J, McLean WH, & Lane EB (1996). Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat Genet, 13(4), 450–457. 10.1038/ng0896-450 [DOI] [PubMed] [Google Scholar]
- Sonnenberg A, & Liem RK (2007). Plakins in development and disease. Exp Cell Res, 313(10), 2189–2203. 10.1016/j.yexcr.2007.03.039 [DOI] [PubMed] [Google Scholar]
- Steinbock FA, & Wiche G (1999). Plectin: a cytolinker by design. Biol Chem, 380(2), 151–158. 10.1515/BC.1999.023 [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Rouan F, Uitto J, Ishida-Yamamoto A, Iizuka H, Owaribe K, Tanigawa M, Ishii N, Yasumoto S, & Hashimoto T (2005). Plectin deficient epidermolysis bullosa simplex with 27-year-history of muscular dystrophy. J Dermatol Sci, 37(2), 87–93. 10.1016/j.jdermsci.2004.11.003 [DOI] [PubMed] [Google Scholar]
- Takizawa Y, Shimizu H, Rouan F, Kawai M, Udono M, Pulkkinen L, Nishikawa T, & Uitto J (1999). Four novel plectin gene mutations in Japanese patients with epidermolysis bullosa with muscular dystrophy disclosed by heteroduplex scanning and protein truncation tests. J Invest Dermatol, 112(1), 109–112. 10.1046/j.1523-1747.1999.00461.x [DOI] [PubMed] [Google Scholar]
- Thorolfsdottir RB, Sveinbjornsson G, Sulem P, Helgadottir A, Gretarsdottir S, Benonisdottir S, Magnusdottir A, Davidsson OB, Rajamani S, Roden DM, Darbar D, Pedersen TR, Sabatine MS, Jonsdottir I, Arnar DO, Thorsteinsdottir U, Gudbjartsson DF, Holm H, & Stefansson K (2017). A Missense Variant in PLEC Increases Risk of Atrial Fibrillation. J Am Coll Cardiol, 70(17), 2157–2168. 10.1016/j.jacc.2017.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu WT, Chen PC, Hou PC, Huang HY, Wang JY, Chao SC, Lee JY, McGrath JA, Natsuga K, & Hsu CK (2020). Plectin Missense Mutation p.Leu319Pro in the Pathogenesis of Autosomal Recessive Epidermolysis Bullosa Simplex. Acta Derm Venereol, 100(15), adv00242. 10.2340/00015555-3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitto J (2004). Compound heterozygosity of unique in-frame insertion and deletion mutations in the plectin gene in a mild case of epidermolysis bullosa with very late onset muscular dystrophy. J Invest Dermatol, 122, A86. [DOI] [PubMed] [Google Scholar]
- Uitto J, Saeidian AH, Youssefian L, & Vahidnezhad H (2021). Interpretation of genomic sequence variants in heritable skin diseases: A primer for clinicians. J Am Acad Dermatol. 10.1016/j.jaad.2021.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Daneshpazhooh M, Mahmoudi H, Kariminejad A, Fischer J, Christiansen J, Schneider H, Guy A, Liu L, McGrath JA, Has C, & Uitto J (2019). Biallelic KRT5 mutations in autosomal recessive epidermolysis bullosa simplex, including a complete human keratin 5 “knock-out”. Matrix Biol, 83, 48–59. 10.1016/j.matbio.2019.07.002 [DOI] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Saeidian AH, Mahmoudi H, Touati A, Abiri M, Kajbafzadeh AM, Aristodemou S, Liu L, McGrath JA, Ertel A, Londin E, Kariminejad A, Zeinali S, Fortina P, & Uitto J (2018). Recessive mutation in tetraspanin CD151 causes Kindler syndrome-like epidermolysis bullosa with multi-systemic manifestations including nephropathy. Matrix Biol, 66, 22–33. 