

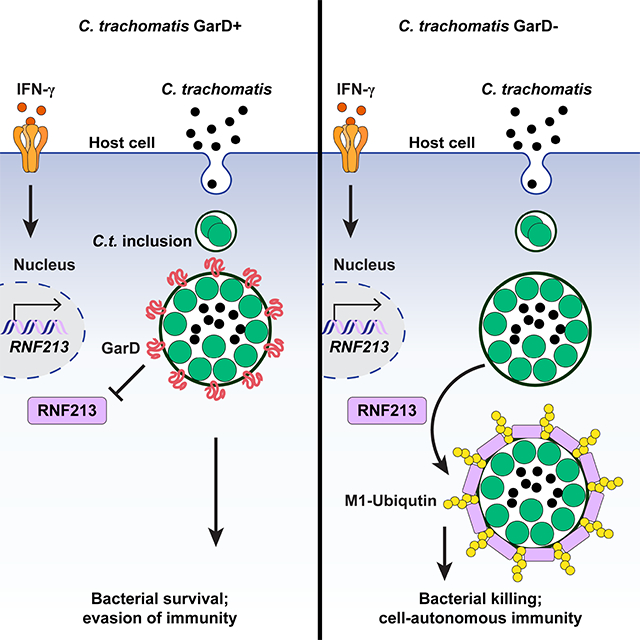
SUMMARY
Chlamydia trachomatis is the leading cause of sexually transmitted bacterial infections and a major threat to women’s reproductive health in particular. This obligate intracellular pathogen resides and replicates within a cellular compartment termed an inclusion, where it is sheltered by unknown mechanisms from gamma-Interferon (IFNγ)-induced cell-autonomous host immunity. Through a genetic screen we uncovered the Chlamydia inclusion membrane protein GarD (gamma resistance determinant) as a bacterial factor protecting inclusions from cell-autonomous immunity. In IFNγ-primed human cells, inclusions formed by garD loss-of-function mutants become decorated with linear ubiquitin and are eliminated. Leveraging cellular genome-wide association data, we identified the ubiquitin E3 ligase RNF213 as a candidate anti-Chlamydia protein. We demonstrate that IFNγ-inducible RNF213 facilitates the ubiquitylation and destruction of GarD-deficient inclusions. Furthermore, we show GarD operates as a cis-acting stealth factor barring RNF213 from targeting inclusions, thus functionally defining GarD as an RNF213 antagonist essential for chlamydial growth during IFNγ-stimulated immunity.
Graphical Abstract
eTOC BLURB
Chlamydia trachomatis causes widespread bacterial infection, yet the components underlying its pathogenicity are not fully understood. Walsh et. al. describe a new host-pathogen axis involving the bacterial effector GarD as a key to avoid immune detection and destruction driven by the human protein RNF213.
INTRODUCTION
Many intracellular bacterial pathogens establish customized vacuolar compartments for residence. These pathogen-containing vacuoles (PVs) not only allow for bacterial replication but also protect their microbial inhabitants from cytosolic defense programs (Anand et al., 2020; Omotade and Roy, 2019). To combat infections with vacuolar pathogens, the mammalian innate immune system evolved immune surveillance strategies that detect molecular patterns associated with PVs (Coers, 2013; Liehl et al., 2015). Following their immune detection, PVs are contained or eliminated by xenophagy or related antimicrobial programs, processes often dependent on host-driven PV ubiquitylation (Deretic, 2021; Huang and Brumell, 2014; Tripathi-Giesgen et al., 2021). However, the full repertoire of PV-ubiquitylating enzymes is not established, and whether microbial virulence factors exist that inhibit the function of specific PV-ubiquitylating enzymes is unknown.
Chlamydia trachomatis is a clinically important pathogen that resides within PVs termed inclusions. This human-adapted obligate intracellular pathogen is responsible for the most common sexually transmitted bacterial infection, which is linked to pelvic inflammatory disease, ectopic pregnancy, and female infertility (Brunham and Paavonen, 2020; Elwell et al., 2016). A central player in anti-Chlamydia host defense is the lymphocyte-derived cytokine IFNγ (Dockterman and Coers, 2021; Finethy and Coers, 2016), a potent inducer of cell-autonomous immunity. IFNγ priming of mammalian cells results in the ubiquitylation and destruction of inclusions formed by non-host-adapted Chlamydia species (Haldar et al., 2015). However, in its adapted human host, C. trachomatis is resistant to IFNγ-induced cell-autonomous immunity (Haldar et al., 2016), suggesting that C. trachomatis employs virulence factors to block IFNγ-induced inclusion ubiquitylation in human cells. To identify such bacterial factors, we conducted a genetic screen for C. trachomatis mutants unable to grow inside IFNγ-primed human cells and identified the inclusion membrane protein CTL0390/ GarD (gamma resistance determinant), as a mediator of bacterial resistance to IFNγ-induced cell-autonomous immunity. Additionally, we demonstrate that the IFNγ-inducible human ubiquitin E3 ligase RNF213 targets and ubiquitylates GarD-devoid inclusions, and that growth of garD mutants is restored in RNF213-deficient human cells. Therefore, this potent RNF213-orchestrated antibacterial response is evaded by Chlamydia through GarD, implicating its central role in the ability of C. trachomatis to withstand IFNγ-mediated human immunity.
RESULTS
A genetic screen identifies C. trachomatis mutants attenuated for growth inside IFNγ-primed human epithelial cells
Whereas rodent-adapted Chlamydia muridarum is susceptible to IFNγ-induced cell-autonomous immunity in human epithelial cells, the human pathogen C. trachomatis is resistant ((Haldar et al., 2016) and Fig. 1A), suggesting the existence of C. trachomatis virulence factors that subvert the function of IFNγ-inducible human antimicrobials. To identify bacterial IFNγ resistance determinants through an unbiased approach, we screened a previously engineered arrayed library of chemically mutagenized C. trachomatis serovar L2 variants (Kokes et al., 2015; Sixt et al., 2017) for mutants specifically defective for growth inside IFNγ-primed human A549 epithelial cells (Fig. 1B). For each arrayed mutant strain, we determined its infectivity ratio (IR) defined as its relative growth in IFNγ-primed cells over its growth in naïve cells. Bacterial growth was determined by quantifying absolute numbers of inclusions (#inc) or percentage of infected cells (%inf) using a high content analysis platform. Thirty-one out of 1272 mutants displayed a Z-score of −2 or less for IR (#inc), or IR (%inf), or an average score of the two (Figs. 1C and S1A) and were classified as statistical outliers or ‘hits.’ Our analysis failed to identify any mutants displaying notable IFNγ-induced changes in inclusion size (area) or shape (curvature) (Fig. S1B), suggesting that our screen exclusively identified mutants susceptible to IFNγ-driven inclusion elimination.
Figure 1. A genetic screen identifies C. trachomatis L2 CTL0390 mutants attenuated for growth inside IFNγ-primed cells.

(A) C. trachomatis (C.t, gray.) serovar L2 and C. muridarum (C.m., blue) burden in A549 cells infected at a multiplicity of infection (MOI) of 2 was measured by high-content imaging at 24 hours post-infection (hpi). Cells were primed with indicated concentration of IFNγ overnight. Data show the mean ± S.D. of three independent experiments. (B) Schematic of screen design using chemically mutagenized C. t. serovar L2 (CTL2M) library. (C) Dot blot depicting infectivity ratio (IR) (#inc), and IR (%inf) Z-score of 1272 screened mutants. Purple dots represent 27 screen hits and green dots represent 4 additional hits harboring SNVs in CTL0390. (D) Secondary validation assay (n = 2 independent experiments) showing IR values for one plaque-purified clonal isolate per each of 14 selected screen hits and two randomly selected CTL2M clones (beige dots). Asterisks mark clones with IRs statistically different from parental C.t. RifR strain. (E) Schematic diagram of domain structure and AlphaFold structure prediction of CTL0390 depicting mutations found in four of the 14 sequenced hits. Insert with predicted membrane topology model. Statistical significance was evaluated by 2-way ANOVA followed by Turkey’s multiple comparison test (A) or 1- way ANOVA followed by Dunnett’s multiple comparison test (D). *= p <0.05, **= p <0.01, *** = p <0.005, **** = p <0.0001; n.s. = not significant.
To identify causative mutations, we selected 14 of the 31 outliers for whole genome sequencing. Sequencing indicated that some of the 14 strains consisted of two or more genetically distinct clones. Because phenotypically wild-type (WT) clonal contaminants could mask the severity of mutant phenotypes, we plaque purified the 14 sequenced hits and phenotypically analyzed 3 to 5 isolates per strain. As predicted, several plaque-purified isolates displayed more severe IFNγ susceptibility phenotypes than their corresponding polyclonal library strains, and in total we found that plaque-purified isolates from 11 out of 14 library hits were significantly more susceptible to IFNγ-mediated host defense than their parental WT strain (RifR, Fig. 1D). Notably, two mutants were as attenuated for growth inside IFNγ-primed A549 cells as C. muridarum (Fig. 1D), a Chlamydia species considered avirulent in humans.
Non-synonymous and nonsense mutations in CTL0390 are associated with IFNγ susceptibility
Library mutants contain an average of approximately 20 chromosomal mutations per strain (Kokes et al., 2015; Sixt et al., 2017). In accordance with the general composition of the library, sequencing of 14 selected outlier strains identified a total of 279 single nucleotide variants (SNVs), composed of 64.9% non-synonymous, 24.0% synonymous, 9.3% non-coding, and 1.8% nonsense mutations (Fig. S1C). To prioritize mutated ORFs for further investigation, we ranked ORFs based on their aggregate number of SNVs within the population of the 14 sequenced strains. Most ORFs contained only a single SNV and only two ORFs, CTL0390 and CTL0859, contained four SNVs (Fig. S1D). In addition to SNV frequency we considered the likelihood of any given SNV to impact gene function. Although all four CTL0859 SNVs resulted in amino acid substitutions, three of the four CTL0859 SNVs were classified functionally neutral by SNAP2 analysis (Hecht et al., 2015) (Fig. S1E), and are therefore less likely to be causative mutations. All four CTL0390 SNVs on the other hand were predicted to impact protein function and included a nonsense mutation truncating CTL0390 protein at amino acid 150 (Fig. S1E). As confirmed by Sanger sequencing, the four CTL0390 SNVs were distributed across four distinct strains, which included the top two confirmed hits with the most pronounced IFNγ susceptibility phenotypes (Fig. 1D).
To further evaluate whether the three non-synonymous CTL0390 mutations were likely to interfere with protein function, we applied protein membrane topology programs and the machine-learning based AlphaFold platform (Jumper et al., 2021a) to predict the topology and 3D structure of CTL0390 (Fig. 1E and S1F). As predicted previously (Dehoux et al., 2011), CTL0390 contains a bilobed transmembrane domain consisting of two hydrophobic domains (HD3 and HD4) separated by a short linker (Fig. 1E), classifying CTL0390 as a member of the Chlamydia inclusion membrane (Inc) protein family (Bannantine et al., 2000). Notably, the G253D mutation localizes to HD4 of the bilobed transmembrane domain. Our screen retrieved no variants containing mutations in the cytosolic C-terminal portion of CTL0390. Instead, we obtained two mutations, T102A and G136E, mapping to a predicted cleft formed between the helical HD2 and an adjacent α-helix connected to HD2 by a positively charged loop. While determination of the exact membrane topology awaits experimental evaluation, we favor a model in which the HD2-containing protein cleft is located on the cytosolic face of the inclusion membrane (Fig. 1E), possibly interacting with inclusion membrane components. Overall, all identified CTL0390 mutations were predicted to impact protein function, which rendered CTL0390 a strong candidate for further study.
Insertional inactivation of GarD (CTL0390) renders C. trachomatis susceptible to IFNγ-primed host defense
In agreement with a previous report showing the subcellular localization of epitope-tagged CTL0390 (Weber et al., 2015), a customized anti-CTL0390 antibody detected protein localization to inclusion membranes (Fig. 2A). As expected, no inclusion membrane staining was observed for the S150* nonsense mutant lacking the C-terminal epitope recognized by anti-CTL0390 (Fig. S1G), thereby confirming the specificity of the anti-CTL0390 antibody. Monitoring the impact of the three recovered non-synonymous mutations on CTL0390 expression, we found that the frequency of inclusion staining was diminished to different degrees for the T102A and G136E mutants but was comparable to WT for the G253D mutant (Fig. S1G), suggesting that the latter mutation impacts protein function without affecting its stability or proper localization.
Figure 2. Inactivation of Inc protein GarD (CTL0390) renders C. trachomatis susceptible to IFNγ-primed host defense.
