

Abstract
The nuclear corepressors NCOR1 and NCOR2 interact with transcription factors involved in B cell development and potentially link these factors to alterations in chromatin structure and gene expression. Herein we demonstrate that Ncor1/2 deletion limits B cell differentiation via impaired recombination, attenuates pre-BCR-signaling, and enhances STAT5-dependent transcription. Furthermore, NCOR1/2-deficient B cells exhibited derepression of EZH2-repressed gene modules, including the p53 pathway. These alterations resulted in aberrant Rag1 and Rag2 expression and accessibility. Whole-genome sequencing of Ncor1/2 DKO B cells identified increased number of structural variants with cryptic recombination signal sequences. Finally, deletion of Ncor1 alleles in mice facilitated leukemic transformation, while human leukemias with less NCOR1 correlated with worse survival. NCOR1/2 mutations in human leukemia correlated with increased RAG expression and number of structural variants. These studies illuminate how the corepressors NCOR1/2 regulate B cell differentiation and provide insights into how NCOR1/2 mutations may promote B cell transformation.
Editor summary:
Farrar and colleagues perform an extensive analysis of Ncor1/2 function in B cell development. Loss of both genes results in defective pre-BCR signaling, increased accessibility of STAT5 chromatin motifs and inappropriate Rag gene expression, leading to accelerated leukemic transformation.
The adaptive B cell response is dependent on a diverse repertoire of mature B cells. To ensure that sufficient quantity and quality of mature B cells are generated, developing B cells undergo alternating cycles of proliferation and differentiation. This process requires specific integration of cytokine and antigen receptor signaling pathways to drive appropriate proliferation, and to establish developmental checkpoints after recombination that ensure functional, non-self-reactive, BCRs are generated1. These diverse and complex actions impinge on B-lymphoid transcription factors that shape B cell development. For example, the divergent proliferation pathways dependent on cytokine signaling versus antigen receptor signaling are preserved via the opposing regulation of IL-7R/STAT5 and pre-BCR/IKZF1/NFκB-signaling pathways2. Thus, strict regulation of these pathways is critical, as perturbations and defects lead to B-cell acute lymphoblastic leukemias (B-ALLs)3–5.
B-lymphoid transcription factors regulate the epigenetic and transcriptional landscape via the recruitment of transcriptional cofactors. Co-repressors play a critical role in this process by linking histone deacetylases to transcription factors, thereby allowing them to drive gene repression. However, the role that specific corepressors play in modulating development, and the mechanism by which they segregate proliferation and differentiation, remain incompletely understood. The nuclear corepressors NCOR1 and NCOR2 interact with multiple transcription factors6. This suggests that NCOR1/2 may have ubiquitous, yet stage-specific roles during development. The corepressors NCOR1/2 interact with multiple histone deacetylases to repress target gene transcription, although they appear particularly critical for driving activity of HDAC36,7. Thus, NCOR1/2 likely play key roles in translating transcription factor binding into alterations in histone acetylation. Finally, mutations and deletions in NCOR1/2 have been found in B cell malignancies, suggesting that NCOR1/2 may play important tumor suppressor roles8,9, yet their role in malignant transformation remain unknown. To understand how the co-repressors NCOR1/2 are involved in governing various stages of B cell development, and the role they play in B-ALL, we generated Cd79a-Cre × Ncor1FL/FL and Cd79a-Cre × Ncor1FL/FL × Ncor2 FL/FL mice. Our studies demonstrate that NCOR1/2 play non-redundant roles in B cell development that affect STAT5, FOXO1, and p53-dependent signaling pathways, RAG gene expression, chromatin stability, and B cell transformation.
Results
Nuclear corepressors are required during B cell development
Nuclear corepressors NCOR1 and NCOR2 are critical for fetal development as deletion of either protein alone is embryonically lethal10,11. This suggests unique contributions of NCOR1 and NCOR2 during fetal development. However, NCOR1 and NCOR2 also share high (~40%) amino acid sequence homology with conserved functional domains6, suggesting similar biological functions. To overcome embryonic lethality of NCOR-knockouts, we utilized Ncor1FL/FL mice12, and generated a conditional Ncor2 knockout mouse using the flip-excision system13 (Extended Data Figure 1a). To understand the role of NCOR1 and NCOR2 during B cell development, we generated B cell conditional knockouts of Ncor1 (Cd79a-Cre × Ncor1FL/FL; NCOR1 KO), Ncor2 (Cd79a-Cre × Ncor2FL/FL; NCOR2 KO), and Ncor1/Ncor2 double-knockouts (Cd79a-Cre × Ncor1FL/FL × Ncor2FL/FL; NCOR1/2 DKO). We also generated mice lacking Ncor2 and one allele of Ncor1 (Cd79a-Cre × Ncor1FL/+ × Ncor2FL/FL; NCOR1 het/NCOR2 KO) or Ncor1 and one allele of Ncor2 (Cd79a-Cre × Ncor1FL/FL × Ncor2FL/+; NCOR1 KO/NCOR2 het) in B cells to assess gene dosage relationships. Using flow cytometry, we found efficient deletion of NCOR1 in NCOR1 KO and NCOR1/2 DKO B cells in the bone marrow and spleen (Extended Data Figure 1b,c). Since specific antibodies for NCOR2 are unavailable, we also used qPCR to assess NCOR1/2 deletion efficiency. The NCOR1/2 DKO B cells in the bone marrow exhibited virtually complete reduction in Ncor1 expression and ~90% reduction in Ncor2 expression; splenic B cells also exhibited complete reduction in Ncor1 expression but only a moderate reduction in Ncor2 expression. Thus, B cells in the spleen of NCOR1/2 DKO mice retain some expression of Ncor2 (Extended Data Figure 1d). To validate deletion of Ncor1 and inversion of the Ncor2 gene trap at a genetic level we performed whole-genome sequencing on NCOR1/2 DKO bone marrow B cells. We confirmed the absence of reads spanning the deleted region in Ncor1 (Extended Data Figure 1e) and the inversion of the Ncor2 gene trap (Extended Data Figure 1f), indicating efficacious Cre-mediated recombination of both Ncor1 and Ncor2 in developing bone marrow B cells. Next, we used flow cytometry to subset B cell development into the B220+CD43+ fractions consisting of pre-pro-B (CD24−BP1−), pro-B (CD24+BP1−) and pro-B/pre-B cells (CD24+BP1+)14(Extended Data Figure 1g). The number of pro-B and pro-B/pre-B progenitor B cells was unchanged in NCOR1 KO, NCOR2 KO, NCOR1 het/NCOR2 KO and NCOR1 KO/NCOR2 het mice (Figure 1a,b). However, deleting both NCOR1 and NCOR2 (NCOR1/NCOR2 DKO) led to a dramatic reduction in pro-B and pro-B/pre-B proliferating B cells (Figure 1a,b). Loss of NCOR1 or NCOR2 had no effect on CD43−IgM− pre-B cells, immature B cells or mature B cells. In contrast, the number of immature B cells (B220lowCD43−IgM+) from NCOR1 KO/NCOR2 het mice was significantly reduced, while pre-B cells were unaffected (Figure 1c,d). Notably, NCOR1 KO/NCOR2 het B cells have a stronger defect than the NCOR1 het/NCOR2 KO B cells, indicating that developing B cells may be more sensitive to Ncor1 gene dosage. This is consistent with higher Ncor1 than Ncor2 transcript expression in B cells reported in the Immgen database15. Consistent with the strong block observed in proliferating pro-B cells, NCOR1/ NCOR2 DKO mice had the largest reductions in small pre-B, immature and mature B cells (Figure 1c,d). NCOR1 KO B cells had a modest reduction in splenic B cells and exhibited a Ncor gene dosage-dependent effect with stepwise decreases in B cells when additional Ncor alleles were deleted (Figure 1e,f). These results suggest differential sensitivity to Ncor1/2 gene dosage during proliferation of B cells and late-stage B cell differentiation and indicate that Ncor1 and Ncor2 are functionally redundant as deletion of both corepressors is required for a strong B cell developmental block.
Figure 1. B cell development requires nuclear corepressors NCOR1 and NCOR2 and is sensitive to their gene dosage.
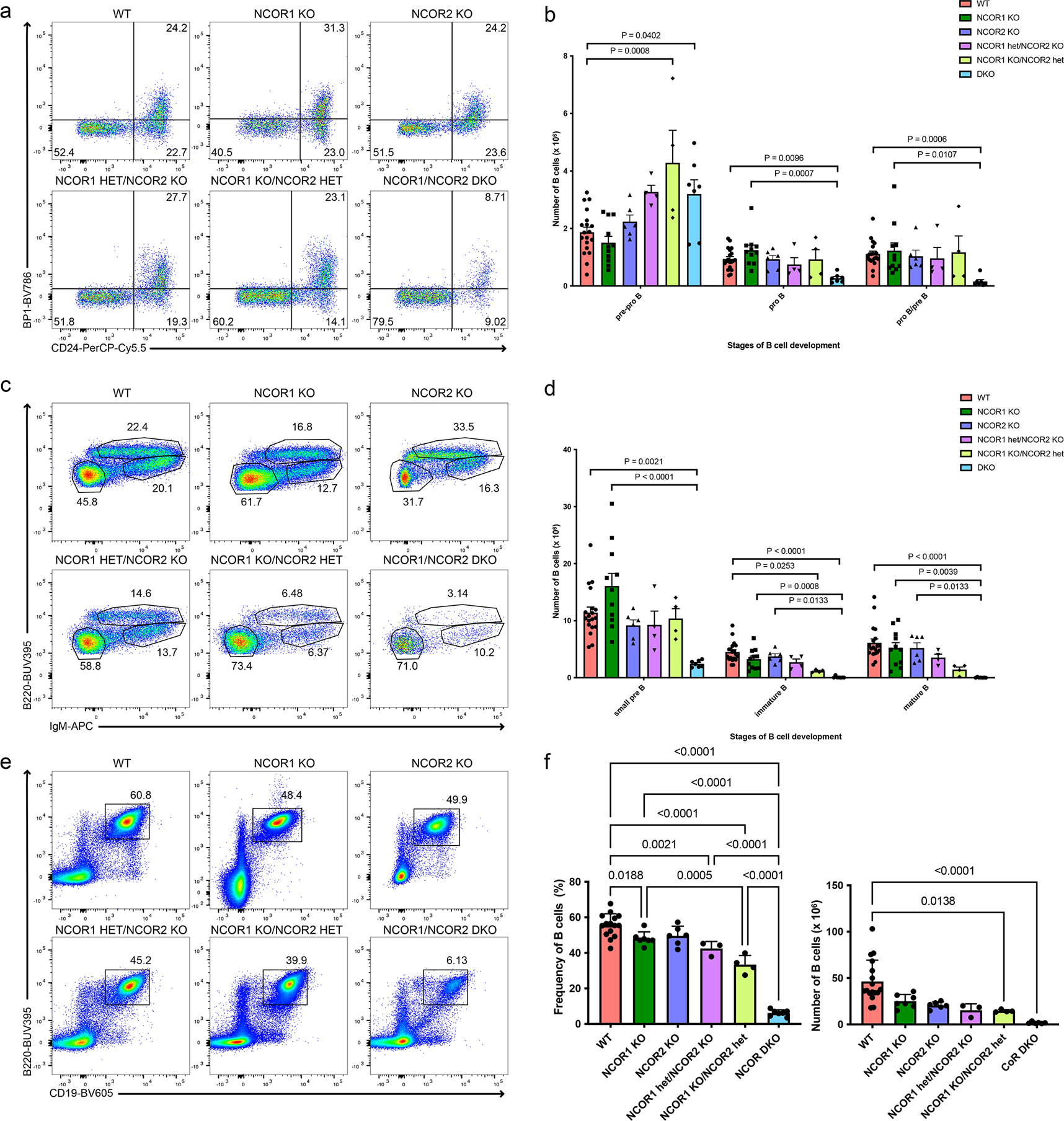
a. Representative flow cytometry plots of pre-pro-B, pro-B and pro-B/pre-B cells in wildtype (WT), NCOR1 KO, NCOR2 KO, NCOR1 het/NCOR2 KO, NCOR1 KO/NCOR2 het, and NCOR1/2 DKO. Cells were pre-gated on B220+CD43+ cells. Numbers in the flow cytometry plot represent the frequency of B220+CD43+ cells. b. Cell number of pre-pro-B, pro-B and pro-B/pre-B cells in the bone marrow among different NCOR knockouts (WT; n=19, NCOR1 KO; n=11, NCOR2 KO; n=6, NCOR1 het/NCOR2 KO; n=4, NCOR1 KO/NCOR2 het; n=4, NCOR1/2 DKO; n=7). An ordinary one-way ANOVA with Tukey’s multiple comparison was performed for the pre-pro-B cell population. A Kruskal-Wallis one-way ANOVA with Dunn’s multiple comparison was performed for the pro-B and pro-B/pre-B populations. c. Representative flow cytometry plots of small pre-B, immature B and mature B cells in different NCOR-knockouts. Numbers in the flow cytometry plot represent the frequency of B220+CD43− cells. d. Cell number of small pre-B, immature B, and mature B in the bone marrow among different NCOR knockouts (n for each genotype is same as in panel b). A Kruskal-Wallis one-way ANOVA with Dunn’s multiple comparison was performed for the small pre-B, immature B and mature B cell populations. e. Representative flow cytometry plots of splenic CD19+B220+ B cells in different NCOR-knockouts. f. Frequency (left) of splenic CD19+B220+ among different NCOR knockouts (WT; n=15, NCOR1 KO; n=7, NCOR2 KO; n=6, NCOR1 het/NCOR2 KO; n=3, NCOR1 KO/NCOR2 het; n=4, NCOR1/2 DKO; n=7) and cell number (right) of splenic CD19+B220+ cells among different knockouts (WT; n=17, NCOR1 KO; n=7, NCOR2 KO; n=6, NCOR1 het/NCOR2 KO; n=3, NCOR1 KO/NCOR2 het; n=4, NCOR1/2 DKO; n=6). An ordinary one-way ANOVA with Tukey’s multiple comparison was performed for statistical analysis of frequency and a Kruskal-Wallis one-way ANOVA with Dunn’s multiple comparison was performed for cell numbers. Number in each flow cytometry gate represents the frequency for the representative sample. Error bars represent standard deviation. All measures of centers indicate mean. Data was obtained from ≥ three independent experiments for all panels.
We next assessed the relationship between NCORs and proliferation by measuring the expression of NCOR1 and Ki67. Proliferating cells with high Ki67 also had high NCOR1 expression (Figure 2a). When NCOR1 expression was broken down by the stages of B cell development, the cycling pro-B and pro-B/pre-B cells had the highest NCOR1 expression (Figure 2b). Given the correlation between NCOR1 and Ki67 expression, we assessed whether Ki67 expression was impacted in NCOR1 KO and NCOR1/2 DKO B cells. Among the pre-pro-B, pro-B and pro-B/pre-B cells, Ki67 expression was lower in the NCOR1/2 DKO progenitor B cells (Figure 2c), suggesting that proliferation may be directly impaired in the absence of nuclear corepressors, or secondarily due to effects on other biological processes. We then assessed the role of nuclear corepressors in recombination by determining the ratio of kappa to lambda light chain usage in mature splenic B cells. In mice, the majority of BCRs utilize the kappa chain (>90%, Figure 2d). Deletion of NCOR1/2 corepressors led to a decrease in the frequency of cells with the kappa light chain, and a corresponding increase in lambda chain usage (Figure 2d,e). Thus, NCOR1/2-deficiency impairs kappa light chain rearrangement. To determine whether the defect in NCOR1/2 DKO was due to defects in assembling a functional BCR, we crossed NCOR1/2 DKO mice MD4-transgenic mice that carry a rearranged in-frame BCR16. The MD4 transgene only partially rescued the frequency of B cells in both the bone marrow and spleen (Figure 2f), suggesting that recombination defects alone were not the reasons for the developmental block we observed. Together, these results suggest that nuclear corepressors have ubiquitous and critical roles during both proliferation and differentiation phases of B cell development.
Figure 2. Nuclear corepressors regulate both proliferation and differentiation during B cell development.
