

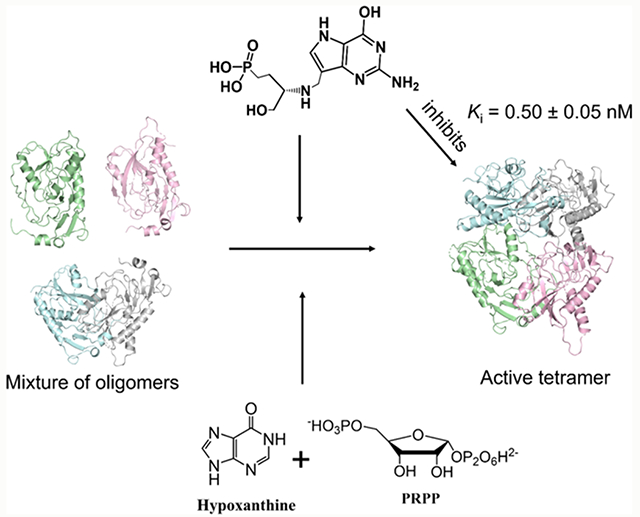
Abstract
Plasmodium falciparum hypoxanthine–guanine–xanthine phosphoribosyltransferase (PfHGXPRT) is essential for purine salvage of hypoxanthine into parasite purine nucleotides. Transition state analogue inhibitors of PfHGXPRT are characterized by kinetic analysis, thermodynamic parameters, and X-ray crystal structures. Compound 1, 9-deazaguanine linked to an acyclic ribocation phosphonate mimic, shows a kinetic Ki of 0.5 nM. Isothermal titration calorimetry (ITC) experiments of 1 binding to PfHGXPRT reveal enthalpically driven binding with negative cooperativity for the binding of two inhibitor molecules in the tetrameric enzyme. Crystal structures of 1 bound to PfHGXPRT define the hydrogen bond and ionic contacts to complement binding thermodynamics. Dynamics of ribosyl transfer from 5-phospho-α-d-ribosyl 1-pyrophosphate (PRPP) to hypoxanthine were examined by 18O isotope exchange at the bridging phosphoryl oxygen of PRPP pyrophosphate. Rotational constraints or short transition state lifetimes prevent torsional rotation and positional isotope exchange of bridging to nonbridging oxygen in the α-pyrophosphoryl group. Thermodynamic analysis of the transition state analogue and magnesium pyrophosphate binding reveal random and cooperative binding to PfHGXPRT, unlike the obligatory ordered reaction kinetics reported earlier for substrate kinetics.
Graphical Abstract
INTRODUCTION
De novo purine synthesis is essential in maintaining human pools of adenylate and guanylate compounds for DNA and RNA synthesis.1,2 Protozoan parasites, including Plasmodium falciparum, lack de novo purine synthesis and require the purine salvage pathway as an essential alternative. As a purine auxotroph, P. falciparum requires the salvage of purine bases from the host erythrocytes to perform purine nucleotide and nucleic acid synthesis. Purine salvage enzymes are attractive targets in the development of novel therapeutics for malaria. In P. falciparum, hypoxanthine–guanine–xanthine phosphoribosyltransferase (PfHGXPRT) is an essential component of this pathway.3–5
Malaria is the leading cause of death in Sub-Saharan Africa and regions of Southeast Asia. Global malaria cases have remained over 200 million yearly with approximately 400 thousand malaria-associated deaths.6–9 Malaria disproportionately affects children under 5 years old. Although several species of Plasmodium parasites cause the disease, P. falciparum accounted for 50% of malaria infections in Southeast Asia and over 99% of cases in Sub-Saharan Africa in 2019.8 A substantial reduction in malaria infections has been achieved by public health measures in Southeast Asia over the past two decades. However, malaria still plagues Sub-Saharan Africa, accounting for greater than 90% of the global malaria burden.7,10 Resistance to the current WHO-approved first-line therapy, artemisinin-based combination therapy (ACT), has been reported in Southeast Asia and, more recently, in Sub-Saharan Africa.11,12 Growing resistance has prompted several efforts to develop new therapies to malaria in addition to a vaccine prevention strategy.13
The essential enzyme PfHGXPRT incorporates hypoxanthine (as well as guanine and xanthine), a key precursor of nucleotide biosynthesis, into the nucleotide pool of the parasite.14,15 Hypoxanthine and inosine are formed in host erythrocytes and are available to the parasites through the P. falciparum purine base and nucleoside transporters in the parasite membrane. 6-Oxopurine nucleosides are in equilibrium with 6-oxopurine bases by the action of purine nucleoside phosphorylases (PNP) present both in erythrocytes and in P. falciparum.16–18 We and others have previously reported PfHGXPRT to be a target for the development of novel antimalarials and demonstrated that transition state analogue (TSA) inhibitors and their lipophilic prodrugs of PfHGXPRT block incorporation of extracellular purines and inhibit the growth of P. falciparum parasites in culture.14,15,19 Other reports have demonstrated the validity of the PfHGXPRT target by inhibition with nucleoside phosphonates.20,21 Targeting PNP with transition state analogues in cells and Aotus primates blocks hypoxanthine formation and results in antiparasitic activity.5 However, parasite-specific PfHGXPRT inhibitors are proposed to be an improvement over PNP inhibition, as a PNP blockade can be bypassed by metabolic conditions with increased hypoxanthine.22 TSA inhibitors immucillin-H 5′-phosphate (ImmHP), immucillin-G 5′-phosphate (ImmGP), and the acyclic nucleoside phosphonates (AIP) have been reported for PfHGXPRT, with dissociation constants of 0.65–14 nM. ImmHP and ImmGP were the first generation of transition state analogues for PfHGXPRT. Although they are potent enzymatic inhibitors, the anionic phosphomonoesters are membrane-impermeable, susceptible to phosphatases, and exhibit low selectivity for the parasite PfHGXPRT relative to the human HGPRT (HsHGPRT).19,23 The AIP compounds provide a biologically stable phosphonate group to mimic the phosphate group of the substrates and have greater selectivity for PfHGXPRT relative to HsHGPRT. Phosphonate groups are established precursors for conversion to membrane-permeable prodrugs.19,24
PfHGXPRT is a magnesium-dependent bi-substrate enzyme, utilizing the purine bases hypoxanthine, guanine, and xanthine with phosphoribosyl pyrophosphate (PRPP) to form the corresponding nucleotide monophosphates. The kinetic mechanism of PfHGXPRT has been reported to be ordered with PRPP binding first followed by purine base binding.25 Steady-state kinetic analyses of PfHGXPRT are complicated by a lag in reaction rates before reaching the steady state, a phase attributed to a conversion from inactive monomers to the active tetrameric form. Tetramer formation is influenced by the binding of PRPP, also supporting the ordered kinetic mechanism of PfHGXPRT.25,26 Additional studies of PfHGXPRT report slow interconversion of the ternary complexes with ordered release of the product (IMP released first) and an alternative ping-pong mechanism.27,28 Human HGPRT is reported to have a similar sequential kinetic mechanism with PRPP binding before binding of the base.29
Here, we use protein cross-linking, inhibitor structure–activity relationships, catalytic site titration, isothermal titration calorimetry (ITC), crystal structures, isotope trapping, and positional isotope exchange (PIX) experiments as definitive tools to probe the mechanism and inhibition of the enzyme.30,31 PfHGXPRT catalyzes its N-ribosyl transferase activity with an SN1-like ribooxocarbenium transition state structure where the leaving group and attacking nucleophile have low bond orders to the anomeric carbon of PRPP and the ribosyl group is a well-developed ribocation, a design element for transition state analogues.32,33
We extend the design, synthesis, and testing of novel TSAs and characterize the structural and thermodynamic interactions by kinetics, ITC, and X-ray crystallography. New insights into the ternary species and chemical intermediates of PfHGXPRT are provided by PIX and isotope trapping experiments. Conditions for the formation of the active tetrameric enzyme from inactive monomers are defined by protein chemical cross-linking.
RESULTS AND DISCUSSION
PfHGXPRT Activation to the Tetramer.
Steady-state kinetic analysis with PfHGXPRT displayed a lag phase prior to product formation (Figure 1A–C). The lag phase disappeared following enzyme activation by incubation with hypoxanthine, PRPP, and MgCl2 (substrate activated). Following activation, the enzyme could be diluted 60-fold into reaction mixtures containing the desired substrate and inhibitor concentrations to initiate steady-state kinetic analysis, thereby avoiding the lag phase (Figure 1A).
Figure 1.
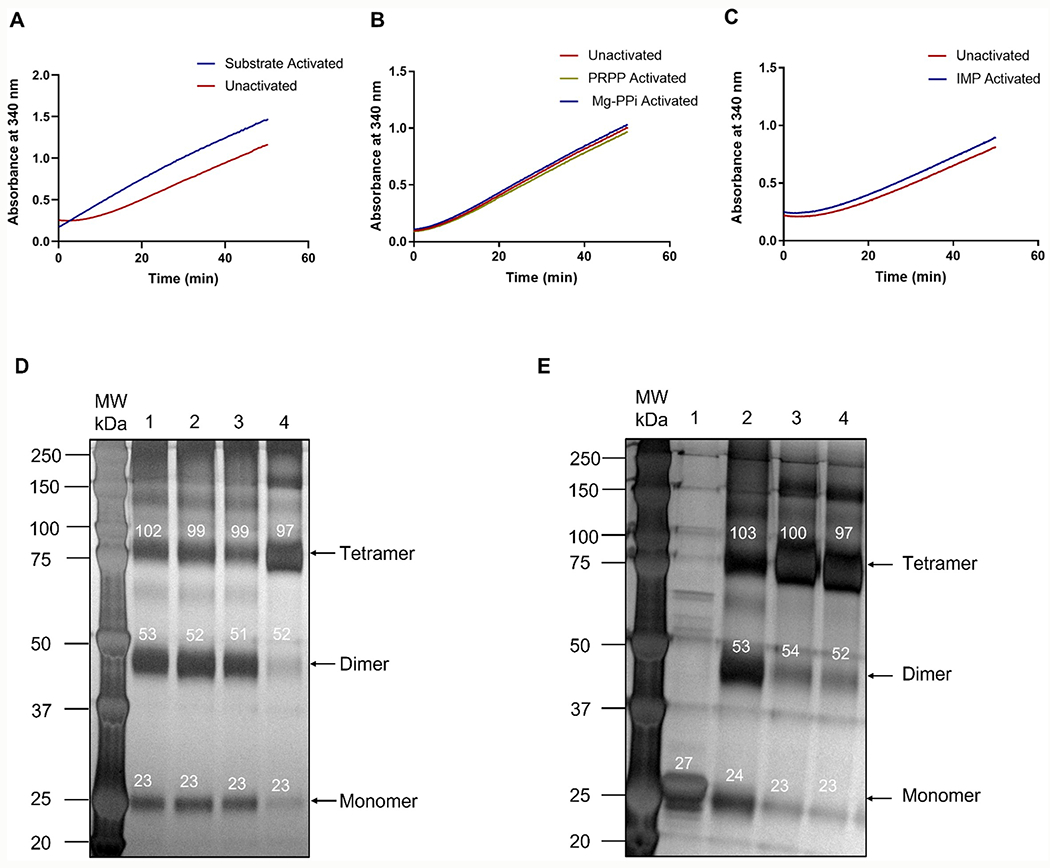
Activation of PfHGXPRT by (A) substrates, (B) PRPP or magnesium pyrophosphate (Mg-PPi), and (C) IMP. Preincubation of PfHGXPRT creates the right condition for the formation of active tetramers and the fully active enzyme. (D,E) Cross-linking gel electrophoresis for activating PfHGXPRT. Gel-scan molecular weights are shown above the relevant bands in white. (D) 1—IMP activated; 2—Mg-PPi activated; 3—PRPP activated; and 4—substrate activated. (E) 1—PfHGXPRT in buffer not cross-linked; 2—PfHGXPRT in buffer, cross-linked; 3—PfHGXPRT incubated with ImmHP; and 4—PfHGXPRT incubated with compound 1 (Figure 4). Conditions for activation are described in the methods.
