

Abstract
BACKGROUND & AIMS:
Non-alcoholic fatty liver disease (NAFLD) is highly associated with obesity and progresses to non-alcoholic steatohepatitis when the liver develops overt inflammatory damage. While removing adenosine in the purine salvage pathway, adenosine kinase (ADK) regulates methylation reactions. We aimed to study whether hepatocyte ADK functions as an obesogenic gene/enzyme to promote excessive fat deposition and liver inflammation.
METHODS:
Liver sections of human subjects were examined for ADK expression using immunohistochemistry. Mice with hepatocyte-specific ADK disruption or overexpression were examined for hepatic fat deposition and inflammation. Liver lipidomics, hepatocyte RNAseq, and single cell RNAseq for liver non-parenchymal cells (NPC) were performed to analyze ADK regulation of hepatocyte metabolic responses and hepatocyte-NPC crosstalk.
RESULTS:
While NAFLD patients showed increased hepatic ADK levels, mice with hepatocyte-specific ADK disruption displayed decreased hepatic fat deposition under a chow diet and were protected from diet-induced excessive hepatic fat deposition and inflammation. In contrast, mice with hepatocyte-specific ADK overexpression displayed increased body weight and adiposity and elevated degrees of hepatic steatosis and inflammation compared with control mice. RNAseq and epigenetic analyses indicated that ADK increased hepatic DNA methylation and decreased hepatic Ppara expression and fatty acid oxidation. Lipidomic and scRNAseq analyses indicated that ADK-driven hepatocyte factors, due to mitochondrial dysfunction, enhanced macrophage proinflammatory activation in manners involving increased expression of stimulator of interferon genes.
CONCLUSIONS:
Hepatocyte ADK functions to promote excessive fat deposition and liver inflammation through suppressing hepatocyte fatty acid oxidation and producing hepatocyte-derived proinflammatory mediators. Therefore, hepatocyte ADK is a therapeutic target for managing obesity and NAFLD.
Keywords: Insulin resistance, mitochondrial DNA
Graphical Abstract
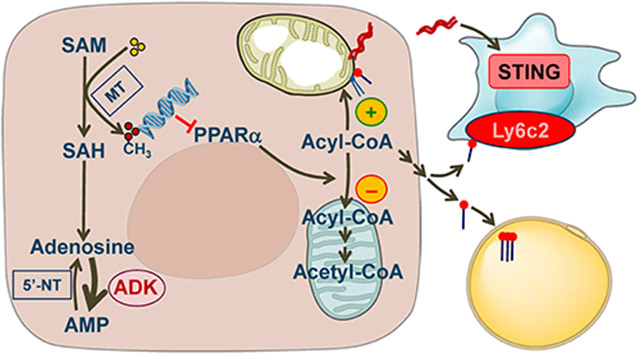
Hepatocyte ADK links DNA methylation and decreased Ppara expression, leading to decreased fatty acid oxidation and increased aberrant subcellular fat deposition and generation of endogenous fats, as well as a consequent increase in adipocyte fat deposition. Moreover, hepatocyte ADK-driven inflammatory mediators promote dysfunctional hepatocyte-macrophage crosstalk to underlie liver inflammation in manners involving STING and Ly6c2.
Lay Summary
The present study provides compelling evidence supporting a critical role for hepatocyte adenosine kinase in the pathogenesis of non-alcoholic fatty liver disease by promoting excessive fat deposition and liver inflammation.
INTRODUCTION
As a primary organ that synthesizes endogenous fats through de novo lipogenesis and formation of very low-density lipoproteins, as well as fatty acid uptake and oxidation, the liver plays a central role in regulating fat metabolic homeostasis 1, 2. Pathologically, excessive accumulation of fats in hepatocytes results in increased inflammatory responses and impaired insulin signaling, which in turn promotes the development of various metabolic diseases including non-alcoholic fatty liver disease (NAFLD) and type 2 diabetes 2-4. NAFLD has been documented as a causal factor of a wide variety of chronic diseases including atherogenic cardiovascular disease and certain forms of cancers 5, 6. However, precisely how the liver regulates fat metabolic homeostasis and hepatic inflammation remains to be elucidated.
Adenosine kinase (ADK) is the enzyme that phosphorylates adenosine to generate adenosine monophosphate. This enables ADK to play two important and distinct functions. First, due to preventing intracellular accumulation of adenosine, ADK facilitates methionine cycle and has been shown to increase methylation reactions 7, 8. This role is exemplified by the finding that genetic ADK knockdown increases intracellular adenosine and, consequently, reduces the activation of transmethylation pathway to attenuate the endothelial inflammatory responses 9. Similarly, ablation of ADK in myeloid cells results in significantly reduced DNA methylation of the ATP-binding cassette transporter G1, thereby leading to decreased foam cell formation 10. Second, due to removing intracellular adenosine 11, ADK functions to keep the extracellular adenosine at a low range through cooperating with enzymes and transporters involved in the processes to metabolize extracellular adenosine. At increased levels, extracellular adenosine activates its receptors, mainly adenosine 2A receptor (A2AR), thereby protecting against injury and inflammatory responses 12-15. Because of this, increasing adenosine levels by ADK inhibition is a therapeutic strategy for neurological disorders 16-18.
Currently, only three studies describe a link between ADK deficiency and liver dysfunctions 7, 19, 20 and only one recent study suggested a role for the ADK in endothelial cells in regulating fat metabolism and systemic insulin sensitivity. Of note, the study by Boison et al. revealed increased liver fat content in neonatal mice upon global ADK disruption 19, whereas the study by Xu et al. showed decreased severities of high-fat diet (HFD)-induced obesity and hepatic steatosis in mice upon endothelial ADK disruption 21. However, it is completely unknown whether and how hepatocyte ADK enables the liver to play a central role in regulating fat metabolism as it relates to systemic insulin sensitivity. Here, we present novel and compelling evidence to demonstrate that hepatocyte ADK functions as an obesogenic gene/enzyme to promote obesity and liver inflammation and elucidate the underlying mechanisms.
MATERIALS AND METHODS
Materials including liver samples from patients with NAFLD and mice with hepatocyte-specific ADK disruption or overexpression and the reagents are detailed in the Supplemental Information (SI). The assays related to mouse phenotypes, along with histological, biochemical, molecular assays, and lipidomics, are also detailed in SI. Primary hepatocytes and liver non-parenchymal cells (NPCs) were isolated from mice and subjected to RNAseq and single cell RNA sequencing (scRNAseq), respectively, as detailed in SI.