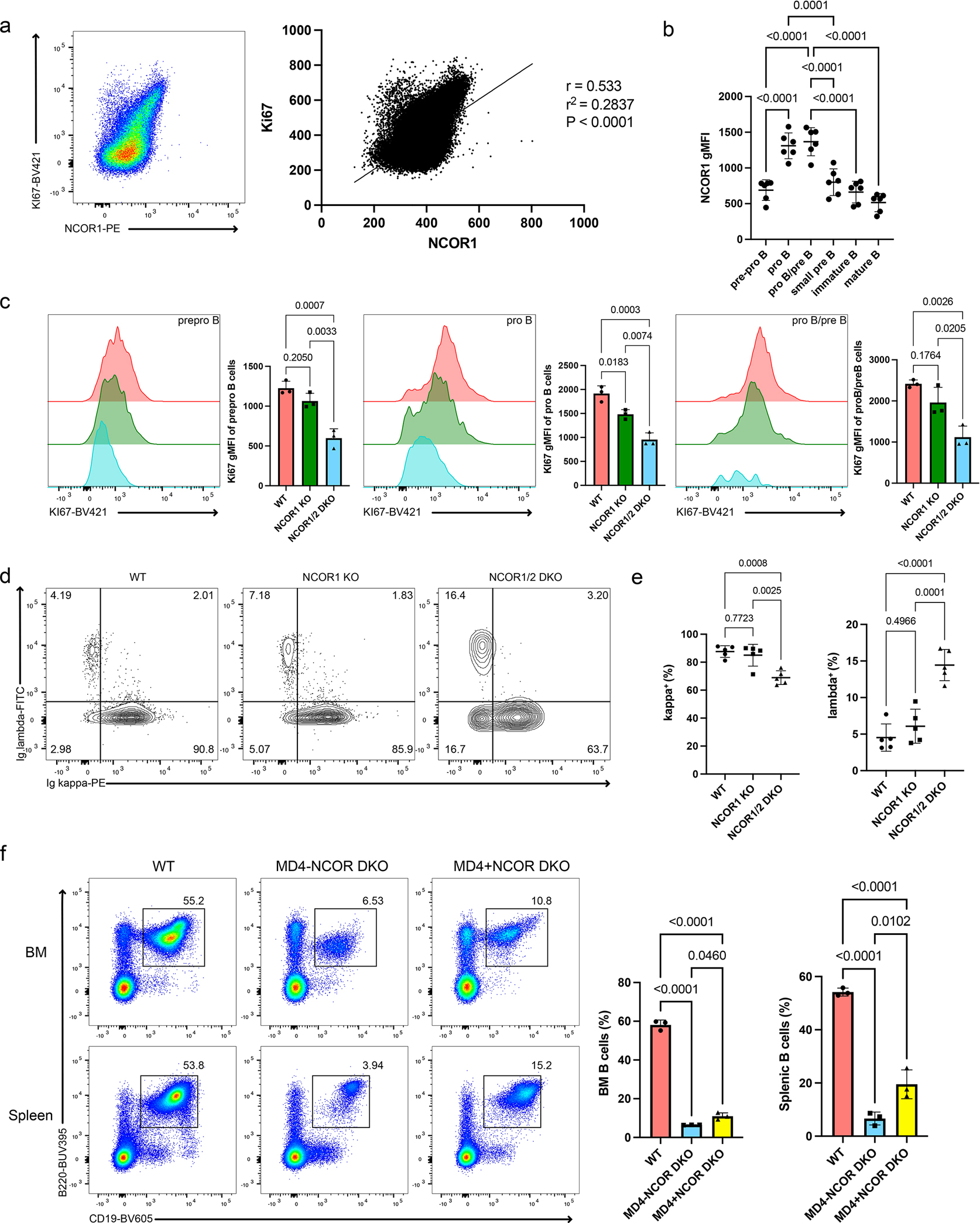
a. NCOR1 expression regulation in proliferating and non-proliferating cells. Representative flow cytometry plot of CD19+B220+ bone marrow B cells and the correlation between Ki67 and NCOR1 expression (left). Pearson correlation test (r and r2) between Ki67 and NCOR1 expression (right). Flow cytometry plot is representative of 6 mice from three independent experiments. b. NCOR1 gMFI expression in different B cell development stages. Datapoints were obtained from three independent experiments. c. Representative Ki67 expression differences between WT, NCOR1 KO, and NCOR1/2 DKO B cells in pre-pro-B, pro-B and pro-B/pre-B cells. Cells were pre-gated on B220+CD43+ B cells, which comprise the majority of proliferating progenitor B cells. Corresponding Ki67 gMFI is plotted on the right. Flow cytometry plot represents n=3 for each genotype derived from two independent experiments. d.e. Representative flow cytometry plots of the kappa:lambda ratio of splenic CD19+B220+ B cells (d) and frequency of Ig κ and Ig λ between WT, NCOR1 KO and NCOR1/2 DKO (e). Data represent n=5 mice for each genotype from three independent experiments. f. Bypassing recombination defects via MD4 BCR-transgene expression. CD19+B220+ B cell frequency in the bone marrow and spleen represent n=3 for each genotype from three independent experiments. An ordinary one-way ANOVA with Tukey’s multiple comparison was performed for statistical analysis; error bar represents standard deviation. All measure of centers indicate mean.
Ncor1/Ncor2 deletion alters pre-BCR and STAT5 signaling
To assess the transcriptional and epigenetic networks that are regulated by NCOR1/2, we performed single-cell RNA-seq (scRNA-seq) and single-cell ATAC-seq (scATAC-seq). For single-cell RNA-sequencing, B cells from wildtype (n=2), NCOR1 KO (n=2) and NCOR1/2 DKO (n=2) were indexed with hashtag antibodies and stained with 6 CITE-seq antibodies (B220, CD19, CD93, CD43, IgM, CD25), sorted at a 1:1 ratio of CD43+ and CD43− cells to enrich for the earlier progenitor B cells and captured together (Figure 3a). scATAC-seq samples were captured separately from WT (n=1), NCOR1 KO (n=1), and NCOR1/2 DKO mice (n=1) and B cell subsets enriched using the same flow sorting scheme as for scRNA-seq. For scRNA-seq, multiplets were removed based on hashtag antibodies (Extended Data Figure 2a), resulting in 23,897 singlets. Cluster identification of the different B cell development stages was performed based on our previous characterization of WT B cells (Extended Data Figure 2b,c)17. Similar to our flow cytometry analysis, the CD43− fractions showed reductions in immature and mature B cells in mice deficient in NCOR1/2 (Figure 3b,c). Notably, the frequency of pre-BCR-dependent stage pre-B cells was significantly reduced in NCOR1/2 DKOs (Figure 3b,c). We next assessed genes associated with the pre-BCR-dependent stage to identify transcriptional changes associated with the lack of nuclear corepressors. Wildtype, NCOR1 KO, and NCOR1/2 DKO B cells had comparable levels of Ybx1, Ybx3, and Myc expression, which define the pre-BCR dependent cluster. The other pre-BCR dependent subset defining gene Nrgn exhibited a stepwise decrease in expression in NCOR1 KO and NCOR1/2 DKOs (Figure 3d, Supplementary Table 1). Similarly, Ebf1 expression, which is repressed during the pre-BCR-dependent stage17, was derepressed in the NCOR1/2 DKOs. These results demonstrate both quantitative and qualitative defects in the pre-BCR-dependent subset. To corroborate this effect, we measured the expression of IKZF1, a B-lymphoid transcription factor involved in terminating pre-B cell proliferation18. IKZF1 expression was significantly lower in small pre-B cells from the NCOR1/2 DKO versus WT mice (Figure 3e). This finding suggests that defective pre-BCR signaling in NCOR1/2 DKO pre-B cells results in a failure to upregulate IKZF1.
Figure 3. Absence of nuclear corepressors results in defective pre-BCR signaling and in turn promotes STAT5-mediated transcription.
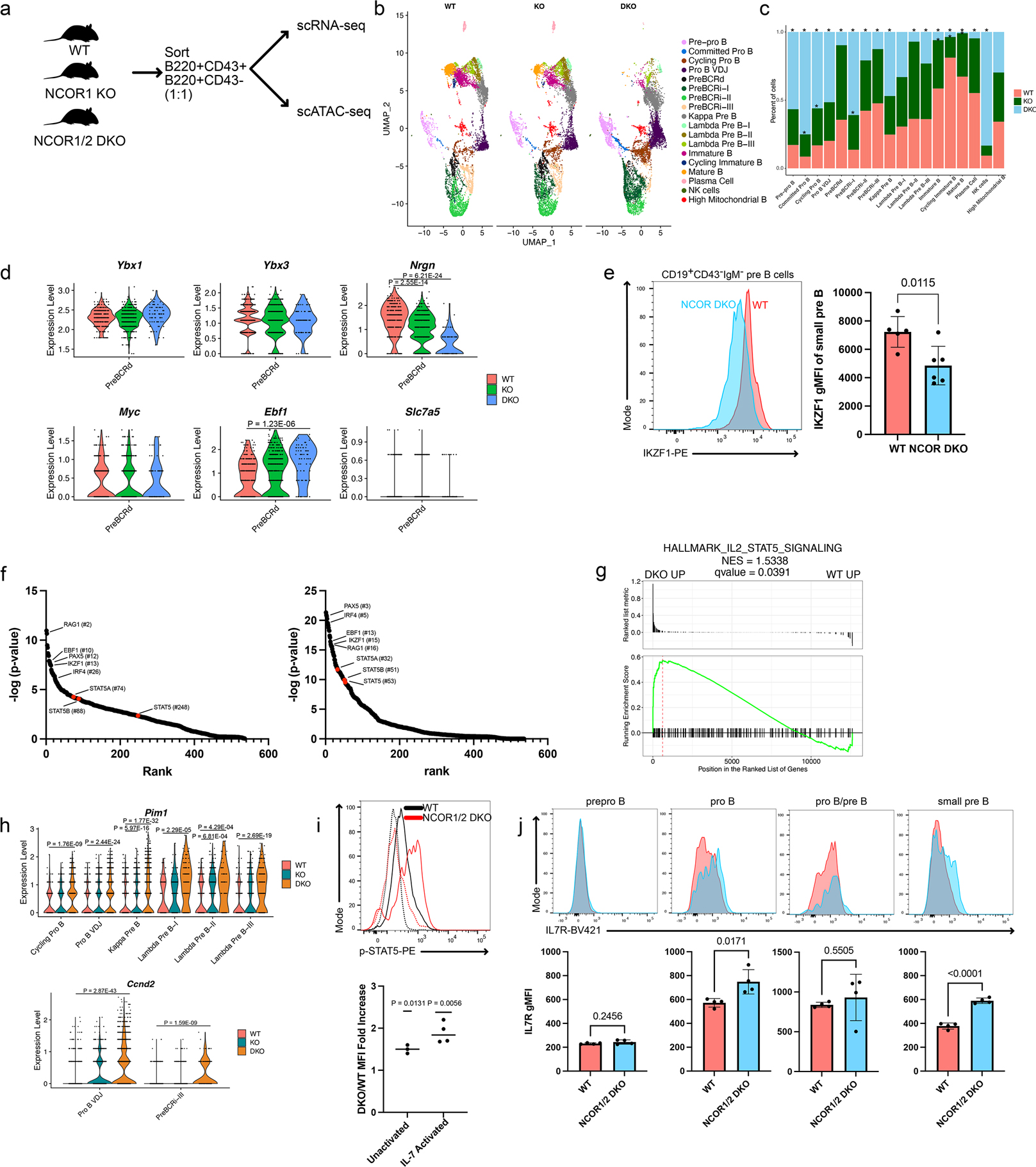
a. Schematic for scRNAseq/scATACseq in WT, NCOR1 KO and NCOR1/2 DKO B cells. b. scRNA-seq feature plot split by genotype. c. Distribution of the cells from each genotype that fall in different clusters. Asterisk (* = FDR <0.05) above the bar indicates statistically significant distribution differences when compared to the wildtype sample distribution derived from a MonteCarlo permutation test. d. Violin plot of genes that are differentially regulated in the Pre-BCR-dependent cluster. Data represents WT (n =301 cells), NCOR1 KO (n=463 cells) and NCOR1/2 DKO (n=82 cells). e. IKZF1 expression in small pre-B cells between WT (n = 5) and NCOR1/2 DKO (n = 6) B cells from 3 independent experiments. A two-sided t-test was used to calculate statistical significance. f. LISA-based prediction of transcriptional regulators of significantly upregulated genes in either WT (left) or NCOR1/2 DKO (right) kappa pre-B cells. Number in parentheses indicates rank. g. Gene set enrichment analysis of kappa pre-B cells between WT and NCOR1/2 DKO B cells. h. Expression differences of STAT5-target genes Pim1 and Ccnd2 between WT, NCOR1 KO and NCOR1/2 DKO. Data represents WT (n=7454 cells), NCOR1 KO (n=7806 cells) and NCOR1/2 DKO (n=8637 cells). i. p-STAT5 expression of CD19lowB220low progenitor B cells from wildtype (black line) and NCOR1/2 DKO (red line). Activated (solid line) and non-activated (dashed line) levels are shown in the representative flow cytometry plot (top) and the fold change difference in p-STAT5 MFI expression between DKO and WT (bottom). A two-tailed one-sample t-test was performed to determine statistical significance. Data points represent three (unactivated; n=3) or four (activated; n=4) independent experiments. j. IL-7R expression in pre-pro-B, pro-B, pre-B and small pre-B cells. P-values were determined by a two-tailed t-test. Data points were derived from WT (n=4) and NCOR1/2 DKO (n=4) B cells from two independent experiments. All centers of measure indicate mean, except panel i, which indicates median. All error bars indicate standard deviation. A two-sided Wilcoxon rank-sum test was used to compute p-values in panels d and h.
We previously demonstrated that STAT5 and pre-BCR-signaling antagonize each other, as defects in pre-BCR signaling components cooperated with STAT5 to drive progenitor B cell transformation2. Recent evidence also suggests that pre-BCR-related ERK signaling and IL-7R/STAT5 cytokine signaling promote distinct proliferation pathways that when present together subvert leukemic transformation19. Therefore, we assessed whether STAT5 signaling was altered. We first performed a Landscape In Silico Analysis (LISA), which predicts transcription factor regulators of differentially expressed gene sets. Genes upregulated in wildtype kappa-expressing pre-B cells had low STAT5A, STAT5B, and STAT5 signature ranks (74, 88, 248, respectively, Figure 3f). In contrast, genes upregulated in the NCOR1/2 DKO kappa+ pre-B cells had higher STAT5A, STAT5B, and STAT5 signature ranks (32, 51, 53, respectively; Figure 3f). We also performed a gene set enrichment analysis (GSEA) on differentially regulated genes from kappa+ pre-B cells. The upregulated genes in NCOR1/2 DKO small pre-B cells were enriched for a STAT5 gene expression signature (Figure 3g). We next examined known STAT5 target genes, Pim1 and Ccnd2, that are directly activated by STAT52,20. In the case of Pim1, kappa+ pre-B and lambda+ pre-B-II clusters had increased Pim1 expression in NCOR1 KO B cells (Figure 3h). NCOR1/2 DKO B cells had higher Pim1 expression in multiple clusters, including cycling pro-B, pro-B VDJ, and light-chain rearranging clusters (Figure 3h). Likewise, Ccnd2 was upregulated in NCOR1/2 DKO B cells (Figure 3h). To assess the presence of heightened STAT5 signaling, we measured p-STAT5 expression in WT and NCOR1/2 DKO B cells at steady-state or after ex vivo stimulation with IL-7. NCOR DKO B cells had significantly higher expression of p-STAT5 than wildtype B cells in both steady state (unactivated) and upon ex vivo stimulation with IL-7 (10 ng/μL) (Figure 3i). Increased p-STAT5 levels in NCOR DKO B cells upon IL-7 stimulation may be partially attributed to increased IL-7R expression on pro-B and pre-B cells in DKO B cells (Figure 3j). Overall, these results indicate that NCOR1/2 are critical for segregating divergent pathways of proliferation (pre-BCR versus cytokine dependent signaling pathways), with the absence of NCOR1/2 resulting in defective pre-BCR signaling and upregulation of STAT5 pathways.
NCORs govern the epigenetic landscape of developing B cells
Using scATAC-seq, we queried how the epigenetic landscape was altered with NCOR-deficiency in B cells. We identified B cell developmental stages by correlating gene accessibility patterns with gene expression levels from scRNA-Seq data (Figure 4a,b, Extended Data Figure 2b). We first evaluated the differences in transcription factor motifs found in open chromatin between WT, NCOR1 KO, and NCOR1/2 DKO cells. Transcription factor motifs enriched in pre-pro-B cells, such as RUNX, TCF and SPI motifs, were significantly higher in NCOR DKO B cells than WT B cells throughout development, whereas motifs enriched in mature B cells, such as POU, were decreased in NCOR DKO B cells (Figure 4c, Supplementary Table 2). WT B cells had a significant stepwise enrichment of CTCF motifs in open chromatin compared to NCOR1 KO and NCOR1/2 DKO cells, respectively (Figure 4d, Supplementary Table 2). As CTCF is important for Igh locus contraction, depletion of CTCF motifs may contribute to VDJ recombination defects. We also assessed whether accessibility in the immunoglobulin regions contributes to recombination defects in NCOR-knockout B cells. While the heavy chain locus in pro-B VDJ cells and the lambda light chain locus in Kappa/Lambda pre-B cells showed minimal changes in accessibility, the kappa light chain locus in the Kappa/Lambda pre-B cells had significantly decreased accessibility (Figure 4e). This decrease in accessibility likely explains the decreased kappa light chain usage in NCOR DKO B cells.
Figure 4. Single-cell ATAC-seq reveals the epigenetic landscape regulated by nuclear corepressors.
