

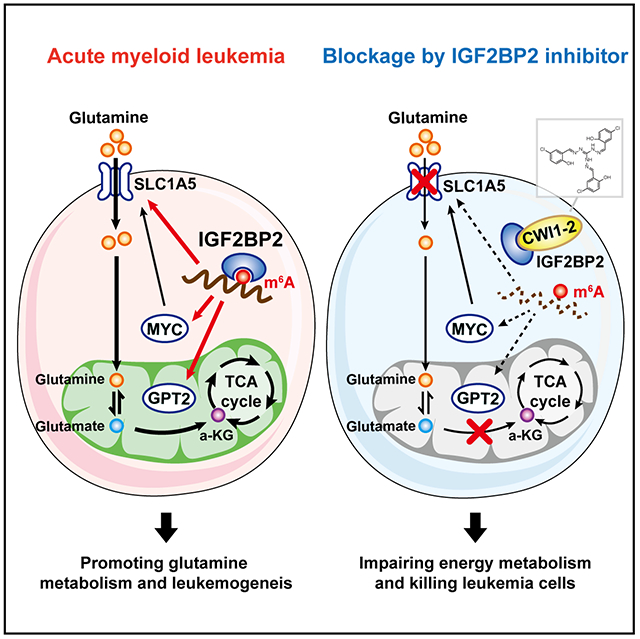
SUMMARY
N6-methyladenosine (m6A) modification and its modulators play critical roles and show promise as therapeutic targets in human cancers, including acute myeloid leukemia (AML). IGF2BP2 was recently reported as an m6A binding protein that enhances mRNA stability and translation. However, its function in AML remains largely elusive. Here we report the oncogenic role and the therapeutic targeting of IGF2BP2 in AML. High expression of IGF2BP2 is observed in AML and associates with unfavorable prognosis. IGF2BP2 promotes AML development and self-renewal of leukemia stem/initiation cells by regulating expression of critical targets (e.g., MYC, GPT2, and SLC1A5) in the glutamine metabolism pathways in an m6A-dependent manner. Inhibiting IGF2BP2 with our recently identified small-molecule compound (CWI1-2) shows promising anti-leukemia effects in vitro and in vivo. Collectively, our results reveal a role of IGF2BP2 and m6A modification in amino acids metabolism, and highlight the potential of targeting IGF2BP2 as a promising therapeutic strategy in AML.
Graphical Abstract
eTOC Blurb
Weng et al. report that IGF2BP2 is highly expressed in leukemia stem cells (LSCs) and promotes AML development/maintenance by regulating glutamine metabolism as an m6A reader. Targeting IGF2BP2 by the newly developed compound represents promising therapeutic strategy for AML therapy.
Introduction
N6-methyladenosine (m6A) modification plays critical roles in regulating mRNA fate, and has been implicated in various physiological and pathological processes, including tumorigenesis 1, 2, 3, 4. The m6A marks on mRNAs are deposited co-transcriptionally mainly by the methyltransferase (‘writer’) complex consisting of the METTL3/METTL14/WTAP core and other cofactors 5, 6, 7, 8, and could be removed by demethylase (‘eraser’) proteins such as FTO or ALKBH5 9, 10. These m6A modulator proteins have been shown by us and others to play critical oncogenic roles in various types of cancers 3, 4, including acute myeloid leukemia (AML) 11, 12, 13, 14, 15.
Similar to DNA and histone epigenetic modifications, the effect of m6A modification relies on effector proteins, i.e., m6A “reader” proteins 16, 17, 18, 19, 20. As the first identified m6A reader, YTHDF2 destabilizes m6A-modified mRNAs 18 and is required for AML initiation and propagation 21. Nonetheless, by analyzing our m6A-seq data upon FTO overexpression or METTL14 knockdown (KD), we found that many downregulated transcripts are also m6A-hypomethylated 11, 13, suggesting that these transcripts are recognized by readers other than YTHDF2. Indeed, we have identified a new class of m6A reader proteins, the IGF2BP protein family, which stabilize m6A-containing mRNAs and promote their translation through their K Homology (KH) domains 17. The oncogenic roles of IGF2BPs were mainly studied in solid tumors. Little is known about the functions and therapeutic potential of IGF2BP proteins, especially as m6A readers, in AML.
As a hallmark of cancer, reprogrammed energy metabolism sustains the growth and proliferation of tumor cells 22. Being an important energy source, Glutamine (Gln) is transported into cells and converted into glutamate (Glu), which is then used for the generation of α-ketoglutarate (α-KG) to fuel the TCA cycle or as precursor for other amino acids, glutathione, and nucleotides 23. As the most fatal subtype of leukemia that has the lowest 5-year overall survival rate (<30%), AML is addicted to Gln 24, 25. Inhibiting Gln uptake and metabolism appears to be an attractive strategy for treating AML cells in vitro and in mouse models 25, 26, 27. However, whether m6A regulates Gln metabolism in AML remains unknown.
Here, we report that IGF2BP2 and m6A modification participate in the regulation of Gln uptake and metabolism in AML. IGF2BP2 is overexpressed in AML, especially in leukemia stem/initiating cells (LSCs/LICs) and its increased expression is associated with poor prognosis in AML patients. Silencing of IGF2BP2, or depletion of METTL3 or METTL14, causes a remarkable reprogramming of cellular metabolites, especially those involved in the Gln and Glu metabolism, resulting in the inhibition of mitochondria activity and ATP production of AML cells. IGF2BP2-mediated stabilization and enhanced translation of m6A-modified target transcripts in the Gln metabolism pathway, including MYC, SLC1A5, and GPT2, is critical for LSC/LIC and AML development. Moreover, we have developed an effective small-molecule inhibitor (namely CWI1-2) that preferentially binds to IGF2BP2 and inhibits its interaction with m6A-modified target transcripts. Targeting IGF2BP2 with CWI1-2 alone, or in combination with other agents, such as daunorubicin (DNR) or homoharringtonine (HHT), shows promising therapeutic efficacy in treating AML.
Results
IGF2BP2 is highly expressed in AML, especially in LSCs/LICs, and its increased expression is associated with poor prognosis
By analyzing The Cancer Genome Atlas (TCGA) datasets, we noticed that IGF2BP2 had the highest mRNA level among the three IGF2BP family genes in AML and was highly expressed in AML compared to the vast majority of other types of cancer (Fig. S1A–B). Using qPCR assays, we showed that IGF2BP2 level was significantly elevated in AML patients carrying various chromosomal abnormalities, especially those carrying t(11q23)/MLL-rearrangements (MLLr), as compared to healthy controls (Fig. 1A). Higher expression of IGF2BP2 was also detected in normal karyotype patients with both NPM1 and FLT3-ITD mutations (often associated with unfavorable outcome) than those with only NPM1 mutation (often associated with favorable outcome) (Fig. S1C). In agreement, analysis of IGF2BP2 expression in the TCGA cohort also confirmed higher expression levels of IGF2BP2 in unfavorable-risk (including intermediate-risk and adverse-risk) AML patients than in favorable-risk ones (Fig. 1B). Such data suggest that high expression of IGF2BP2 may be associated with poor survival. Indeed, in the GSE14468 cohort, higher level of IGF2BP2 correlates with shorter overall survival in AML patients (Fig. 1C). After control for other factors such as age, sex, and molecular alterations, higher level of IGF2BP2 expression remains associated with poor prognosis (Tables S1) and thus represents a potential prognosis marker. Consistent with the RNA expression, the protein level of IGF2BP2 was also elevated in AML patients compared to healthy controls (Fig. 1D).
Figure 1. Elevated IGF2BP2 level correlates with a poor prognosis in AML patients.
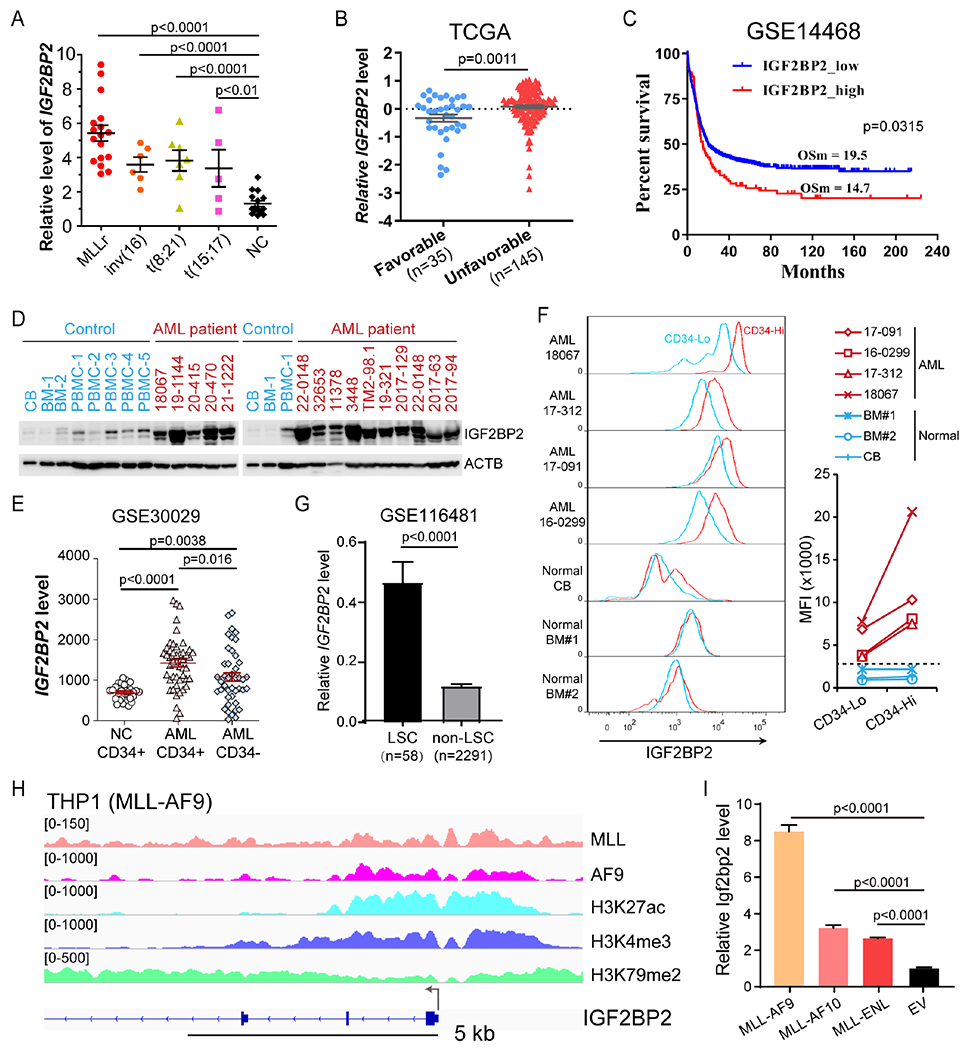
(A) qPCR showing expression of IGF2BP2 in AML patients and healthy donors. (B) Normalized IGF2BP2 RNA level in AML patients with favorable and unfavorable risk from TCGA. (C) Kaplan–Meier survival analyses of AML patients based on their IGF2BP2 expression level. OSm, medium overall survival. nlow = 425, nhigh = 93. The X-tile software 52 was used to determine the optimal cutoff values for predicting survival. (D) Western blotting using normal CB, BM or peripheral blood mononuclear cells (PBMC) from healthy control (blue) or AML patients (red). (E) Expression of IGF2BP2 in CD34+ and CD34− fractions from BM of healthy donors (NC) or AML patients as detected by microarray. (F) Flow cytometry analysis of IGF2BP2 expression based on the expression of CD34 in MNCs from AML patients or normal CB or BM. Mean fluorescence intensity (MFI) was compared between CD34 low (Lo) and high (Hi) populations. (G) Expression of IGF2BP2 in LSCs and non-LSCs from scRNA-seq data. (H) IGV tracks of ChIP-seq data in THP1 cells. (I) Relative expression of Igf2bp2 mRNA in mouse HSPCs transduced with empty vector (EV) or MLL-fusions. Data are represented as mean ± SD. See also Figure S1 and Table S1.
Interestingly, in analysis of the GSE30029 dataset, we observed a significant higher level of IGF2BP2 in CD34+ AML cells, representing the LSC/LIC population, than in CD34− bulk AML cells and normal CD34+ cells (Fig. 1E). To validate this observation, we used bone marrow (BM) samples collected from 4 AML patients, along with three healthy control donor samples, for intracellular staining of IGF2BP2. Notably, CD34-high populations displayed higher level of IGF2BP2 than CD34-low populations in these AML samples, but not in cells from normal BM or cord blood (Fig. 1F). We also sorted CD34-high and CD34-low cells from these AML samples for colony-forming/replating assays (CFAs), which revealed that CD34-high cells, but not their CD34-low counterparts, could form many colonies in methylcellulose medium (Fig. S1D), suggesting that the CD34-high populations in these samples indeed represent LSCs/LICs. Furthermore, analysis of published single cell RNA-seq data (GSE116481) from AML patients 28 showed significantly higher expression level of IGF2BP2 in phenotypically defined LSCs (CD34+CD38−Lin−CD123+) than in non-LSCs (Fig. 1G and Fig. S1E). These data together indicate that IGF2BP2 may play a role in AML pathogenesis and self-renewal of LSCs/LICs.
To understand the mechanisms underlying the upregulation of IGF2BP2 in AML, we analyzed a published ChIP-seq dataset (GSE79899) generated from a substrain of THP1 cells that expresses both wildtype (WT) MLL and MLL-AF9 (MA9) but not WT AF9. MA9-binding peaks were defined as those MLL-binding sites that also show a high AF9 signal. A cluster of high-confidence MA9-binding peaks around the transcription start site (TSS) of IGF2BP2 was observed, accompanied by the presence of H3K27ac, H3K4me3, and H3K79me2 modifications; Fig. 1H), which are epigenetic signatures of MLL-fusion target genes. Similar results were also obtained in the ChIP-seq data of the MV4-11 cell line (Fig. S1F). In line with these findings, our CFA and subsequent qPCR assays showed that ectopic expression of MLL-fusions (including MA9, MLL-AF10, and MLL-ENL) significantly increased Igf2bp2 expression in mouse hematopoietic stem/progenitor cells (HSPCs) (Fig. 1I), implying IGF2BP2 as a direct target of MLL-fusions. Thus, AML-associated oncoproteins (e.g., MLL fusions) may directly or indirectly up-regulate expression of IGF2BP2.
IGF2BP2 promotes AML initiation/progression and LSC/LIC self-renewal as an m6A reader
Through CFA, we showed that forced expression of either human or murine IGF2BP2 significantly enhanced MA9-mediated cell immortalization in vitro (Fig. S2A). Notably, this enhancement was remarkably eliminated by mutations of GXXG in the IGF2BP2 KH3-4 domain (MUT, Fig. 2A), which is indispensable for m6A recognition 17 In addition, the ability of IGF2BP2 in promoting MA9-mediated cell transformation was largely abolished in the absence of Mettl14 in BM cells collected from Mettl14 conditional knockout (KO) mice 13 (Fig. 2B). Such data indicates that METTL14-mediated m6A modification is indispensable for the promotion of cell immortalization mediated by IGF2BP2. On the other hand, KD of IGF2BP2 remarkably inhibited colony formation mediated by MA9 (Fig. S2B and Fig. 2C). Consistently, heterozygous or homozygous deletion of Igf2bp2 dramatically impaired MA9-mediated growth of HSPCs in liquid culture and colony-formation/immortalization of HSPCs in methylcellulose medium (Fig. 2D–E and Fig. S2C–E).
Figure 2. IGF2BP2 promotes AML initiation/development and maintenance as an m6A reader.
