

Abstract
The immunosuppressive tumor microenvironment in some cancer types, such as luminal breast cancer, supports tumor growth and limits therapeutic efficacy. Identifying approaches to induce an immunostimulatory environment could help improve cancer treatment. Here, we demonstrate that inhibition of cancer-intrinsic EZH2 promotes antitumor immunity in estrogen receptor α-positive (ERα+) breast cancer. EZH2 is a component of the PRC2 complex, which catalyzes trimethylation of histone H3 at lysine 27 (H3K27me3). A 53-gene PRC2 activity signature was closely associated with the immune responses of ERα+ breast cancer cells. The stimulatory effects of EZH2 inhibition on immune surveillance required specific activation of type I interferon (IFN) signaling. Integrative analysis of PRC2-repressed genes and genome-wide H3K27me3 landscape revealed that type I IFN ligands are epigenetically silenced by H3K27me3. Notably, the transcription factor STAT2, but not STAT1, mediated the immunostimulatory functions of type I IFN signaling. Following EZH2 inhibition, STAT2 was recruited to the promoters of IFN-stimulated genes even in the absence of the cytokines, suggesting the formation of an autocrine IFN-STAT2 axis. In patients with luminal breast cancer, high levels of EZH2 and low levels of STAT2 were associated with the worst antitumor immune responses. Collectively, this work paves the way for the development of an effective therapeutic strategy that may reverse immunosuppression in cancer.
Introduction
Dysregulation of immune-associated genes due to epigenetic alterations in cancer cells or surrounding immune cells is one of the mechanisms for immune escape and cancer progression (1). The histone methyltransferase EZH2 has been repeatedly demonstrated to play an important role in epigenetic regulation of genes involved in immunoediting (2). It forms the polycomb-repressive complex 2 (PRC2) with other core subunits such as SUZ12 and EED (3,4) and catalyzes methylation of histone H3 at lysine 27 (H3K27me), which is associated with gene silencing (5). Although EZH2 is generally demonstrated to elicit a tumor immune-tolerant phenotype, the exact mechanisms underlying PRC2-mediated immune escape are quite controversial and context dependent. On one hand, cancer-intrinsic EZH2 promotes cancer immunoediting that ultimately constrains the immune system. It does so by either silencing genes that evoke antitumor immunity via H3K27me3, such as the T-cell-attracting chemokines in ovarian and colon cancer cells (6,7), or upregulating the expression of genes essential for cancer cells to escape from immune surveillance, such as PD-L1 in lung cancer cells in a methyltransferase-independent manner (8). On the other hand, EZH2 expressed in immune cells plays an important role in determining their fates. For example, T cell development, differentiation and activation can be regulated by EZH2 as a canonical subunit of the PRC2 complex or as a noncanonical methyltransferase for non-histone substrates (9). All these results highlight the complexity of EZH2’s effects on cancer immunity. However, the underlying mechanisms are very dynamic and contribution of the epigenetic mark H3K27me3 to the immunomodulatory role of EZH2 warrants further investigation.
Although immunotherapy including immune checkpoint blockade has been a paradigm shift in cancer treatment, many types of cancer are found non-immunogenic, limiting the clinical applications of immunotherapeutic approaches. Insensitivity of cancer cells to immunotherapy is usually associated with the presence of an immunosuppressive tumor microenvironment characterized by a low mutational load, inefficient cytotoxic T-lymphocyte infiltration, defective antigen presentation, etc (10–12). In breast cancer, checkpoint inhibitors show the greatest benefits in triple-negative subtype, whereas other subtypes, especially the luminal, hormone receptor-positive one, are innately insensitive to these immunotherapy drugs (13). Thus, it is clinically urgent to better understand the basis of immune escape, aiming to regenerate an antitumor immunity in these immunologically inert cancer types. Accumulated evidence suggests that selective inhibitors of EZH2 methyltransferase activity hold great promise for overcoming immunotherapy resistance in a wide range of cancers (14). Unfortunately, whether EZH2-targeting drugs will boost immune surveillance in luminal breast cancer has never been investigated.
In the present study, we demonstrated that pharmacological and genetic inhibition of cancer-intrinsic EZH2 stimulates an antitumor immunity by activating interferon (IFN) signaling in estrogen receptor α-positive (ERα+) breast cancer. Intriguingly, type I IFN ligands are specifically silenced by the PRC2 complex and selective inhibitors of PRC2 lead to the activation of an autocrine IFN-STAT2 axis. Our findings have laid the foundation for an anticancer therapeutic strategy that will potentiate immune responses in luminal breast cancer by targeting the epigenetic regulator EZH2.
Materials and Methods
Cell culture
Human breast cancer cell lines MCF-7 and T-47D were originally purchased from the American Type Culture Collection (ATCC). They were cultured in DMEM and RPMI 1640, respectively. HEK293T cells were also from ATCC and they are always maintained in DMEM. Murine breast cancer cell line 67NR was a gift generously provided by Ann Richmond (Vanderbilt University) and cultured in DMEM. All the culture media were supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (P/S). All the cells are incubated at 37°C with 5% CO2 and routinely tested for mycoplasma contamination using Mycoplasma Detection Kit-DigitalTest v2.0 (BioTool, Cat#B39132). Cell lines used in this study are not in the list of commonly misidentified cell lines that is curated by the International Cell Line Authentication Committee (ICLAC).
Antibodies and reagents
Antibodies used for immunoblotting analysis in this study include: αEZH2 (BD Biosciences, #612667), αActin (Sigma-Aldrich, #A5441), αH3K27me3 (Cell Signaling, #9733), αH3 (Cell Signaling, #9715), αpSTAT2 (Cell Signaling, #88410), αSTAT2 (Santa Cruz, #sc-1668 and Cell Signaling, #72604), αpSTAT1 (Cell Signaling, #9167), αSTAT1 (Cell Signaling, #9172) and αGAPDH (Santa Cruz, #sc-365062). Antibodies that were applied in other assays, such as ChIP, fluorescent immunohistochemistry and flow cytometry, are specified in the corresponding sections.
In this study, the following inhibitors of the PRC2 activity were included: GSK503 (Selleckchem, #S7804), GSK126 (Xcessbio, #60071), EPZ-6438 (Xcessbio, #60122) and EED226 (Selleckchem, #S8496). The interferon (IFN) ligands used in this study are: IFNβ, IFNγ and IL-29, and their details are provided in the section “Neutralization of IFN ligands” below.
Plasmids expressing shRNAs or sgRNAs
Sequences of shRNAs against non-specific control sequences (shCtrl) and EZH2 (shEZH2–4 and -A) were referred to the previous publication (15). shRNAs targeting mouse Ezh2 (shEzh2, TRCN0000304506), human STAT1 (shSTAT1, TRCN0000004264) or human STAT2 (shSTAT2–1, TRCN0000364398 and shSTAT2–2, TRCN0000364400) were purchased from Sigma-Aldrich (St. Louis, Missouri). The lentiviral vectors co-expressing Cas9 and sgRNA against non-specific sequences (sgCtrl), Ezh2 or Stat2 were generated based on the pLentiCRISPRv2-puro plasmid (Addgene, #98290). Sense and anti-sense oligos were annealed using primers that are listed in Supplemental Table 1 and then ligated into pLentiCRISPRv2-puro plasmid digested with BsmBI.
Lentivirus production and generation of stable cell lines
Lentivirus-mediated establishment of stable clones were performed as previously described (15). Briefly, lentiviral vectors carrying shRNA sequences were co-transfected with packaging (pCMV delta R8.9 or psPAX2) and envelope (VSV-G) plasmids into HEK293T cells. The lentiviral particles were harvested twice at 48- and 72-hour time point after transfection, passed through a membrane filter with 0.45 μm pore size and concentrated with Lenti-X concentrator (TAKARA). Host cells were infected with the lentiviral particles in the presence of 5 μg/mL polybrene (Sigma-Aldrich), then selected using 1.5 μg/mL puromycin (Sigma-Aldrich) 24 hours after infection for at least 3 days, and finally propagated in fresh medium supplemented with the same concentration of puromycin for at least two passages, which are about 4–6 days depending on the host breast cancer cell lines.
Protein lysis and Western blots
Immunoblotting analysis was performed as previously described (15). Human cells or allograft tumors were collected and lysed in the RIPA lysis buffer [1% IGEPAL CA-630, 0.5% sodium deoxycholate, 1% sodium dodecyl sulphate (SDS), 150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 10 mM sodium fluoride (NaF), 2 mM sodium orthovanadate (Na3VO4), 1 mM phenylmethanesulfonyl fluoride (PMSF) and 1×protease inhibitor (Roche)] for at least 30 minutes at 4°C. For H3K27me3 detection, cells were lysed in EBC buffer [0.5% IGEPAL CA-630, 120 mM NaCl, 1 mM EDTA (pH 8.0), 50 mM Tris-HCl (pH 7.5)] followed by sonication for 3–4 minutes in Q800R sonicator (QSONICA). Protein lysates were subjected to SDS-PAGE electrophoresis and finally analyzed using Western blots with indicated antibodies.
Animal experiments
All the animal protocols were approved, and ethical compliance was enforced by UTHSCSA Institutional Animal Care and Use Committee (IACUC). Briefly, 1×105 67NR cells, either regular or engineered lineages (control and Ezh2-depeleted stable clones), were suspended in 50 μL of FBS-free DMEM and 50 μL of Matrigel Membrane Matrix HC (Corning). The mixture was injected into the mammary fat pads of female immunocompetent BALB/c mice (Charles River) or NSG mice (Jackson Laboratory) at 5–7 weeks of age. Tumor sizes were measured twice a week by caliper and calculated using the formula length×width2/2.
For the animal studies of assessing the efficacy of EZH2 inhibitor GSK503, when tumors reached a size ≥ 150 mm3, mice were randomly assigned to vehicle- or GSK53-treated group. GSK503 was dissolved in 20% Captisol (CyDex Pharmaceuticals) solution to make a stock solution with a final concentration of 15 mg/mL and delivered into the animal by intraperitoneal injection at a dosage of 150 mg/kg daily until termination of the experiment (approximately 10 days). The same volume of 20% Captisol solution was administered to mice in vehicle-treated group via the same route.
Mass cytometry (CyTOF) and flow cytometry
Allograft tumors collected from the syngeneic mouse models of breast cancer were first minced into small fragments and then digested in dissociation buffer [1 mg/mL Collagenase IV (Worthington Biochemical) and 20 μg/mL DNase I (Sigma Aldrich) in RPMI 1640 containing 10% FBS] for 20 minutes at 37°C with rotation. The cell suspension passed through a filter with 70 μM pore size to remove clumps and the remaining red blood cells were lysed in RBC lysis buffer (BioLegend, #420302). Single cells were counted and blocked with anti-CD16/32 (BioLegend, #156603) for 15 minutes on ice.
Staining of cell surface markers was performed with the indicated antibodies [anti-CD45 (BioLegend, #103116), anti-CD4 (BioLegend, #100510), anti-CD8 (BioLegend, #100708), and anti-CD3 (BioLegend, #100236)] for 20–30 minutes on ice. Antibodies used in CyTOF were listed in Supplemental Table 2. For intracellular markers, cells were washed, fixed and permeabilized using Foxp3/Transcription Factor Staining Buffer (eBiosciecne) and Maxpar® Fix I/Perm-S buffer (Fluidigm) for flow cytometry and CyTOF, respectively, and finally subject to immunostaining with anti-FoxP3 (eBioscience, #17-5773-82) for 30 minutes on ice. For CyTOF analysis, samples were run on a CyTOF instrument (Fluidigm) in the BASiC Core Facility at UTHSCSA, and the data were analyzed with Cytobank software. For flow cytometry, samples were read on LSR II (BD Biosciences) or Celesta (BD Biosciences) in the institutional Flow Cytometry Facility, and the data were analyzed using FlowJo software.
For flow cytometry analysis of the human or murine major histocompatibility complex (MHC) proteins, 1×106 cells per condition were stained with appropriate antibodies [anti-human HLA-A/B/C (BioLegend, #311426), anti-human β2M (BioLegnd, #316304), anti-mouse H-2Kd (BioLegend, #116605), or anti-mouse β2m (BioLegend, #154506)] in FACS buffer [5% (v/v) FBS and 0.05% (w/v) sodium azide in 1×PBS] for 20 minutes on ice.
For double-stranded RNA (dsRNA) staining, cells were fixed with 4% formaldehyde for 20 minutes at room temperature (RT) and permeabilized by 0.1% (v/v) Triton X-100 in 1×PBS for 15 minutes before sequential incubation with anti-dsRNA monoclonal antibody (SCICONS, #10010200) and Alexa Fluor 488 Goat anti-Mouse IgG secondary antibody (Invitrogen, #A11001). Matched Fluorescence Minus One (FMO) controls were included for each condition.
Fluorescent immunohistochemistry
Allograft tissues from the syngeneic mice were collected and fixed in 10% (v/v) formalin overnight and embedded in paraffin. Formalin-fixed paraffin embedded (FFPE) tissue samples were sectioned onto microscope slides, which were dewaxed and rehydrated sequentially in a graded ethanol series (100%, 90%, 70% and 50%) and finally in 1×PBS for 3 minutes per wash. Antigen retrieval was performed using antigen unmasking solution (Vector Laboratories, #H-3300). Slides were incubated with anti-CD3 (Cell Signaling, #78588) or anti-CD8 (Cell Signaling, #98941) antibody overnight at 4°C, followed by Alexa Fluor 488 Goat anti-Rabbit IgG secondary antibody (Invitrogen, #A11034) for 1 hour at RT. After washing with 1×PBS for three times, tissues were mounted by ProLong Gold Antifade Mountant with DAPI (Invitrogen, #P36931). Images were captured on a confocal laser scanning microscope (Carl Zeiss, LSM710).
