

Abstract
Flavin‐dependent halogenases (FDHs) natively catalyze selective halogenation of electron rich aromatic and enolate groups. Nearly all FDHs reported to date require a separate flavin reductase to supply them with FADH2, which complicates biocatalysis applications. In this study, we establish that the single component flavin reductase/flavin dependent halogenase AetF catalyzes halogenation of a diverse set of substrates using a commercially available glucose dehydrogenase to drive its halogenase activity. High site selectivity, activity on relatively unactivated substrates, and high enantioselectivity for atroposelective bromination and bromolactonization was demonstrated. Site‐selective iodination and enantioselective cycloiodoetherification was also possible using AetF. The substrate and reaction scope of AetF suggest that it has the potential to greatly improve the utility of biocatalytic halogenation.
Keywords: Atroposelectivity, C−H Functionalization, Flavin Dependent Halogenase, Halocyclization, Iodinase
The single component flavin reductase/flavin dependent halogenase AetF catalyzes halogenation of a diverse set of substrates. High site selectivity, activity on relatively unactivated substrates, and high enantioselectivity for atroposelective bromination and bromolactonization was demonstrated. Site‐selective iodination and enantioselective cycloiodoetherification was also possible using AetF.

Flavin‐dependent halogenases (FDHs) natively catalyze site‐selective halogenation of electron rich aromatic and enolate groups in a diverse range of halogenated natural products.[ 1 , 2 , 3 ] This unique capability has led to extensive efforts to understand FDH mechanism and the origins of their site selectivity.[ 4 , 5 ] Early studies established that FDH catalysis initially mirrors flavoprotein monooxygenase catalysis in that an enzyme‐bound, reduced flavin adenine dinucleotide (FADH2) cofactor reacts with O2 to generate a hydroperoxy flavin intermediate. [6] In FDHs, this intermediate reacts with bound halide, typically bromide or chloride, to generate HOX, which migrates through the enzyme to a substrate binding pocket.[ 7 , 8 , 9 ] Most evidence now suggests that hydrogen bonding by a key active site lysine residue activates HOX for electrophilic halogenation, and precise substrate binding leads to site‐selective halogenation by this species.[ 5 , 10 , 11 ]
Nearly all FDHs reported to date require a separate flavin reductase to supply FADH2,[ 6 , 12 ] and this enzyme is typically driven by a glucose/glucose dehydrogenase cofactor regeneration system for biocatalysis applications (Figure 1A). [13] The need for a separate flavin reductase complicates biocatalysis since these enzymes are not widely available and are typically produced in‐house, they add to the protein waste that must be removed during product isolation, and they can lead to undesired background reactions. [14] Previously, our group demonstrated that genetically fusing the flavin reductase RebF to the FDH RebH improved halogenation yields from whole‐cell biocatalysis, suggesting that increased local concentration of FADH2 can improve the efficiency of biocatalysis relative to the free enzymes (Figure 1A). [15] A recent family‐wide sequence/activity analysis of FDHs did not reveal any naturally occurring flavin reductase/FDH fusions, [16] but Moore reported that the FDH Bmp5 adopts a more elegant solution to this problem by combining both reductase and halogenase activities in a single chain enzyme capable of binding both FAD and NADP (Figure 1B). [17] Bmp5 catalyzes halogenation and decarboxylative halogenation of 4‐hydroxybenzoic acid, but because this substrate and the others examined were halogenated at electronically activated sites, it is not clear that the operational simplicity of this enzyme would extend to less activated sites/substrates that comprise much of the biocatalytic value of FDHs.
Figure 1.