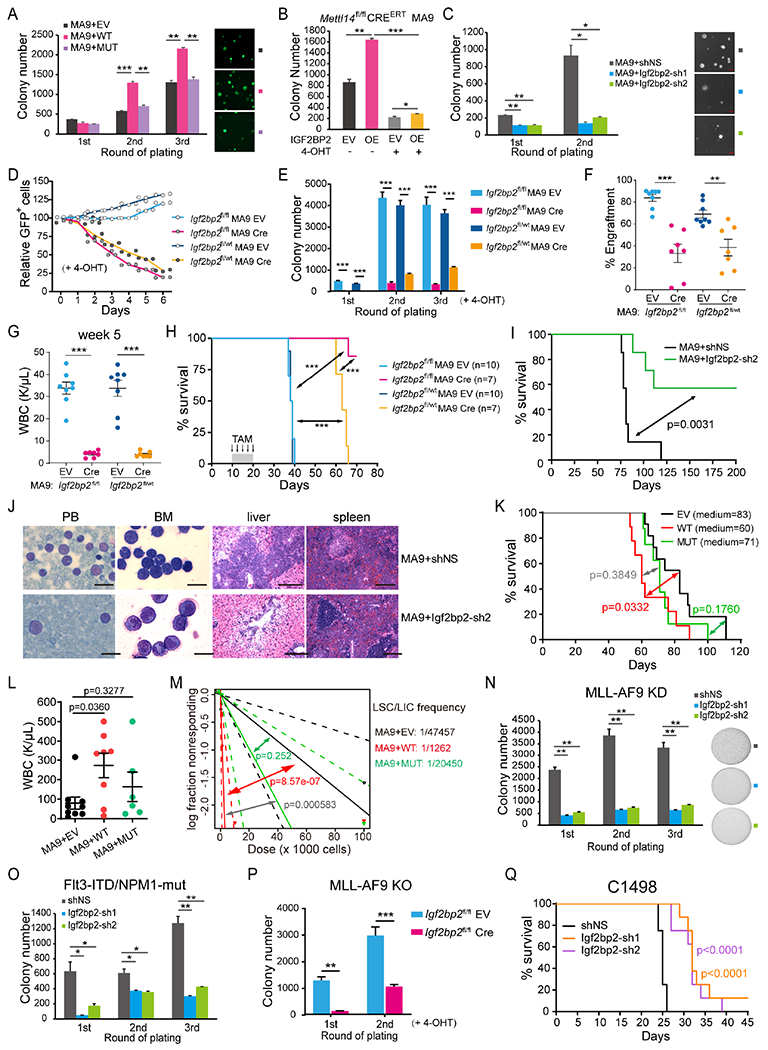
(A) CFA using mouse HSPCs transduced with MSCVneo-MLL-AF9 (MA9) plus MSCV-PIG (EV), IGF2BP2-WT (WT), or IGF2BP2-KH3-4 (MUT) retroviruses. Colony numbers and photos of GFP-expressing colonies are shown. (B) CFA using HSPCs from Mettl14fl/fl-CREERT mice. 4-Hydroxytamoxifen (4-OHT, 1 μM) was added at the first round of plating to induce Mettl14 KO. OE, overexpression. (C) CFA using mouse HSPCs transduced with MA9 plus shRNA targeting Igf2bp2 or negative control (shNS). (D) Growth competition assays. MA9-transduced Igf2bp2fl/fl or Igf2bp2fl/wt HSPCs were infected with Cre-ERT2-IRES-GFP (Cre) or ERT2-IRES-GFP (EV) lentiviruses, followed by 4-OHT treatment. Percentages of GFP+ cells were measured by flow cytometry. (E) CFA using Igf2bp2fl/fl or Igf2bp2fl/wt HSPCs transduced with MA9 retrovirus and Cre or EV lentivirus. (F, G) Donor cell percentages (F) and WBCs (G) in PB of recipient mice 5 weeks after BMT using cells collected from first plating of CFA in (E). (H, I) Kaplan–Meier curves showing the effect of Igf2bp2 KO (H) or KD (I) on MA9-induced de novo leukemogenesis in lethally irradiated recipient mice. TAM, tamoxifen. (J) Wright–Giemsa staining of PB and BM, and hematoxylin and eosin (H&E) staining of liver (LV) and spleen (SP) of the primary BMT recipient mice at the end point. Bar= 20 μm (for PB and BM) or 200 μm (for SP and LV). (K) Kaplan–Meier curves showing the effect of IGF2BP2 overexpression on MA9-induced de novo leukemogenesis in lethally irradiated recipient mice. (L) WBC counts of mice from (K). (M) LDA using BM leukemia cells from BMT mice in (K). (N, O) CFA using BM cells from MA9 (N) or Flt3-ITD/NPM1-mut (O) leukemia mice. (P) CFA using Cre or EV transduced BM cells from Igf2bp2fl/fl MA9 EV leukemia mice. (Q) Kaplan–Meier curves showing the effect of IGF2BP2 KD on C1498 induced AML onset and progression in recipient mice. Data are represented as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; t test. See also Figure S2.
Bone marrow transplantation (BMT) assays were then conducted to determine the pathological role of IGF2BP2 in vivo. Consistent with the in vitro results, conditional KO of Igf2bp2 significantly suppressed engraftment of MA9-transduced donor cells, reduced white blood cells (WBCs) in the peripheral blood (PB), significantly delayed the initiation/development of AML and improved the survival in recipient mice (Fig. 2F–H and Fig. S2F). Similarly, KD of Igf2bp2 significantly delayed leukemogenesis mediated by MA9 and substantially prolonged the survival of recipient mice (Fig. 2I and Fig. S2G), associated with a remarkable decrease of immature blast cell population in PB and BM and reduced leukemia cell infiltration in spleens and livers (Fig. 2J and Fig. S2H).
Conversely, overexpression of WT rather than MUT IGF2BP2 significantly promoted MA9-mediated leukemogenesis, shortened survival of primary BMT recipient mice, and increased WBCs in PB (Fig. 2K–L). Given that IGF2BP2 is highly expressed in LSCs/LICs, we conducted limiting dilution assays (LDA) using BM cells harvested from primary BMT recipient mice to evaluate the effect of IGF2BP2 overexpression on the frequency of LSC/LIC. Notably, the WT IGF2BP2-overexpressing group had a significantly higher LSC/LIC frequency than the control or KH3-4 mutant-overexpressing group (Fig. 2M), demonstrating the role of IGF2BP2 in promoting the self-renewal of LSCs/LICs.
To further determine the role of IGF2BP2 in AML maintenance, we performed CFAs with leukemic BM cells collected from primary AML mice induced by MA9 or FLT3-ITD/NPM1-mut. As expected, Igf2bp2 KD or KO significantly inhibited colony-forming/replating capacity in BM cells from both leukemia models (Fig. 2N–P). Secondary BMT assay with C1498, a murine AML cell line that arose spontaneously in a C57BL/6 mouse and grows aggressively in syngeneic or congenic mice 29, further demonstrated that KD of Igf2bp2 significantly inhibited the maintenance and progression of AML in vivo (Fig. 2Q).
Amino acids metabolism is promoted by IGF2BP2
In multiple human AML cell lines, KD of IGF2BP2 with 3 individual shRNAs substantially suppressed cell grow/proliferation, induced terminal myeloid differentiation and apoptosis (Fig. 3A–C and Fig. S3A–B). Conversely, ectopic expression of WT rather than MUT IGF2BP2 significantly promoted the growth of AML cells (Fig. 3D and Fig. S3C). In addition, growth inhibition induced by IGF2BP2 KD could be rescued by WT but not MUT IGF2BP2 (Fig. 3E). Thus, the pathological role of IGF2BP2 in AML is dependent on its m6A reader capacity.
Figure 3. IGF2BP2 regulates glutamine uptake and metabolism in AML cells as an m6A reader.
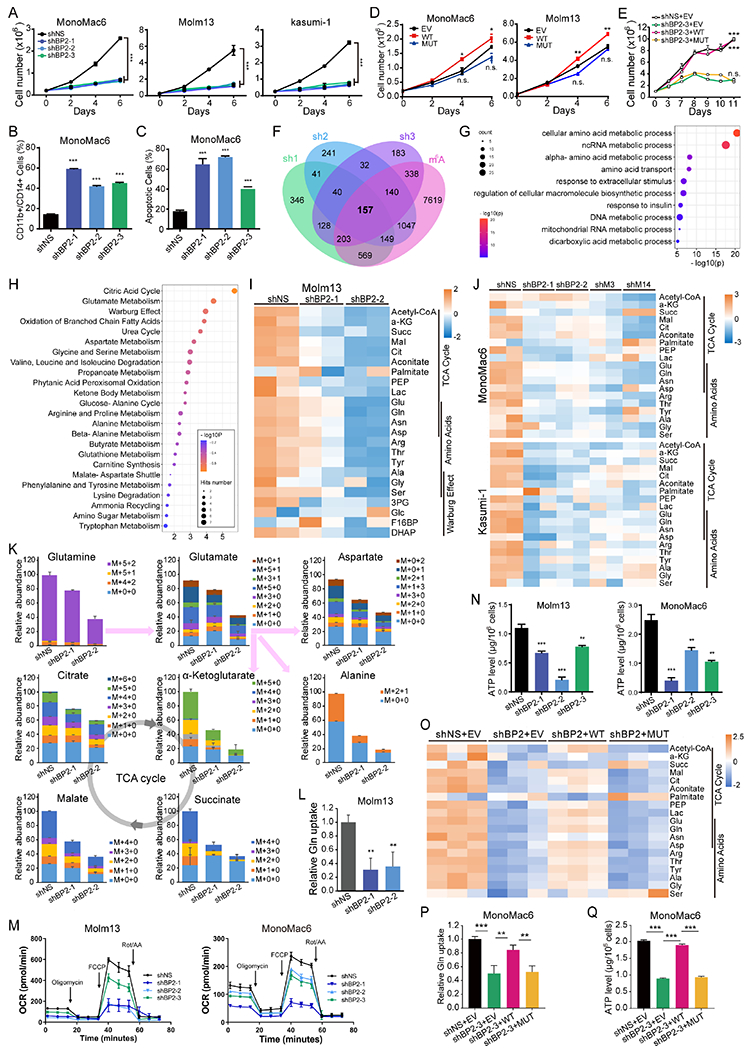
(A) Viable cell counting of AML cell lines with or without IGF2BP2 KD. (B, C) Flow cytometric analyses of differentiated (CD11b+ or CD14+, B) and apoptotic (Annexin V+, C) cells after IGF2BP2 KD. (D) Viable cell counting of AML cells lentivirally transduced with WT or MUT IGF2BP2 or empty vector (EV). (E) Viable cell counting of MonoMac6 cells transduced with shRNA (shBP2-3) targeting 3’ UTR of IGF2BP2 plus EV, WT, or MUT IGF2BP2-overexpression vector. (F) Venn diagram showing overlapping of significantly (FC<0.667, p<0.0.5) downregulated genes in MonoMac6 cells transduced with different shRNAs targeting IGF2BP2. Information regarding existence of m6A signal or not in MonoMac6 cells (GSE97408) in each transcript was included for analysis. (G) Bubble diagram showing GO enrichment of the 157 candidate targets of IGF2BP2 in (F). (H) Bubble diagram showing enrichment of metabolic pathways by the metabolites with reduced level after IGF2BP2 KD in Molm13 cells. (I, J) Heatmap showing levels of representative metabolites after KD of IGF2BP2 (I), METTL3, or METTL14 (J) in indicated AML cells. (K) Total levels and isotopologue distribution (M+x+y; x, numbers of 13C; y, numbers of 15N) of TCA cycle intermediates and indicated amino acids measured by LC-MS in Molm13 cells transduced with IGF2BP2 shRNAs or shNS and grown in media containing 13C5;15N2-glutamine. (L) Effect of IGF2BP2 KD on glutamine uptake. (M, N) OCR (M) and ATP level (N) changes in AML cells upon IGF2BP2 KD. (O) Heatmap showing levels of representative metabolites in the TCA cycle in IGF2BP2 KD MonoMac6 cells rescued with WT or MUT IGF2BP2. (P, Q) Glutamine uptake (P) and ATP levels (Q) in IGF2BP2 KD cells rescued with WT or MUT IGF2BP2. Data are represented as mean ± SD. **p < 0.01; ***p < 0.001; n.s., not significant; t test. See also Figure S3.
To explore the mechanism underlying the role of IGF2BP2 in AML, we performed RNA sequencing (RNA-seq) in AML cells with or without IGF2BP2 KD (Fig. 3F). As IGF2BP2 protein was reported to stabilize m6A-modified mRNA 17, we focused on the genes positively regulated by IGF2BP2. The mRNA level of 197 protein-coding genes was significantly downregulated in all 3 shRNA groups compared to the control MonoMac6 cells; among them, 157 transcripts were m6A methylated according to our published m6A-seq data of MonoMac6 cells 13 (Fig. 3F), and thus represented potential m6A-dependent targets of IGF2BP2. Gene ontology (GO) analysis of these candidate genes revealed an enrichment in cell metabolic pathways, especially in the metabolism and transport of amino acids (Fig. 3G). The down-regulation of representative target genes from these pathways upon IGF2BP2 KD was validated by qPCR (Fig. S3D, left). Moreover these target transcripts were all downregulated when METTL14 was knocked down (Fig. S3D, right), further suggesting that they were regulated in an m6A-dependent manner and that m6A modification is likely involved in the regulation of amino acids metabolism. Notably, the rest 40 protein-coding genes downregulated by 3 shRNAs but without m6A modifications were also enriched in the amino acids metabolism pathway (Fig. S3E), suggesting that IGF2BP2 participates in amino acids metabolism through direct or indirect regulation of the expression of amino acids metabolism-related genes.
We therefore performed metabolites profiling in AML cells. In order to detect early changes, we virally infected Molm13 cells, selected transduction-positive cells with puromycin for one day, and incubated cells with stable isotope labelled Gln (13C5;15N2-glutamine) overnight for metabolites profiling by liquid chromatography-mass spectrometry (LC-MS) analysis. Enrichment analysis revealed that metabolites involved in citric acid cycle, Glu metabolism, and Warburg effect were affected greatly by IGF2BP2 KD (Fig. 3H), with most of the metabolites in these pathways being reduced in IGF2BP2 KD cells (Fig. 3I). Similar results were observed in MonoMac6 and Kasumi-1 cells when IGF2BP2, METTL3, or METTL14 was knocked down (Fig. 3J), confirming an impact of m6A modification on cell metabolism. Interestingly, Glu metabolism again appeared to be on the top 3 pathways affected by modulation of these m6A modulators (Fig. S3F), implying that m6A modification has a profound effect on Glu metabolism.
Further analysis of the LC-MS data showed that the incorporation of labelled Gln into Glu metabolism was reduced, and the overall levels of most Glu-derived amino acids and TCA cycle intermediates were decreased in IGF2BP2 KD cells (Fig. 3K). Notably, the intracellular level of overall and labelled Gln was decreased in IGF2BP2 KD cells (Fig. 3K), suggesting that the uptake of Gln was inhibited when IGF2BP2 was depleted. Indeed, we observed a decrease of Gln uptake in AML cells upon IGF2BP2 KD (Fig. 3L). Consistent with the decrease of TCA cycle intermediates, IGF2BP2 KD resulted in reduced oxygen consumption during both basal and maximal respiration (Fig. 3M), and eventually led to less ATP production (Fig. 3N). Furthermore, IGF2BP2 KD-induced metabolites reprogramming could be at least partially rescued by ectopic expression of WT but not MUT IGF2BP2 (Fig. 3O). Specifically, reduction of Gln uptake and ATP production upon IGF2BP2 KD could be almost completely restored by WT but not MUT IGF2BP2 (Fig. 3P–Q). Thus, these data suggest that the role of IGF2BP2 in regulating amino acids metabolism is largely dependent on its role as an m6A reader.
IGF2BP2 fine tunes Gln uptake and metabolism through increasing expression of MYC, GPT2, and SLC1A5 in an m6A-dependent manner
According to our RNA-seq and metabolite profiling data, GPT2 and SLC1A5, two critical genes in the Gln metabolism pathway, were chosen as potential targets of IGF2BP2. GPT2 encodes a critical enzyme in Gln metabolism that catalyzes the reversible conversion of pyruvate and Glu to alanine andαKG in the mitochondria 30, while SLC1A5 encodes the primary transporter of Gln in cancer cells 31. Additionally, the MYC oncogene, which was reported by us as a direct target of METTL14 13 and IGF2BP2 17 and was shown to be downregulated by IGF2BP2 KD (Fig. S3D), participated in Gln transport by regulating SLC1A5 32. Thus, MYC was also included as a potential target of IGF2BP2 in AML metabolism for further studies. Consistent with the change of their mRNA level (Fig. S3D), expression of GPT2, SLC1A5, and MYC at the protein level was also evidently reduced in AML cells upon depletion of IGF2BP2 (Fig. 4A and Fig. S4A). Of note, KD of METTL2 or METTL14 also resulted in the reduction of GPT2, SLC1A5, and MYC protein level (Fig. 4A). In addition, protein level of these three target genes could be elevated by forced expression of WT but not MUTIGF2BP2 (Fig. 4B). Further, IGF2BP2 KD-induced decrease of MYC, GPT2, and SLC1A5 at the mRNA and protein levels could be at least partially rescued by forced expression of WT but not MUT IGF2BP2 (Fig. 4C–D and Fig. S4B–C). These data together implicate an m6A-dependent regulatory mechanism.
