

Abstract
BACKGROUND & AIMS:
Intestinal fibrosis is a significant complication of Crohn’s disease (CD). Gut microbiota reactive Th17 cells are crucial in the pathogenesis of CD; however, how Th17 cells induce intestinal fibrosis is still not completely understood.
METHODS:
In this study, T cell transfer model with wild-type (WT) and Areg−/− Th17 cells and dextran sulfate sodium (DSS)-induced chronic colitis model in WT and Areg−/− mice were used. CD4+ T cell expression of Areg was determined by qRT-PCR and ELISA. The effect of Areg on proliferation/migration/collagen expression in human intestinal myofibroblasts was determined. Areg expression was assessed in healthy controls and CD patients with or without intestinal fibrosis.
RESULTS:
Although Th1 and Th17 cells induced intestinal inflammation at similar levels when transferred into Tcrβxδ−/− mice, Th17 cells induced more severe intestinal fibrosis. Th17 cells expressed higher levels of Areg than Th1 cells. Areg−/− mice developed less severe intestinal fibrosis compared with WT mice upon DSS insults.Transfer of Areg−/− Th17 cells induced less severe fibrosis in Tcrβxδ−/− mice compared with WT Th17 cells. IL-6 and IL-21 promoted Areg expression in Th17 cells by activating Stat3. Stat3 inhibitor suppressed Th17-induced intestinal fibrosis. Areg promoted human intestinal myofibroblast proliferation, motility, and collagen I expression, which was mediated by activating mTOR and MEK. Areg expression was increased in intestinal CD4+ T cells in fibrotic sites compared with non-fibrotic sites from CD patients.
CONCLUSIONS:
These findings reveal that Th17-derived Areg promotes intestinal fibrotic responses in experimental colitis and human CD patients. Thereby, Areg might serve as a potential therapeutic target for fibrosis in CD.
Keywords: Effector CD4+ T cells, Inflammatory bowel diseases, Intestinal inflammation, intestinal myofibroblasts
Graphical Abstract
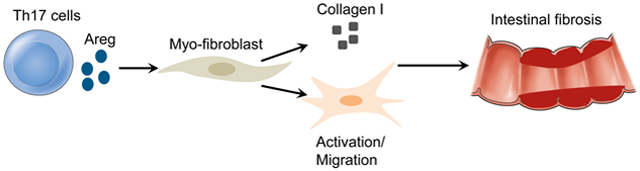
Lay Summary
Th17 cells induce more severe intestinal fibrosis through the production of Areg, which promotes human intestinal myofibroblast proliferation and migration.
Introduction
Crohn’s disease (CD) is a chronic intestinal inflammatory disease[1], and intestinal fibrosis is a significant complication of CD[2, 3]. Increased numbers of intestinal myofibroblasts (MFs) that are classically believed to have a mesenchymal origin and excessive amount of the extracellular matrix (ECM) proteins are hallmarks of intestinal fibrosis[4]. There are currently no effective drugs with anti-fibrotic mechanisms in treating CD patients with fibrosis. Endoscopic or surgical intervention remains the only long-term effective treatment option for CD patients with fibrosis.
Accumulating evidence supports the role of inflammation in the induction of intestinal fibrosis in CD[5–7]. Inflammation is currently believed to be pivotal in the initiation and development of intestinal fibrosis by activating MFs and ECM production. An intestinal injury usually follows acute inflammation of the intestines. Thereafter, healing of the damaged tissue and recovery of intestinal structure and function occur under normal physiological conditions. Conversely, chronic inflammation in the intestine is characterized by recurrent damage and repair events, which lead to the initiation and progression of intestinal fibrosis. It has been shown that MFs play crucial roles in tissue fibrosis, particularly for CD patients[8]. Immune responses, including innate and adaptive immune responses, which play a pivotal role in chronic intestinal inflammation, participate in transferring the normal healing process toward fibrogenesis. Innate and adaptive immune responses are involved in the activation of ECM-producing MFs and subsequently lead to the initiation and progression of intestinal fibrosis[9]. Gut microbiota antigen-specific T cells, especially Th1 and Th17 cells, have been implicated in the pathogenesis of CD[10–12]. An overlap between inflammation and fibrosis was observed in resection specimens of CD patients with fibrosis[2, 13]. In addition to the induction of intestinal inflammation, T cells have been implicated in the early stages of fibrosis. Mechanisms other than inflammation have also been reported in the regulation of intestinal fibrosis. It is often observed that intestinal fibrosis develops despite the removal of an inflammatory stimulus and elimination of inflammation, including through the use of anti-TNF therapy[14]. However, how T cells, especially Th17 cells, induce intestinal fibrosis remains unclear.
In this report, we investigated the cellular and molecular mechanisms that regulate Th17 cell induction of intestinal fibrosis. We found that although Th1 and Th17 cells induced intestinal inflammation at similar levels, Th17 cells induced more severe fibrosis when transferred into Tcrβxδ−/− mice. Th17 cells produced higher levels of amphiregulin (Areg) which is crucial in the induction of intestinal fibrosis. Importantly, Areg expression was increased in CD patients with fibrosis compared with that in healthy controls and CD patients without fibrosis.
Methods and materials
Mice
Specific pathogen-free C57BL/6 WT mice and Tcrβx δ−/− mice were purchased from the Jackson Laboratory and maintained in the animal facilities of the University of Texas Medical Branch (UTMB). CBir1 TCR transgenic (Tg) mice were bred in the animal facilities of UTMB. Areg−/− mice were provided by Dr. Jingwu Xie from Indiana University School of Medicine, which are originally on the B6.129 background and have been backcrossed with C57BL/6 for ten generations. Stat3−/− mice were provided by Dr. Defu Zeng from The Beckman Research Institute of City of Hope. Both male and female mice were used. All experiments were reviewed and approved by the Institutional Animal Care and Use Committees of UTMB.
Human tissue and cells
CD patients with or without fibrosis were recruited at the Gastroenterology Department of the First Affiliated Hospital of Nanjing Medical University. CD was diagnosed based on clinical, endoscopic, and histologic criteria. Mucosal biopsies from CD patients with intestinal fibrosis were obtained from stenotic regions and adjacent non-stenotic regions of the ileum. The presence of intestinal stenosis was confirmed by a radiologist and an endoscopist. The area of stenosis was identified by the pathologist by macroscopic evaluation and was confirmed by histology. Intestinal biopsies and peripheral blood were collected from CD patients with fibrosis, CD patients without fibrosis, and healthy controls. The basic information for CD patients and healthy controls is illustrated in Supplementary Table 1.
Intestinal lamina propria cells and MFs were isolated from full thickness tissue specimens from CD patients obtained from the Department of Surgery at Penn State Health Medical Center The purity of isolated CD90+ MFs (98–99%) was confirmed by flow cytometry. Studies were performed with primary MF isolates in passages 3–9[15]. MFs were cultured at 37°C in 5% CO2 atmosphere in complete MEM media with 10% FBS (Sigma-Aldrich). Tissues were from the PennState Inflammatory Bowel and Colorectal Diseases Biobank and procured in compliance with the protocol approved by the College of Medicine at Penn State Health Medical Center Institutional Review Board (#HY98-057EP-A).
Statistical analysis
Graphpad Prism 9.0 (GraphPad software, USA) was used for statistical analysis. Data are shown as the mean ± standard error of the mean (SEM). Paired/unpaired student’s t-test or one-way analysis of variance (ANOVA) were used for the analysis of differences between two groups or more than two groups. Mann–Whitney U test was used for the analysis of pathological scores. P value less than 0.05 was considered statistically significant.
Results
1. Th17 cells induce more severe intestinal fibrosis in Tcrβxδ−/− mice
Gut microbiota-specific Th1 and Th17 cells have been implicated in the pathogenesis of inflammatory bowel diseases. To investigate the roles of Th1 and Th17 cells in the induction of intestinal fibrosis, we used a gut microbiota antigen-specific T cell transfer colitis model. We cultured CD4+T cells isolated from CBir1 TCR transgenic (CBir1 Tg) mice, which are specific for an immunodominant microbiota antigen CBir1 flagellin[16], under Th1 and Th17-polarization conditions and transferred them into Tcrβxδ−/− mice. The mice were sacrificed 6 weeks after cell transfer, and disease severity was measured by histopathology. Tcrβxδ−/− mice transferred with Th17 cells developed colitis at levels similar to that in the mice that received Th1 cells, as evidenced by histopathology and pathological scores (Figure 1A). Meanwhile, we determined cytokine production using ELISA in the supernatants of colonic organ cultures and found that there were no differences in TNF-α and IL-6 production between the mice reconstituted with Th1 and Th17 cells (Figures 1B–C).
Figure 1. Th17 cells induce more severe intestinal fibrosis in Tcrβxδ−/− mice.