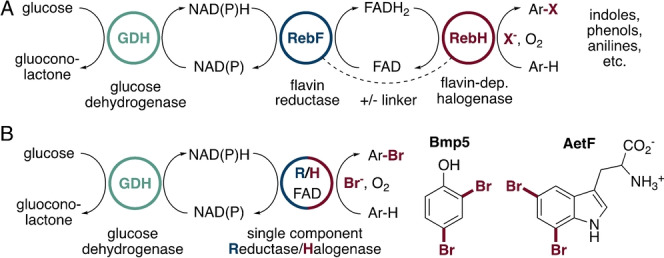
A) Halogenation of electron rich aromatic compounds has been reported using either two‐component or fused flavin reductase/halogenase systems like RebH/RebF. [4] B) Native products of the two known single component flavin reductase/halogenases (R/H), Bmp5 and AetF.[ 17 , 18 ]
We became intrigued by more recent reports describing a second single component FDH, AetF, from A. hydrillicola. [19] AetF was first characterized by Wilde and Niedermeyer as a tryptophan halogenase that catalyzes dibromination of tryptophan to generate the 5,7‐dibromo‐L‐tryptophan core of aetokthonotoxin (Figure 1B). Investigating activity on the native tryptophan substrate, the Moore group showed that AetF is a unique single component flavin reductase/FDH. [18] Inspired by the potential benefits of a single component FDH with the unique catalyst‐controlled selectivity of tryptophan FDHs, we set out to evaluate the catalytic efficiency of AetF on non‐native substrates. We find that AetF has broad substrate scope, halogenating not only electron rich aromatics, but also relatively electron deficient substrates and heterocycles that are not halogenated efficiently or at all by any FDHs that we have studied to date.[ 16 , 20 ] AetF also provides good levels of conversion and selectivity toward substrates that undergo enantioselective halogenation processes via topologically distinct mechanisms.[ 21 , 22 , 23 ] Finally, we find that AetF catalyzes iodination of aromatic compounds at sites that are less activated than those that have been characterized using other FDHs and enables enantioselective cycloiodoetherification. These findings suggest that AetF has great potential for expanding the scope of biocatalytic halogenation.
Initial analysis of AetF substrate tolerance focused on electron rich indoles, anilines, and phenols similar to those that we and others have previously used to probe the activity of different FDHs.[ 1 , 3 , 16 , 20 ] Reactions were conducted using purified AetF and a commercially available glucose dehydrogenase for NADPH regeneration under conditions analogous to those that we typically use for FDH biocatalysis. Specifically, excess glucose is needed to account for decoupling between flavin reduction and product formation, [24] and glutathione and catalase are added to scavenge HOBr and H2O2 generated by the enzyme. [21] Conversions were obtained by UPLC/MS, and isolated yields for select compounds were obtained from preparative reactions (≈5 mg scale). Selective bromination of several substrates was observed (Figure 2, 1 a–5 a), showing that the AetF has robust activity on non‐native substrates. Importantly, halogenation did not always occur at sites with the highest calculated halenium affinity (HalA) [25] values, meaning that the enzyme, rather than substrate electronics, was controlling site selectivity in these cases (e.g. 1 a and 2 a).
Figure 2.
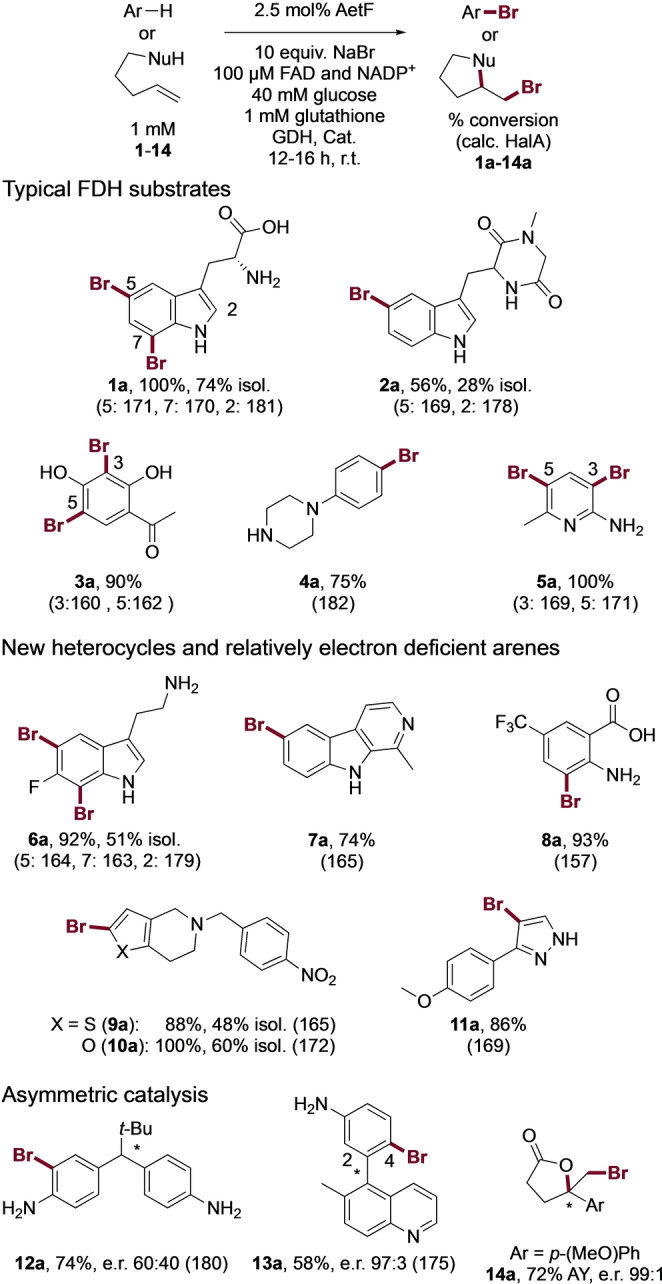
Representative examples of substrates brominated by AetF (see also Figure S1 and Table S1). Conversions were determined from relative integration values for starting material and product for reactions conducted on 75 μL scale; selected isolated yields (isol.) and assay yields (AY) relative to authentic internal standards were also obtained for reactions conducted on 20–30 mL and 75 μL scale, respectively. Halenium affinity (HalA) values [25] were calculated as previously reported. [20] GDH=glucose dehydrogenase, Cat.=catalase.