Oligomeric analysis of PfHGXPRT before and after activation used glutaraldehyde cross-linking followed by SDS-PAGE to view the oligomeric species under different ‘activating’ conditions. Purified PfHGXPRT without activation appears predominantly as the ~26 kDa monomer. Incubation with the substrates, hypoxanthine, PRPP, and MgCl2 gave predominantly the tetrameric enzyme (Figure 1D,E). Activation conditions stabilizing the tetramer eliminated the lag phase of the initial rate curves (Figure 1A). Tetrameric PfHGXPRT was also formed when the enzyme was preincubated with Mg-PPi and the transition state analogue inhibitors, ImmHP and compound 1 (Figure 1E). Preincubation of the enzyme with PRPP, IMP, or magnesium pyrophosphate (Mg-PPi) as single agents resulted in a mixture of enzyme oligomeric species (Figure 1B–D). Full activation of PfHGXPRT for kinetic activity requires binding of all substrates or filling the catalytic site with a transition state analogue. Substrate activation of PfHGXPRT was carried out prior to all steady-state kinetic and inhibition measurements.
ImmHP is a transition state analogue of PfHGXPRT, and its activation of the enzyme captures the enzyme in a configuration near the transition state, consistent with stabilizing the active tetrameric form of the enzyme (Figure 1E). Because ImmHP is a powerful inhibitor of PfHGXPRT, its slow release from the enzyme makes it incompatible as an activating agent.23,34 Earlier reports for PfHGXPRT activation involved incubation of (1) 30 μM PfHGXPRT with 60 μM IMP, (2) 1 mM PRPP and 12 mM MgCl2, in 5 mM DTT and 10 mM phosphate buffer pH 7.0 at 0 °C for 3 h, or (3) 1 mM MgCl2, 1 mM pyrophosphate, and 5 mM DTT in 10 mM phosphate buffer pH 7.0 at 0 °C for 3 h.25,26,35 In our hands, these protocols provided an equilibrium of monomeric, dimeric, and tetrameric species in the cross-linking analysis (Figure 1). PfHGXPRT from Escherichia coli extracts copurifies with substoichiometric amounts of purine and may account for the results obtained by activation with PRPP as a single agent.25,26,32,35
Activated PfHGXPRT gave Km values for hypoxanthine and PRPP of 10 ± 1 and 185 ± 14 μM, respectively, with a kcat of 0.12 ± 0.01 s−1. Kinetic assays with unactivated PfHGXPRT gave Km values for hypoxanthine and PRPP of 2.7 ± 0.3 and 55 ± 11 μM, respectively, and a kcat of 0.020 ± 0.002 s−1. The sixfold increase in the kcat resulted in a 1.6-fold increase in catalytic efficiency (kcat/Km) for the activated enzyme.
Forward Commitment.
The kinetic mechanism of PfHGXPRT has been reported to be steady-state ordered, requiring the binding of PRPP prior to hypoxanthine (Hx) and IMP as the first product released (Scheme 1).25,36 We tested the kinetic mechanism and reversibility from the Michaelis complex using activated PfHGXPRT in forward commitment experiments.
Scheme 1.
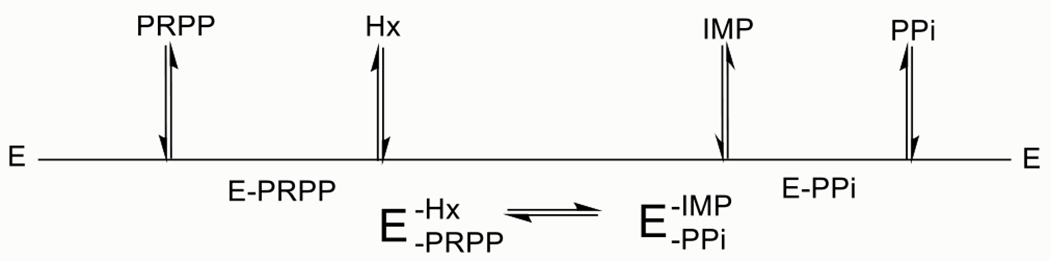
Compulsory Ordered Reaction Mechanism of PfHGXPRT Proposed from Earlier Reports25,36
The ternary Michaelis complex was formed in a modified isotope trapping experiment using hypoxanthine and [5-14C]PRPP in a pre-steady-state incubation serving to initiate tetramer formation and form a competent catalytic complex (see the Methods section). The chemical fate of [5-14C]PRPP in the E-Hx-[5-14C]PRPP complex was determined by dilution of the complex with excess unlabeled PRPP and hypoxanthine followed by reaction monitoring for the labeled and total IMP product (Figure 2). In a compulsory ordered mechanism (Scheme 1), saturation with the second substrate (hypoxanthine) traps the first substrate ([5-14C]PRPP) where it is converted to the product without equilibration with the unbound PRPP pool (Scheme 1). This extension of the approach reported by Rose30 also permits the estimation of a forward commitment (Cf), a partition coefficient for the relative probability of bound [5-14C]PRPP being converted to the product relative to its release as unreacted [5-14C]PRPP. A high probability of a bound substrate being converted to the product (Cf > 1.0) is predicted from the proposed mechanism. In contrast, the measured Cf was 0.017 ± 0.003 [Figure 2 ( )], establishing that bound [14C]PRPP can escape from the Michaelis complex even when near-saturating hypoxanthine is present. For the purpose of comparison, the dashed line of Figure 2 would be expected for a modest Cf = 0.5. The total IMP formed during the initial 8 s continued at the same rate following dilution with unlabeled PRPP and hypoxanthine [Figure 2 (
)], establishing that bound [14C]PRPP can escape from the Michaelis complex even when near-saturating hypoxanthine is present. For the purpose of comparison, the dashed line of Figure 2 would be expected for a modest Cf = 0.5. The total IMP formed during the initial 8 s continued at the same rate following dilution with unlabeled PRPP and hypoxanthine [Figure 2 ( ). Escape of [14C]PRPP from the ternary complex is inconsistent with a compulsory ordered mechanism and supports random binding of substrates. The Cf obtained here is similar to the small Cf previously reported for PfHGXPRT.32 However, the previously reported value was obtained with undefined activation of the enzyme, resulting in low catalytic efficiency. The Cf reported here does not alter the intrinsic kinetic isotope effect (KIE) values or transition state structure reported earlier.
). Escape of [14C]PRPP from the ternary complex is inconsistent with a compulsory ordered mechanism and supports random binding of substrates. The Cf obtained here is similar to the small Cf previously reported for PfHGXPRT.32 However, the previously reported value was obtained with undefined activation of the enzyme, resulting in low catalytic efficiency. The Cf reported here does not alter the intrinsic kinetic isotope effect (KIE) values or transition state structure reported earlier.
Figure 2.
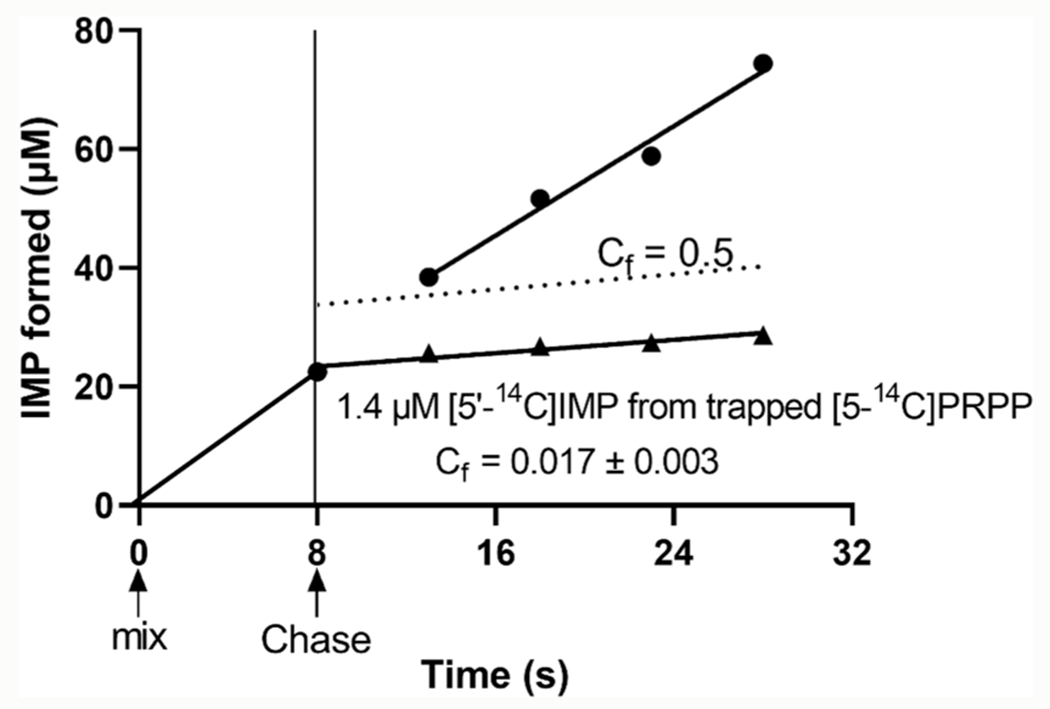
Forward commitment (Cf value) from isotope trapping of [5-14C]PRPP in the ternary complex of PfHGXPRT. PfHGXPRT was incubated briefly with hypoxanthine and [5-14C]PRPP to form the Michaelis complex (8 s of mix to Chase). Rapid dilution with excess unlabeled PRPP and hypoxanthine (Chase) allowed analysis of the [5′-14C]IMP produced from trapped [5-14C]PRPP (▲). The amount of IMP formed during the 8 s mix continued at the same rate during the Chase with excess unlabeled PRPP and hypoxanthine (·). The ordinate displacement at 8 s (1.4 μM) is the amount of [5′-14C]IMP formed from the E-[5-14C]PRPP-Hx complex existing at the time of the Chase. The dashed line for Cf = 0.5 would be observed if bound [5-14C]PRPP partitions 2:1 in favor of substrate release relative to product formation. The ordered mechanism in Scheme 1 predicts a Cf value > 1.0.