RESULTS
Hepatic ADK amount is increased in patients with NAFLD and mice with diet-induced obesity, hepatic steatosis, and systemic insulin resistance
Dysfunctional ADK may underlie liver dysfunction in human subjects 7, 20. We examined hepatic ADK amounts in patients with NAFLD including non-alcoholic steatohepatitis (NASH). Compared with control, liver sections from NAFLD patients displayed significantly increased ADK expression, predominately in hepatocytes (Figure 1A). Next, we analyzed ADK expression pattern in wild-type (WT) C57BL/6J mice and showed that ADK, both short- and long-forms (~ 40-42 kDa), was expressed at the highest level in the liver (Figure 1B). We then examined the pathophysiological relevance of ADK in obesity and metabolic dysregulation using WT mice upon HFD (60% fat calories) feeding for 12 weeks. Compared with mice fed a low-fat diet (LFD, 10% fat calories), HFD-fed mice displayed significantly increased hepatic amount of ADK, accompanied with increased body weight, excessive hepatic fat deposition, elevated circulating levels of triglycerides, and systemic insulin resistance (Figure 1C-H). At the transcriptional level, hepatic Adk mRNAs in mice fed an HFD for 12 weeks were insignificantly decreased relative to those in LFD-fed mice (Figure 1D). This likely reflects a counter-regulatory response to excessive hepatocyte fat deposition since hepatic Adk mRNAs were increased in mice fed an HFD for 3 weeks, when mice did not gain much fat. These results suggest that ADK amount is positively associated with obesity-related excessive fat deposition and insulin resistance.
Figure 1. Hepatic ADK amount is increased in the patients with NAFLD and mice with diet-induced obesity and hepatic steatosis.

(A) Liver sections from patients with NAFLD were stained with H&E (top row) and for ADK expression (bottom two rows). (B) Immunoblot for ADK tissue distribution in mice. (C,D,E,F,G,H) Hepatic ADK amount correlates with diet-induced metabolic dysregulation. C, representative immunoblots for liver ADK amount. Bar graph, quantification of blots. n = 4. D, Hepatic Adk mRNAs. E, Body weight before and after dietary feeding. F, H&E staining of liver sections. G, plasma triglyceride levels. n = 7. H, Insulin tolerance tests. n = 10 - 12. For C - H, male C57BL/6J mice, at 5 - 6 weeks of age, were fed a high-fat diet (HFD, 60% fat calories) or low-fat diet (LFD, 10% fat calories) for 3 or 12 weeks. For bar and line graphs, data are means ± SEM. **, P < 0.01 HFD vs. LFD (in C and G) after HFD feeding (in E) for the same time point (in D and H). The scale bar is 75 μm for 10× images or 50 μm for 20× images.
Hepatocyte-specific ADK deficiency decreases hepatic fat deposition and proinflammatory signaling
To establish a cause-and-effect relationship between hepatocyte ADK and obesity-related phenotype, we generated hepatocyte-specific ADK-disrupted mice (Figure 2A). Both male and female Hep-ADK−/− (ADKF/F-AlbCre+) mice stopped gaining body weight and size after weaning (Supplemental Figure S1A,B). Also, all Hep-ADK−/− mice died at 5 to 7 weeks of age (Supplemental Figure S1C). We then performed glucose tolerance tests (GTTs) in mice immediately after weaning and showed that Hep-ADK−/− mice displayed hypoglycemia at 0 time point during the GTTs and had enhanced glucose clearance compared with any group of control mice (Supplemental Figure S1D). Histological analyses confirmed significant liver injuries in both male and female Hep-ADK−/− mice (Supplemental Figure S2). Notably, Hep-ADK−/− mice displayed significantly decreased hepatic fat deposition and glycogen content compared with ADKF/F (ADKF/F-AlbCre−), Hep-ADK+/+ (ADK+/+-AlbCre+), or Hep-ADK+/− (ADKF/+-AlbCre+) mice (Figure 2B; Supplemental Figure S3). When analyzed for RNA transcriptome in livers from 3-week-old male mice, hepatic expression of carnitine palmitoyltransferase 1a (Cpt1a), a ratedetermining enzyme of fatty acid oxidation, and Ppara and 3-hydroxybutyrate dehydrogenase type 2 (Bdh2), which increase fatty acid oxidation and ketogenesis, were significantly increased (Supplemental Figure S4).
Figure 2. Hepatocyte-specific ADK-deficiency or maturity-onset hepatocyte ADK deficiency decreases liver fat content and/or inflammation while decreasing body weight.

(A) Immunoblot validation of hepatocyte-specific ADK disruption. NS, non-specific bands. (B) Liver sections were stained with oil red O. For A and B, homozygous hepatocyte-specific ADK deficient (Hep-ADK−/−) mice, heterozygous hepatocyte-specific ADK-disrupted (Hep-ADK+/−), and AlbCre+ control (Hep-ADK+/+) mice, as well as ADK/F/F control mice (described in Supplemental Figure S1), were fed a chow diet and harvested at 5 weeks of age. (C) Immunoblot validation of hepatocyte-specific ADK deficiency. (D,E) Body weight (D) and body composition (E). (F) Plasma triglyceride levels. (G) Plasma levels of acetoacetate (AcAc) and 3-hydorbutyrate (BOH). (H,I) Liver sections were stained with H&E (H) and oil red Ο (I). (J) Liver proinflammatory signaling. Bar graph, quantification of blots. For C - J, male ADKF/F mice, at 11 - 12 weeks of age, were treated with adeno-associated viral vector (AAV) to generate maturity-onset hepatocyte-specific ADK deficiency (AAV-Hep-ADK−/−). n = 7 - 9. For bar graphs in D - G, and J, data are means ± SEM. *, P < 0.05 and **, P < 0.01 AAV-Hep-ADK−/− vs. AAV-Hep-ADK+/+ (in F) after AAV treatment (in D) for the same type of fat (in E), ketone body (in G), or protein (in J). The scale bar is 75 μm for 10× images.
Since Hep-ADK−/− caused early lethality, we sought to treat ADKF/F mice, at 11 - 12 weeks of age, with AAV8.TBG.PI.Cre.rBG (adeno-associated viral vector for liver-specific expression of Cre) to create maturity-onset homozygous hepatocyte-ADK deficient (AAV-Hep-ADK−/−) mice. AAV8.TBG-PI.Null.bGH-treated ADKF/F mice (AAV-Hep-ADK+/+) served as the control. Because AAV-Hep-ADK−/− mice died at 3 weeks post AAV treatment due to liver damage resulting from excessive ketogenesis, we decided to harvest the mice at 18 d post AAV treatment in separate experiments. Hepatic ADK deletion was verified and accompanied with decreased body weight, adiposity, plasma triglyceride levels, and hepatic fat deposition, along with increased plasma levels of ketone bodies without causing significantly altering liver morphology (Figure 2C-I). Additionally, AAV-Hep-ADK−/− mice displayed significantly decreased hepatic inflammatory signaling through Jnk p46 and Nfκb p65 compared with control mice (Figure 2J). These results indicate that hepatocyte-specific ADK deficiency is a primary cause of decreased hepatic fat deposition and proinflammatory responses.