We next challenged the enzyme with a broader range of heterocycles and aromatic compounds bearing electron withdrawing groups. Notably, AetF catalyzes site‐selective halogenation of a diverse set of substrates that are poorly reactive toward several FDHs obtained from previous engineering [13] and genome mining [16] efforts (Figure 2, 6 a–11 a). This set includes furan, thiophene, and pyrazole heterocycles lacking additional electron donating groups. Calculated HalA values for some of these substrates show that the sites halogenated are lower than those for typical indole and aniline substrates and the HalA cutoff of 164 kcal mol−1 [20] that we previously established as the minimum required for halogenation by the tryptophan halogenase RebH.
Given our ongoing interest in asymmetric FDH catalysis, we evaluated the activity of AetF on substrates capable of undergoing desymmetrization, [22] atroposelective dynamic kinetic resolution, [23] and halolactonization [21] via bromination. Again, AetF provides good conversion and modest‐to‐high enantioselectivity on each of the substrates examined (Figure 2, 12 a‐‐14 a). The observed activity is significantly higher than that provided by any of the WT enzymes that were examined in our original studies. For example, while low enantioselectivity was observed for 12 a, only chlorination of 12 was previously reported, [22] so the observed bromination is notable. None of our previously reported FDHs could provide brominated biaryl 13 a, but essentially a single atropisomer of this compound is provided by AetF. While a variant of RebH was able to catalyze formation of 14 a with up to 96 : 4 e.r., [21] that variant had six mutations relative to WT.
Despite the broad scope of AetF toward bromination, only the native substrate, tryptophan, underwent chlorination to a significant extent (≈20 % conversion, Figure S2), mirroring previous examples of FDHs with a preference toward bromide.[ 16 , 26 ] On the other hand, we were surprised to observe quantitative conversion of tryptophan to 5,7‐diiodotryptophan in the presence of sodium iodide (Figure 3A). Formation of iodinated products has only been reported for reactions involving a few FDHs, including Bmp5, [17] PltM, [27] and VirX1. [28] A more recent study showed that electronically activated indoles and phenols can also be iodinated by HOI generated from I− and H2O2 that is produced by flavin reductases alone. [14] This reactivity could be eliminated by including catalase in the iodination reactions, and, as noted above, we have similarly shown that undesired reactivity of free HOBr could be eliminated using glutathione as a scavenger. [21] Selective diiodination of the benzene ring of tryptophan by AetF in the presence of both catalase and glutathione is therefore notable. We found that 6‐fluorotryptamine also undergoes 5,7‐diiodination, pyrazole 11 undergoes selective monoiodination, and several other substrates undergo iodination based on LC/MS analysis (Figure S3). Finally, AetF catalyzes cycloiodoetherification to give tetrahydrofuran 15 a in good yield and 99 : 1 e.r. This reaction is not possible using other FDHs in our laboratory, [29] and its high enantioselectivity provides strong evidence that AetF iodinase activity is not due to free HOI (Figure 3A).