10.1016/j.matbio.2017.11.003 [DOI] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Saeidian AH, Mozafari N, Barzegar M, Sotoudeh S, Daneshpazhooh M, Isaian A, Zeinali S, & Uitto J (2016). KRT5 and KRT14 Mutations in Epidermolysis Bullosa Simplex with Phenotypic Heterogeneity, and Evidence of Semidominant Inheritance in a Multiplex Family. J Invest Dermatol, 136(9), 1897–1901. 10.1016/j.jid.2016.05.106 [DOI] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Saeidian AH, Touati A, Pajouhanfar S, Baghdadi T, Shadmehri AA, Giunta C, Kraenzlin M, Syx D, Malfait F, Has C, Lwin SM, Karamzadeh R, Liu L, Guy A, Hamid M, Kariminejad A, Zeinali S, McGrath JA, & Uitto J (2019). Mutations in PLOD3, encoding lysyl hydroxylase 3, cause a complex connective tissue disorder including recessive dystrophic epidermolysis bullosa-like blistering phenotype with abnormal anchoring fibrils and type VII collagen deficiency. Matrix Biol, 81, 91–106. 10.1016/j.matbio.2018.11.006 [DOI] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Saeidian AH, Touati A, Sotoudeh S, Abiri M, Barzegar M, Aghazadeh N, Mahmoudi H, Norouz-Zadeh S, Hamid M, Zahabiyon M, Bagherian H, Zeinali S, Fortina P, & Uitto J (2017). Multigene Next-Generation Sequencing Panel Identifies Pathogenic Variants in Patients with Unknown Subtype of Epidermolysis Bullosa: Subclassification with Prognostic Implications. J Invest Dermatol, 137(12), 2649–2652. 10.1016/j.jid.2017.07.830 [DOI] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Saeidian AH, Touati A, Sotoudeh S, Jazayeri A, Guy A, Lovell PA, Liu L, Kariminejad A, McGrath JA, Zeinali S, & Uitto J (2018). Next generation sequencing identifies double homozygous mutations in two distinct genes (EXPH5 and COL17A1) in a patient with concomitant simplex and junctional epidermolysis bullosa. Hum Mutat, 39(10), 1349–1354. 10.1002/humu.23592 [DOI] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Saeidian AH, & Uitto J (2019). Phenotypic Spectrum of Epidermolysis Bullosa: The Paradigm of Syndromic versus Non-Syndromic Skin Fragility Disorders. J Invest Dermatol, 139(3), 522–527. 10.1016/j.jid.2018.10.017 [DOI] [PubMed] [Google Scholar]
- Vahidnezhad H, Youssefian L, Saeidian AH, Zeinali S, Touati A, Abiri M, Sotoudeh S, Norouz-Zadeh S, Amirinezhad N, Mozafari N, Daneshpazhooh M, Mahmoudi H, Hamid M, Bradfield JP, Kim CE, Hakonarson H, & Uitto J (2019). Genome-wide single nucleotide polymorphism-based autozygosity mapping facilitates identification of mutations in consanguineous families with epidermolysis bullosa. Exp Dermatol, 28(10), 1118–1121. 10.1111/exd.13501 [DOI] [PubMed] [Google Scholar]
- Valari M, Theodoraki M, Loukas I, Gkantseva-Patsoura S, Karavana G, Falaina V, Lykopoulou L, Pons R, Athanasiou I, Wertheim-Tysarowska K, Kanaka-Gantenbein C, & Kiritsi D (2019). Novel PLEC Variant Causes Mild Skin Fragility, Pyloric Atresia, Muscular Dystrophy and Urological Manifestations. Acta Derm Venereol, 99(13), 1309–1310. 10.2340/00015555-3317 [DOI] [PubMed] [Google Scholar]
- Villa CR, Ryan TD, Collins JJ, Taylor MD, Lucky AW, & Jefferies JL (2015). Left ventricular non-compaction cardiomyopathy associated with epidermolysis bullosa simplex with muscular dystrophy and PLEC1 mutation. Neuromuscul Disord, 25(2), 165–168. 10.1016/j.nmd.2014.09.011 [DOI] [PubMed] [Google Scholar]