Quantitative reverse transcription PCR (RT-qPCR)
Total RNAs were isolated using TRIzol Reagent (Thermo Fisher Scientific) and quantified by Cytation 5 Cell Imaging Multi-Mode Reader (BioTek). 1–2 μg of RNAs were reverse transcribed into cDNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) and subject to qPCR reactions in the SYBR™ Green qPCR Master Mix (Bio-Rad) using the Bio-Rad CFX96 Real-time PCR Detection System. Levels of tested transcripts were determined with the 2−ΔΔCt method and normalized to the expression of GAPDH, which serves as the internal control. The primers used in RT-qPCR were listed in Supplemental Table 3.
RNA-seq in allograft tissues or human cancer cell lines
67NR-grafted tumors were collected from control or GSK503-treated mice at the endpoint. Tumor tissue was processed into single cell suspension as described above in the section “Mass cytometry (CyTOF) and flow cytometry”. Total RNAs were extracted from the suspension using TRIzol Reagent (Thermo Fisher Scientific), and four to five independent tumors per condition were included as biological replicates. MCF-7 cells were infected with either control shRNA or EZH2-targeting shRNA (shEZH2–4). Total RNAs were extracted from these stable clones using TRIzol Reagent (Thermo Fisher Scientific). 1 μg of total RNAs were consumed to construct the mRNA-seq libraries using KAPA stranded mRNA Seq kit, which were sequenced on HiSeq 3000 at Genome Sequencing Facility of UTHSCSA.
Chromatin immunoprecipitation (ChIP)-qPCR and ChIP-seq
ChIP was performed with a dual cross-linking protocol that starts with 2 mM disuccinimidyl glutarate (DSG) (CovaChem) for 30 minutes followed by 1% formaldehyde (Sigma-Aldrich) for 10 minutes at RT. Cross-linking was quenched with 0.125 M Glycine for 5 minutes at RT. Nuclei were isolated sequentially using buffers LB1 [50 mM HEPES-KOH (pH 7.5), 140 mM NaCl, 1 mM EDTA (pH 8.0), 10% (v/v) glycerol, 0.5% (v/v) NP-40, 0.25% (v/v) Triton X-100 and 1×complete protease inhibitor (Roche)], LB2 [10 mM Tris-HCl (pH 8.0), 200 mM NaCl, 1 mM EDTA (pH 8.0), 0.5 mM EGTA (pH 8.0) and 1×complete protease inhibitor (Roche)] and LB3 [10 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA (pH 8.0), 0.5 mM EGTA (pH 8.0), 0.1% (w/v) Na-deoxycholate, 0.5% (w/v) N-lauroylsarcosine and 1×complete protease inhibitor (Roche)]. Isolated chromatin was fragmented in Q800R sonicator (QSONICA) and incubated with appropriate amounts of antibodies [1 μg H3K27me3 antibody (Cell Signaling, #9733), 10 μg EZH2 antibody (Proteintech, #21800–1-AP), 3 μg STAT2 antibody (Cell Signaling, #72604) or 3 μg phosphorylated STAT2 antibody (Cell Signaling, #88410)] that have been conjugated to Pierece™ Protein G magnetic beads overnight at 4°C. The immunoprecipitates were washed 5 times in RIPA wash buffer [50 mM HEPES-KOH (pH 7.5), 500 mM LiCl, 1 mM EDTA, 1% (v/v) NP40, 0.7% (w/v) sodium deoxycholate and 1×complete protease inhibitor (Roche)], twice in 1×TE buffer [10 mM Tris-HCl (pH 8.0) and 1 mM EDTA (pH 8.0)], and finally eluted in the elution buffer [1% (w/v) SDS, 10 mM Tris-HCl (pH 8.0) and 10 mM EDTA (pH 8.0)]. ChIP’d DNA was reverse cross-linked at 65°C overnight, purified by the treatment with RNase A and proteinase K, and finally subject to either qPCR reactions or ChIP-seq library construction. The ChIP-qPCR primers were listed in Supplemental Table 4. ChIP-seq libraries were prepared using ThruPLEX DNA-seq kit (Rubicon) and sequenced on HiSeq 3000 at Genome Sequencing Facility of UTHSCSA.
ELISA
Control and experimental (EZH2-knockdown or EZH2 inhibitor-treated) cells were propagated until they reached 70–80% confluency. The culture media were replaced 24–48 hours before collection of the replenished media. Type I interferon cytokines were analyzed using human IFN alpha DuoSet ELISA and human IFN beta DuoSet ELISA kits (R&D systems) according to the manufacturer’s instructions, and the absorbance was measured using Cytation 5 Cell Imaging Multi-Mode Reader (BioTek).
Neutralization of IFN ligands
Neutralizing antibodies were added into the culture media at the following final concentrations: 2 μg/mL for IgG isotype control [(R&D Systems, #AB-108-C) or (Invitrogen, #31903)], 0.5 μg/mL for anti-IFNα (R&D Systems, #21100–1), 2 μg/mL for anti-IFNβ (R&D Systems, # AF814), 2 μg/mL for anti-IFNγ (R&D Systems, #MAB2851), 4 μg/mL for anti-IL-29 (R&D Systems, #MAB15981) and 1 μg/mL for anti-IL-28A (R&D Systems, # MAB1587). Cells were incubated with the control or neutralizing antibodies for 24 hours with no intervening replenishments. Successful neutralization was confirmed by treating the cells with 250 pg/mL IFNβ (R&D Systems, #8499-IF-010), 250 pg/mL IFNγ (R&D Systems, #285-IF-100) or 50 ng/mL IL-29 (R&D Systems, #1598-IL-025). The recombinant human interferon ligands were administered 24 hours before total RNAs were extracted for gene expression analysis.
RNA-seq data analysis
Sequencing reads were aligned to human genome (hg19) using STAR 2.5.2b (16). After removing reads that were mapped to rRNAs, read counting for each gene was conducted by featureCounts package with default parameter (17). Genes with less than one read in at least 2 samples of the same condition were discarded. DESeq2 1.14.1 was used to call differentially expressed genes with fold change (FC)>=1.3 and false discovery rate (FDR)<= 0.05 as the statistical cutoffs (18). The clinical gene expression data were retrieved from TCGA (19) and METABRIC (20) datasets available on cBioportal website (https://www.cbioportal.org/faq).
ChIP-seq data analysis
Reads were aligned to human genome (hg19) using bowtie (21). Only uniquely mapped and non-redundant reads were retained for peak calling, which was fulfilled using MACS2 with a statistical cutoff of q value at 1E-3 (22). When calling peaks in H3K27me3 ChIP-seq data, the ‘broad’ mode was turned on. Peaks overlapped with the UCSC blacklist regions were removed. Motif enrichment analysis was performed using Homer (23) based on the top 1000 peaks in the ChIP-seq data of total STAT2 under GSK126 treatment condition or ChIP-seq data of phosphorylated STAT2 in EZH2-knockdown cells.
Definition of the repressive activity of EZH2
Genes upregulated upon knockdown of EZH2 in MCF-7 cells were ranked based on the fold changes compared to the control cells, and the top 200 genes that show the strongest upregulation were further considered. Among these 200 genes, only 53 of them were chosen to indicate the repressive activity of EZH2, as there are at least one H3K27me3 peak within the 5 kb window size, both upstream and downstream, from the transcriptional start sites (TSS) of these selected genes. Details of these 53 signature genes were listed in Supplementary Table 5.
We used Gene Set Variation Analysis (GSVA) (24) to calculate the signature score of these 53 signature genes, which represents the repressive activity of EZH2, in every individual patient with breast cancer from the TCGA (19) or METABRIC (20) dataset. In the analysis, expression of each signature gene in TCGA dataset was log2 transformed, and the Gaussian kernel was used to estimate the cumulative distribution function of the expression across different patients.
Identification of cancer hallmarks associated with EZH2 repressive activity in patients with different subtypes of breast cancer
Patients with the same subtype (luminal, HER2-positive or triple-negative) of breast cancer in the TCGA (19) or METABRIC (20) dataset were separated into quartiles based on the GSVA scores of the 53 signature genes. Patients in Quartile 1 (upper) have the highest and those in Quartile 4 (bottom) have the lowest GSVA scores, which indicate the weakest and strongest repressive activity of EZH2, respectively. Gene Set Enrichment Analysis (GSEA) was performed using genes highly expressed in patients in Quartiles 1 (red bars) and 4 (blue bars) were identified and used as the input for the against the cancer hallmarks collected in the Molecular Signatures Database.
Correlation of EZH2 repressive activity with immune response in patients with luminal breast cancer
GSVA scores of the 53 signature genes in patients with luminal breast cancer from the TCGA (19) or METABRIC (20) dataset were correlated with the following parameters that have been associated with active immune responses of cancer cells: expression of APP genes (25), T scores (26), MImm scores (27) and CYT scores (28–30). Eleven key genes involved in antigen processing and presentation (APP) pathway (B2M, HLA-A, HLA-B, HLA-C, TAP1, TAP2, TAPBP, ERAP1, ERAP2, PSMB8 and PSMB9) were selected (31) and the Z-score transformed expression of all these APP genes was summed in each patient with ERα+ breast cancer from the TCGA (19) or METABRIC (20) dataset. The T score was calculated based on the weighted expression sum of 18 genes (TNFRSF8, IDO1, NLRC5, CD3E, CCL5, GZMK, CD2, HLA-DRA, CXCL13, IL2RG, NKG7, HLA-E, CXCR6, LAG3, TAGAP, CXCL10, STAT1 and GZMB) in every patient with luminal breast cancer. As defined in a previous study (32), these 18 genes were obtained by overlapping the IFNγ-responsive genes and T cell-associated inflammatory genes. MImm score can be used to measure the magnitude of leukocyte infiltration and to predict response of cancer cells to checkpoint inhibition (26). In order to determine the MImm scores in patients with luminal breast cancer, we first transformed the expression of 141 immune-related genes that were defined in a published study (33) in each patient with luminal breast cancer to standard normal deviates using the “inverse normal transformation” method that is commonly applied in regression analyses (27), and then simply summed the standard normal deviates of each gene. Thus, the sum represents the MImm score for that particular patient. Cytolytic activity (CYT) score is considered as a new index of anti-cancer immune response, which is positively associated with cytotoxic T cell infiltration/activity and good clinical outcomes in a variety of cancers (28–30). Based on its original definition (34), we obtained the CYT score in individual patients with luminal breast cancer by taking the geometric mean of the mRNA levels of two genes, granzymes A (GZMA) and perforin (PRF1).
Association of EZH2 repressive activity with the copy number alterations of EZH2 gene in patients with luminal breast cancer
Information about the copy-number levels of EZH2 gene was obtained from cBioportal (https://www.cbioportal.org/faq). Patients with ERα+ breast cancer in the TCGA (19) or METABRIC (20) dataset were grouped into the following four groups based on the copy-number analysis algorithms that are used in cBioportal: (1). “−1”, meaning Shallow Deletion or a possible heterozygous deletion; (2). “0”, representing Diploid or cases with normal copy numbers of the gene; (3). “1”, indicating a low-level gain or a few additional, often broad, copies; and (4). “2”, suggesting Amplification or say more, often focal, copies. GSVA scores of the 53 signature genes were then calculated in each above-mentioned group of patients.
Data collection and visualization
All the ChIP-seq data were visualized in the Integrative Genomics Viewer (IGV) (35). All the genomic data sets were normalized to 10 million reads per sample.
Statistical analysis
All the experiments were repeated with at least three biological and technical replicates. Results were shown as mean ± standard error of mean (SEM). P values were calculated using two-tailed unpaired t-test or the appropriate statistical methods as mentioned in the figure legends.
Data and materials availability:
All the genome-wide datasets generated in this study, which include RNA-seq (in allograft tissues collected from control and GSK503-treated mice or in control and EZH2-knockdown MCF-7 cells) and ChIP-seq (H3K27me3, EZH2, total STAT2 and phosphorylated STAT2 in MCF-7 cells), have been deposited at the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) with an accession number GSE183459.
Results
EZH2 inhibition stimulates antitumor immunity.
To evaluate the impact of EZH2 on tumor microenvironment in luminal breast cancer, we set up an allograft model by injecting the ERα-positive (ERα+) murine breast cancer cells 67NR into the immunocompetent female mice orthotopically. When the tumors became palpable, we treated the mice with either vehicle or GSK503, a highly potent and specific small-molecule inhibitor of EZH2 methyltransferase activity (36). We found that EZH2 inhibitor treatment significantly retarded 67NR tumor growth (Fig. 1A) accompanied by a noticeable reduction of total H3K27me3 levels (Fig. 1B). EZH2-targeting drug was well tolerated since the body weights of animals in the control and treatment groups were comparable (Supplementary Fig. S1A). Since EZH2 inhibitors have been shown to directly block proliferation of cancer cells by inducing apoptosis or cell cycle arrest (37–39), we injected 67NR cells into highly immunodeficient NSG mice and found that the inhibitory effect of GSK503 was remarkably dampened (Supplementary Fig. S1B). We did not detect apoptosis in control and GSK503-treated allografts (Supplementary Fig. S1C). Results obtained in the NSG mice highlight the essentiality of intact immunity in the potency of EZH2 inhibitors and also implicate that the antitumor microenvironment we observed in the syngeneic mice upon GSK503 treatment is not likely to be triggered due to the apoptotic signals in breast cancer cells, which has been associated with robust infiltration and activation of cytolytic T cells in breast cancer (40). Furthermore, as the effect of EZH2 inhibitors may be attributed to the function of the methyltransferase expressed in cancer cells or in immune cells (41), we depleted Ezh2 in 67NR by either CRISPR-Cas9 technique or the specific short hairpin RNA (shRNA). No tumors were even formed upon complete knockout of Ezh2 (Fig. 1C). Residual Ezh2 in 67NR cells after partial knockdown was sufficient to maintain the growth of allografted tumors, albeit significantly slower compared to those in mice bearing the control cells (Supplementary Fig. S1D). Taken together, these results suggested that genetic and pharmacological inhibition of EZH2 in ERα+ breast cancer cells display better efficacy in abrogating tumor growth in immune intact system.