Hepatocyte-specific ADK disruption alleviates diet-induced hepatic steatosis and inflammation, as well as systemic insulin resistance
Hep-ADK+/− mice displayed comparable characteristics relative to Hep-ADK+/+ mice on a chow diet. Next, we examined HFD-induced metabolic dysregulation in these mice. While showing no significant differences in plasma alanine aminotransferase levels (Figure 3A) and hepatic mRNAs for markers of liver damage (not shown), HFD-fed Hep-ADK+/− mice did not differ significantly from HFD-fed Hep-ADK+/+ mice in body weight and fat mass (not shown). However, both male and female Hep-ADK+/− mice displayed decreased severity of HFD-induced hepatic fat deposition and the proinflammatory responses (Figure 3B,C), as well as systemic insulin resistance compared with their respective control (Figure 3D). Next, we examined the effect of hepatocyte ADK disruption on diet-induced NASH. Upon feeding a methionine- and choline-deficient diet (MCD) for 5 weeks, Hep-ADK+/+ mice displayed overt NASH phenotypes, evidenced by hepatic steatosis, inflammation (increased macrophage aggregations and proinflammatory signaling), and fibrosis. However, the severity of MCD-induced NASH in Hep-ADK+/− mice was significantly lower than that in Hep-ADK+/+ mice (Figure 3E,F). These results indicate that hepatocyte-specific ADK disruption protects mice from diet-induced NAFLD/NASH phenotypes.
Figure 3. Hepatocyte-specific ADK disruption alleviates diet-induced hepatic steatosis and inflammation and systemic insulin resistance.

(A) Plasma ALT levels. (B) Liver sections were stained with H&E. (C) Liver proinflammatory signaling (male mice only). (D) Insulin tolerance tests. n = 7 - 10 (male) and 10 - 14 (female). (E) Liver histology. (F) Liver proinflammatory signaling. For A - D, male and/or female Hep-ADK+/− and Hep-ADK+/+ mice, at 5 - 6 weeks of age, were fed an HFD for 12 weeks. For E and F, male mice, at 11 - 12 weeks of age, were fed a MCD diet for 5 weeks. For bar graphs in C and F, quantification of blots. n = 4 (C) or 6 (F). For A, C, D, and F, numeric data are means ± SEM. *, P < 0.05 and **, P < 0.01 Hep-ADK+/− vs. Hep-ADK+/+ for the same protein (C and F) or time point (D). The scale bar is 75 μm for 10× images.
Hepatocyte-specific ADK overexpression increases body weight, adiposity, liver fat deposition and inflammation while promoting systemic insulin resistance
To further verify a role for hepatocyte ADK in promoting obesity-related metabolic dysregulation, we generated hepatocyte-specific ADK-overexpressing (AlbCre+-ADK transgenic, Hep-ADKTg) mice (Supplemental Figure S5A,B). Under a chow diet, both male and female Hep-ADKTg mice displayed marked increases in body weight and fat mass, as well as systemic insulin resistance, compared with their respective control (Hep-ADKWT) mice without showing much difference in energy expenditure (Figure 4A,B and Supplemental Figure S5C-G) and food intake (not shown). Next, we analyzed feeding efficiency and the expression of intestinal genes for nutrient absorption. Compared with their respective control mice, both male and female Hep-ADKTg mice displayed significantly increased feeding efficiency (Supplemental Figure S5H), suggesting increased nutrient absorption when food consumption and energy expenditure were comparable with those in control mice. Consistently, intestinal expression of key genes for nutrient absorption in Hep-ADKTg mice was significantly increased compared with that in Hep-ADKWT mice (Supplemental Figure S5I). Hep-ADKTg mice also displayed significantly increased circulating levels of triglycerides and degrees of hepatic fat deposition compared with Hep-ADKWT mice (Figure 4C,D) while revealing significantly decreased hepatic expression of Cpt1a (Figure 4E). Regarding the inflammatory responses, livers of Hep-ADKTg mice displayed significantly increased macrophage infiltration and phosphorylation states of Jnk p46 and Nfκb p65 compared with those of Hep-ADKWT mice (Figure 4F,G). When white adipose tissue (WAT) phenotype was analyzed, Hep-ADKTg mice displayed increased adipocyte size and macrophage infiltration relative to Hep-ADKWT mice. Consistently, Hep-ADKTg mice showed decreased WAT expression of hormone-sensitive lipase and increased WAT expression of proinflammatory mediators compared with Hep-ADKWT mice (Supplemental Figure S5J,K). Interestingly, female Hep-ADKTg mice displayed a comparable phenotype in body weight and fat mass, as well as systemic insulin sensitivity compared with male Hep-ADKWT mice (Supplemental Figure S6). These results, together with those from Hep-ADK−/− mice, AAV-Hep-ADK−/− mice, and HFD- or MCD-fed Hep-ADK+/− mice, validate hepatocyte ADK as a primary cause in promoting hepatic fat deposition and increasing liver inflammation. In addition, hepatocyte ADK overexpression caused secondary changes in extrahepatic tissues (e.g., WAT), which likely contributed to metabolic phenotype in Hep-ADKTg mice.
Figure 4. Hepatocyte-specific ADK overexpression promotes obesity, hepatic steatosis, and liver inflammation.

(A,B) Body weight (A) and body composition (B). n = 10. (C) Plasma triglyceride levels. n = 7. (D) Liver sections were stained with oil red O. (E) Hepatic expression of metabolic genes. n = 8. (F) Liver sections were stained for F4/80. (G) Liver proinflammatory signaling (immunoblots). For A - G, both male and female Hep-ADKTg and Hep-ADKWT (control) mice were maintained on a chow diet for 7 months. For C - G, samples were from male mice only. For bar graphs in A - C, and E, data are means ± SEM. *, P < 0.05; **, P < 0.01; and ***, P < 0.001 Hep-ADKTg vs. Hep-ADKWT (in C) within the same sex (in A) for the same mass (in B) or for the same gene (in E). The scale bar is 75 μm for 10× images or 50 μm for 20× images.