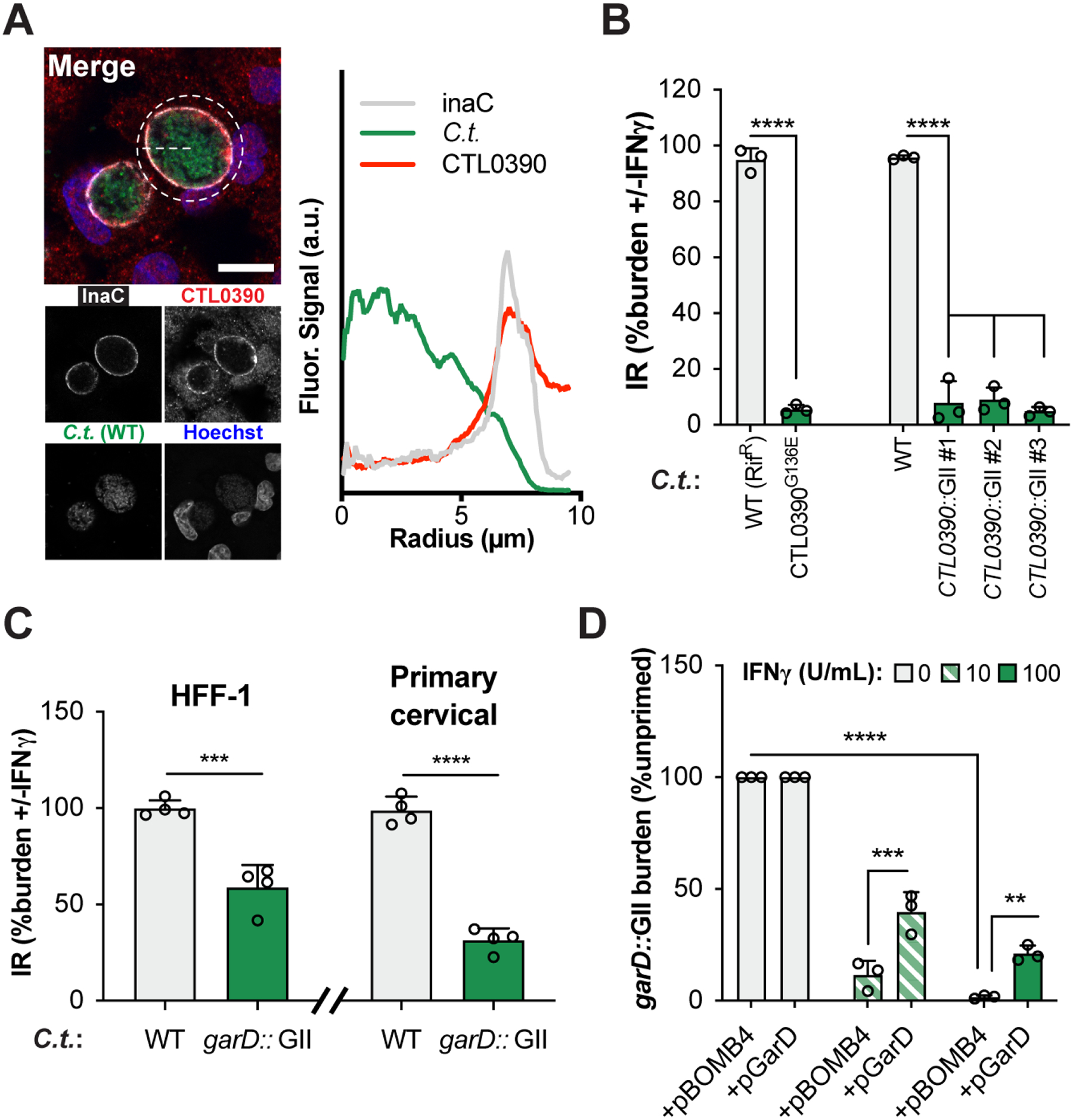
(A) Immunostaining of endogenous GarD and radial line tracing demonstrates its colocalization with inclusion membrane marker InaC in naïve A549 cells at 20 hpi. Scale bar = 10 μm. (B) IR values reflecting IFNγ-mediated suppression of bacterial growth are shown for garD mutant CTL0390G136E, three independent GII mutants (CTL0390::GII), and their corresponding WT controls. CTL0390::GII#1 was renamed garD::GII and selected for subsequent studies. (C) IFNγ-mediated growth restriction of WT and garD::GII C.t. serovar L2 was measured in HFF-1 and primary cervical epithelial cells using Cellomics high content imaging. (D) Complementation of CTL0390::GII partially restores bacterial growth inside IFNγ-primed A549 cells. Data show the means ± S.D. of three independent experiments. 1-way (B) or 2-way ANOVA (D) followed by Tukey’s multiple comparison test, or an unpaired t-test (C) were performed to assess statistical significance. **= p <0.01, *** = p <0.005, **** = p <0.0001; a.u. = arbitrary units.
To directly explore the potential anti-immunity function of CTL0390, we generated CTL0390-deficient strains on a WT genetic background using group II intron (GII) insertional inactivation (Fig. S2A–B). Three independent CTL0390-deficient strains were severely compromised for growth inside IFNγ-primed A549 cells, comparable to screen mutant hit G136E (Fig. 2B). Therefore, CTL0390 is essential for escaping IFNγ-activated cell-autonomous immunity and was renamed gamma resistance determinant (GarD). Restriction of garD::GII growth was also observed in IFNγ-primed human foreskin fibroblasts (HFF-1) and primary cervical epithelial cells (Fig. 2C), expanding the role for GarD in escaping IFNγ-triggered host defense to additional cell types including those relevant to human infections.
As expected, anti-GarD antibody failed to immunostain garD::GII inclusion membranes (Fig. S2C), further confirming the successful disruption of the garD locus. Ectopic expression of GarD in the mutant background restored anti-GarD staining at the inclusion membrane (Fig. S2C), but also resulted in deformed inclusions (Fig. S2D) and enhanced host cell death (Fig. S2E), indicative of inclusion rupture (Giebel et al., 2019; Weber et al., 2017). Toxicity resulting from GarD overexpression, as also observed by others (Bishop and Derré, 2022; Yang et al., 2021), was reflected in the rapid loss of a GarD expression plasmid in the absence of positive antibiotic marker selection (Fig. S2D). Endogenous expression of GarD, on the other hand, had no detrimental effects on bacterial fitness, as evidenced by the fact that garD::GII produced infectious progeny at levels comparable to WT bacteria in unprimed Vero cells (Fig. S2F), which are defective for autocrine type I IFN signaling (Desmyter et al., 1968). Despite the reduced bacterial fitness caused by ectopic GarD expression, complementation of garD::GII partially restored bacterial growth in IFNγ-primed A549 cells (Fig. 2D), confirming that loss of GarD expression is responsible for the IFNγ susceptibility of garD::GII mutants.
GarD blocks inclusion ubiquitylation in cis
C. trachomatis employs an unknown virulence factor to block IFNγ-induced inclusion ubiquitylation (Haldar et al., 2016). Our discovery of GarD as a potent inhibitor of IFNγ-induced cell-autonomous immunity led us to hypothesize that GarD could function as an inclusion ubiquitylation antagonist. To test this hypothesis, we primed cells with IFNγ at 3 hours post-infection (hpi) and immunostained at 20 hpi, a protocol optimized to visualize the dynamic delivery of host defense proteins to inclusions in human epithelial cells (Haldar et al., 2016). We found that approximately half of all garD::GII inclusions were decorated with ubiquitin in IFNγ-primed cells, whereas inclusions harboring WT or the complemented garD::GII mutant bacteria remained devoid of ubiquitin (Fig. 3A). An antibody specific for linear (M1-linked) ubiquitin (Noad et al., 2017) co-stained with the pan-ubiquitin antibody FK2, marking about half of all inclusions at 20 hpi (Fig. 3B). K48- or K63-linked ubiquitin on the other hand was not detectable (Fig. 3B), suggesting that M1-linked ubiquitin is the main ubiquitin species delivered to garD::GII inclusions. Linear ubiquitylation of inclusions and anti-Chlamydia host defense occurred independent of the ubiquitin E3 ligases HOIL-1 or HOIP (Fig. S3A–C), two essential components of the linear ubiquitin assembly complex (LUBAC) (Dittmar and Winklhofer, 2019), indicating the existence of a non-canonical pathway of M1-linked ubiquitylation at the inclusion membrane.
Figure 3. GarD acts in cis to protect C. trachomatis inclusions from ubiquitylation.

Percentages of inclusions positive for (A) total (FK2) or (B) K48-, K63-, or M-linked (linear) ubiquitin assessed by immunofluorescence in IFNγ-primed (100U/mL) and naïve A549s at 20 hours post infection (hpi). (C) Co-infection of IFNγ-primed (100U/mL) A549s with indicated C.t. strains to distinguish between trans- and cis-acting activity of GarD in blocking inclusion ubiquitylation at 20hpi. Representative confocal images show inclusions within IFNγ-primed cells. Scale bars = 10 μm. Diagram created using BioRender. Data show the means ± S.D. of three independent experiments. 2-way ANOVA followed by Tukey’s multiple comparison test was performed. *= p <0.05, **** = p <0.0001; n.s. = not significant.
Next, we asked whether GarD was executing its anti-ubiquitylation activity in cis, i.e. directly at the inclusion membrane on which it resides. To answer this question, we capitalized on the ability of C. trachomatis inclusions to fuse with each other, a process that requires the bacterial IncA protein (Hackstadt et al., 1999). Co-infections of garD mutants with WT C. trachomatis resulted in fused inclusions containing a mix of WT and garD mutant bacteria and an accompanying reduction in inclusion ubiquitylation (Fig. 3C). Inclusions formed by garD::GII in cells co-infected with fusion-deficient incA−, on the other hand, continued to be ubiquitylated at high frequency (Fig. 3C). Collectively, these data demonstrated that GarD operates in cis, shielding inclusion membranes from the activity of an unknown ubiquitin E3 ligase.
GarD mutant inclusions become decorated with ubiquitin adaptors and delivered into LAMP1+ compartments
Host cells can eliminate intracellular pathogens through autophagy-related xenophagy involving adaptor proteins capturing ubiquitylated microbes for lysosomal delivery (Deretic, 2021; Tripathi-Giesgen et al., 2021). Indicating that a xenophagy-like process could be involved in garD mutant destruction, we observed pronounced recruitment of the high affinity M1-linked ubiquitin-binding adaptor proteins OPTN, NDP52, and TAX1BP1 (Hu et al., 2018; Miyashita et al., 2021; Nakazawa et al., 2016; Tumbarello et al., 2015) to garD::GII inclusions, whereas WT and complemented garD::GII inclusions remained largely devoid of these markers (Fig. 4A). The additional ubiquitin adaptors p62 and NBR1 also localized to garD::GII but not WT inclusions (Fig. S4), albeit less frequently than the M1-linked ubiquitin adaptors. Similarly, the autophagosome-associated ubiquitin-like proteins LC3 and GABARAPs, and lysosomal LAMP1 co-localized with garD::GII, but not WT or complemented garD::GII inclusions upon IFNγ-priming (Fig. 4B–C). Although the specific role of each individual ubiquitin adaptor in anti-Chlamydia host defense will need to be investigated further, these data establish that GarD prevents the recruitment of autophagy adaptors to inclusions and blocks the delivery of C. trachomatis into LAMP1+ compartments.
Figure 4. GarD-deficient inclusions become decorated with ubiquitin adaptor proteins and delivered into LC3+/GABARAPs+ and LAMP1+ compartments.

Immunofluorescence-based quantification of inclusions decorated with (A) ubiquitin-binding proteins OPTN, NDP52, or TAX1BP1, (B) ubiquitin-like LC3 and GABARAP family proteins, and (C) lysosomal LAMP1 in naïve and IFNγ-primed (100U/mL) A549 cells at 20 hours post infection (hpi). Representative confocal images show inclusions in IFNγ-primed cells. Scale bars = 5 μm. Data show the means ± S.D. of at least three independent experiments. 2-way ANOVA followed by Tukey’s multiple comparison test was performed. *= p <0.05, **** = p <0.0001; n.s. = not significant.