Positional Isotope Exchange (PIX).
Transition state analysis of PfHGXPRT reports a cationic riboxocarbenium transition state structure.32 Transition state analysis by KIE gives information at the transition state but does not provide temporal information of the riboxocarbenium lifetime. The possibility of an enzyme-stabilized riboxocarbenium reaction intermediate was explored with PIX. The conversion of [1-13C, bridge-18O]PRPP to IMP was monitored continuously by NMR (Figure 3B,C). A PIX effect is observed when 18O in the bridging position of PRPP scrambles to the non-bridging position in the PRPP pool.
Figure 3.
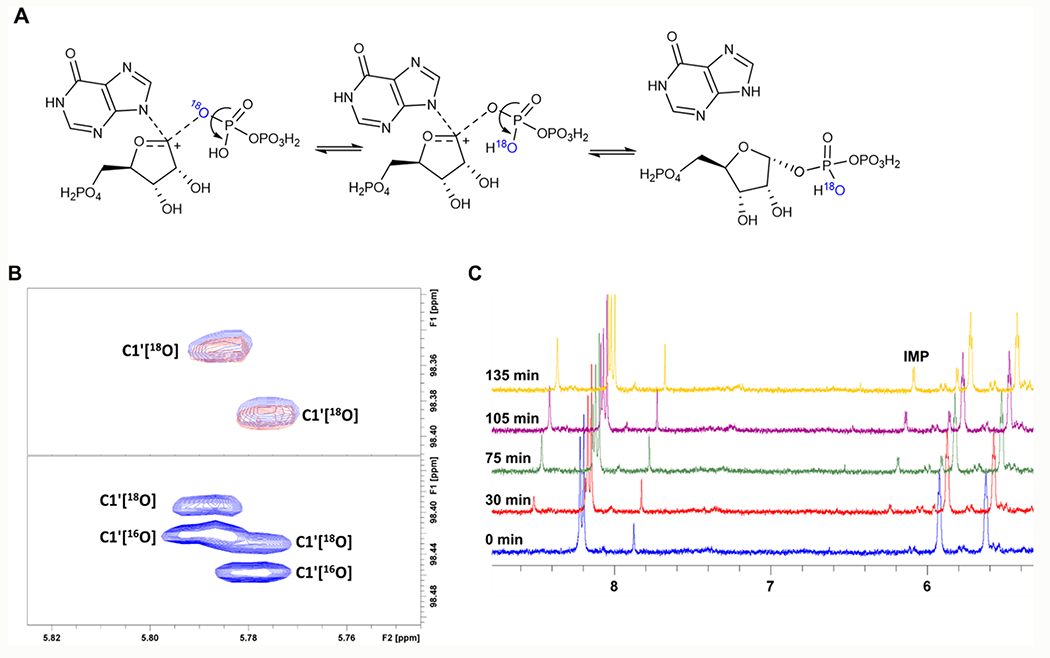
(A) Reaction scheme for PIX of PfHGXPRT. Isotope scrambling from the bridging oxygen happens when the riboxocarbenium intermediate forms, and there is free torsional rotation of the pyrophosphate ion. Reformation of the substrate from the breakdown of the Michaelis complex results in PRPP with 18O in the non-bridging position. (B) Upper panel, 2D 1H–13C HSQC NMR monitoring isotope scrambling; lower panel: an overlay of natural abundance PRPP and [1-13C,18O]-PRPP. No isotope scrambling was observed. (C) 1D 1H NMR monitoring reaction progress and the formation of IMP at approximately 6.2 ppm.
This occurs if a stabilized ribocation has a lifetime sufficient to permit rotation of the α-phosphoryl group, reformation of [1-13C, bridge-16O]PRPP, and release to the substrate PRPP pool. No isotope scrambling in PRPP was observed by monitoring the 13C chemical shift as influenced by the adjacent bridge-18O (Figure 3B). The growth of the IMP peak at 6.2 ppm established that no PIX occurred in the presence of ongoing product formation (Figure 3C). A reversible riboxocarbenium intermediate with phosphoryl freedom of rotation in the PfHGXPRT reaction can be eliminated by the lack of PIX.
PIX isotopic scrambling is expected if a reversible riboxocarbenium intermediate is formed during the reaction (Figure 3A), the Michaelis complex releases the substrate faster than is converted to products, and there is free torsional rotation of the pyrophosphate α-phosphoryl in the active site.37 A new downfield resonance signal corresponding to 16O in the bridging position would be observed by NMR.38 Taking into consideration the low forward commitment factor and the SN1-like mechanism from transition state analysis,23 the lack of an 16O–18O PIX demonstrates constrained torsional rotation of the pyrophosphate. Strong coordination of the pyrophosphate leaving group to the divalent magnesium ions or a transition state lifetime shorter than phosphoryl rotation could prevent phosphoryl rotation to prevent isotope scrambling.39,40 The crystal structure of PfHGXPRT with ImmGP at the catalytic site shows that two magnesium ions coordinated four oxygens of the pyrophosphate, bridging the pyrophosphate to the vicinal iminoribitols of the inhibitor.40 In contrast, inhibitor 1 has a single magnesium ion in contact with O2, O6, and O7 of the pyrophosphate (see below). These Mg-PPi contacts are candidates to immobilize the pyrophosphate within the active site.19
PfHGXPRT Inhibition.
The kinetic activity of PfHGXPRT in the physiological direction of IMP formation (Figure 4A) was monitored by coupling to IMP dehydrogenase (IMPDH) and the production of NADH at 340 nm (see the Methods section). For the tightest-binding inhibitors, the dissociation constants were calculated using the Morrison equation41 with a Km for hypoxanthine of 10 μM.
Figure 4.
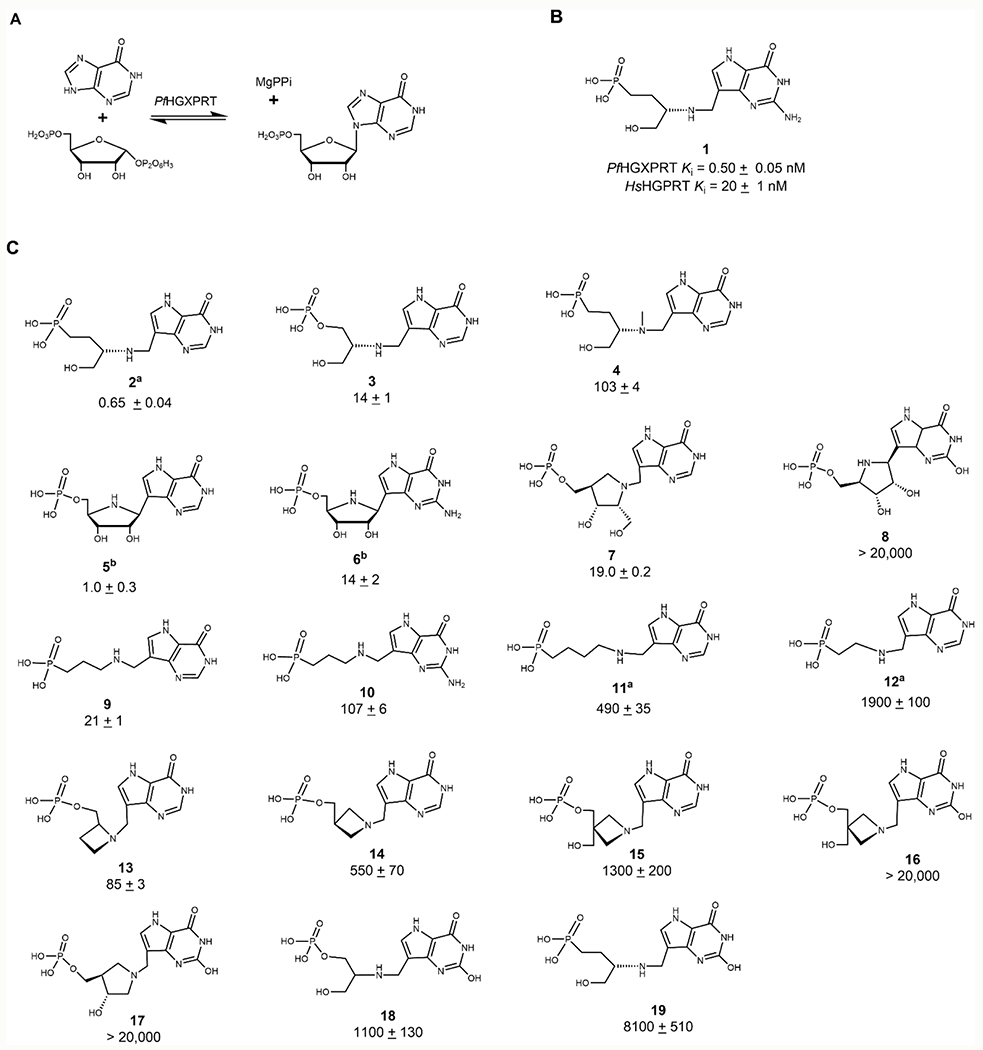
(A) Reaction catalyzed by PfHGXPRT. IMP formation was assayed by coupling to the formation of xanthine monophosphate (XMP) by inosine 5′-monophosphate dehydrogenase (IMPDH) with NADH absorbance monitored at 340 nm. (B) A transition state analogue inhibitor of PfHGXPRT demonstrates selectivity for the parasite enzyme (top) relative to the human HGPRT (bottom). (C), Serinol, pyrrolidine, and azetidine transition state analogues synthesized and tested for inhibitory activity against PfHGXPRT. Morrison Ki values reported as nanomolar concentrations. Compounds 5 and 6 represent ImmHP and ImmGP, respectively. 9-Deazaxanthine analogue molecules generally have weak or no inhibitory activity against PfHGXPRT. aValues reported in ref 24, and bvalues reported in ref 23. Errors reported are the standard errors (SE) of at least two replicates. Inhibitors with Ki >1 μM were tested once, and errors represent SE of fits to inhibition as a function of inhibitor concentration.