ADK alteration of lipid profile is accompanied by hepatic mitochondrial stress and increased macrophage STING expression
Excessive fat deposition promotes hepatocyte generation of mediators that stimulate the proinflammatory activation of liver macrophages 22, 23. Next, we performed liver lipidomics. Hep-ADKTg mice displayed significant increases in total amount and most phosphatidic acid species (Figure 5A), consistent with increased hepatic fat deposition. Also, Hep-ADKTg mice displayed significantly increased levels of multiple bis-monoacylglycerol phosphate and lysophosphatidylcholine species and significantly decreased levels of tetralinoleoyl cardiolipin (18:2-18:2-18:2-18:2) (Figure 5B-D), indicative of increased oxidative stress and mitochondrial dysfunction that contribute to the proinflammatory responses 24. Consistently, hepatic levels of mitochondrial DNA (mtDNA, D-loops 1 through 3) were significantly increased in Hep-ADKTg mice compared with Hep-ADKWT mice and were accompanied with increased expression of stimulator of interferon genes (STING), which is activated in response to mtDNA, in liver macrophages (Figure 5E,F).
Figure 5. ADK alteration of lipid profile is accompanied by hepatic mitochondrial stress and increased macrophage STING expression.

(A,B,C,D) Hepatic lipid profile. (E) Hepatic mtDNA levels. (F) Liver sections were co-stained for STING and F4/80 using immunofluorescent histochemistry. For A - F, male Hep-ADKTg and Hep-ADKWT mice were maintained on a chow diet for 5 months. For A - E, data are means ± SEM. n = 4 (A - D) or 6 (E). *, P < 0.05; **, P < 0.01; and ***, P < 0.001 Hep-ADKTg vs. Hep-ADKWT for the same lipid species (A - D) or mtDNA (E). The scale bar is 50 μm for 20× images.
ADK overexpression alters hepatocyte transcriptome in a manner promoting fat deposition and methylation
To gain mechanistic insights of ADK actions, we isolated primary hepatocytes from chow diet-fed Hep-ADKTg and Hep-ADKWT mice and examined hepatocyte fat deposition. Compared with control cells, Hep-ADKTg hepatocytes displayed a significant increase in palmitate-induced fat deposition (Figure 6A). Next, we examined hepatocyte RNA transcriptomes using RNAseq and performed differential expression (DE) analysis between Hep-ADKTg and Hep-ADKWT hepatocytes. Of the 18,630 genes retained for the DE analysis, 2,105 (11.3%) were found to be differentially expressed genes (DEGs) with an absolute value of fold change ∣FC∣ > 1.5 and Benjamini-Hochberg-corrected false discovery rate (FDR) < 0.05. There were 1,783 genes whose expression was significantly decreased in Hep-ADKTg hepatocytes compared with those in Hep-ADKWT hepatocytes. These down-regulated DEGs were found to be enriched in pathways including the PPAR signaling and the fatty acid, triacylglycerol, and ketone body metabolism (Figure 6B,C and Supplemental Figure S7A). These DEGs included cytochrome P450 omega-hydroxylase 4A14 (Cyp4a14), which plays a role in promoting liver fat deposition and inflammation 25, and enoyl-CoA hydratase and 3-hydroxyacyl CoA dehydrogenase (Ehhadh), which encodes an enzyme of peroxisomal fatty acid oxidation pathway that responds to activation of peroxisome proliferator 26 and is required to inhibit liver inflammation induced by toxic dicarboxylic acids 27. The down-regulated DEGs also included acetyl-Coenzyme A acyltransferase 1B (Acaa1b), which is predicted to enable the activities of C-acyltransferase, acetate CoA-transferase, palmitoyl-CoA oxidase, and acyl-CoA synthetase long chain family member 1 (Acsl1), which functions to promote mitochondrial fatty acid oxidation.
Figure 6. ADK overexpression alters hepatocyte transcriptomes in a manner promoting fat deposition and methylation.

(A) Hepatocyte fat deposition. (B,C,D,E) Hepatocyte RNA transcriptome. (F) Liver sections were stained for 5-mC. (G) Hepatic methylation status of Ppara (left) and hepatocyte Ppara mRNAs (right). (H) PPARα overexpression reversal of ADK-induced decreases in the expression of genes for fatty acid oxidation. n = 6 - 8. For A - E, G (right panel), and H, primary hepatocytes were isolated from male Hep-ADKTg and Hep-ADKWT mice, at 5 months of age. For A, hepatocytes were supplemented with palmitate (250 μM) for 24 hr and stained with oil red O for the last 1 hr. For B - E and G (right), the isolated hepatocytes were analyzed for RNA transcriptome using RNAseq. n = 3. For B and C, pathways (B) enriched with ADK-driven down-regulation of genes for fatty acid oxidation (C). Cyp4a14, cytochrome P450 omega-hydroxylase 4A14; Ehhadh, enoyl-CoA hydratase and 3-hydroxyacyl CoA dehydrogenase; Acaa1b, acetyl-Coenzyme A acyltransferase 1B; Acsl1, acyl-CoA synthetase long chain family member 1. For D and E, pathways (D) enriched with ADK-driven up-regulation of genes for methylation (E). Bhmt, betaine-homocysteine S-methyltransferase; Mat2a, methionine adenosyltransferase 2A; and Dmgdh, dimethylglycine dehydrogenase. For bar graphs in C, E, G, and H, data are means ± SEM. *, P < 0.05; **, P < 0.01; and ***, P < 0.001 Hep-ADKTg vs. Hep-ADKWT (in C, E and G) or Hep-ADKTg/Ad-PPARα vs. Hep-ADKTg/Ad-GFP for the same gene (in H); P < 0.001 Hep-ADKTg/Ad-GFP vs. Hep-ADKWT/Ad-GFP for the same gene (in H). †††, The scale bar is 75 μm for 10× images or 50 μm for 20× images.
There were 322 DEGs whose expression was increased (FDR < 0.05) in Hep-ADKTg hepatocytes compared with those in Hep-ADKWT hepatocytes (Figure 6D,E and Supplemental Figure S7B). These DEGs were enriched in the pathways including the methylation, the interferon α/β signaling and the fibrinolysis pathways. Example genes included betaine-homocysteine S-methyltransferase (Bhmt), methionine adenosyltransferase 2A (Mat2a), and dimethylglycine dehydrogenase (Dmgdh), which all encode key enzymes that promote methylation reactions.