Figure 4. MYC, GPT2, and SLC1A5 are direct targets of IGF2BP2.
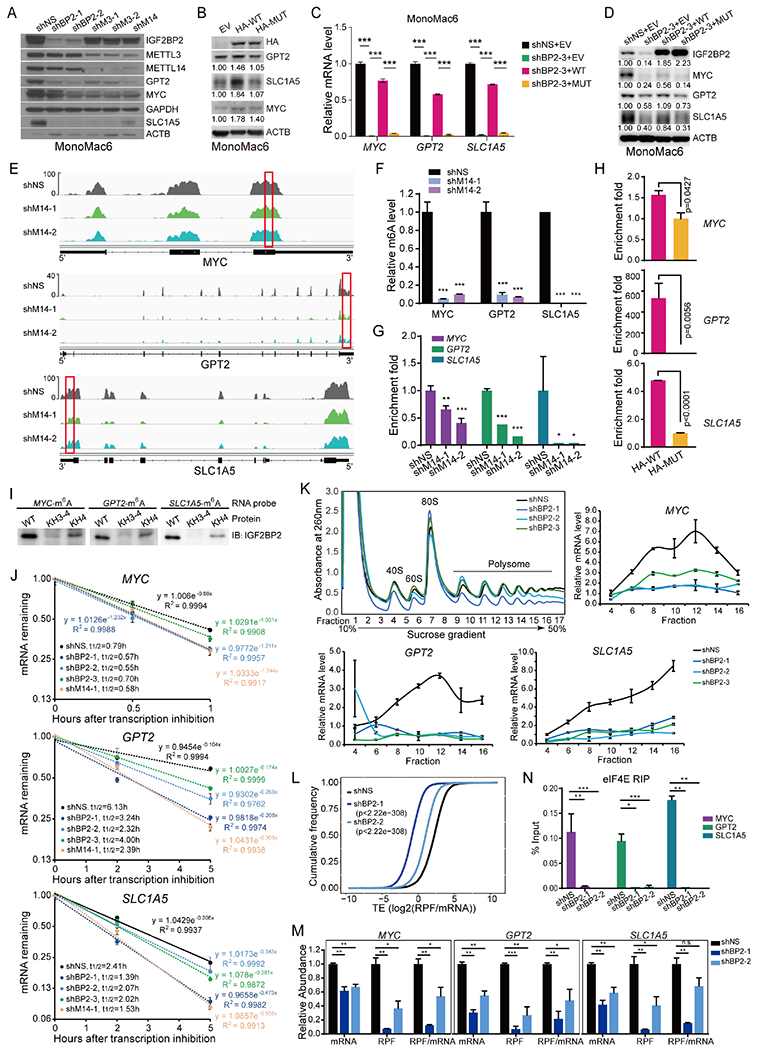
(A, B) Western blot showing expression change of MYC, GPT2, and SLC1A5 after KD of IGF2BP2, METTL3, or METTL14 (A), or overexpression of HA-tagged wildtype (HA-WT) or KH3-4 mutated (HA-MUT) IGF2BP2 (B). Band intensity in (B) was quantified by ImageJ2. (C, D) mRNA (C) and protein (D) levels of MYC, GPT2, and SLC1A5 in IGF2BP2 KD cells rescued with WT or MUT IGF2BP2. (E) IGV tracks showing the m6A distribution in indicated transcripts in MonoMac6 cells. Red rectangles depict high-confidence m6A regions for RIP-qPCR validation. (F) Evaluation of relative m6A abundance changes at annotated specific loci in MonoMac6 cells by utilizing Bst DNA polymerase-mediated cDNA extension and qPCR assays. (G) RIP assays using IGF2BP2 antibody were followed by qPCR in METTL14 KD or control MonoMac6 cells. (H) RIP assays using HA antibody were followed by qPCR in U937 cells with ectopically expressed WT or MUT IGF2BP2. (I) RNA pulldown assays using recombinant WT, KH3-4, or KH4 mutated IGF2BP2 protein with m6A-modified RNA oligos corresponding to IGF2BP2 binding sites in MYC, GPT2, and SLC1A5 mRNAs. (J) The mRNA half-life (t1/2) of target genes in MonoMac6 cells with KD of IGF2BP2 or METTL14. (K) Polysome fractionation of MonoMac6 cell lysates (top left) and subsequent qPCR assays. Actb mRNA was used as a reference in qPCR. (L) Cumulative frequency of global translation efficiency (TE) changes upon IGF2BP2 KD in MonoMac6 cells. (M) Relative abundance of MYC, GPT2, and SLC1A5 mRNA, ribosome protected fragment (RPF), and RFP/mRNA in control (shNS) and IGF2BP2 KD MonoMac6 cells from Ribo-seq. (N) RIP assays using eIF4E antibody were followed by qPCR in IGF2BP2 KD or control Molm13 cells. Data are represented as mean ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001. t test. See also Figure S4.
According to our published m6A-seq data (GSE97408), MYC, GPT2, and SLC1A5 transcripts all have high abundance of m6A modification that decreased uponMETTL14 KD (Fig. 4E). High-confidence m6A modification sites were predicted based on the m6A-seq data as well as the prediction from SRAMP (www.cuilab.cn) (Fig. S4D), and then Bst DNA polymerase-mediated cDNA extension and qPCR assays were utilized to validate the abolishment of m6A marks on these transcripts uponMETTL14 KD (Fig. 4F). In addition, RNA immunoprecipitation (RIP) assays showed that the binding of endogenous IGF2BP2 to such m6A-modified regions was significantly impaired when METTL14 was knocked down (Fig. 4G). In HA-IGF2BP2 ectopically expressed U937 cells (Fig. S4E), RIP assays using HA antibody revealed that WT IGF2BP2 is more enriched in the m6A-modified regions of MYC, GPT2, and SLC1A5 transcripts than does MUT IGF2BP2 (Fig. 4H). Furthermore, we performed in vitro RNA pulldown assays using synthesized RNA oligos with or without m6A modifications and purified recombinant IGF2BP2 proteins in the WT or MUT form. The results confirmed that WT IGF2BP2 bound to m6A-modified RNA oligos of MYC, GPT2, and SLC1A5, whereas this binding was impaired by mutation of the KH4 domain and became even weaker by mutation of the KH3-4 di-domain (Fig. 4I). These data together strongly demonstrate that GPT2 and SLC1A5, as well as MYC, were directly bound and regulated by IGF2BP2 in an m6A-dependent manner.
IGF2BP2 was previously identified as an m6A reader that stabilizes and promotes translation of target mRNAs 17. Consistent with the decrease of mRNA level, a shortening of mRNA half-life of MYC, GPT2, and SLC1A5 was observed upon KD of IGF2BP2 or METTL14 (Fig. 4J). Co-immunoprecipitation (co-IP) revealed that IGF2BP2 interacts with the mRNA stabilizer PABPC1 (Fig. S4F). In addition, KD of PABPC1, similar to IGF2BP2 KD, decreased mRNA level of MYC, GPT2, and SLC1A5 but not non-target genes of IGF2BP2 (Fig. S4G). Thus, while IGF2BP2 itself determines target gene specificity, it recruits generic mRNA stabilizers (e.g., PABPC1) to stabilize its targets. Notably, IGF2BP2 has limited overlapped target genes with YTHDF2 (Fig. S4H). IGF2BP2 does not compete with YTHDF2 for binding sites in MYC, GPT2, and SLC1A5 transcripts and vice versa (Fig. S4I), and KD of YTHDF2 has no or marginal effect on the mRNA level of MYC, GPT2, and SLC1A5 (Fig. S4J).
Further polysome fractionation showed that KD of IGF2BP2 resulted in a decrease of MYC, GPT2, and SLC1A5 mRNAs in polysomes (Fig. 4K), implying that IGF2BP2 may regulate both mRNA stability and translation of these target transcripts. Ribosome profiling (Ribo-seq) was then conducted, which confirmed that KD of IGF2BP2 inhibits global protein translation (Fig. 4L). This could be explained by the enrichment of ribosome biogenesis and cellular amino acid metabolic process as the two mostly significantly changed processes affected by IGF2BP2 KD (Fig. S4K). Specifically, ribosome occupancy of MYC, GPT2, and SLC1A5, but not of IGF2BP2 non-target genes, was significantly suppressed in IGF2BP2 KD cells (Fig. 4M and Fig. S4L). To understand the underlying mechanism, we performed co-IP and found that IGF2BP2 interacted with components of the eukaryotic translation initiation factor (eIF) complexes, such as eIF4E and eIF3A (Fig. S4M). In addition, KD of IGF2BP2 significantly inhibited binding of eIF4A to MYC, GPT2, and SLC1A5 mRNAs (Fig. 4N), suggesting that IGF2BP2 recruits eIF proteins to its target mRNAs to fine tune their translation.
MYC, GPT2, and SLC1A5 mediate the effects of IGF2BP2 in AML cells
Loss-of-function assays were conducted to evaluate the roles of these target genes of IGF2BP2 in AML. Consistent with their functions in the Glu metabolism and TCA cycle, KD of MYC, GPT2, or SLC1A5 inhibited Gln uptake, oxygen consumption and ATP production (Fig. S5A–B and Fig. 5A–B), and resulted in inhibition of AML cell growth and proliferation as well as induction of AML cell differentiation and apoptosis (Fig. 5C–E). During MA9-mediated cell immortalization of mouse HSPCs, KD of Gpt2, Slc1a5, or Myc inhibited colony formation capacity (Fig. S5C and Fig. 5F), mimicking the effect of Igf2bp2 KD (Fig. 2C). In addition, silencing of these genes in leukemic BM blast cells collected from MA9 leukemic mice suppressed colony forming ability (Fig. 5G). Given that IGF2BP2 plays critical roles in promoting LSCs/LICs, we performed in vitro LDA to evaluate the effects of target gene KD on LSC/LIC frequency. As expected, KD of these target genes phenocopied Igf2bp2 KD in reducing LSC/LIC frequency in MA9-transformed mouse HSPCs (Fig. 5H). In line with these findings, expression of GPT2, SLC1A5, and MYC is higher in CD34+ populations than in CD34− counterparts in AML patient samples, similar to the pattern of IGF2BP2 (Fig. S5D).
Figure 5. MYC, GPT2, and SLC1A5 are functionally important targets of IGF2BP2 in AML cells.
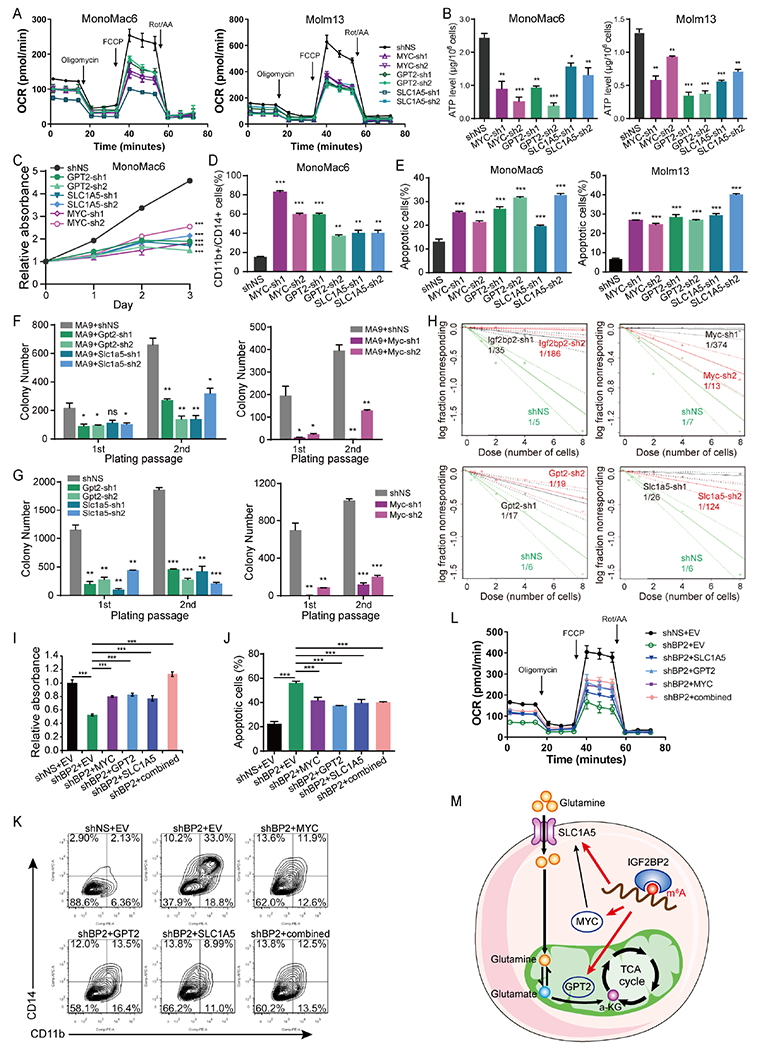
(A, B) Effects of target genes KD on OCR (A) and ATP levels (B) in AML cells. (C-E) Effects of the depletion of target genes on cell growth (C), differentiation (D), and apoptosis (E) in AML cells. (F) Effect of the depletion of Gpt2, Slc1a5 or Myc on the colony-forming/replating capacity of mouse HSPCs immortalized by MA9. (G) CFA using BM cells fromMA9 leukemic mice. (H) In vitro LDA. Logarithmic plots showing the percentage of nonresponding wells at different doses of mouse HSPCs cotransduced with MA9 and shNS or shRNAs targeting Gpt2, Slc1a5, or Myc. Nonresponding wells are wells without colonies. (I) MonoMac6 cells were transduced with shNS or IGF2BP2-sh1, and with or without lentiviruses encoding GPT2, SLC1A5, or MYC alone or in combination. Cells were seeded for MTT assays after puromycin selection, and the absorbance was measured at day 3. (J, K) Flow cytometry analyses showing the rescue effect of overexpression of individual target genes alone or in combination on apoptosis (J) and differentiation (K) of MonoMac6 cells induced by IGF2BP2 KD. (L) OCR in MonoMac6 cells with indicated treatment. (M) Proposed working model of IGF2BP2 in regulating glutamine metabolism in an m6A-dependent manner. Data are represented as mean ± SD. n.s., not significant; *p < 0.05; **p < 0.01; ***p < 0.001; t test. See also Figure S5.
Moreover, ectopic expression of GPT2, SLC1A5, or MYC could at least partially reverse IGF2BP2 KD-mediated AML cell growth inhibition, differentiation, and apoptosis (Fig. S5E and Fig. 5I–K). In addition, ectopic expression of these individual target genes or in combination at least partially recovered mitochondrial oxygen consumption in IGF2BP2 KD cells (Fig. 5L). Collectively, our data uncover that MYC, GPT2, and SLC1A5 are functionally essential targets of IGF2BP2 and their post-transcriptional regulation by IGF2BP2 (in an m6A-dependent manner) contributes to the overall function of IGF2BP2 in Gln metabolism and AML biology (Fig. 5M).
IGF2BP2 is a promising therapeutic target in AML
The above data together demonstrated that IGF2BP2, through regulating Gln metabolism pathways, is critical for maintaining AML cell survival and stemness. Considering the critical roles of glutaminolysis in AML, we then tested whether IGF2BP2 could be used as a therapeutic target in AML. Consistent with the results from cell lines, similar inhibitory effect of IGF2BP2 KD on cell growth was observed in primary leukemic blast cells from AML patients (Fig. 6A). In addition, colony-forming ability of AML patient blasts in methylcellulose medium was remarkably inhibited when IGF2BP2 was knocked down (Fig. 6B), further highlighting a role of IGF2BP2 in the self-renewal of LSCs/LICs. Importantly, although it was recently reported that Igf2bp2 KO induces quiescence loss and impairs HSC function in mice, the differentiation and maturation of all blood cell types in PB and BM were not affected 33, suggesting that targeting of IGF2BP2 is tolerant in vivo. As a proof of concept, we performed xeno-transplantation experiments and found that silencing of IGF2BP2 greatly inhibited the engraftment of human AML cells in BM of recipient mice and significantly prolonged the overall survival of the recipients (Fig. 6C and Fig. S6A). Such data suggest IGF2BP2 as a promising therapeutic target for AML.