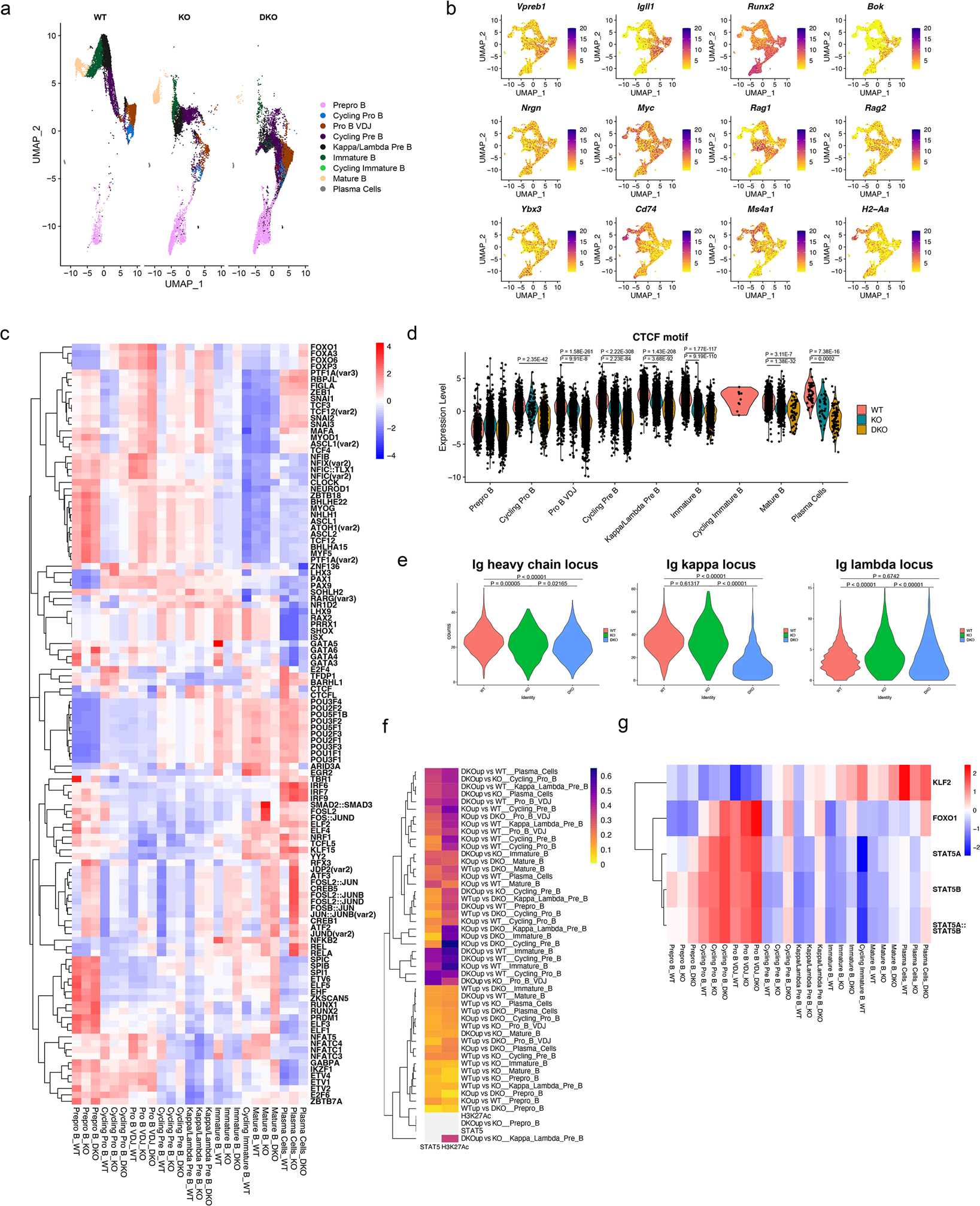
a. scATAC-seq feature plot split by genotype. b. Accessibility of stage-defining genes shown as a feature plot. c. Heatmap of transcription factor motifs that are differentially present in the WT, NCOR1 KO and NCOR1/2 DKO cells in different clusters. Transcription factor motif analysis was performed using chromVar. Scale for the heatmap represents row z-scores. d. chromVar motif score for CTCF in different clusters split by the different genotypes. A two-tailed Wilcoxon rank sum test was used to compute the p-value for pairwise comparisons with at least a log2 fold change of 0.25. Data represents WT (n=8852 cells), NCOR1 KO (n=6008 cells) and DKO (n=8574 cells) from a single biologically replicate derived from three separate captures. e. Accessibility of the immunoglobulin heavy chain locus in pro-B VDJ cluster, and κ and λ chain locus accessibility in the Kappa/Lambda Pre-B cell cluster amongst wildtype, NCOR1 KO and NCOR1/2 DKO cells. A one-way ANOVA was performed to compute the statistical significance in differences. f. Heatmap denoting the fraction of overlap between differentially accessible regions and H3K27Ac and STAT5 sites. H3K27Ac and STAT5 ChIP-seq data were obtained from GSM463433 and GSM2309799, respectively. Heatmap was clustered on rows. Color scale represents the fraction of overlap. g. Heatmap of chromvar motif score for KLF2, FOXO1 and STAT5 motifs in different clusters. Color scale represents row z-score.
NCORs interact with HDAC3, which regulates the H3K27Ac landscape. We examined whether differentially accessible peaks found in NCOR-knockouts overlapped with H3K27Ac sites21,22. Since NCORs regulate STAT5 function, we also examined whether the differentially accessible peaks found in the NCOR-knockouts overlapped with STAT5 binding sites. Sites with increased accessibility in NCOR-knockout B cells (ex. DKOup vs WT, KOup vs WT, DKOup vs KO) shared a higher degree of overlap with STAT5 binding sites and H3K27Ac regions, compared to the sites that were differentially upregulated in the NCOR-intact B cells (ex. WTup vs DKO, WTup vs KO, KOup vs DKO) (Figure 4f). Finally, we compared the corresponding clusters for overlap between genes that were upregulated in expression (Supplementary Table 1) and accessibility (Supplementary Table 3) upon NCOR1/2 deletion. In accordance with the increased STAT5 signature in NCOR-knockout B cells, STAT5 target genes (Socs2, Ccnd2, Pim1, Mcl1, Gimap6, and Rhoh) were upregulated in expression and accessibility (Extended Data Figure 3). In addition, we found increased expression and accessibility in V(D)J recombination genes (Foxo1, Rag1, and Dntt), DNA damage and repair genes (Shisa5, Asf1a, Btg1, Btg2, and Apobec3), and cellular localization and egress genes (Klf2 and Cd69) (Extended Data Figure 3). To determine whether increased expression and accessibility of transcription factors (STAT5, FOXO1, and KLF2) resulted in increased transcription factor activity, we assessed motif enrichment across the different genotypes. The NCOR-knockout B cells had increased motif enrichments for STAT5, FOXO1, and KLF2 across B cell developmental stages (Figure 4g). Therefore, NCORs repress both the transcriptional and epigenetic landscape of critical B cell development mechanisms including, STAT5 signaling, V(D)J recombination and cellular egress.
NCOR1/2-deficiency upregulates p53 and KLF2 target genes
The top ranked transcriptional regulator predicted from LISA for NCOR1/2 DKO proliferating pro-B cells and pre-BCR-dependent cells was EZH2 (Extended Data Figure 4a). EZH2 is expressed in pro-B and pre-B cells23,24 and prevents expression of Cdkn1a and Cdkn2a, and stabilization of p53 in developing B cells24. We hypothesized that NCOR1/2 deletion leads to upregulation of the p53 pathway during proliferation. GSEA analysis of cycling pro-B cell and pre-BCR-dependent clusters between WT and NCOR1/2 DKO mice demonstrated that loss of NCOR1/2 leads to activation of the p53 pathway (Extended Data Figure 4b). In addition, cycling pro-B cells deficient in NCOR1/2 were enriched for senescence and DNA repair gene sets. Likewise, the pre-BCR-dependent proliferating NCOR DKO B cells were enriched for senescence but negatively associated with fatty acid metabolism and oxidative phosphorylation (Extended Data Figure 4b). When assessing gene expression differences related to the p53 pathway, Cdkn1a expression was low in proliferating wildtype B cells but exhibited significantly higher expression in proliferating NCOR1/2 DKO B cells (Extended Data Figure 4c). Likewise, other p53 pathway genes, such as Btg225 and Trp53inp126, had higher expression in NCOR1/2 DKO B cells compared to wildtype B cells, while Btg2 gene accessibility was increased (Extended Data Figure 4c). p53 has been shown to release the pro-apoptotic factor BIM from sequestering complexes such as MCL-127. Furthermore, NCOR1 has been shown to directly repress BIM expression in developing T cells28,29. We found that BIM expression was modestly elevated in the NCOR1/2 DKO pro-B and pre-B cells (Extended Data Figure 4d). In accordance with this, Bcl2l11 (Bim) displayed increased accessibility in pro-B and pre-B cells (Extended Data Figure 4e), suggesting NCOR1/2 have modest effects on apoptosis.
Next, we assessed whether cellular localization and egress mechanisms were perturbed in the NCOR-knockout B cells. KLF2 is critical for driving thymic egress of developing T cells30 and also regulates expression of genes required for B cell trafficking31. For example, KLF2 activates transcription of Cd69 and Sell (L-selectin; CD62L)32 and increases L-selectin protein expression in B cells31. NCOR-knockout B cells demonstrated increased expression and accessibility of Klf2, Cd69, and Sell (Extended Data Figure 5a,b). To determine whether increased expression and accessibility of Sell led to increased protein expression, we stained for CD62L expression in the NCOR DKO B cells. NCOR1/2 DKO pro-B/pre-B, small pre-B, and immature B cell subsets had an increased frequency of CD62L+ B cells (Extended Data Figure 5c). Thus, heightened KLF2 expression correlates with increased target gene expression and accessibility in NCOR DKO B cells and may indicate aberrant cellular localization of developing B cells that could explain the increased frequency of Ig kappa−lambda− B cells in the periphery.
NCOR1/2 limits Rag expression and genome instability
Rag1/2 expression is tightly segregated to non-proliferating cells, as these enzymes can pose a threat to the genome if they are expressed in dividing cells33. When breaking down Rag1 expression among different subsets and genotypes, both proliferating (Cycling Pro-B, Pre-BCRi-I, II, II) and differentiation clusters (Pro-B VDJ, Kappa Pre-B, and Lambda Pre-B-I, II, and III) had higher Rag1 expression in NCOR1/2 DKO cells (Figure 5a). Likewise, Rag2 expression trended higher in the NCOR1 KO and NCOR1/2 DKO B cells (Figure 5a). Similarly, Rag1 and Rag2 gene accessibility was significantly increased in NCOR DKO B cells (Figure 5b). The Rag locus, including the promoters and enhancers, becomes mostly inaccessible at the immature B in WT B cells but remained accessible in NCOR KO B cells (Figure 5b,c). To assess Rag1 expression in proliferating B cells, we identified cells co-expressing Rag1 and Mki67. Wildtype B cells had few cells expressing both Mki67 and Rag1 transcripts, especially cells expressing high levels of both transcripts. In contrast, NCOR1 KO B cells had increased density of dual-Mki67/Rag1 expressing cells, while NCOR1/2 DKO B cells had many cells that were strongly positive for both Mki67 and Rag1 transcripts (Figure 5d), suggesting desegregation of Rag1 expression and proliferation. Other recombination-related genes, such as Dntt, Xrcc6 and Top1, were also aberrantly expressed in multiple B cell subsets in NCOR1/2 DKO mice (Figure 5e). To understand whether aberrant regulation of Rag1/2 expression contributed to genome instability, we performed whole-genome sequencing on genomic DNA extracted from sorted WT (n=2) and NCOR DKO (n=3) B cells (CD19+B220+). NCOR DKO B cells had an increased number of structural variants (Figure 5f), with primary differences coming from the number of deletions. When assessing for possible RAG-mediated effects, we investigated deletions that were found affecting the genes Ercc2 and Stag2 (Supplementary Table 4). Deletions in Ercc2 were present in all three NCOR DKO B cells but not found in WT B cells, while Stag2 deletion was present in one of the NCOR DKO B cell samples but not in any other samples (Supplementary Table 4). We found cryptic heptamer recombination signal sequences present within the deleted regions of Ercc2 and Stag2, suggesting possible RAG off-target effects (Figure 5g). Overall, NCOR1/2 play an important role in restricting Rag1/2 chromatin accessibility and gene expression, while dysregulation of Rag1/2 correlated with increased structural variants that may be attributed to RAG off-target effects.
Figure 5. Nuclear corepressors segregate RAG expression from proliferating cells and protect the genome integrity.
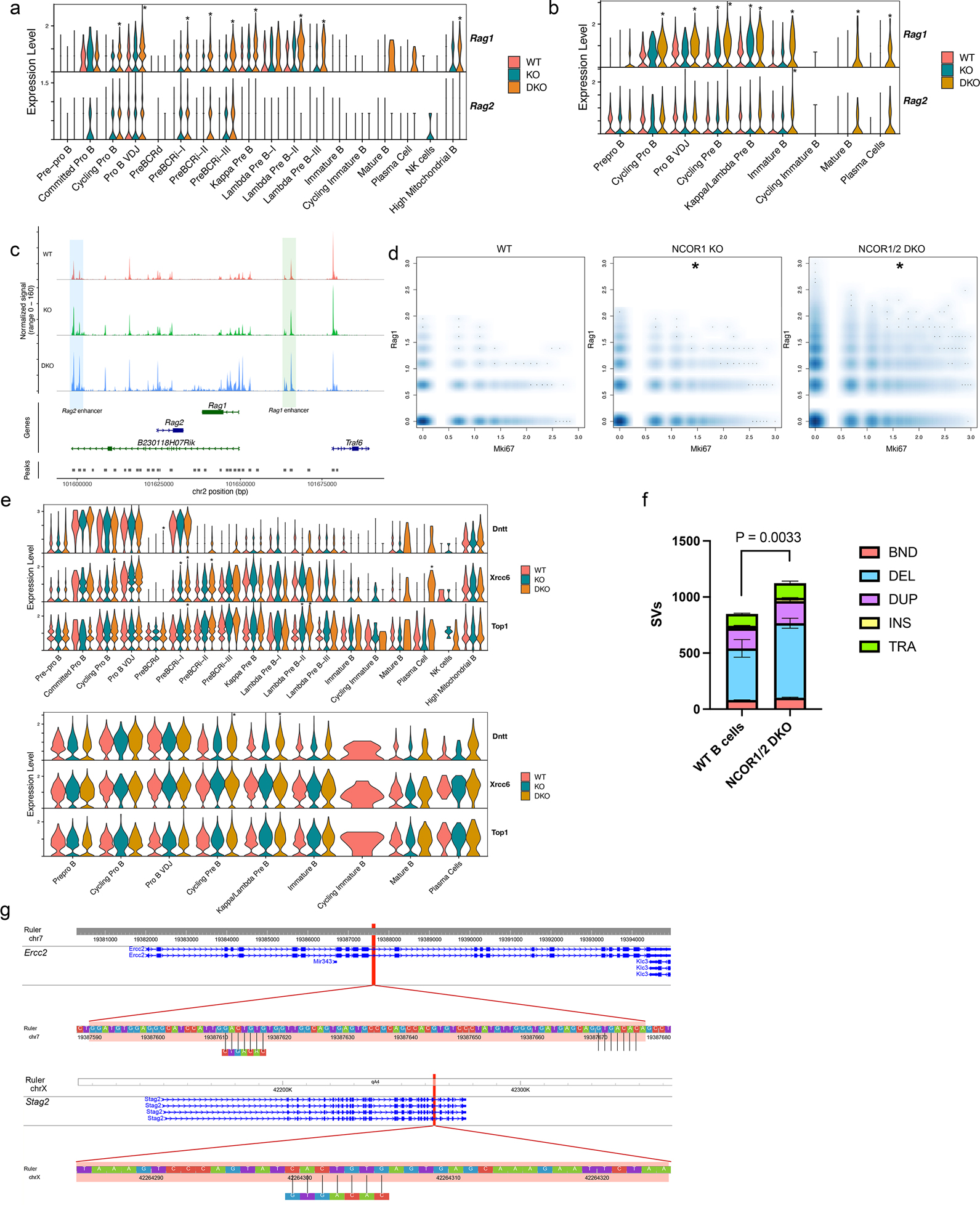
a. Rag1 and Rag2 RNA expression differences between WT, NCOR1 KO and NCOR1/2 DKO B cells derived from scRNA-seq. b. Rag1 and Rag2 scATAC-seq accessibility differences between WT, NCOR1 KO and NCOR1/2 DKO B cells across different clusters. c. Rag1 and Rag2 track locus of immature B cells and accessibility profiles split by genotype. The blue highlighted area indicates the Rag2 enhancer, whereas the green highlighted area indicates the Rag1 enhancer. The y-axis is identical for all three plots (range from 0–160). d. Density scatter plot of Mki67 and Rag1 transcript between WT, NCOR1 KO, and NCOR1/2 DKO B cells. Asterisk (*) indicates statistical significance with FDR <0.05 with at least a log2 fold change > 0.58 when compared to the frequency of wildtype B cells. A permutation test was performed to calculate the statistical significance of frequency changes of Mki67+Rag1+ cells in the NCOR1 KO and NCOR1/2 DKO compared to the wildtype cells. The package scProportiontest was used to perform the statistical test. e. Recombination-related (Dntt, Xrcc6 and Top1) gene expression and accessibility differences in different clusters. f. Number of structural variants in wildtype (n=2) and NCOR1/2 DKO (n=3) B220+CD19+ B cells. BND; breakend, DEL; deletion, DUP; duplication, INS; insertion, TRA; translocation. Structural variants were called using Smoove. A two-tailed t-test was performed to compute the statistical significance. g. Cryptic heptamer recombination sequences found in deleted regions of Ercc2 and Stag2 genes from NCOR DKO B cells.
Ncor1 deficiency promotes leukemic transformation
NCOR1 mutations and deletions are often found in in human B-cell malignancies8,9. To understand whether loss of Ncor1 alleles promotes transformation, we used constitutively active STAT5b (Stat5b-CA) mice to generate Stat5b-CA × Cd79a-Cre × Ncor1FL/+ (n=9) and Stat5b-CA × Cd79a-Cre × Ncor1FL/FL (n=5) mice. Stat5b-CA mice have low leukemic penetrance (2–10%)2,34,35 (Figure 6a) and typically require cooperating mutations that enhance STAT5 signaling to increase leukemic penetrance36; when one or both alleles of Ncor1 was deleted, there was an increase in transformation and worse survival (Figure 6a). We evaluated expression of different B cell development markers via flow cytometry in these B cell leukemias. Stat5b-CA × Cd79a-Cre × Ncor1FL/+ leukemic mice generated an admixture of B cell (62.5%) and non-B cell leukemias (37.5%) (Figure 6b,c). B cell leukemias had variable CD19 expression and were CD93+ while non-B cell leukemias were CD90.2+ but were mostly CD3− and expressed variable amounts of B220 (Figure 6b). Furthermore, we found that the majority of Stat5b-CA × Cd79a-Cre × Ncor1FL/FL leukemic cells expressed increased IL-7R levels when compared to Stat5b-CA × Cd79a-Cre × Ncor1FL/+ leukemic cells or wildtype B cells. In contrast, no significant difference was seen in the expression of another surface marker CD25 (Figure 6c). Our results suggest that NCOR1 safeguards against transformation, as loss of Ncor1 cooperates with STAT5 activation to initiate leukemic transformation.
Figure 6. NCOR1 deficiency promotes leukemic transformation with phenotypic heterogeneity.