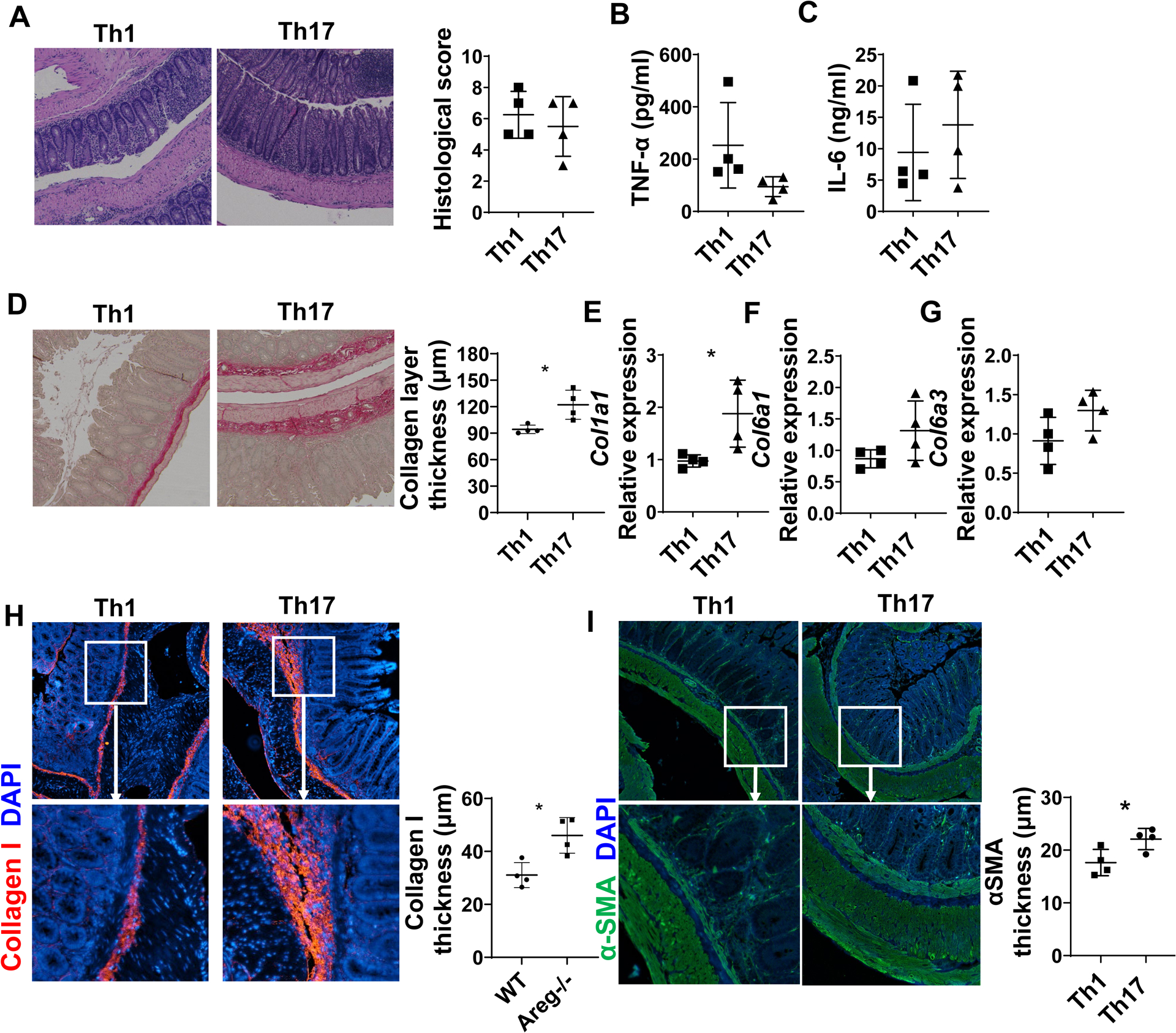
CD4+T cells were isolated from CBir1 TCR transgenic (CBir1 Tg) mice and cultured under Th1 and Th17 conditions and then transferred into Tcrβxδ−/− mice (n=4/group). The mice were sacrificed 6 weeks after cell transfer. (A) Disease severity was measured by histopathology. (B-C) TNF-α (B) and IL-6 (C) levels in supernatants of colonic organ cultures were measured by ELISA. (D) Colon tissues were stained with Sirius Red, and collagen layer thickness were measured. (E-G) Col1a1, Col6a1, and Col6a3 levels in colonic tissues were measured by RT-PCR. (H-I) Colon tissues were stained with immunofluorescence. Collagen I thickness and αSMA layer thickness were analyzed. Representative data from 3 independent experiments. (A) Mann–Whitney U test; (B-I) unpaired Student’s t-test. *p < .05.
The increase of collagen deposition, the most abundant fibrous protein in the ECM, is one of the characteristics of intestinal fibrosis. To determine whether Th1 and Th17 cells induce intestinal fibrosis differentially, we measured the collagen levels in the intestine by Sirius Red staining which bind to all type of collagen. We found that Th17 cells induced more collagen deposition than Th1 cells in Tcrβxδ−/− mice (Figure 1D). Because increase deposition of several collagens, including type I and VI collagen was associated with fibrosis in CD[17], we analyzed expression of genes involved in these collagen synthesis. We observed that, both mRNA and protein levels of collagen type I were increased in the colon of mice receiving Th17 cells (Figures 1E and H). We also found that collagen type VI, col6a1, and col6a3, showed an increased tendency in Tcrβxδ−/− recipients of Th17 cells (Figures 1F and G). Furthermore, αSMA, a marker for activated MFs and smooth muscle cells involved in fibrosis[18], was increased in the mucosal lamina propria and its adjacent smooth muscle layer in the colon of Tcrβxδ−/− mice reconstituted with Th17 cells compared with mice reconstituted with Th1 cells (Figure 1I), suggesting that Th17 cells induce MFs activation. However, there were no differences in Il-36a, Il36g, and Il1rl2 expression in the colon (Supplementary figures 1A–C), which has been reported to be involved in the intestinal fibrosis[19]. Overall, these data indicated although gut microbiota-specific Th1 and Th17 cells induce similar levels of intestinal inflammation, Th17 cells induce more severe intestinal fibrosis than Th1 cells.
To confirm the difference in intestinal fibrosis induced by Th1 and Th17 cells, we also used WT Th1 and Th17 cells to induce colitis in Tcrβxδ−/− mice. Similar to CBir1-specific Th17 cells, polyclonal Th17 cells induced an increase in both collagen deposition and α-SMA expression, and showed more severe intestinal fibrosis than polyclonal Th1 cells (Supplementary figures 2A–C).
2. Th17 cells produce high levels of Areg
To explore the genes that drive Th17 cell induction of more severe intestinal fibrosis, we analyzed gene expressions in Th1 and Th17 cells using a microarray. CD4+T cells were cultured under neutral (Th0), Th1, and Th17-polarization conditions for microarray analysis. Many genes were differentially expressed between Th1 and Th17 cells, including their signature cytokines and transcription factors, i.e., Il17a and Rorc in Th17 cells and Ifng and Tbx21 in Th1 cells (Figures 2A and B). In addition to pro-inflammatory cytokines and transcription factors, Areg, a member of the epidermal growth factor (EGF) family and has been found to induce airway fibrosis[20], was 2.35 times higher in Th17 cells than Th1 cells (Figure 2B) and 2.06 times higher in Th17 cells than Th0 cells (Supplementary figure 3A). There was no difference in Areg expression between Th0 and Th1 cells (Supplementary figure 3B).
Figure 2. Th17 cells produce high levels of Areg.