Inhibitors to mimic the riboxocarbenium transition state structure of PfHGXPRT were designed, synthesized, and tested for inhibitory activity (Figure 4B,C). The most potent of these was compound 1, with a Ki value of 0.50 ± 0.05 nM. Compound 1 is a weaker inhibitor of HsHGPRT with a Ki of 20 ± 0.3 nM, therefore displaying a selectivity index (SI) of 40 for the P. falciparum enzyme (Figure 4B and Table 1). Inhibition by the 9-deazahypoxanthine analogue (2) was similar to 1, reflecting the preference of PfHGXPRT for hypoxanthine and guanine as substrates (Figure 4C). Although the substrate specificity of PfHGXPRT would suggest that the 9-deazahypoxanthine phosphate derivative (3) should share similar affinity, it does not, with the affinity decreased by 30-fold (Figure 4B,C). PfHGXPRT presents an unusual case where a phosphonate improves binding relative to the phosphate monoester.
Table 1.
Transition State Analogues of PfHGXPRT and Their Selectivity Indices for HsHGPRT
| inhibitor | PfHGXPRT (Ki, nM) | HsHGPRT (Ki, nM) | selectivity index (SI) |
|---|---|---|---|
| 1 | 0.50 ± 0.05 | 20 ± 0.3 | 40 |
| 4 | 103 ± 4 | 4600 ± 70 | 43 |
| 7 | 19.0 ± 0.2 | 4500 ± 110 | 220 |
| 8 | >20,000 | >20,000 | |
| 9 | 21 ± 1 | 1350 ± 30 | 64 |
| 10 | 107 ± 12 | 1000 ± 5 | 9.3 |
| 13 | 85 ± 3 | >20,000 | >240 |
| 19 | 8000 ± 510 | >20,000 | >3 |
The crystal structures of the complex provide additional insights into inhibitor affinity (see below). Methylation of the serinol nitrogen of 2 forms 4 which retains its cationic character in solution but introduces steric interference for ion pair formation between the inhibitor, serinol nitrogen, and an oxygen of bound pyrophosphate, reducing affinity by 210-fold (Figure 4B,C) (see structural analysis below). Iminoribitol 5′-phosphates demonstrate a 14-fold preference for the 9-deazahypoxanthine relative to 9-deazaguanine (5 and 6) (Figure 4C). Linear cationic phosphonate derivatives of 9-deazapurines bind with higher affinity when the cation and phosphonate are separated by a propyl bridge (9, 11, and 12). In this configuration, 9-deazahypoxanthine (9) is preferred to 9-deazaguanine (10) by a factor of 6 (Figure 4C). Azetidine phosphate derivatives of 9-deazahypoxanthine (13 and 14) and an additional hydroxyl group (15) decreased affinity. All 9-deazaxanthine derivatives were poor inhibitors for PfHGXPRT (16–19) (Figure 4C).
Most active-site residues are conserved between HsHGPRT and PfHGXPRT with similar contacts to the inhibitor. The difference in the binding of compound 1 from other acyclic transition state analogue inhibitors might be attributed to the differences in the energetic contribution of N7 to hydrogen bonding in the enzymes, as revealed by NMR analysis of this interaction,42 or to interactions with the exocyclic N2 of 9-deazaguanine. Many of the best inhibitors for PfHGXPRT also demonstrate a high selectivity index for PfHGXPRT of 9 to >240 (Table 1). Structural analysis explored these interactions.
Crystal Structure of 1 with PfHGXPRT.
PfHGXPRT was co-crystallized with 1 in the presence of magnesium and pyrophosphate. The structure was determined by molecular replacement and refined to 1.62 Å resolution (Table 2). PfHGXPRT co-crystallized with compound 1 as a homotetramer (four PfHGXPRT monomers in the asymmetric unit; Figure 5A), consistent with previous reports.19,39,40 Tetramerization of PfHGXPRT buries a total 11,660 Å2 of solventexposed surface area, demonstrating the tetramer stabilization by catalytic site filling. The tertiary structures for each of the four subunits are similar with an average rmsd of <0.6 Å over 180 aligned Cα atoms with a typical PRTase-like fold (Figures 5A,B and S4A). Small differences between subunits are observed in regions that are involved in crystal contacts. Catalytic sites are at the base of the central β-sheet structure composed of six parallel β-strands (Figure 5B). The four distinct but equivalent active sites in the assembly are distal from the tetramer interface (Figure 5A). Electron densities in the active sites were identified as co-crystallized transition state analogue, compound 1, pyrophosphate, and Mg2+ ions (Figure S4B). Interactions between PfHGXPRT and the catalytic site ligands are nearly identical in all four subunits of the tetrameric assembly. The subsequent structural analysis is taken from the atomic contacts of subunit 1 (Figure 5A,B).
Table 2.
Crystallographic Data Statistics of the PfHGXPRT-Compound 1 Complex
| data collection | |
|---|---|
|
| |
| source | APS ID-31 |
| wavelength (Å) | 0.97931 |
| temperature (K) | 100 |
| number of crystals | 1 |
| space group | C2221 |
| cell dimensions a, b, c (Å) | 105.4, 111.2, 173.7 |
| Vm (Å3/Da) | 2.3 |
| number of subunits in the asymmetric unit | 4 |
| resolution (Å) | 173.65–1.62 (1.65–1.62) |
| total reflections | 950,288 (37,965) |
| unique reflections | 128,959 (6298) |
| Rmerge (%)c | 6.6 (106.9) |
| CC1/2 (%) | 99.9 (74.7) |
| I/σ(I)d | 16.9 (1.8) |
| completeness (%) | 99.9 (99.8) |
| multiplicity | 7.4 (6.0) |
| Wilson B-factor (Å2) | 20.4 |
|
| |
| refinementa | |
|
| |
| Rwork (%)e | 17.0 |
| Rfree (%)f | 19.5 |
| number of atoms | 8671 |
| protein | 7604 |
| ligand | 88 |
| water | 979 |
|
| |
| model quality r.m.s.d. | |
|
| |
| bond length (Å) | 0.006 |
| bond angle (deg) | 1.4 |
|
| |
| average B-factor (Å2) | |
|
| |
| protein | 26.5 |
| ligand (Å2) | 20.9 |
| water (Å2) | 36.3 |
|
| |
| MOLPROBITYb | |
|
| |
| favored regions | 96.5% (770 aa) |
| allowed | 100% (798 aa) |
| outlier | none |
| clash score | 100th percentile |
| MOLPROBITY score | 100th percentile |
| RCSB code | 7TUX |
Statistics are calculated using REFMAC5,43 values in highest resolution shells indicated in parentheses.
Calculated with the program MOLPROBITY.44
Rmerge = (ΣhklΣi|Ii(hkl) − ⟨I(hkl)⟩|)/ΣhklΣi⟨Ii(hkl)⟩, where Ii(hkl) is the intensity of the ith measurement of reflection (hkl) and ⟨I(hkl)⟩ is the mean intensity.
I is the integrated intensity, and σ(I) is the estimated standard deviation.
Rwork = (Σhkl∣Fo − Fcl)/ΣhklFo where Fo and Fc are the observed and calculated structure factors, respectively.
Rfree is calculated as for Rwork but from a randomly selected subset of the data (5%), excluded from the refinement calculation.
Figure 5.
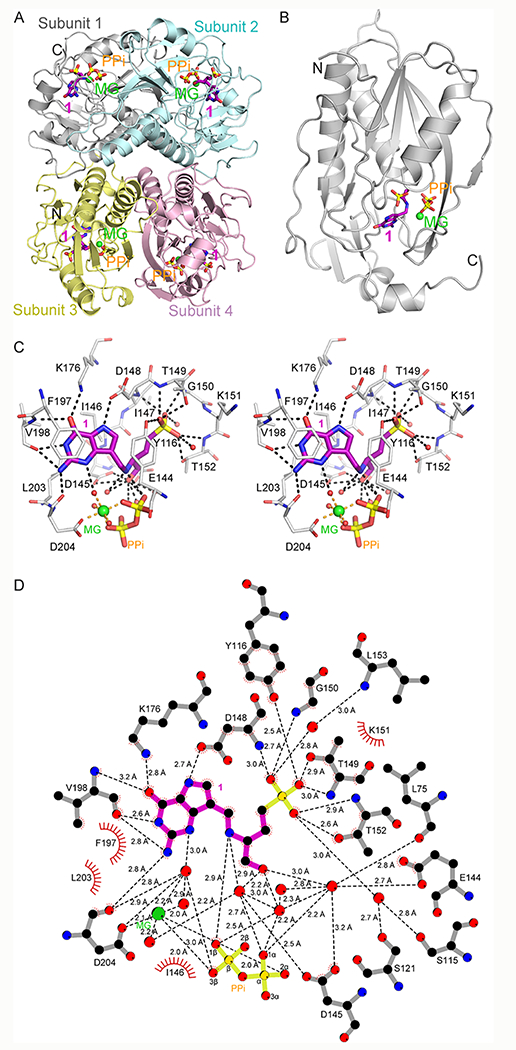
Crystal structure and assembly of compound 1 bound to PfHGXPRT. (A) PfHGXPRT tetramer (subunit 1 in gray, subunit 2 in cyan, subunit 3 in yellow, and subunit 4 in pink) shown as ribbons for helices and arrows for β strands. Bound compound 1 (magenta) and pyrophosphate (orange) are shown in stick representation, and the magnesium ion is shown as a green sphere. (B) Subunit 1 of the tetramer shown as in panel (A). (C) Stereoview of the active site of the PfHGXPRT- 1 complex (subunit 1). Selected residues from PfHGXPRT are shown in stick representation (gray). Compound 1, pyrophosphate, and the magnesium ion are as in panel (A). Waters are shown in red spheres. Distances (Angstroms) are indicated by dashed lines. (D) 2-D ligand contact map depicts interactions of compound 1 and magnesium with the enzyme, pyrophosphate, and water. Not all pyrophosphate contacts are shown.