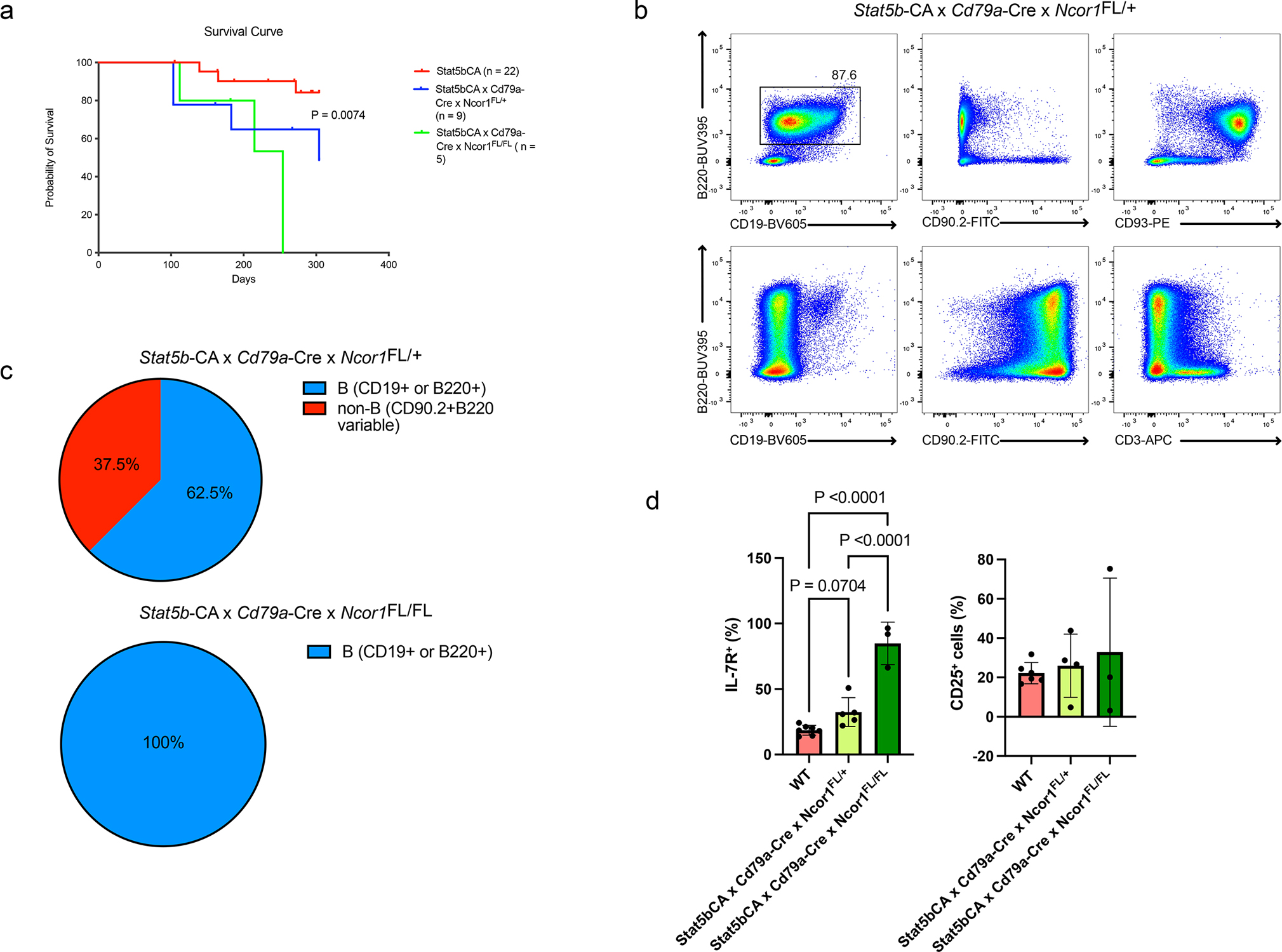
a. Survival curve of constitutively active Stat5b mice (Stat5b-CA, red) and Stat5b-CA × Cd79a-Cre × Ncor1FL/+ (blue, n=9) or Stat5b-CA × Cd79a-Cre × Ncor1FL/FL (green, n=5) mice. P-value was derived using a log-rank test for trend. b. Representative flow cytometry plots of Stat5b-CA × Cd79a-Cre × Ncor1FL/+ leukemias with B cell phenotype (top) and non-B cell (CD90.2+) phenotype. c. Distribution of B and non-B cell (CD90.2+) leukemias from Stat5b-CA × Cd79a-Cre × Ncor1FL/+ and Stat5b-CA × Cd79a-Cre × Ncor1FL/FL mice. d. Frequency of IL-7R+ and CD25+ expressing cells in wildtype, Stat5b-CA × Cd79a-Cre × Ncor1FL/+ and Stat5b-CA × Cd79a-Cre × Ncor1FL/FL leukemias. An ordinary one-way ANOVA with Tukey’s multiple comparison was performed for statistical analysis; error bar represents standard deviation. Data derived from WT (n=7), Stat5b-CA × Cd79a-Cre × Ncor1FL/+ (n=5) and Stat5b-CA × Cd79a-Cre × Ncor1FL/FL leukemias (n=3), from at least three independent experiments. All measure of centers indicate mean.
NCOR defects correlate with genome instability in B-ALLs
Characterization of the genomic and transcriptional landscape of human B-ALL samples has identified various B-ALL subtypes37. Using St Jude’s ProteinPaint database, we assessed the types of mutations that are present in the NCOR1 and NCOR2 genes. Mutations, including damaging frameshift and nonsense mutations, were present in the NCOR1 gene, while some missense mutations were found in NCOR2 (Figure 7a). Among 28 human B-ALL samples with NCOR1 mutations (Supplementary Table 5), 16 samples had intrachromosomal structural variants, whereas 3 out of 7 NCOR2-mutant samples had intrachromosomal structural variants (Figure 7b, Supplementary Table 5). Similarly, 17 out of 28 NCOR1-mutated samples and 4 out of 7 NCOR2-mutated samples had interchromosomal structural variants (Figure 7c). Among the samples with interchromosomal structural variants, we observed the presence of IGH, TCF3::PBX1 and ETV6 translocations (Supplementary Table 5). IGH and TCF3::PBX1 translocations are associated with inappropriate RAG activity at recombination signal sequences or deaminated CpG nucleotides, respectively38,39. While RAG enzymes are not directly responsible for ETV6::RUNX1 translocations, RAG activity is paramount for additional oncogenic rearrangements and deletions in ETV6::RUNX1 expressing cells that leads to transformation40. We also found that NCOR1 or NCOR2-defective B-ALLs had significantly elevated expression of FOXO1 and FOXO1 target genes (RAG1, RAG2, and IL7R) compared to B-ALLs with wildtype NCOR1/2 (Figure 7d). As NCORs are recruited to repress target gene transcription, the deletion or mutation of NCOR1 or NCOR2 in B-ALLs may lead to increased accessibility and vulnerability to RAG-mediated DNA damage. For example, ETV6::RUNX1 and DUX4-rearranged subtypes, which have RAG-associated deletions in BTG140 or ERG41, respectively, also can have NCOR1 or NCOR2 mutations. Using our scRNA-seq and scATAC-seq dataset, we found Btg1 and Erg expression to be elevated in the NCOR-knockouts (Extended Data Figure 6a). Similarly, the gene locus of Btg1 and Erg displayed increased genomic accessibility (Extended Data Figure 6b), suggesting that RAG proteins may have higher chances to disrupt these genes in NCOR-defective B-ALLs. In accordance with increased RAG1 and RAG2 expression, the NCOR1/2-defective human B-ALL samples had increased numbers of structural variants, compared to the NCOR1/2-wildtype leukemias (Figure 7e). Finally, we observed that patients with below-median expression of NCOR1 had worse overall survival compared to patients with above-median expression (Figure 7f), although this may be due to the association of NCOR expression levels with other covariates. Similarly, bottom quantile expression of NCOR1 led to worse event-free survival in pediatric B-ALL patients (Figure 7f). These results highlight the diversity of NCOR mutations present in human B-ALL samples and demonstrate that NCOR mutations correlate with high RAG expression and potential RAG-mediated DNA damage.
Figure 7. Human B-ALL patients with nuclear corepressor mutations harbor structural variants with high RAG expression.
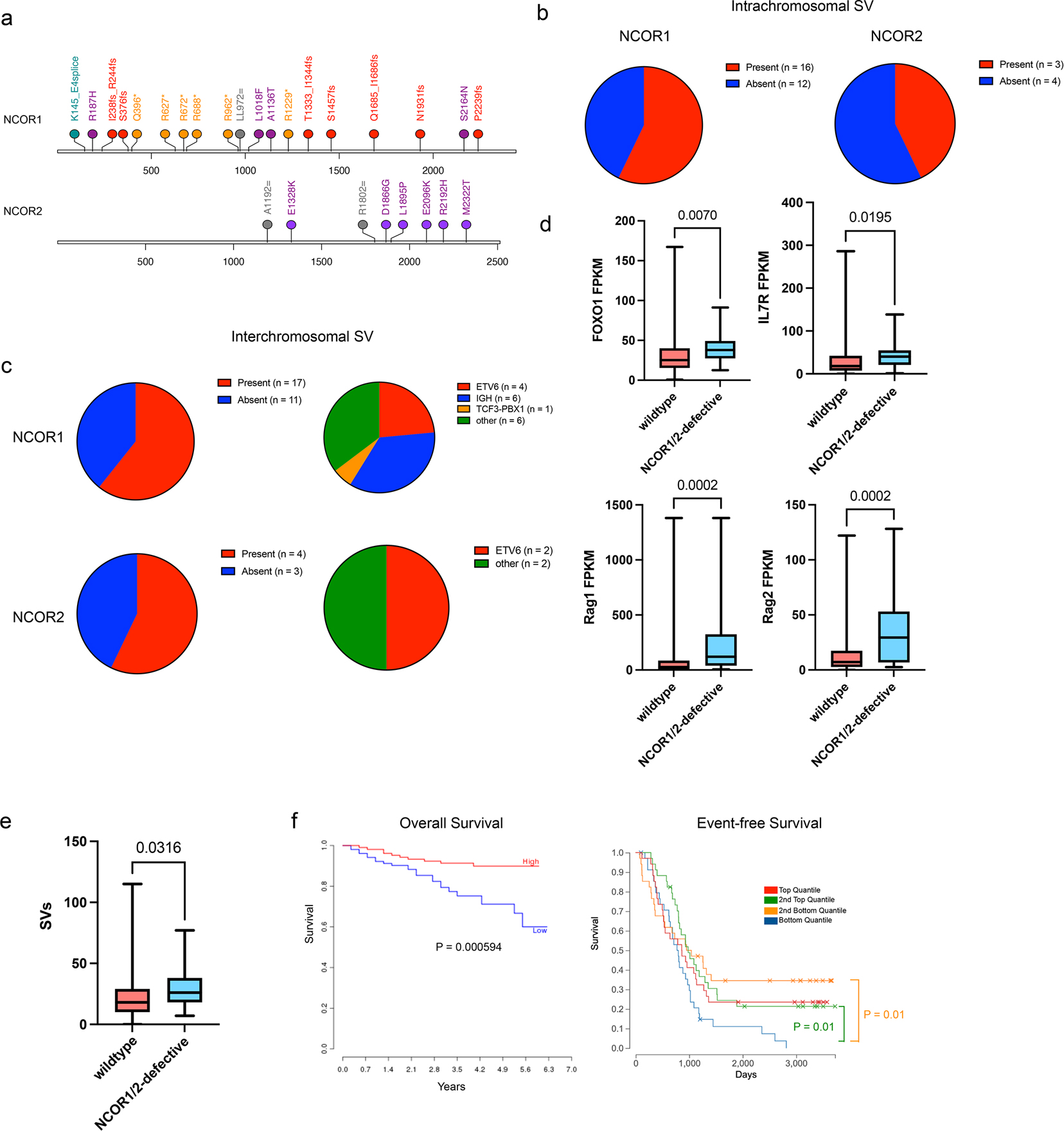
a. Lollipop plot of NCOR1 and NCOR2 mutations found in human B-ALL patients. Orange/*; nonsense mutations; Red/fs, frameshift mutation; green/=; silent mutations; purple, missense mutations. b. Presence of intrachromosomal structural variants in B-ALL patients with NCOR1 mutations (left) or NCOR2 mutations (right). c. Presence of interchromosomal structural variants in B-ALL patients with NCOR1 mutations (top left) or NCOR2 mutations (bottom left). Breakdown of chromosome translocation types in those that had interchromosomal structural variants with NCOR1 mutations (top right) or NCOR2 mutations (bottom right). d. FOXO1, IL7R, RAG1 and RAG2 expression in NCOR1/NCOR2 wildtype (n=709) NCOR1 or NCOR2-defective B-ALLs (n=2). P-value was derived from a two-tailed unpaired t-test. e. Number of structural variants in NCOR1/NCOR2 wildtype B-ALLs (n=539) and NCOR1 or NCOR2-defective human B-ALLs (n=15). P-value was derived from a one-tailed unpaired t-test. f. NCOR1 expression correlation with B-ALL prognosis of overall survival (left; years) and event-free survival (right; days). Low and high correspond to below-median and above-median expression, respectively (left) and p-value was generated using a log-rank test. For event-free survival, blue indicates bottom quantile, orange is 2nd bottom quantile, green is 2nd top quantile, and red is top quantile (right) with p-values generated using a log-rank test. Mutation profile, presence of structural variants, and RAG1/RAG2 expression from Human B-ALL data was obtained from St Jude ProteinPaint. The correlation of NCOR1 expression with overall survival was derived from PRECOG, and quantile NCOR1 expression correlation with event-free survival was obtained from St Jude ProteinPaint. Box plot center line represents median, with the box extending from 25th to 75th percentile. The whiskers represent the minimum and maximum value.
Discussion
Here we describe the mechanisms by which the nuclear corepressors NCOR1 and NCOR2 orchestrate proliferation and differentiation during B cell development, and establish how reduced expression of NCOR1/2, or NCOR1/2 mutations, promote leukemic transformation. The nuclear corepressors NCOR1 and NCOR2 are partially redundantly required during B cell development, where they govern the balance between cytokine and antigen receptor signaling, V(D)J recombination, the p53 pathway, and Klf2 expression, and protect against aberrant activation of Rag1/2 and other components of the recombination machinery during B cell proliferation. Furthermore, deletion of 1 or 2 Ncor1 alleles cooperated with constitutively active Stat5b to promote transformation. Overall, these mechanistic insights illuminate how the nuclear corepressors NCOR1/2 orchestrate appropriate B cell differentiation and protect the integrity of the genome during B cell development.
Our results highlight different mechanisms by which B cell leukemias may exploit defects in nuclear corepressors. Cytokine-mediated IL-7R/STAT5 signaling and antigen receptor-mediated pre-BCR signaling are antagonistic2 and signaling via both pathways subverts the development of leukemia19. NCOR1/2 deletion led to quantitative and qualitative defects in pre-BCR-dependent pre-B cells, characterized by reduced IKZF1 expression and heightened STAT5 signaling. Consistent with this observation, studies in T cells found that NCOR1 is important in integrating signals downstream of TCR signaling28,29. Likewise, STAT5 cooperates with defects in pre-BCR-signaling modules to drive leukemia development2. In accordance with this, Ncor1 deletions led to increased transformation in Stat5b-CA mice. This likely reflects increased STAT5 activation upon NCOR deletion, as transformation requires greater STAT5 activation than provided by the Stat5b-CA transgene alone36. Furthermore, Foxo1 expression and accessibility was increased in murine NCOR-knockout B cells and in NCOR-defective human B-ALL. FOXO1 can induce IL-7R expression in B cells42. Consistent with this observation we found increased IL-7R protein expression in NCOR DKO B cells and increased IL7R expression in NCOR-defective human B-ALLs. Thus, loss of NCOR1/NCOR2 may initiate leukemia by inducing hyperactive IL-7R/STAT5 and by limiting pre-BCR dependent signaling.
Our data also suggested that the deletion of Ncor1 and Ncor2 led to increased expression and accessibility of DNA damage repair genes. We observed derepression of the p53 pathway including increased expression of Cdkn1a, Btg2, and Trp53inp1. Activation of the p53 pathway may be related to aberrant RAG expression and resultant DNA damage in proliferating progenitor B cells. Collectively, p53 pathway activation and putative DNA damage responses from aberrant expression of NCORs may lead to a senescence-like phenotype that could impact therapy response, and contribute to clonal evolution and subsequent relapse43.
The absence of NCOR1/2 resulted in elevated expression of Rag1 and Rag2, especially in B cell subsets that should not express these genes. For example, we found high concomitant expression of Mki67 and Rag1 and increased Rag gene accessibility and expression in NCOR1/2 KO B cells, suggesting a failure to silence this locus in the absence of NCORs. This correlated with an increase in structural variants and the presence of cryptic heptamer sequences in deleted regions found in the NCOR DKO B cells. Other structural variants observed in our study may arise via RAG-independent mechanisms. For example, previous work in Hdac3−/− fibroblasts demonstrated increased DNA damage due to increased H4K5 acetylation44. Deletion of Ncor1 and Ncor2 in B cells may recapitulate the Hdac3-null mechanism of DNA damage and impaired DNA repair in fibroblasts, leading to structural variants during B cell differentiation. Finally, high RAG1 expression correlates with poor prognosis and has been shown to drive clonal evolution and transformation of B cells45. Thus, temporal regulation of Rag1/2 is critical to minimize RAG-induced genome instability. However, what causes elevated Rag expression in B-ALL patient samples with intra- and interchromosomal structural variants is unclear. Interestingly, IgH translocations, TCF3::PBX1 and ETV6 translocations occasionally coexist with NCOR1 or NCOR2 mutations. The immunoglobulin heavy chain locus has strong recombination signal sequence signatures that allows for VDJ recombination. Similarly, TCF3::PBX1 translocations arise from RAG-induced damage at deaminated CpG nucleotides38. Thus the emergence of RAG-driven IGH or TCF3::PBX1 translocations can potentially be attributed to NCOR-mediated dysregulation of RAG1/2. On the other hand, ETV6::RUNX1 translocations are not thought to arise due to RAG-related activity40. However, the presence of ETV6::RUNX1 translocations alone is not sufficient to drive leukemic transformation46; further mutations are acquired through RAG activity to promote leukemogenesis40. Similarly, DUX4-rearranged B-ALL subtypes, concomitantly harbor ERG deletions, which are secondary events that arise from RAG-mediated deletions41. Therefore, NCOR mutations or copy number loss present in ETV6::RUNX1 or DUX4-rearranged subtypes may promote aberrant RAG expression that drives mutations which promote transformation.