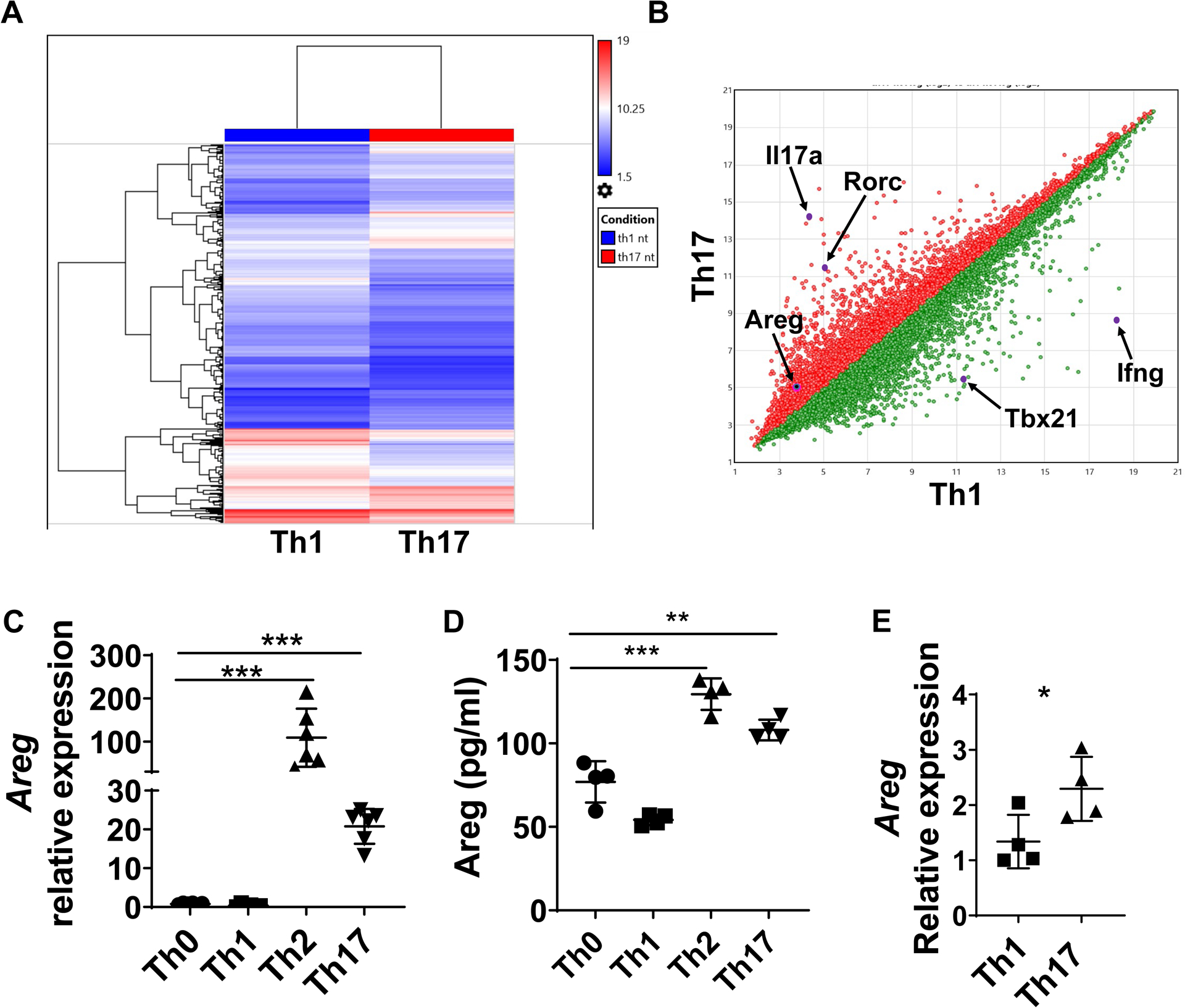
(A-B) WT spleen CD4+T cells were activated with α-CD3 and α-CD28 mAb under Th1 and Th17-polarization conditions for 24h for analysis of gene differences by microarray analysis. (A) The expression of different genes is shown in the heatmap. (B) Scatterplot displaying the log2 fold change in expression between the two groups. (C-D) Splenic CD4+T cells of CBir1 Tg mice were cultured with irradiated APCs and CBir1 peptide under neutral, Th1, Th2, and Th17 polarization conditions for 5 days. Areg expression was detected by qRT-PCR (C). Areg production was determined by ELISA (D). (E) Intestinal Areg expression in CBir1 Tg Th1-recipient mice and Th17-recipient mice was determined by qRT-PCR. Representative data from 3 independent experiments. (C-D) One-way ANOVA; (E) unpaired Student’s t-test. *p < .05; **p < .01; ***p < .001; ****p < .0001.
Areg expression has been detected in various cells, including epithelial cells, T cells, dendritic cells, MFs, and keratinocytes[20–24]. It has been shown that Areg is highly expressed in Th2 cells and absent in Th1 cells[23], which could contribute to Th2 cell induction of airway fibrosis[20]. However, Areg expression in Th17 cells is still unclear We next sought to verify Areg expression in Th17 cells. CBir1 CD4+T cells were cultured under Th0, Th1, Th2, and Th17 conditions, and Areg expression was determined by qRT-PCR and ELISA. Consistent with a previous report[23], Th2 cells highly expressed Areg, whereas Th1 and Th0 cells expressed low levels of Areg at both mRNA and protein levels (Figures 2C and D). Interestingly, Th17 cells highly expressed Areg at both mRNA and protein levels (Figures 2C and D). We also measured Areg expression in the intestines of colitic Tcrβxδ−/− mice received Th1 and Th17 cells and found that Areg expression was higher in Th17-recipient mice than in Th1-recipient mice (Figure 2E).
As Th17 cells produce high levels of Areg, we then investigated whether CD4+T cells can also respond to Areg to regulate Th17 cell differentiation. The expression of EGFR was detectable on different subsets of CD4+T cells (Supplementary figure 4A). Notably, Th1, Th2, and Th17 cells expressed higher levels of EGFR than Th0 cells. Areg induced phosphorylation of EGFR in T cells (Supplementary figure 4B). Next, we cultured WT and Areg−/− CD4+T cells under Th17 conditions. There was no difference in IL-17+ cells between WT and Areg−/− CD4+T cells (Supplementary figure 4C). Furthermore, Areg treatment did not affect T cell differentiation and proliferation (Supplementary figures 4D–E). These data indicated that Areg does not affect T cell differentiation and proliferation.
3. Areg−/− Th17 cells induce less severe intestinal fibrosis
To investigate the role of Areg in intestinal fibrosis, we induced intestinal fibrosis in WT and Areg−/− mice using a chronic DSS-induced colitis model. We treated WT and Areg−/− mice with DSS in the drinking water for 7 days, followed by an additional 7 days with drinking water alone, and repeated 2 times for a total of 3 cycles of DSS treatment. When mice were sacrificed, we found that Areg−/− mice developed more severe colitis compared with WT mice (Figure 3A). However, intestinal fibrosis was less severe in Areg−/− mice, in that although the collagen genes expressions, including Col1a1, Col6a1, and Col6a3 (Figures 3B–D), were not significantly changed, collagen layer thickness (Figure 3E) and collagen I thickness (Figure 3F) were decreased in the colon of Areg−/− mice, indicating that deficiency of Areg suppressed collagen deposition in the colon. In addition, αSMA was decreased in Areg−/− mice. These data suggest that Areg promotes intestinal fibrosis.
Figure 3. The deficiency of Areg induces less severe intestinal fibrosis.
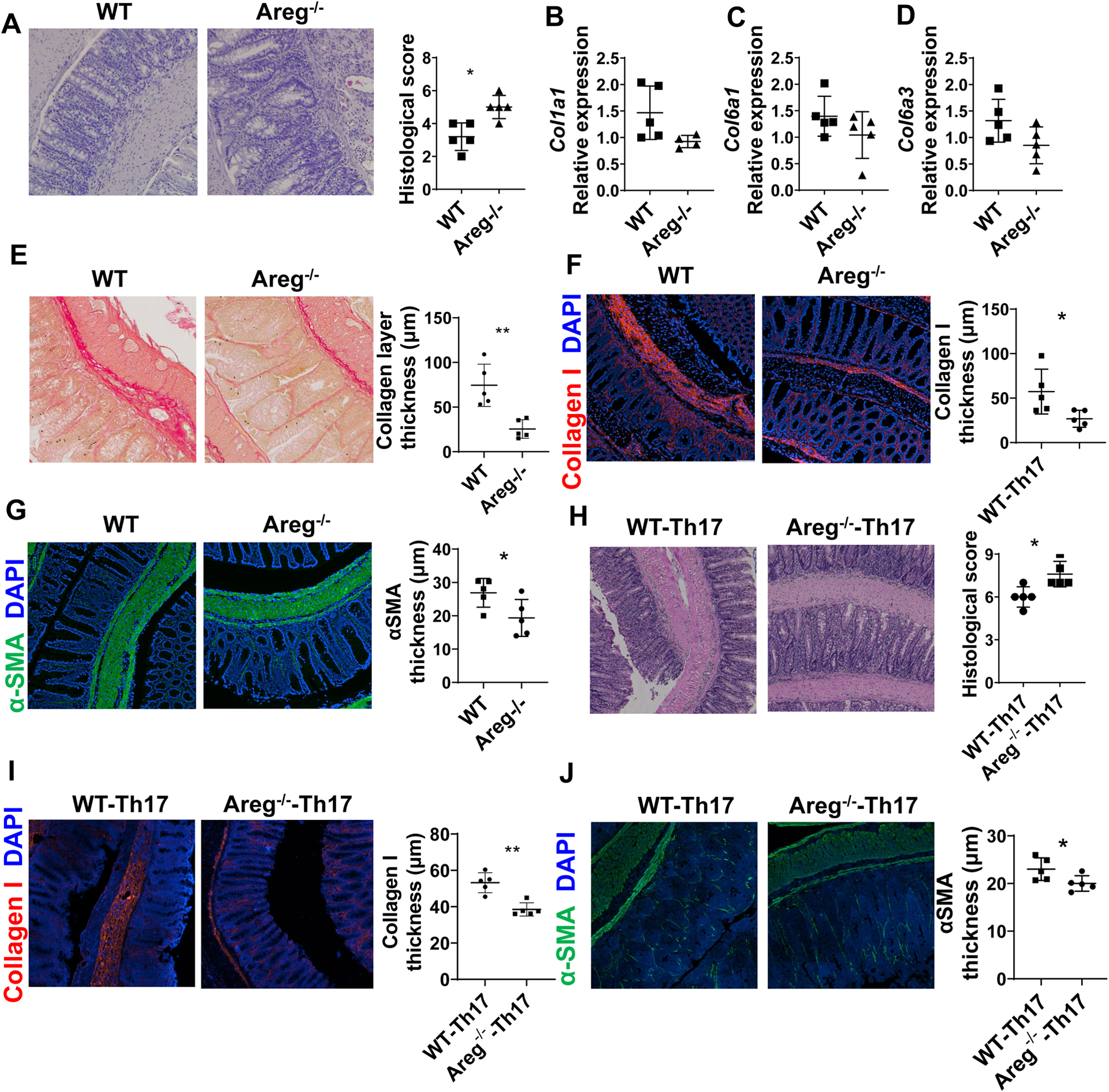
(A-G) WT mice and Areg−/− mice (n=5/group) were administered with 3 cycles of DSS insults. In each cycle, mice were given 2.0% DSS (w/v) in drinking water for 7 days and control drinking water for 7 days. (A) Colitis severity was measured by histopathology and pathological scores. (B-D) Col1a1, Col6a1, and Col6a3 levels in colonic tissues were measured by RT-PCR. (E-G) Colon tissues were stained with (E) Sirius Red and (F-G) immunofluorescence. Collagen layer thickness, Collagen I thickness, and αSMA layer thickness were analyzed. (H-J) WT and Areg−/− T cells were cultured under Th17 conditions for 5 days and then transferred into Tcrβxδ−/− mice (n=5/group). The mice were sacrificed 4 weeks later. (H) Colitis severity was measured by histopathology and pathological scores. (I-J) Intestinal fibrosis was determined by Collagen I staining (I) and αSMA staining (J). Representative data from 2 independent experiments with similar results. (A and H) Mann–Whitney U test; (B-G and I-J) unpaired Student’s t-test. *p < .05; **p < .01.