Figure 3.
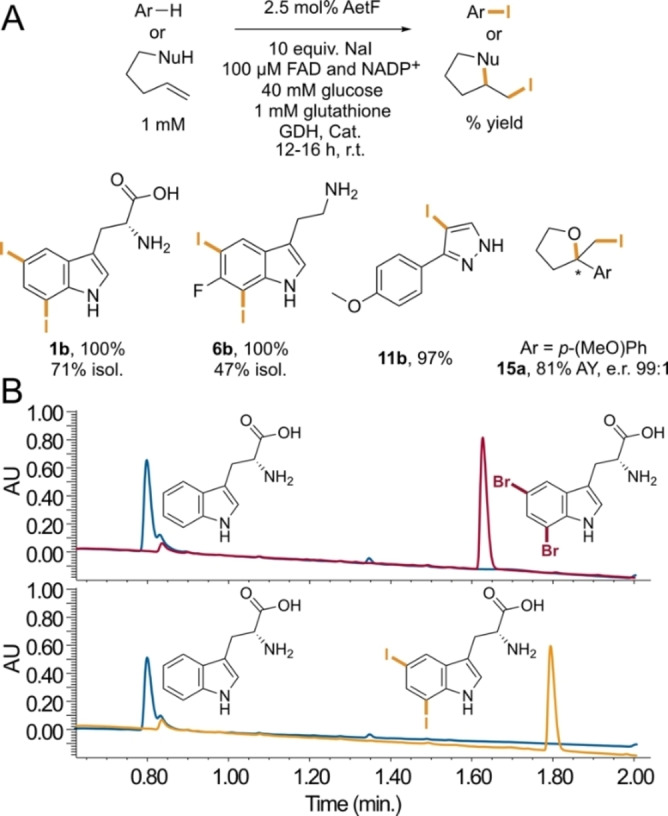
A) AetF‐catalyzed aromatic iodination and cycloiodoetherification. Conversions were determined from relative integration values for starting material and product for reactions conducted on 75 μL scale; selected isolated yields (isol.) and assay yields (AY) relative to authentic internal standards were also obtained for reactions conducted on 20–30 mL scale. B) Bromination and iodination of tryptophan catalyzed by AetF (red and orange traces, respectively) and AetF K258A (blue traces), showing no conversion to either halogenated product using the latter enzyme. GDH=glucose dehydrogenase, Cat.=catalase.
Given the unique reactivity of AetF, we sought to provide initial insights into its structure and mechanism. Nearly all FDHs reported to date possess an active site lysine that is believed to activate HOX toward electrophilic attack.[ 5 , 10 , 11 ] Because substrates typically bind in a manner that projects the site of halogenation toward this residue, [11] identifying it can aid targeted mutagenesis efforts aimed at altering FDH activity and selectivity. We used AlphaFold2 [30] to generate a model of AetF (Figure 4A). While AetF has low sequence homology to characterized FDHs or flavoprotein monooxygenases (FPMOs), [18] several FPMOs with high structural homology to the AetF model were identified using the DALI protein structure comparison server. [31] The highest homology was observed for predicted ancestral FPMOs that were engineered and evaluated in a study aimed at understanding extant mammalian FPMOs. [32] The structures of these enzymes show the NADP and FAD cofactors proximal to one another in the FPMO active site. Both cofactors were docked into the AetF model using Rosetta [33] and are predicted to bind in orientations analogous to those observed in the reconstituted ancestral FPMOs. Notably, however, the AetF model shows a lysine residue that is absent in the FPMOs (K258) directly over the isoalloxazine ring of the bound FAD (Figure 4B). Aligning the terminal C−C−N fragment of the active site lysine residue in the AetF model (K258) with that in the crystal structure of the FDH RebH (K79) [34] suggests that tryptophan can bind to AetF in a manner that projects a site of halogenation toward lysine, just as it does in RebH and other FDHs. [11] Suspecting that K258 might therefore function in analogy to the conserved lysine residues typically observed in FDHs,[ 8 , 11 ] we mutated this residue to alanine. AetF K258A was inactive toward both bromination and iodination (Figures 3B, S4, and S5), supporting the hypothesis that K258 acts in analogy to the active site lysine residues in other FDHs to activate HOX and providing a preliminary model to guide initial AetF engineering efforts. This result, analogous to that also shown for the K79A variant of VirX1, [28] provides further evidence that the observed iodination is not due to the release of free HOI into solution.