Figure 1. EZH2 inhibition abrogates tumor growth and triggers an antitumor immunity in syngeneic mouse model of ERα-positive (ERα+) breast cancer.
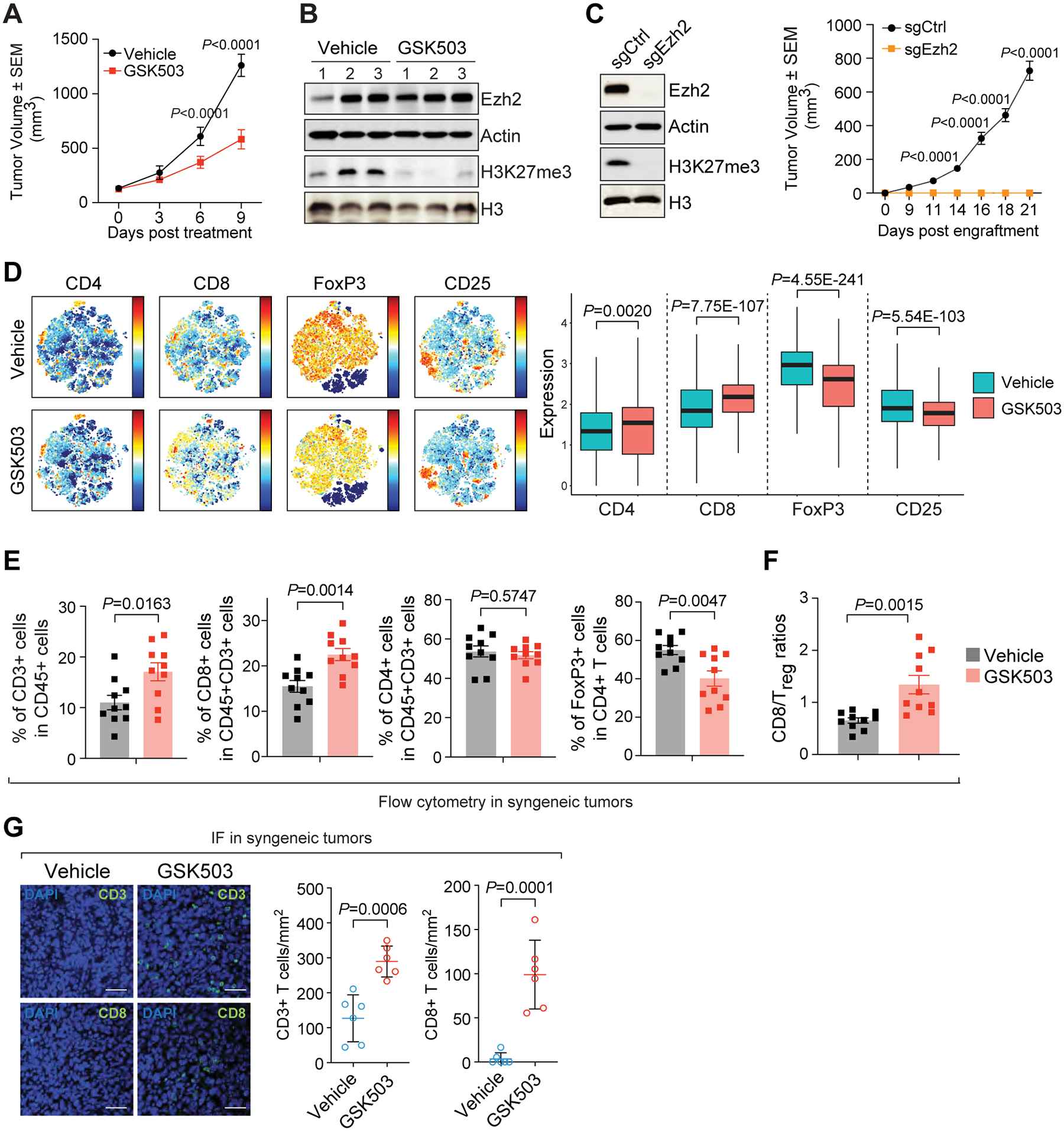
(A- B) Growth (A, n=12 per group) and immunoblotting analysis (B) of allograft tumors in 67NR-derived syngenetic mouse model upon the treatment with vehicle or 150 mg/kg GSK503. Numbers in (B), triplicates of independent allograft tumors. (C) Growth (n=14 per group) of allograft tumors established by the control (sgCtrl) or Ezh2-knockout (sgEzh2) 67NR cells. Left panel, Western blot in control and Ezh2-knockout 67NR cells with indicated antibodies. (D-F) CyTOF analysis (D) and flow cytometry analysis (E) of infiltrated lymphocytes with indicated markers and ratios of CD8+ T cells to Treg cells (F) in the allograft tumors treated with vehicle or 150 mg/kg GSK503. Left and right panel in (D), viSNE results and expression of the marker, respectively. (G) Representative images (left panel) and quantification (right panel) of fluorescent immunohistochemistry of CD3+ and CD8+ T cells in vehicle- and GSK503-treated allograft tumors. Six random microscopic views were examined in each biological replicate.
To explore any GSK503-induced changes in tumor microenvironment, we performed mass cytometry (CyTOF) analysis of the immune cells in the allografts collected from control and GSK503-treated mice. While fractions of most tumor-infiltrating leukocytes were unaffected by GSK503 treatment, there was a significant increase in CD4+ and CD8+ cells, but a significant decrease in CD25+ and FoxP3+ cells (Fig. 1D). These changes indicate that tumor immune surveillance was enhanced upon EZH2 inhibitor treatment, as evidenced by the infiltration of cytotoxic T lymphocytes (CTLs) recognized by CD8 marker and absence of regulatory T cells (Treg) characterized by FoxP3 expression. Flow cytometry analysis further confirmed that compared to the control tumors, GSK503-treated allografts were infiltrated by more CD3+CD8+ but fewer FoxP3+ T lymphocytes, while the portion of CD3+CD4+ T cells was moderately changed (Fig. 1E and Supplementary Fig. S1E). Quantification of the flow cytometry data corroborated these findings and further showed that the ratio of CD8+ to Treg tumor-infiltrating lymphocytes (CD8/Treg), which indicates the ability of immune system to fight cancer (42) and associates with favorable clinical outcomes in multiple tumor types (43), was significantly increased upon GSK503 treatment (Fig. 1F). In allografts generated from Ezh2-knockdown 67NR cells, similar patterns of tumor-infiltrating lymphocytes were obtained (Supplementary Fig. S1F) and again the CD8/Treg ratios were significantly elevated (Supplementary Fig. S1G). Consistently, fluorescent immunohistochemical staining of the surface markers CD3 and CD8 clearly showed augmented penetration of CD3+ and CD8+ cells into the tumor microenvironment following EZH2 inhibitor treatment (Fig. 1G). All these data suggest that EZH2 inhibition induces a shift of the tumor microenvironment in luminal breast cancer in favor of an antitumor immunity.
PRC2 inhibition induces interferon response in luminal breast cancer cells.
To investigate the mechanism by which EZH2 in ERα+ breast cancer cells promotes the immunosuppressive microenvironment, we conducted gene expression profiling in vehicle- and GSK503-treated allograft tumors. Much more genes were significantly upregulated by GSK503, in agreement with the transcriptional repressive activity of PRC2-catalyzed H3K27me3 (Fig. 2A). Meanwhile, we performed a genome-wide transcriptome analysis in ERα+ human breast cancer cells MCF-7, where EZH2 was silenced by its specific shRNA. Genes that were differentially expressed upon GSK503 treatment in 67NR-derived syngeneic mouse model were also changed in EZH2-knockdown MCF-7 cells, not only to a large extent but also in the same direction (Fig. 2B). In particular, genes that are upregulated in allograft tumors from GSK503-treated mice were significantly overlapped with genes that are elevated in human breast cancer MCF-7 cells upon EZH2 knockdown (Supplementary Fig. S2A). Thus, we identified a group of genes that may represent the authentic targets of the PRC2 complex in luminal breast cancer cells. They are mainly transcriptionally silenced by EZH2, they are not species biased and their expression is dependent on the methyltransferase activity of EZH2. Interestingly, gene set enrichment analysis (GSEA) showed that multiple biological processes known to regulate immune responses were particularly enriched especially for EZH2-repressed genes, and the most prominent one is the interferon (IFN) signaling (Fig. 2C). Indeed, in two luminal breast cancer cell lines MCF-7 (Supplementary Fig. S2B) and T-47D (Supplementary Fig. S2C), knockdown of EZH2 by two independent shRNAs significantly elevated multiple IFN signaling genes like interferon regulatory factors (IRFs) and STAT transcription factors, and therefore the downstream interferon-stimulated genes (ISGs) such as IFIT1 and SP100 were remarkably upregulated. We also treated MCF-7 (Fig. 2D) and T-47D (Supplementary Fig. S2D) cells with two different EZH2 inhibitors, GSK126 and EPZ-6438, both of which competitively block the association of the methyl donor S-adenosylmethionine (SAM) with the binding pocket on the EZH2 catalytic domain (44,45). Both compounds completely abolished total H3K27me3 signals without affecting EZH2 protein level and significantly upregulated the expression of IFN-activated transcription factors as well as downstream ISGs. These results confirmed that EZH2-mediated inactivation of the interferon signaling is dependent on its enzymatic activity. To further validate the role of PRC2’s activity in transcriptional regulation of these genes, we treated MCF-7 cells with EED226, a selective allosteric inhibitor of the PRC2 complex by impairing the interaction between H3K27me3 and EED protein (46). Again, we found that all the tested IFN-responsive transcription factors and ISGs were significantly increased (Fig. 2E). Taken together, our results suggest that the PRC2 complex ensures the inert activity of interferon signaling in ERα+ breast cancer cells, which is likely to be mediated by the histone methyltransferase activity of EZH2.
Figure 2. Interferon signaling is suppressed by PRC2 complex.
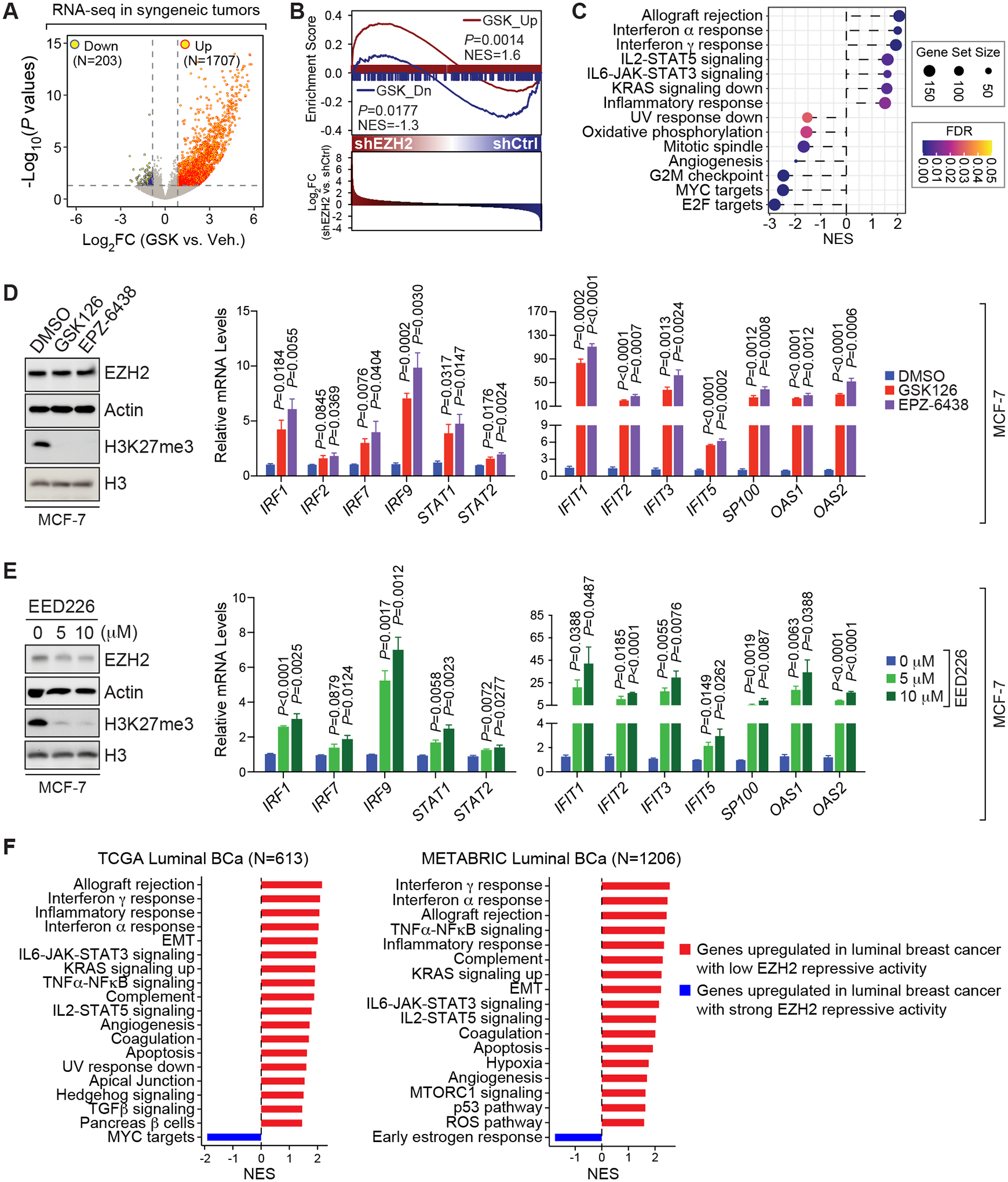
(A) Volcano plot showing genes that are significantly increased (Up) or decreased (Down) in GSK503 (GSK)-treated allograft tumors compared to vehicle (Veh.)-treated ones, which were established from 67NR cells. Horizontal and vertical dashed lines, log2-transformed fold change (Log2FC) at ±log2(1.3) and log10-transformed P values [−Log10 (P values)] at −log10(0.05), respectively; N, numbers of significantly changed genes. (B) Gene Set Enrichment Analysis (GSEA) of GSK503-upregulated (GSK_Up, red line) or -downregulated (GSK_Dn, blue line) genes from the syngeneic mouse model of breast cancer in the gene expression profiling performed in control (shCtrl) and EZH2-knockdown (shEZH2) MCF-7 cells. NES, normalized enrichment score; P values were determined by two-way ANOVA corrected for multiple comparisons. (C) Functional annotations of GSK503-upregulated (NES>0) or -downregulated (NES<0) genes identified in (A). (D-E) Immunoblotting analysis (left panel) and expression of selected interferon-stimulated genes (ISGs) (right panel) in MCF-7 cells upon treatment with 5 μM EZH2 inhibitor (GSK126 and EPZ6438) (D) or upon treatment with 0, 5 or 10 μM EED226 (E) for 7 days. (F) Top cancer hallmarks enriched in genes upregulated in patients with luminal breast cancer (Luminal BCa) displaying the lowest (red bars) or the highest (blue bars) EZH2 repressive activity. Patient information was retrieved from TCGA (19) (left panel) and METABRIC (20) (right panel) dataset. NES, normalized enrichment score; N, numbers of patients with luminal breast cancer.