We then determined the role of Areg in Th17 cell induction of intestinal fibrosis. We transferred WT and Areg−/− Th17 cells into Tcrβxδ−/− mice. The mice were sacrificed 4 weeks after cell transfer, and disease severity and the levels of fibrosis were determined. The recipient mice that received Areg−/− Th17 cells showed more severe colitis than those receiving WT Th17 cells (Figure 3H). Interestingly, Areg−/− Th17 cells induced less severe fibrosis in Tcrβxδ−/− mice than WT Th17 cells, as evidenced by decreased collagen I deposition and αSMA thickness in the colon (Figures 3I–J). These data indicate that Areg mediates Th17 cell induction of intestinal fibrosis in an inflammation-independent manner.
4. IL-6 and IL-21 promote Areg expression through activation of Stat3 in Th17 cells.
We then investigated the mechanisms driving the Th17 cell expression of Areg. RORγt has been identified as the major transcription factor driving Th17 cell differentiation[25]. To determine whether high levels of Areg expression require the development of Th17 cells, we cultured CD4+T cells under Th17 conditions in the presence or absence of the RORγt inhibitor, GSK805. As expected, the addition of the RORγt inhibitor suppressed Th17 cell differentiation (Supplementary figure 5). Interestingly, the RORγt inhibitor inhibited Areg expression (Figure 4A), verifying that Th17 polarization promotes Areg expression in T cells.
Figure 4. IL-6 and IL-21 promote Areg expression through activation of Stat3 in CD4+ T cells.
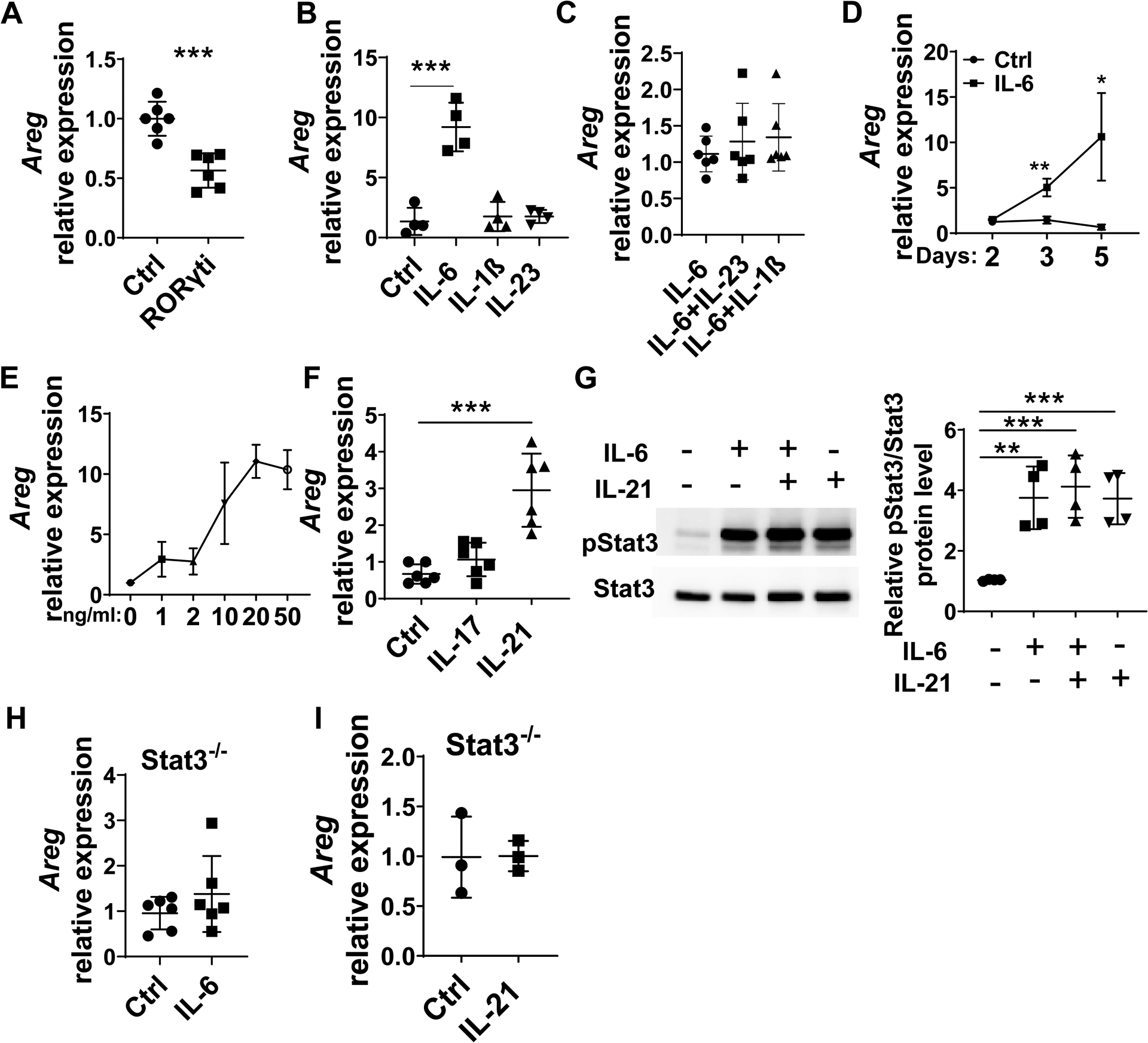
(A) CD4+T cells were cultured under Th17-polarization conditions with or without the RORγt inhibitor. Areg expression was detected by qRT-PCR. (B) CD4+T cells were cultured with or without 10 ng/ml IL-6, 10 ng/ml IL-23, or 10 ng/ml IL-1β for 5 days. Areg expression was detected by qRT-PCR. (C) CD4+T cells were cultured with IL-6 alone or together with IL-23 and IL-1β for 5 days. Areg expression was detected by RT-PCR. (D) CD4+T cells were cultured with 10 ng/ml IL-6 for indicated days. Areg expression was detected by qRT-PCR. (E) CD4+T cells were cultured with IL-6 at indicated dose for 5 days. Areg expression was detected by qRT-PCR. (F) CD4+T cells were cultured in the presence of IL-17 or IL-21 for 5 days. Areg expression was detected by qRT-PCR. (G) CD4+T cells were cultured with or without IL-6 and IL-21 for 5 minutes. Phosphorylated Stat3 and total Stat3 were detected by western blot. (H-I) CD4+T cells were isolated from Stat3−/− mice and treated with IL-6 or IL-21 for 5 days. Areg expression was detected by qRT-PCR. Representative data from 2–3 independent experiments with similar results. (A, D, H, and I) Unpaired Student’s t-test; (B, C, F, and G) one-way ANOVA. *p < .05; **p < .01; ***p < .001.