Human genetic diversity reveals RNF213 as candidate anti-Chlamydia E3-ubiquitin ligase
Our results demonstrating GarD-mediated inhibition of inclusion ubiquitylation implies the presence of a host E3-ubiquitin ligase that targets C. trachomatis for destruction. Leveraging natural human genetic diversity revealed RNF213 as a highly plausible candidate for carrying out this role. We used a previously published cellular genome-wide association study of C. trachomatis infection (H2P2; (Wang et al., 2018)) to screen for natural human genetic differences in E3 ubiquitin ligase genes that are associated with intracellular C. trachomatis burden. In this study, 528 lymphoblastoid cell lines (LCLs) from diverse individuals were assayed for susceptibility to infection with GFP-tagged C. trachomatis by measuring GFP median intensity in infected cells. A stratified QQ plot (Schork et al., 2013), focusing our genetic search space to single nucleotide polymorphisms (SNPs) in 363 annotated E3 ubiquitin ligase genes in the human genome (Medvar et al., 2016), revealed no inflation of the test statistic (lambda = 1.0047) but two SNPs with p-values lower than expected by chance (Fig. 5A). Both rs12051852 and rs12051748 fall within an intron of the RNF213 gene (Fig. 5B). The T allele for rs12051852 is associated with lower median GFP fluorescence, suggesting greater C. trachomatis restriction (p = 2.4 ×10−7 by QFAM-parents family-based association test in PLINK) (Fig. 5C). This relationship (T < G for median fluorescence) was observed with all LCLs combined but also when each of the four human populations used in H2P2 were examined separately (Fig. S5). Thus, out of 175589 SNPs in E3 ubiquitin ligase genes, only two SNPs in RNF213 showed an association with C. trachomatis restriction at significance more than expected by chance, although we do not know if and how these SNPs might impact RNF213 function.
Figure 5. Human genetic diversity identifies RNF213 as candidate anti-Chlamydia E3-ubiquitin ligase.

(A) Stratified QQ plots examining SNPs associated with median C. trachomatis serovar L2 GFP fluorescence at 27 hpi. Empirical P-values were calculated from family-based association analysis using QFAM-parents in PLINK. (B) Regional Manhattan plot around rs12051852 demonstrates that SNPs associated with C. trachomatis burden overlap only the RNF213 gene. SNPs are plotted by position on chromosome 17 and -log(p-value) and color-coded by r2 value to rs12051852 from 1000 Genomes European data. (C) Median C. trachomatis GFP fluorescence in infected cells at 27 hpi plotted by rs12051852 genotype. Each dot represents a single lymphoblastoid cell line (LCL), averaged between three independent experiments. Line marks the median and box indicates the first and third quartiles.
RNF213 translocates to and ubiquitylates garD::GII inclusions
Robust inclusion ubiquitylation requires IFNγ priming, suggesting that the responsible E3 ligase itself may be an IFNγ-inducible protein. In agreement with this hypothesis and the H2P2-based prediction that RNF213 exerts an anti-Chlamydia activity, we detected IFNγ-inducible RNF213 protein expression in infected and uninfected A549, primary cervical epithelial and HFF-1 cells (Fig. S6A) and, importantly, observed frequent targeting of RNF213 to garD::GII but not WT or complemented inclusions in all three cell lines (Figs. 6A–B). Nearly all ubiquitin+ garD::GII inclusions co-localized with RNF213 (Figs. 6A–B), further implicating RNF213 in inclusion ubiquitylation. RNF213 also decorated inclusions formed by a garD (CT135) loss-of-function mutant in C. trachomatis serovar D (Fig. S6B), pointing to a conserved function for GarD across chlamydial subspecies. To further interrogate the role of RNF213 in anti-Chlamydia host defense, we generated three independent pools of RNF213-deficient (KO) A549 cells (Fig. S6C). Loss of RNF213 eliminated inclusion ubiquitylation, labeling of inclusions with linear ubiquitin, and the targeting of the ubiquitin binding protein OPTN, NDP52, and TAX1BP1 to garD::GII inclusions (Figs. 6C–E and S6D–F), thus defining RNF213 as the critical ubiquitin E3 ligase orchestrating IFNγ-inducible inclusion ubiquitylation.
Figure 6. GarD protects inclusions against attack by the anti-Chlamydia host ubiquitin E3 ligase RNF213.

Immunostaining demonstrates co-localization of RNF213 with ubiquitin on garD::GII inclusions in (A) A549, (B) HFF-1 and primary cervical epithelial cells at 20 hours post infection (hpi). In (A), radial line tracing of fluorescent intensity is shown for RNF213 and ubiquitin. (C-E) Co-localization with ubiquitin in IFNγ-primed and naïve A549 cells with the adaptor proteins (C) OPTN, (D) NDP52, and (E) TAX1BP1. (F) RNF213 localization to inclusions formed during co-infection of IFNγ-primed and naive A549 cells with garD::GII and non-fusogenic incA− or fusogenic WT at 20hpi. (G) IR for WT and garD::GII C.t. in WT and three independent CRISPR-generated RNF213 KO pools at 24 hpi. Representative confocal images show inclusions within IFNγ-primed cells. Scale bars = 10 μm. All data depict the mean ± S.D. from at least three independent experiments. 2-way ANOVA followed by Tukey’s multiple comparison test was used to determine significance. *= p <0.05, **= p <0.01, **** = p <0.0001; n.s. = not significant. (A-E) Statistical comparisons shown for “Both” groups (orange bars).
GarD protects inclusions against RNF213-executed cell-autonomous immunity
GarD operates in cis to protect inclusions from ubiquitylation (Fig. 3C). In a similarly designed experiment, we demonstrated that inclusion membrane-resident GarD prevents RNF213 from docking on inclusion membranes (Fig. 6F), supporting a model in which GarD obscures an inclusion-associated molecular pattern recognized by RNF213. Because RNF213 operates as a sensor of ISGylated proteins (Thery et al., 2021), we investigated the role of ISGylation in RNF213-dependent destruction of garD::GII mutants. We found that ISGylation was dispensable for anti-Chlamydia host defense and the translocation of RNF213 to C. trachomatis (Fig. S6G–I), suggesting that RNF213 detects a novel molecular pattern associated with C. trachomatis inclusions. Lastly, we observed that growth of garD::GII serovar L2 as well as garD− (CT135−) serovar D under IFNγ priming conditions was restored in RNF213-deficient cells (Figs. 6G and S6J), illustrating that GarD specifically interferes with RNF213-mediated host defense to subvert human immunity.
DISCUSSION
C. trachomatis is a stealth pathogen that can establish persistent human infections despite a robust IFNγ-skewed type 1 immune response (Labuda and McSorley, 2018; Poston and Darville, 2018; Wong et al., 2019). C. trachomatis salvages genital microbiome-derived indole to subdue IFNγ-induced nutritional immunity (Aiyar et al., 2014), but other mechanisms by which C. trachomatis overcomes IFNγ-mediated host defense are unknown. Here, we show that the secreted bacterial effector GarD safeguards C. trachomatis from IFNγ-driven inclusion ubiquitylation and associated cell-autonomous immunity. We identify RNF213 as the human ubiquitin E3 ligase responsible for inclusion ubiquitylation and demonstrate that garD mutant growth is restored in RNF213-deficient epithelial cells. Overall, our study defines GarD as an RNF213 antagonist obstructing cell-autonomous immunity in human cells.
RNF213, also known as mysterin, is a giant ubiquitin E3 ligase associated with Moyamoya disease, a progressive cerebrovascular disorder of unknown etiology (Ahel et al., 2020; Liu et al., 2011). Recently, RNF213 was also implicated in host defense to bacterial and viral pathogens (Houzelstein et al., 2021; Martina et al., 2021; Otten et al., 2021; Thery et al., 2021). RNF213 directly associates with cytosolic bacterial pathogens, suggesting it detects surface-exposed bacterial molecules. Supporting this hypothesis, RNF213 binds to and ubiquitylates bacterial lipid A (Otten et al., 2021). Because lipid A is an outer membrane component unique to Gram-negative bacteria, RNF213 translocation to cytosolic Gram-positive bacteria (Thery et al., 2021) or to PV membranes, as shown here, must be accomplished through alternative mechanisms.
One PV membrane-associated pattern recognized by distinct host defense proteins is loss of PV membrane integrity (Feeley et al., 2017; Thurston et al., 2012). Therefore, to avoid immune recognition, C. trachomatis and other bacterial pathogens secrete effectors to stabilize their surrounding PV membranes (Anand et al., 2020; Andersen et al., 2021). Defects in any of at least three secreted Inc proteins, i.e. CpoS, IncC, and CT383, result in premature inclusion lysis, activation of host cell death pathways, and early termination of the chlamydial developmental growth cycle (Sixt et al., 2017; Weber et al., 2017). Arguing against a similar inclusion-stabilizing role for Inc protein GarD, we found that loss of GarD expression did not notably alter bacterial fitness or levels of host cell death during culture in naïve host cells. Accordingly, it is less likely that loss of membrane integrity is the primary signal promoting the recruitment of RNF213 to GarD-deficient inclusions.
Whether RNF213 directly detects molecular patterns on GarD-deficient inclusion membranes or is recruited to inclusions by auxiliary host factors is one of many open questions emanating from our study. One known interaction partner of RNF213 is the IFN-inducible ISG15 (Thery et al., 2021), a ubiquitin-like antimicrobial protein covalently linked to various protein substrates (Perng and Lenschow, 2018). However, our data exclude ISGylation as a potential mechanism directing RNF213 towards inclusions. Nonetheless, other bona-fide pattern recognition receptors previously shown to localize to inclusion membranes (Finethy and Coers, 2016) could play important roles in the delivery of RNF213 to inclusions formed by garD mutants.
As already alluded to, an attractive alternative model posits that RNF213 itself acts as a pattern recognition receptor detecting an unknown molecular pattern defining the inclusion membrane as non-self or aberrant. The precedent of RNF213 binding to and ubiquitylating bacterial lipid A (Otten et al., 2021) justifies speculations that RNF213 may directly detect microbe- or host-derived lipids enriched in the inclusion membrane. Within the framework of this model, we put forward the hypothesis that GarD conceals putative RNF213 binding substrates resident in the inclusion membrane to block immune detection by RNF213.
Future work is needed not only to determine the mechanism by which RNF213 detects inclusions but also to determine the substrates ubiquitylated by RNF213 on the inclusion membrane. Although RNF213 generates K63-linked polyubiquitin chains in vitro (Habu and Harada, 2021), we failed to detect K63-linked ubiquitin at the inclusion membrane. Instead, we detected M1-linked ubiquitin chains, which remained at WT levels in cells lacking essential components of LUBAC, the only E3 ligase complex known to mediate M1-linked ubiquitin chain formation (Dittmar and Winklhofer, 2019). These unexpected findings indicate that LUBAC-independent inclusion ubiquitylation is mechanistically distinct from LUBAC-dependent M1-linked ubiquitylation of Salmonella (Noad et al., 2017; Otten et al., 2021), although both processes require RNF213. The precise nature of the LUBAC-independent linear ubiquitylation pathway occurring at inclusion membranes will require further investigation.
In concurrence with the presence of M1-linked ubiquitin at GarD-deficient inclusion membranes, we observed prominent recruitment of the linear ubiquitin adaptor proteins OPTN, NDP52, and TAX1BP1 (Hu et al., 2018; Miyashita et al., 2021; Nakazawa et al., 2016; Tumbarello et al., 2015) in an RNF213-dependent manner. Co-staining experiments provide some insights into the temporal hierarchy of events occurring at the inclusion membrane. For example, ubiquitin+ inclusions universally stained positive for RNF213, whereas a marked percentage of RNF213+ inclusions lacked ubiquitin, agreeing with a model in which inclusion ubiquitylation occurs downstream from RNF213 recruitment. In the same vein, virtually all OPTN+ and TAX1BP1+ inclusions were also ubiquitin+ but not the other way around, indicating that inclusion ubiquitylation occurs upstream from the recruitment of these two adaptor proteins to the inclusion. In contrast to OPTN and TAX1BP1, a subset of NDP52+ inclusions remained ubiquitin-negative, suggesting that NDP52 can be recruited to PVs through both ubiquitin-dependent and ubiquitin-independent mechanism, similar to what was previously reported (Thurston et al., 2012). The degree to which individual ubiquitin adaptor proteins contribute to host defense and the role of the autophagic machinery in the delivery of C. trachomatis into LAMP1+ compartments await further exploration.
Different Chlamydia species and serovars acquire loss-of-functions mutations in garD following serial passage in cell culture (Bonner et al., 2015; Borges et al., 2013; Borges et al., 2015; Ramsey et al., 2009; Russell et al., 2011; Sturdevant et al., 2010). Because GarD overexpression destabilizes inclusion membrane integrity and triggers host cell death ((Bishop and Derré, 2022; Yang et al., 2021) and this study), it was reasonable to speculate that garD loss-of-function mutations could convey a fitness advantage to bacteria grown in cell culture (Borges et al., 2013; Sturdevant et al., 2010). Alternatively, a lack of positive selection to maintain garD gene function could also explain the acquisition of garD mutations with serial passage. In refute of the former but support of the latter hypothesis, we found that an engineered loss-of-function garD mutation failed to convey any fitness advantage to bacteria. While further work is required to understand how GarD impacts inclusion membrane properties, all observations align with a ‘Goldilocks model.’ According to this model, GarD expression at lower physiological levels shields an unknown inclusion membrane component from recognition by RNF213 and thereby promotes bacterial survival inside IFN-primed host cells. Ectopic GarD overexpression, on the other hand, is detrimental to bacterial growth due to excess GarD interactions with components of the inclusion membrane, ultimately resulting in membrane destabilization. While we favor a model in which GarD directly alters inclusion membrane features for intracellular camouflage, we cannot exclude alternative models of GarD activity (Bishop and Derré, 2022; Borges et al., 2015; Yang et al., 2021).