To evaluate the clinical relevance of our finding, we defined a 53-gene signature that may reflect the activity of EZH2 in epigenetic gene silencing (thereafter referred to as “the repressive activity of EZH2”). These genes were selected from the top 200 genes that were commonly upregulated by EZH2 inhibitor and EZH2 knockdown in our RNA-seq data, and they also contain H3K27me3 at their promoters. We performed gene set variation analysis (GSVA) of these 53 signature genes in TCGA (19) and METABRIC (20) datasets, and found that the repressive activity of EZH2 represented by the GSVA scores was associated with neither the expression (Supplementary Fig. S3A) nor the copy number variations (Supplementary Fig. S3B) of EZH2 gene in patients with luminal breast cancer. These results suggest that the repressive activity of EZH2 is not merely an indicator of EZH2 levels, which is reasonable considering the reported alternative mechanisms underlying the oncogenic effect of EZH2 in addition to its role as a histone methyltransferase (47–49). By ranking from the lowest to highest GSVA scores, patients with luminal breast cancer were distributed into quartiles with patients in Quartile 1 having the strongest while patients in Quartile 4 having the weakest repressive activity of EZH2. In contrast to genes overly abundant in patients in Quartile 1, genes highly expressed in patients in Quartile 4 were functionally enriched in IFN signaling (Fig. 2F), implying an association between low gene silencing activity of EZH2 and active status of IFN signaling pathways in clinical scenarios. When we performed similar analyses in HER2-positive and triple-negative breast cancer, IFN signaling is also enriched in genes upregulated in patients in Quartile 4, albeit not consistently in different clinical datasets, which may be possibly due to the relatively small sample sizes for these two subtypes of breast cancer (Supplementary Fig. S3C–S3D). Taken together, our data demonstrated that EZH2 silences multiple immune-related genes in luminal breast cancer, especially those involved in the IFN signaling, which is reliant on its methyltransferase activity toward H3K27.
EZH2 inhibition stimulates immune surveillance.
It is well accepted that IFN signaling plays an immune-modulatory role in various types of cancer (50). Several IFN-inducible gene products are known to attract CD8+ effector T cells and stimulate cancer immune surveillance (51,52). We found that genes involved in antigen processing and presentation (APP), such as those encoding the major histocompatibility complex (MHC) class I molecules (B2M, HLA-A, HLA-B, and HLA-C) and the peptide transporters (TAP1 and TAP2), were all significantly elevated upon EZH2 knockdown (Fig. 3A) or EZH2 inhibitor treatment (Fig. 3B) in MCF-7 cells. Chemokines that are important for the activation of antitumor immune response, for example, CXCL9, CXCL10 and CXCL11, were also significantly upregulated. Upon addition of EED226 in MCF-7 cells (Supplementary Fig. S4A), expression of all the tested APP or chemokine genes was increased, further supporting that transcriptional regulation of IFN-responsive genes is dependent on the enzymatic activity of the PRC2 complex. To further indicate the impact of EZH2-mediated regulation of IFN signaling on immune response, we detected the levels of β2M and MHC class I proteins on the surface of MCF-7 cells and found them significantly augmented when EZH2 was silenced (Fig. 3C) or pharmacologically inhibited (Fig. 3D). Similar expression patterns of β2M and MHC class I proteins were observed on the surface of T-47D cells upon genetic and pharmacological inhibition of EZH2 (Supplementary Fig. S4B). In 67NR-established allograft tumors, we obtained the same results: GSK503 treatment (Fig. 3E) and Ezh2 knockdown (Supplementary Fig. S4C) both elevated the expression of APP genes (H2d1, H2k1, and Tap1), IFN-activated TFs (Stat1 and Stat2), ISGs (Ifit1, Oas1b, and Oas2) and chemokine genes (Cxcl9 and Cxcl11). On the surface of mouse breast cancer 67NR cells, flow cytometry analysis demonstrated higher amounts of murine β2m and MHC class I antigen (H-2kd) upon GSK503 treatment (Fig. 3F). Altogether, we concluded that EZH2 epigenetically silences IFN-responsive genes that help evoke an antitumor immune response, and therefore the biological effect of IFN signaling on immune surveillance is restrained by the functional PRC2 complex.
Figure 3. EZH2 inhibition enhances the capability of interferon signaling to induce immune surveillance in ERα+ breast cancer cells.
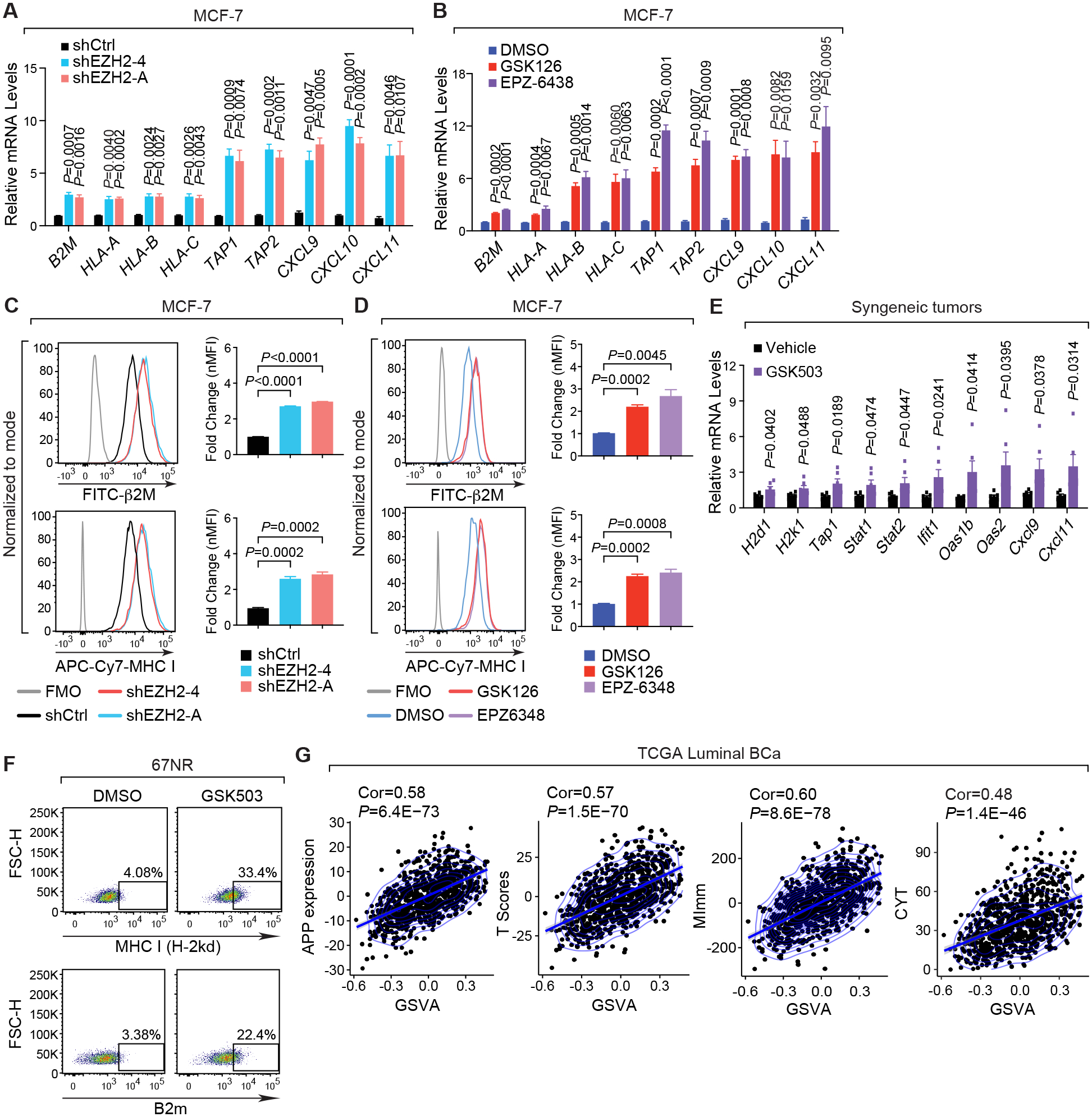
(A-B) Expression of exemplary genes in MCF-7 cells where endogenous EZH2 was silenced by two specific shRNAs (shEZH2–4 and -A) (A) or inhibited by 5 μM inhibitors (GSK126 and EPZ-6438) for 8 days (B). Control shRNA (shCtrl) and DMSO serve as controls in (A) and (B), respectively. (C-D) Representative histograms (left panels) and normalized mean fluorescence intensity (nMFI) (right panels) of flow cytometry results showing the levels of MHC class-I (MHC I) and β2M proteins on the surface of MCF-7 cells upon knockdown of EZH2 (C) or treatment with EZH2 inhibitors (D) as described in (A-B). (E) Expression of selected genes in 67NR-derived allograft tumors collected from vehicle- and GSK503-treated mice. (F) Flow cytometry analysis of MHC class-I [MHC I (H-2kd)] and β2m proteins on the surface of 67NR cells treated with DMSO or 10 μM GSK503 for 8 days. (G) Correlation of EZH2 repressive activity represented by the Gene Set Variation Analysis (GSVA) scores of the 53 signature genes with the expression sum of genes involved in antigen processing and presentation (APP), T scores, MImm scores and CYT scores in patients with luminal breast cancer (Luminal BCa). Clinical information was retrieved from TCGA dataset (19).
To verify the negative association between EZH2 repressive activity and favorable immune responses in clinical cases, we correlated the GSVA scores of the 53 signature genes we pinpointed with four separate parameters that have been demonstrated to predict the activation of antineoplastic immune reaction in a variety of cancers (25,26,29). These immune-related parameters are the expression sum of APP genes, T score derived from the levels of a common subset of T cell-inflamed and IFNγ-responsive genes to predict clinical outcome for anti-PD-1/PD-L1 antibodies (32), CYT score that is calculated from the mRNA levels of two key genes mediating cytolysis, GZMA and PRF1, to quantify immune effector activity (34), and MImm score that is used to assess the magnitude of leukocyte infiltration based on the expression of the immune-cell markers (27). First of all, we confirmed that our signature genes are not overlapped with any groups of signature genes that are used to calculate these four immune-related parameters, indicating that the 53-gene signature we identified is an independent set of genes (Supplementary Fig. S4D). Based on the clinical data from women with ERα+ breast cancer in TCGA (19) (Fig. 3G) and METABRIC (20) (Supplementary Fig. S4E) datasets, we found that the GSVA scores, which inversely reflects the repressive activity of EZH2, is positively correlated with all the above-mentioned parameters. Low repressive activity of EZH2, indicated by the high GSVA scores of the 53-gene signature, is always associated with high levels of all these parameters predictive of immune response, meaning that interfered function of the PRC2 complex in gene silencing links with an active antitumor immunity in luminal breast cancer. All these results further highlight the clinical significance of the epigenetic repression of IFN signaling by EZH2.