IL-6, IL-23, and IL-1β are the critical cytokines for Th17 development in addition to TGFβ. To determine whether these cytokines regulate T cell production of Areg, we cultured CD4+T cells in the presence or absence of the IL-6, IL-23, or IL-1β for 5 days. IL-6, but not IL-23 or IL-1β, induced Areg expression (Figure 4B). In addition, neither IL-23 nor IL-1β facilitated IL-6 induction of Areg expression in T cells (Figure 4C). Furthermore, IL-6 induced T cell Areg expression in a time and dose-dependent manner (Figures 4D and E).
To explore whether Th17 signature cytokines, IL-17 and IL-21, mediate the production of Areg in T cells, we cultured CD4+T cells with or without IL-17 and IL-21. Treatment with IL-21, but not IL-17, upregulated Areg expression (Figure 4F).
We then investigated the underlying mechanisms by which IL-6 and IL-21 induce Th17 cell expression of Areg. It has been shown both IL-6 and IL-21 activate Stat3, which is necessary for IL-6 induction of Th17 cells and IL-21 amplification of Th17 differentiation[26]. To determine whether IL-6 and IL-21 induce Areg production in Th17 cells through activation of Stat3, we cultured CD4+T cells with IL-6 or/and IL-21. IL-6 and IL-21 treatment activated Stat3 (Figure 4G). Furthermore, IL-6 or IL-21 did not induce Areg expression in Stat3−/− T cells (Figures 4H–I). Together, these data indicated that Stat3 is critical for IL-6 and IL-21 induction of Areg in Th17 cells.
5. Blockade of Stat3 pathway suppresses intestinal fibrosis.
Given that Stat3 is critical in regulating Th17 expression of Areg, we then investigate whether Stat3 mediates Th17 induction of intestinal fibrosis. To this end, we transferred Th17 cells into Tcrβxδ−/− mice and treated the mice with or without HJC0152[27], a Stat3 inhibitor, every other day. When mice were sacrificed 4 weeks after cell transfer, we found that treatment with Stat3 inhibitor suppressed intestinal inflammation (Figure 5A) and inhibited intestinal TNFα but not IL-6 production (Figures 5B–C). In addition, treatment with Stat3 inhibitor suppressed intestinal fibrosis, as evidenced by a thinner collagen layer (Figure 5D), decreased expression of Co1a1 and Col6a1 (Figures 5E–F), and decreased expression of Collagen I and αSMA in the colon (Figures 5G–H), indicating a role of Stat3 in Th17 induction of intestinal fibrosis.
Figure 5. Stat3 inhibitor attenuates intestinal fibrosis induced by Th17 cells.
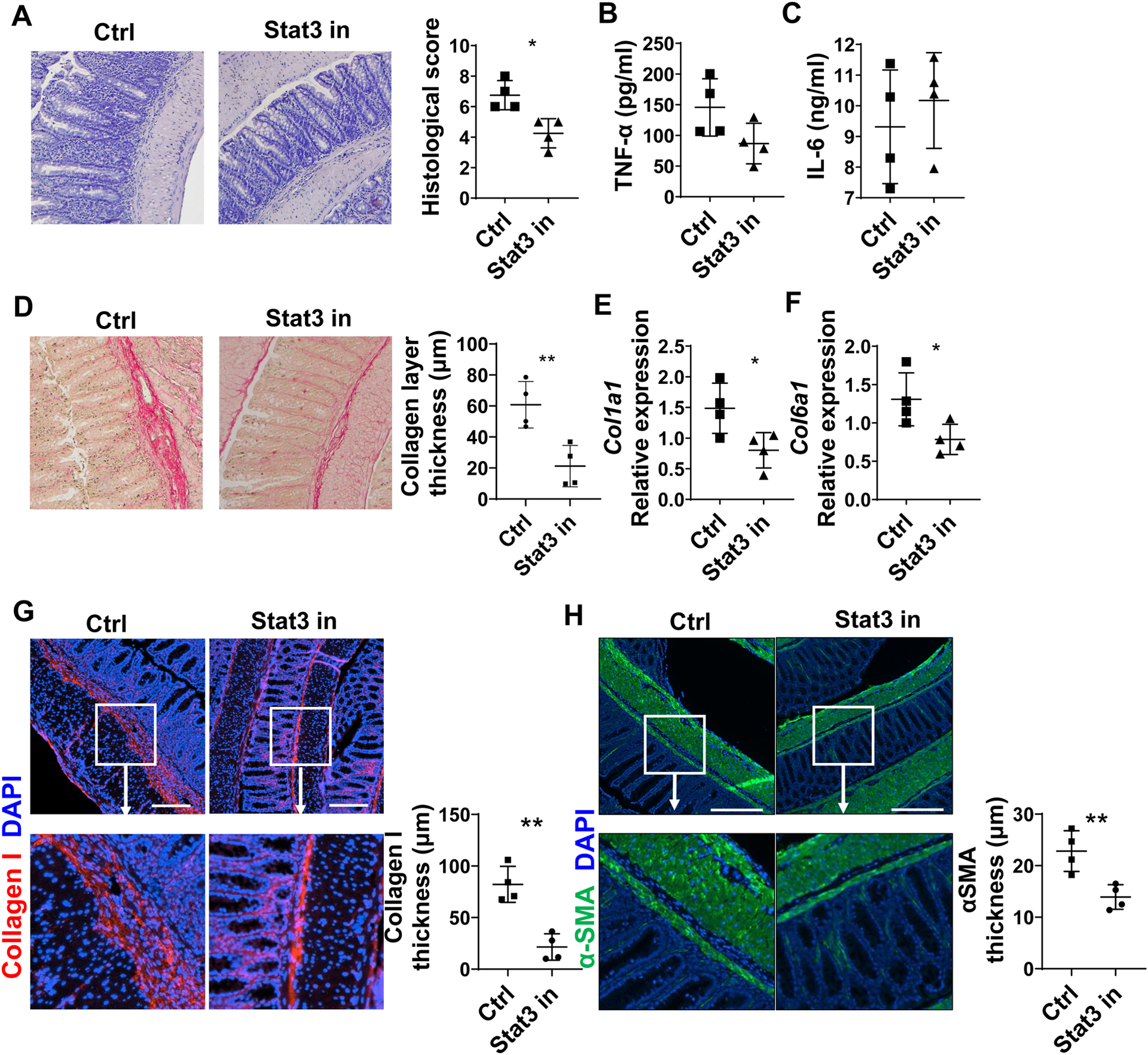
CBir1 Th17 cell were transferred into Tcrβxδ−/− mice (n=4/group). Mice were injected with or without Stat3 inhibitor (10 mg/kg, HJC0152) every other day starting from the day of cell transfer. (A) Disease severity was measured by histopathology and pathological scores. (B) TNF-α and (C) IL-6 levels in supernatants of colonic organ cultures were measured by ELISA. (D) Colon tissues were stained with Sirius Red, and Collagen layer thickness were analyzed. (E-F) Col1a1 and Col6a1 levels in colonic tissues were measured by RT-PCR. (G-H) Colon tissues were stained with immunofluorescence. Collagen I thickness (G) and αSMA layer thickness (H) were measured. Representative data from 2 independent experiments with similar results. (A) Mann–Whitney U test; (B-H) unpaired Student’s t-test. *p < .05; **p < .01.
6. Areg promotes human intestinal myofibroblast proliferation and motility through activation of mTOR
Intestinal MF proliferation and migration are two critical events in the development of intestinal fibrosis. To determine whether Areg affects fibroproliferative responses in intestinal fibrosis, we investigated the direct effects of Areg on intestinal MFs. Human intestinal MFs were treated with Areg and stained with Ki67, a marker specifically detecting cycling cells. The percentage of Ki67+ MFs in the Areg treatment group was higher than that in the control group (Figure 6A), indicating that Areg treatment promotes the proliferation of human intestinal MFs. We then investigated whether Areg affects human intestinal MF motility using a scratch wound-healing assay. The residual wound gap was measured after human intestinal MF monolayers were scratch wounded. Areg treatment enhanced the rate and extent of wound gap reduction relative to untreated control cells (Figure 6B, Supplementary videos 1 and 2), indicating that Areg enhances intestinal MF motility. We also found that Areg treatment increased Col1a1 expression in MFs (Figure 6C).
Figure 6. Areg promotes human intestinal myofibroblast proliferation and motility through activation of mTOR and MEK.