Intact garD is maintained in clinical isolates (Bonner et al., 2015) indicating that GarD is essential in vivo to overcome aspects of IFNγ-induced cell-autonomous immunity during human genital infections. In contrast to these human studies, C. trachomatis serovar D garD frameshift mutants were recovered after passage through mice (Sturdevant et al., 2010), indicating that mouse infections do not confer substantial selective pressure against garD mutant alleles. Similarly, recent work demonstrated that mice clear C. trachomatis WT and a garD loss-of-function mutant with comparable kinetics (Kamada et al., 2011; Yang et al., 2021). We offer two non-mutually exclusive explanations to account for these observations. First, while undergoing an evolutionary arms race with its human host, C. trachomatis may have evolved a GarD protein that efficiently blocks human RNF213 but not mouse RNF213 from binding to inclusion membranes. Second, anti-chlamydial host defense mediated by RNF213 may be obsolete in mice due to the presence of an additional, highly potent IFNγ-induced cell-autonomous immune response executed by murine immunity related GTPases (IRG). The IRG system is compromised in humans (Bekpen et al., 2005) but functional in mice, which employ IRG immunity to rapidly clear infections with human-adapted C. trachomatis (Bernstein-Hanley et al., 2006; Coers et al., 2008; Coers et al., 2011). Therefore, RNF213-mediated immunity and evasion thereof by GarD is expected to play a more prominent role in shaping C. trachomatis pathogenesis in the IRG-deficient human host than in the IRG-competent murine host.
In conclusion, our study establishes GarD as a C. trachomatis virulence factor subverting human IFNγ-mediated cell-autonomous immunity. We show that GarD protects inclusions from recognition by the human ubiquitin E3 ligase RNF213, demonstrating the existence of microbial virulence factors mediating escape from RNF213-driven host defense. An understanding of the mechanism underlying RNF213-executed pathogen detection and corresponding microbial counter-immunity is expected to provide future opportunities for therapeutic interventions.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jörn Coers (jorn.coers@duke.edu).
Materials availability
All unique materials generated in this study are available from the lead contact upon request and without restriction.
Data and Code availability
Raw files for whole-genome sequencing of Chlamydia library mutants have been deposited to the Sequence Read Archive (NCBI, SRA: PRJNA865907) and will be available at the time of publication. Plasmid maps or 3D structure predictions reported in this paper will be shared by the lead contact upon request. Original images used for microscopy and western blotting have been deposited to Mendeley Data (doi: 10.17632/hkvx8gx2ft.1) and will be available at the time of publication. Genome-wide association studies (GWAS) data is publicly available from a previous publication (Wang et al., 2018).
Original code used to generate Quantile-Quantile plots (Figure 5A) is included at the end of the STAR Methods (Methods S1).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
A549 (ATCC #CCL-185) and Vero cells (ATCC #CCL-81) were cultured in Dulbeco’s Modified Eagle Medium (DMEM; Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Omega Scientific), 1% MEM Non-essential Amino Acids (NEAA; Gibco) and 55 μM 2-Mercaptoethanol (BME; Gibco). HFF-1 cells (ATCC #SCRC-1041) were cultured in DMEM supplemented with 15% heat-inactivated FBS, NEAA, BME and 1% Glutamax (Gibco). Primary cervical epithelial cells (HCerEpiC, ScienCell, 7060) were kindly provided by Dr. Carolyn Coyne (Duke University) and cultured in EpiCM media (ScienCell, 4101) supplemented with 10% heat-inactivated FBS. A549s are lung epithelial cells derived from a 58-year-old Caucasian male with lung cancer; HFF-1s are foreskin fibroblasts derived from two male children; primary cervical cells (lot #16983) were isolated from female uterine tissue. ISG15 knockout (KO) cells and their corresponding parental A549 cell line were kindly provided by Dr. David Sibley (Washington University in St. Louis; (Bhushan et al., 2020)). All cells were grown at 37°C in 5% CO2 and were routinely PCR tested for Mycoplasma contamination. Cell Line Authentication (CLA) for A549s was performed by the Duke University DNA Analysis Facility using GenePrint 10 (Promega).
Chlamydia strains
All new Chlamydia trachomatis strains generated de novo in this study are serovar LGV-L2/434/Bu. The Chlamydia trachomatis L2 mutant (CTL2M) library and its corresponding parental, rifampin-resistant strain (C.t. RifR) have been used in previous studies (Kokes et al., 2015; Nguyen and Valdivia, 2012). Targeted inactivation of the CTL0390/garD gene (garD::GII) was achieved using a modified TargeTron platform adapted specifically for C. trachomatis (Johnson and Fisher, 2013; Lowden et al., 2015) while complementation strains (garD::GII pGarD) were constructed via cloning and transformation of pBOMB4 (Bauler and Hackstadt, 2014). Subsequent cloning and transformation are described below.
C. trachomatis strains were transformed with the plasmid pGFP::SW2 (Wang et al., 2011) to facilitate microscopic visualization (Haldar et. al., 2016). A fluorescent C. muridarum MoPn strain harboring pGFP::CM was also used (Liu et al., 2014). C. trachomatis L2 strains used in co-infection studies include a wildtype and incA nonsense mutant (Kokes et al., 2015; Sixt et al., 2017) transformed with p2TK2-IncDProm-mCherry-IncDTerm (Agaisse and Derre, 2013).
Two density-gradient purified elementary body (EB) preps of C. trachomatis serovar D/UW-3/Cx strains (C.t. D; wildtype and C.t. D CT135−) were a generous gift from Dr. Harlan Caldwell (National Institutes of Health; Bethesda, MD). These strains are isogenic with the exception of a single premature stop codon (R201*) in the CT135 locus (Yang et al., 2021).
METHOD DETAILS
Chlamydia propagation and tissue culture infections
Chlamydia samples were propagated in Vero cells and collected by washing samples once with ddH2O (Invitrogen), and lysing cells in ddH2O with periodic scraping and pipetting at room temperature for 20 minutes. Following hypotonic lysis, lysates were mixed with 5X SPG buffer (1X concentrations 0.5 g/L KH2PO4, 1.2 g/L Na2PO4, 0.72 g/L L-glutamic acid, 75 g/L sucrose, pH 7.5) and stored at −80°C. Seed preps of Chlamydia strains were used for all infections unless otherwise stated. Bacterial titers, or inclusion forming units (IFUs), were determined by serial dilutions of Chlamydia onto fresh Vero cells.
Infections were performed as described previously (Haldar et al., 2016). All infections were performed at a multiplicity of infection (MOI) of 2 unless otherwise stated. Diluted Chlamydia was added to fully confluent cell monolayers in cold DMEM supplemented with excess L-tryptophan (100 μg/mL; Sigma). Plates were centrifuged at 4°C, 3000 rpm for 30 minutes and returned to a 37°C incubator until the indicated timepoints for each assay. Time of infection (0 hours post-infection, hpi) was defined as the halfway point (15 minutes) of the centrifugation step, unless otherwise stated.
Co-infections (Figures 3 and 6) involving two strains were performed in the same manner, but with an MOI of 3 for garD::GII-GFP and an MOI of 5 for incA− and WT strains.
Interferon Response (IR) and burden assays
Bacterial burden was assessed by infectivity assays as previously described (Giebel et al., 2019; Haldar et al., 2016). Cells were seeded in black 96-well flat clear-bottom plates (Corning) at a density of 1.2*104 cells per well. After 24 hours, cells were stimulated with human interferon gamma (IFNγ; Millipore, IF005) at a concentration of 100 U/mL (5 ng/mL) or 10 U/mL (0.5 ng/mL) in DMEM supplemented with L-Trp (100μg/mL), unless otherwise stated. At 20h post-priming, cells were infected in technical duplicate or triplicate at an MOI of 2 (4*104 IFUs per well) in cold DMEM supplemented with L-Trp. At 24–26 hpi (or 42 hpi for serovar D strains, Figure S6J) cells were fixed at room temperature with either cold 4% paraformaldehyde (PFA; Sigma, pH 7.4) in phosphate-buffered saline (Gibco) for 20 minutes or ice-cold methanol (Fisher) for 1 to 2 minutes.
Samples fixed with PFA were stained for DNA using Hoechst 33258 (Invitrogen) diluted 1:1000 in PBS and incubated for 10 minutes.
Methanol-fixed samples were stained for inclusions and nuclei by blocking with 3% bovine serum albumin (BSA; VWR) in PBS, followed by incubation with mouse anti-Chlamydial LPS sera (EVI-H1, 1:20 or 1:250 in blocking buffer). Nuclear and secondary staining were done with Hoechst and goat anti-mouse AlexaFluor 488 (1:1000; Invitrogen).
Samples were stored in 200 μL of PBS and sealed using aluminum adhesive (Thermo). Plates were processed using a CellInsight CX5 High Content Screening platform (Thermo). Nine fields of view corresponding to the middle of an individual well were imaged for inclusions (GFP channel) and nuclei (DAPI channel). A customized protocol identified and counted the total numbers of fluorescent objects based on size, intensity and object roundness. Infectivity was calculated as the total number of inclusions divided by the number of non-Chlamydial nuclear objects per well. Differences in bacterial burden were assessed using one of two parameters: i) the “interferon response” (IR) ratio, calculated by dividing the infectivity of the stimulated well (+IFNγ) by the infectivity of the unstimulated well (−IFNγ); or via normalizing the infectivity of each sample to an unstimulated wildtype cell control.
Screen for IFNγ-sensitive C.t. mutants
Screening for candidate genes conferring resistance to human IFNγ leveraged the C. trachomatis LGV-L2 434/Bu Mutant Library (CTL2M) (Kokes et al., 2015). To amplify the library, pre-existing library plates were thawed on ice and 5 μL of seed was infected into confluent Vero cells. At 42 to 48 hpi, Chlamydia were harvested by hypotonic lysis and stored in 200 μL SPG buffer at −80°C. A549 cells were seeded in black 96-well plates and stimulated with 100 U/mL human IFNγ as described above. On the day of infection, CTL2M plates were thawed on ice and infected at a final dilution of 1:1000 from the seed plate. At 24 to 26 hpi, cells were fixed with ice-cold methanol and immunostained for Chlamydial LPS (EVI-H1, 1:20) and processed by CellInsight CX5 as described above. Mutant hits were defined as displaying an IR Z-score of less than −2 for either total numbers of inclusions (#inc), percentage of cells infected (%inf) or an average of these two parameters (Fig. 1C and S1A). Three mutants displaying elevated Z-scores (top right quadrant of Fig. 1C) were treated as artifacts and are not shown.
Whole-genome sequencing
Enrichment of Chlamydia genomic DNA (gDNA), preparation of sequencing libraries and analysis of single nucleotide variants (SNVs) were performed as previously described (Kokes et al., 2015; Kędzior and Bastidas, 2019). Confluent Vero cells were infected with individual strains of Chlamydia at an MOI of 3. Between 44 and 48 hpi, cells were harvested by hypotonic lysis and briefly sonicated. Bacteria were pelleted by spinning lysates at 14,000 rpm and 4°C for 15 minutes. To remove host gDNA and enrich bacterial gDNA, pellets were resuspended in 100 μL of buffer supplemented with 4 units of DNAse I (NEB, M303S) and incubated at 37°C for 30 minutes. An additional 4 units of DNAse I were added and incubated for an additional 30 minutes at 37°C. Pellets were centrifuged again, resuspended in buffer and processed using a Qiagen DNEasy Blood and Tissue kit along with RNAse A digestion (Qiagen). gDNA concentrations were measured using a Qubit fluorometer (Thermo) and stored at −80°C. For quality control, 5 μL of gDNA were checked on a 1% agarose gel for potential degradation.
Sequencing libraries (DNA-Seq) were prepared from 500 ng - 1μg of gDNA using the Kapa Hyper Prep kit (Roche) according to manufacturer’s instructions by the Duke GCB Sequencing and Genomic Technologies Share Resource. Next-generation sequencing of paired-end reads was achieved using the NovaSeq 6000 S-prime system (Illumina).
Raw sequencing files (SRA: PRJNA865907) were assembled into genomes according to the C. trachomatis serovar L2/434/Bu reference genome (NCBI GenBank #NC_010297.1) using Geneious Prime 1.0 software (Biomatters Ltd). Parameters to call single nucleotide variants (SNVs) within these genomes included a minimum variant frequency of 10%, maximum P-value of 10^−6, average PHRED quality score >30 and a strand bias of <65%. As an additional quality control, all sequenced mutants were confirmed to contain the same 11 point mutations as the rifampin-resistant (RifR) parental strain published previously (Kokes et al., 2015; Nguyen and Valdivia, 2012). Mutations found in a previously individually sequenced mutant (CTL2M0007 or a cpoS nonsense mutant; Kokes et al., 2015; Sixt et al., 2017) were included in our analysis, but the individual strain was not re-sequenced for this study. Of the 14 sequenced strains, 11 contained the same non-synonymous mutation at position 1014167, within the rpoD gene (CT615). This mutation did not show any obvious predicted effect by SNAP2 analysis (described below) and was previously described as a neutral mutation in a separate report (Soules et al., 2020). Therefore, this SNV was regarded as a background mutation of the in-house RifR parental strain and was removed from subsequent analyses.