Figure 6. Identification of CWI1-2 as an IGF2BP2 inhibitor.
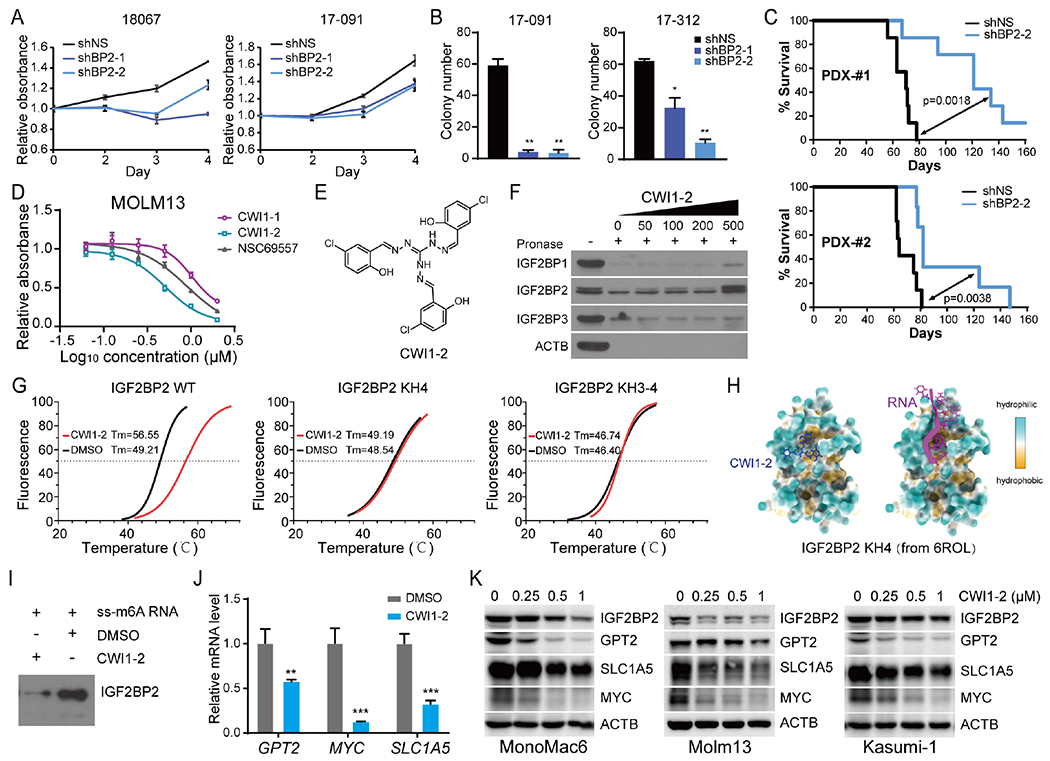
(A, B) MTT assays (A) and CFA (B) of patient-derived AML cells with or without IGF2BP2 KD. (C) Kaplan-Meier survival curves showing effect of IGF2BP2 KD on leukemia-inducing capacity of patient-derived AML cells in NRGS immunodeficient mice. (D) MTT assays at 48 hours after drug treatment. (E) Structure of the compound CWI1-2. (F) DARTS assays with Molm13 cell lysates in the presence of indicated concentration of CWI1-2. (G) Boltzmann fitting curves of the cell-free thermal shift assay showing binding of CWI1-2 (12 μM) with fluorescence-labeled recombinant IGF2BP2 proteins as indicated. (H) Docking model of CWI1-2 (left) and RNA (right) on KH4 of IGF2BP2 based on the 6ROL structure from the Protein Data Bank. (I) RNA pulldown assays. (J) qPCR showing effect of CWI1-2 (0.5 μM, 24 hours) treatment on expression of MYC, GPT2, and SLC1A5 in MonoMac6 cells. 18S rRNA was used as a reference. (K) Western blot showing effect of CWI1-2 on the protein level of IGF2BP2 targets in AML cells. Data are represented as mean ± SD. **p < 0.01; ***p < 0.001; t test. See also Figure S6.
Virtual screening identifies an IGF2BP2 inhibitor
Therefore, we sought to identify IGF2BP2 inhibitor(s) for AML therapy. We conducted a structure-based virtual screening to identify potential IGF2BP2 inhibitors from approximately 260,000 compounds of the National Cancer Institute Developmental Therapeutics Program (NCI DTP) library. A total of 128 compounds from the top 150 candidate compounds with high scores of docking to the IGF2BP KH3-4 domain were available and obtained from the NCI and screened for anti-leukemic activity. Among them, 7 compounds, as represented by NSC69557, exhibited potent growth inhibitory effects on Molm13 in the initial screen and were further validated in 3 additional AML cell lines (Fig. S6B). Drug affinity responsive target stability (DARTS) assays 34 were then conducted (Fig. S6C), which suggested that NSC69557 could robustly bind to IGF2BP2 protein in vitro. Importantly, binding of NSC69557 provided a better protective effect against protease digestion than that of BTYNB (Fig. S6C), a reported IGF2BP1 inhibitor 35 which was shown by us to exhibit anti-leukemia effect and bind to IGF2BP2 (Fig. S6D–E).
However, mass spectrum (MS) analysis of NSC69557 indicated that the compound was not pure but rather a mixture, with NSC69557-representing compound being a minor component and two other compounds being major components (Fig. S6F). These two additional compounds, with molecular weight of 381 and 519, respectively, are structurally related to NSC69557 as their fragmentation pattern closely resembles that of NSC69557 in MS. We then purified these two compounds with HPLC, which were named CWI1-1 and CWI1-2. Of them, CWI1-2 demonstrated a greater inhibitory effect on AML cell viability/growth than the mixture, whereas CWI1-1 was less effective (Fig. 6D). By analyzing the MS and NMR data, we speculated the structure of CWI1-2 and synthesized the compound (Fig. 6E). Subsequent MTT assays showed that this synthesized compound recapitulated the inhibitory efficacy of CWI1-2 purified from the mixture (Fig. S6G), suggesting that it is indeed CWI1-2 and represents the major bioactive component in the NSC69557 mixture.
Cellular thermal shift assay (CETSA) 36 and DARTS assay were then applied to evaluate the potential binding of CWI1-2 with IGF2BP2. Notably, compared to IGF2BP1 or IGF2BP3, IGF2BP2 could be better protected by CWI1-2 against high temperature-induced denaturation (Fig. S6H) or protease-mediated degradation (Fig. 6F), suggesting that IGF2BP2 is a preferential target of CWI1-2. The direct interaction of CWI1-2 with IGF2BP2 protein was further confirmed by the split-luciferase CETSA (Split-Luc CETSA) 37 in 293T cells (Fig. S6I) and the cell-free fluorescence protein thermal shift assay (FTSA) 38 (Fig. 6G). Intriguingly, molecular docking showed that CWI1-2 docks to the hydrophobic pocket within KH4 near the RNA binding site in IGF2BP2 (Fig. 6H), suggesting potential competitive binding between CWI1-2 and RNA to IGF2BP2. Indeed, our cell-free FTSA demonstrated that the direct binding of CWI1-2 with IGF2BP2 was completely disturbed by mutation of KH4 or KH3-4 in IGF2BP2 (Fig. 6G). In addition, RNA pull-down assays showed that the binding of recombinant IGF2BP2 protein with m6A-modified single stranded RNA (ss-m6A) was largely abrogated in the presence of CWI1-2 (Fig. 6I). These data together indicate that CWI1-2 binds directly to IGF2BP2 to competitively inhibit its binding to RNA targets.
Similar to IGF2BP2 KD, CWI1-2 treatment of AML cells resulted in decreased RNA and protein levels of IGF2BP2 targets (Fig. 6J–K and Fig. S6J). RNA-seq of CWI1-2 treated cells revealed that 59.2% of differentially expressed genes (DEGs) upon IGF2BP2 KD are also dysregulated in the presence of CWI1-2 (Fig. S6J), and that cellular amino acid metabolic process is also among the most enriched pathways in CWI1-2 treated cells (Fig. S6K). It is noticeable that CWI1-2 (Fig. 6K), similar to BTYNB (Fig. S6L), also reduced IGF2BP2 protein level, which may be attributed to decreased stability of IGF2BP2 protein when its binding to target transcripts was inhibited.
CWI1-2 exhibits promising anti-leukemia efficacy in vitro and in vivo
The anti-leukemia efficacy of CWI1-2 was then evaluated by various in vitro assays. Noticeable, compared to their counterparts isolated from healthy donors, CD34+ cells from AML patients were more sensitive to CWI1-2 treatment (Fig. 7A), indicating a therapeutic window. In line with this result, the IC50 values of CWI1-2 in IGF2BP2-high AML cells were lower than that in IGF2BP2-low leukemia cells, such as U937 and SUP-B16 (Fig. 7B and Fig. S7A). Treatment of IGF2BP2-high cells with CWI1-2 induced dramatic cell differentiation and apoptosis in a concentration-dependent manner, whereas minimal changes were observed in IGF2BP2-low cells (Fig. S7B). Consistent with its inhibitory effect on the expression of IGF2BP2 target genes involved in Gln metabolism, CWI1-2 treatment reduced Gln uptake and impaired mitochondria function, leading to decreased ATP production in AML cells (Fig. 7C–E). In addition, the presence of CWI1-2 remarkably inhibited colony-forming ability of blast cells from MAP-induced leukemia mice and greatly compromised self-renewal of LSCs/LICs (Fig. 7F–G).
Figure 7. CWI1-2 exhibits potent anti-leukemia efficacy in vitro and in vivo.
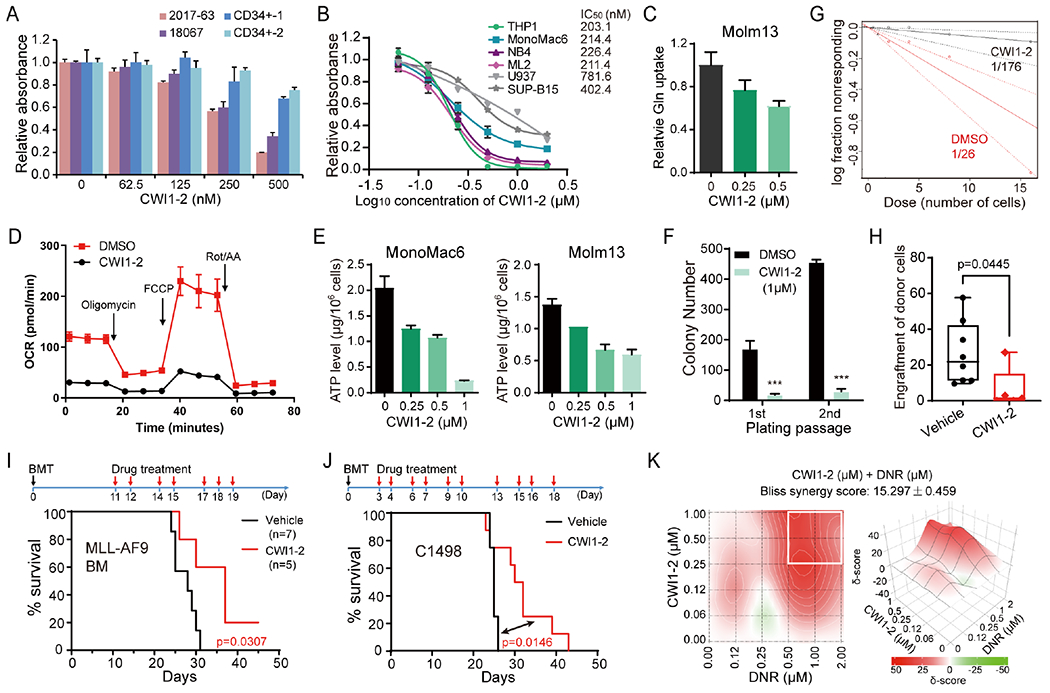
(A) MTT assays of patient-derived AML cells or CD34+ CB cells from healthy controls in the presence of CWI1-2. (B) MTT assays and the IC50 values of CWI1-2 in leukemia cell lines after 48 hours of treatment. (C-E) AML cell lines were treated with indicated concentration of CWI1-2 for 24 hours and subjected to the examination of glutamine uptake (C), OCR (D), and ATP levels (E). (F) Effect of CWI1-2 on the colony-forming/replating capacity of MA9-immortalized mouse HSPCs. (G) The in vitro LDA using MA9-immortalized mouse HSPCs. (H) Engraftment of donor cells in the PB of recipient mice after transplanting with BM cells from MA9-induced leukemic mice and subsequent i.v. injection of CWI1-2 or vehicle control. (I) Kaplan-Meier survival curves of recipient mice in (H). (J) Kaplan-Meier survival curves of mice transplanted with C1498 cells and treated with CWI1-2 or vehicle control. Days of BMT and drug treatment were shown in the upper panels in (I) and (J), with black arrows indicating BMT while red arrows indicating drug treatment. (K) Synergistic effect of CWI1-2 with DNR on inhibition of the survival/growth of C1498 cells as determined by the Bliss independent model. Drug combinations with the strongest synergistic effects are outlined with white squares. δ-score represents the percentage of response beyond expectation due to drug interactions. Data are represented as mean ± SD. ***p < 0.001; t test. See also Figure S7.
To test the in vivo efficacy of CWI1-2, we conducted proof-of-concept animal model studies. Pharmacokinetic studies showed that the half-life of CWI1-2 is approximately 18.702 hours when given intravenously at a single dose corresponding to 5 mg/kg in mice (Fig. S7C). Secondary BMTs were then performed using blast cells from MAP-induced leukemia mice or C1498 murine AML cells, followed by intravenous (i.v.) injection of CWI1-2 at 5 mg/kg once per day for 7-10 days. PB bleeding and subsequent flow cytometric analysis revealed a significant inhibition of leukemia engraftment in CWI1-2 treated mice (Fig. 7H). Consequently, CWI1-2 treatment significantly delayed leukemia onset and prolonged the survival of BMT recipient mice (Fig. 7I–J). Importantly, monitoring of body weight from the day of drug treatment for 12-18 days did not show any reduction of body weight upon CWI1-2 treatment (Fig. S7D). These data together suggest that CWI1-2 is an effective and largely safe leading compound targeting IGF2BP2 for AML treatment.
Many of the targeted therapeutic agents for AML, including those approved by FDA such as FLT3 inhibitor (midostaurin) and BCL2 inhibitor (venetoclax), exhibited much stronger efficacy when used in combination than used alone 39, 40. Aspired by these findings, we tested whether combined use of CWI1-2 with other AML therapeutic agents could provide better AML killing effect. Indeed, we found that CWI1-2 exhibited synergistic effects with daunorubicin (DNR) or homoharringtonine (HHT) in inhibiting AML cell viability/growth, but not with decitabine (DAC) (Fig. 7K and Fig. S7E), which shed light for the future optimization of CWI1-2-based treatment of AML.
Discussion
Although m6A modification has been shown to play important roles in the pathogenesis of various types of cancers 3, 4, 41, whether and how it regulates cancer metabolism, besides glycolysis 42, remain largely unknown. In this study, we report that m6A modification plays a key role in the regulation of Gln metabolism in AML, in which IGF2BP2 mediates the effect of m6A modification on the expression of critical genes, e.g., MYC, GPT2, and SLC1A5. By positively regulating mRNA stability and translation of these transcripts, IGF2BP2 fuels the TCA cycle and maintains mitochondria activity, allowing for the rapid proliferation and survival of AML cells. Together with our previous report 42, our studies indicate that m6A modification influences many aspects in cancer metabolism.