- Walker GD, Woody M, Orrin E, Mellerio JE, & Levy ML (2017). Epidermolysis Bullosa with Pyloric Atresia and Significant Urologic Involvement. Pediatr Dermatol, 34(1), e61–e64. 10.1111/pde.13026 [DOI] [PubMed] [Google Scholar]
- Walko G, Vukasinovic N, Gross K, Fischer I, Sibitz S, Fuchs P, Reipert S, Jungwirth U, Berger W, Salzer U, Carugo O, Castanon MJ, & Wiche G (2011). Targeted proteolysis of plectin isoform 1a accounts for hemidesmosome dysfunction in mice mimicking the dominant skin blistering disease EBS-Ogna. PLoS Genet, 7(12), e1002396. 10.1371/journal.pgen.1002396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter MC, Reilich P, Krause S, Hiebeler M, Gehling S, Goebel HH, Schoser B, & Abicht A (2021). Congenital myopathy and epidermolysis bullosa due to PLEC variant. Neuromuscul Disord, 31(11), 1212–1217. 10.1016/j.nmd.2021.09.009 [DOI] [PubMed] [Google Scholar]
- Wiche G, & Winter L (2011). Plectin isoforms as organizers of intermediate filament cytoarchitecture. Bioarchitecture, 1(1), 14–20. 10.4161/bioa.1.1.14630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter L, Turk M, Harter PN, Mittelbronn M, Kornblum C, Norwood F, Jungbluth H, Thiel CT, Schlotzer-Schrehardt U, & Schroder R (2016). Downstream effects of plectin mutations in epidermolysis bullosa simplex with muscular dystrophy. Acta Neuropathol Commun, 4(1), 44. 10.1186/s40478-016-0314-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Ren Y, Lin Z, Wang H, Zhou Y, & Yang Y (2015). Compound heterozygous PLEC mutations in a patient of consanguineous parentage with epidermolysis bullosa simplex with muscular dystrophy and diffuse alopecia. Int J Dermatol, 54(2), 185–187. 10.1111/ijd.12655 [DOI] [PubMed] [Google Scholar]
- Yiu EM, Klausegger A, Waddell LB, Grasern N, Lloyd L, Tran K, North KN, Bauer JW, McKelvie P, Chow CW, Ryan MM, & Murrell DF (2011). Epidermolysis bullosa with late-onset muscular dystrophy and plectin deficiency. Muscle Nerve, 44(1), 135–141. 10.1002/mus.22076 [DOI] [PubMed] [Google Scholar]
- Youssefian L, Saeidian AH, Palizban F, Bagherieh A, Abdollahimajd F, Sotoudeh S, Mozafari N, Farahani RA, Mahmoudi H, Babashah S, Zabihi M, Zeinali S, Fortina P, Salas-Alanis JC, South AP, Vahidnezhad H, & Uitto J (2021). Whole-Transcriptome Analysis by RNA Sequencing for Genetic Diagnosis of Mendelian Skin Disorders in the Context of Consanguinity. Clin Chem, 67(6), 876–888. 10.1093/clinchem/hvab042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wang Z, Mi Z, Sun L, Fu X, Yu G, Pang Z, Liu H, & Zhang F (2021). Epidermolysis Bullosa in Chinese Patients: Genetic Analysis and Mutation Landscape in 57 Pedigrees and Sporadic Cases. Acta Derm Venereol, 101(7), adv00503. 10.2340/00015555-3843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Chen G, Dang Y, Liao H, Zhang J, & Lan D (2017). Novel compound heterozygous PLEC mutations lead to earlyonset limbgirdle muscular dystrophy 2Q. Mol Med Rep, 15(5), 2760–2764. 10.3892/mmr.2017.6309 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data associated with this study are presented in the paper. All novel variants have been submitted to the LOVD database (https://www.lovd.nl)