SNVs were classified into four groups: non-coding, synonymous, non-synonymous and nonsense/frameshift (Figure S1C). Candidate genes for follow-up study were selected based on enriched frequency in the latter two groups.
Individual mutations that mapped to CTL0390 among four CTL2M mutant hits were confirmed by PCR amplification of the C.t. locus using Phusion polymerase (NEB), PCR purification (Qiagen) and Sanger Sequencing of plaque-purified clones (GeneWiz).
SNAP2 analysis
The effects of amino acid changes caused by non-synonymous mutations were predicted using SNAP2 program (Screening for non-acceptable polymorphisms; https://rostlab.org/services/snap2web/) (Hecht et al., 2015). SNAP2 uses a neural-network based classification system to make a two-state prediction (effect/neutral) on whether an amino acid change will impact overall protein structure.
Plaque purifications
Plaque purification of CTL2M mutant hits was performed as previously described (Kędzior and Bastidas, 2019). Confluent Vero cells in 6-well plates (Corning) were infected with 10-fold serial dilutions of C.t. seed preps. At 2 hpi, cells were covered with a DMEM-agar overlay, made from a filtered solution of DMEM powder (US Biological) and 22 μM sodium bicarbonate (Sigma) supplemented with 1% MEM non-essential amino acids (Gibco), 10% FBS (Omega Scientific), 0.025 mg/mL gentamycin (Gibco), 100 μg/mL L-Trp and 0.54% SeaKem LE agarose (Lonza). Between 10 to 20 days post-infection, individual clones were cored using a sterile p1000 tip, re-infected into a fresh monolayer of Vero cells, titered and stored in SPG buffer at −80°C.
An initial IR assay tested three to five clones for all plaque purified strains (except one hit, which yielded a single purified clone). An independent follow-up IR assay tested the two most restricted clones from the first experiment. The average of these two experiments is shown in Fig. 1D.
3D Structure Predictions
The 3D structure for CTL0390 was predicted from the primary amino acid sequence (UniProt Accession #A0A0H3MBI2) into AlphaFold Colab, a simplified version of AlphaFold v2.1.0 (Jumper et al., 2021b) (https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb). The resulting protein data bank (PDB) file was visualized with PYMOL 2.4.2. The structure containing per-residue estimations of its confidence based on Local Distance Difference Test (LDDT) is shown in Fig. S1F.
Plasmid Constructions
Targeted gene inactivation of CTL0390 was achieved through cloning of the vector pDFTT3aadA (Lowden et al., 2015), which allows for group II (GII) intron mutagenesis (TargeTron) with spectinomycin resistance. Design of TargeTron compatible sites for CTL0390 was achieved using TargeTronics (http://www.targetrons.com/). Cloning of three independent TargeTron vectors (pDFTT3aadA-CTL0390-1 to −3) was performed following a recent publication (Wang et al., 2011; Weber and Faris, 2019). All relevant primers and plasmids are listed in the Supplementary Table S1, and predicted insertion sites are shown in Fig. S2A. Ligated constructs were transformed into DH5α E. coli (NEB) and resulting plasmids were termed “pKO-390-1 to −3”.
Complementation plasmids were generated using pBOMB4-MCI (GenBank accession no. KF790907), a cloning vector derived from pGFP::SW2 that harbors mCherry and a beta-lactamase (bla) resistant marker (Bauler and Hackstadt, 2014). The CTL0390 ORF plus native upstream and downstream genomic regions were PCR amplified (pBOMB_CTL0390_for/pBOMB_CTL0390_rev primers) and purified. The intergenic regions between CTL0389/CTL0390 (+57bp) and CTL0390/CTL0391 (89bp) were used as predicted “promoter” and “terminator” regions, respectively. pBOMB4-MCI was digested using PstI and NotI (NEB) and ligated with the CTL0390 genomic region using InFusion cloning and transformation into Stellar Competent E. coli according to the manufacturer’s instructions (TaKaRa). Resulting constructs were termed “pGarD”.
Correct plasmid clones were verified using a combination of restriction digest enzymes (NEB) and Sanger Sequencing (GeneWiz). Plasmid amplification was achieved by culturing of transformants in LB medium followed by plasmid isolation with Plasmid Maxi Kits (Qiagen).
Customized anti-CTL0390 (anti-GarD) antibody
A customized antibody against C. trachomatis GarD (CTL0390) was developed by Thermo Scientific (Project 1VC2780). Two rabbits were immunized with a short peptide (DAQAYSFLSVSPLDARIE) corresponding to the last 18 amino acids of the protein. Sera from both animals was collected at day 72 and used for affinity purification. The affinity-purified eluents were mixed with bovine serum albumin (1% w/v; VWR), aliquoted and stored at −80°C.
Chlamydia transformations
Transformations were carried out as previously described (Agaisse and Derre, 2013; Wang et al., 2011). A total of 1*108 IFUs of C. trachomatis seed were mixed with 20μg of plasmid DNA in 500μL of transformation buffer (10mM Tris, 50mM CaCl2, pH 7.5) and incubated for 30 minutes at room temperature. The transformation mixture was then mixed with cold DMEM and added to a confluent monolayer of Vero cells. Plates were spun at 10°C, 2000 rpm for 25 minutes and returned to a 37°C CO2 incubator. At 14 or 18hpi, fresh DMEM supplemented with either 150μg/mL spectinomycin (for pKO-390 plasmids) or 1U/mL penicillin G (0.6μg/mL; for pGarD and pGFP::SW2 plasmids), respectively, was added to the cells. At 42–46hpi, cells were harvested by hypotonic lysis, collected by centrifugation, and re-infected into a fresh monolayer of Vero cells. Each passage was visually inspected for the presence of inclusions, which typically occurred at passage two. After four passages, Chlamydia were stored at −80°C in SPG and titered by serial dilutions. gDNA isolated from transformed strains was used to genotype both the chromosomal CTL0390 locus and presence of the pGarD plasmid (Fig. S2A). Insertional mutants for CTL0390, containing an aadA spectinomycin-resistant cassette, are referred to as “garD::GII”.
One-step growth curve by IFU assay
Vero cells were seeded in 24-well plates (Corning) at a density of 2*105 cells per well and incubated overnight. The next day, cells were infected at an MOI of 0.8 and centrifuged for 30 minutes at 3000 rpm and 4°C. The time of infection (0hpi) was defined as the end of the spin. Samples collected at 1 hpi, 6 hpi, 12 hpi, 24 hpi, 36 hpi and 48 hpi were harvested by hypotonic lysis and stored at −80°C in SPG. Inclusion forming units (IFUs), were quantified by re-infecting a fresh monolayer of Vero cells with serial dilutions of the lysates and processed using the CellInsight CX5 as described above.
Immunocytochemistry
Immunocytochemistry was performed as described before (Haldar et al., 2016). Briefly, the day prior to the infection cells were seeded on glass coverslips and tissue-culture treated 24-well plates (Corning) at a density of 2*105 cells per well. The next day, cells were infected as described above. At 3 hpi, media was replaced with fresh DMEM supplemented with L-Tryptophan (100 μg/mL). Half the wells were treated with 100 U/mL human IFNγ (Millipore, IF005). At 20 hpi (or 42 hpi for serovar D strains, Fig S6B and S6F), cells were fixed at room temperature with either cold 4% (w/v) PFA for 20 minutes or ice-cold methanol for 1 minute. Cells fixed with PFA were permeabilized with one of the following: ice-cold methanol for 1 minute, or 0.05% saponin (Amresco) added to blocking buffer. Cells were blocked in PBS supplmeneted with 5% BSA and 2.2% glycine (VWR) for 30 minutes. The following primary antibodies were diluted in blocking buffer and incubated with samples for 1 hour:
Mouse monoclonal anti-Chlamydia LPS serum (1:20 to 1:250; EVI-H1)
Mouse monoclonal anti-inaC/CT813 (Chen et al., 2006)
Rabbit polyclonal anti-GarD/CTL0390 (1:100; Thermo, AB3075)
Mouse monoclonal anti-Ubiquitin, FK2 (1:100; Cayman Chemical, 14220)
Rabbit monoclonal anti-K48 ubiquitin, clone Apu2 (1:100; Sigma, ZRB2150)
Rabbit monoclonal anti-K63 ubiquitin, clone Apu3 (1:100; Sigma, 05-1308)
Rabbit monoclonal anti-Linear ubiquitin, clone 1E3 (1:250; Sigma, MABS199 and Sigma, ZRB2114)
Rabbit polyclonal anti-p62 (1:500; MBL, PM045)
Mouse monoclonal anti-NBR1 (1:100; Abcam, ab55474)
Rabbit polyclonal anti-NDP52 (1:200; Abnova, H00010241-D01)
Rabbit polyclonal anti-Optineurin (1:200; ProteinTech, 10837-1-AP)
Rabbit polyclonal anti-TAX1BP1 (1:250; Bethyl Laboratories, A303-791A)
Mouse monoclonal anti-LAMP1 (1:500; DSHB, H4A3-c)
Rabbit polyclonal anti-LC3 (1:500; MBL, PM036)
Rabbit monoclonal anti-GABARAP+GABARAPL1+ GABARAPL2 (1:100; Abcam, ab109364)
Rabbit polyclonal anti-RNF213 (1:500; Sigma, HPA003347)
Samples were stained with Alexa Fluor conjugated secondary antibodies (1:1000; Invitrogen) and Hoechst (see Key Resources table for specific information).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Chlamydia LPS serum | Provided by Dr. Daniel Rockey (Oregon State University) | EVI-H1 |
| Mouse monoclonal anti-inaC (CT813) | Chen et al., 2006 | N/A |
| Rabbit polyclonal anti-GarD/CTL0390 | This study, Thermo | AB3075, Project 1VC2780 |
| Mouse monoclonal anti-Ubiquitin, FK2 | Cayman Chemical | Cat#14220 |
| Rabbit monoclonal anti-Linear Ubiquitin, 1E3 | Sigma | Cat#MABS199, RRID:AB_2576212 |
| Rabbit monoclonal anti-Linear Ubiquitin, 1E3 | Sigma | Cat#ZRB2114 |
| Rabbit monoclonal anti-K48 ubiquitin, Apu2 | Sigma | Cat#ZRB2150 |
| Rabbit monoclonal anti-K63 ubiquitin, Apu3 | Sigma | Cat#05-1308, RRID:AB_1587580 |
| Rabbit polyclonal anti-HOIP | Abcam | Cat#ab46322, RRID:AB_945269 |
| Mouse monoclonal anti-HOIL-1, clone 2E2 | Sigma | Cat#MABC576, RRID:AB_2737058 |
| Rabbit polyclonal anti-p62 (SQSTM1) | MBL International | Cat#PM045, RRID:AB_1279301 |
| Mouse monoclonal anti-NBR1, 5C3 | Abcam | Cat#ab55474, RRID:AB_2149404 |
| CALCOCO2 MaxPab rabbit polyclonal antibody (D01; anti-NDP52) | Abnova | Cat#H00010241-D01, RRID:AB_10633399 |
| Rabbit polyclonal anti-Optineurin | ProteinTech | Cat#10837-1-AP, RRID:AB_2156665 |
| Rabbit polyclonal anti-TAX1BP1 | Bethyl Laboratories | Cat#A303-791A, RRID:AB_11218189 |
| Mouse monoclonal anti-LAMP1 | DSHB | Cat#H4A3, RRID:AB_2296838 |
| Rabbit polyclonal anti-LC3 | MBL International | Cat#PM036, RRID:AB_2274121 |
| Rabbit monoclonal anti-GABARAP+GABARAPL1+GABARAPL2 | Abcam | Cat#ab109364, RRID:AB_10861928 |
| Rabbit polyclonal anti-RNF213 | Sigma | Cat#HPA003347, RRID:AB_1079204 |
| Rabbit polyclonal anti-GAPDH | Abcam | Cat#ab9485, RRID:AB_307275 |
| Rabbit polyclonal anti-SLC1 (CT043) | Saka et al., 2011 | N/A |