Given a link between hepatic methylation reactions and the activities of PPARα pathway, we examined liver DNA methylation and hepatic PPARα expression. In loss-of-function studies, liver sections of AAV-Hep-ADK−/− mice displayed significantly decreased intensity of 5-methylcytosine (5-mC) staining compared with those of AAV-Hep-ADK+/+ mice. This decrease in DNA methylation was accompanied by significantly increased liver amount of PPARα (Supplemental Figure S8A-C). In contrast, the intensity of 5-mC staining in liver sections of Hep-ADKTg mice was significantly stronger than that of Hep-ADKWT mice (Figure 6F) and was associated with decreased levels of S-adenosylmethionine and S-adenosylhomocysteine (Supplemental Figure S8D). This suggests an inverse relationship between increased liver DNA methylation and hepatocyte activities of PPARα pathway (e.g., fatty acid oxidation). Consistently, DNA methylation of Ppara at the promoter and exon1 (Supplemental Figure S8E) in livers of Hep-ADKTg mice was significantly higher than that in Hep-ADKWT mice and accompanied by significantly decreased expression of Ppara (Figure 6G). Next, we examined the effect of forced PPARα expression on altering gene expression related to fat deposition in Hep-ADKTg hepatocytes. Consistent with increased fat deposition (Figure 6A), the mRNA levels of Cpt1a and Ppard, which both function to increase fatty acid oxidation, were significantly lower in hepatocytes from Hep-ADKTg mice than their respective levels in hepatocytes from Hep-ADKWT mice (Figure 6H, lower panel). Upon PPARα overexpression, the decrease in Cpt1a and Ppard mRNAs in Hep-ADKTg hepatocytes was reversed. PPARα overexpression also significantly increased the mRNA levels of pyruvate dehydrogenase kinase 4 (Pdk4), which promotes fatty acid oxidation, in Hep-ADKTg hepatocytes compared with control-treated Hep-ADKTg hepatocytes (Figure 6H). These results indicate that ADK promotes hepatocyte fat deposition in a manner involving suppression of Ppara, through increasing DNA methylation.
Hepatocyte-specific ADK disruption reverses the deleterious effects of hepatocyte-specific adenosine 2A receptor deficiency on hepatocyte fat deposition, liver inflammation, and systemic insulin resistance
ADK disruption or inhibition increases intracellular adenosine 8, 9, whose release into extracellular space functions to activate A2AR. The latter has been implicated to protect against diet-induced NAFLD phenotype and systemic insulin resistance 15. When the relationship between hepatic ADK and A2AR was analyzed, the mRNA levels of Adora2a in hepatocytes from Hep-ADKTg mice were lower than those in hepatocytes from Hep-ADKWT mice (P = 0.06, Supplemental Figure S9A). In livers of A2AR-disrupted (A2AR−/−) mice, liver amount of ADK was significantly increased compared with that of WT mice (Supplemental Figure S9A). This suggests an inverse relationship between ADK and A2AR. Next, we sought to examine the effect of hepatocyte-specific ADK disruption on alleviating metabolic phenotypes associated with hepatocyte-specific A2AR deficiency. In hepatocytes isolated from mice with heterozygous ADK disruption and homozygous A2AR deficiency (Hep-A2AR−/−-ADK+/− or Hep-DKO), the degrees of fat deposition were significantly lower than those in hepatocytes from Hep-A2AR−/− mice under both basal and palmitate-stimulated conditions (Supplemental Figure S9B). When liver inflammation was analyzed in mice fed an HFD, the phosphorylation states of Jnk p46 and Nfκb P65 in livers from Hep-DKO mice were significantly lower than their respective levels in livers from Hep-A2AR−/− mice (Supplemental Figure S9C). Moreover, Hep-A2AR−/− mice displayed significantly increased systemic insulin resistance compared with Hep-A2AR+/+ mice (Supplemental Figure S9D). This metabolic phenotype of hepatocyte-specific A2AR deficiency, however, was completely reversed upon hepatocyte-specific ADK disruption (Supplemental Figure S9D,E). Together with the results in Figure 6, these results suggest that ADK regulation of hepatocyte fat deposition and proinflammatory responses, as well as systemic insulin resistance is attributable to ADK promotion of methylation reaction and independent of A2AR in hepatocytes.
Hepatocyte ADK promotes proinflammatory changes in the RNA transcriptomes of liver non-parenchymal cells
Hepatocyte ADK levels positively correlate with liver inflammation. We then examined the direct effect of ADK-driven hepatocyte mediators on macrophage activation. Compared with macrophages treated with conditioned media (CM) of Hep-ADKWT hepatocytes, macrophages treated with CM of Hep-ADKTg hepatocytes displayed significantly increased mRNA levels of Il1b, Il6, Il10, and Tnfa, which all are indictive of macrophage proinflammatory activation, under both basal and LPS-stimulated conditions (Figure 7A). Next, we performed scRNAseq analysis to gain insights into how ADK-driven hepatocyte mediators regulate the RNA transcriptome of liver NPCs. Among NPCs isolated from chow diet-fed Hep-ADKTg and Hep-ADKWT mice, nine types of cells, additional to hepatocytes, were verified based on their respective markers (Figure 7B and Supplemental Figure S10). Additionally, ADK overexpression in hepatocytes did not cause significant shifts in cell types and populations of NPCs (Figure 7C). Considering the role for macrophages in regulating liver inflammation 28, 29, we chose macrophages as our first target and performed scRNAseq DE analysis on the 539 macrophages grouped by the Hep-ADKTg and the Hep-ADKWT genotypes (Supplemental Figure S11A,B). Among the 9,107 genes retained in the analysis, 53 genes were significantly up-regulated, and 363 genes were significantly down-regulated in liver macrophages in response to ADK overexpression [for all DE genes, ∣log2(FC)∣ > log2(1.2), FDR < 0.05; Supplemental Figure S11C]. While suggesting decreased fatty acid betaoxidation, DEGs indicated increased proinflammatory activation of macrophages from livers of Hep-ADKTg mice relative to those from livers of Hep-ADKWT mice (Figure 7D and Supplemental Figure S11C). Top DEGs, collectively indicating macrophage proinflammatory activation, included the phosphoglycerate mutase 2 (Pgam2), annexin A2 (Anxa2), the lymphocyte antigen 6 complex, locus A (Ly6a), the lymphocyte antigen 6 complex, locus C2 (Ly6c2), the lymphocyte antigen 6 complex, locus D (Ly6d), and the C-C motif chemokine receptor 9 (Ccr9) (Figure 7D). Because monocyte-derived macrophages are the main source of proinflammatory macrophages during liver inflammation 30, we performed a scRNA-seq DE analysis on the 1,339 monocytes grouped by the Hep-ADKTg and the Hep-ADKWT genotypes. Of the 10,110 genes retained in the analysis, 618 genes were identified as DE genes [∣log2(FC)∣ > log2(1.2), FDR < 0.05]. Among the DEGs, 194 were up-regulated and 393 were down-regulated in liver monocytes upon hepatocyte ADK overexpression (Supplemental Figure S12). Functions of down-regulated DEGs suggested a decrease in the fatty acid oxidation level in monocytes. Furthermore, upon comparing the transcriptomes of macrophages and monocytes, we identified multiple common DEGs shared between the two cell types (Supplemental Figure S13). We speculated Ly6c2 as one of the top candidate genes that stimulate macrophage proinflammatory activation in response to ADK-driven hepatocyte mediators. Consistently, treatment of macrophages with palmitate, an inflammatory fatty acid, significantly increased Ly6c2 expression, which was accompanied with increased expression of Il1b, Il6, Il10, and Tnfa (Figure 7E).