Catalytic Site Interactions.
Four functional regions of the catalytic site for PfHGXPRT include the (i) purine binding site, (ii) ribose binding site, (iii) 5′-phosphate binding site, and (iv) magnesium pyrophosphate binding site.19 The purine binding site is a combination of hydrophobic and hydrogen bond interactions. The 9-deazapurine of compound 1 stacks between the sidechains of Tyr116 and Ile146 with additional hydrophobic interactions to the sidechains of Phe197, Val198, and Leu203 (Figure 5C,D). The N1 and exocyclic N2 atoms of 9-deazaguanine form hydrogen bonds with the carbonyl oxygen of Val198 (2.6 Å and 2.8 Å, respectively; Figure 5D). The exocyclic N2 atom also interacts with the carbonyl oxygen of Asp204 (2.8 Å). The sidechain of Lys176 forms a hydrogen bond with the purine O6 (2.8 Å), while the amide nitrogen of Val198 is more distant (3.2 Å). The purine N7 atom forms a hydrogen bond with the carboxyl group of Asp148 (2.7 Å). A weaker water-mediated interaction is observed with the deazapurine N3 atom (3.0 Å) (Figure 5C,D). The ribose binding site of the enzyme is formed by the sidechains of Tyr116, Glu144, and Asp145 (Figure 5C,D). The acyclic amino alcohol phosphonate of 1 replaces the ribose, with the serinol cationic nitrogen as a mimic of the cationic transition state. The serinol nitrogen ion pairs with the nucleophilic pyrophosphate anion (2.9 Å), a proposed mimic of the riboxocarbenium ion transition state structure. The 5′-phosphate binding site involves hydrogen bonds with the sidechains of Tyr116 (2.5 Å), Thr149 (2.9 Å), and Thr152 (2.6 Å) and the amide nitrogens of Asp148 (3.0 Å), Thr149 (3.0 Å), Gly150 (2.7 Å), and Thr152 (2.9 Å). The phosphate monoester binding site is remarkable by the lack of ion pairs to the dianionic phosphate. Pyrophosphate in the active site of PfHGXPRT is coordinated by the sidechains of Arg112 (2.6 Å) and Arg210 (2.9 Å), the backbone amide nitrogens from Lys77 (2.8 Å), Gly78 (3.1 Å), Ser115 (2.9 Å), and Tyr116 (2.9 Å), and the bidentate magnesium ion forming 2.0 Å ionic bonds with both phosphoryl groups of the pyrophosphate (Figures 5D and S5). The bound magnesium ion is octahedrally coordinated with the carboxyl of Asp204 (2.1 Å), two oxygens of the pyrophosphate (both 2.0 Å), and three water molecules (2.0–2.2 Å) (Figures 5C and S5). Compound 1 interactions with PfHGXPRT active site residues are similar to the interactions reported for the complex with the 9-deazahypoxanthine analogue of 1 (RCSB ID 3OZG)19 with a key difference by the added hydrogen bonds between the exocyclic N2 of 1 and the carbonyl oxygens of Val198 and Asp204, absent in the complex with RCSB ID 3OZG. This additional interaction with 1 is proposed to contribute to the greater affinity of 9-deazaguanine inhibitors.
Interactions of Inhibitors with PfHGXPRT.
Purified PfHGXPRT is an equilibrium of subunit forms in the presence of Mg-PPi (Figure 1D). Catalytic site titration of PfHGXPRT with compound 1 in the presence of Mg-PPi activates the enzyme twofold as the inhibitor binds to a single subunit of the PfHGXPRT tetramer (Figure 6A). This is a stoichiometric “one per tetramer” interaction and is interpreted as tetramer formation causing activation of subunits not filled with compound 1. Inhibition occurs as compound 1 is titrated to the tetramer (Figure 6A). Therefore, titrations of the inhibitor into the enzyme and Mg-PPi involve activation to tetramers followed by binding and inhibition of the tetramer.
Figure 6.
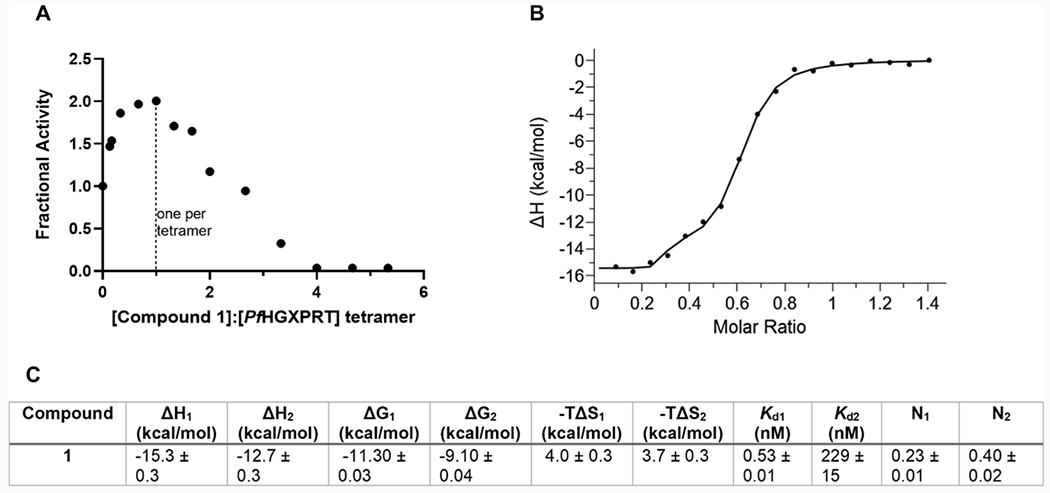
Interaction of compound 1 with PfHGXPRT. (A) Catalytic site titration of PfHGXPRT with compound 1. The enzyme was preincubated with substoichiometric amounts of compound 1. The catalytic activity of enzyme-inhibitor mixes was compared to the no inhibitor control to determine the fractional activity. (B) ITC binding isotherm of compound 1 binding showing large favorable enthalpy. PfHGXPRT (20 μM) was titrated with 150 μM compound 1 in 2 μL injections at 25 °C in 1.4 mM Mg-PPi, 0.5 mM TCEP, and 50 mM HEPES, pH 7.4. (C) Thermodynamic parameters from ITC. ΔH values, Kd1, Kd2, and binding ratios (N1 and N2) are reported directly from the ITC and are the average of four experimental titrations. ΔG and −TΔS values were determined from eqs 1 and 2, respectively, using the Kd1 and Kd2 (see methods) with standard errors determined from the average of four replicates.
Stoichiometry of Compound 1 Binding by ITC.
Transition state analogues convert the energy for catalysis into thermodynamic binding energy.45 The 0.5 nM inhibition constant of compound 1 in kinetic assays is readily explained by the 12 hydrogen bonds between the inhibitor and the catalytic site (≤3.0 Å) and an ion pair between bound pyrophosphate and the serinol nitrogen of 1 (Figure 5A). All four catalytic sites are saturated at the high inhibitor concentration used in crystallization studies. Thermodynamic and stoichiometric contributions of inhibitor binding were quantitated by ITC. ITC isotherms were fit to a two-site binding model using MicroCal PEAQ-ITC analysis software. Binding the first equivalent of compound 1 to PfHGXPRT (N = 0.23 ± 0.01 per monomer, approximately one per tetramer) in the presence of Mg-PPi revealed a large change in enthalpy (ΔH) of −15.3 ± 0.3 kcal/mol, assigned to 0.5 nM on the basis of kinetic experiments (Figure 6B,C). Subsequent binding of compound 1 involves approximately half the catalytic sites (N = 0.4 ± 0.02 per monomer, approximately two per tetramer) with a Kd2 of 229 ± 15 nM, 500-fold weaker binding affinity than the 0.5 nM Ki associated with the first subunit binding event (Kd1).
Kinetic analysis of catalytically active tetrameric PfHGXPRT indicates tight binding of compound 1 with a Ki of 0.5 nM. Binding of compound 1 by ITC in the presence of Mg-PPi is consistent not only with a tight interaction but also a second subunit catalytic site filling with negative cooperativity, a more weakly bound interaction. At high compound 1 concentrations, all sites are filled as observed in X-ray crystallography (Figure 5).
The thermodynamic values for the initial tight binding (0.5 nM) of compound 1 from ITC have a ΔH of −15.3 ± 0.3 kcal/mol and a ΔG of −11.30 ± 0.03 kcal/mol, giving a small entropic penalty of 4.0 ± 0.3 kcal/mol (Figure 6C). Thus, compound 1 is dominated by enthalpy, reflecting the multiple hydrogen and ionic bond contributions involved in inhibitor binding.
CONCLUSIONS
PfHGXPRT is essential in the purine salvage pathway of P. falciparum and a target for development of novel antimalarials. The catalytic mechanism, inhibitor kinetics, structure, and thermodynamics are defined for the novel high-affinity binding of transition state analogues. Inhibitors with iminoribitol and serinol ribocation scaffolds are nanomolar inhibitors and demonstrate the dynamic linkage between the catalytic site protein elements, the 9-deazapurines common to the inhibitors, the ribocationic mimics, and the phosphate or phosphonate binding sites. Forward commitment, inhibitor and pyrophosphate binding experiments establish a random, rather than obligatory sequential binding mechanism for PfHGXPRT. The transition state has a lifetime too short or too constrained to permit reversible phosphoryl group rotation. Bound PRPP in the ternary complex of PfHGXPRT-hypoxanthine-PRPP freely equilibrates with unbound substrates more rapidly than the chemical steps. This new information for the catalytic mechanism and inhibitor design defines precursors for improved candidates as antimalarials. Inhibitors for PfHGXPRT are phosphate- or phosphonate-containing compounds. As anions present membrane permeability issues, prodrug approaches to protect the phosph(on)ate are likely to be required. Synthesis of neutral compounds using prodrugs or phosphate bio-isosteres will yield the next generation of inhibitors of PfHGXPRT suitable for testing in biological systems.
METHODS
Expression and Purification of PfHGXPRT, HsHGPRT, and SpIMPDH.