Expression of type I IFN ligands was silenced by EZH2-catalyzed H3K27me3 in luminal breast cancer cells.
Next, we sought to elucidate how EZH2 transcriptionally represses IFN-responsive genes. First, we examined the chromatin-binding intensities of H3K27me3 at the promoter regions of all ISGs that are repressed by EZH2 and found that they are much lower than the H3K27me3 signals around genes harboring the repressive histone mark and rather comparable to the background signals (Fig. 4A), suggesting that transcriptional repression of ISGs is not mediated directly by the action of the PRC2 complex on chromatin. Second, since a recent study in prostate cancer cells reported that EZH2 inhibition led to enhanced generation of double-stranded RNA (dsRNA) (26), a key mediator of IFN induction (53), we measured the levels of dsRNA in ERα+ breast cancer cells upon EZH2 inhibitor treatment (Supplementary Fig. S5A). There was little change in dsRNA expression in treated MCF-7 or T-47D cells, excluding the possibility that activation of IFN signaling by EZH2 inhibition is indirectly induced due to derepression of dsRNA expression. Considering all these results and the profound effect of EZH2 on the expression of ISGs, we speculated that some upstream signaling molecules of IFN pathways are epigenetically silenced by PRC2.
Figure 4. Type I interferon ligands are directly silenced by H3K27me3 in ERα+ breast cancer cells.
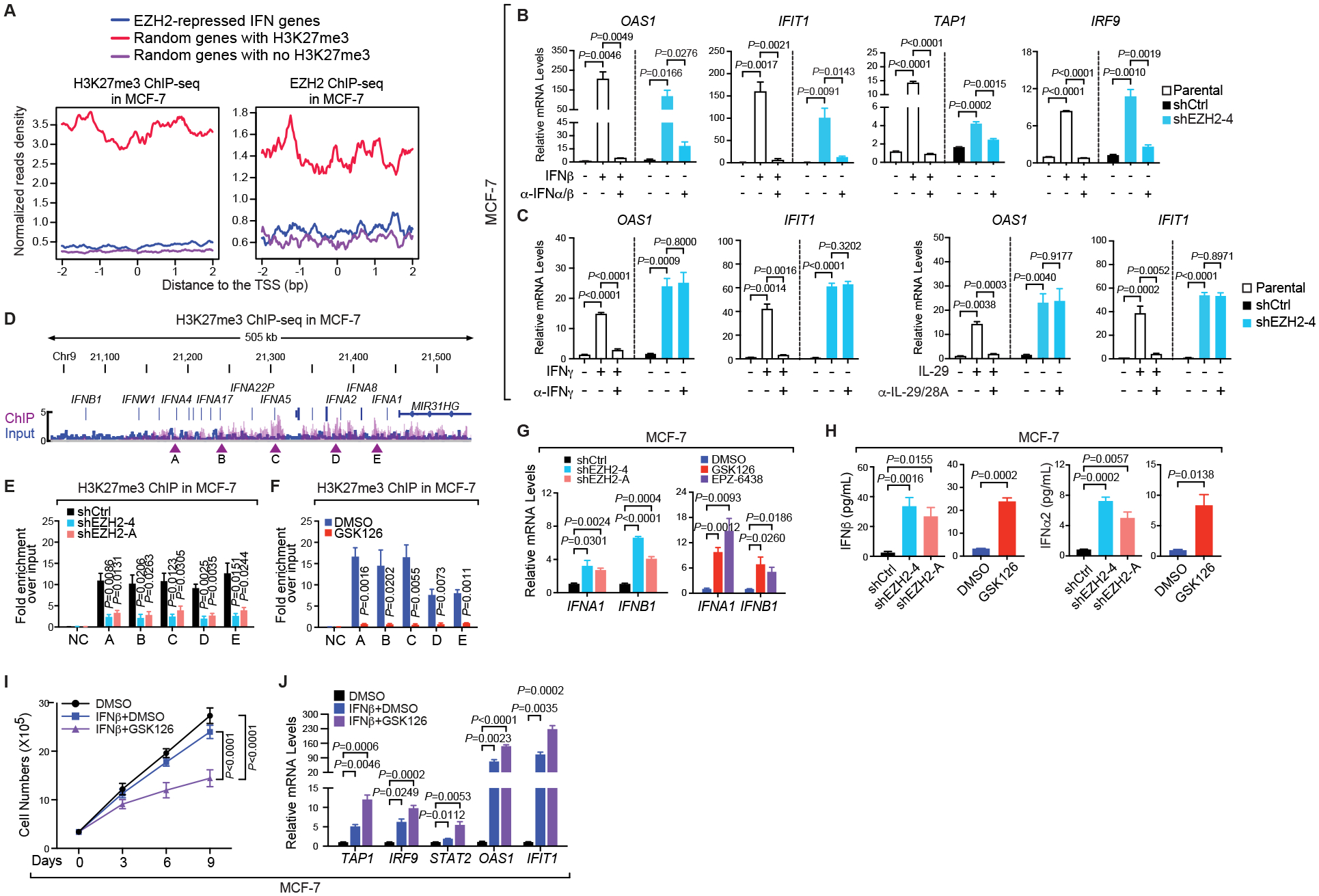
(A) Aggregation plots showing H3K27me3 (left panel) and EZH2 (right panel) ChIP-seq signals around the transcription start sites (TSS) of indicated groups of genes in MCF-7 cells. The same numbers of genes were randomly picked from genes bound by H3K27me3 (red line) or void of the histone modification (purple line), serving as positive and negative controls respectively. (B-C) Expression of specified genes in parental, control (shCtrl) or EZH2-knockdown (shEZH2–4) MCF-7 cells treated with (+) or without (–) the indicated IFN ligands for 7 days [250 pg/mL IFNβ in (B) and 250 pg/mL IFNγ or 50 ng/mL IL-29 in (C)] in the absence (–) and presence (+) of the corresponding neutralizing antibody for 24 hours [2.5 μg/mL α-IFNα/β in (B) and 2 μg/mL α-IFNγ or 4 μg/mL α-IL-29/28A in (C)]. (D) Snapshot of Integrative Genomics Viewer (IGV) showing the enrichment of H3K27me3 ChIP-seq signals at the type I IFN gene cluster in MCF-7 cells. Arrowheads, five chromatin sites selected for ChIP-qPCR confirmation. (E-F) ChIP of H3K27me3 in MCF-7 cells infected with control shRNA (shCtrl) or EZH2-specific shRNAs (shEZH2–4 and -A) (E) or treated with DMSO or 5 μM GSK126 for 7 days (F). NC, negative control using the promoter of KIAA0066. (G-H) Expression of IFNA1 and IFNB1 genes (G) and levels of IFNα2 and IFNβ proteins in the culture medium detected by ELISA (H) in MCF-7 cells where EZH2 was silenced by two independent shRNAs (shEZH2–4 and -A) or inhibited by 5 μM inhibitors [GSK126 and EPZ-6438 in (G) and GSK126 in (H)] for 7 days. (I-J) Growth (I) and expression of specified genes (J) in MCF-7 cells treated with DMSO or 200 pg/mL IFNβ, either alone or together with 3 μM GSK126, for up to 9 days.
Three types of IFN ligands have been identified, which control the transcription of common and unique subsets of immune-related genes (50). To find out exactly which class of IFN underlying the immunomodulatory role of EZH2 in luminal breast cancer, we added neutralizing antibodies against the dominant IFN ligands of each type, which are IFNα/β (type I), IFNγ (type II) and IL-29/28A (type III, also known as IFN-λ1/λ2), to MCF-7 cells where the endogenous EZH2 was knocked down. As a control, we treated the parental MCF-7 cells with the IFN ligand in the presence and absence of the corresponding antibody. Addition of the IFN ligand activated the transcription of target genes, which was impaired by the specific neutralizing antibody, suggesting that the antibody works as intended. Consistent with above results, expression of all the paradigm ISGs was elevated in EZH2 knockdown cells even without exogenous IFNs, which was drastically diminished only in the presence of the neutralizing antibody against type I IFN ligand (Fig. 4B), but not those against type II or III ligands (Fig. 4C). We repeated this assay in T-47D cells and again confirmed that upregulation of ISGs upon EZH2 depletion was significantly mitigated when the type I IFN ligands were neutralized (Supplementary Fig. S5B). All these results imply that the immunosuppressive effect of EZH2 could be mediated through inactivation of IFN signaling triggered by type I IFNs.
The next question is which signaling molecules that uniquely initiate type I IFN pathway are silenced by H3K27me3. Interestingly, we found dense H3K27me3 peaks around the cluster of type I IFN ligand genes in MCF-7 cells (Fig. 4D). Intensities of H3K27me3 at five selected chromatin sites across the entire gene cluster were significantly reduced upon knockdown of EZH2 (Fig. 4E) or GSK126 treatment (Fig. 4F). Similar results were observed when T-47D cells were treated with GSK126 (Supplementary Fig. S5C). These data suggest that type I IFN ligands are direct targets of the repressive histone mark H3K27me3. Concordantly, expression of the tumor cell-intrinsic genes encoding type I IFN ligands, such as IFNA1 and IFNB1, was remarkably increased upon genetic or pharmacological inhibition of EZH2 in MCF-7 (Fig. 4G) and T-47D (Supplementary Fig. S5D). Furthermore, enzyme-linked immunosorbent assay (ELISA) showed that secretion of type I IFN proteins were stimulated in both EZH2-knockdown and GSK126-treated MCF-7 (Fig. 4H) or T-47D (Supplementary Fig. S5E) cells. All these data suggest that EZH2-catalyzed H3K27me3 downregulates tumor-intrinsic and secreted levels of type I IFN ligands in ERα+ breast cancer cells, blocking the activation of IFN signaling in an autocrine fashion.
Given the fact that IFNs show important immune-modulatory and antineoplastic properties (54), we asked whether EZH2 inhibition enhances the potency of type I IFN signaling. Indeed, amalgamation with GSK126 enhanced the potency of IFNβ to retard growth of MCF-7 (Fig. 4I) and T-47D (Supplementary Fig. S6A) at a concentration of the cytokine that was not effective when alone. IFNβ-induced expression of ISGs was also boosted following GSK126 cotreatment in MCF-7 (Fig. 4J) and T-47D (Supplementary Fig. S6B) cells. Moreover, combination of both EZH2 inhibitor and IFNβ potentiated the immunostimulatory effect of type I IFN signaling, as when GSK126 was present, expression of MHC class I molecules inside (Supplementary Fig. S6C) or on the surface (Supplementary Fig. S6D) of MCF-7 cells became detectable even at low concentrations of IFNβ, which normally are insufficient to elicit any biological effects. Collectively, these results suggest that EZH2-mediated transcriptional silencing of type I IFN ligand genes has a far-reaching impact on the biological function of IFN signaling.
STAT2 mediates IFN signaling activated upon EZH2 inhibition.
The classical transcription factors that mediate the cellular responses to the IFN ligands are STAT1 and STAT2. Following association of cytokines with their distinct receptors, both STAT proteins are phosphorylated and activated, translocate to the nucleus, bind to the specific DNA motif called interferon-stimulated response elements (ISREs) and activate transcription of target genes (55). We found that knockdown of EZH2 or treatment with EZH2 inhibitors increased overall levels and phosphorylation of STAT1 and STAT2 in MCF-7 (Fig. 5A) and T-47D (Supplementary Fig. S7A), suggesting that this signaling cascade is indeed activated when EZH2 is inhibited. Neutralizing antibody against IFNβ ligand mitigated GSK126-induced upregulation of the STAT transcription factors (Fig. 5B), confirming that the effect of EZH2 on the signal transducers is mediated through the action of type I IFN like IFNβ. To highlight the contribution of each STAT protein to the biological effects of EZH2 inhibitors, we knocked down STAT1 (Fig. 5C) or STAT2 (Fig. 5D) in MCF-7 cells treated with GSK126. Both transcription factors were efficiently silenced by their specific shRNAs. It is worthy of note that STAT1 was also concomitantly diminished when STAT2 was depleted, although not vice versa. Surprisingly, knockdown of STAT1, a predominant effector protein of the IFN signaling, hardly impacted the effects of GSK126 on cell proliferation (Fig. 5E) or expression of ISGs (Fig. 5F), even though it was still required for IFNγ-stimulated upregulation of these genes (Supplementary Fig. S7B). In contrast, upon loss of STAT2 in MCF-7 cells, inhibitory effect of GSK126 on cell growth was significantly alleviated (Fig. 5G) and selected ISGs became less responsive to the compound (Fig. 5H). Requirement of STAT2 in GSK126-induced upregulation of ISGs was also seen in T-47D cells (Supplementary Fig. S7C). All these results suggest that STAT2, rather than STAT1, plays a critical role in mediating type I IFN signaling that is activated upon EZH2 inhibition.
Figure 5. STAT2, but not STAT1, mediates the type I interferon signaling that is activated upon EZH2 inhibition.