| Rabbit polyclonal anti-ISG15 | ProteinTech | Cat#15981-1-AP, RRID:AB_2126302 |
| Goat anti-rabbit AlexaFluor 488 | Invitrogen | Cat#A11034, RRID:AB_2576217 |
| Goat anti-mouse AlexaFluor 488 | Invitrogen | Cat#A11029, RRID:AB_138404 |
| Donkey anti-rabbit AlexaFluor 568 | Invitrogen | Cat#A10042, RRID:AB_2534017 |
| Goat anti-mouse AlexaFluor 568 | Invitrogen | Cat#A11004, RRID:AB_2534072 |
| Goat anti-rabbit AlexaFluor 660 | Invitrogen | Cat#A21073, RRID:AB_10374435 |
| Goat anti-mouse AlexaFluor 660 | Invitrogen | Cat#A21054, RRID:AB_1500635 |
| Goat anti-rabbit IgG, HRP | Invitrogen | Cat#65-6120, RRID:AB_2533967 |
| Goat anti-mouse IgG, HRP | Invitrogen | Cat#62-6520, RRID:AB_2533947 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant human interferon gamma (IFN-γ) | Millipore | Cat#IF005 |
| Critical Commercial Assays | ||
| SYTOX™ Green Nucleic Acid Stain | Invitrogen | Cat#S7020 |
| Deposited Data | ||
| Raw sequencing data for whole genomes of C. trachomatis mutant library hits (Sequence Read Archive) | This study | SRA: PRJNA865907 |
| Original western blot and emicroscopy images (Mendeley Data) | This study | ; doi: 10.17632/hkvx8gx2ft.1 |
| H2P2 database and web portal | L. Wang et al., 2018 | http://h2p2.oit.duke.edu/H2P2Home/ |
| ESBL database of Human E3 Ubiquitin Ligases | Medvar et al., 2016 | https://esbl.nhlbi.nih.gov/Databases/KSBP2/Targets/Lists/E3-ligases/ |
| Experimental Models: Cell Lines | ||
| Vero cells | ATCC | CCL-81, RRID:CVCL_0059 |
| A549 cells | ATCC | CCL-185, RRID:CVCL_0023 |
| HFF-1 cells | ATCC | SCRC-1041, RRID:CVCL_3285 |
| Primary cervical epithelial cells (HCerEpiC) | ScienCell | Cat#7060, lot #16983 |
| HOIP KO A549s, clone #1 | This study | N/A |
| HOIP KO A549s, clone #2 | This study | N/A |
| HOIL-1 KO A549s, clone #1 | This study | N/A |
| HOIL-1 KO A549s, clone #2 | This study | N/A |
| RNF213 KO A549s, pool #1 | This study | N/A |
| RNF213 KO A549s, pool #2 | This study | N/A |
| RNF213 KO A549s, pool #3 | This study | N/A |
| ISG15 knockout A549s | Bhushan et al., 2020 | N/A |
| Experimental Models: Organisms/Strains | ||
| Chlamydia trachomatis, serovar LGV-L2/434/Bu | Haldar et al., 2016 | N/A |
| Chlamydia trachomatis, serovar LGV-L2/434/Bu + pGFP::SW2 | Haldar et al.; 2016, Wang et al., 2011 | N/A |
| Chlamydia muridarum MoPn + pGFP::CM | Haldar et al., 2016; Liu et al., 2014 | N/A |
| Chlamydia trachomatis, serovar LGV-L2/434/Bu RifR | Kokes et al., 2015; Nguyen & Valdivia, 2012 | N/A |
| Chlamydia trachomatis, serovar LGV-L2/434/Bu chemical mutant library (CTL2M library) | Kokes et al., 2015 | N/A |
| Chlamydia trachomatis, serovar LGV-L2/434/Bu incA− (CTL2M0923, CTL0374 R197*) + p2TK2-SW2-incDprom-mCherry-incDterm | Kokes et al., 2015; Sixt et al., 2017 | N/A |
| Chlamydia trachomatis, serovar LGV-L2/434/Bu + p2TK2-SW2-incDprom-mCherry-incDterm | Sixt et al., 2017 | N/A |
| Chlamydia trachomatis, serovar D/UW-3/Cx (CtD) | Yang et al., 2021 | N/A |
| Chlamydia trachomatis, serovar D/UW-3/Cx CT135R201* (CtD CT135−) | Yang et al., 2021 | N/A |
| Chlamydia trachomatis, serovar LGV-L2/434/Bu CTL0390::bla (garD::GII strain #1) | This study | N/A |
| Chlamydia trachomatis, serovar LGV-L2/434/Bu CTL0390::bla (garD::GII strain #2) | This study | N/A |
| Chlamydia trachomatis, serovar LGV-L2/434/Bu CTL0390::bla (garD::GII strain #3) | This study | N/A |
| Chlamydia trachomatis, serovar LGV-L2/434/Bu CTL0390::bla +pGFP::SW2 (garD::GII+ pGFP::SW2) | This study | N/A |
| Chlamydia trachomatis, serovar LGV-L2/434/Bu + pBOMB4-MCI | Bauler et al. 2014; This study | N/A |
| Chlamydia trachomatis, serovar LGV-L2/434/Bu + pGarD | This study | N/A |
| Chlamydia trachomatis, serovar LGV-L2/434/Bu garD::GII +pBOMB4-MCI | This study | N/A |
| Chlamydia trachomatis, serovar LGV-L2/434/Bu garD::GII +pGarD | This study | N/A |
| Oligonucleotides | ||
| *see Supplementary Table S1 for primer information* | ||
| Recombinant DNA | ||
| pGFP::SW2 | Wang et al., 2011 | N/A |
| pDFTT3aadA | Lowden et al., 2015 | N/A |
| pDFTT3aadA-CTL0390-1 (pKO-390-1) | This study | N/A |
| pDFTT3aadA-CTL0390-2 (pKO-390-2) | This study | N/A |
| pBOMB4-MCI | Bauler et al., 2014 | GenBank accession no. KF790907 |
| pDFTT3aadA-CTL0390-3 (pKO-390-3) | This study | N/A |
| pBOMB4-ctl0390prom-CTL0390-ctl0390term (pGarD) | This study | N/A |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | Ran et al., 2013 | RRID:Addgene_62988 |
| Software and Algorithms | ||
| Prism 9.4 | GraphPad Software, LLC | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ 1.53k | NIH | https://imagej.nih.gov/ij/ |
| Fiji 2.3.0 | ImageJ | https://fiji.sc/ |
| Geneious Prime 1.0 | Biomatters Ltd., Dotmatics | https://www.geneious.com/prime/ |
| Alphafold v2.1.0 | Jumper et al., 2021 | (https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb |
| PLINK v1.9 | Chang et al., 2015; Purcell et al., 2007 | https://www.cog-genomics.org/plink/ |
| Other | ||
| SNAP2 web tool | Hecht et al., 2015 | https://www.rostlab.org/services/snap/ |
| BioRender | BioRender.com | https://biorender.com/ |
| R script to make Quantile-Quantile plot and statistics in Figure 5A | This study | See Methods S1 or (https://github.com/lw157/PopulationGenetics/blob/master/fast_qqplot.R) |
Goat anti-rabbit AlexaFluor 488 (Invitrogen, A11034)
Goat anti-mouse AlexaFluor 488 (Invitrogen, A11029
Donkey anti-rabbit AlexaFluor 568 (Invitrogen, A10042)
Goat anti-mouse AlexaFluor 568 (Invitrogen, A11004)
Goat anti-rabbit AlexaFluor 660 (Invitrogen, A21073)
Goat anti-mouse AlexaFluor 660 (Invitrogen, A21054)
Coverslips were mounted on glass slides (Fisher) using a 1:9 mixture of p-phenylenediamine (PPD) and Mowiol 4–88 and cured at room temperature overnight. Samples were blinded either prior to image acquisition using tape, or during digital quantitation using the Blind Analysis Tool (https://imagej.net/plugins/blind-analysis-tools). Some samples were not blinded.
Images were acquired on either a Zeiss Axio Observer.Z1 epifluorescent microscope or a Zeiss 880 Airyscan Fast Inverted Confocal microscope using AxioVision 4.8 or ZEN software (Zeiss). For each sample, at least five randomly chosen microscopic fields of view (63x magnification) and more than 100 C.t. inclusions were imaged.
Image processing and quantitation was performed using ImageJ and Fiji softwares. Representative confocal images shown in this study were minimally adjusted by brightness and contrast for visibility. Images for quantitation of targeting were left unadjusted. C.t. strains harboring mCherry fluorescent markers (i.e. pBOMB4-MCI and pGarD) were false-colored to green to align with the more commonly used GFP-positive strains. Targeting was defined as the presence of intense protein signal enveloping >50% of the C.t. membrane (GarD, inaC, Ubiquitin, LAMP1 and RNF213) or the presence of one or more intense, curved line segments of host protein on the inclusion membrane (all other proteins). Relative fluorescent signal was quantified as an average of several radial “slices” using the Radial Profile tool in ImageJ (https://imagej.nih.gov/ij/plugins/radial-profile-ext.html).
Western Blotting
Cells were plated in 24-well plates (Corning) at a density of 1.2*105 cells per well. The next day, cells were stimulated with 100 U/mL IFNγ and incubated for 24 hours. Protein samples were harvested on ice in 100 μL RIPA buffer (Sigma) supplemented with 1% protease inhibitor (Sigma) and 1% DNAse I (NEB) and incubated at 4°C for 30 minutes. Lysates were centrifuged for 10 min at 20,000 rcf and 4°C and mixed with 4X reducing buffer made of Laemmli Buffer (BioRad) supplemented with beta-mercaptoethanol (Amresco; final concentration 2.5%). Samples were heated at 95°C for 10 minutes and stored at −20°C. Protein lysates for RNF213 were heated at 56°C for 10 minutes to prevent aggregation of very large proteins.
Sample collection for western blotting of Chlamydia-infected samples was performed as described above with some modifications previously described (Chen et al., 2012). Briefly, confluent A549 and Vero cells were infected with Chlamydia at an MOI of 2. Samples were collected with the same protocol as above, but substituting RIPA buffer with 8M urea (Amresco) plus 325U/mL Benzonase nuclease (Sigma).
Gel electrophoresis was performed using 10% or gradient 4–20% Mini-PROTEAN TGX polyacrylamide gels (BioRad) in running buffer (25mM Tris, 192mM glycine, 0.1% SDS). Semidry transfer to PVDF membranes (BioRad) was performed for GarD, HOIP, HOIL-1 and ISG15 using Trans-Blot Turbo system (BioRad), while wet transfers to PVDF membranes for RNF213 were performed overnight at 4°C in Towbin buffer (25mM Tris, 192mM glycine, 10–20% methanol, 0.1% SDS). Blocking buffer was composed of either 5% BSA or 5% nonfat dry milk (BioRad) in Tris-buffered saline supplemented with 0.1% Tween-20 (TBST). The following antibodies were used for primary antibody incubation:
Rabbit polyclonal anti-GAPDH (1:5000; Abcam ab9485)
Rabbit polyclonal anti-Slc1 (CT043) (1:10,000; (Saka et al., 2011)
Rabbit polyclonal anti-CTL0390 (1:1000; Thermo, AB3075)
Rabbit polyclonal anti-HOIP (1:1000; Abcam ab46322)
Mouse monoclonal anti-HOIL-1, clone 2E2 (1:1000; Sigma, MABC576)
Rabbit polyclonal anti-RNF213 (1:1000; Sigma, HPA003347)
Rabbit polyclonal anti-ISG15 (1:1000; ProteinTech, 15981-1-AP)
Goat anti-rabbit IgG HRP (1:5000; Invitrogen, 65-6120) or Goat anti-mouse IgG HRP (1:4000; Invitrogen 62-6520) were used as secondary antibodies. Chemiluminescent blot imaging was performed with Clarity ECL Substrate (BioRad) or Amersham ECL Prime Western Blotting Detection Reagent (Cytiva) and imaged using an Azure 500 Western Blot imager (Azure Biosystems).
SYTOX Green Cell death assay
To assess apparent cell death caused by ectopic overexpression of GarD, pBOMB4-MCI and pGarD plasmids were transformed into WT and garD::GII backgrounds as described above. A549 cells were seeded in black-96-well clear-bottom plates at a density of 2*104 cells per well. After 24 hours, cells were infected with indicated strains at an MOI of 2. Time of infection (0 hpi) was defined as the end of the centrifugation step. At 3 hpi, media was replaced with DMEM supplemented with L-Trp and 0.5 nM SYTOX Green (Invitrogen). Signal generated by SYTOX Green was measured using an EnSpire 2300 Multilabel Plate Reader (Perkin Elmer). Untreated cells and a “mock infection” (equivalent volume of lysed Vero cells in SPG) were used as negative controls. All samples were normalized to signal produced by cells incubated with DMEM plus 1% Triton-X (Amresco) at each timepoint (%Maximum).