Figure 7. Hepatocyte ADK overexpression promotes the proinflammatory activation of liver NPCs through altering RNA transcriptomes.

(A) Stimulation of macrophage expression of proinflammatory cytokines by ADK-driven hepatocyte factors. (B,C) Cell types identified during scRNAseq. (D) Hepatocyte ADK-driven up-regulation of macrophage genes. (E) Upregulation of macrophage Ly6c2 expression by palmitate. (F) Ligand-receptor pathways between hepatocytes and liver NPCs. For A and E, wild-type bone marrow-derived macrophages (BMDMs) were incubated with media mixed with or without conditioned media (CM) from Hep-ADKTg and/or Hep-ADKWT hepatocytes for 24 hr. Prior to harvest, BMDMs were treated with LPS (20 ng/mL) for 6 hr (A) or palmitate (Pal, 250 μM) for 24 hr (E). Data are means ± SEM. n = 6 - 8. *, P < 0.05; **, P < 0.01; and ***, P < 0.001 BMDM/ADKTg Hep-CM vs. BMDM/ADKWT Hep-CM for the same gene (A) or Pal-treated cells vs. control-treated cells (BSA) (E).
In addition to monocytes/macrophages, neutrophils, lymphocytes, and other liver NPCs critically regulate liver inflammation. Next, we analyzed cell-cell interactions among hepatocytes and all liver NPCs and identified strong interactions between hepatocytes and any one type of cells among monocytes, macrophages, neutrophils, B cells, T cells, endothelial cells, and hepatic stellate cells (HSCs) (Supplemental Figure S14). The pathway-level function of CellChat 31 was used to estimate the combined strength of interactions of multiple ligand-receptor pairs when the genes belong to the same signaling pathways. We identified 15 pathways with significant different interaction strengths (p-value < 0.05). Among these pathways, eight were found to have stronger interactions in Hep-ADKTg than Hep-ADKWT and seven showed the opposite pattern (Figure 7F). Notably, interactions through the insulin-like growth factor (IGF) signaling pathway and the protease-activated receptors (PARs) pathway were enhanced in response to hepatocyte-specific ADK overexpression (Supplemental Figure S15). How ADK-driven factors upregulate these two pathways, as well as their functions, remains to be elucidated.
DISCUSSION
ADK assists in removing intracellular adenosine and enhances methylation reactions. Here, we provide the primary evidence validating that hepatic ADK amount was increased in subjects with NAFLD and positively correlated with obesity-associated metabolic phenotypes in HFD-fed WT mice. Furthermore, using loss-of-function studies, we established a causal role for hepatocyte-ADK disruption in decreasing hepatocyte fat deposition, although hepatocyte ADK deficiencydriven excessive fatty acid oxidation or ketogenesis in mice complicated the pathogenesis of obesity and related metabolic dysregulation. In contrast, using gain-of-function studies, we provided compelling evidence establishing a causal role for forced hepatocyte ADK expression in promoting hepatic steatosis and metabolic dysregulation in mice. Notably, hepatocyte-specific ADK overexpression significantly increased body weight, adiposity, and hepatocyte fat deposition in both male and female mice even when mice were maintained on a chow diet. Additionally, sex differences did not completely superimpose the role played by ADK because female hepatocyte ADK-overexpressing mice displayed comparable degrees, but female control mice displayed smaller degrees, in body weight and insulin sensitivity relative to age-matched male control mice. Collectively, these lines of evidence strongly support hepatocyte ADK as an obesogenic gene (enzyme), which has high translational implication in managing obesity and related metabolic diseases.
While ADK promotes hepatocyte fat deposition, an interesting question to ask is how ADK, as an enzyme that phosphorylates adenosine, regulates fat metabolic fluxes. Considering that maturity-onset hepatocyte ADK deficiency increased ketogenesis, fatty acid oxidation is then speculated as the key fat flux altered by hepatocyte ADK. Instead of using classic assays to quantify metabolic fluxes, the present study analyzed ADK regulation of hepatocyte RNA transcriptome with hope to elucidate the mechanisms underlying ADK promotion of hepatocyte fat deposition. Notably, two exciting findings pointed to a link between decreased Ppara expression and increased DNA methylation in response to ADK overexpression. This link was also confirmed by the finding that modulating hepatocyte ADK amount caused a positive correlation between hepatic amount or expression of PPARα and hepatic amount of hepatic 5-mC, indicative of DNA methylation. Specifically, hepatocyte ADK overexpression significantly increased the methylation of both promoter and exon regions of Ppara gene, resulting in decreased Ppara expression. As a result of reduced Ppara expression and PPARα signaling, hepatocyte fat acid oxidation was reduced, thereby contributing to increased hepatocyte fat deposition. Consistent with this mechanism, hepatocyte overexpression of PPARα caused a reversal of down-regulation of the expression of genes for fatty acid oxidation including Cpt1a, Ppard, and Pdk4. Because of this, the present study provides the primary evidence to support a role for hepatocyte ADK in promoting hepatocyte fat deposition through increasing Ppara methylation to decrease the expression of genes for fatty oxidation. To be noted, ADK inhibition is shown to increase intracellular adenosine 9, whose release into extracellular space brings about an anti-inflammatory effect largely through Adora2a 32. This led to the postulation that activation of Adora2a, which functions to protect against hepatocyte fat deposition 15, contributes to ADK disruption-driven decrease in hepatocyte fat deposition; however, this was not the case. Hepatocyte-specific Adora2a deficiency increased the severity of HFD-induced hepatic steatosis and systemic insulin resistance, which was completely reserved by hepatocyte-specific ADK disruption. This serves as additional evidence validating that ADK promotion of DNA methylation, but not suppression of Adora2a signaling, accounts for increased hepatocyte fat deposition. Additionally, in response to hepatocyte ADK overexpression, extrahepatic tissues, e.g., intestine and WAT, displayed aspects indicative of increased nutrient absorption and WAT dysfunction. This suggests that ADK-driven communications between the liver and extrahepatic tissues were involved in obesity and metabolic dysregulation.