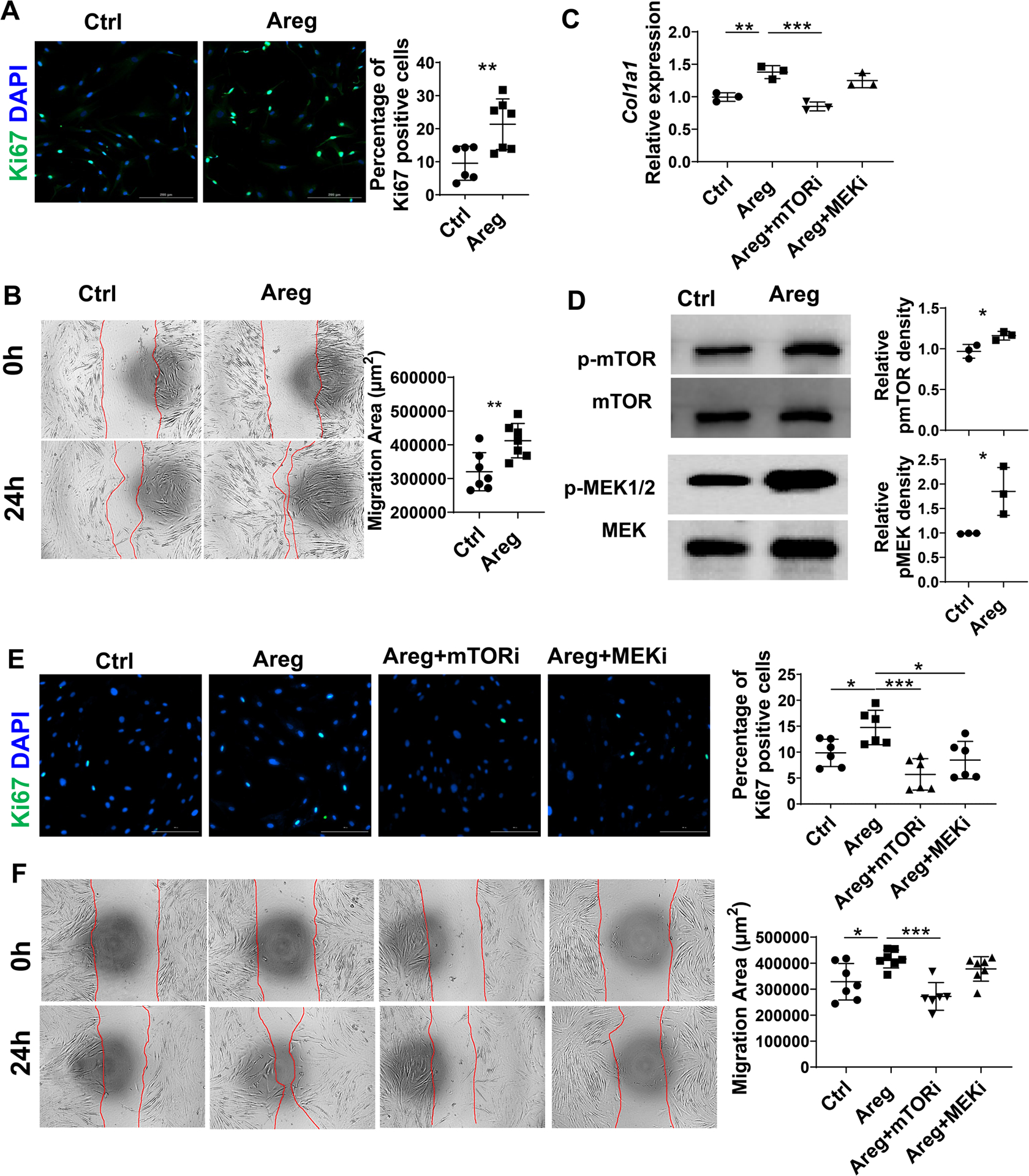
(A) Intestinal MFs isolated from CD patients were treated with or without 100 ng/ml Areg for 48h, and Ki67+ cells were analyzed by immunofluorescence. (B) Human intestinal MFs were wounded and treated with the 100 ng/ml Areg. Images were recorded with Biotek Cytation 5. (C) Human intestinal MFs were treated with or without the 100 ng/ml Areg in the presence or absence of Rapamycin or U0126 for 48h, and Col1a1 expression was determined by qRT-PCR. (D) Intestinal MFs were treated with Areg for 48h, and phosphorylation of mTOR and MEK was determined by western blot. (E) Human intestinal MFs were treated with or without 100 ng/ml Areg in the presence or absence of Rapamycin (mTOR inhibitor) or U0126 (MEK inhibitor) for 48h, and Ki67+ cells were analyzed by immunofluorescence. (F) Human intestinal MFs were wounded and treated with or without the 100 ng/ml Areg in the presence or absence of Rapamycin or U0126, and images were recorded by Biotek Cytation 5. Representative data from 2–3 independent experiments with similar results. (A-B, and D) Unpaired Student’s t-test; (C, and E-F) one-way ANOVA. *p < .05; **p < .01; ***p < .001; ****p < .0001.
Several signaling pathways have been shown to regulate cell proliferation and migration, including Stat3, mTOR, and MEK. We first asked whether Areg promotes activation of Stat3, mTOR, and MEK in intestinal MFs. Areg treatment enhanced activation of MEK and mTOR, but not Stat3 (Figure 6D and Supplementary figure 6A). To determine whether these pathways mediate Areg induction of intestinal MF proliferation, migration, and collagen, we added mTOR inhibitor (Rapamycin) or MEK inhibitor (U0126) in the intestinal MF culture with or without Areg. The addition of either mTOR inhibitor or MEK inhibitor suppressed Areg induction of proliferation (Figure 6E). In contrast, only mTOR inhibitor decreased the intestinal MF migration (Figure 6F and Supplementary videos 3–6) and Col1a1 expression (Figure 6C) induced by Areg. To determine whether the EGF pathway serves as a potential downstream signaling mediator, we measured EGFR phosphorylation in MFs treated with or without Areg. Areg promoted EGFR phosphorylation in MFs (Supplementary figure 6B). These data suggest that activation of mTOR and MEK differentially mediates Areg induction of human intestinal MF proliferation, migration, and collagen expression.
7. Areg expression is increased in CD patients with fibrosis
To determine whether Areg expression plays a role in intestinal fibrosis in human CD patients with intestinal fibrosis, we first collected intestinal tissues from healthy controls and CD patients with or without intestinal fibrosis, where the inflammation levels were similar (Figure 7A) and measured the Areg expression in intestinal tissues. Areg expression in intestinal biopsies of CD patients with fibrosis was higher than that in healthy controls and CD patients without intestinal fibrosis (Figure 7B), indicating that higher levels of Areg are associated with fibrosis in CD patients. We also determined the expression of Areg in non-fibrotic and fibrotic sites from the same patients. We found that Areg expression was higher in fibrotic sites than in non-fibrotic sites (Figure 7C).
Figure 7. Areg expression is increased in CD patients with fibrosis.
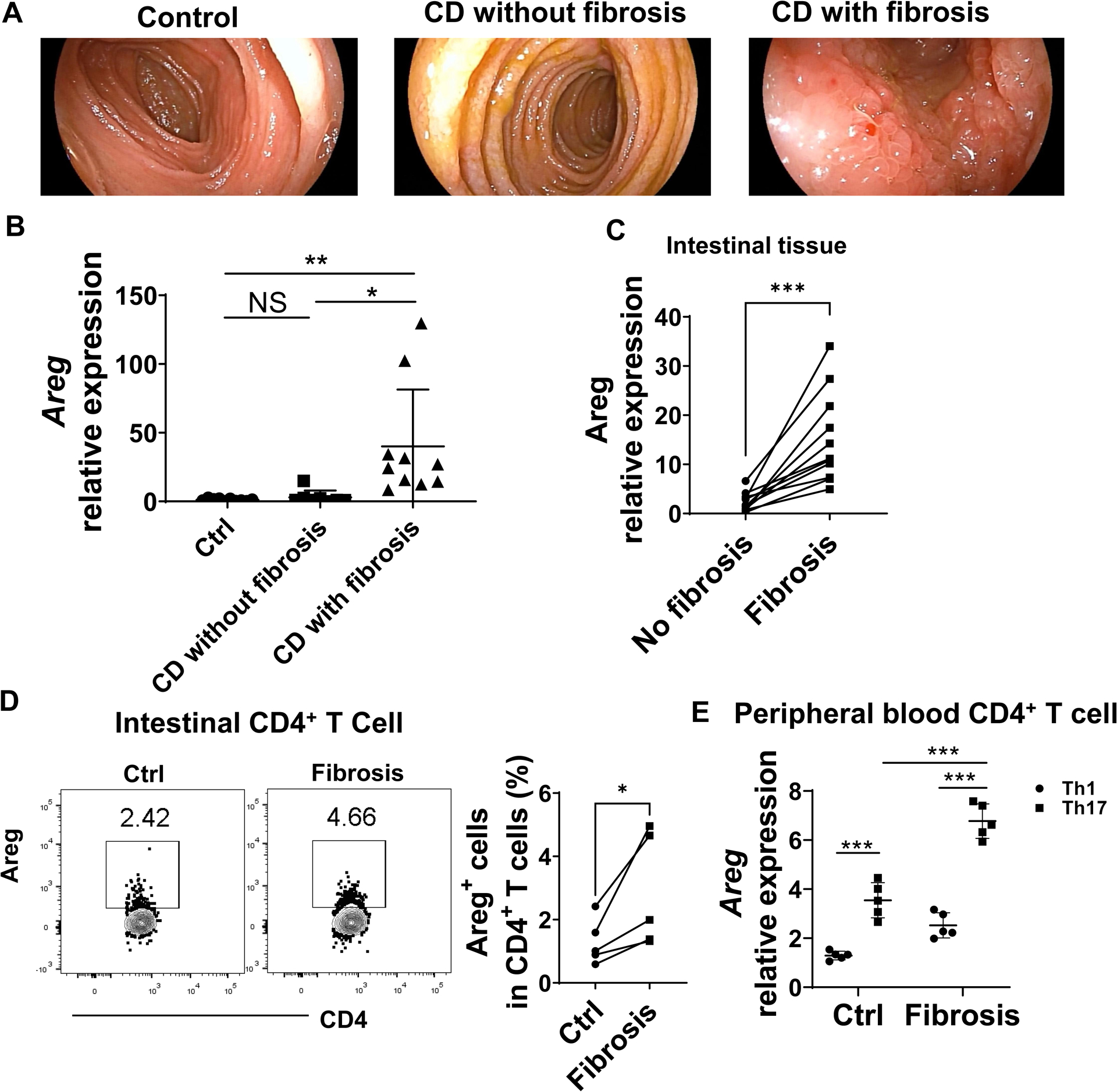
(A) Endoscopic images of the intestinal tract of healthy control, CD patients with or without intestinal fibrosis. (B) Intestinal biopsies were taken from healthy controls (Control, n=10), CD patients without fibrosis (n=8), and CD patients with fibrosis (n=10). The expression of Areg in intestinal biopsies was measured by qRT-PCR. (C) Areg expression in non-fibrotic (Ctrl) and fibrotic sites from the same CD patients (n=11) was determined by qRT-PCR. (D) Intestinal lamina propria lymphocytes were isolated from fibrotic and non-fibrotic sites of the same CD patients (n=5). Areg+ CD4+ T cells were determined by flow cytometry. (E) Peripheral blood CD4+ T cells were isolated from healthy controls (n=5) and CD patients with intestinal fibrosis (n=5) and then cultured under Th1 or Th17 conditions. Areg expression in T cells was measured by qRT-PCR. (B) One-way ANOVA; paired Student’s t-test(C-D); unpaired Student’s t-test (E). *p < .05; ***p < 0.001.