PfHGXPRT was expressed and purified as previously published.22 Briefly, a thrombin-cleavable N-terminal six-histidine tag was cloned into pDEST-14. The plasmid was overexpressed in E. coli BL21-AI competent cells (Invitrogen) to an OD600 of 0.6 at 37 °C, and the temperature was decreased to 18 °C for 45 min. The culture was induced with 0.2% l-arabinose overnight at 18 °C and harvested by centrifugation at 5000g for 20 min at 4 °C. All protein purification was performed at 4 °C. Cells were suspended in 20 mM Tris-HCl, 5 mM imidazole, 500 mM NaCl, and 1 mM DTT, pH 7.9, with DNase I, lysozyme (Sigma), and protease inhibitor tablet (Roche) for 30 min, lysed by sonication, and centrifuged at 20,000g for 20 min. Ni-NTA resin (Qiagen) was washed with 10 column volumes of cell suspension buffer after which the clarified lysate was added and incubated for 45 min with rocking. The mixture was transferred to a gravity column and washed with 10 column volumes of cell suspension buffer. A stepwise elution was performed in 50–500 mM imidazole. Collected fractions were analyzed by SDS-PAGE. Fractions highly enriched in PfHGXPRT (~26 kDa) were pooled and dialyzed in 50 mM potassium phosphate, 5 mM imidazole, 150 mM KCl, 1 mM DTT, and 10% glycerol, pH 7.5. The enzyme solution was concentrated to approximately 300 μM, aliquoted, frozen in liquid nitrogen, and stored at −80 °C. HsHGPRT was purified and stored as previously described.19
A six-histidine tagged protein sequence with a TEV cleavage site of Streptococcus pyogenes IMP dehydrogenase (SpIMPDH) was designed and cloned into pET-28b(+) by GenScript. The plasmid was overexpressed in E. coli BL21 (DE3) to an OD600 of 0.6 at 37 °C and induced with 1 mM IPTG overnight at 18 °C. All protein purification steps were performed at 4 °C. Cells were suspended in 50 mM potassium phosphate, 500 mM KCl, 5 mM imidazole, and 0.5 mM TCEP, pH 8.0, with added protease inhibitor tablet (Roche), DNase I, and lysozyme (Sigma) for 30 min. Cells were lysed by sonication and clarified by centrifugation at 20,000 g for 20 min. The supernatant was added to washed Ni-NTA resin and incubated for 45 min with shaking. Protein was eluted in a stepwise imidazole concentration from 50 to 250 mM and analyzed by SDS-PAGE. Fractions highly enriched in SpIMPDH (protein band at 52.8 kDa) were pooled and dialyzed in 50 mM HEPES, 100 mM KCl, and 20% glycerol, pH 7.9. The enzyme was aliquoted, frozen rapidly, and stored at −80 °C. Yield of SpIMPDH was 600 mg from a 4 L culture.
PfHGXPRT Activation and Cross-Linking.
PfHGXPRT was activated by preincubating 30 μM PfHGXPRT with 1 mM hypoxanthine, 2 mM PRPP, 10 mM MgCl2, 1 mM DTT, and 50 mM potassium phosphate pH 7.4 in a 50 μL reaction mix for 30 min at 25 °C. Activation by magnesium pyrophosphate, IMP, and PRPP was performed according to the method outlined by Roy et al.25,26,35 A 60-fold dilution of the activation mix was made into assay mixtures described below to determine the efficiency of activation. For activation by ImmHP or compound 1, 30 μM PfHGXPRT was preincubated with 30 μM ImmHP or compound 1, 1.4 mM MgCl2, 1.4 mM pyrophosphate, and 50 mM potassium phosphate pH 7.4 in a 50 μL reaction mix for 30 min at 25 °C.
Glutaraldehyde cross-linking was performed by incubating activation mixes with 1% glutaraldehyde in a total volume of 100 μL for 5 min at 37 °C. The cross-linking reaction was quenched by adding 10 μL of 1M Tris-HCl pH 8.0. Samples were analyzed by SDS-PAGE to determine the protein oligomeric states. Molecular weight analysis of the PfHGXPRT bands was performed using Image Lab software by comparison to molecular weight standards (Biorad).
Synthesis of Transition State Analogue Inhibitors.
Compounds 1, 4, 8, and 19,46 2, 3, and 9-12,24 and 5 and 623 were synthesized as reported. The synthetic procedures and NMR spectra for all new compounds are in the Supporting Information. The NMR spectra reported earlier for 19, 9–12, and 6 were for the same batch of compounds used here.
PfHGXPRT Kinetics and Inhibition Assays.
Steady-state kinetics and inhibition of PfHGXPRT and human enzyme (HsHGPRT) were measured spectrophotometrically using an IMPDH coupled assay. The IMP product produced by PfHGXPRT and HsHGPRT from hypoxanthine and PRPP was coupled to the formation of XMP, and the spectral change of NAD+ to NADH was measured at 340 nm. All assays were performed in 50 mM HEPES pH 7.4 containing 500 nM PfHGXPRT or 78 nM HsHGPRT, 3 μM SpIMPDH, 1 mM hypoxanthine, 2 mM PRPP, 5 mM NAD+, 10 mM MgCl2, and 1 mM DTT. For inhibition assays, inhibitor concentration was varied between 0 and 100 μM in reaction mixtures containing 1.4 mM pyrophosphate. Inhibitors were tested against both PfHGXPRT and HsHGPRT to measure selectivity of the inhibitors. Selectivity indices were calculated as the ratio of the inhibitory constants of HsHGPRT to PfHGXPRT. Data were analyzed using Prism 9 software by fitting to the Michaelis–Menten equation for steady-state calculations and to the Morrison equation for calculation of the inhibition constants (Ki) for tight-binding inhibitors.41
Co-crystallization of PfHGXPRT in Complex with Compound 1.
PfHGXPRT (190 μM) was mixed with compound 1 + pyrophosphate + MgCl2 at a 1:2:5:5 molar ratio and incubated on ice for 2 h. Initial screening of the complex was performed using 1 μL (protein/mother liquor = 1:1) sitting drops by vapor diffusion at 22 °C against 70 μL of reservoir solution in 96-well Intelli plates with a Crystal Gryphon (Art Robbins Instruments) utilizing MCSG (1-4, Microlytic) and Crystal Screen HT (Hampton Research) sparse matrix crystallization suites. Diffraction quality crystals were obtained within 2 weeks against well solutions containing 0.2 M lithium acetate 20% (w/v) PEG 3350. Crystals were cryo-preserved by flash-cooling in liquid nitrogen.
Data Collection and Processing, Structure Refinement, and Analysis.
Data from the crystals of the complex were collected with a CCD Pixel Dectris Pilatus 6M detector, with a wavelength of 0.97931 Å, on the ID-31 (LRL-CAT) beamline at the Argonne National Laboratory (Table 2). Single crystal data were integrated and scaled using iMOSFLM and AIMLESS,47 respectively. Diffraction was consistent with the orthorhombic space group C2221 (unit cell dimensions are in Table 2) and extended to 1.62 Å resolution with four molecules (chain A, B, C, and D) in the asymmetric unit. Initial phases were determined by molecular replacement (MR) with PHASER48 using refined coordinates of PfHGXPRT (RCSB ID 1CJB);42 the initial MR coordinate was manually inspected and corrected using COOT.49 The model was refined with PHENIX-REFINE.50 During initial rounds of refinement, inhibitor molecules were not included in the model, and after incorporating water molecules into the model, inhibitor molecules were built into their respective electron densities. Analyses of the structures were performed in COOT and evaluated using MOLPROBITY;44 B-factors were calculated using the BAVERAGE program in the CCP4 suit. The crystallographic model exhibited excellent geometry with no residues in disallowed regions of the Ramachandran plot.51 Crystallographic statistics and RCSB accession codes are provided in Table 2. All figures depicting the structure were generated with PyMol,52 unless stated otherwise.
Catalytic Site Titration.
PfHGXPRT catalytic sites were titrated with compound 1 at varying ratios of the inhibitor to tetrameric enzyme. Incubation mixtures contained 30 μM PfHGXPRT, compound 1 from 0 to 40 μM, 1.4 mM MgCl2, 1.4 mM pyrophosphate, and 50 mM potassium phosphate buffer pH 7.4. Incubations were carried out for 30 min. Following incubation, samples were diluted into an assay mix containing 100 μM guanine, 1 mM PRPP, 10 mM MgCl2, and 1 mM DTT in 50 mM HEPES pH 7.4. The final concentration of the enzyme in the assays was 500 nM. The conversion of guanine to GMP was followed by the absorbance change at 258 nm (extinction coefficient = 5800 M−1·cm−1).
Isothermal Titration Calorimetry (ITC).
Purified PfHGXPRT was dialyzed against buffer containing charcoal to remove purines that co-purify with the enzyme. Size exclusion chromatography (AKTA-FPLC) was used to purify the protein followed by dialysis in 50 mM HEPES, 1.4 mM MgCl2, 1.4 mM pyrophosphate, and 0.5 mM TCEP, pH 7.4. Compound 1 was prepared in the protein dialysate (150 μM), loaded into the syringe, and titrated into 20 μM PfHGXPRT in the cell. Titration was performed at 25 °C using a series of 2 μL individual injections for a total of 19 titrations spaced at 150 s intervals. ITC experiments used a MicroCal PEAQ-ITC. The sample cell was filled with approximately 300 μL of PfHGXPRT, and the injection syringe was loaded with approximately 40 μL of the ligand solution. The reference cell was filled with 300 μL of filtered (Millipore 0.2 μm) Milli-Q water. Binding isotherms were fit to the two-site binding model using MicroCal PEAQ-ITC analysis software. The Kd1 of the first binding site (Kd1) was estimated as the Ki for compound 1 determined from kinetic assays in the fitting model. ITC binding experiments were performed four times to obtain average changes in enthalpy values (ΔH), binding ratio (N), and Kd2 of the second binding site. The Gibbs free energy (ΔG1 and ΔG2) values were determined from the Kd1 and Kd2 respectively, using eq 1. The entropic terms (−TΔS1 and −TΔS2) were calculated from the respective ΔG values using eq 2. Control titrations of the inhibitor into the ITC buffer were performed and the heats were subtracted from the inhibitor: enzyme titrations during data analysis.
| (1) |
| (2) |
[1-13C, 1-18O]PRPP Synthesis.
[1-13C, 1-18O]PRPP was synthesized chemoenzymatically from D-[1-13C]ribose (Sigma) and 18O water (Cambridge Isotope Laboratories). D-[1-13C]ribose was converted to D-[1-13C, 1-18O]ribose by 18O exchange in 18O water and 5 mM potassium phosphate pH 7.0 at 61 °C overnight. SpeedVac evaporation of the solvent yielded D-[1-13C, 1-18O]ribose. [1-13C, 1-18O]PRPP was synthesized from 2 mg of D-[1-13C, 18O]ribose, 500 μM ATP, 5 mg/mL phosphoenolpyruvate (PEP), 5 mM MgCl2, 4.5 μM ribokinase, 2 U PRPP synthase, 2 U pyruvate kinase, and 3 U myokinase in a 1 mL reaction of 50 mM phosphate buffer pH 7.4 at 25 °C (Figure S1). The reaction was stopped after 1 h by the addition of 5 mM EDTA, and proteins were removed using an Amicon ultrafiltration tube, centrifuged at 14,000 g for 1 h at 4 °C. The filtrate was frozen and stored at −80 °C prior to additional purification.