Glucose and Gln are two major energy sources of proliferating tumor cells by supplying cells with bioenergy and intermediates for macromolecular synthesis, mainly through glycolysis and mitochondrial oxidative phosphorylation (OXPHOS) 43, 44 Despite the Warburg effect referring to the finding that cancer cells are more dependent on glycolysis than OXPHOS for ATP production 45, increasing studies in recent years have revealed that cancer stem cells, including LSCs/LICs, are dependent on OXPHOS for survival 46, 47, 48, 49. Being an essential component of OXPHOS, TCA cycle requires intermediates catabolized from amino acids, fatty acids, and glucose. Our findings that IGF2BP2 is highly expressed in LSCs/LICs and promotes AML progression and LSC/LIC self-renewal through regulating Gln metabolism provide further evidence supporting the critical roles of Gln metabolism and OXPHOS in AML. Interestingly, IGF2BP2 was previously shown to affect mitochondrial energy production in glioblastoma cancer stem cells, likely through providing mRNA delivery to mitochondria and regulating respiratory complex assembly 46. Our data suggest a distinct mechanism in AML cells, in which IGF2BP2 acts as an m6A reader to stabilize and promote translation of mRNAs that encode proteins involved in Gln uptake and metabolism. Whether this m6A-related mechanism is conserved among different cancer types remains to be explored. Nonetheless, since LSCs/LICs are considered as the origin and the key therapeutic target of AML, the metabolic vulnerability of reliance on Gln metabolism provides a potential therapeutic target for future AML therapies.
IGF2BP2 is an oncofetal gene that is lowly expressed in normal adult human tissues, including bone marrow, colon, kidney, salivary gland, small intestine, and testis compared to fetal liver 50, and is overexpressed in a various types of cancer, such as glioblastoma, liver cancer, and breast cancer, therefore making it a promising therapeutic target in cancer 46, 51. We show here that IGF2BP2 is elevated in AML and has an even higher expression level in LSCs/LICs than in bulk AML cells. Moreover, silencing of IGF2BP2 dramatically induced terminal differentiation of AML cells and inhibited the engraftment of patient-derived AML cells and AML progression in immunodeficient mice, but showed minor effects on cytokine-induced in vitro differentiation of human normal CD34+ cord blood cells, highlighting a therapeutic window of targeting IGF2BP2 for AML treatment. Moreover, our proof-of-concept studies led to the discovery of a lead compound, CWI1-2, that directly binds IGF2BP2 and exhibits promising anti-leukemia efficacy both in vitro and in vivo by suppressing IGF2BP2’s m6A reader activity, while showing minimal side effects. Further investigations on the optimization of CWI1-2 and on the testing of its combinational therapeutics with other therapeutic agents are warranted.
In summary, our studies demonstrate the vital role of IGF2BP2, as an m6A binding protein, in controlling Gln metabolism during the pathogenesis of AML, and highlight that targeting of IGF2BP2 is an effective therapeutic strategy for AML and other types of IGF2BP2-overexpressing cancers.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact Jianjun Chen (jianchen@coh.org).
Materials availability
Plasmids generated in this study will be made available upon request and completion of a Material Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice and Animal Housing
The Mettl14 inducible knockout mouse (Mettl14fl/flCreERT mice) in C57BL/6 background was kindly provided by Dr. Chuan He (University of Chicago). The Igf2bp2 floxed mouse (Igf2bp2fl/fl) was purchased from The Jackson Laboratory. C57BL/6 (CD45.2) mice were purchased from Envigo (Indianapolis, IN, USA) or GemPharmatech Co. Ltd (Nanjing, China), and B6.SJL (CD45.1) mice were purchased from the Charles River Laboratories (Wilmington, MA, USA). NRGS mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All laboratory mice were maintained in the animal facility at City of Hope (COH), University of Cincinnati (UC), or Guangzhou Institute of Biomedicine and Health, Chinese Academy of Sciences (GIBH). All animal experiments were approved by Institutional Animal Care and Use Committee (IACUC) of COH, UC, or GIBH.
Mouse bone marrow transplantation (BMT)
These assays were conducted as described previously 13 with some modifications. Briefly, colony cells were transplanted via tail vein injection into lethally (900 cGy, 96 cGy/min) irradiated 7- to 9-week-old B6.SJL (CD45.1) or C57BL/6 (CD45.2) recipient mice. For each recipient mouse, 0.1-0.2x106 donor cells from CFA assays and a radioprotective dose of whole bone marrow cells (1x106) freshly harvested from a B6.SJL (CD45.1) or C57BL/6 (CD45.2) mouse were transplanted. For secondary BMT, BM cells from primary leukemic mice were transplanted into sublethally (400-480 cGy, 96 cGy/min) irradiated 7- to 9-week-old B6.SJL (CD45.1) recipient mice. For BMT using mouse C1498 cells, C57BL/6 mice were transplanted with 0.5-2 x106 C1498 via tail vein injection. In some cases, two doses of busulfan (20 mg/Kg) were given to recipient mice before C1498 cells were injected to promote engraftment. For BMT using Lin− Igf2bp2fl/fl BM cells co-transduced with MA9 and Cre-ERT2-IRES-GFP vectors (Cre) or ERT2-IRES-GFP (EV; as a negative control), 1.0 x106 donor cells from CFA assays and a radioprotective dose of whole bone marrow cells (1x106) freshly harvested from a B6.SJL (CD45.1) were transplanted. 10 days post-transplantation, Tamoxifen (TAM, 75 mg/kg) was injected intraperitoneally to induce Igf2bp2 KO every other day for 5 times in total. Leukemic mice were euthanized by CO2 inhalation when they showed signs of systemic illness. BM cells were isolated from both tibia and femur, and 50,000 cells were loaded for cytospin preparation. BM cytospin and blood smear slides were stained with Wright-Giemsa (Polysciences). Portions of the spleen and liver from leukemic mice were collected, fixed in formalin and embedded in paraffin before they were sectioned and stained with hematoxylin and eosin (H&E).
Leukemic patient samples and normal hematopoietic cell samples
The leukemic samples were obtained with informed consent at time of diagnosis or relapse at Cincinnati Children’s Hospital Medical Center (CCHMC) and COH, and were approved by the corresponding institutional/hospital review board. Leukemia blasts and mononuclear cells (MNCs) were purified using NycoPrep 1.077A (Axis-Shield, Oslo, Norway) or Ficoll-Paque PLUS (GE Healthcare Life Sciences). Normal MNC, CD34+ hematopoietic stem/progenitor cells (HSPCs), and CD34− cells were purified from cord blood of healthy donors and leukemic patients from CCHMC or COH using Ficoll-Paque PLUS and CD34+ beads (Miltenyi Biotec). The sample size of leukemic samples was not pre-determined. Samples were allocated to different groups according to their cytogenetic characteristics.
Cell culture
U937, Kasumi-1, HEK293T were all purchased from American Type Culture Collection (ATCC). U937, MonoMac6, and MOLM13 cells were authenticated by Short Tandem Repeat (STR) analysis. U937, Kasumi-1, and MOLM13 cells were maintained in regular RPMI-1640 medium (Invitrogen, Carlsbad, CA) containing 10% FBS, 1% HEPES and 1% penicillin-streptomycin. For MonoMac6 cells, 2 mM L-Glutamine, 1 3 Non-Essential Amino Acid, 1 mM sodium pyruvate, and 9 mg/ml insulin (Invitrogen) were added to the regular RPMI-1640. HEK293T cells were grown in DMEM medium supplemented with 10% FBS and 1% penicillin-streptomycin. Primary CD34+ blasts cells from AML patients or patient-derived AML cells were cultured in IMDM medium supplemented with 20% FBS and 10 ng/mL of SCF, TPO, Flt-3L, IL-3, and IL-6.
METHODS DETAILS
Plasmid construction
Wild-type and KH3-4 domain mutant human IGF2BP2 sequences were PCR-amplified from corresponding pcDNA3-based vectors gifted by Hüttelmaier (Martin Luther University, Germany) with an HA-tag coding sequence inserted before the start codon, and subcloned into the pCDH lentiviral vector (CD513B-1, System Biosciences) through the XbaI and EcoRI enzymatic sites or into the pSIN4 vector. GPT2 cDNA ORF with an N-tenninal HA tag (HG22770-NY) was purchased from Sino Biological and subcloned into the pCDH lentiviral vector through the XbaI and NotI enzymatic sites. Human SLC1A5 coding sequence was reverse transcribed and PCR-amplified from 293T total RNA, with an N-tenninal HA tag being added during PCR, and cloned into the pCDH lentiviral vector through the XbaI and EcoRI enzymatic sites. shRNA vectors targeting human or mouse IGF2BP2, MYC, GPT2, and SLC1A5 were either purchased from GE Dharmacon or constructed by synthesizing shRNA-encoded DNA oligos and cloning into the pLKO.1 vector (Addgene). Mature antisense sequences of shRNAs were listed in Supplementary Table S1.
Cell proliferation/growth, apoptosis and differentiation assays
The cell proliferation/growth was assessed by MTT (G4000, Promega, Madison, WI) or by cell counting using Trypan blue staining or AO/PI staining. For growth competition assays, cells were transduced with GFP expression vector to obtain ~50% GFP+ cell population. Cells were then subjected to flow cytometric analysis at indicated days to determine the percentage of GFP+ cells overtime. For apoptosis assays, FITC Annexin V apoptosis Detection Kit 1 (BD Biosciences, San Diego, CA) was used following the manufacturer’s manuals. Apoptosis was examined by flow cytometry on a BD LSRFortessa or FACSAria III analyzer (BD Biosciences) after staining cells with FITC-Annexin V and propidium iodide (PI). For differentiation assays, cells were harvested and washed with chilled PBS, followed by staining with APC anti-CD 14 (17-0149-41, eBioscience; or 367118, BioLegend) and PE anti-CD11b (101208, BioLegend) or Pacific Blue anti-CD11b (101224, BioLegend) and analyzed on a BD LSRFortessa or FACSAria III analyzer (BD Biosciences).
Virus preparation and infection
These assays were conducted as described previously 8, 13. Briefly, retroviruses or lentiviruses were produced in 293T cells by co-transfection of individual expression construct with the pCL-Eco packaging vector (IMGENEX, San Diego, CA) or the pMD2.G:pMDLg/pRRE:pRSV-Rev packaging mix (individually purchased from Addgene), respectively. The virus particles were harvested at 48 and 72 hours after transfection and added to cells with or without concentrating, and one or two rounds of “spinoculation” were performed to allow the infection of viruses. In some cases, when co-infecting AML cells with overexpression and shRNA viruses, overexpression viruses were spinned down first in RetroNectin-coating plates before cells and shRNA viruses were added for spinoculation.
In vitro colony-forming and replating (CFA) assay
Bone marrow (BM) cells were collected from 5- to 7-week-old wild-type, Mettl14fl/flCreERT, or Igf2bp2fl/fl mice, and BM progenitor (HSPC, i.e., Lin− or c-kit+) cells were enriched with the Mouse Lineage Cell Depletion Kit or the CD117 microbeads (Miltenyi Biotec). BM progenitor cells were co-transduced with different combinations of retroviruses or lentiviruses as indicated through two rounds of “spinoculation”, then plated into mouse methylcellulose medium (1201, ReachBio or HSC006, R&D) supplemented with 10 ng/ml of murine recombinant IL-3, IL-6, GM-CSF and 30 ng/ml of murine recombinant SCF. For HSPCs from Igf2bp2fl/fl mice, Cre-ERT2-IRES-GFP (Cre) or ERT2-IRES-GFP (EV; as a negative control) lentiviruses were used additionally to transduce cells. 4-OHT (1 μg/mL. Sigma-Aldrich), G418 (1.0 mg/mL, GIBCO BRL, Gaithersburg, MD) and/or puromycin (2.5 mg/mL, Sigma-Aldrich) were added when necessary. Cultures were incubated at 37°C in a humidified atmosphere of 5% CO2 for 6 to 7 days and serial replating was performed by collecting colony cells and replating them in methylcellulose medium every 5-7 days. Colonies were counted for each passage.
For CFA assays using human primary cells, freshly sorted CD34+/CD34− cells or CD34+ HSPCs transduced with lentivirus were seeded into MethoCult H4434 Classic medium (StemCell Technologies) or Human Methylcellulose Complete Media (HSC003, R&D) with the addition of 2.5 mg/mL puromycin when necessary. Cultures were incubated at 37C in a humidified atmosphere of 5% CO2 for 7-10 days before counting.
Limiting dilution assay (LDA)
For in vivo LDAs, frozen BM cells collected from primary BMT mice with IGF2BP2 overexpression that developed full-blown leukemia were thawed and injected into lethally irradiated wild-type mice through tail vein with four different doses (0.1×106, 0.01×106, 0.001×106, 0.0001×106) of donor cells for each group. The number of recipient mice developed full-blown leukemia within eight weeks post-transplantation was counted for each group with each dose of donor cells. ELDA software 68 was used to estimate the frequency of leukemia stem/initiating cells (LSCs/LICs).
For in vitro LDAs, HSPCs from wild-type C57BL/6 mouse transduced with MLL-AF9 retroviruses together with shRNA lentiviruses targeting mouse Igf2bp2, Myc, Gpt2, or Slc1a5 were seeded into Mouse Methylcellulose Base Media (HSC006, R&D) supplied with 10 ng/mL of human recombinant IL-6 (PeproTech), murine recombinant IL-3 (PeproTech), GM-CSF (PeproTech) and 30 ng/ml of murine recombinant SCF (PeproTech), along with 2 μg/ml of puromycin (Sigma-Aldrich). Four days later, the colony cells were collected and replated into 96-well plates with six different doses of cell number for each group. The number of wells with MLL-AF9 colonies was counted 7 to 10 days later, and the estimated frequencies of LSCs/LICs were calculated using the EFDA software.
RNA extraction and real time quantitative PCR (RT-qPCR) analysis
Total RNA was isolated using the miRNeasy mini kit (Qiagen) or the TRIzol reagent (Thermo fisher Scientific) according to the manufacturer’s instructions. For cDNA synthesis, 500-1,000 ng of total RNA was reverse-transcribed into cDNA in a total reaction volume of 10 μL with the QuantiTect Reverse Transcription Kit (Qiagen) or in a 20 μL reaction volume using HiScript III RT SuperMix for qPCR (+gDNA wiper) (Vazyme, China) according to the manufacturer’s instructions. Quantitative real-time PCR (qPCR) was then performed in a 10 μL reaction volume system containing 0.5 μL diluted cDNA using Maxima SYBR green qPCR master mix (Thermo Fisher) or Applied Biosystems PowerUp SYBR Green Master Mix (Thermo Fisher) on the QuantStudio 7 Flex PCR system (Thermo Fisher Scientific). ACTB was used as internal control for gene expression evaluation, while 18S rRNA was used as internal control for RNA stability assays.
RNA-seq and data analysis
Total RNA from MonoMac6 cells with or without IGF2BP2 or YTHDF2 knockdown, or from DMSO or CWI1-2 treated MonoMac6 cells was isolated using the TRIzol reagent (Thermo fisher Scientific). Fibrary construction of 1 μg RNA per sample was made using the NEBNext® Ultra™ RNA Fibrary Prep Kit for Illumina® (NEB, USA) following manufacturer’s recommendations. PCR products were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2100 system. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. The library preparations were sequenced on an Illumina NovaSeq platform and 150 bp paired-end reads were generated. The raw RNA-seq reads were quality checked by FastQC (version 0.11.5) and then adaptors trimmed by Cutadapt (version 3.4). The processed reads were mapped to human transcriptome built with gene annotations (hg38, GENCODE v37) 69 by STAR (version 2.7.2b) 59 with parameters recommended by ENCODE project. The aligned results were then passed to RSEM (version 1.2.31) 57 to estimate the read count of genes. Finally, normalization and differential gene expression (DEG) were done using DESeq2 (v1.32.0) with the following parameters: sfType=poscounts, test=Wald and lfcShrink(type=none).
RNA immunoprecipitation (RIP)
RNA immunoprecipitation was performed as previously described with some modifications. In brief, 10 million cells were harvested and washed with cold PBS, followed by 254 nm UV crosslinking. Cells were lysed with 1 mL of RIP buffer (150 mM KCl, 25 mM Tris, pH7.4, 5 mM EDTA, 0.5 mM DTT, 0.5% IGEPAL,100 U/mL RNAase inhibitor SUPERase•in™, and cocktail proteinase inhibitors), and sonicated for 10 cycles with 30s on and 30s off for each cycle using Bioruptor (Diagenode). 10% of lysate was saved as input. The rest of the lysate was pre-cleared with protein A/G magnet beads, and then subjected to immunoprecipitation using anti-IGF2BP2 or anti-HA antibodies at 4°C for 2 h, followed by incubation with protein A/G magnet beads at 4°C overnight with rotation. Beads were washed with RIP buffer for three times. Immunoprecipitated samples were subjected to DNase and Proteinase K digestion, followed by total RNA isolation (in parallel with the input samples) using the TRIzol reagent.