RAG activity is dependent on the global epigenetic environment. We found that chromatin accessibility sites regulated by NCORs overlap with H3K27ac sites, suggesting NCORs act via effects on H3K27Ac. Likewise, we observed a stepwise depletion of CTCF binding motifs in accessible regions of NCOR1 KO and NCOR1/2 DKO B cells. CTCF marks the boundaries between active and repressed chromatin47,48. Thus, the shift in balance between active and repressed chromatin in NCOR1/2 DKO B cells could result in altered CTCF binding globally. Changes in CTCF activity also have significant implications for RAG activity, as CTCF is important for Igh locus contraction49 and CTCF-dependent RAG linear scanning across large chromatin loops50. While differential CTCF binding and changes in three-dimensional chromatin interactions in NCOR-deficient B cells need experimental validation, the depletion of CTCF binding motifs in NCOR-deficient B cells raises the possibility of abnormal chromatin loop formation and alterations in the susceptibility to off-target RAG activity. RAG has been shown to have widespread binding sites across the genome but RAG binding is heavily dependent on chromatin accessibility33. This is evidenced by RAG1 primarily binding at active enhancers and promoters33,40 and the association with H3K4me333. Considering the role of nuclear corepressors in recruiting histone deacteylases to repress target genes, a deficiency in nuclear corepressors may lead to changes that promote increased chromatin accessibility and thus more vulnerable targets of RAG activity. In accordance with this idea, genes that are associated with RAG-mediated deletion in human B-ALLs, such as BTG1 and ERG, were significantly upregulated or had increased accessibility in NCOR-knockout B cells, which may enhance RAG-mediated deletions at these gene loci. We propose that nuclear corepressors directly repress Rag gene expression while preventing off-target RAG activities by strictly controlling chromatin accessibility. In this context, NCOR1/2 would play critical roles in maintaining genomic integrity throughout B cell differentiation, thereby promoting normal B cell differentiation, and preventing transformation.
Methods
Animals
All animals used in this study were bred and housed at University of Minnesota in pathogen-free facilities. Breeding and experimental protocols for mice were approved by the Institutional Animal Care and Use Committee (IACUC 2010–38515A and IACUC 1904–36975A) at the University of Minnesota. All animals were housed in a dark/light cycle of 10 hrs/14 hrs. Light cycle was from 6 AM to 8 PM. Ambient temperature was maintained at 22°C and the humidity of the room ranged from 30–40%. Ncor1FL/FL mice12 were provided by E. Olson (UT-Southwestern) and J. Auwerx (École Polytechnique Fédérale, Lausanne, Switzerland). Tg(IghelMD4)4Ccg/J (MD4-transgene) mice were provided by M. Jenkins (Univ. of Minnesota). Cd79a-Cre mice were obtained from T. Bender (University of Virginia) and backcrossed for 30 generations to the C57Bl/6 background. Stat5b-CA mice were previously described34. Ncor2 conditional knockout mice were generated using a flip-excision system13. A retroviral vector containing a splice acceptor followed by lacZ reporter gene and poly A sequence and flanked by FRT and loxP sequences was used to insert the gene trap vector between exon 1 and 2 of the Ncor2 gene in mouse embryonic stem cells; embryonic stem cells were injected into blastocysts to make gene trapped mice. These mice were bred with mice expressing constitutive FLPe, inverting the cassette, allowing for normal gene splicing. Mice with the inverted cassette were bred to Cd79a-Cre, allowing for conditional knockout of Ncor2 in B cells. A schematic describing this mouse model is shown in Extended Data Figure 1a. For all NCOR-knockout phenotyping experiments, animals used were between 6- to 13-week old C57BL/6 male and female mice with appropriate age and sex matched controls. For the single-cell RNA-seq experiment, 8-week-old male C57BL/6 mice were used for WT (n=2), NCOR1 KO (n=2), and NCOR1/2 DKO (n=2) samples. For the single-cell ATAC-seq experiment, 9–12 week old male and female C57BL/6 mice were used for WT (n=1), NCOR1 KO (n=1) and NCOR1/2 DKO (n=1) samples. Stat5b-CA, Stat5b-CA × Cd79a-Cre × Ncor1FL/+, and Stat5b-CA × Cd79a-Cre × Ncor1FL/FL mice ranged from 103–304 days old at time of analysis and included male and female C57BL/6 mice.
Tissue Processing and Cell Preparation
Bilateral femurs, tibias and the spleen were harvested. The femurs and tibias were flushed with FACS buffer (1X PBS with 2% fetal bovine serum (Sigma Aldrich, 12133C), 0.1% sodium azide (Ricca, 7144.8–16) and 0.5 mM ethylenediaminetetraacetic acids (EDTA; Fisher Scientific, S3113), pH 7.4). The spleen was mechanically grinded between the rough sides of frosted glass slides. Cells were filtered through a 70 μm mesh filter and centrifuged at 350 × g for 5 minutes. Cells were incubated and centrifuged at 350 × g for 5 minutes in ACK lysis buffer (0.15M ammonium chloride (Fisher, A661), 10 mM potassium bicarbonate (Fisher, P184), 1mM EDTA (Fisher Scientific, S3113)). Cells were washed and centrifuged at 350 × g and resuspended in 2 mL of FACS buffer. Counting was performed on a hemocytometer (Fisher Scientific. 02–671–10).
Flow Cytometry and Antibodies
Single-cell suspensions obtained from bone marrow and spleen samples were stained with different FACS antibodies. Antibodies used include B220-BUV395 (RA3–6B2, BDBiosciences, 563793, 1:100), B220-Pacific Blue (RA3–6B2, BDBiosciences, 558108, 1:100), CD11c-APCef780 (N418, ThermoFisher, 47–0114-82, 1:100), GhostRed780 (Tonbo Biosciences, 13–0865, 1:1000), Ter119-APCef780 (TER-119, ThermoFisher, 47–5921-82, 1:100), NK1.1-APCef780 (PK136, ThermoFisher, 47–5941-82, 1:100), Ly6G-APCef780 (RB6–8C5, ThermoFisher, 47–5931-82, 1:100), CD4-APCef780 (GK1.5, ThermoFisher, 47–0041-82, 1:100), CD8-APCef780 (53–6.7, ThermoFisher, 47–0081-82, 1:100), CD43-Biotin (S7, BDBiosciences, 553269, 1:100), CD43-FITC (S7, BDBiosciences, 561856, 1:100), CD19-BV605 (6D5, BioLegend, 115540, 1:100), CD93-PE (12–5892-82, eBioscience, 1:100), IgM-APC (115–136-075, Jackson Immuno Research, 1:100), IgM-APC (RMM-1, BioLegend, 406509, 1:100), IgM-FITC (eB121–15F9, eBioscience, 11–5890-85, 1:100), CD62L-APC (MEL-14, Tonbo Biosciences, 20–0621-U100, 1:100), CD25-BV421 (BDB562606, BDBiosciences, 1:100), CD24-PerCP-Cy5.5 (562360, BDBiosciences, 1:100), CD249/BP1-BV786, (740882, BDBiosciences, 1:100), CD90.2-FITC (35–0903-U100, Tonbo Biosciences, 1:100), CD3-APC (20–0031-U100), Ig Lambda-FITC (1175–02, SouthernBiotech, 1:100), Ig Kappa-PE (1175–09, SouthernBiotech, 1:100), IKAROS-PE (653304, BioLegend, 1:100), Ki67-BV421 (652411, BioLegend, 1:100), NCOR1 (5948S, Cell Signaling, 1:100), BIM (2933T, Cell Signaling, 1:100), p-STAT5 (A17016B.Rec, BioLegend, 936904, 1:100), donkey anti-Rabbit IgG-PE (12–4739-81, eBioscience, 1:100), goat anti—Rabbit IgG-PE (ab72465, Abcam, 1:100), IL-7R-BV421 (A7R34, BioLegend, 135023, 1:100), Streptavidin-BV421 (BioLegend, 405225, 1:100). For surface staining, antibodies at appropriate dilutions in FACS buffer were mixed with cells and stained for 20 minutes at 4°C. Cells were washed in FACS buffer and centrifuged at 350 × g. Cells were resuspended and analyzed on the cytometer or underwent intracellular staining. For intracellular staining, surface-stained cells were fixed and permeabilized with the eBioScience Transcription Factor staining kit (eBioscience, 00–5523-00) for 30 minutes at 22°C. Fixed cells were washed in 2 mL of FACS buffer and centrifuged at 650 × g. Intracellular staining antibodies were mixed with permeabilization buffer at the appropriate dilution and stained at 22°C for 30 minutes. Cells were washed and resuspended in FACS buffer for analysis. For intracellular staining of antibodies without a conjugated fluorochrome (ex. NCOR1), an additional staining step with anti-rabbit IgG-PE was performed for 30 minutes at 22°C, washed, and resuspended before flow cytometry analysis. For p-STAT5 staining, bone marrow cells harvested from WT or NCOR1/2 DKO mice were incubated in the presence or absence of IL-7 (10 ng/μL). Cells were fixed and permeabilized using True-Phos Perm Buffer (425401, BioLegend) and Fixation Buffer (420801, BioLegend) according to the manufacturer protocol from BioLegend. The protocol was modified by reducing the volumes to accommodate staining in a 96 well plate. Flow cytometry was performed on a BD Fortessa (BD Biosciences) or BD LSR II (BD Biosciences) with data collected using FACSDiva 8.0 (BD Biosciences) and data obtained was subsequently analyzed in FlowJo software (Tree Star).
Cell Sorting and Single-Cell RNA sequencing
For single-cell RNA-sequencing, we obtained suspensions of bone marrow samples from wild type (n=2), NCOR1 KO (n=2), and NCOR1/2 DKO (n=2) mice (8-week old males). Each sample was uniquely labelled with 1 μg of a hashtag antibody, which includes TotalSeq A0301 (M1/42, BioLegend, 155801, 1:50), TotalSeqA0302, M1/42, BioLegend, 155803, 1:50), TotalSeq A0303 (M1/42, BioLegend, 155805, 1:50), TotalSeq A0304 (M1/42, BioLegend, 155807, 1:50), TotalSeq A0305 (M1/42, BioLegend, 155809, 1:50), TotalSeq A0306 (M1/42, BioLegend, 155811, 1:50) representing the six samples we had. Samples were stained in FACS buffer with 1 μg of CITE-Seq antibodies: B220 (TotalSeq-A0103, RA3–6B2, BioLegend, 103263, 1:50), CD19 (TotalSeq A0093, 6D5, BioLegend, 115559, 1:50), CD25 (TotalSeq A0097, PC61, BioLegend, 102055, 1:50), CD93 (TotalSeq A0113, AA4.1, BioLegend, 136513, 1:50), and IgM (TotalSeq A0450, RMM-1, BioLegend, 406535, 1:50). Surface-stained samples were washed and resuspended in 1 μg of streptavidin-PE (TotalSeq A0113, BioLegend, 405251, 1:50) containing 1X PBS with 2% fetal bovine serum (FBS), 2 mM ethylenediaminetetraacetic acids, pH 7.4 buffer (sort buffer) for both sorting and as a CITE-Seq antibody for CD43-Biotin and stained for 30 minutes. Cells were washed and resuspended in sort buffer. B220+CD43+ and B220+CD43− cells were sorted at a 1:1 ratio, resulting in 20,000 cells per mouse. Cells sorted from each mouse were combined and resuspended in sort buffer. Cell sorting was performed on a BD FACSAria sorter (BD Biosciences). Sorted cells were then captured using the 10X Chromium platform. Reverse transcription PCR and library preparation was performed on the RNA, ADT/CITE-Seq, and hashtag libraries using Single Cell 3’ v3 protocol (10X Genomics) per manufacturer’s recommendations. After library prep, quantification was performed using a bioanalyzer (Agilent 2100 Bioanalyzer; Agilent Technologies). The RNA library was sequenced on a MiSeq (Illumina) to obtain an estimate of number of cells captured and assess quality of the library. All libraries were then sequenced on a NovaSeq 6000 with 2×150 bp paired-end reads (Illumina).
Single-Cell RNA-seq analyses
Processing and filtering was performed as previously described17. In short, the CellRanger pipeline from 10X Genomics was used to demultiplex the RNA, ADT, HTO libraries and derive counts after aligning reads to mm10 mouse genome. Seurat v3.0.3 and v4.0.1 were used to analyze all downstream applications51,52. The dataset was filtered on parameters of number of genes expressed and proportion of mitochondrial RNA (top 0.5% of all GEMs). The corresponding HTO and ADT counts were added to the remaining cells and normalized by a centered-log ratio method. Multiplets containing more than one hashtag antibodies in a GEM were identified and removed using the HTODemux Seurat function. The raw RNA counts were transformed using the Seurat function “SCTransform”53 including the percent of mitochondria expression as a regression factor. This normalized SCTransform dataset was used as input for principal component analysis (PCA), where the top 30 principal components were used to perform dimensional reduction by UMAP. The FindClusters function at a resolution of 0.4 was used for clustering and cluster-defining markers were derived using the FindAllMarkers function. Differential expression of individual clusters between the wildtype, NCOR1 KO, and NCOR1/2 DKO samples was performed using the FindMarkers function, comparing wildtype vs NCOR KO, wildtype vs. NCOR1/2 DKO, and NCOR1 KO vs NCOR1/2 DKO. These differential gene lists between wildtype and NCOR1/2 DKO clusters were used as input for landscape in silico analysis (LISA)54 and gene set enrichment analysis (GSEA v4.0). For pathway analysis, we used preranked (by log2 fold change) DE gene lists of all possible genes with the GSEA function from the clusterProfiler R package (v 3.14.3) to identify significantly enriched gene sets from the Molecular Signature Databases (human orthologs converted to mouse symbols by the msigdbr R package, v 7.0.1)55,56.
Single-Cell ATAC sequencing
For single-cell ATAC-sequencing, we harvested bone marrow from wildtype (n=1, male), NCOR1 KO (n=1, female) and NCOR1/2 DKO (n=1, female) that ranged between 9–12 weeks old. The cell sorting scheme was identical as the single-cell RNA-seq sorting scheme, except we sorted 50,000 B220+CD43+ cells and 50,000 B220+CD43− cells, for a total of 100,000 cells for each genotype. Sorted cells were centrifuged at 300 × g for 5 minutes at 4°C. Cells were resuspended in 100 μl of chilled lysis buffer, which consists of 10mM Tris-HCl pH 7.4 (T2914, Sigma-Aldrich), 10mM NaCl (S5150, Sigma), 3mM MgCl2 (M1028, Sigma-Aldrich), 0.1% Tween 20 (BP337, Fisher Scientific), 0.1% nonidet P40 substitute (74385, Sigma-Aldrich), 0.01% digitonin (BN2006, ThermoFisher), 1% BSA (130–091-376, Miltenyi Biotec), and nuclease-free water, and incubated on ice for 5 minutes. After incubation, 1 mL of chilled wash buffer, including 10mM Tris-HCl pH 7.4 (T2914, Sigma-Aldrich), 10mM NaCl (S5150, Sigma), 3mM MgCl2 (M1028, Sigma-Aldrich), 0.1% Tween 20 (BP337, Fisher Scientific), 1% BSA (130–091-376, Miltenyi Biotec), and nuclease-free water, was added to lysed cells, and centrifuged at 500 × g for 5 minutes at 4°C. Cells were resuspended in diluted nuclei buffer and nuclei permeabilization was determined using a Countess II (ThermoFisher). Cells were captured on the 10X Chromium and library prep was generated using the Chromium Next GEM Single Cell ATAC library kit v1.1 (PN-1000164, 10X Genomics), following the manufacturer’s recommendations. Post-library quality control was performed for sizing and quantification using the Agilent Bioanalyzer (Agilent 2100 Bioanalyzer; Agilent Technologies) and KAPA library quantification kit for Illumina platforms (KR0405, Roche). All libraries were sequenced on a NovaSeq 6000 with 2×150 bp paired-end reads (Illumina)
Single-Cell ATAC-seq analyses
After sequencing, the reads were demultiplexed using bcl2fastq (Illumina) and fastqs were processed for each library using the ‘cellranger-atac count’ tool (10X Genomics). The outputs of the initial ‘cellranger-atac count’ analysis for each library were used as inputs for the 10X ‘cellranger-atac aggr’ function to aggregate the three libraries into one feature barcode matrix. In this aggregation step, depth-based normalization was used wherein reads were subsampled until all three libraries had, on average, an equal number of median unique fragments per cell. The normalized fragment count and feature barcode matrix was imported into R and analyzed with Signac v1.5.057. Cells were filtered based on multiple QC metrics, including peak region fragments, percentage of reads in peaks, nucleosome signal, and TSS enrichment. Gene annotations were derived from the ‘EnsDb.Mmusculus.v79’ package (R package version 2.99.0). Cell type predictions were added to the scATAC-Seq Seurat object from the scRNA-Seq Seurat object using the functions ‘FindTransferAnchors’,’TransferData’ and ‘AddMetaData’. The cycling pre-B cell subsets were merged into one cluster labeled “Cycling Pre-B” and the Kappa and Lambda pre-B cell subsets were merged into one cluster labeled “Kappa/Lambda Pre-B”. The ‘GeneActivity’ function was used to compute the counts within the immunoglobulin heavy chain, kappa and lambda light chain locus. The ‘pro-B VDJ’ cells were subset to assess accessibility of immunoglobulin heavy chain. The ‘Kappa/Lambda Pre-B’ cluster was subset to assess accessibility of immunoglobulin kappa and lambda light chain. To account for sex differences between the WT and KO/DKO samples we identified genes that would be differentially expressed in male vs female mouse B cells based on publicly available IMMGEN RNA-Seq count data (GSE124829). The only genes that were significantly differentially accessible (FDR < 0.05 and no fold change threshold) were on ChrX and ChrY (Xist, Eif2s3y, Ddx3y, Uty, and Kdm5d). Since no autosomal genes were differentially accessible, we removed all peaks associated with Chr X and Chr Y regions before proceeding with differential accessibility analysis. Differential accessibility was calculated using the ‘FindMarkers’ function and the ‘ClosestFeature’ function to assign the closest genes to differential accessible regions. Transcription factor activity was inferred using chromVAR v1.1658. The positional weight matrix was obtained from JASPAR2020 database59. Motif activity was computed using the ‘RunChromVAR’ function from Signac. Differential motif activity between genotypes was calculated using the ‘FindMarkers’ function.