[1-13C, 1-18O]PRPP was purified by HPLC with a 5 mL HiTrap QFF column (GE Life Sciences) in a water (solvent A) and 1.5 M ammonium acetate (solvent B) gradient. The HPLC gradient was varied from 0 to 100% solvent B in 65 min and maintained for 5 min and 100–0% solvent B in 1 min and maintained for 35 min. Purified [1-13C, 1-18O]PRPP was frozen and lyophilized. Excess ammonium acetate was removed by several rounds of water dilution, freezing, and lyophilizing. Solid [1-13C, 1-18O]PRPP was stored at −80 °C prior to use. Purity and concentration of [1-13C, 1-18O]PRPP were determined by NMR (Figure S2).
Positional Isotope Exchange (PIX).
PIX was monitored by measuring 16O/18O isotope scrambling at C-1 of [1-13C, 1-18O]PRPP by 2D 1H–13C HSQC NMR. A reaction mixture containing 4.8 mM [1-13C, 1-18O]PRPP, 1 mM hypoxanthine, 10 mM MgCl2 and 2 μM PfHGXPRT, and 50 mM HEPES pH 7.4 in a final volume of 200 μL was dissolved in D2O and put into a 3 mm NMR tube. NMR data were acquired at 25 °C using a Bruker AVIII 600 MHz spectrometer running TopSpin 3.6 and equipped with a 5 mm H/F-TCI cryogenic probe. The 1-13C of PRPP was monitored in a high-resolution 2D 1H–13C HSQC to resolve the cross peak belonging to the 1-13C bonded to 18O from that bonded to 16O expected to appear if PIX occurred during the enzymatic reaction. Each HSQC timepoint was run for 13 min with eight scans acquired for each increase using a spectral width of 14 ppm and 1 ppm for 1H and 13C, respectively, and a recycle delay of 1.1 s. The data sets were collected using 4096 and 32 complex points for 1H and 13C, respectively, and the time-domain NMR data were linear predicted in the indirect dimension and multiplied with a shifted sine-bell function (SSB = 3) in each dimension prior to Fourier transformation. The reaction was monitored for a total of 135 min during which a total of 10 HSQC spectra were collected. Control reactions measured the effect of MgCl2 on spectral acquisition by measuring the 1-13C chemical shifts of PRPP in natural abundance PRPP in the presence and absence of MgCl2 (Figure S3). Additional controls mixed natural abundance PRPP with [1-13C, 1-18O]PRPP to establish the 18O-induced shifts by 2D 13C NMR. These controls ensured the reliable detection of 18O to 16O shifts during the PIX reaction.
Forward Commitment.
[5-14C]PRPP was synthesized from [5-14C]AMP according to a previously published method.32 The forward commitment (Cf) was measured for PfHGXPRT using the isotope trapping method developed by Rose30 with some modifications to ensure that a catalytically active complex was formed during enzyme loading with [5-14C]PRPP and hypoxanthine. Briefly, 114 μM PfHGXPRT was preincubated for 8 s in a 320 μL reaction mixture containing 214 μM [5-14C]PRPP, 1 mM hypoxanthine, 1.4 mM MgCl2, 0.1 mM DTT, and 50 mM HEPES, pH 7.4. This period involves binding of substrates, oligomerization toward active tetramers, and catalysis. During the 8 s, 22.5 ± 0.02 μM [5-14C]PRPP was converted to [5′-14C]IMP, establishing the presence of the functional enzyme-[5-14C]PRPP-hypoxanthine complex. The reaction mixture was diluted at 8 s with excess unlabeled PRPP and hypoxanthine by the addition of 180 μL containing 2.5 mM hypoxanthine, 2 mM unlabeled PRPP, 1.4 mM MgCl2, 0.1 mM DTT, and 50 mM HEPES pH 7.4. At dilution, the enzyme-bound [5-14C]PRPP partitions to the IMP product (forward reaction) or releases to the PRPP pool (dissociation). The partition ratio determines the Cf. Reaction samples (120 μL) were quenched with 5 mM EDTA at 5, 10, 15, and 20 s following isotopic dilution and flash-frozen in liquid nitrogen. The product [5′-14C]IMP from each timepoint was purified by HPLC using a Luna C18(2) 15 μm, 100 Å, 250 mm × 4.6 mm column (Phenomenex).22 Radiolabeled analyses were performed on samples dried by vacuum centrifugation, dissolved in 500 μL of H2O, mixed with 10 mL of scintillation fluid, and counted to estimate [5′-14C]IMP formed at the various timepoints. [5′-14C]IMP formed following the dilution is based on the PRPP present in the initial ternary complex. [5′-14C]IMP formed from the E-[5-14C]PRPP-Hx complex following the chase with excess unlabeled PRPP and hypoxanthine at 8 s was determined from the graphical analysis. The Km value of PRPP for activated PfHGXPRT was used to estimate the fraction of PfHGXPRT saturated with [5-14C]PRPP. The Cf is defined as the probability for a bound PRPP molecule in the E-PRPP-Hx to convert to the product relative to being released as the unreacted substrate where equal probability is Cf = 1.0.
Supplementary Material
ACKNOWLEDGMENTS
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. The use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly and Company, which operates the facility.
Funding
This work was supported by NIH research grants (AI127807 and GM041916), the NIH training grant (T32 AI070117) for geographic medicine and emerging infectious diseases, and the New Zealand Endeavour Fund grant E3352. The Bruker 600 MHz NMR instrument in the Structural NMR Resource at the Albert Einstein College of Medicine was purchased using funds from NIH award 1S10OD016305 and was supported by a Cancer Center Support Grant (P30 CA013330). The Albert Einstein College of Medicine Crystallographic Core X-ray diffraction facility is supported by NIH Shared Instrumentation Grant S10 OD020068.
ABBREVIATIONS
- PfHGXPRT
Plasmodium falciparum hypoxanthine–guanine–xanthine phosphoribosyltransferase
- HsHGPRT
human hypoxanthine–guanine phosphoribosyltransferase
- SpIMPDH
Streptococcus pyogenes inosine monophosphate dehydrogenase
- IMP
inosine monophosphate
- PRPP
5-phosphoribosyl 1-pyrophosphate
- PIX
positional isotope exchange
- ITC
isothermal titration calorimetry
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00546.
Molecular weight analysis for cross-linked PfHGXPRT, synthesis of [5-14C]PRPP, NMR spectra for PRPP-18O exchange experiments, crystallography details for PfHGXPRT interactions, synthetic procedures for inhibitor synthesis, and NMR spectra for new inhibitors and intermediates (PDF)
NMR spectra for all new compounds (PDF)
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.2c00546
Contributor Information
Yacoba V. T. Minnow, Department of Biochemistry, Albert Einstein College of Medicine, Bronx, New York 10461, United States
Kajitha Suthagar, Ferrier Research Institute, Victoria University of Wellington, Lower Hutt 5010, New Zealand.
Keith Clinch, Ferrier Research Institute, Victoria University of Wellington, Lower Hutt 5010, New Zealand.
Rodrigo G. Ducati, Department of Biochemistry, Albert Einstein College of Medicine, Bronx, New York 10461, United States
Agnidipta Ghosh, Department of Biochemistry, Albert Einstein College of Medicine, Bronx, New York 10461, United States.