Co-immunoprecipitation (co-IP)
Molm13 cells were lysed with 1 mL NP40 buffer (150 mM NaCl, 1.5 mM MgCl2, 0.5% NP40, 50 mM Tris-HCl at pH8.0) supplemented with proteinase inhibitors. After sonication (Qsonica Q700: 40% Amplitude, 4°C, 5s on, 10s off, 48 cycles), 1 mg of cell lysates were used for immunoprecipitation with 1 μg of IGF2BP2 (#14672S, CST) antibody or the corresponding IgG (rabbit IgG from CST, #2729S). Proteins were conjugated to Protein A/G Magnetic Beads (Thermo Fisher Scientific) by incubation at 4 °C overnight. Beads were then washed for 3 times with IP washing buffer (10 mM Tris-HCl pH7.5, 1 mM EDTA, 1 mM EGTA, 150 mM NaCl, 1% Triton-X, 0.2 mM sodium orthovanadate). Proteins were dissolved in 1×SDS buffer and analyzed by western blotting.
RNA stability assays
MonoMac6 cells with or without METTL14 or IGF2BP2 knockdown were treated with actinomycin D (A9415, Sigma-Aldrich) at a final concentration of 5 mg/mL and collected at indicated time points. Total RNA was extracted by EastepTM Super Total RNA Extraction Kit (Promega) and analyzed by qPCR assays. The half-life of mRNA was calculated as previously reported.
Polysome fractionation
Polysome fractionation was performed following the reported protocols with some modifications. Briefly, 100 μg/mL cycloheximide (CHX) (Sigma-Aldrich) was added into MonoMac6 cells with or without IGF2BP2 knockdown for 15 min before collection. Cells were harvested and washed with cold PBS containing 100 μg/mL CHX twice, and then lysed with lysis buffer for 30 min on ice, followed by subsequent ultracentrifugation on 5%-50% sucrose cushion. The samples was then fractionated into 17 fractions and analyzed with ECONOUV monitor (Biocomp) and Piston Gradient Fractionator (Biocomp). Equal volume of TRIzol reagent was added to the 17 fractions and RNA was isolated and subjected to qPCR analysis. ACTB was used as internal control in the qPCR analysis.
Ribosome profiling (Ribo-seq)
Cells were treated with 100 μg/mL CHX for 15 min before collection. A total of 107 cells was washed twice with ice-cold PBS containing 100 μg/mL CHX. Ribosome profiling was performed using Epi™ Ribosome Profiling Kit (Epibiotek, R1814) by Guangzhou Epibiotek Co.,Ltd.. RPFs (ribosome-protected RNA fragments) were extracted using RNA clean&ConcentratorTM-5 kit (ZYMO, R1016). EpiTM RiboRNA Depletion Kit (Human/Mouse/Rat) (Epibiotek, R1805) was used for rRNA depletion. Sequencing libraries were constructed using QIAseq miRNA Library kit (QIAGEN, 1103679). Parallel mRNA sequencing was performed for same batch of cells without CHX treatment by using the VAHTS Stranded mRNA-seq Library Prep Kit for Illumina V2 (Vazyme Biotech, NR612-02) for library preparation. Prepared libraries were sequenced on an Illumina NovaSeq platform. The raw reads of ribo-seq were first quality checked by FastQC and then adaptors trimmed by Cutadapt with the following parameters: -a AACTGTAGGCACCATCAAT -m 25 -M 34. Any processed reads that mapped to tRNA, snRNA and rRNA were discarded by using bowtie and then the remained reads were mapped to the human transcriptome by use of STAR with following parameters: --outFilterMultimapNmax 20 --outFilterMismatchNoverReadLmax 0.08. The aligned results were then passed to RSEM to estimate the read counts of genes. To obtain the raw read count and background expression levels of genes measured with Transcript Per Million (TPM), we processed the mRNA sequencing reads by mapping with STAR and estimating with RSEM, which were similar with RNA-seq analysis.
To avoid the over-normalization, we then normalized the read count of genes from ribo-seq and RNA-seq by using the sequencing depth of libraries and the median-of-ratio method, respectively. Finally, we estimated the translational efficiency of each protein coding gene as the following formula:
ChIP-seq analysis
The raw sequencing data of public ChIP-seq dataset (GSE79899) was downloaded and decompressed from GEO database by using SRAtoolkit (v3.0.0). The raw sequencing reads were then firstly quality controlled with FastQC and mapped to the human genome (hg38) by using bowtie2 70 (version 2.3.2). After alignments, we used Picard software (version 2.21.1) to mark and discard the PCR duplicates from aligned results. We then estimated the fragment sizes of ChIP-seq libraries with phantompeakqualtools 63 as the parameter “--extsize” of MACS2 and analyzed the significant binding or histone modification regions by utilizing MACS2 56 (version 2.2.7.1) with following parameters: -g hs --mfold 5 50 -p 0.01 --nomodel --shift 0 --keep-dup all -B --SPMR. Finally, the peaks with p-values less than 0.01 were recognized as significantly enriched regions and kept for downstream analysis. The normalized signals of aligned ChIP-seq tags were calculated by deeptools 64 (version 3.5.1).
Intracellular staining and flow cytometric analysis of IGF2BP2 expression
Patient-derived AML cells, primary AML cells, or cord blood MNCs were incubated with zombie yellow solution before staining with anti-human PE-CD34 antibody. Cells were then washed with PBS, fixed, and permeabilized, followed by incubation with primary anti-IGF2BP2 antibody (GTX33256, GeneTex) or isotype control. After washing with PBS and resuspending cell with permeabilization buffer, cells were incubated with Alexa Flour 488 conjugated anti-rabbit secondary antibody and subjected to flow cytometric analysis on a BD LSRFortessa™ X-20 Cell Analyzer. Zombie yellow negative cells (representing live cells) were divided into 2 groups according to the level of CD34, and IGF2BP2 protein level in each group was analyzed.
Western blot assay
Cells were counted and washed twice with PBS, followed by direct lysis in 1×SDS buffer (100 μL for 1 million cells) at 95 °C for 10 min. Equal volumes of cell lysates were separated by SDS-PAGE and transferred onto PVDF membranes. The membranes were blocked with 5% non-fat milk, incubated sequentially with primary and secondary antibodies, and detected by immunoblotting with the Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific).
Metabolite extraction and mass-spectrometry-based metabolomics analysis
Cells with different treatments (e.g., transduced with shRNAs against IGF2BP2, METTL3, METTL14, or scramble control, or co-transduced with IGF2BP2-sh3 and WT or MUT IGF2BP2) were cultured in their regular culture medium without glutamine but supplemented with 13C5;15N2-glutamine (2mM, Cambridge Isotope Laboratories, Inc.). 24 hours later, 1 × 106 cells were collected and rinsed with 150 mmol/L ammonium acetate (pH 7.3), and 100 μL cold 80% methanol (Optima* LC/MS, Fisher Scientific) was added to cells. 5 nmol norvaline (Sigma-Aldrich) was added to each sample as internal standard. Samples were then vortexed every 5 minutes for three times and spun down at top speed for 5 minutes. The supernatant was transferred to a new Eppendorf tube, and pellet was resuspended in 50 μL cold 80% methanol for second extraction. Combined 150 μL supernatant was dried on Vacufuge Plus (Eppendorf) at 30°C. Extracted metabolite samples were stored at −80°C. The mass spectrometry-based analysis of extracted metabolites was conducted at UCLA Metabolomics Center. Metabolites were resuspended in 50% acetonitrile (ACN), and one tenth of the sample was analyzed with a Thermo Scientific Q Exactive mass spectrometer coupled to the UltiMate 3000 or the Vanquish Flex UPLC chromatography systems. The chromatographic separation was performed with 5 mM NH4AcO (pH 9.9, mobile phase A) and ACN (mobile phase B) at a flow rate of 200 μL/min on a Phenomenex Luna 3u NH2 100A (150×2.0 mm) column. Maven (v8.1.27.11; http://maven.princeton.edu/index.php) was used to quantify targeted metabolites by Top Area using accurate mass measurements (±5 ppm) and prior established retention times. Data were normalized to the cell number. T-test was used to test the significance of abundance changes of metabolites between control and knockdown groups, and the compounds with p < 0.05 were kept for downstream analysis. MetaboAnalystR R package (v2.0) was then used to analyze the enriched metabolic pathways of significantly changed metabolites with default parameters.
Seahorse XF Cell Mito Stress Test Profiling
ORC were measured using a Seahorse XF Cell Mito Stress Test Kit (Agilent Technologies) according to the manufacturer’s instructions. Briefly, MonoMac6 (0.06 million cells/well) or Molm13 (0.1 million cells/well) cells were washed with XF RPMI medium (Agilent Technologies) supplemented with 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose, seeded in XF96 Cell Culture Microplate coated with Cell-Tak (Corning), and incubated in XF RPMI medium (Agilent Technologies) supplemented with 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose for 30 min to 45 min at 37 °C (non-CO2 incubator). OCR was determined in the presence of the mitochondrial inhibitor oligomycin (1.5 μM), mitochondrial uncoupling compound carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (0.5 μM for MonoMac6 cells and 1.0 μM for Molm13 cells), and respiratory chain inhibitor Rotenone & antimycin A mixture (Rot/AA) (0.5 μM).
Glutamine (Gln) uptake assay
Glutamine level in the culture media was measured using Glutamine/Glutamate-Glo™ Assay kit (Promega) according to the manufacturer’s instructions. In brief, 0.2 million/mL of cells incubated with fresh complete medium were plated onto 96-well plate. Aliquot of fresh complete medium was saved for measurement of glutamine level at the starting time point (T0). After 24 h, the supernatant was collected (T24). 25 μl of the supernatant at T0 or T24 was mixed with 25 μl of glutaminase, followed by the addition of 50 μl of Glutamate Detection Reagent in a 96-well white plate. Glutamate concentrations were measured in the presence or absence of glutaminase Enzyme solution. The glutamine concentration was calculated by total glutamate subtracting the glutamate in the absence of Glutaminase Enzyme solution, and glutamine uptake was calculated by subtracting glutamine concentration at T24 from that at T0.
Cellular ATP level measurement
ATP levels were measured by the ApoSENSOR ATP Cell Viability Bioluminescence Assay Kit (K254, BioVision) according to the manufacturer’s instructions. Brief, 10 μl of indicated cells (containing 5000 cells) was transferred into luminometer plate, followed by the addition of 100 μl of the Nucleotide Releasing Buffer. Then, 10 μl ATP Monitoring Enzyme was added to the cell lysate, and the plate was read within ~ 1-2 min in a Cytation 5 luminometer (BioTek Instruments, Inc) or a GloMax® Navigator (Promega).
Bst DNA polymerase-mediated cDNA extension and qPCR (BST-qPCR)
Total RNA was isolated from MonoMac6 cells with or without MKTTL14 depletion. BST DNA polymerase-mediated cDNA extension was performed at 60 °C for 40 min in a 25 μL reaction containing 200 ng of total RNA, 2.5 μl of 10× Isothermal Amplification Buffer II, 1.5 μL MgSO4 (100 mM), 3.5 μL dNTP Mix (10 mM), 1 μL site-specific reverse primer, and 1.0 μL Bst 3.0 DNA polymerase (8000 U/mL, New England Biolabs). The cDNAs were used for qPCR detection, with cDNAs transcribed from non-m6A site primer being used as internal controls.
Virtual screening
To identify possible inhibitors for IGF2BP2, our in-house developed virtual screening method LiVS was performed to obtain the initial compounds from NCI DTP library for experimental validations. The structural model of KH3-4 domain of human IGF2BP2 was built by using its chicken homology (PDB id 2n8l) with sequence identity of about 95%. LiVS is a full-coverage and multiple-stage structure-based virtual screening pipeline that has been successfully employed to develop inhibitors for proteins related to cancers 71, 72, 73 and HIV 74. The whole NCI DTP compound library with about 260,000 small molecules was fully docked to human IGF2BP2 by using Glide 75 in Schrodinger software. The three Glide docking precision modes (HTVS, SP, and XP) were utilized in tandem to reduce computation time. First, each compound in NCI DTP library was docked by using the lowest precision but fastest HTVS mode. Then the top 10,000 compounds with highest docking scores were picked to re-dock at SP mode with higher precision. At the third docking stage, the top 1,000 compounds resulted from SP mode were selected to re-score at XP mode with highest precision but most time-consuming. A final list of 150 compounds was produced from LiVS by a comprehensive consideration of druglikeness, ADMET properties and other factors. Among them, 128 compounds were available to order from NCI for validation.
Molecular docking
The crystal structure of IGF2BP2 was downloaded from the Research Collaboration for Structural Bioinformatics Protein DataBank (RCSB-PDB) (PDBID: 6rol) and opened by MOE (Molecular Operating Environment) software. The IGF2BP2 crystal structure was obtained by removing the small molecules (PEG, glycerol, etc.) from the complex. Chemdraw was used to draw the molecular structure of CWI1-2. Molecular docking was performed using MOE with the following settings: docking site = AllAtoms; docking pocket = amino acid within 4.5 Å of the ligand molecule; docking mode = RigidReceptor; Score = GBVI/WSAΔG; Poses = 5. The docking patterns were observed in the 3D rendering area of the MOE software and the ligand-receptor interactions were analyzed one by one.
Cellular thermal shift assay (CETSA)
CETSA was conducted with intact Molm13 cells as published previously (Jafari et al., 2014). Briefly, 8 × 106 Molm13 cells were pretreated with 50 μM DMSO or compound for 2 hours. Cells from each group were collected, washed once with cold PBS, and re-suspended in 0.8 ml PBS supplemented with protease inhibitor cocktail. The cell suspension was then aliquoted into 14 different 0.2 ml PCR tubes with 50 μl in each tube, heat shocked in the Bio-Rad T100 Thermal Cycler at indicated temperatures for 3 minutes to denature proteins, and immediately cooled down to room temperature for another 3 minutes. Finally, the samples were subjected to three freeze-thaw cycles with dry ice and a Thermal Cycler set at 25°C to lyse cells, and centrifuged at 20,000 g for 20 min at 4°C. The supernatant was boiled with 5× SDS loading buffer for western blot analysis. The band intensities of the target protein were quantified using TotalLab software and plotted.
Drug affinity responsive target stability (DARTS) assays
DARTS was conducted to evaluate the direct binding of IGF2BP2 protein with compounds according to a published protocol (Lomenick et al., 2009) with modifications. Briefly, 5×107 cells were lysed in 1 mL M-PER buffer (78501, Thermo Fisher Scientific) for 10 minutes on ice. After centrifuging at 18000×g for 10 min at 4 °C, the supernatant was transferred into a fresh tube and mixed with one ninth volume of the 10×TNC buffer (500 mM Tris-HCl, pH 8.0, 500 mM NaCl, 100 mM CaCl2) before determining protein concentration by BCA assays. 300 μg of the lysate was incubated with DMSO or compound at indicated concentrations for 1 hour at room temperature, followed by Pronase (10165921001, Roche, 1:400-1:500) digestion for an additional 30 minutes at room temperature. The reaction was quenched by adding protein inhibitor cocktail and immediately placing the samples on ice. SDS loading buffer was then added and samples were subjected to western blot analysis. ACTB, GAPDH, or DHFR were used as negative controls.