Whole Genome Sequencing
To perform whole genome sequencing, we harvested bone marrow from two wildtype and three NCOR1/2 DKO mice (both males and females that ranged between 7–9 weeks). We flow sorted 300,000 CD19+B220+ B cells from each sample; CD19+B220+ B cells were subject to DNA isolation using a DNA extraction kit (11796828001, Roche). Nucleic acid quantification was performed using both a nanodrop (ND-ONE-W, ThermoFisher) and Quanti-iT PicoGreen dsDNA assay kit (P11496, ThermoFisher). Library preparation for whole genome sequencing was performed using the TruSeq Nano DNA kit (20015964, Illumina). Sequencing was performed on a NovaSeq 6000 with 2×150 bp paired-end reads (Illumina).
Whole Genome Sequencing Analyses
Adapter sequences were trimmed from raw FASTQ files using Trimmomatic (v 0.39) and aligned to the mouse genome (GRCm38/mm10) using bwa mem software (0.7.17). Duplicate reads were removed using Picard MarkDuplicates (v 2.25.5). Structural variants were called using Smoove (0.2.8) software with default settings60. The original SVTYPE classification was preserved for each variant. VCFs from all samples were imported into R, where the original SVTYPE classification was preserved, and SV support metrics were extracted from the metadata tags (including the number of split reads, discordant reads, and SV length). The number of variants were tallied by SVTYPE and sample.
ChIP-seq and scATAC-seq Peak Overlap Analyses
Publicly available STAT5 and H3K27ac ChIP-seq peaks bed files were downloaded from the Gene Expression Omnibus (GSE86878) or Cistrome.org (GSM1463433) repositories, respectively. scATAC-seq differential accessibility (DA) peaks from this study (see Methods) were generated for each cluster, identifying DA peaks by sample type (e.g. WT vs. DKO). The DA peaks for each comparison were split into up and down sets (i.e. peaks up in one sample vs. the other) and converted to bed files. Each DA peaks bed file was compared against the STAT5 or H3K27ac ChIP-seq bed files using IntervalStats software (v 1.0.1)61. The tool reports exact P-values for each query (ATAC-seq peak) against the closest reference (ChIP-seq peak). An overlap summary statistic is reported as the fraction of query P-values below 0.05, where a high fraction indicates strong overlap of the query peaks against reference peaks.
Quantitative real-time PCR
Using flow cytometry, we sorted 200,000 CD19+B220+ cells from WT and NCOR1/2 DKO B cells from bone marrow and spleen. RNA was isolated using the RNeasy micro kit (Qiagen). First strand cDNA was synthesized using 100–200 ng of RNA and the high-capacity cDNA Reverse Transcription Kit (4368814, Thermo Fisher). Realtime PCR was performed using SYBR Green Master (1725271, Bio-rad) on the LightCycler 480 (Roche). Amplification was performed using 95°C for 5 minutes, 40 cycles of 95°C for 10 seconds, 60°C for 10 seconds, and 72°C for 10 seconds. NCOR2 expression was normalized as described using the 2−ΔΔCT method62. NCOR1 primers used were CTGGTCTTTCAGCCACCATT for forward primer and CCTTCATTGGATCCTCCATC for reverse primer. NCOR2 qPCR primer was obtained from IDT Primetime assay ID: Mm.PT.58.23639430.
NCOR1 and NCOR2 analysis of human B-ALL samples
To investigate the NCOR1 and NCOR2 mutation landscape, we used the St Jude ProteinPaint database (https://pecan.stjude.cloud/proteinpaint)63. The trackviewer package (v1.30.0)64 was used to construct a lollipop mutation plot of NCOR1 and NCOR2. The disco plots of each patient sample containing NCOR1 or NCOR2 mutations were manually inspected to assess the presence of intrachromosomal or interchromosomal structural variants. Among NCOR1 or NCOR2 mutated or copy number loss patient samples that had available RNA-sequencing data, the FOXO1, RAG1, RAG2, and IL7R FPKM was derived and compared with NCOR-wildtype human B-ALL samples. The correlation of NCOR1 expression with overall survival and event-free survival was obtained from PRECOG65 and St Jude ProteinPaint63, respectively.
Statistical Analysis
No statistical methods were used to pre-determine sample sizes, but our sample sizes are similar to those reported in our previous publications. Mice for phenotyping were selected based on their genotypes for age-matched cohorts. Otherwise, animals and samples were randomly selected for experiments. Data normality was assessed using a Shapiro-Wilk test. For comparisons that passed the normality test between two groups that were unpaired, an unpaired student t-test was performed. For those that did not pass the normality test, a Mann-Whitney nonparametric t-test was performed. For comparisons between more than two groups, an ordinary one-way ANOVA was performed along with a Tukey’s multiple comparison test (normal distribution), or a Kruskal-Wallis one-way ANOVA with Dunn’s multiple comparison test (non-normal) was performed. Statistical analyses were performed using Prism 9 (GraphPad) and datapoints are shown as mean +/− standard deviation. For survival analysis, a log-rank test for trend was used. For determining the correlation between Ki67 and NCOR1 expression, a two-tailed Pearson correlation-coefficient test was performed. Landscape in silico analysis (LISA) used a one-sided Wilcoxon rank-sum test to assess statistical significance. To determine the significance of p-STAT5 expression differences in WT and NCOR DKO B cells, a one sample t-test was performed. To calculate statistical significance of proportion differences of clusters between different genotypes and frequency differences in Mki67+Rag1+ cells between genotypes, scProportiontest package (https://github.com/rpolicastro/scProportionTest) was used, which utilizes a permutation test to calculate the p-value for each cluster and generate a confidence interval using a bootstrapping method. Barplot was generated using the R package dittoSeq66.
Data collection and data exclusion
Data collection and analyses were not performed blind to the condition of the experiments. Excluded data includes single-cell RNA-seq and single-cell ATAC-seq data with filtered cells that did not meet minimum quality thresholds or hashtag-based doublets.
Data Availability Statement
The data supporting this study are available within the paper and supplementary information file. A reporting summary is also available. Mouse genome mm10 was used as reference sequence (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/). Single-cell RNA-Seq and single-cell ATAC-seq data were deposited at Gene Expression Omnibus, with the following accession code: GSE208656 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE208656). Whole genome sequencing data was deposited at SRA with the following accession code: PRJNA860179 (http://www.ncbi.nlm.nih.gov/bioproject/860179). STAT5 ChIP-Seq peaks data were obtained from GSE86878 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE86878) and H3K27ac ChIP-seq peaks data were obtained from cistrome.org via project GSM1463433. IMMGEN RNA-Seq count data was from GSE124829. Human B-ALL NCOR1 and NCOR2 mutation and RNA-seq data were obtained from StJude ProteinPaint (https://pecan.stjude.cloud/proteinpaint). Source data are provided with this paper.
Code Availability Statement
Only publicly available software tools were used for analysis (e.g. CellRanger, Seurat, Signac, WGS, GSEA, etc.). No custom software was used or developed in this study, but analysis scripts will be provided upon request.
Extended Data
Extended Data Fig. 1. Ncor2 conditional knockout mouse model and flow cytometry gating scheme.
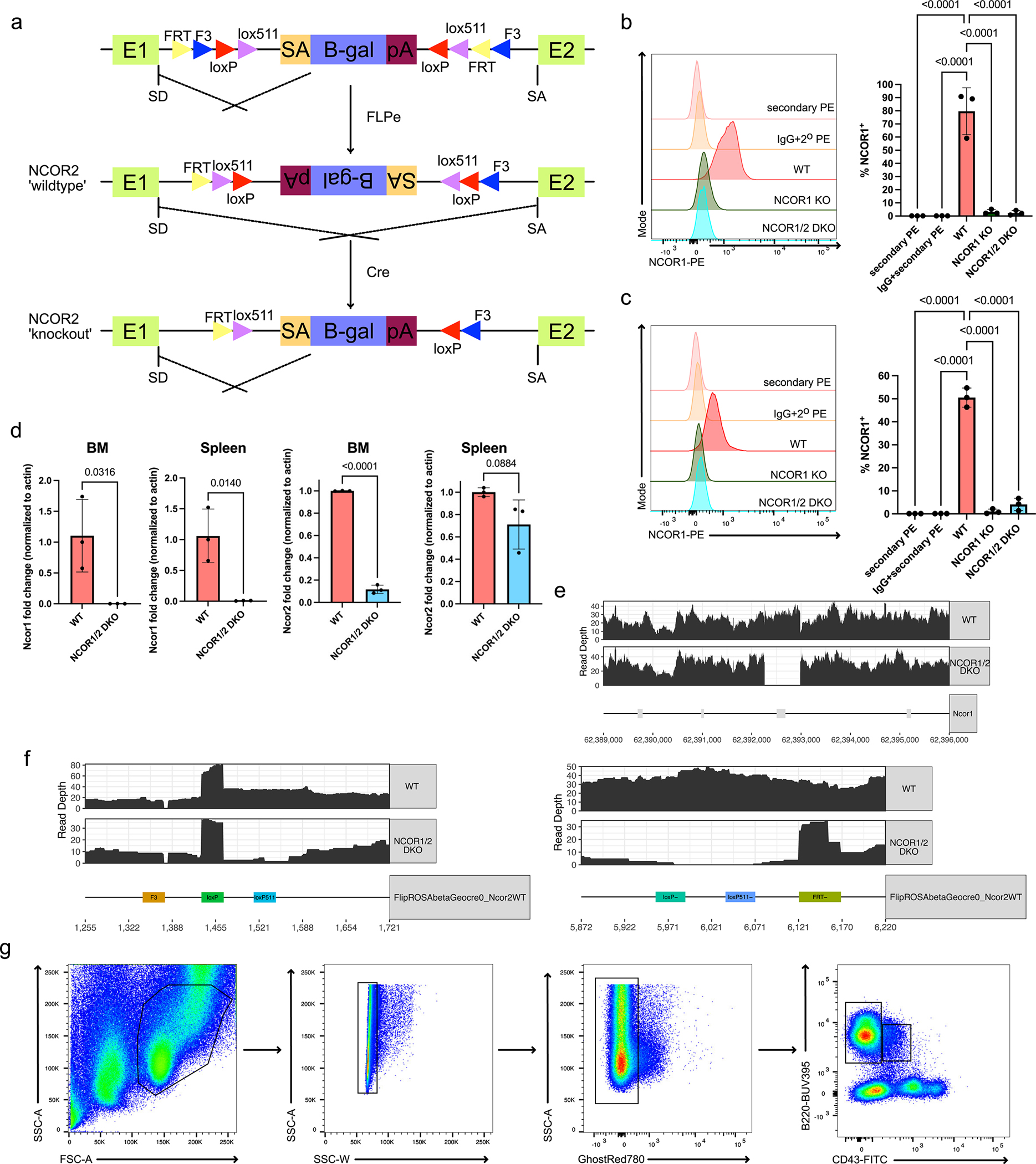
a. Schematic for generating Ncor2 flox mice using the flip-excision system (FlEx). A retrovirus vector containing a strong splice acceptor site followed by a lacZ reporter gene was flanked by FRT and loxP sites. This gene trap was targeted in the intronic region between exon 1 and 2. Mice harboring the gene trap were bred to FLPe constitutive expressing mice, inverting the gene trap and allowing for normal splicing of Ncor2. Subsequent breeding with Cd79a-Cre mice then allows for conditional deletion of Ncor2 in developing B cells. b. Representative NCOR1 protein expression in CD19+B220+ bone marrow B cells with isotype controls (left) and quantification of NCOR1-positive B cells (right). Each genotype represents n=3, from three independent experiments. Center of measure indicates mean and error bars indicate standard deviation. A one-way ANOVA was used to calculate statistical significance. c. Representative NCOR1 protein expression in CD19+B220+ splenic B cells with isotype controls (left) and quantification of NCOR1-positive B cells (right). Each genotype represents n=3, from three independent experiments. Center of measure indicates mean and error bars indicate standard deviation. A one-way ANOVA was used to calculate statistical significance. d. Quantification of Ncor1 and Ncor2 expression via qPCR in wildtype and NCOR1/2 double-knockout B cells from the bone marrow and the spleen. Each genotype represents n=3, from two independent experiments. Center of measure indicates mean and error bars indicate standard deviation. A two-tailed t-test was performed to calculate statistical significance. e. Whole-genome sequencing read depth coverage across the Ncor1 gene for wildtype and NCOR1/2 DKO B cells. f. Whole-genome sequencing mapping of reads across the Ncor2 gene trap for wildtype (Cre- littermate) and NCOR1/2 DKO B cells; left and right panels indicate 5’ and 3’ inversion breakpoints, respectively. g. Lymphocytes are gated using FSC-A, SSC-A, and then subsequently on SSC-A and SSC-W for singlets. Dead cells are excluded using the GhostRed780 Live/dead dye and gated on B220 and CD43. CD43+ and CD43− cells are gated on to assess Hardy fractions A-C and D-F, respectively.
Extended Data Fig. 2. Single-cell RNA-sequencing multiplet removal and cluster identification.
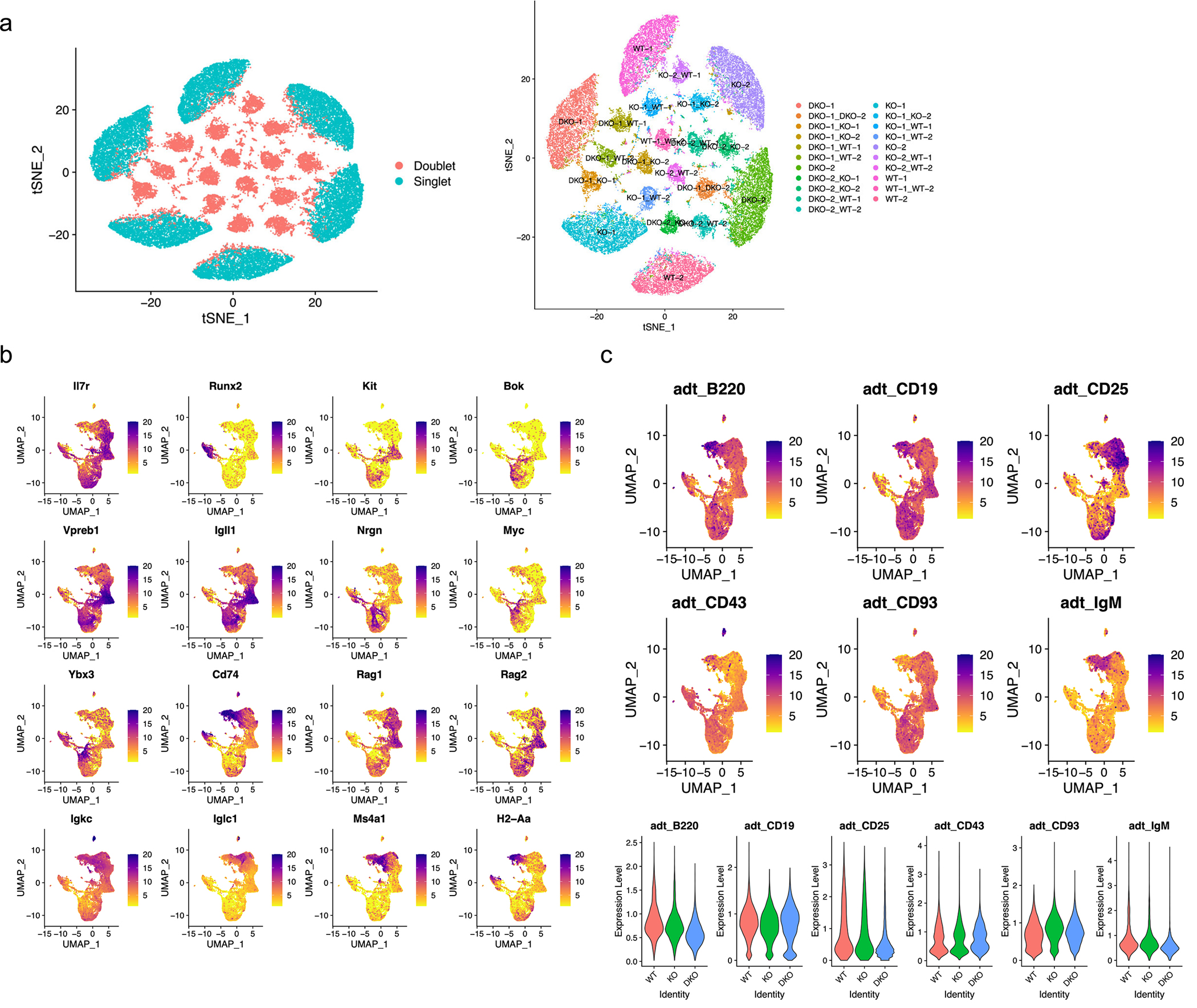
a. Multiplet identification using HTOdemux at a resolution of k=22 (left). Multiplet and singlet classification based on genotype is demonstrated on the right. b. Feature plots of temporally regulated genes during B cell development. c. Feature plot of CITE-Seq antibodies, B220, CD19, CD25, CD43, CD93, and IgM (top) and violin plot of the CITE-seq antibodies (bottom) are shown. These gene expression profiles and CITE-seq antibody expression patterns were used to characterize different subsets of B cell development.