Excessive fat deposition in hepatocytes is a main cause of liver inflammation that contributes to the pathogenesis of local (hepatic) and systemic insulin resistance 33, 34. Upon ADK deficiency or overexpression only in hepatocytes in mice, the present study provided complementary evidence to support a critical role for hepatocyte ADK in promoting liver inflammation. Since ADK overexpression occurred in hepatocytes, but not liver immune cells, it is speculated that ADK-driven changes in hepatic lipid profile are triggers contributing to liver inflammation. As supporting evidence, the levels of most hepatic bis(monoacylglycero)phosphate and lysophosphatidylcholine species were increased whereas tetralinoleoyl cardiolipin levels were decreased upon hepatocyte ADK overexpression, indicating oxidative stress and mitochondrial dysfunction 24. To be noted, hepatic levels of mtDNA were increased likely acting through cyclic GMP-AMP synthase (cGAS)-STING signaling 35 to stimulate macrophage STING, as indicated by the results upon co-staining STING and F4/80 in livers sections. This suggests that ADK-driven dysfunctional hepatocyte-macrophage crosstalk, due to, in large part, hepatocyte mitochondrial dysfunction, accounts for liver inflammation. Consistently, treatment of macrophages with conditioned media from ADK-overexpressing hepatocytes significantly increased the expression of proinflammatory cytokines under both basal and LPS-stimulated conditions. Because of this, it is conceivable that ADK-driven proinflammatory mediators act via paracrine manners to activate liver immune cells, thereby elevating liver inflammation. In agreement with this notion, ADK overexpression in hepatocytes altered RNA transcriptome of liver macrophages in a pattern favoring its proinflammatory activation. Similar changes were also observed in the RNA transcriptome of liver monocytes, which gave rise to most proinflammatory macrophages in the liver. Notably, Ly6c2 is among several common genes whose expression was increased in both macrophages and monocytes. This points to the involvement of Ly6c2 in macrophage proinflammatory activation in response to hepatocyte ADK overexpression. In other words, Ly6c2 responded to ADK-driven hepatocyte mediators to increase macrophage activation. In support of this, treatment of macrophages with palmitate significantly increased Ly6c2 expression, along with increased expression of key proinflammatory cytokines. The results of RNA transcriptomic studies of liver NPCs also indicated the involvement of neutrophils, lymphocytes, and endothelial cells in the development of liver inflammation in response to elevated ADK in hepatocytes. Hepatocyte ADK-driven cell-cell communications were mediated at least, in part, through IFGF and PAR signaling, whose functions warrant further studies.
In summary, we demonstrated a role for hepatocyte ADK in promoting obesity and liver inflammation while validating the translational implication of hepatocyte ADK in subjects with NAFLD. The mechanisms through which hepatocyte ADK functions as an obesogenic enzyme are attributable to ADK suppression of hepatocyte expression of genes in the Ppara pathway through promoting DNA methylation, resulting in decreased fatty acid oxidation that contributes to increased hepatocyte fat deposition. Moreover, the ADK in hepatocytes promotes the proinflammatory activation of liver NPCs through ADK-driven hepatocyte mediators via manners involving macrophage STING and Ly6c2. Accordingly, hepatocyte ADK is a viable target for management of obesity and related metabolic disease.
Supplementary Material
What You Need to Know.
BACKGROUND & CONTEXT
The role for hepatocyte adenosine kinase (ADK) in the pathophysiology of non-alcoholic fatty liver disease (NAFLD) is unknown
NEW FINDINGS
Hepatocyte ADK triggers or exacerbates the pathogenesis of NAFLD by promoting excessive fat deposition and liver inflammation.
LIMITATIONS
The study did not examine the extent to which the stimulator of interferon genes (STING) in macrophages mediators the effects of ADK-driven hepatocyte factors on promoting liver inflammation.
BASIC RESEARCH RELEVANCE
The study validates the feasibility of ADK inhibition as a novel therapeutic strategy for NAFLD.
ACKNOWLEDGEMENTS
This work was supported in whole or in part by grants from the National Institutes of Health (DK095862 and DK124854 to C.W.), and the Hickam Endowed Chair, Gastroenterology, Medicine, Indiana University, the Indiana University Health – Indiana University School of Medicine Strategic Research Initiative to G.A. and H.F., the SRCS (IK6 BX004601) and VA Merit award to G. A. (5I01BX000574), and RCS (IK6BX005226) and VA Merit award to H.F. (1I01BX003031) from the United States Department of Veteran’s Affairs, Biomedical Laboratory Research and Development Service and NIH grants DK108959 and DK110421 (HF), DK054811, DK115184, DK076898, DK107310, DK110035, and AA028711 to G.A. and S.G. NIH P30 AG044271 and P30 AG013319 support the Functional Lipidomics Core at Barshop Institute for Longevity and Aging Studies. Also, C.W. is supported by the Hatch Program of the National Institute of Food and Agriculture (NIFA).
Abbreviations
- A2AR
adenosine 2A receptor
- Acc1
acetyl-CoA carboxylase 1
- ADK
adenosine kinase
- BMDM
bone marrow-derived macrophages
- BSA
bovine serum albumin
- CM
conditioned media
- Cpt1a
carnitine palmitoyltransferase 1a
- DEGs
differentially expressed genes
- DMEM
Dulbecco’s modified Eagle’s medium
- Fas
fatty acid synthase
- FBS
fetal bovine serum
- FFA
free fatty acids
- G6pase
glucose-6-phosphatase
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- Gk
glucokinase
- GTT
glucose tolerance test
- H&E
hematoxylin and eosin
- HFD
high-fat diet
- HSCs
hepatic stellate cells
- LFD
low-fat diet
- Il1b
interleukin 1β
- I16
interleukin 6
- IMDM
Iscove’s Modified Dulbecco’s medium
- ITT
insulin tolerance test
- Ly6c2
lymphocyte antigen 6C2 precursor
- LPS
lipopolysaccharide
- JNK
c-Jun N-terminal kinases
- NAFLD
non-alcoholic fatty liver disease
- NFκB
nuclear factor kappa B
- NASH
non-alcoholic steatohepatitis
- PBS
phosphate-buffered saline
- Pp65
phosphorylated p65 subunit of NFκB
- Pp46
phosphorylated JNK1 (p46)
- PPARα
peroxisome proliferator activated receptor alpha
- PPARδ
peroxisome proliferator activated receptor delta
- RQ
respiratory quotient
- STING
stimulator of interferon genes
- TG
triglycerides
- Tnfa
tumor necrosis factor α
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST: The authors declare that they do not have conflict of interest. The content of this article does not involve any materials/information applicable to US export control.