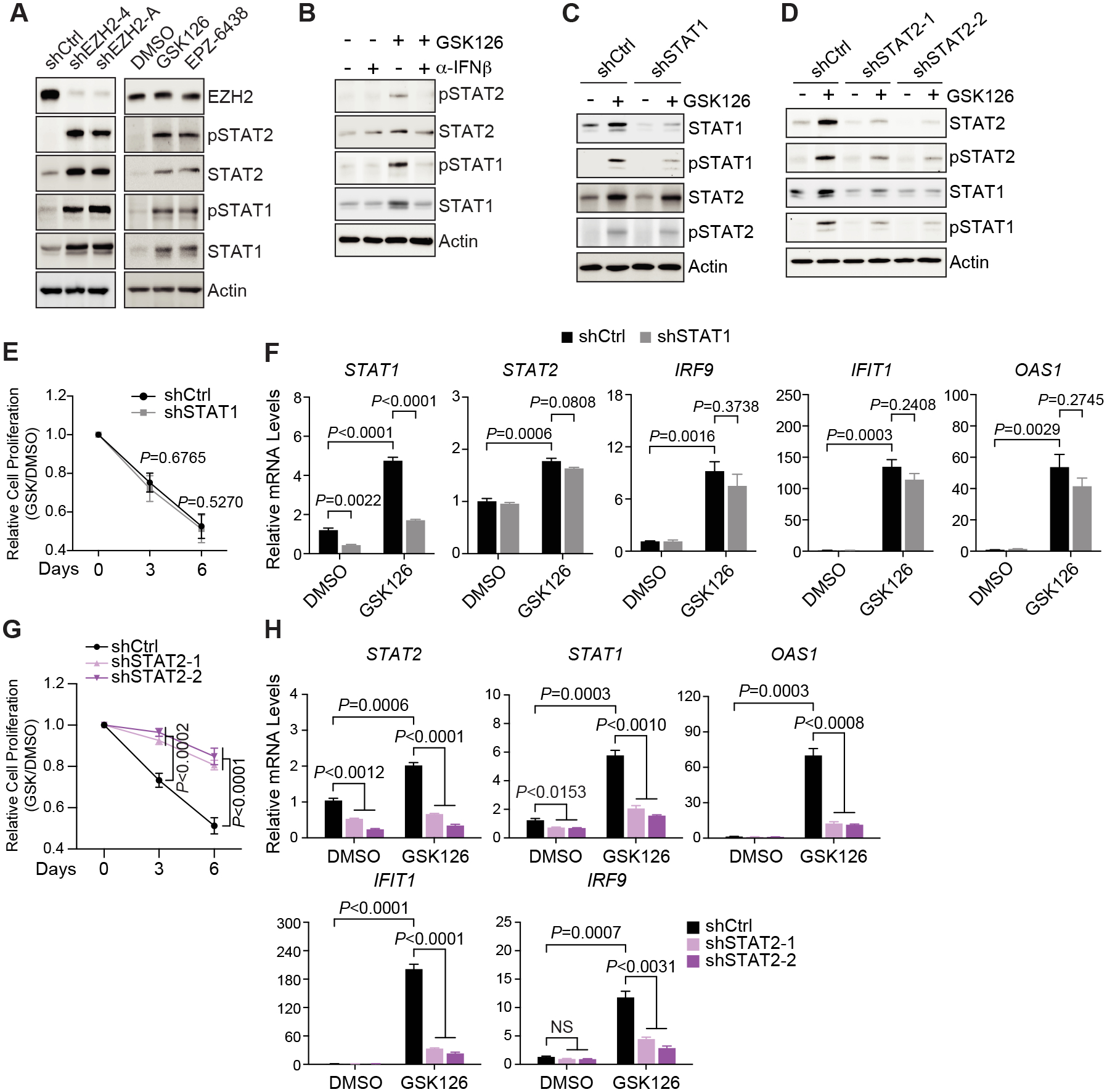
(A-B) Western blot with specified antibodies in MCF-7 cells stably expressing two EZH2-specific shRNAs (shEZH2–4 and -A) or treated with 5 μM EZH2 inhibitors (GSK126 and EPZ-6438) for 7 days (A) or in the presence of 5 μM GSK126, either alone (–) or with (+) 2 μg/mL IFNβ-specific neutralizing antibody (α-IFNβ), for 7 days (B) (C-H) Immunoblotting analysis (C-D), cell proliferation (E and G) and expression of exemplary genes (F and H) in MCF-7 cells upon knockdown of STAT1 (shSTAT1, C and E-F) or STAT2 (shSTAT2–1 and −2, D and G-H) in the presence of 5 μM GSK126 for indicated period of time (E and G) or up to 7 days (F and H).
Based on these results, we mapped genome-wide chromatin-binding sites of total STAT2 in GSK126-treated MCF-7 cells. To prove the authenticity of potentially identified peaks, we also conducted ChIP-seq of phosphorylated STAT2 upon EZH2 knockdown. Strikingly, chromatin recruitment of total STAT2 or the active form of STAT2 was remarkably enhanced upon EZH2 inhibition even without addition of the exogenous IFNs (Fig. 6A), and their binding regions were mostly overlapped (Fig. 6B). Motif analysis revealed ISRE as one of the top enriched DNA sequence patterns in both ChIP-seq datasets (Supplementary Fig. S8A), suggesting that when EZH2 is inhibited, STAT2 is recruited to the canonical cis-regulatory elements that it would bind to upon activation by the IFN ligands. More importantly, genes that are bound by total STAT2 or phosphorylated STAT2 upon EZH2 inhibition are functionally enriched in type I IFN signaling in the GSEA analysis (Fig. 6C) and they were mostly upregulated when EZH2 was knocked down in MCF-7 cells (Fig. 6D). Furthermore, chromatin association of STAT2 upon EZH2 knockdown in MCF-7 cells was almost completely diminished by addition of the neutralizing antibody against IFNβ to the cells (Fig. 6E). Since we have demonstrated the presence of an autocrine type I IFN signaling loop upon EZH2 inhibition in ERα+ breast cancer cells, all these data strongly support the conclusion that enhanced secretion of type I IFN ligands induced by EZH2 inhibition leads to activation of transcription factor STAT2, which subsequently binds to specific chromatin loci and transactivates downstream target genes.
Figure 6. STAT2 is recruited to classical interferon-sensitive response elements around the target genes of type I interferon signaling upon EZH2 inhibition in ERα+ breast cancer cells.
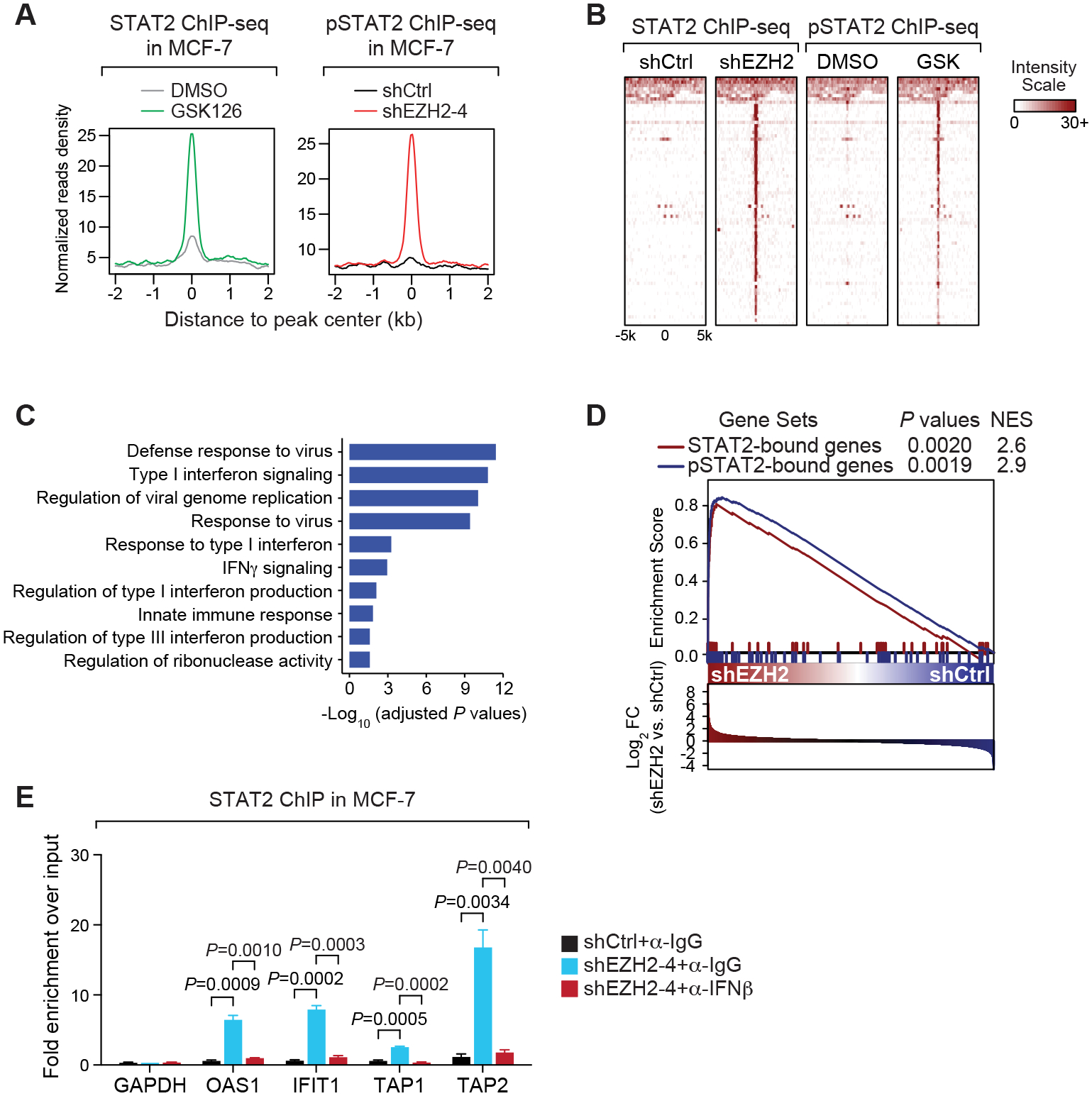
(A-B) Aggregation plots (A) and heat maps (B) showing ChIP-seq signals of total STAT2 in MCF-7 cells treated with 5 μM GSK126 (GSK) for 7 days or ChIP-seq signals of Tyr690-phosphorylated STAT2 (pSTAT2) in EZH2-knockdown (shEZH2–4) MCF-7 cells. (C) Gene Ontology (GO) analysis of genes containing at least one STAT2 peak upon GSK126 treatment or at least one pSTAT2 peak upon EZH2 knockdown within 1 kb from the transcription start sites. (D) GSEA analysis of genes defined in (C) in gene expression profiling carried out in MCF-7 cells infected with control shRNA (shCtrl) or EZH2-specific shRNA (shEZH2). (E) STAT2-targeted ChIP in control (shCtrl) and EZH2-knockdown (shEZH2–4) MCF-7 cells in the presence of 2.0 μg/mL IgG isotype control (α-IgG) or 2.0 μg/mL IFNβ-specific neutralizing antibody (α-IFNβ) for 3 days. Promoter of GAPDH servers as the negative control.
Indeed, we observed prominent peaks of total STAT2 or phosphorylated STAT2 at the promoters of EZH2-repressed genes downstream of type I IFN signaling, which only happened in EZH2-knockdown or EZH2 inhibitor-treated MCF-7 cells (Supplementary Fig. S8B). This was further confirmed by targeted ChIP of STAT2 in MCF-7 treated with EZH2 inhibitors (Supplementary Fig. S8C) and in T-47D cells where EZH2 was genetically or pharmacologically inhibited (Supplementary Fig. S8D). In addition, we detected much enhanced DNA-binding signals of Stat2 around the exemplary ISG genes in GSK503-treated murine breast cancer 67NR cells (Supplementary Fig. S8E). Interestingly, although in the canonical model, STAT2 is reported to form a heterodimer with STAT1 for its optimal transcriptional activity in response to type I IFN ligands (56), GSK126-induced chromatin recruitment of STAT2 seems to be independent of STAT1, as STAT2 binds to the targeted chromatin loci with an equivalent, or even stronger, affinity in STAT1-knockdown cells compared to the control cells (Supplementary Fig. S8F).
STAT2 mediates EZH2 inhibition-driven antitumor immunity.
With the essentiality of STAT2 in the transmission of type I IFN signals upon EZH2 inhibition, we wondered whether STAT2 determines the potency of EZH2 inhibitor as an antitumor immunotherapy in luminal breast cancer. To this end, we knocked out Stat2 in 67NR cells using CRISPR-Cas9 technology. Both basal and GSK503-elevated levels of Stat2 protein were remarkably diminished in the knockout cells (Fig. 7A), and therefore GSK503-induced upregulation of the selected ISGs was severely dampened (Fig. 7B). Consistently, EZH2 inhibitor-induced accumulation of MHC class I proteins on the cell surface was severely blocked in Stat2-knockout cells compared to the control counterparts (Fig. 7C). All these results confirmed STAT2 contributes to the immunomodulatory potential of EZH2 inhibitor.
Figure 7. STAT2 is essential for the inhibitory effects of EZH2 inhibitors on tumor growth and immune escape in luminal breast cancer.