Extended Data Fig. 3. Overlap between scRNA-seq and scATACseq differentially upregulated expression and accessibility genes.
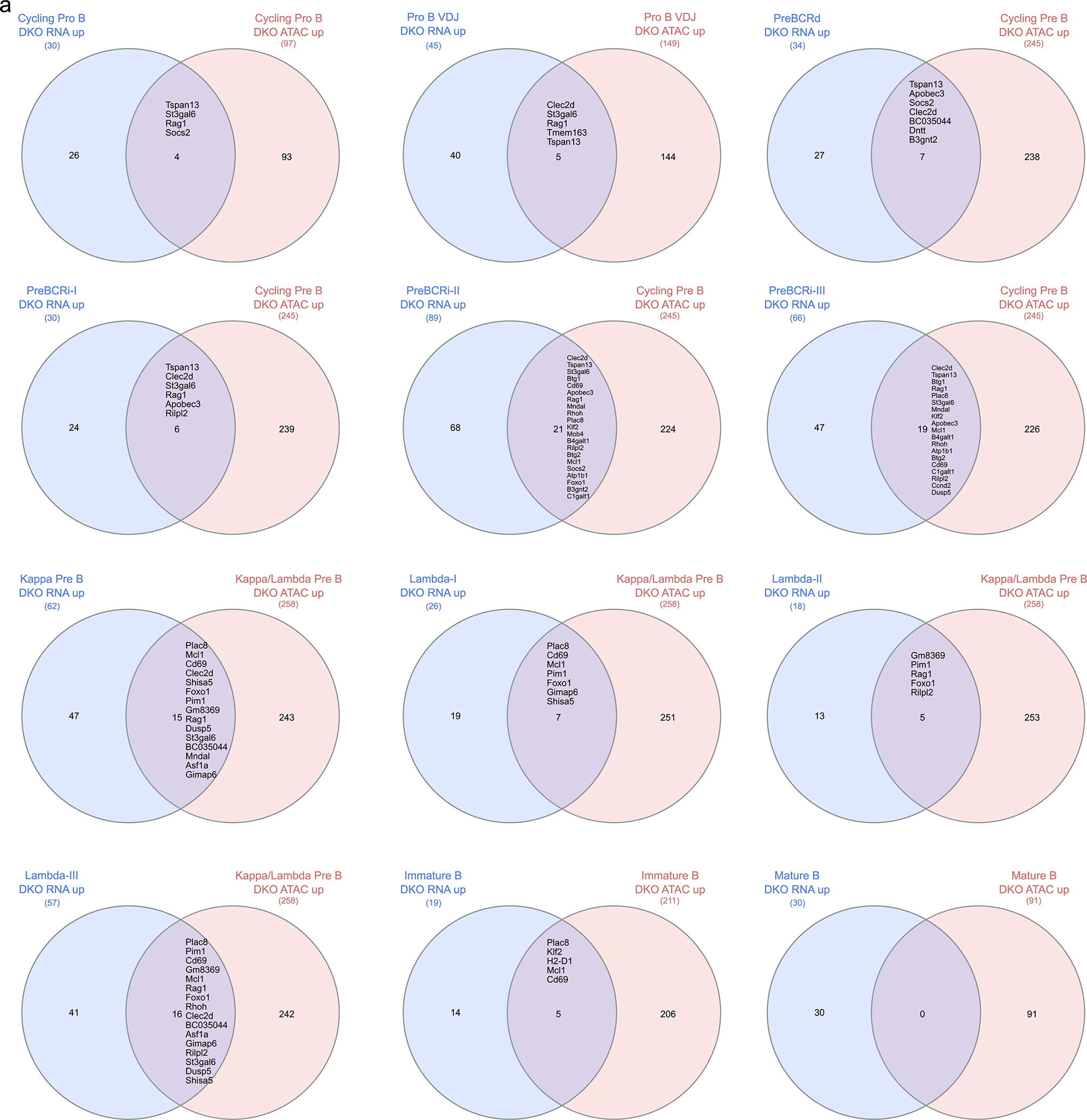
a. Cluster comparison between scRNAseq and scATACseq genes that are differentially upregulated in expression and accessibility in the NCOR1/2-knockout (DKO) B cells.
Extended Data Fig. 4. Nuclear corepressors regulate the p53 pathway and repress BIM expression.
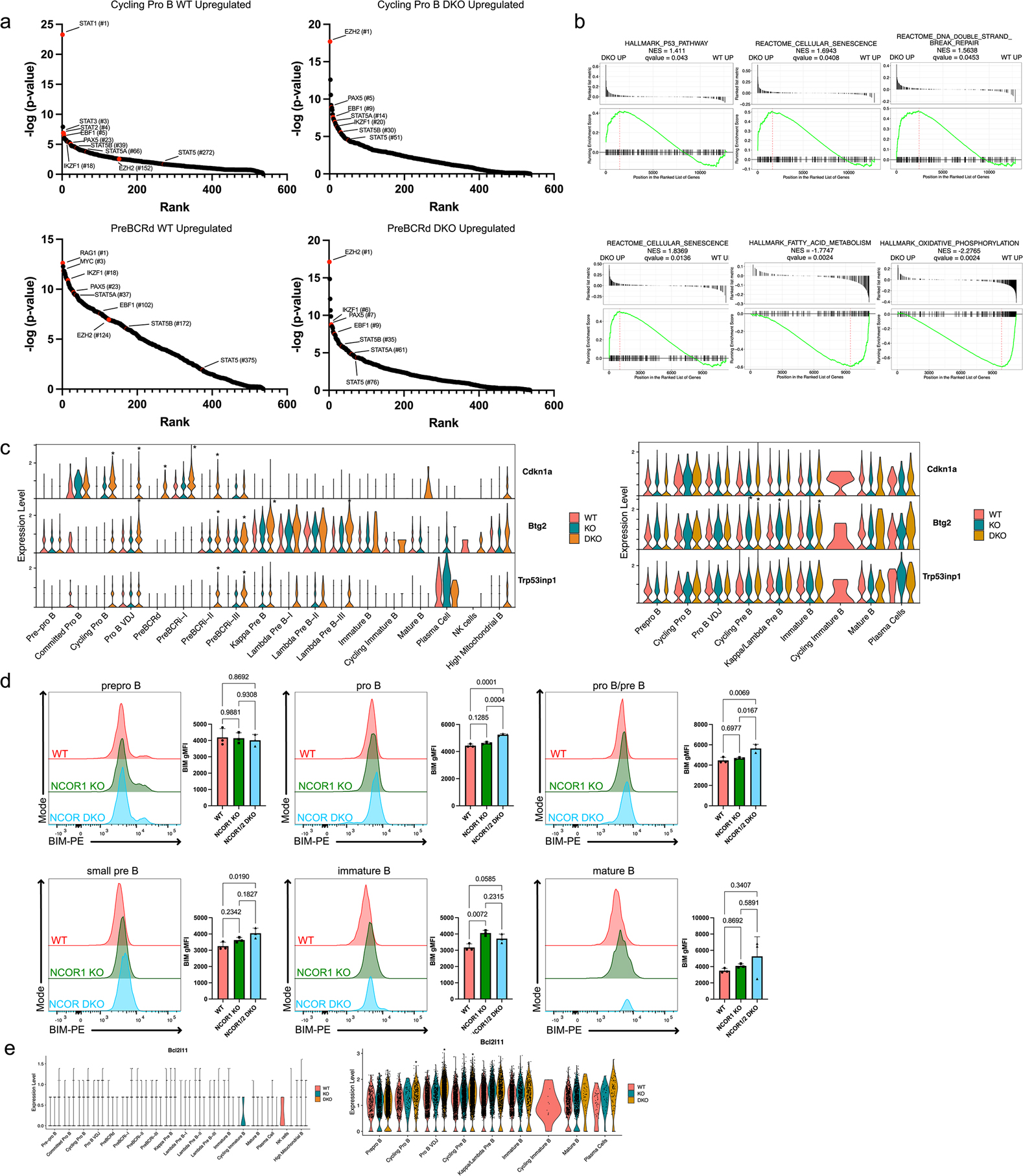
a. Landscape in silico analysis (LISA)-based prediction of transcriptional regulators of significantly upregulated genes in either WT (left) or NCOR1/2 DKO (right) of cycling pro B cells (top) or pre-BCR-dependent cells (bottom). Number in parentheses indicates rank. A two-tailed wilcoxon rank-sum test was used to calculated statistical significance. b. GSEA of p53 pathway and senescence pathway in NCOR1/2 DKO cells in both cycling pro B (top) and pre-BCR-dependent cells (bottom). c. Changes in expression of p53 pathway genes, Cdkn1a, Btg2, and Trp53inp1, between WT, NCOR1 KO and NCOR1/2 DKO B cell clusters. d. Pro-apoptosis factor, BIM expression of WT and NCOR1/2 DKO in pre-pro B, pro B, pro B/pre B, small pre B, immature B and mature B cells. Plot represents n=3 of each genotype from two independent experiments. Error bar indicates standard deviation. A one-way ANOVA was used to calculate statistical significance. e. BIM (Bcl2l11) gene expression and accessibility differences in wildtype, NCOR1 KO and NCOR1/2 DKO B cells. Asterisk indicates comparisons with at least a log2 fold change of 0.25 that have a p-value <0.05 when compared to wildtype. RNA expression of Bcl2l11 (left) represents WT (n=7454 cells), NCOR1 KO (n=7806 cells), NCOR1/2 DKO (n=8637 cells) from two biological replicates for each genotype from a single capture. Accessibility of Bcl2l11 (right) represents WT (n=8552 cells), NCOR1 KO (n=6008 cells), and NCOR1/2 DKO (n=8574 cells) from a single biological sample derived from three separate captures. A two-tailed Wilcoxon rank-sum test was used to calculate statistical significance. All measure of centers indicate mean.
Extended Data Fig. 5. KLF2 and its target genes are aberrantly expressed in NCOR1/2-deficient B cells.
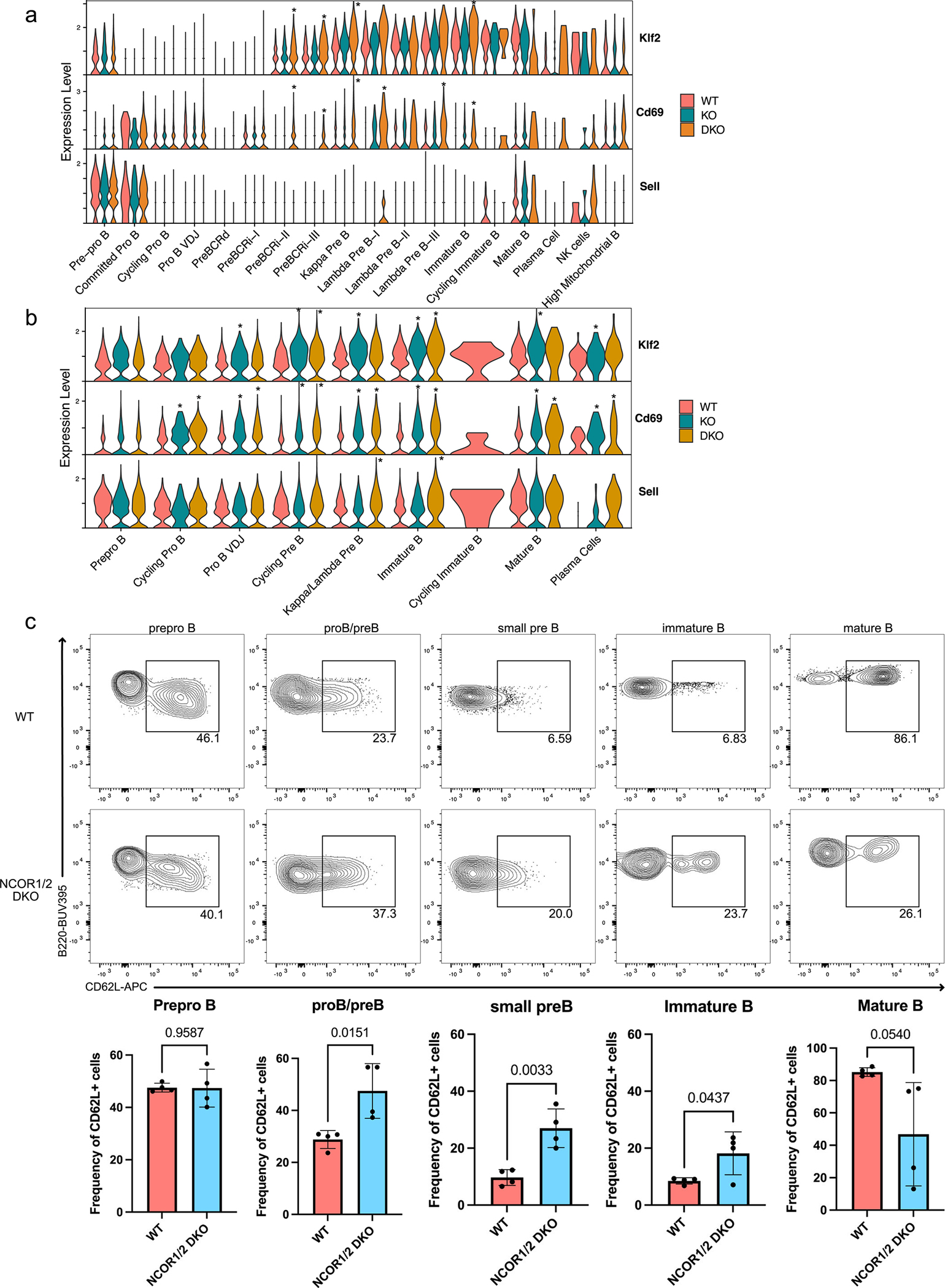
a. RNA expression of Klf2 and its target genes Cd69 and Sell (CD62L; L-selectin) in wildtype (WT), NCOR1-knockout (KO) and NCOR1/2-knockout (DKO) B cells. Asterisk (*; P<0.05) indicates statistically significant expression changes compared to wildtype cells for Klf2 in PreBCRi-II (DKO; P=4.24E-15), preBCRi-III (DKO; P=1.43E-9), and immature B (DKO; P=1.34 E-8), for Cd69 in PreBCRi-II (DKO; P=1.86E-39), PreBCRi-III (DKO; P=2.74E-8), Kappa Pre B (DKO; P=1.47E-37), Lambda Pre B-I (DKO; P=1.69E-12), Lambda Pre B-III (DKO; P=9.14E-14), and Immature B (DKO; P=1.89E-7). b. Gene activity of Klf2 and its target genes Cd69 and Sell (CD62L; L-selectin)in WT, KO and DKO B cells. Asterisk (*; P< 0.05) indicates statistically significant accessibility changes compared to wildtype cells for Klf2 in Pro B VDJ (KO; P=1.23E-9), Cycling Pre B (KO; P=2.94E-36, DKO; P=4.97E-24), Kappa/Lambda Pre B (KO; P=5.31E-33), Immature B (KO; P=1.22E-14, DKO; P=6.40E-11), mature B (KO; P=4.94E-13), plasma cells (KO; P=4.94E-13, DKO; P=5.39E-7) and for Cd69 in Cycling Pro B (KO; P=1.74E-6, DKO; P=2.12E-12), Pro B VDJ (KO; P=4.20E-13, DKO; P=2.47E-30), Cycling Pre B (KO; P=1.29E-27, DKO; P=6.87E-84), Kappa/Lambda Pre B(KO; P=1.96E-57, DKO; P=6.92E-46), Immature B (KO; P=3.72E-27, DKO; P=3.13E-22), mature B (KO; P=1.68E-20, DKO;P=5.39E-7), plasma cells (KO; P=1.68E-20, DKO; P=5.39E-7) and for Sell in Kappa/Lambda Pre B (DKO; P=4.27E-7), and immature B (DKO;P=1.47E-16). c. CD62L expression of different B cell development stages in wildtype (WT) and NCOR1/2-knockout (DKO) B cells. Top represents representative flow cytometry plots for CD62L expression and bottom represents summarized frequency of CD62L+ cells for prepro B (B220+CD43+CD24−CD19−, n=4), proB/preB (B220+CD43+CD24+CD19+, n=4), small pre B (B220+CD43+CD19+IgM−, n=4), immature B (B220lowCD43+gM+, n=4), and mature B cells (B220highCD43+gM+, n=4) from three independent experiments. A two-tailed t-test was used to calculate statistical significance. Center of measure indicates mean and error bars indicate standard deviation.
Extended Data Fig. 6. Transcriptional and accessibility changes in genes associated with Rag-mediated genomic changes in human B-ALLs.