Split-luciferase cellular thermal shift assay (splitLuc-CETSA)
The split-Luc tag was cloned into the C terminal of IGF2BP2 in the pCDH-based vector using Infusion cloning reagents (Takara) as previously described 76. 293T cells were transduced with IGF2BP2-splitLuc through lentivirus infection. After selection, cells were harvested and resuspended at 1 × 106cells/mL in CETSA buffer containing DPBS and supplemented with 1 g/L glucose and 1× Halt protease inhibitor cocktail. Samples were aliquoted to PCR strips at 30 μL/tube, and certain amount of compound or vehicle was added and incubated at 37 °C for 1 h. Samples were then heated for 5 min using a pre-heated thermal cycler (the temperature range is 37 °C-41 °C-45 °C-49 °C-53 °C-57 °C-61 °C-65 °C), allowed to equilibrate to room temperature, and 6 μL of 6% NP40 was added to each well. The 11S part of luciferase (GenScript) and furimazine substrate (Promega) were added to the samples at final concentrations of 100 nM and 0.5×, respectively. Samples were transferred to 96 half-area white plate and the luminescence intensity was read using the Synergy Neo2 Multi-Mode Microplate Reader (Biotek).
Fluorescence protein thermal shift assay
The fluorescence protein thermal shift assay was conducted according to a previous repot38. The recombinant IGF2BP2 protein was purified from 293T cells using the Mammalian Protein Purification System (Promega). The thermal shift assay was conducted in the QuantStudio 12K Flex Real-Time PCR System (ThermoFisher) using the Protein Thermal Shift kit (ThermoFisher) following the manufacturer’s instructions. Briefly, we used 5 μg purified IGF2BP2 protein in a 20 μl thermal shift reaction with indicated concentration of CWI1-2 or vehicle control. The plate was heated from 25 to 99 °C with a heating rate of 0.05 °C/s in the Real-Time PCR System. The fluorescence intensity was measured with Excitation: x4 (580±10) and Emission: m4 (623±14). The fluorescence Tm values were calculated by Boltzmann fitting using the Protein Thermal Shift™ Software v1.4 (ThermoFisher).
RNA pulldown assays
RNA pulldown was conducted as previously reported with some modifications. Biotin-labelled RNA oligonucleotides with m6A modification was denatured and immobilized onto streptavidin magnetic beads. RNA-conjugated streptavidin beads were washed, resuspended in binding buffer (20 mM Tris, 200 mM NaCl, 6 mM EDTA, 5 mM potassium fluoride, 5 mM β-glycerophosphate, 2μg/mL aprotinin at pH 7.5), and equally aliquoted. Recombinant Flag tagged IGF2BP2 protein and compound at indicated concentration were added and incubated overnight at 4 °C with rotation. After extensive washing, RNA-protein complexes were dissolved in 1× SDS buffer and subjected for western blot analysis using the anti-Flag antibody.
Pharmacokinetics
We set three groups (6 rats/group) of SD rats with a single intravenous bolus injection of different doses of CWI1-2: high dose with 10.302 mg/kg, medium dose with 3.434 mg/kg, and low dose with 1.145 mg/kg, corresponding to the dosage of 15 mg/kg, 5 mg/kg, and 1.667 mg/kg in mice, respectively. Peripheral blood samples were collected from the orbital plexus at the following 12 time points: 5 min, 15 min, 30 min, 1 hour, 2 hours, 4 hours, 8 hours, 12 hours, 24 hours, 48 hours, 72 hours, and 96 hours after administration. Then, the plasma concentrations of CWI1-2 were determined by high performance liquid chromatography (HPLC) using an Agilent 1260 HPLC with a GP-C18 reversed-phase column with the following parameters: column chamber temperature: 30°C; liquid phase conditions: 0 min methanol:water = 50:50, 5 min methanol:water = 60:40, 20 min methanol:water = 95:5; detection wavelengths of 230, 254 and 280 nm. Only rats in the medium group showed a reasonable level and a time-dependent decrease of plasma concentration of CWI1-2, and thus were used for determining pharmacokinetic data of CWI1-2. The pharmacokinetic analysis software (DAS2.0) was applied to draw the Drug Concentration-Time Curve according to the least square method, and to calculate the pharmacokinetic parameters with the weight coefficient being set as 1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed and presented as mean±SD. Two-tailed Student’s t-test was used to compare means between groups as indicated; p < 0.05 was considered significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOUCE | SOUCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Rabbit anti-IGF2BP1 antibody | Cell Signaling Technology | Cat.: #8482, clone D33A2 |
| Rabbit anti-IGF2BP2 antibody | Cell Signaling Technology | Cat.: #14672, clone D4R2F |
| Rabbit anti-IGF2BP3 antibody | Bethyl Laboratories | Cat.: A303-426A, RRID: AB_10951696 |
| GPT2 (G-7) mouse mAb | Santa Cruz Biotechnology | Cat.: sc-398383 |
| c-Myc (D3N8F) Rabbit mAb | Cell Signaling Technology | Cat# 13987, RRID:AB_2631168 |
| ASCT2 Rabbit mAb | Cell Signaling Technology | Cat# 8057S, clone D7C12 |
| Asct2 Rabbit mAb | Cell Signaling Technology | Cat# 5345S, clone V501 |
| beta-Actin (8H10D10) Mouse mAb | Cell Signaling Technology | Cat# 3700, RRID:AB_2242334 |
| GAPDH antibody (0411) | Santa Cruz Biotechnology | Cat# sc-47724, RRID: AB_627678 |
| HA-Tag Rabbit mAb | Cell Signaling Technology | Cat# 3724, clone C29F4 |
| Anti-METTL14 antibody, rabbit | Sigma-Aldrich | HPA038002, RRID:AB_10672401 |
| Anti-METTL3 antibody [EPR18810] | Sigma | Cat# ab195352 |
| Donkey anti-rabbit IgG-HRP | Santa Cruz Biotechnology | Cat# sc-2077, RRID: AB_631745 |
| Goat anti-mouse IgG-HRP | Santa Cruz Biotechnology | Cat# sc-2055, RRID: AB_631738 |
| Anti-rabbit IgG (H+L), F(ab′)2 Fragment (Alexa Fluor® 488) | Cell Signaling Technology | Cat# 4412S |
| IGF2BP2 antibody | GeneTex | Cat# GTX33256 |
| CD45 antibody PE | BD Biosciences | Cat# 555483, RRID:AB_395875 |
| CD33 antibody FITC | BD Biosciences | Cat# 555626, RRID:AB_395992 |
| PE anti-mouse/human CD11b | eBioscience | Cat# 101208, RRID: AB_312791 |
| CD14 Monoclonal Antibody (61D3), APC | eBioscience | Cat# 17-0149-41, RRID: AB_10670627 |
| Anti-mouse CD45.2-PE | eBioscience | Cat# 12-0454-82, RRID:AB_465678 |
| Anti-Mouse CD45.1 PE | eBioscience | Cat# 12-0453-82, RRID:AB_465675 |
| Anti-Mouse CD45.2 APC | eBioscience | Cat# 17-0454-82, RRID:AB_469400 |
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
| Stbl3 E. coli | Transgene | Cat# CD521-02 |
| One Shot® Stbl3™ Chemically Competent E. coli | Life Technologies | Cat# C737303 |
| 5-alpha Competent E. coli | Transgene | Cat# CD201-02 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| Recombinant Human IGF2BP2 protein | Origene | Cat# TP305673 |
| L-Glutamine (200mM) | Thenno Fisher Scientific | Cat# 25030-081 |
| 13C5;15N2-glutamine | Cambridge Isotope Laboratories, Inc. | Cat# CNLM-1275-H-PK |
| MEM Non Essential Amino Acids Solution (100×) | Thermo Fisher Scientifific | Cat# 11-140-050 |
| Sodium Pyruvate (100mM) | Thermo Fisher Scientifific | Cat# 11360-070 |
| glucose | Thermo Fisher Scientifific | Cat# A2494001 |
| Halt protease inhibitor cocktail | Thermo Fisher Scientifific | Cat# 78442 |
| DPBS | Thermo Fisher Scientifific | Cat# 14040117 |
| 11S part of luciferase | GenScript | |
| Puromycin dihydrochloride | Sigma-Aldrich | Cat# P8833 |
| G418 | Thermo Fisher Scientific | Cat# 11811-031 |
| Actinomycin D | Sigma-Aldrich | Cat# A9415 |
| cycloheximide | Sigma-Aldrich | Cat# C1988 |
| Recombinant Human IL-6 | PeproTech | Cat# 200-06 |
| Recombinant Human IL-3 | PeproTech | Cat# 200-03 |
| Recombinant Human Flt3-Ligand | PeproTech | Cat# 300-19 |
| Recombinant Human SCF | PeproTech | Cat# 300-07 |
| Recombinant Human TPO | PeproTech | Cat# 300-18 |
| Recombinant mouse IL-3 | PeproTech | Cat# 213-13 |
| Insulin, human recombinant zinc solution | Thermo Fisher Scientifific | Cat# 12585014 |
| Penicillin Streptomycin | Thermo Fisher Scientifific | Cat# 15-140-122 |
| Recombinant mouse SCF | PeproTech | Cat# 250-03 |
| Recombinant murine GM-CSF | PeproTech | Cat# 315-03 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 4693159001 |
| Amersham ECL Prime Western Blotting Detection Reagent | Thermo Fisher Scientific | Cat# 45-010-090 |
| Pierce Protein A/G Magnetic Beads | Thermo Fisher Scientific | Cat# 88803 |
| Wright-Giemsa Stain | Polysciences | Cat# 24985 |
| Wright-Giemsa Stain/Buffer | Polysciences | Cat# 24984 |
| Cycloheximide solution | Sigma-Aldrich | Cat# C4859 |
| Bst 3.0 DNA Polymerase | New England Biolabs | Cat# M0374S |
| Busulfan | Cayman Chemical Company | Cat# 14843 |
| XF 1.0 M Glucose Solution | Seahorse Bioscience | Cat# 103577 |
| XF 100 mM Pyruvate Solution | Seahorse Bioscience | Cat# 103578 |
| XF 200 mM Glutamine Solution | Seahorse Bioscience | Cat# 103579 |
| CWI1-2 | TC Scientific Inc. | |
| BTYNB | Cayman Chemical Company | Cat# 25623 |
| NSC69557 | NCI | |
| DMSO | Sigma | Cat# D2650 |
| ss-m6A RNA | IDT | |
| Pronase from Streptomyces griseus | Roche | Cat# 10165921001 |
| Solutol HS-15 | MedChem Express | Cat# HY-Y1893 |
| IGF2BP2 WT protein | This study | |
| IGF2BP2 KH4 protein | This study | |
| IGF2BP2 KH3-4 protein | This study | |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Glutamine/Glutamate-Glo Assay | Promega | Cat# J8021 |
| EastepTM Super Total RNA Extraction Kit | Promega | Cat# LS1040 |
| Seahorse XF Cell Mito Stress Test Kit | Seahorse Bioscience | Cat# 103015-100 |
| ClonExpress Ultra One Step Cloning Kit | Vazyme | Cat# C115-02 |
| In-Fusion HD Cloning Plus CE | Takara | Cat# 638916 |
| CD117 MicroBead Kit, mouse | Miltenyi Biotec | Cat# 130-091-224 |
| Maxima SYBR Green qPCR Master Mix | ThermoFisher | Cat# K0253 |
| ApoSENSOR ATP Cell Viability Bioluminescence Assay Kit | BioVision | Cat# K254 |
| X-tremeGENE HP DNA Transfection Reagent | Roche | Cat# 6366236001 |
| FITC Annexin V Apoptosis Detection Kit I | BD Biosciences | Cat# 556547 |
| MTT Cell Proliferation and Cytotoxicity Assay Kit | Beyotime | Cat# C0009M |
| CD34 MicroBead Kit, human | Miltenyi Biotec | Cat# 130-046-702 |
| Lineage Cell Depletion Kit | Miltenyi Biotec | Cat# 130-090-858 |
| HiScript III RT SuperMix for qPCR (+gDNA wiper) | Vazyme Biotech | Cat# R323-01 |
| QIAGEN Plasmid Mini Kit | QIAGEN | Cat# 12125 |
| Mammalian Protein Purification System | Promega | Cat# G6790 |
| Protein Thermal Shift kit | ThennoFisher | Cat# 4462263 |
| Epi™ Ribosome Profiling Kit | Epibiotek | Cat# R1814 |
| RNA clean&ConcentratorTM-5 kit | Zymo | Cat# R1016 |
| EpiTM RiboRNA Depletion Kit | Epibiotek | Cat# R1805 |
| QIAseq miRNA Library kit | QIAGEN | Cat# 1103679 |
| VAHTS Stranded mRNA-seq Library Prep Kit for Illumina V2 | Vazyme Biotech | Cat# NR612-02 |
|
| ||
| Deposit Data | ||
|
| ||
| RNA-seq (Raw and analyzed data) | This study | GSE199204 |
|
| ||
| Ribo-seq (Raw and analyzed data) | This study | GSE211618 |
|
| ||
| Experimental Models: Cell lines | ||
|
| ||
| HEK293T | ATCC | CRL-3216 |
| Molm13 | DSMZ ACC-554 | RRID: CVCL_2119 |
| U937 | ATCC | CRL-1593.2 |
| MonoMac 6 | DSMZ | ACC-124 |
| NB4 | DSMZ | ACC-207 |
| THP1 | ATCC | TIB-202; RRID: CVCL_0006 |
| ML2 | DSMZ | ACC-15 |
| Kasumi-1 | ATCC | CRL-2724; RRID: CVCL_0589 |
| C1498 | ATCC | TIB-49 |
| SUP-B15 | ATCC | CRL-1929 |
| HL60 | ATCC | CCL-240 |
| Nomo-1 | DSMZ | (ACC-542); RRID:CVCL_1609 |
| MV4-11 | ATCC | (CRL-9591); RRID:CVCL_0064 |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| NRGS mouse | The Jackson Laboratory | Stock No: 024099 |
| C57BL/6 mice | Harlan Laboratories | Stock No: 057 |
| C57BL/6 mice | GemPharmatech Co. Ltd | Stock No: N000013 |
| B6.SJL-PtprcaPepcb/BoyCrCrl mice | Charles River Laboratories | Strain code: 564 |
| Mettl14fl/flCREERT | From Dr. Chuan He | |
| Igf2bp2 fl/fl | The Jackson Laboratory | Stock No: 032494 |
| SD rat | SPF (Beijing) Biotechnology Co., Ltd. | |
|
| ||
| Recombinant DNA | ||
|
| ||
| pCDH-IGF2BP2-HA | This study | N/A |
| pCDH-Igf2bp2-HA | This study | N/A |
| pCDH-IGF2BP2-KH3-4 | This study | N/A |
| MSCV-PIG--IGF2BP2-HA | This study | N/A |
| MSCV-PIG-Igf2bp2-HA | This study | N/A |
| pLKO.1-shIGF2BP2-1 | This study | N/A |
| pLKO.1-shIGF2BP2-2 | This study | N/A |
| pLKO.1-shIGF2BP2-3 | This study | N/A |
| pLKO.1-shIgf2bp2-1 | This study | N/A |
| pLKO.1-shIgf2bp2-2 | This study | N/A |
| pLKO.1-shGPT2-1 | This study | N/A |
| pLKO.1-shGPT2-2 | This study | N/A |
| pLKO.1-shSLC1A5-1 | This study | N/A |
| pLKO.1-shSLC1A5-2 | This study | N/A |
| pLKO.1-shMYC-1 | This study | N/A |
| pLKO.1-shMYC-2 | This study | N/A |
| pLKO.1-shGpt2-1 | This study | N/A |
| pLKO.1-shGpt2-2 | This study | N/A |
| pLKO.1-shSlc1a5-1 | This study | N/A |
| pLKO.1-shSlc1a5-2 | This study | N/A |
| pLKO.1-shMyc-1 | This study | N/A |
| pLKO.1-shMyc-2 | This study | N/A |
| pLKO.1-shYTHDF2 | This study | N/A |
| pLKO.1-shPABPC1-1 | This study | N/A |
| pLKO.1-shPABPC1-2 | This study | N/A |
| pCDH-HA-GPT2 | This study | N/A |
| pCDH-HA-SLC1A5 | This study | N/A |
| MSCV-ERT2-IRES-GFP | This study | N/A |
| MSCV-Cre-ERT2-IRES-GFP | This study | N/A |
| psi-LVRU6MH- shIGF2BP2-3 | This study | N/A |
| pSIN4- IGF2BP2 | This study | N/A |
| pSIN4- IGF2BP2-KH3-4 | This study | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| DNA primers or RNA oligos, see Table S2 | Integrated DNA Technologies (IDT) | |
|
| ||
| Software and Algorithms | ||
|
| ||
| Bowtie2-2.3.2 | 53 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Cufflinks-2.2.1 | 54 | http://cole-trapnell-lab.github.io/cufflinks/ |
| Homer-4.8 | 55 | http://homer.ucsd.edu/homer/ |
| MACS2-2.2.7.1 | 56 | https://github.com/taoliu/MACS |
| RSEM-1.2.31 | 57 | https://deweylab.github.io/RSEM/ |
| samtools-1.3.1 | 58 | http://samtools.sourceforge.net/ |
| STAR-2.7.2b | 59 | http://code.google.com/p/rna-star/ |
| jvenn −1.6.x | 60 | http://jvenn.toulouse.inra.fr/app/index.html |
| Bedtools-2.25.0 | 61 | http://bedtools.readthedocs.io/en/latest/ |
| DESeq2-1.32.0 | 62 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Cutadapt (version 3.4) | http://code.google.com/p/cutadapt/ | |
| R-3.3.3 | https://www.r-project.org/ | |
| phantompeakqualtools | 63 | http://encodeproject.org/ENCODE/deeptools.ie-freiburg.mpg.de |
| Deeptools (version 3.5.1) | 64 | |
| igv-2.3.72g | 65 | http://software.broadinstitute.org/software/igv/ |
| GSEA-2.2.3 | 66 | http://software.broadinstitute.org/gsea/index.jsp |
| MetaboAnalystR R package-2.0 | 67 | https://github.com/xia-lab/MetaboAnalystR |
| ELDA | 68 | http://bioinf.wehi.edu.au/software/elda/ |
| X-tile | 52 | https://medicine.yale.edu/lab/rimm/research/software/ |
| Maven (v8.1.27.11) | http://maven.princeton.edu/index.php | |
Highlights.