Figure 4.
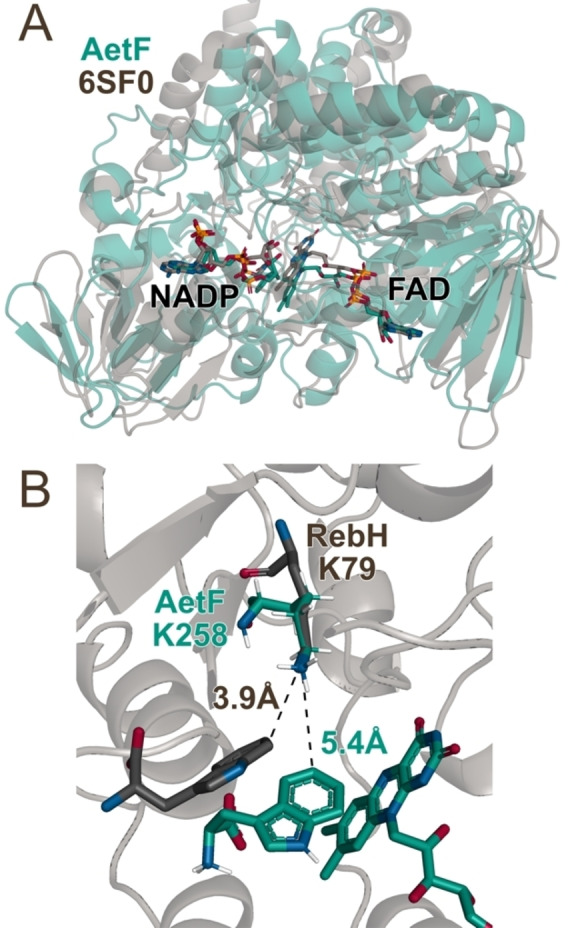
Overlays of A) a AetF⋅FAD⋅NADP model constructed using AlphaFold with cofactors docked in using Rosetta Ligand (green) with the crystal structure of a predicted ancestral FPMO (PDB ID 6SF0, grey) [32] and B) the AetF⋅FAD⋅Trp model active site (green residues) with K79 and bound Trp substrate from the FDH RebH (PDB ID 2OA1, grey residues). [34]
In summary, this study establishes that the single component flavin reductase/flavin dependent halogenase AetF catalyzes selective bromination and iodination of a diverse set of substrates. High site selectivity was observed for halogenation of several aromatic substrates, and activity on relatively unactivated substrates and heterocyclic substrates not previously reported for other FDHs was demonstrated. High enantioselectivity was observed for several transformations, including an atroposelective bromination and a cycloiodoetherification reaction that were not possible using enzymes available in our laboratory. In short, AetF can recapitulate and even exceed the catalytic capabilities of FDH variants generated via several directed evolution efforts. Computational modeling was used to confirm that a putative active site lysine residue is critical for AetF catalysis, just as it is for conventional FDHs. The unique approach required to identify this residue was necessitated by the low sequence identity of AetF to other characterized FDHs and highlights the growing utility of de novo structure prediction and structure homology tools to inform targeted mutagenesis studies. The remarkable substrate and reaction scope of AetF, along with the fact that it requires only a commercially available glucose dehydrogenase to drive its selective halogenase activity, suggest that it and related single component halogenases have the potential to greatly improve the utility of biocatalytic halogenation. Directed evolution and cascade biocatalysis involving such enzymes will be particularly advantageous relative to two‐component halogenase/reductase systems since only a single enzyme must be adapted to desired process conditions. The unique active site architecture observed in the AetF model might also minimize decoupling pathways characterized in conventional halogenases, [24] but additional structural and kinetic analysis will be required to explore this possibility.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This study was supported by the NIH (R01GM115665 to J.C.L.) and by Pfizer, Inc. Y.S.Z. was supported by the IUB Chester Davis Fellowship in Inorganic Chemistry. D.M. was supported by the Precision Health Initiative of Indiana University. NMR data were acquired on a spectrometer funded by the NSF (MRI CHE‐1920026) using a Prodigy probe that was partially funded by the Indiana Clinical and Translational Sciences Institute. MS data were acquired on a spectrometer funded by NSF Grant CHE1726633. We thank Ariana Vargas for development of chiral chromatography methods for establishing the enantioselectivity of relevant FDH reactions.