Next, we compared Areg expression in the intestine of healthy controls and CD patients with intestinal fibrosis using immunofluorescence staining, and found that Areg+CD4+T cells were increased in intestinal tissues from CD patients with intestinal fibrosis compared with those in healthy controls (Supplementary figure 7A). Then, we assessed the Areg expression in intestinal lamina propria cells in non-fibrotic and fibrotic sites from the same CD patients. CD4+ T cells showed increased expression of Areg in fibrotic sites compared with non-fibrotic sites (Figure 7D), while there was no difference in Areg expression in other cell types, including CD8+ T cells and B cells (Supplementary figure 7B). In addition, we found that both Th1 and Th17 cells were increased in fibrotic sites, while there was no difference in Th2 cells and Treg cells (Supplementary figure 7C).
To compare the Areg expression between human Th1 cells and Th17 cells, we isolated peripheral blood CD4+ T cells from healthy controls and CD patients with fibrosis and then cultured them under Th1 and Th17 conditions. We found that Th17 cells expressed a higher level of Areg than Th1 cells either from healthy controls or CD patients with fibrosis (Figure 7E). In addition, Areg expression was much higher in Th17 cells from CD patients with fibrosis than from healthy controls (Figure 7E).
Discussion
Both Th1 and Th17 cells have been implicated in the pathogenesis of fibrosis during intestinal inflammation[6, 28], which is mainly attributed to their proinflammatory cytokine production. In this report, we demonstrated that Th17 cells induced more severe intestinal fibrosis than Th1 cells, which was mediated by Th17 cell production of Areg. Areg expression was higher in CD4+T cells in CD patients with fibrosis, which promoted intestinal MF proliferation and motility.
Both Th1 and Th17 cells are crucial in the pathogenesis of IBD[12, 29]. Despite the progress made regarding T cells in intestinal inflammation, how they induce intestinal fibrosis is still poorly defined. One major argument on the role of T cells in driving intestinal fibrosis in CD is that Th1 and Th17 cells induce severe intestinal inflammation, which causes fibrosis[30]. However, we found that although gut microbiota-specific Th1 and Th17 cells induced similar levels of intestinal inflammation in our setting, Th17 cells induced more severe fibrosis than Th1 cells, indicating that although inflammation plays a critical role in driving fibrosis, other factors in addition to pro-inflammatory cytokines also contribute to the development of fibrosis in Th17 cell induction of fibrosis. IL-17 has been reported to be increased in the intestinal fibrosis in CD patients[31]. In this study, we found that Th17 cells expressed higher levels of Areg than Th1 cells. Interestingly, Areg−/− Th17 cells induced less severe fibrosis than WT Th17 cells, indicating that Th17 expression of Areg contributes to the development of intestinal fibrosis. However, Areg−/− Th17 cells produced similar levels of IL-17 compared with WT Th17 cells, indicating that Areg and IL-17 might contribute to intestinal fibrosis independently.
Areg is a member of the EGF family, and its receptor is EGFR[22, 24]. A variety of cells, including epithelial cells, leukocytes, dendritic cells, MFs, and keratinocytes, express Areg[20–24], which is critical in protection against intestinal inflammation[32]. ILC2-derived Areg protects against intestinal inflammation in a Dextran sulfate sodium (DSS) model[33]. Neutrophil-derived Areg is important in the maintenance of IEC barrier function and homeostasis[34]. It has been reported that various T cells, including Th2[23] and Treg cells[35], also highly express Areg. Areg promotes Treg cell development and enhances the function of Treg cells to maintain tissue homeostasis and improve muscle repair in acutely injured skeletal muscle[24, 28, 36]. Areg has also been associated with Th2 cell induction of airway fibrosis[20]. We demonstrated that Th17 cells express Areg at levels similar to Th2 cells, and Areg at least partially mediates Th17 cell induction of intestinal fibrosis. More importantly, the analysis of peripheral blood CD4+ T cells, intestinal biopsies, and LPL revealed increased expression of Areg in CD patients with fibrosis. Although both Th1 and Th17 cells were increased in fibrotic sites, there was no difference in Th2 cells and Treg cells in CD patients with or without fibrosis, indicating a crucial role of Th17 production of Areg in the induction of intestinal fibrosis in CD patients. However, our study does not exclude the possibility of other cells-derived Areg in mediating intestinal fibrosis. We thus provide novel insights into how Th17 cells induce intestinal fibrosis through the production of Areg, a potential target for the treatment of fibrosis in CD patients.
Our study demonstrated that Th17 polarization promotes Areg expression in that inhibition of RORγt, a transcription factor for Th17 cell development, inhibited Th17 cell production of Areg. Among cytokines promoting Th17 cell differentiation, IL-6 but not IL-23 or IL-1β promoted Th17 cell production of Areg, which was mediated by Stat3. Interestingly, IL-21, a Th17 cell signature cytokine that in turn promotes Th17 cell differentiation in a positive loop[26], also promoted Areg production by Th17 cells, indicating that the cytokines that drive Th17 cell differentiation can induce Areg production in Th17 cells by initiating Th17 cell differentiation (IL-6) or amplifying Th17 cells (IL-21).
Although Areg has been shown to regulate cell proliferation and migration of MFs through the activation of MAPK and mTOR pathways in the lung and skin[22, 37–39], the mechanisms involved in intestinal fibrosis are still not far more understood. In our study, we found that Areg treatment induced a significant increase in proliferation and migration of intestinal MFs, which was differentially mediated by activation of Mtor and MEK. A recent report found that mTOR activation was enhanced in a TNBS-induced colitis mouse model, and mTOR inhibitor rapamycin treatment ameliorated intestinal fibrosis[40], further supporting the role of mTOR in driving fibrosis specifically in the intestines. We found that Areg promoted EGFR phosphorylation in myofibroblast cells, suggesting the EGFR pathway is upstream of mTOR and MEK in myofibroblasts treated with Areg.
In summary, our current study demonstrated that gut microbiota-specific Th17 cell production of Areg contributes to the development of intestinal fibrosis, in which the IL-6/IL-21-Stat3-Areg-mTOR/MEK axis plays a crucial role in the pathology of fibrosis. Our study thus provided a novel avenue for the development of targeted therapeutic strategies for CD-associated intestinal fibrosis.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT:
Intestinal fibrosis is a significant complication of Crohn’s disease (CD). Gut microbiota antigen-specific T cells have been implicated in the pathogenesis of CD and early stages of fibrosis.
NEW FINDINGS:
Th17 cells induce more severe intestinal fibrosis through the production of Areg. Areg promotes human intestinal myofibroblast proliferation and migration through activation of mTOR and MEK.