REFERENCES
- 1.Doege H, Baillie RA, Ortegon AM, et al. Targeted deletion of FATP5 reveals multiple functions in liver metabolism: alterations in hepatic lipid homeostasis. Gastroenterology 2006;130:1245–1258. [DOI] [PubMed] [Google Scholar]
- 2.Kotronen A, Juurinen L, Tiikkainen M, et al. Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology 2008;135:122–130. [DOI] [PubMed] [Google Scholar]
- 3.Samuel VT, Liu Z-X, Qu X, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem 2004;279:32345–32353. [DOI] [PubMed] [Google Scholar]
- 4.Lyu K, Zhang Y, Zhang D, et al. A Membrane-Bound Diacylglycerol Species Induces PKCϵ-Mediated Hepatic Insulin Resistance. Cell Metabolism 2020;32:654–664.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biddinger SB, Hernandez-Ono A, Rask-Madsen C, et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metabolism 2008;7:125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Younossi ZM, Otgonsuren M, Henry L, et al. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015;62:1723–1730. [DOI] [PubMed] [Google Scholar]
- 7.Bjursell Magnus K, Blom Henk J, Cayuela Jordi A, et al. Adenosine kinase deficiency disrupts the methionine cycle and causes hypermethioninemia, encephalopathy, and abnormal liver function. Am J Hum Genet 2011;89:507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murugan M, Fedele D, Millner D, et al. Adenosine kinase: An epigenetic modulator in development and disease. Neurochemistry International 2021;147:105054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Y, Wang Y, Yan S, et al. Regulation of endothelial intracellular adenosine via adenosine kinase epigenetically modulates vascular inflammation. Nature Communications 2017;8:943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang M, Zeng X, Yang Q, et al. Ablation of Myeloid ADK (Adenosine Kinase) Epigenetically Suppresses Atherosclerosis in ApoE−/− (Apolipoprotein E Deficient) Mice. Arteriosclerosis, Thrombosis, and Vascular Biology 2018;38:2780–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boison D Adenosine kinase: exploitation for therapeutic gain. Pharmacol Rev 2013;65:906–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gnad T, Scheibler S, von Kugelgen I, et al. Adenosine activates brown adipose tissue and recruits beige adipocytes via A2A receptors. Nature 2014;516:395–399. [DOI] [PubMed] [Google Scholar]
- 13.Imarisio C, Alchera E, Sutti S, et al. Adenosine A2a receptor stimulation prevents hepatocyte lipotoxicity and nonalcoholic steatohepatitis (NASH) in rats. Clin Sci 2012;123:323–32. [DOI] [PubMed] [Google Scholar]
- 14.Antonioli L, Blandizzi C, Csoka B, et al. Adenosine signalling in diabetes mellitus - pathophysiology and therapeutic considerations. Nat Rev Endocrinol 2015;11:228–241. [DOI] [PubMed] [Google Scholar]
- 15.Cai Y, Li H, Liu M, et al. Disruption of adenosine 2A receptor exacerbates NAFLD through increasing inflammatory responses and SREBP1c activity. Hepatology 2018;68:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li T, Ren G, Lusardi T, et al. Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Invest 2008;118:571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen H-Y, Singer P, Lytle N, et al. Adenosine augmentation ameliorates psychotic and cognitive endophenotypes of schizophrenia. J Clin Invest 2012;122:2567–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boison D, Aronica E. Comorbidities in Neurology: Is adenosine the common link? Neuropharmacology 2015;97:18–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boison D, Scheurer L, Zumsteg V, et al. Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc Natl Acad Sci USA 2002;99:6985–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shakiba M, Mahjoub F, Fazilaty H, et al. Adenosine kinase deficiency with neurodevelopemental delay and recurrent hepatic dysfunction: A case report. Adv Rare Dis 2016;3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu J, Yang Q, Zhang X, et al. Endothelial adenosine kinase deficiency ameliorates diet-induced insulin resistance. J Endocrinol 2019;242:159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakamura S, Takamura T, Matsuzawa-Nagata N, et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J Biol Chem 2009;284:14809–14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma L, Li H, Hu J, et al. Indole Alleviates Diet-induced Hepatic Steatosis and Inflammation in a Manner Involving Myeloid Cell PFKFB3. Hepatology 2020;72:1191–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He Q, Han X. Cardiolipin remodeling in diabetic heart. Chem Phys Lipids 2014;179:75–81. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Li S, Zhou Y, et al. Ablation of cytochrome P450 omega-hydroxylase 4A14 gene attenuates hepatic steatosis and fibrosis. Proc Natl Acad Sci 2017;114:3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qi C, Zhu Y, Pan J, et al. Absence of spontaneous peroxisome proliferation in enoyl-CoA Hydratase/L-3-hydroxyacyl-CoA dehydrogenase-deficient mouse liver. Further support for the role of fatty acyl CoA oxidase in PPARalpha ligand metabolism. J Biol Chem 1999;274:15775–15780. [DOI] [PubMed] [Google Scholar]
- 27.Ding J, Loizides-Mangold U, Rando G, et al. The Peroxisomal Enzyme L-PBE Is Required to Prevent the Dietary Toxicity of Medium-Chain Fatty Acids. Cell Reports 2013;5:248–258. [DOI] [PubMed] [Google Scholar]
- 28.Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nature Reviews Immunology 2017;17:306. [DOI] [PubMed] [Google Scholar]
- 29.Luo X, Li H, Ma L, et al. Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology 2018;155:1971–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krenkel O, Puengel T, Govaere O, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018;67:1270–1283. [DOI] [PubMed] [Google Scholar]
- 31.Jin S, Guerrero-Juarez CF, Zhang L, et al. Inference and analysis of cell-cell communication using CellChat. Nature communications 2021;12:1088–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Idzko M, Ferrari D, Riegel A-K, et al. Extracellular nucleotide and nucleoside signaling in vascular and blood disease. Blood 2014;124:1029–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Savage DB, Choi CS, Samuel VT, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest 2006;116:817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirsova P, Ibrahim SH, Krishnan A, et al. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology 2016;150:956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen L, Dong J, Liao S, et al. Loss of Sam50 in hepatocytes induces cardiolipin-dependent mitochondrial membrane remodeling to trigger mtDNA release and liver injury. Hepatology 2022;00:1–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.