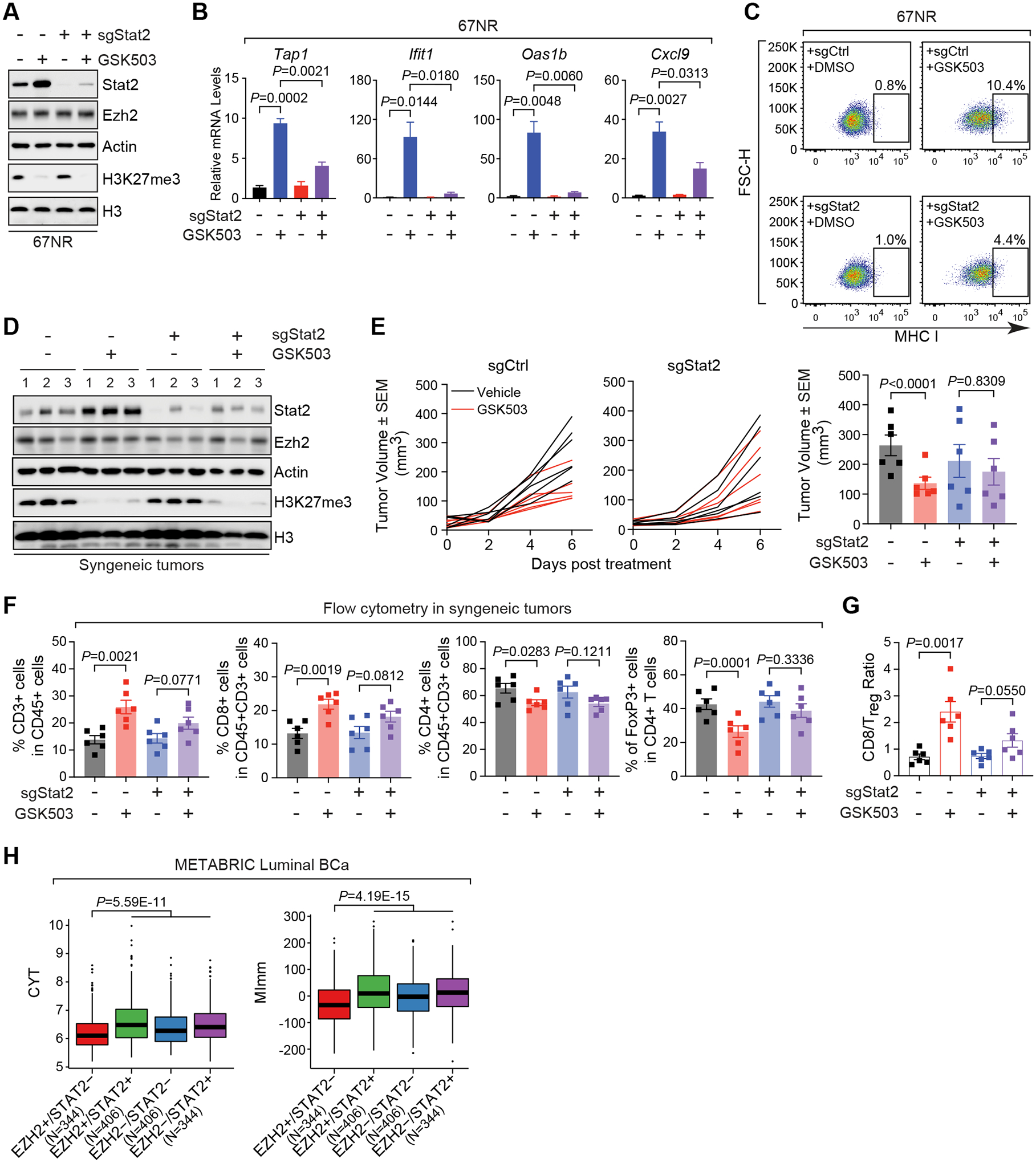
(A-C) Western blot (A), expression of specified genes (B) and flow cytometry analysis of cell surface levels of MHC class-I (MHC I) proteins (C) in 67NR cells stably expressing control sgRNA (sgCtrl) or Stat2-targeting sgRNA (sgStat2) in the presence (+) and absence (–) of 10 μM GSK503 for 8 days. (D-E) Immunoblotting analysis (D) and growth (E) of allograft tumors established by inoculating control (sgCtrl) or Stat2-knockout (sgStat2) 67NR cells into immunocompetent BALB/c mice receiving vehicle or 150 mg/kg GSK503. Numbers in (D), triplicates of independent allograft tumors. Right panel in (E), Quantification of tumor sizes at the endpoint (F-G) Quantification of flow cytometry analysis of T cells with indicated markers (F) and the ratio of CD8+ T cells to Treg cells (G) in the final allograft tumors expressing Stat2-specific sgRNA (sgStat2) upon the treatment with 150 mg/kg GSK503. (H) Association of high (+) or low (–) expression of EZH2 and STAT2 genes, when considered concurrently, with CYT and MImm scores in patients with luminal breast cancer (Luminal BCa). Clinical information was retrieved from METABRIC (20) dataset. N, numbers of patients with luminal breast cancer in each specified group of EZH2 and STAT2 expression.
Afterwards, we moved to the syngeneic mouse model by inoculating Stat2-knockout 67NR cells into immunocompetent mice and administering GSK503 when the grafted tumors became palpable. Although the enzymatic activity of EZH2 was equally impaired by GSK503 in the control and knockout cells, as demonstrated by the substantial loss of H3K27me3 (Fig. 7D), the capability of this compound in retarding the tumor growth was significantly compromised upon Stat2 knockout (Fig. 7E), supporting that STAT2 is essential for the efficacy of EZH2 inhibitors in luminal breast cancer. Most importantly, flow cytometry analysis of the allograft tumors clearly showed that depletion of STAT2 nullified the effect of EZH2 inhibitor on the distribution pattern of tumor-infiltrating lymphocytes (Fig. 7F), leading to a lower CD8/Treg ratio compared to the control mice receiving GSK503 treatment (Fig. 7G). These results further confirm indispensability of STAT2 in the generation of an antitumor immunity induced by EZH2 inhibitors in vivo.
To indicate the clinical relevance of our findings, we retrieved the clinical information from METABRIC (20) dataset and showed that high levels of EZH2 but low levels of STAT2 are associated with the lowest CYT and MImm scores in patients with luminal breast cancer (Fig. 7H), suggesting inhibition of cancer immune surveillance in clinical cases expressing high levels of EZH2 but at the same time low levels of STAT2. In summary, our results indicate that activation of type I IFN-STAT2 axis upon EZH2 inhibition plays a critical role in establishing an antitumor microenvironment in luminal breast cancer.
Discussion
As an important epigenetic enzyme, EZH2 has been shown to promote immune escape through epigenetic abnormalities that change the expression of immune-related genes in either cancer cells or surrounding immune cells (41). However, such an effect of EZH2 on the tumor microenvironment in breast cancer is underappreciated, especially whether current EZH2-targeting drugs can activate antitumor immunity in this disease has not been systematically explored. In this study, we suggested a new working model that explains how dysregulation of an EZH2-catalyzed epigenetic program reverses immunosuppression in luminal breast cancer (Fig. 8). We found that type I interferon (IFN) signaling is specifically activated upon inhibition of EZH2 methyltransferase activity in ERα+ breast cancer cells. Integrative analysis of EZH2-repressed genes and H3K27me3 global pattern revealed that type I IFNs, including IFNα/β, but not type II or III IFNs are direct targets of this repressive histone mark. We further demonstrated that STAT2 is the main transcription factor mediating the immunomodulatory effects of type I IFN signaling activated upon EZH2 inhibition. Thus, impairing the activity of the PRC2 complex by the pharmacological inhibitors of EZH2 leads to elevation of secreted IFN ligands and a constitutive formation of an autocrine type I IFN signaling loop, activates STAT2 and the downstream immune-related target genes, and retards the growth of ERα+ mammary tumors, through reprogramming the local microenvironment to kill the tumor cells. Taken together, our work has elucidated a new mechanism underlying the action of an EZH2-catalyzed epigenetic program in regulating the breast cancer tumor microenvironment and laid the foundation for clinical application of EZH2-targeting drugs to improve immune response in luminal breast cancer.
Figure 8. Model depicting the function of PRC2-catalyzed H3K27me3 in inactivating type I IFN-STAT2 axis in luminal breast cancer.
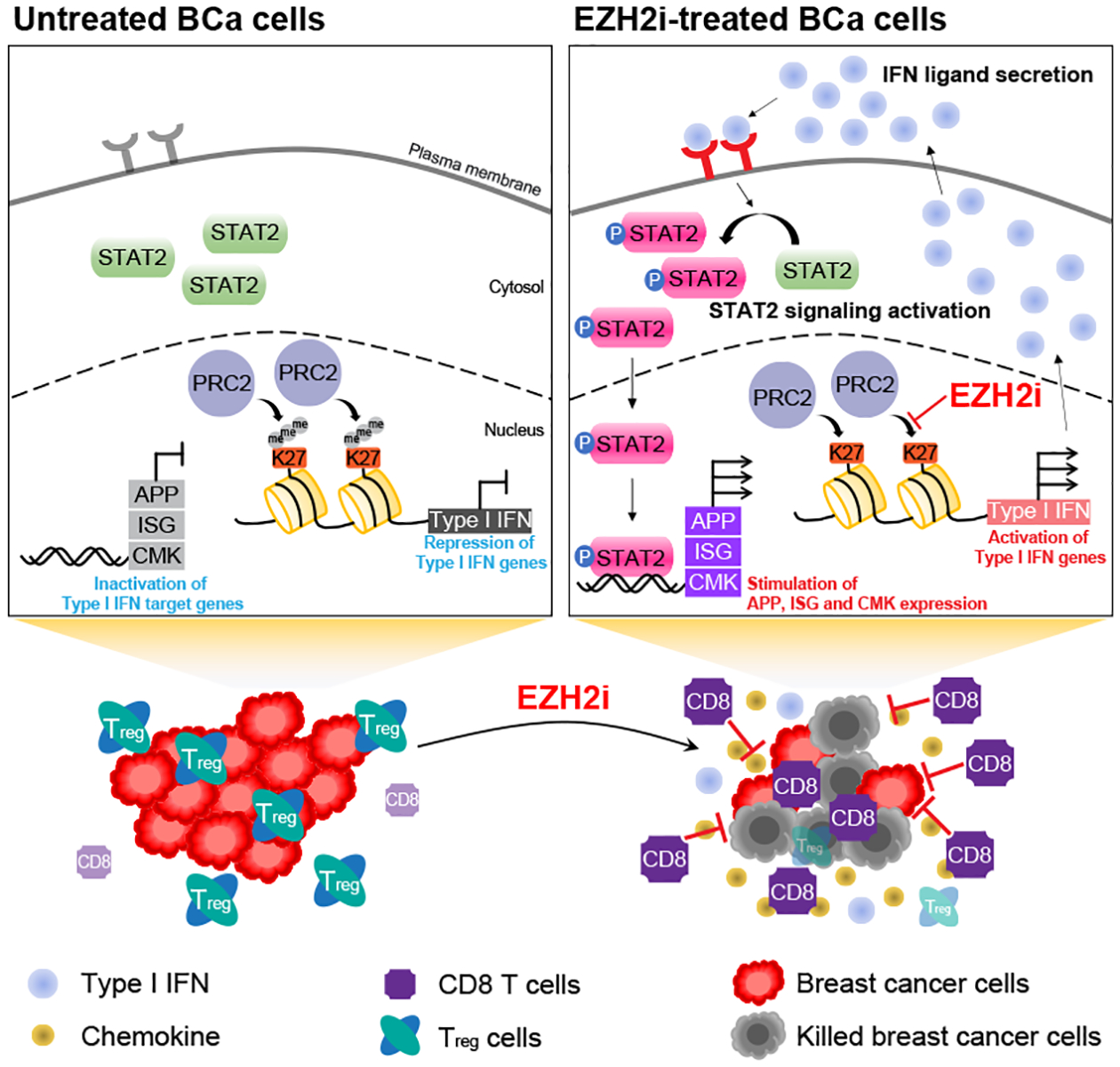
Pharmacological inhibition of PRC2 complex (EZH2i) releases the epigenetic silencing of genes encoding type I IFN ligands, induces an autocrine activation of the IFN signaling mediated by STAT2 and subsequently generates an antitumor microenvironment in ERα+ breast cancer (BCa) cells.
Epigenetic regulation of IFN signaling has long been suggested, and distinct signaling molecules may be transcriptionally activated or repressed by certain epigenetic modifications (57). In our study, we found that type I IFN cytokines are directly silenced by PRC2-catalyzed H3K27me3 in ERα+ breast cancer cells. Such regulation may be cancer type specific and context dependent. It has been reported that the activity of EZH2 is required for the signaling transduction from the type I IFN ligands in systemic lupus erythematosus (58). Therefore, EZH2 inhibitor hinders the activation of this particular type of IFN signaling, which is opposite to the effect of EZH2 we observed in breast cancer cells. On the other hand, EZH2 also suppresses total intracellular levels of double-stranded RNAs (dsRNAs) in some cancer types, such as sarcoma (59) and prostate cancer (26), and therefore EZH2 inhibitors induce an antiviral response by activating dsRNA-driven type I IFN signaling. However, we demonstrated that this dsRNA-involved mechanism does not account for the inhibitory action of EZH2 on type I IFN signaling in luminal breast cancer cells. Taken together, although regulation of type I IFN signaling by EZH2-mediated epigenetic program may be a common event, the underlying mechanisms and biological consequences appear to be context-dependent and require investigation on a case-by-case basis.