IGF2BP2 is overexpressed in AML (especially LSCs) and associated with poor outcome
IGF2BP2 promotes AML initiation/development and maintenance as an m6A reader
IGF2BP2 controls glutamine metabolism in AML by targeting GPT2, SLC1A5, and MYC
Pharmacological inhibition of IGF2BP2 represents a new strategy for AML therapy
ACKNOWLEDGEMENTS
This work was supported in part by the National Key R&D Program of China 2020YFA0112403 (H.W.), 82270168 (H.W.) and 82173058 (H.H.) from National Natural Science Foundation of China, 2021A1515010425 (H.H.) from Guangdong Basic and Applied Basic Research Foundation, and the National Institutes of Health (NIH) Grants R01 CA271497 (J.C.), R01 CA243386 (J.C.), R01 CA236399 (J.C.), DK124166 (J.C.), and P30CA033572 (D.H.), as well as the Simms/Mann Family Foundation (J.C.) and PhRMA Foundation Postdoctoral Fellowship inTranslational Medicine (W.L.). J.C. is a Leukemia & Lymphoma Society (LLS) Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplementary Information includes seven figures and two tables and can be found with this article online.
COMPETING FINANCIAL INTERESTS
A U.S. patent (No. 17/794,922) has been filed, with J.C., H.W., H.H., D.H., Y.M., H.L., and X.D. as inventors. J.C. is a Scientific Advisor for Race Oncology. All the other authors have no competing interests.
DATA AND CODE AVAILABILITY
Data of RNA-seq and Ribo-seq obtained in this study have been deposited in the gene expression omnibus (GEO) repository under the accession numbers GSE199204 and GSE211618, respectively.
REFERENCES
- 1.Shi H Wei J He C Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol Cell 2019; 74: 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang Y Hsu PJ Chen YS Yang YG Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res 2018; 28: 616–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng X Su R Weng H Huang H Li Z Chen J RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res 2018; 28: 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang H Weng H Chen J m(6)A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 2020; 37: 270–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J Yue Y Han D Wang X Fu Y Zhang L Jia G Yu M Lu Z Deng X et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 2014; 10: 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ping XL Sun BF Wang L Xiao W Yang X Wang WJ Adhikari S Shi Y Lv Y Chen YS et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 2014; 24: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwartz S Mumbach MR Jovanovic M Wang T Maciag K Bushkin GG Mertins P Ter-Ovanesyan D Habib N Cacchiarelli D et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep 2014; 8: 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang H Weng H Zhou K Wu T Zhao BS Sun M Chen Z Deng X Xiao G Auer F et al. Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature 2019; 567: 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jia G Fu Y Zhao X Dai Q Zheng G Yang Y Yi C Lindahl T Pan T Yang YG et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 2011; 7: 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng G Dahl JA Niu Y Fedorcsak P Huang CM Li CJ Vagbo CB Shi Y Wang WL Song SH et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 2013; 49: 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Z Weng H Su R Weng X Zuo Z Li C Huang H Nachtergaele S Dong L Hu C et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell 2017; 31: 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vu LP Pickering BF Cheng Y Zaccara S Nguyen D Minuesa G Chou T Chow A Saletore Y MacKay M et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med 2017; 23: 1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weng H Huang H Wu H Qin X Zhao BS Dong L Shi H Skibbe J Shen C Hu C et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell Stem Cell 2018. 22: 191–205 el99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen C Sheng Y Zhu AC Robinson S Jiang X Dong L Chen H Su R Yin Z Li W et al. RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell 2020; 27: 64–80 e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J Li Y Wang P Han G Zhang T Chang J Yin R Shan Y Wen J Xie X et al. Leukemogenic Chromatin Alterations Promote AML Leukemia Stem Cells via a KDM4C-ALKBH5-AXL Signaling Axis. Cell Stem Cell 2020; 27: 81–97 e88. [DOI] [PubMed] [Google Scholar]
- 16.Hsu PJ Zhu Y Ma H Guo Y Shi X Liu Y Qi M Lu Z Shi H Wang J et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res 2017; 27: 1115–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang H Weng H Sun W Qin X Shi H Wu H Zhao BS Mesquita A Liu C Yuan CL et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol 2018; 20: 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X Lu Z Gomez A Hon GC Yue Y Han D Fu Y Parisien M Dai Q Jia G et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014; 505: 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y Li Y Toth JI Petroski MD Zhang Z Zhao JC N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 2014; 16: 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao W Adhikari S Dahal U Chen YS Hao YJ Sun BF Sun HY Li A Ping XL Lai WY et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell 2016; 61: 507–519. [DOI] [PubMed] [Google Scholar]
- 21.Paris J Morgan M Campos J Spencer GJ Shmakova A Ivanova I Mapperley C Lawson H Wotherspoon DA Sepulveda C et al. Targeting the RNA m(6)A Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell 2019; 25: 137–148 e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanahan D Weinberg RA Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 23.Lu W Pelicano H Huang P Cancer metabolism: is glutamine sweeter than glucose? Cancer Cell 2010; 18: 199–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samudio I Konopleva M Asparaginase unveils glutamine-addicted AML. Blood. 2013; 122: 3398–3400. [DOI] [PubMed] [Google Scholar]
- 25.Willems L Jacque N Jacquel A Neveux N Maciel TT Lambert M Schmitt A Poulain L Green AS Uzunov M et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013; 122: 3521–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gregory MA Nemkov T Park HJ Zaberezhnyy V Gehrke S Adane B Jordan CT Hansen KC D’Alcssandro A DeGregori J Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin Cancer Res 2019; 25: 4079–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gallipoli P Giotopoulos G Tzelepis K Costa ASH Vohra S Medina-Perez P Basheer F Marando L Di Lisio L Dias JML et al. Glutaminolysis is a metabolic dependency in FLT3(ITD) acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood. 2018; 131: 1639–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pollyea DA Stevens BM Jones CL Winters A Pei S Minhajuddin M D’Alessandro A Culp-Hill R Riemondy KA Gillen AE et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med 2018; 24: 1859–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mopin A Driss V Brinster C A Detailed Protocol for Characterizing the Murine C1498 Cell Line and its Associated Leukemia Mouse Model. J Vis Exp 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim M Gwak J Hwang S Yang S Jeong SM Mitochondrial GPT2 plays a pivotal role in metabolic adaptation to the perturbation of mitochondrial glutamine metabolism. Oncogene 2019; 38: 4729–4738. [DOI] [PubMed] [Google Scholar]
- 31.Ni F Yu WM Li Z Graham DK Jin L Kang S Rossi MR Li S Broxmeyer HE Qu CK Critical role of ASCT2-mediated amino acid metabolism in promoting leukaemia development and progression. Nat Metab 2019; 1: 390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wise DR DeBerardinis RJ Mancuso A Sayed N Zhang XY Pfeiffer HK Nissim I Daikhin E Yudkoff M McMahon SB et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 2008; 105: 18782–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yin R Chang J Li Y Gao Z Qiu Q Wang Q Han G Chai J Feng M Wang P et al. Differential m(6)A RNA landscapes across hematopoiesis reveal a role for IGF2BP2 in preserving hematopoietic stem cell function. Cell Stem Cell 2022; 29: 149–159 e147. [DOI] [PubMed] [Google Scholar]
- 34.Lomenick B Hao R Jonai N Chin RM Aghajan M Warburton S Wang J Wu RP Gomez F Loo JA et al. Target identification using drug affinity responsive target stability (DARTS). Proc Natl Acad Sci U S A 2009; 106: 21984–21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahapatra L Andruska N Mao C Le J Shapiro DJ A Novel IMP1 Inhibitor, BTYNB, Targets c-Myc and Inhibits Melanoma and Ovarian Cancer Cell Proliferation. Transl Oncol 2017; 10: 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jafari R Almqvist H Axelsson H Ignatushchenko M Lundback T Nordlund P Martinez Molina D The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 2014; 9: 2100–2122. [DOI] [PubMed] [Google Scholar]
- 37.Martinez NJ Asawa RR Cyr MG Zakharov Urban DJ Roth JS Wallgren E Klumpp-Thomas C Coussens NP Rai G et al. A widely-applicable high-throughput cellular thermal shift assay (CETSA) using split Nano Luciferase. Sci Rep 2018; 8: 9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lo MC Aulabaugh A Jin G Cowling R Bard J Malamas M Ellestad G Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem 2004; 332: 153–159. [DOI] [PubMed] [Google Scholar]
- 39.Stone RM Mandrekar SJ Sanford BL Laumann K Geyer S Bloomfield CD Thiede C Prior TW Dohner K Marcucci G et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 2017; 377: 454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Othman TA Azenkot T Moskoff BN Tenold ME Jonas BA Venetoclax-based combinations for the treatment of newly diagnosed acute myeloid leukemia. Future Oncol 2021; 17: 2989–3005. [DOI] [PubMed] [Google Scholar]
- 41.Dong S Wu Y Liu Y Weng H Huang H N6 - methyladenosine Steers RNA Metabolism and Regulation in Cancer. Cancer Commun 2021; 41: 538–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qing Y Dong L Gao L Li C Li Y Han L Prince E Tan B Deng X Wetzel C et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m(6)A/PFKP/LDHB axis. Mol Cell 2021; 81: 922–939 e929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deberardinis RJ Sayed N Ditsworth D Thompson CB Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev 2008; 18: 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones CL Inguva A Jordan CT Targeting Energy Metabolism in Cancer Stem Cells: Progress and Challenges in Leukemia and Solid Tumors. Cell Stem Cell 2021; 28: 378–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Warburg O On respiratory impairment in cancer cells. Science 1956; 124: 269–270. [PubMed] [Google Scholar]
- 46.Janiszewska M Suva ML Riggi N Houtkooper RH Auwerx J Clement-Schatlo V Radovanovic I Rheinbay E Provero P Stamenkovic I Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev 2012; 26: 1926–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lagadinou ED Sach A Callahan K Rossi RM Neering SJ Minhajuddin M Ashton JM Pei S Grose V O’Dwyer KM et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013; 12: 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sriskanthadevan S Jeyaraju DV Chung TE Prabha S Xu W Skrtic M Jhas B Hurren R Gronda M Wang X et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 2015; 125: 2120–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones CL Stevens BM D’Alessandro A Reisz JA Culp-Hill R Nemkov T Pei S Khan N Adane B Ye H et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2019; 35: 333–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hammer NA Hansen T Byskov AG Rajpert-De Meyts E Grondahl ML Bredkjaer HE Wewer UM Christiansen J Nielsen FC Expression of IGF-II mRNA-binding proteins (IMPs) in gonads and testicular cancer. Reproduction 2005; 130: 203–212. [DOI] [PubMed] [Google Scholar]
- 51.Lu S Han L Hu X Sun T Xu D Li Y Chen Q Yao W He M Wang Z et al. N6-methyladenosine reader IMP2 stabilizes the ZFAS1/OLA1 axis and activates the Warburg effect: implication in colorectal cancer. J Hematol Oncol 2021. 14: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Camp RL Dolled-Filhart M Rimm DL X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res 2004; 10: 7252–7259. [DOI] [PubMed] [Google Scholar]
- 53.Langmead B Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 2012. 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trapnell C Williams B Pertea G Mortazavi A Kwan G Baren M J. v. Salzberg SL Wold BJ Pachtcr L Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010; 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heinz S Benner C Spann N Bertolino E Lin YC Laslo P Cheng JX Murre C Singh H Glass CK Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010; 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y Liu T Meyer CA Eeckhoute J Johnson DS Bernstein BE Nussbaum C Myers RM Brown M Li W et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol 2008. 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li B Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011; 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H Handsaker B Wysoker A Fennell T Ruan J Homer N Marth G Abecasis G Durbin R Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dobin A Davis CA Schlesinger F Drenkow J Zaleski C Jha S Batut P Chaisson M Gingeras TR STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bardou P Mariette J Escudie F Djemiel C Klopp C jvenn: an interactive Venn diagram viewer. BMC Bioinformatics 2014; 15: 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quinlan AR BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics 2014; 47: 11 12 11–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Love MI Huber W Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Landt SG Marinov GK Kundaje A Kheradpour P Pauli F Batzoglou S Bernstein BE Bickel P Brown JB Cayting P et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 2012; 22: 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramirez F Ryan DP Gruning B Bhardwaj V Kilpert F Richter AS Heyne S Dundar F Manke T deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44: W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thorvaldsdottir H Robinson JT Mesirov JP Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration Br Bioinform 2013; 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Subramanian A Tamayo P Mootha VK Mukherjee S Ebert BL Gillette MA Paulovich A Pomeroy SL Golub TR Lander ES et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-wide Expression Profiles. Proc Natl Acad Sci U S A 2005; 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chong J Yamamoto M Xia J MetaboAnalystR 2.0: From Raw Spectra to Biological Insights. Metabolites 2019; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hu Y Smyth GK ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 2009; 347: 70–78. [DOI] [PubMed] [Google Scholar]
- 69.Frankish A Diekhans M Ferreira AM Johnson R Jungreis I Loveland J Mudge JM Sisu C Wright J Armstrong J et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 2019; 47: D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langmead B Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012; 9: 357–U354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu W Zhou M Li Z Li H Polaczek P Dai H Wu Q Liu C Karanja KK Popuri V et al. A Selective Small Molecule DNA2 Inhibitor for Sensitization of Human Cancer Cells to Chemotherapy. EBioMedicine 2016; 6: 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Singh AK Zhao B Liu X Wang X Li H Qin H Wu X Ma Y Horne D Yu X Selective targeting of TET catalytic domain promotes somatic cell reprogramming. Proc Natl Acad Sci U S A 2020; 117: 3621–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Su R Dong L Li Y Gao M Han L Wunderlich M Deng X Li H Huang Y Gao L et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer cell 2020; 38: 79–96 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tian L Kim MS Li H Wang J Yang W Structure of HIV-1 reverse transcriptase cleaving RNA in an RNA/DNA hybrid. Proc Natl Acad Sci U S A 2018; 115: 507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Friesner RA Banks JL Murphy RB Halgren TA Klicic JJ Mainz DT Repasky MP Knoll EH Shelley M Perry JK et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004; 47: 1739–1749. [DOI] [PubMed] [Google Scholar]
- 76.Martinez NJ Asawa RR Cyr MG Zakharov A Urban DJ Roth JS Wallgren E Klumpp-Thomas C Coussens NP Rai G et al. A widely-applicable high-throughput cellular thermal shift assay (CETSA) using split Nano Luciferase. Sci Rep 2018; 8: 9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data of RNA-seq and Ribo-seq obtained in this study have been deposited in the gene expression omnibus (GEO) repository under the accession numbers GSE199204 and GSE211618, respectively.