Knockout Cell line generation
Knockout (KO) A549 cell lines for HOIP, HOIL-1 and RNF213 were generated by the Duke Functional Genomics core using CRISPR/Cas9 technology as previously described (Haldar et al., 2016). Single guide RNAs (sgRNAs) targeting early exonic regions of each gene were designed using CHOPCHOP (Labun et al., 2019) and Cas-OFFinder (Bae et al., 2014) and cloned into PX459 V2 (Addgene #62988; (Ran et al., 2013). A549s were transfected with sgRNAs using Lipofectamine 3000 (ThermoFisher) according to manufacturer’s instructions. 24 hours post-transfection, cells were selected with 2 μg/mL puromycin (Sigma) for three days, followed by growth as CRISPR-edited pools. For HOIP and HOIL-1 KO pools, cells were diluted to single cell concentrations, expanded, screened by light microscopy and genotyped by western blotting (Fig. S3A).
For RNF213 KOs, pooled cells exhibited highly efficient genome editing (>95%) as measured by PCR, and pooled cells were thus used for all subsequent experiments. Knockout status of all three RNF213 pools was confirmed by western blotting on three separate occasions (between cell passage 2 and 8) to confirm genetic drift towards a predominantly unedited cell population did not occur.
QUANTIFICATION AND STATISTICAL ANALYSIS
Cellular GWAS for Chlamydia infection and stratified QQ analysis
Cellular GWAS summary statistics for Chlamydia infection were obtained from a previous study (Wang et al., 2018). In that study, median C. trachomatis GFP fluorescence in infected cells after 27-hour infection was measured in 528 lymphoblastoid cell lines (LCLs) and GWAS was performed using the family-based association method for quantitative traits (QFAM) implemented in PLINK v1.9 (Chang et al., 2015; Purcell et al., 2007). Aiming to evaluate the impact of E3 ubiquitin ligases on intracellular Chlamydia, we focused on the 377 ubiquitin ligases in the human genome based on the ESBL website (https://esbl.nhlbi.nih.gov/Databases/KSBP2/Targets/Lists/E3-ligases/). As H2P2 was conducted on autosomal genes, X chromosomal genes were removed. Finally, 363 genes were included in the subsequent analysis. SNPs located within those genes and the surrounding regions (+/−10,000 bp) were extracted. The p values of selected SNPs were visualized using a Quantile-Quantile (QQ) plot, and genomic lambda was also calculated to investigate any potential inflation. The QQ plot and lambda value were generated using an in-house R script (https://github.com/lw157/PopulationGenetics/blob/master/fast_qqplot.R).
Regional Manhattan plot was made using LocusZoom (Pruim et al., 2010), with association data for SNPs with C. trachomatis 27 hour Median GFP (Wang et al., 2018) for genomic region chr17:78076360-78476068 in HG19 coordinates. Genotypic median plots were made in R (Team, 2020).
Statistics and Data Visualization
Graphs and statistics were generated and calculated in GraphPad Prism 9. All data are presented as the mean ± standard deviation (S.D.) of at least three independent experiments, unless otherwise stated. Infectivity assays were performed in technical duplicate or triplicate for each infection condition. Z-score calculations among C. trachomatis mutants (Figure 1C and S1A) were performed in Microsoft Excel in two discrete sets (plates1–6, n = 531 library mutants; and plates 7–16, n = 741 library mutants) based on two batches to complete the screen, with a Z-score of less than −2 defined as a threshold for significance. All other figures show “n” of at least 3, where n is the number of independent experiments performed. Statistical significance for GWAS data presented in Figure 5 is described above. All other significant results were defined as having a p-value less than 0.05 using a Student’s t-test, One-way- or Two-way-ANOVA followed by Dunnett’s or Tukey’s multiple comparison tests. Statistical details (e.g. p-values, sample sizes, analysis type) for individual experiments are listed in Figure legends. Not all statistical comparisons are shown. Figures were prepared using Adobe Illustrator. Additional illustrations in Figure 3 were prepared using BioRender.com.
Supplementary Material
Table S1. Primers used in this study, related to STAR Methods.
HIGHLIGHTS.
A screen for interferon resistance in C. trachomatis (C.t.) reveals CTL0390/GarD
Interferon stimulation leads to ubiquitylation and clearance of GarD-devoid C.t.
Ubiquitin E3 ligase RNF213 is required for host defense against C.t. garD mutants
GarD shields the C.t. vacuole from RNF213 translocation and ubiquitylation in cis
ACKNOWLEDGEMENTS
We would like to thank Drs. Carolyn Coyne, David Sibley, Dan Rockey, and Harlan Caldwell for sharing reagents, members of the Duke University Light Microscopy Core and Functional Genomics core facilities for technical support, and members of the labs of Drs. Clare Smith, Edward Miao, and David Tobin for helpful discussions. This work was supported by National Institutes of Health grant A030801 (to JC), AI150106 (to SCW), AI140019 (to RJB), and U19-AI084044 subproject 6652 to DCK. JC holds an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing financial or professional interests.
REFERENCES
- Agaisse H, and Derre I (2013). A C. trachomatis cloning vector and the generation of C. trachomatis strains expressing fluorescent proteins under the control of a C. trachomatis promoter. PLoS One 8, e57090. 10.1371/journal.pone.0057090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahel J, Lehner A, Vogel A, Schleiffer A, Meinhart A, Haselbach D, and Clausen T (2020). Moyamoya disease factor RNF213 is a giant E3 ligase with a dynein-like core and a distinct ubiquitin-transfer mechanism. Elife 9. 10.7554/eLife.56185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiyar A, Quayle AJ, Buckner LR, Sherchand SP, Chang TL, Zea AH, Martin DH, and Belland RJ (2014). Influence of the tryptophan-indole-IFNgamma axis on human genital Chlamydia trachomatis infection: role of vaginal co-infections. Front Cell Infect Microbiol 4, 72. 10.3389/fcimb.2014.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand I, Choi W, and Isberg RR (2020). The vacuole guard hypothesis: how intravacuolar pathogens fight to maintain the integrity of their beloved home. Curr Opin Microbiol 54, 51–58. 10.1016/j.mib.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen SE, Bulman LM, Steiert B, Faris R, and Weber MM (2021). Got mutants? How advances in chlamydial genetics have furthered the study of effector proteins. Pathog Dis 79. 10.1093/femspd/ftaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Park J, and Kim JS (2014). Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475. 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannantine JP, Griffiths RS, Viratyosin W, Brown WJ, and Rockey DD (2000). A secondary structure motif predictive of protein localization to the chlamydial inclusion membrane. Cell Microbiol 2, 35–47. 10.1046/j.1462-5822.2000.00029.x. [DOI] [PubMed] [Google Scholar]
- Bauler LD, and Hackstadt T (2014). Expression and targeting of secreted proteins from Chlamydia trachomatis. J Bacteriol 196, 1325–1334. 10.1128/jb.01290-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekpen C, Hunn JP, Rohde C, Parvanova I, Guethlein L, Dunn DM, Glowalla E, Leptin M, and Howard JC (2005). The interferon-inducible p47 (IRG) GTPases in vertebrates: loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol 6, R92. 10.1186/gb-2005-6-11-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein-Hanley I, Coers J, Balsara ZR, Taylor GA, Starnbach MN, and Dietrich WF (2006). The p47 GTPases Igtp and Irgb10 map to the Chlamydia trachomatis susceptibility locus Ctrq-3 and mediate cellular resistance in mice. Proc Natl Acad Sci U S A 103, 14092–14097. 10.1073/pnas.0603338103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan J, Radke JB, Perng YC, McAllaster M, Lenschow DJ, Virgin HW, and Sibley LD (2020). ISG15 Connects Autophagy and IFN-γ-Dependent Control of Toxoplasma gondii Infection in Human Cells. mBio 11. 10.1128/mBio.00852-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop RC, and Derré I (2022). The Chlamydia trachomatis Inclusion Membrane Protein CTL0390 Mediates Host Cell Exit via Lysis through STING Activation. Infect Immun 90, e0019022. 10.1128/iai.00190-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner C, Caldwell HD, Carlson JH, Graham MR, Kari L, Sturdevant GL, Tyler S, Zetner A, and McClarty G (2015). Chlamydia trachomatis virulence factor CT135 is stable in vivo but highly polymorphic in vitro. Pathog Dis 73, ftv043. 10.1093/femspd/ftv043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges V, Ferreira R, Nunes A, Sousa-Uva M, Abreu M, Borrego MJ, and Gomes JP (2013). Effect of long-term laboratory propagation on Chlamydia trachomatis genome dynamics. Infect Genet Evol 17, 23–32. 10.1016/j.meegid.2013.03.035. [DOI] [PubMed] [Google Scholar]
- Borges V, Pinheiro M, Antelo M, Sampaio DA, Vieira L, Ferreira R, Nunes A, Almeida F, Mota LJ, Borrego MJ, and Gomes JP (2015). Chlamydia trachomatis In Vivo to In Vitro Transition Reveals Mechanisms of Phase Variation and Down-Regulation of Virulence Factors. PLoS One 10, e0133420. 10.1371/journal.pone.0133420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunham RC, and Paavonen J (2020). Reproductive system infections in women: upper genital tract, fetal, neonatal and infant syndromes. Pathog Dis 78. 10.1093/femspd/ftaa023. [DOI] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, and Lee JJ (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7. 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AL, Johnson KA, Lee JK, Sütterlin C, and Tan M (2012). CPAF: a Chlamydial protease in search of an authentic substrate. PLoS Pathog 8, e1002842. 10.1371/journal.ppat.1002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Chen D, Sharma J, Cheng W, Zhong Y, Liu K, Jensen J, Shain R, Arulanandam B, and Zhong G (2006). The hypothetical protein CT813 is localized in the Chlamydia trachomatis inclusion membrane and is immunogenic in women urogenitally infected with C. trachomatis. Infect Immun 74, 4826–4840. 10.1128/iai.00081-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coers J (2013). Self and non-self discrimination of intracellular membranes by the innate immune system. PLoS Pathog 9, e1003538. 10.1371/journal.ppat.1003538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coers J, Bernstein-Hanley I, Grotsky D, Parvanova I, Howard JC, Taylor GA, Dietrich WF, and Starnbach MN (2008). Chlamydia muridarum evades growth restriction by the IFN-gamma-inducible host resistance factor Irgb10. J Immunol 180, 6237–6245. 10.4049/jimmunol.180.9.6237. [DOI] [PubMed] [Google Scholar]
- Coers J, Gondek DC, Olive AJ, Rohlfing A, Taylor GA, and Starnbach MN (2011). Compensatory T cell responses in IRG-deficient mice prevent sustained Chlamydia trachomatis infections. PLoS Pathog 7, e1001346. 10.1371/journal.ppat.1001346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehoux P, Flores R, Dauga C, Zhong G, and Subtil A (2011). Multi-genome identification and characterization of chlamydiae-specific type III secretion substrates: the Inc proteins. BMC Genomics 12, 109. 10.1186/1471-2164-12-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V (2021). Autophagy in inflammation, infection, and immunometabolism. Immunity 54, 437–453. 10.1016/j.immuni.2021.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmyter J, Melnick JL, and Rawls WE (1968). Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J Virol 2, 955–961. 10.1128/JVI.2.10.955-961.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmar G, and Winklhofer KF (2019). Linear Ubiquitin Chains: Cellular Functions and Strategies for Detection and Quantification. Front Chem 7, 915. 10.3389/fchem.2019.00915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockterman J, and Coers J (2021). Immunopathogenesis of genital Chlamydia infection: insights from mouse models. Pathog Dis 79. 10.1093/femspd/ftab012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell C, Mirrashidi K, and Engel J (2016). Chlamydia cell biology and pathogenesis. Nat Rev Microbiol 14, 385–400. 10.1038/nrmicro.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeley EM, Pilla-Moffett DM, Zwack EE, Piro AS, Finethy R, Kolb JP, Martinez J, Brodsky IE, and Coers J (2017). Galectin-3 directs antimicrobial guanylate binding proteins to vacuoles furnished with bacterial secretion systems. Proc Natl Acad Sci U S A 114, E1698–E1706. 10.1073/pnas.1615771114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finethy R, and Coers J (2016). Sensing the enemy, containing the threat: cell-autonomous immunity to Chlamydia trachomatis. FEMS Microbiol Rev 40, 875–893. 10.1093/femsre/fuw027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebel AM, Hu S, Rajaram K, Finethy R, Toh E, Brothwell JA, Morrison SG, Suchland RJ, Stein BD, Coers J, et al. (2019). Genetic Screen in Chlamydia muridarum Reveals Role for an Interferon-Induced Host Cell Death Program in Antimicrobial Inclusion Rupture. mBio 10. 10.1128/mBio.00385-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habu T, and Harada KH (2021). UBC13 is an RNF213-associated E2 ubiquitin-conjugating enzyme, and Lysine 63-linked ubiquitination by the RNF213-UBC13 axis is responsible for angiogenic activity. FASEB Bioadv 3, 243–258. 10.1096/fba.2019-00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackstadt T, Scidmore-Carlson MA, Shaw EI, and Fischer ER (1999). The Chlamydia trachomatis IncA protein is required for homotypic vesicle fusion. Cell Microbiol 1, 119–130. 10.1046/j.1462-5822.1999.00012.x. [DOI] [PubMed] [Google Scholar]