In classical type I IFN signaling, the transcription factor STAT1 plays a very important role in eliciting the cellular responses to the cytokines. Once activated by type I IFN ligands, STAT1 forms a homodimer or heterodimer with STAT2 and engages with chromatin to stimulate the transcription of target genes (55). Therefore, our finding that STAT1 is not required for EZH2 inhibition-activated type I IFN signaling seems atypical. However, STAT1-independent function of STAT2 has been occasionally implicated (60–62). In all these cases, STAT2 is recruited to chromatin containing ISRE sequences to transactivate ISGs in cancer cells that lack STAT1 (60). Since STAT2 binds DNA poorly by itself (63), the interferon regulatory factor 9 (IRF9), a DNA-binding subunit of all ISRE-associated transcription complexes (64), is shown to assist with chromatin recruitment of STAT2 and facilitate its action on transcriptional regulation in the absence of STAT1 (61). Therefore, it is conceivable that IRF9 is also involved in the type I IFN signaling transduction that is triggered upon EZH2 inhibition. In addition, we noticed that STAT1 expression was decreased after STAT2 knockdown, whereas STAT2 levels were unchanged by depletion of STAT1. The fact that STAT1 is positively regulated by STAT2 but not the other way around may also help explain why STAT2 is the decisive factor in mediating the IFN signaling in luminal breast cancer cells.
Our work has several clinical implications. First, although inhibition of EZH2 can directly abrogate tumor growth by inducing apoptosis or blocking cell cycle progression in cancer cells (37–39), our results of assessing the efficacy of EZH2 inhibitors in immunodeficient and immunocompetent mice suggest that the intact immune system may improve the potency of EZH2-targeting drugs. Upon the treatment with EZH2 inhibitor for a relatively short period of time, growth retardation of 67NR-derived allograft tumors was already pronounced in the syngeneic mice, but not obvious in immunocompromised NSG mice. This is consistent with the relatively mild effect of EZH2 inhibitors as a monotherapy in human tumor xenograft models (65,66), and further highlight the important roles of the tumor microenvironment in executing the inhibitory effect of EZH2 inhibitors. Second, EZH2 inhibition could sensitize luminal breast tumors, which are typically immunologically cold, to FDA-approved immunotherapy. In our syngeneic mouse model of breast cancer, pharmacological inhibitors of the PRC2 complex induce type I IFN production, enhance antigen presentation and facilitate lymphocytic infiltration, all of which have been demonstrated to improve the anticancer efficacy of anti-PD-1/PD-L1 therapy approved for TNBC (67–69). Therefore, immunotherapies such as the immune checkpoint inhibitors, which usually show limited effectiveness against ERα+ breast cancer (70), may become clinically beneficial when combined with approved EZH2-targeting drugs. Third, based on EZH2-repressed IFN-responsive genes and H3K27me3 landscape, we pinpointed a 53-gene signature that reflects the gene silencing activity of EZH2. Interestingly, expression of our signature genes is highly associated with the parameters that are currently used to assess the functions of tumor microenvironment, although they are not overlapped with any list of genes used to calculate these parameters. These analyses suggest that the 53-gene signature we identified may serve as an independent parameter to predict the active immune response in luminal breast cancer. Lastly, we also found enrichment of IFN signaling pathways in HER2-postive and triple-negative breast cancer with low repressive activity of EZH2, suggesting that the effects of EZH2 on tumor immunity we observed in luminal breast cancer may represent a common mechanism underlying action of EZH2 in breast cancer. Therefore, it is clinically relevant to confirm whether EZH2 inhibition represents a viable treatment strategy to boost the anticancer immunity in all subtypes of breast cancer.
In summary, our study reveals a new working model for the regulation of immunity in breast cancer by EZH2-catalyzed epigenetic program. It also provides the scientific foundation for the development of promising anticancer strategy that combines EZH2 inhibitors and immunotherapy.
Supplementary Material
Significance.
Inhibition of EZH2 activates a type I interferon-STAT2 signaling axis and provides a therapeutic strategy to stimulate antitumor immunity and therapy responsiveness in immunologically cold luminal breast cancer.
Acknowledgments
We sincerely thank Dr. Ann Richmond at Vanderbilt University for sharing their 67NR cell line with us. Some of the results presented in this study are based upon data generated by the TCGA Research Network (https://www.cancer.gov/tcga).
This work was supported by grants from CPRIT award (RR140072 to K.X.), Voelcker Fund Young Investigator award (to K.X.), V Foundation Translational Award (T2017-010 to K.X.), and Susan Komen Award (CCR19605875 to K.X.).
Footnotes
Conflict of interest statement: The authors declare no potential conflicts of interest.
References
- 1.Dai E, Zhu Z, Wahed S, Qu Z, Storkus WJ, Guo ZS. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol Cancer 2021;20:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gan L, Yang Y, Li Q, Feng Y, Liu T, Guo W. Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomark Res 2018;6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell 2004;15:57–67 [DOI] [PubMed] [Google Scholar]
- 4.Cao Q, Wang X, Zhao M, Yang R, Malik R, Qiao Y, et al. The central role of EED in the orchestration of polycomb group complexes. Nat Commun 2014;5:3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002;298:1039–43 [DOI] [PubMed] [Google Scholar]
- 6.Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015;527:249–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res 2016;76:275–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res 2005;65:1089–96 [PubMed] [Google Scholar]
- 9.Vasanthakumar A, Xu D, Lun AT, Kueh AJ, van Gisbergen KP, Iannarella N, et al. A non-canonical function of Ezh2 preserves immune homeostasis. EMBO Rep 2017;18:619–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang S, He Z, Wang X, Li H, Liu XS. Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction. Elife 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li F, Li C, Cai X, Xie Z, Zhou L, Cheng B, et al. The association between CD8+ tumor-infiltrating lymphocytes and the clinical outcome of cancer immunotherapy: A systematic review and meta-analysis. EClinicalMedicine 2021;41:101134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strickler JH, Hanks BA, Khasraw M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin Cancer Res 2021;27:1236–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denkert C The immunogenicity of breast cancer--molecular subtypes matter. Ann Oncol 2014;25:1453–5 [DOI] [PubMed] [Google Scholar]
- 14.Kang N, Eccleston M, Clermont PL, Latarani M, Male DK, Wang Y, et al. EZH2 inhibition: a promising strategy to prevent cancer immune editing. Epigenomics 2020;12:1457–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012;338:1465–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923–30 [DOI] [PubMed] [Google Scholar]
- 18.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas N Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012;486:346–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009;10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010;38:576–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reeves E, James E. Antigen processing and immune regulation in the response to tumours. Immunology 2017;150:16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morel KL, Sheahan AV, Burkhart DL, Baca SC, Boufaied N, Liu Y, et al. EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat Cancer 2021;2:444–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robinson DR, Wu YM, Lonigro RJ, Vats P, Cobain E, Everett J, et al. Integrative clinical genomics of metastatic cancer. Nature 2017;548:297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wakiyama H, Masuda T, Motomura Y, Hu Q, Tobo T, Eguchi H, et al. Cytolytic Activity (CYT) Score Is a Prognostic Biomarker Reflecting Host Immune Status in Hepatocellular Carcinoma (HCC). Anticancer Res 2018;38:6631–8 [DOI] [PubMed] [Google Scholar]
- 29.Hu Q, Nonaka K, Wakiyama H, Miyashita Y, Fujimoto Y, Jogo T, et al. Cytolytic activity score as a biomarker for antitumor immunity and clinical outcome in patients with gastric cancer. Cancer Med 2021;10:3129–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balli D, Rech AJ, Stanger BZ, Vonderheide RH. Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clin Cancer Res 2017;23:3129–38 [DOI] [PubMed] [Google Scholar]
- 31.Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol 2008;8:607–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160:48–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013;23:677–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang JP, Ling K. EZH2 and histone deacetylase inhibitors induce apoptosis in triple negative breast cancer cells by differentially increasing H3 Lys(27) acetylation in the BIM gene promoter and enhancers. Oncol Lett 2017;14:5735–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pawlyn C, Bright MD, Buros AF, Stein CK, Walters Z, Aronson LI, et al. Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J 2017;7:e549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li HS, Xu Y. Inhibition of EZH2 via the STAT3/HOTAIR signalling axis contributes to cell cycle arrest and apoptosis induced by polyphyllin I in human non-small cell lung cancer cells. Steroids 2020;164:108729. [DOI] [PubMed] [Google Scholar]
- 40.Murthy V, Oshi M, Tokumaru Y, Endo I, Takabe K. Increased apoptosis is associated with robust immune cell infiltration and cytolytic activity in breast cancer. Am J Cancer Res 2021;11:3674–87 [PMC free article] [PubMed] [Google Scholar]
- 41.Qiu J, Sharma S, Rollins RA, Paul TA. The complex role of EZH2 in the tumor microenvironment: opportunities and challenges for immunotherapy combinations. Future Med Chem 2020;12:1415–30 [DOI] [PubMed] [Google Scholar]
- 42.Kareva I. Metabolism and Gut Microbiota in Cancer Immunoediting, CD8/Treg Ratios, Immune Cell Homeostasis, and Cancer (Immuno)Therapy: Concise Review. Stem Cells 2019;37:1273–80 [DOI] [PubMed] [Google Scholar]
- 43.deLeeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res 2012;18:3022–9 [DOI] [PubMed] [Google Scholar]
- 44.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012;492:108–12 [DOI] [PubMed] [Google Scholar]
- 45.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 2013;110:7922–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qi W, Zhao K, Gu J, Huang Y, Wang Y, Zhang H, et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat Chem Biol 2017;13:381–8 [DOI] [PubMed] [Google Scholar]
- 47.Jiao L, Shubbar M, Yang X, Zhang Q, Chen S, Wu Q, et al. A partially disordered region connects gene repression and activation functions of EZH2. Proc Natl Acad Sci U S A 2020;117:16992–7002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013;23:839–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, et al. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol Cell 2011;43:798–810 [DOI] [PubMed] [Google Scholar]
- 50.Fu X, De Angelis C, Schiff R. Interferon Signaling in Estrogen Receptor-positive Breast Cancer: A Revitalized Topic. Endocrinology 2021 [DOI] [PubMed] [Google Scholar]
- 51.Padovan E, Spagnoli GC, Ferrantini M, Heberer M. IFN-alpha2a induces IP-10/CXCL10 and MIG/CXCL9 production in monocyte-derived dendritic cells and enhances their capacity to attract and stimulate CD8+ effector T cells. J Leukoc Biol 2002;71:669–76 [PubMed] [Google Scholar]
- 52.Gessani S, Conti L, Del Corno M, Belardelli F. Type I interferons as regulators of human antigen presenting cell functions. Toxins (Basel) 2014;6:1696–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gantier MP, Williams BR. The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev 2007;18:363–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saleiro D, Platanias LC. Interferon signaling in cancer. Non-canonical pathways and control of intracellular immune checkpoints. Semin Immunol 2019;43:101299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen K, Liu J, Cao X. Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J Autoimmun 2017;83:1–11 [DOI] [PubMed] [Google Scholar]
- 56.Li X, Leung S, Qureshi S, Darnell JE Jr., Stark GR. Formation of STAT1-STAT2 heterodimers and their role in the activation of IRF-1 gene transcription by interferon-alpha. J Biol Chem 1996;271:5790–4 [DOI] [PubMed] [Google Scholar]
- 57.Selinger E, Reinis M. Epigenetic View on Interferon gamma Signalling in Tumour Cells. Folia Biol (Praha) 2018;64:125–36 [DOI] [PubMed] [Google Scholar]
- 58.Wu L, Jiang X, Qi C, Zhang C, Qu B, Shen N. EZH2 Inhibition Interferes With the Activation of Type I Interferon Signaling Pathway and Ameliorates Lupus Nephritis in NZB/NZW F1 Mice. Front Immunol 2021;12:653989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jayabal P, Ma X, Shiio Y. EZH2 suppresses endogenous retroviruses and an interferon response in cancers. Genes Cancer 2021;12:96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walter KR, Balko JM, Hagan CR. Progesterone receptor promotes degradation of STAT2 to inhibit the interferon response in breast cancer. Oncoimmunology 2020;9:1758547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lou YJ, Pan XR, Jia PM, Li D, Xiao S, Zhang ZL, et al. IRF-9/STAT2 [corrected] functional interaction drives retinoic acid-induced gene G expression independently of STAT1. Cancer Res 2009;69:3673–80 [DOI] [PubMed] [Google Scholar]
- 62.Sarkis PT, Ying S, Xu R, Yu XF. STAT1-independent cell type-specific regulation of antiviral APOBEC3G by IFN-alpha. J Immunol 2006;177:4530–40 [DOI] [PubMed] [Google Scholar]
- 63.Bluyssen HA, Levy DE. Stat2 is a transcriptional activator that requires sequence-specific contacts provided by stat1 and p48 for stable interaction with DNA. J Biol Chem 1997;272:4600–5 [DOI] [PubMed] [Google Scholar]
- 64.Paul A, Tang TH, Ng SK. Interferon Regulatory Factor 9 Structure and Regulation. Front Immunol 2018;9:1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kong Y, Zhang Y, Mao F, Zhang Z, Li Z, Wang R, et al. Inhibition of EZH2 Enhances the Antitumor Efficacy of Metformin in Prostate Cancer. Mol Cancer Ther 2020;19:2490–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ratz L, Brambillasca C, Bartke L, Huetzen MA, Goergens J, Leidecker O, et al. Combined inhibition of EZH2 and ATM is synthetic lethal in BRCA1-deficient breast cancer. Breast Cancer Res 2022;24:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016;167:397–404 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garcia-Lora A, Algarra I, Garrido F. MHC class I antigens, immune surveillance, and tumor immune escape. J Cell Physiol 2003;195:346–55 [DOI] [PubMed] [Google Scholar]
- 69.Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol 2016;17:e542–e51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goldberg J, Pastorello RG, Vallius T, Davis J, Cui YX, Agudo J, et al. The Immunology of Hormone Receptor Positive Breast Cancer. Front Immunol 2021;12:674192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.