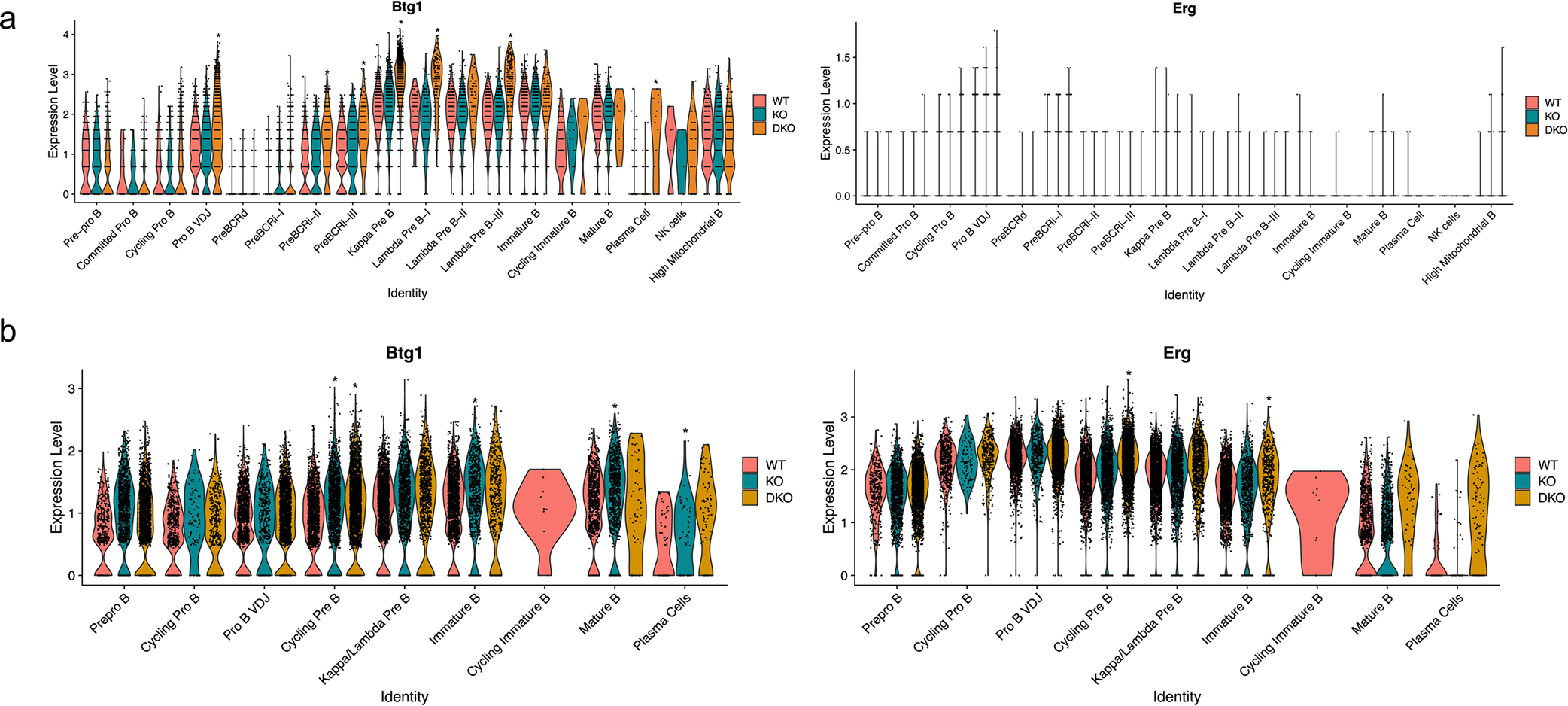
a. Transcriptional changes between WT, NCOR1 KO, and NCOR1/2 DKO derived from scRNA-seq. b. Accessibility changes between WT, NCOR1 KO, and NCOR1/2 DKO derived from scATAC-seq. Asterisk (*) indicates comparisons with at least a log2 fold change of 0.25 that have a p-value <0.05 when compared to wildtype. RNA expression (left) data represents WT (n = 7454 cells), NCOR1 KO (n = 7806 cells), NCOR1/2 DKO (n = 8637 cells) from two biological replicates for each genotype from a single capture. ATAC accessibility data (right) represents WT (n = 8552 cells), NCOR1 KO (n = 6008 cells), and NCOR1/2 DKO (n = 8574 cells) from a single biological sample derived from three separate captures. A two-tailed Wilcoxon rank-sum test was used to calculate statistical significance.
Supplementary Material
Supplementary Table 1. Pairwise comparison of differentially expressed genes between WT, NCOR1 KO and NCOR1/2 DKO. A two-tailed Wilcoxon rank-sum test was performed to calculate statistical significance. Cluster labels correspond to the following cell type as following: Cluster 0 - Kappa Pre B, Cluster 1 - Pro B VDJ, Cluster 2 - PreBCRi-I, Cluster 3 - PreBCRi II, Cluster 4 - Prepro B, Cluster 5 - Immature B, Cluster 6 - Cycling Pro B, Cluster 7 - Mature B, Cluster 8 - PreBCRi-III, Cluster 9 - Lambda Pre B-II, Cluster 10 - Lambda Pre B-III, Cluster 11 - PreBCR-d, Cluster 12 - High mitochondrial B, Cluster 13 - Lambda Pre B-I, Cluster 14 - Committed Pro B, Cluster 15 - Plasma Cells, Cluster 16 - Cycling Immature B.
Supplementary Table 2. Pairwise comparison of differential transcription factor motifs present between WT, NCOR1 KO and NCOR1/2 DKO. Transcription factor motifs were derived from the JASPAR database. A two-tailed Wilcoxon rank-sum test was performed to calculate statistical significance.
Supplementary Table 3. Pairwise comparison of differentially accessible genes between WT, NCOR1 KO and NCOR1/2 DKO. Accessibility of the gene was defined as peaks found within the gene body and peaks present within the upstream 2000 bp promoter. A two-tailed Wilcoxon rank-sum test was performed to calculate statistical significance.
Supplementary Table 4. Structural variants found in the wildtype and NCOR DKO B cells. Structural variants including, BND; break-end, DEL; deletion, INS; insertion, TRA; translocation, INV; inversion, and their genomic position and affected genes are listed. Structural variant calling was performed using Smoove.
Supplementary Table 5. Human B-ALL NCOR1 mutation type and the presence of intrachromosomal or interchromosomal structural variants. Types of NCOR1 mutations and structural variants that are present are broken down into intrachromosomal and interchromosomal. Interchromosomal translocations that are B-ALL subtype defining are present. Structural variant information was derived from StJude ProteinPaint.
Acknowledgments
We thank G. Hubbard, A. Rost, and N. Keller, for technical assistance and mouse husbandry, E. Stanley, J. Daniels, and Dr. K. Beckman and the University of Minnesota Genomics Center for 10X genomics single-cell capture and sequencing, J. Motl, R. Arora, and P. Champoux for cell sorting and Flow Cytometry Core Facility maintenance at University of Minnesota (5P01AI035296) and S. Dehm and K. Schwertfeger (University of Minnesota) for comments on the manuscript. We thank E. Olson (UT-Southwestern), W. Ellmeier (Medical University of Vienna) and J. Auwerx (École Polytechnique Fédérale, Lausanne, Switzerland) for providing the Ncor1FL/FL mice. The Minnesota Supercomputing Institute (MSI) at the University of Minnesota provided bioinformatic support and resources that contributed to the research results reported within this paper. We thank the support from the German Gene Trap Consortium for assisting in the generation of the Ncor2FL/FL mice. This work was supported by an individual predoctoral F30 fellowship from the NIH (F30CA232399) and T32 training grant (T32 GM008244) to R.D.L, St Jude Children’s Research Hospital Cancer Center Core Grant CA021765 and NIH grant R35CA197695 to C.G.M. and NIH grants R01AI124512, R01AI147540, R01CA232317 to M.A.F.
Footnotes
Competing Interests Statement
C.G.M. provides consultation and advise for Illumina (compensated), Faze Medicines (compensated), Beam Therapeutics (compensated) and receives research funding from Pfizer and Abbvie. The remaining authors declare no competing interests.
Editor recognition statement (if applicable to your journal):
L. A. Dempsey was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Peer Review Information:
Nature Immunology thanks the anonymous reviewers for their contribution to the peer review of this work.
References
- 1.Melchers F Checkpoints that control B cell development. J. Clin. Invest. 125, 2203–2210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Katerndahl CDS et al. Antagonism of B cell enhancer networks by STAT5 drives leukemia and poor patient survival. Nat. Immunol. 18, 694–704 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mullighan CG et al. Deletion of IKZF1 and Prognosis in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 360, 470–480 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heltemes-Harris LM et al. Identification of mutations that cooperate with defects in B cell transcription factors to initiate leukemia. Oncogene 40, 6166–6179 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heltemes-Harris LM et al. Ebf1 or Pax5 haploinsufficiency synergizes with STAT5 activation to initiate acutelymphoblastic leukemia. J. Exp. Med. 208, 1135–1149 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mottis A, Mouchiroud L & Auwerx J Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 27, 819–835 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.You SH et al. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat. Struct. Mol. Biol. 20, 182–187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mullighan CG et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471, 235–241 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reddy A et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 171, 481 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jepsen K et al. Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell 102, 753–763 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Jepsen K et al. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature 450, 415–419 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Yamamoto H et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell 147, 827–839 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schnütgen F et al. A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat. Biotechnol. 21, 562–565 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Hardy RR, Carmack CE, Shinton SA, Kemp JD & Hayakawa K Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J. Exp. Med. 173, 1213–1225 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heng TSP et al. The immunological genome project: Networks of gene expression in immune cells. Nat. Immunol. 9, 1091–1094 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Goodnow CC et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 334, 676–682 (1988). [DOI] [PubMed] [Google Scholar]
- 17.Lee RD et al. Single-cell analysis identifies dynamic gene expression networks that govern B cell development and transformation. Nat. Commun. 12, 6843 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heizmann B, Kastner P & Chan S Ikaros is absolutely required for pre-B cell differentiation by attenuating IL-7 signals. J. Exp. Med. 210, 2823–2832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan LN et al. Signalling input from divergent pathways subverts B cell transformation. Nature 583, 845–851 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goetz CA, Harmon IR, O’Neil JJ, Burchill MA & Farrar MA STAT5 Activation Underlies IL7 Receptor-Dependent B Cell Development. J. Immunol. 172, 4770–4778 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Stengel KR et al. Histone deacetylase 3 controls a transcriptional network required for B cell maturation. Nucleic Acids Res. 47, 10612–10627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hatzi K et al. A Hybrid Mechanism of Action for BCL6 in B Cells Defined by Formation of Functionally Distinct Complexes at Enhancers and Promoters. Cell Rep. 4, 578–588 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su IH et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat. Immunol. 4, 124–131 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Jacobsen JA et al. EZH2 Regulates the Developmental Timing of Effectors of the Pre–Antigen Receptor Checkpoints. J. Immunol. 198, 4682–4691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rouault JP et al. Identification of BTG2, an antiproliferative p53-dependent component of the DNA damage cellular response pathway. Nat. Genet. 14, 482–486 (1996). [DOI] [PubMed] [Google Scholar]
- 26.Tomasini R et al. p53-dependent expression of the stress-induced protein (SIP). Eur. J. Cell Biol. 81, 294–301 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Han J, Goldstein LA, Hou W, Gastman BR & Rabinowich H Regulation of mitochondrial apoptotic events by p53-mediated disruption of complexes between antiapoptotic bcl-2 members and bim. J. Biol. Chem. 285, 22473–22483 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J et al. NCoR1 restrains thymic negative selection by repressing Bim expression to spare thymocytes undergoing positive selection. Nat. Commun. 8, 959 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Müller L et al. The corepressor NCOR1 regulates the survival of single-positive thymocytes. Sci. Reports 7, 15928 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carlson CM et al. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature 442, 299–302 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Hart GT, Wang X, Hogquist KA & Jameson SC Krüppel-like factor 2 (KLF2) regulates B-cell reactivity, subset differentiation, and trafficking molecule expression. Proc. Natl. Acad. Sci. U. S. A. 108, 716–721 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skon CN et al. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 14, 1285–1293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teng G et al. RAG Represents a Widespread Threat to the Lymphocyte Genome. Cell 162, 751–765 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burchill MA et al. Distinct Effects of STAT5 Activation on CD4+ and CD8+ T Cell Homeostasis: Development of CD4+CD25+ Regulatory T Cells versus CD8+ Memory T Cells. J. Immunol. 171, 5853–5864 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Nakayama J et al. BLNK suppresses pre–B-cell leukemogenesis through inhibition of JAK3. Blood 113, 1483–1492 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heltemes-Harris LM et al. Sleeping Beauty transposon screen identifies signaling modules that cooperate with STAT5 activation to induce B-cell acute lymphoblastic leukemia. Oncogene 35, 3454–3464 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gu Z et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 51, 296–307 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsai AG et al. Human Chromosomal Translocations at CpG Sites and a Theoretical Basis for Their Lineage and Stage Specificity. Cell 135, 1130–1142 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fischer U et al. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat. Genet. 47, 1020 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Papaemmanuil E et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet. 46, 116–125 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang J et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat. Genet. 48, 1481–1489 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abdelrasoul H et al. Synergism between IL7R and CXCR4 drives BCR-ABL induced transformation in Philadelphia chromosome-positive acute lymphoblastic leukemia. Nat. Commun. 11, 3194 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waanders E et al. Mutational Landscape and Patterns of Clonal Evolution in Relapsed Pediatric Acute Lymphoblastic Leukemia. Blood Cancer Discov. 1, 96–111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhaskara S et al. Hdac3 Is Essential for the Maintenance of Chromatin Structure and Genome Stability. Cancer Cell 18, 436–447 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swaminathan S et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat. Immunol. 16, 766–774 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mori H et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc. Natl. Acad. Sci. 99, 8242–8247 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Narendra V et al. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khoury A et al. Constitutively bound CTCF sites maintain 3D chromatin architecture and long-range epigenetically regulated domains. Nat. Commun. 11, 54 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Degner SC et al. CCCTC-binding factor (CTCF) and cohesin influence the genomic architecture of the Igh locus and antisense transcription in pro-B cells. Proc. Natl. Acad. Sci. U. S. A. 108, 9566–9571 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ba Z et al. CTCF orchestrates long-range cohesin-driven V(D)J recombinational scanning. Nature 586, 305–310 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 51.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hafemeister C & Satija R Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qin Q et al. Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol. 21, 32 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramanian A et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liberzon A et al. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stuart T, Srivastava A, Madad S, Lareau CA & Satija R Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schep AN, Wu B, Buenrostro JD & Greenleaf WJ chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fornes O et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 48, D87–D92 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Layer RM, Chiang C, Quinlan AR & Hall IM LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 15, 1–19 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chikina MD & Troyanskaya OG An effective statistical evaluation of ChIPseq dataset similarity. Bioinformatics 28, 607–613 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Livak KJ & Schmittgen TD Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 63.Zhou X et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat. Genet. 48, 4–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ou J & Zhu LJ trackViewer: a Bioconductor package for interactive and integrative visualization of multi-omics data. Nat. Methods 16, 453–454 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Gentles AJ et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 21, 938–945 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bunis DG, Andrews J, Fragiadakis GK, Burt TD & Sirota M dittoSeq: universal user-friendly single-cell and bulk RNA sequencing visualization toolkit. Bioinformatics 36, 5535–5536 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Pairwise comparison of differentially expressed genes between WT, NCOR1 KO and NCOR1/2 DKO. A two-tailed Wilcoxon rank-sum test was performed to calculate statistical significance. Cluster labels correspond to the following cell type as following: Cluster 0 - Kappa Pre B, Cluster 1 - Pro B VDJ, Cluster 2 - PreBCRi-I, Cluster 3 - PreBCRi II, Cluster 4 - Prepro B, Cluster 5 - Immature B, Cluster 6 - Cycling Pro B, Cluster 7 - Mature B, Cluster 8 - PreBCRi-III, Cluster 9 - Lambda Pre B-II, Cluster 10 - Lambda Pre B-III, Cluster 11 - PreBCR-d, Cluster 12 - High mitochondrial B, Cluster 13 - Lambda Pre B-I, Cluster 14 - Committed Pro B, Cluster 15 - Plasma Cells, Cluster 16 - Cycling Immature B.
Supplementary Table 2. Pairwise comparison of differential transcription factor motifs present between WT, NCOR1 KO and NCOR1/2 DKO. Transcription factor motifs were derived from the JASPAR database. A two-tailed Wilcoxon rank-sum test was performed to calculate statistical significance.
Supplementary Table 3. Pairwise comparison of differentially accessible genes between WT, NCOR1 KO and NCOR1/2 DKO. Accessibility of the gene was defined as peaks found within the gene body and peaks present within the upstream 2000 bp promoter. A two-tailed Wilcoxon rank-sum test was performed to calculate statistical significance.
Supplementary Table 4. Structural variants found in the wildtype and NCOR DKO B cells. Structural variants including, BND; break-end, DEL; deletion, INS; insertion, TRA; translocation, INV; inversion, and their genomic position and affected genes are listed. Structural variant calling was performed using Smoove.
Supplementary Table 5. Human B-ALL NCOR1 mutation type and the presence of intrachromosomal or interchromosomal structural variants. Types of NCOR1 mutations and structural variants that are present are broken down into intrachromosomal and interchromosomal. Interchromosomal translocations that are B-ALL subtype defining are present. Structural variant information was derived from StJude ProteinPaint.
Data Availability Statement
The data supporting this study are available within the paper and supplementary information file. A reporting summary is also available. Mouse genome mm10 was used as reference sequence (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/). Single-cell RNA-Seq and single-cell ATAC-seq data were deposited at Gene Expression Omnibus, with the following accession code: GSE208656 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE208656). Whole genome sequencing data was deposited at SRA with the following accession code: PRJNA860179 (http://www.ncbi.nlm.nih.gov/bioproject/860179). STAT5 ChIP-Seq peaks data were obtained from GSE86878 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE86878) and H3K27ac ChIP-seq peaks data were obtained from cistrome.org via project GSM1463433. IMMGEN RNA-Seq count data was from GSE124829. Human B-ALL NCOR1 and NCOR2 mutation and RNA-seq data were obtained from StJude ProteinPaint (https://pecan.stjude.cloud/proteinpaint). Source data are provided with this paper.