- Haldar AK, Foltz C, Finethy R, Piro AS, Feeley EM, Pilla-Moffett DM, Komatsu M, Frickel EM, and Coers J (2015). Ubiquitin systems mark pathogen-containing vacuoles as targets for host defense by guanylate binding proteins. Proc Natl Acad Sci U S A 112, E5628–5637. 10.1073/pnas.1515966112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar AK, Piro AS, Finethy R, Espenschied ST, Brown HE, Giebel AM, Frickel EM, Nelson DE, and Coers J (2016). Chlamydia trachomatis Is Resistant to Inclusion Ubiquitination and Associated Host Defense in Gamma Interferon-Primed Human Epithelial Cells. mBio 7. 10.1128/mBio.01417-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht M, Bromberg Y, and Rost B (2015). Better prediction of functional effects for sequence variants. BMC Genomics 16 Suppl 8, S1. 10.1186/1471-2164-16-S8-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houzelstein D, Simon-Chazottes D, Batista L, Tokuda S, Langa Vives F, Flamand M, Montagutelli X, and Panthier JJ (2021). The ring finger protein 213 gene (Rnf213) contributes to Rift Valley fever resistance in mice. Mamm Genome 32, 30–37. 10.1007/s00335-020-09856-y. [DOI] [PubMed] [Google Scholar]
- Hu S, Wang Y, Gong Y, Liu J, Li Y, and Pan L (2018). Mechanistic Insights into Recognitions of Ubiquitin and Myosin VI by Autophagy Receptor TAX1BP1. J Mol Biol 430, 3283–3296. 10.1016/j.jmb.2018.06.030. [DOI] [PubMed] [Google Scholar]
- Huang J, and Brumell JH (2014). Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol 12, 101–114. 10.1038/nrmicro3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CM, and Fisher DJ (2013). Site-specific, insertional inactivation of incA in Chlamydia trachomatis using a group II intron. PloS one 8, e83989-e83989. 10.1371/journal.pone.0083989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. (2021a). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. (2021b). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada F, Aoki Y, Narisawa A, Abe Y, Komatsuzaki S, Kikuchi A, Kanno J, Niihori T, Ono M, Ishii N, et al. (2011). A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet 56, 34–40. 10.1038/jhg.2010.132. [DOI] [PubMed] [Google Scholar]
- Kokes M, Dunn JD, Granek JA, Nguyen BD, Barker JR, Valdivia RH, and Bastidas RJ (2015). Integrating chemical mutagenesis and whole-genome sequencing as a platform for forward and reverse genetic analysis of Chlamydia. Cell Host Microbe 17, 716–725. 10.1016/j.chom.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kędzior M, and Bastidas RJ (2019). Forward and Reverse Genetic Analysis of Chlamydia. Methods Mol Biol 2042, 185–204. 10.1007/978-1-4939-9694-0_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labuda JC, and McSorley SJ (2018). Diversity in the T cell response to Chlamydia-sum are better than one. Immunol Lett 202, 59–64. 10.1016/j.imlet.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, and Valen E (2019). CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res 47, W171-W174. 10.1093/nar/gkz365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liehl P, Zuzarte-Luis V, and Mota MM (2015). Unveiling the pathogen behind the vacuole. Nat Rev Microbiol 13, 589–598. 10.1038/nrmicro3504. [DOI] [PubMed] [Google Scholar]
- Liu W, Morito D, Takashima S, Mineharu Y, Kobayashi H, Hitomi T, Hashikata H, Matsuura N, Yamazaki S, Toyoda A, et al. (2011). Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS One 6, e22542. 10.1371/journal.pone.0022542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chen C, Gong S, Hou S, Qi M, Liu Q, Baseman J, and Zhong G (2014). Transformation of Chlamydia muridarum reveals a role for Pgp5 in suppression of plasmid-dependent gene expression. J Bacteriol 196, 989–998. 10.1128/JB.01161-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowden NM, Yeruva L, Johnson CM, Bowlin AK, and Fisher DJ (2015). Use of aminoglycoside 3’ adenyltransferase as a selection marker for Chlamydia trachomatis intron-mutagenesis and in vivo intron stability. BMC Res Notes 8, 570. 10.1186/s13104-015-1542-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina L, Asselman C, Thery F, Boucher K, Delhaye L, Maia TM, Dermaut B, Eyckerman S, and Impens F (2021). Proteome Profiling of RNF213 Depleted Cells Reveals Nitric Oxide Regulator DDAH1 Antilisterial Activity. Front Cell Infect Microbiol 11, 735416. 10.3389/fcimb.2021.735416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvar B, Raghuram V, Pisitkun T, Sarkar A, and Knepper MA (2016). Comprehensive database of human E3 ubiquitin ligases: application to aquaporin-2 regulation. Physiol Genomics 48, 502–512. 10.1152/physiolgenomics.00031.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita H, Oikawa D, Terawaki S, Kabata D, Shintani A, and Tokunaga F (2021). Crosstalk Between NDP52 and LUBAC in Innate Immune Responses, Cell Death, and Xenophagy. Front Immunol 12, 635475. 10.3389/fimmu.2021.635475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa S, Oikawa D, Ishii R, Ayaki T, Takahashi H, Takeda H, Ishitani R, Kamei K, Takeyoshi I, Kawakami H, et al. (2016). Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat Commun 7, 12547. 10.1038/ncomms12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen BD, and Valdivia RH (2012). Virulence determinants in the obligate intracellular pathogen Chlamydia trachomatis revealed by forward genetic approaches. Proc Natl Acad Sci U S A 109, 1263–1268. 10.1073/pnas.1117884109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noad J, von der Malsburg A, Pathe C, Michel MA, Komander D, and Randow F (2017). LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-kappaB. Nat Microbiol 2, 17063. 10.1038/nmicrobiol.2017.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omotade TO, and Roy CR (2019). Manipulation of Host Cell Organelles by Intracellular Pathogens. Microbiol Spectr 7. 10.1128/microbiolspec.BAI-0022-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otten EG, Werner E, Crespillo-Casado A, Boyle KB, Dharamdasani V, Pathe C, Santhanam B, and Randow F (2021). Ubiquitylation of lipopolysaccharide by RNF213 during bacterial infection. Nature 594, 111–116. 10.1038/s41586-021-03566-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng YC, and Lenschow DJ (2018). ISG15 in antiviral immunity and beyond. Nat Rev Microbiol 16, 423–439. 10.1038/s41579-018-0020-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poston TB, and Darville T (2018). Chlamydia trachomatis: Protective Adaptive Responses and Prospects for a Vaccine. Curr Top Microbiol Immunol 412, 217–237. 10.1007/82_2016_6. [DOI] [PubMed] [Google Scholar]
- Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, and Willer CJ (2010). LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337. 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, and Sham PC (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575. 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KH, Sigar IM, Schripsema JH, Denman CJ, Bowlin AK, Myers GA, and Rank RG (2009). Strain and virulence diversity in the mouse pathogen Chlamydia muridarum. Infect Immun 77, 3284–3293. 10.1128/IAI.00147-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell M, Darville T, Chandra-Kuntal K, Smith B, Andrews CW Jr., and O’Connell CM (2011). Infectivity acts as in vivo selection for maintenance of the chlamydial cryptic plasmid. Infect Immun 79, 98–107. 10.1128/IAI.01105-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka HA, Thompson JW, Chen YS, Kumar Y, Dubois LG, Moseley MA, and Valdivia RH (2011). Quantitative proteomics reveals metabolic and pathogenic properties of Chlamydia trachomatis developmental forms. Mol Microbiol 82, 1185–1203. 10.1111/j.1365-2958.2011.07877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schork AJ, Thompson WK, Pham P, Torkamani A, Roddey JC, Sullivan PF, Kelsoe JR, O’Donovan MC, Furberg H, Tobacco, et al. (2013). All SNPs are not created equal: genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS Genet 9, e1003449. 10.1371/journal.pgen.1003449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixt BS, Bastidas RJ, Finethy R, Baxter RM, Carpenter VK, Kroemer G, Coers J, and Valdivia RH (2017). The Chlamydia trachomatis Inclusion Membrane Protein CpoS Counteracts STING-Mediated Cellular Surveillance and Suicide Programs. Cell Host Microbe 21, 113–121. 10.1016/j.chom.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soules KR, Dmitriev A, LaBrie SD, Dimond ZE, May BH, Johnson DK, Zhang Y, Battaile KP, Lovell S, and Hefty PS (2020). Structural and ligand binding analyses of the periplasmic sensor domain of RsbU in Chlamydia trachomatis support a role in TCA cycle regulation. Mol Microbiol 113, 68–88. 10.1111/mmi.14401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturdevant GL, Kari L, Gardner DJ, Olivares-Zavaleta N, Randall LB, Whitmire WM, Carlson JH, Goheen MM, Selleck EM, Martens C, and Caldwell HD (2010). Frameshift mutations in a single novel virulence factor alter the in vivo pathogenicity of Chlamydia trachomatis for the female murine genital tract. Infect Immun 78, 3660–3668. 10.1128/IAI.00386-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team, R.C. (2020). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL: http://www.R-project.org/. [Google Scholar]
- Thery F, Martina L, Asselman C, Zhang Y, Vessely M, Repo H, Sedeyn K, Moschonas GD, Bredow C, Teo QW, et al. (2021). Ring finger protein 213 assembles into a sensor for ISGylated proteins with antimicrobial activity. Nat Commun 12, 5772. 10.1038/s41467-021-26061-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, and Randow F (2012). Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418. 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi-Giesgen I, Behrends C, and Alpi AF (2021). The ubiquitin ligation machinery in the defense against bacterial pathogens. EMBO Rep 22, e52864. 10.15252/embr.202152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbarello DA, Manna PT, Allen M, Bycroft M, Arden SD, Kendrick-Jones J, and Buss F (2015). The Autophagy Receptor TAX1BP1 and the Molecular Motor Myosin VI Are Required for Clearance of Salmonella Typhimurium by Autophagy. PLoS Pathog 11, e1005174. 10.1371/journal.ppat.1005174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Pittman KJ, Barker JR, Salinas RE, Stanaway IB, Williams GD, Carroll RJ, Balmat T, Ingham A, Gopalakrishnan AM, et al. (2018). An Atlas of Genetic Variation Linking Pathogen-Induced Cellular Traits to Human Disease. Cell Host Microbe 24, 308–323 e306. 10.1016/j.chom.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, and Clarke IN (2011). Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog 7, e1002258. 10.1371/journal.ppat.1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MM, Bauler LD, Lam J, and Hackstadt T (2015). Expression and localization of predicted inclusion membrane proteins in Chlamydia trachomatis. Infect Immun 83, 4710–4718. 10.1128/IAI.01075-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MM, and Faris R (2019). Mutagenesis of Chlamydia trachomatis Using TargeTron. Methods Mol Biol 2042, 165–184. 10.1007/978-1-4939-9694-0_12. [DOI] [PubMed] [Google Scholar]
- Weber MM, Lam JL, Dooley CA, Noriea NF, Hansen BT, Hoyt FH, Carmody AB, Sturdevant GL, and Hackstadt T (2017). Absence of Specific Chlamydia trachomatis Inclusion Membrane Proteins Triggers Premature Inclusion Membrane Lysis and Host Cell Death. Cell Rep 19, 1406–1417. 10.1016/j.celrep.2017.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WF, Chambers JP, Gupta R, and Arulanandam BP (2019). Chlamydia and Its Many Ways of Escaping the Host Immune System. J Pathog 2019, 8604958. 10.1155/2019/8604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Lei L, Collins JWM, Briones M, Ma L, Sturdevant GL, Su H, Kashyap AK, Dorward D, Bock KW, et al. (2021). Chlamydia evasion of neutrophil host defense results in NLRP3 dependent myeloid-mediated sterile inflammation through the purinergic P2X7 receptor. Nat Commun 12, 5454. 10.1038/s41467-021-25749-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers used in this study, related to STAR Methods.
Data Availability Statement
Raw files for whole-genome sequencing of Chlamydia library mutants have been deposited to the Sequence Read Archive (NCBI, SRA: PRJNA865907) and will be available at the time of publication. Plasmid maps or 3D structure predictions reported in this paper will be shared by the lead contact upon request. Original images used for microscopy and western blotting have been deposited to Mendeley Data (doi: 10.17632/hkvx8gx2ft.1) and will be available at the time of publication. Genome-wide association studies (GWAS) data is publicly available from a previous publication (Wang et al., 2018).
Original code used to generate Quantile-Quantile plots (Figure 5A) is included at the end of the STAR Methods (Methods S1).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.