Joshua N. Buckler, Ferrier Research Institute, Victoria University of Wellington, Lower Hutt 5010, New Zealand
Rajesh K. Harijan, Department of Biochemistry, Albert Einstein College of Medicine, Bronx, New York 10461, United States
Sean M. Cahill, Department of Biochemistry, Albert Einstein College of Medicine, Bronx, New York 10461, United States
Peter C. Tyler, Ferrier Research Institute, Victoria University of Wellington, Lower Hutt 5010, New Zealand
Vern L. Schramm, Department of Biochemistry, Albert Einstein College of Medicine, Bronx, New York 10461, United States
REFERENCES
- (1).Yin J; Ren W; Huang X; Deng J; Li T; Yin Y Potential Mechanisms Connecting Purine Metabolism and Cancer Therapy. Front. Immunol 2018, 9, 1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lane AN; Fan TW Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cassera MB; Hazleton KZ; Riegelhaupt PM; Merino EF; Luo M; Akabas MH; Schramm VL Erythrocytic adenosine monophosphate as an alternative purine source in Plasmodium falciparum. J. Biol. Chem 2008, 283, 32889–32899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Cassera MB; Zhang Y; Hazleton KZ; Schramm VL Purine and pyrimidine pathways as targets in Plasmodium falciparum. Curr. Top. Med. Chem 2011, 11, 2103–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Cassera MB; Hazleton KZ; Merino EF; Obaldia N 3rd; Ho MC; Murkin AS; DePinto R; Gutierrez JA; Almo SC; Evans GB; Babu YS; Schramm VL Plasmodium falciparum parasites are killed by a transition state analogue of purine nucleoside phosphorylase in a primate animal model. PLoS One 2011, 6, No. e26916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).WHO. World Malaria Report 2020; World Health Organization: Geneva, 2020; Vol. license CC BY-NC-SA 3.0 IGO. [Google Scholar]
- (7).WHO. World Malaria Report 2018; World Health Organization: Geneva, 2018. [Google Scholar]
- (8).WHO. World Malaria Report; World Health Organization: Geneva, 2019; Vol. License: CC BY-NC-SA 3.0 IGO. [Google Scholar]
- (9).WHO. World Malaria Report 2021; World Health Organization: Geneva, 2021; Vol. IGO license CC BY-NC-SA 3.0 IGO. [Google Scholar]
- (10).Shah HA; Carrasco LR; Hamlet A; Murray KA Exploring agricultural land-use and childhood malaria associations in sub-Saharan Africa. Sci. Rep 2022, 12, 4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Balikagala B; Fukuda N; Ikeda M; Katuro OT; Tachibana SI; Yamauchi M; Opio W; Emoto S; Anywar DA; Kimura E; Palacpac NMQ; Odongo-Aginya EI; Ogwang M; Horii T; Mita T Evidence of Artemisinin-Resistant Malaria in Africa. N. Engl. J. Med 2021, 385, 1163–1171. [DOI] [PubMed] [Google Scholar]
- (12).Amato R; Pearson RD; Almagro-Garcia J; Amaratunga C; Lim P; Suon S; Sreng S; Drury E; Stalker J; Miotto O; Fairhurst RM; Kwiatkowski DP Origins of the current outbreak of multidrug-resistant malaria in southeast Asia: a retrospective genetic study. Lancet Infect. Dis 2018, 18, 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Duffy PE; Patrick Gorres J Malaria vaccines since 2000: progress, priorities, products. npj Vaccines 2020, 5, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Keough DT; Ng AL; Winzor DJ; Emmerson BT; de Jersey J Purification and characterization of Plasmodium falciparum hypoxanthine-guanine-xanthine phosphoribosyltransferase and comparison with the human enzyme. Mol. Biochem. Parasitol 1999, 98, 29–41. [DOI] [PubMed] [Google Scholar]
- (15).Keough DT; Hocková D; Holý A; Naesens LM; Skinner-Adams TS; Jersey J; Guddat LW Inhibition of hypoxanthine-guanine phosphoribosyltransferase by acyclic nucleoside phosphonates: a new class of antimalarial therapeutics. J. Med. Chem 2009, 52, 4391–4399. [DOI] [PubMed] [Google Scholar]
- (16).Sosa Y; Egbo D; Akabas MH Impact of Field Isolate Identified Nonsynonymous Single Nucleotide Polymorphisms on Plasmodium falciparum Equilibrative Nucleoside Transporter 1 Inhibitor Efficacy. ACS Infect. Dis 2020, 6, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Deniskin R; Frame IJ; Sosa Y; Akabas MH Targeting the Plasmodium vivax equilibrative nucleoside transporter 1 (PvENT1) for antimalarial drug development. Int. J. Parasitol.: Drugs Drug Resist 2016, 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Quashie NB; Ranford-Cartwright LC; de Koning HP Uptake of purines in Plasmodium falciparum-infected human erythrocytes is mostly mediated by the human equilibrative nucleoside transporter and the human facilitative nucleobase transporter. Malar. J 2010, 9, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hazleton KZ; Ho MC; Cassera MB; Clinch K; Crump DR; Rosario I Jr.; Merino EF; Almo SC; Tyler PC; Schramm VL Acyclic immucillin phosphonates: second-generation inhibitors of Plasmodium falciparum hypoxanthine-guanine-xanthine phosphoribosyltransferase. Chem. Biol 2012, 19, 721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hocková D; Holý A; Andrei G; Snoeck R; Balzarini J Acyclic nucleoside phosphonates with a branched 2-(2-phosphonoethoxy)ethyl chain: efficient synthesis and antiviral activity. Bioorg. Med. Chem 2011, 19, 4445–4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Keough DT; Špaček P; Hocková D; Tichý T; Vrbková S; Slavětínská L; Janeba Z; Naesens L; Edstein MD; Chavchich M; Wang TH; de Jersey J; Guddat LW Acyclic nucleoside phosphonates containing a second phosphonate group are potent inhibitors of 6-oxopurine phosphoribosyltransferases and have antimalarial activity. J. Med. Chem 2013, 56, 2513–2526. [DOI] [PubMed] [Google Scholar]
- (22).Kicska GA; Tyler PC; Evans GB; Furneaux RH; Schramm VL; Kim K Purine-less death in Plasmodium falciparum induced by immucillin-H, a transition state analogue of purine nucleoside phosphorylase. J. Biol. Chem 2002, 277, 3226–3231. [DOI] [PubMed] [Google Scholar]
- (23).Li CM; Tyler PC; Furneaux RH; Kicska G; Xu Y; Grubmeyer C; Girvin ME; Schramm VL Transition-state analogs as inhibitors of human and malarial hypoxanthine-guanine phosphoribosyltransferases. Nat. Struct. Biol 1999, 6, 582–587. [DOI] [PubMed] [Google Scholar]
- (24).Clinch K; Crump DR; Evans GB; Hazleton KZ; Mason JM; Schramm VL; Tyler PC Acyclic phosph(on)ate inhibitors of Plasmodium falciparum hypoxanthine-guanine-xanthine phosphoribosyltransferase. Bioorg. Med. Chem 2013, 21, 5629–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Roy S; Nagappa LK; Prahladarao VS; Balaram H Kinetic mechanism of Plasmodium falciparum hypoxanthine-guanine-xanthine phosphoribosyltransferase. Mol. Biochem. Parasitol 2015, 204, 111–120. [DOI] [PubMed] [Google Scholar]
- (26).Roy S; Karmakar T; Prahlada Rao VS; Nagappa LK; Balasubramanian S; Balaram H Slow ligand-induced conformational switch increases the catalytic rate in Plasmodium falciparum hypoxanthine guanine xanthine phosphoribosyltransferase. Mol. Biosyst 2015, 11, 1410–1424. [DOI] [PubMed] [Google Scholar]
- (27).Henderson JF; Brox LW; Kelley WN; Rosenbloom FM; Seegmiller JE Kinetic studies of hypoxanthine-guanine phosphoribosyltransferase. J. Biol. Chem 1968, 243, 2514–2522. [PubMed] [Google Scholar]
- (28).Krenitsky TA; Papaioannou R Human hypoxanthine phosphoribosyltransferase. II. Kinetics and chemical modification. J. Biol. Chem 1969, 244, 1271–1277. [PubMed] [Google Scholar]
- (29).Xu Y; Eads J; Sacchettini JC; Grubmeyer C Kinetic mechanism of human hypoxanthine-guanine phosphoribosyltransferase: rapid phosphoribosyl transfer chemistry. Biochemistry 1997, 36, 3700–3712. [DOI] [PubMed] [Google Scholar]
- (30).Rose IA The isotope trapping method: desorption rates of productive E.S complexes. Methods Enzymol. 1980, 64, 47–59. [DOI] [PubMed] [Google Scholar]
- (31).Rose IA; O’Connell EL; Litwin S; Tana JB Determination of the rate of hexokinase-glucose dissociation by the isotope-trapping method. J. Biol. Chem 1974, 249, 5163–5168. [PubMed] [Google Scholar]
- (32).Ducati RG; Firestone RS; Schramm VL Kinetic Isotope Effects and Transition State Structure for Hypoxanthine-Guanine-Xanthine Phosphoribosyltransferase from Plasmodium falciparum. Biochemistry 2017, 56, 6368–6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tao W; Grubmeyer C; Blanchard JS Transition state structure of Salmonella typhimurium orotate phosphoribosyltransferase. Biochemistry 1996, 35, 14–21. [DOI] [PubMed] [Google Scholar]
- (34).Schramm VL Enzymatic transition states and transition state analogues. Curr. Opin. Struct. Biol 2005, 15, 604–613. [DOI] [PubMed] [Google Scholar]
- (35).Roy S; Karmakar T; Nagappa LK; Prahlada Rao VS; Balasubramanian S; Balaram H Role of W181 in modulating kinetic properties of Plasmodium falciparum hypoxanthine guanine xanthine phosphoribosyltransferase. Proteins 2016, 84, 1658–1669. [DOI] [PubMed] [Google Scholar]
- (36).Karmakar T; Roy S; Balaram H; Prakash MK; Balasubramanian S Product Release Pathways in Human and Plasmodium falciparum Phosphoribosyltransferase. J. Chem. Inf. Model 2016, 56, 1528–1538. [DOI] [PubMed] [Google Scholar]
- (37).Chen CY; Sato Y; Schramm VL Isotope trapping and positional isotope exchange with rat and chicken liver phosphoenolpyruvate carboxykinases. Biochemistry 1991, 30, 4143–4151. [DOI] [PubMed] [Google Scholar]
- (38).Williams L; Zheng R; Blanchard JS; Raushel FM Positional isotope exchange analysis of the pantothenate synthetase reaction. Biochemistry 2003, 42, 5108–5113. [DOI] [PubMed] [Google Scholar]
- (39).Cho YK; Matsunaga TO; Kenyon GL; Bertagnolli BL; Cook PF Isotope Exchange as a Probe of the Kinetic Mechanism of Pyrophosphate-Dependent Phosphofructokinase. Biochemistry 1988, 27, 3320–3325. [DOI] [PubMed] [Google Scholar]
- (40).Herschlag D; Jencks WP The Effect of Divalent Metal Ions on the Rate and Transition-State Structure of Phosphoryl-Transfer Reactions. J. Am. Chem. Soc 1987, 109, 4665–4674. [Google Scholar]
- (41).Morrison JF; Walsh CT The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol 1988, 61, 201–301. [DOI] [PubMed] [Google Scholar]
- (42).Shi W; Li CM; Tyler PC; Furneaux RH; Cahill SM; Girvin ME; Grubmeyer C; Schramm VL; Almo SC The 2.0 A structure of malarial purine phosphoribosyltransferase in complex with a transition-state analogue inhibitor. Biochemistry 1999, 38, 9872–9880. [DOI] [PubMed] [Google Scholar]
- (43).Murshudov GN; Skubak P; Lebedev AA; Pannu NS; Steiner RA; Nicholls RA; Winn MD; Long F; Vagin AA REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Schramm VL Enzymatic transition states: thermodynamics, dynamics and analogue design. Arch. Biochem. Biophys 2005, 433, 13–26. [DOI] [PubMed] [Google Scholar]
- (46).Glockzin K; Kostomiris D; Minnow YVT; Suthagar K; Clinch K; Gai S; Buckler JN; Schramm VL; Tyler PC; Meek TD; Katzfuss A Kinetic Characterization and Inhibition of Trypanosoma cruzi Hypoxanthine–Guanine Phosphoribosyltransferases. Biochemistry 2022, 61, 2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Liebschner D; Afonine PV; Baker ML; Bunkoczi G; Chen VB; Croll TI; Hintze B; Hung LW; Jain S; McCoy AJ; Moriarty NW; Oeffner RD; Poon BK; Prisant MG; Read RJ; Richardson JS; Richardson DC; Sammito MD; Sobolev OV; Stockwell DH; Terwilliger TC; Urzhumtsev AG; Videau LL; Williams CJ; Adams PD Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr., Sect. D: Struct. Biol 2019, 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ramachandran GN; Ramakrishnan C; Sasisekharan V Stereochemistry of polypeptide chain configurations. J. Mol. Biol 1963, 7, 95–99. [DOI] [PubMed] [Google Scholar]
- (52).DeLano WL The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.