Y. Jiang, H. M. Snodgrass, Y. S. Zubi, C. V. Roof, Y. Guan, D. Mondal, N. H. Honeycutt, J. W. Lee, R. D. Lewis, C. A. Martinez, J. C. Lewis, Angew. Chem. Int. Ed. 2022, 61, e202214610; Angew. Chem. 2022, 134, e202214610.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv‐2022‐88xzv).
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Weichold V., Milbredt D., van Pée K.-H., Angew. Chem. Int. Ed. 2016, 55, 6374–6389; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6482–6498. [Google Scholar]
- 2. Gkotsi D. S., Dhaliwal J., McLachlan M. M., Mulholand K. R., Goss R. J., Curr. Opin. Chem. Biol. 2018, 43, 119–126. [DOI] [PubMed] [Google Scholar]
- 3. Latham J., Brandenburger E., Shepherd S. A., Menon B. R. K., Micklefield J., Chem. Rev. 2018, 118, 232–269. [DOI] [PubMed] [Google Scholar]
- 4. Yeh E., Garneau S., Walsh C. T., Proc. Natl. Acad. Sci. USA 2005, 102, 3960–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Flecks S., Patallo E. P., Zhu X., Ernyei A. J., Seifert G., Schneider A., Dong C., Naismith J. H., van Pée K.-H., Angew. Chem. Int. Ed. 2008, 47, 9533–9536; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 9676–9679. [Google Scholar]
- 6. Yeh E., Cole L., Barr E., Bollinger J., Ballou D., Walsh C., Biochemistry 2006, 45, 7904–7912. [DOI] [PubMed] [Google Scholar]
- 7. Dong C., Flecks S., Unversucht S., Haupt C., van Pée K.-H., Naismith J. H., Science 2005, 309, 2216–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhu X., Laurentis W. D., Leang K., Herrmann J., Ihlefeld K., van Pée K.-H., Naismith J. H., J. Mol. Biol. 2009, 391, 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Prakinee K., Phintha A., Visitsatthawong S., Lawan N., Sucharitakul J., Kantiwiriyawanitch C., Damborsky J., Chitnumsub P., van Pée K.-H., Chaiyen P., Nat. Catal. 2022, 5, 534–544. [Google Scholar]
- 10. Ainsley J., Mulholland A. J., Black G. W., Sparagano O., Christov C. Z., Karabencheva-Christova T. G., ACS Omega 2018, 3, 4847–4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Andorfer M. C., Evans D., Yang S., He C. Q., Girlich A. M., Vergara-Coll J., Sukumar N., Houk K. N., Lewis J. C., Chem Catal. 2022, 2, 2658–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moritzer A.-C., Niemann H. H., Protein Sci. 2019, 28, 2112–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Andorfer M. C., Lewis J. C., Annu. Rev. Biochem. 2018, 87, 159–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Y., Chen L., Chen H., Huang T., Shi Q., Wang X., Wang Y., Tang M.-C., Zhou N.-Y., Lin S., Sci. China Chem. 2021, 64, 1730–1735. [Google Scholar]
- 15. Andorfer M. C., Belsare K. D., Girlich A. M., Lewis J. C., ChemBioChem 2017, 18, 2099–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fisher B. F., Snodgrass H. M., Jones K. A., Andorfer M. C., Lewis J. C., ACS Cent. Sci. 2019, 5, 1844–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Agarwal V., Gamal A. A. E., Yamanaka K., Poth D., Kersten R. D., Schorn M., Allen E. E., Moore B. S., Nat. Chem. Biol. 2014, 10, 640–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adak S., Lukowski A. L., Schäfer R. J. B., Moore B. S., J. Am. Chem. Soc. 