LIMITATIONS:
A limitation of the current study is that we did not use an anti-Areg antibody to treat intestinal fibrosis.
IMPACT:
These findings reveal previously unrecognized roles of Th17-produced Areg in promoting intestinal fibrosis and suggest Areg as a potentially druggable target to ameliorate intestinal fibrosis in CD patients.
Acknowledgment:
We appreciate Dr. Sherry Haller of The University of Texas Medical Branch for proofreading the manuscript. We thank PennState Colorectal Diseases Biobank bank supported by Peter and Marshia Carlino Fund for IBD research for providing tissue samples in preparation of intestinal myofibroblasts.
Grant Support:
This work was supported by the National Institutes of Health grants DK112436, DK125011, AI150210, and DK124132, the University of Texas System STARs award (Y.C.).
Abbreviations:
- IBD
Inflammatory bowel disease
- LP
Lamina propria
- CD
Crohn’s disease
- MFs
myofibroblasts
- ECM
extracellular matrix
- EGF
epidermal growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: No authors have conflicting financial, professional, or personal interests.
Microarray data: Microarray data in this study is available in ArrayExpress database at the European Molecular Biology Laboratory-European Bioinformatics Institute under the accession number E-MTAB-10398.
References
- [1].Cosnes J, Gower-Rousseau C, Seksik P, et al. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology 2011;140(6): 1785–94. [DOI] [PubMed] [Google Scholar]
- [2].Rieder F Managing Intestinal Fibrosis in Patients With Inflammatory Bowel Disease. Gastroenterol Hepatol (N Y) 2018;14(2): 120–122. [PMC free article] [PubMed] [Google Scholar]
- [3].Peyrin-Biroulet L, Harmsen WS, Tremaine WJ, et al. Surgery in a population-based cohort of Crohn’s disease from Olmsted County, Minnesota (1970–2004). Am J Gastroenterol 2012;107(11): 1693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rieder F, Fiocchi C. Intestinal fibrosis in inflammatory bowel disease – Current knowledge and future perspectives. J Crohns Colitis 2008;2(4): 279–90. [DOI] [PubMed] [Google Scholar]
- [5].Torle J, Dabir PD, Korsgaard U, et al. Levels of Intestinal Inflammation and Fibrosis in Resection Specimens after Preoperative Anti-Tumor Necrosis Factor Alpha Treatment in Patients with Crohn’s Disease: A Comparative Pilot Study. Surg Res Pract 2020;2020(6085678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lawrance IC, Wu F, Leite AZ, et al. A murine model of chronic inflammation-induced intestinal fibrosis down-regulated by antisense NF-kappa B. Gastroenterology 2003;125(6): 1750–61. [DOI] [PubMed] [Google Scholar]
- [7].Macias-Ceja DC, Ortiz-Masia D, Salvador P, et al. Succinate receptor mediates intestinal inflammation and fibrosis. Mucosal Immunol 2019;12(1): 178–187. [DOI] [PubMed] [Google Scholar]
- [8].Lawrance IC, Rogler G, Bamias G, et al. Cellular and Molecular Mediators of Intestinal Fibrosis. J Crohns Colitis 2017;11(12): 1491–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Latella G, Di Gregorio J, Flati V, et al. Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand J Gastroenterol 2015;50(1): 53–65. [DOI] [PubMed] [Google Scholar]
- [10].Powrie F, Leach MW, Mauze S, et al. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+T cells. Immunity 1994;1(7): 553–62. [DOI] [PubMed] [Google Scholar]
- [11].Jin Y, Lin Y, Lin L, et al. IL-17/IFN-γ interactions regulate intestinal inflammation in TNBS-induced acute colitis. J Interferon Cytokine Res 2012;32(11): 548–56. [DOI] [PubMed] [Google Scholar]
- [12].Imam T, Park S, Kaplan MH, et al. Effector T Helper Cell Subsets in Inflammatory Bowel Diseases. Front Immunol 2018;9(1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rieder F, Zimmermann EM, Remzi FH, et al. Crohn’s disease complicated by strictures: a systematic review. Gut 2013;62(7): 1072–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sorrentino D, Avellini C, Beltrami CA, et al. Selective effect of infliximab on the inflammatory component of a colonic stricture in Crohn’s disease. Int J Colorectal Dis 2006;21(3): 276–81. [DOI] [PubMed] [Google Scholar]
- [15].Johnson P, Beswick EJ, Chao C, et al. Isolation of CD 90+ Fibroblast/Myofibroblasts from Human Frozen Gastrointestinal Specimens. J Vis Exp 2016107): e53691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cong Y, Feng T, Fujihashi K, et al. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci U S A 2009;106(46): 19256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Alexakis C, Caruelle JP, Sezeur A, et al. Reversal of abnormal collagen production in Crohn’s disease intestinal biopsies treated with regenerating agents. Gut 2004;53(1): 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hinz B, Dugina V, Ballestrem C, et al. α-Smooth Muscle Actin Is Crucial for Focal Adhesion Maturation in Myofibroblasts. Molecular Biology of the Cell 2003;14(6): 2508–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Scheibe K, Kersten C, Schmied A, et al. Inhibiting Interleukin 36 Receptor Signaling Reduces Fibrosis in Mice With Chronic Intestinal Inflammation. Gastroenterology 2019;156(4): 1082–1097.e11. [DOI] [PubMed] [Google Scholar]
- [20].Morimoto Y, Hirahara K, Kiuchi M, et al. Amphiregulin-Producing Pathogenic Memory T Helper 2 Cells Instruct Eosinophils to Secrete Osteopontin and Facilitate Airway Fibrosis. Immunity 2018;49(1): 134–150 e6. [DOI] [PubMed] [Google Scholar]
- [21].Ding L, Liu T, Wu Z, et al. Bone Marrow CD11c+ Cell-Derived Amphiregulin Promotes Pulmonary Fibrosis. J Immunol 2016;197(1): 303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Berasain C, Avila MA. Amphiregulin. Semin Cell Dev Biol 2014;28(31–41. [DOI] [PubMed] [Google Scholar]
- [23].Zaiss DM, Yang L, Shah PR, et al. Amphiregulin, a TH2 cytokine enhancing resistance to nematodes. Science 2006;314(5806): 1746. [DOI] [PubMed] [Google Scholar]
- [24].Zaiss DMW, Gause WC, Osborne LC, et al. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 2015;42(2): 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006;126(6): 1121–33. [DOI] [PubMed] [Google Scholar]
- [26].Wei L, Laurence A, Elias KM, et al. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem 2007;282(48): 34605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen H, Yang Z, Ding C, et al. Discovery of O-Alkylamino Tethered Niclosamide Derivatives as Potent and Orally Bioavailable Anticancer Agents. ACS Med Chem Lett 2013;4(2): 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li J, Liu L, Zhao Q, et al. Role of Interleukin-17 in Pathogenesis of Intestinal Fibrosis in Mice. Dig Dis Sci 2020;65(7): 1971–1979. [DOI] [PubMed] [Google Scholar]
- [29].Weaver CT, Elson CO, Fouser LA, et al. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu Rev Pathol 2013;8(477–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Strober W, Zhang F, Kitani A, et al. Proinflammatory cytokines underlying the inflammation of Crohn’s disease. Curr Opin Gastroenterol 2010;26(4): 310–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Biancheri P, Pender SL, Ammoscato F, et al. The role of interleukin 17 in Crohn’s disease-associated intestinal fibrosis. Fibrogenesis Tissue Repair 2013;6(1): 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shao J, Sheng H. Amphiregulin promotes intestinal epithelial regeneration: roles of intestinal subepithelial myofibroblasts. Endocrinology 2010;151(8): 3728–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Monticelli LA, Osborne LC, Noti M, et al. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A 2015;112(34): 10762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chen F, Yang W, Huang X, et al. Neutrophils Promote Amphiregulin Production in Intestinal Epithelial Cells through TGF-β and Contribute to Intestinal Homeostasis. J Immunol 2018;201(8): 2492–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Arpaia N, Green JA, Moltedo B, et al. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 2015;162(5): 1078–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Burzyn D, Kuswanto W, Kolodin D, et al. A special population of regulatory T cells potentiates muscle repair. Cell 2013;155(6): 1282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lee CM, Park JW, Cho WK, et al. Modifiers of TGF-β1 effector function as novel therapeutic targets of pulmonary fibrosis. Korean J Intern Med 2014;29(3): 281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhou Y, Lee JY, Lee CM, et al. Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-β-induced pulmonary fibrosis. J Biol Chem 2012;287(50): 41991–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Seykora JT. Grabbing amphiregulin by the tail to better understand keratinocyte growth. J Invest Dermatol 2010;130(8): 1966–8. [DOI] [PubMed] [Google Scholar]
- [40].Mathur R, Alam MM, Zhao XF, et al. Induction of autophagy in Cx3cr1(+) mononuclear cells limits IL-23/IL-22 axis-mediated intestinal fibrosis. Mucosal Immunol 2019;12(3): 612–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.