2022, 144, 2861–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Breinlinger S., Phillips T. J., Haram B. N., Mareš J., Yerena J. A. M., Hrouzek P., Sobotka R., Henderson W. M., Schmieder P., Williams S. M., Lauderdale J. D., Wilde H. D., Gerrin W., Kust A., Washington J. W., Wagner C., Geier B., Liebeke M., Enke H., Niedermeyer T. H. J., Wilde S. B., Science 2021, 371, eaax9050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andorfer M. C., Grob J. E., Hajdin C. E., Chael J. R., Siuti P., Lilly J., Tan K. L., Lewis J. C., ACS Catal. 2017, 7, 1897–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mondal D., Fisher B. F., Jiang Y., Lewis J. C., Nat. Commun. 2021, 12, 3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Payne J. T., Butkovich P. H., Gu Y., Kunze K. N., Park H.-J., Wang D.-S., Lewis J. C., J. Am. Chem. Soc. 2018, 140, 546–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Snodgrass H. M., Mondal D., Lewis J. C., J. Am. Chem. Soc. 2022, 144, 16676–16682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Phintha A., Prakinee K., Jaruwat A., Lawan N., Visitsatthawong S., Kantiwiriyawanitch C., Songsungthong W., Trisrivirat D., Chenprakhon P., Mulholland A. J., van Pée K.-H., Chitnumsub P., Chaiyen P., J. Biol. Chem. 2021, 296, 100068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ashtekar K. D., Marzijarani N. S., Jaganathan A., Holmes D., Jackson J. E., Borhan B., J. Am. Chem. Soc. 2014, 136, 13355–13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Neubauer P. R., Widmann C., Wibberg D., Schröder L., Frese M., Kottke T., Kalinowski J., Niemann H. H., Sewald N., PLoS One 2018, 13, e0196797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mori S., Pang A. H., Chandrika N. T., Garneau-Tsodikova S., Tsodikov O. V., Nat. Commun. 2019, 10, 1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gkotsi D. S., Ludewig H., Sharma S. V., Connolly J. A., Dhaliwal J., Wang Y., Unsworth W. P., Taylor R. J. K., McLachlan M. M. W., Shanahan S., Naismith J. H., Goss R. J. M., Nat. Chem. 2019, 11, 1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jiang Y., Mondal D., Lewis J. C., ACS Catal. 2022, 12, 13501–13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., Bridgland A., Meyer C., Kohl S. A. A., Ballard A. J., Cowie A., Romera-Paredes B., Nikolov S., Jain R., Adler J., Back T., Petersen S., Reiman D., Clancy E., Zielinski M., Steinegger M., Pacholska M., Berghammer T., Bodenstein S., Silver D., Vinyals O., Senior A. W., Kavukcuoglu K., Kohli P., Hassabis D., Nature 2021, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Holm L., Rosenstrom P., Nucleic Acids Res. 2010, 38, W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nicoll C. R., Bailleul G., Fiorentini F., Mascotti M. L., Fraaije M. W., Mattevi A., Nat. Struct. Mol. Biol. 2020, 27, 14–24. [DOI] [PubMed] [Google Scholar]
- 33. Davis I. W., Baker D., J. Mol. Biol. 2009, 385, 381–392. [DOI] [PubMed] [Google Scholar]
- 34. Bitto E., Huang Y., Bingman C. A., Singh S., Thorson J. S., Phillips G. N., Proteins Struct. Funct. Bioinf. 2008, 70, 289–293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.