

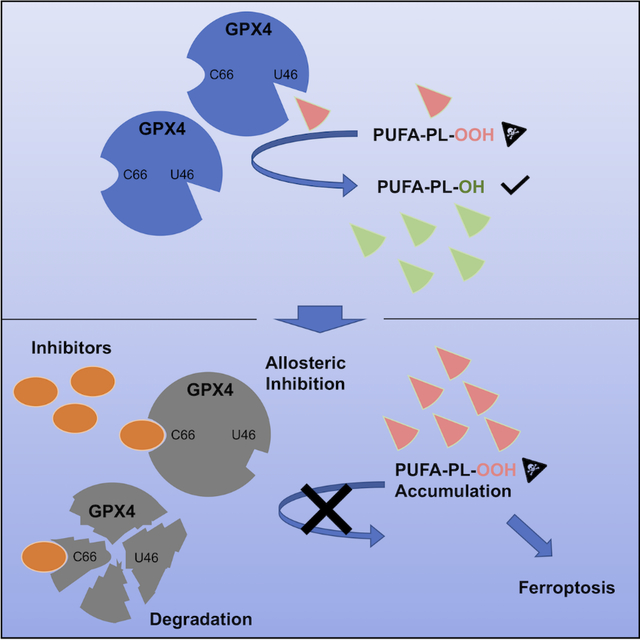
Summary
Encouraged by the dependence of drug-resistant, metastatic cancers on GPX4, we examined biophysical mechanisms of GPX4 inhibition, which revealed an unexpected allosteric site. We found that this site was involved in native regeneration of GPX4 under low glutathione conditions. Covalent binding of inhibitors to this allosteric site caused a conformational change, inhibition of activity, and subsequent cellular GPX4 protein degradation. To verify this site in an unbiased manner, we screened a library of compounds, and identified and validated that an additional compound can covalently bind in this allosteric site, inhibiting and degrading GPX4. We determined co-crystal structures of six different inhibitors bound in this site. We have thus identified an allosteric mechanism for small molecules targeting aggressive cancers dependent on GPX4.
eTOC Blurb
Liu et al. find that covalent binding of RSL3 to an allosteric site inhibits and degrades GPX4. Furthermore, they find this site has a role in the regeneration of GPX4 under low GSH conditions. Co-crystal structures of six distinct compounds covalently binding to the allosteric site are reported.
Graphical Abstract
Introduction
As leading causes of deaths, metastatic and drug-resistant cancers are among the most pressing problems in oncology1,2. A key step in metastasis is epithelial-to-mesenchymal transition (EMT), which increases motility of tumor cells and enables invasion of distant sites by primary tumors. EMT has been reported to render cancer cells resistant to apoptosis and chemotherapy3. During EMT, elevated levels of polyunsaturated-fatty-acid-(PUFA)-containing phospholipids (PUFA-PLs) increase fluidity of cell membranes, as the cis conformation of double bonds in PUFA-PLs hinders efficient stacking of fatty acyl tails4. However, PUFA-PLs are inherently susceptible to peroxidation at bis-allylic positions. Cancer cells that have undergone EMT become more dependent on a key protein, glutathione peroxidase 4 (GPX4), which is the only peroxidase in mammals capable of reducing phospholipid hydroperoxides within cell membranes5–7. When GPX4 activity is impaired, lipid peroxidation causes ferroptosis, a tumor-suppressive form of regulated cell death8,9. Indeed, as cancers evolve into aggressive and drug-resistant states with mesenchymal or other signatures, they simultaneously acquire an exquisite sensitivity to GPX4 inhibition, which indicates a tantalizing possibility that aggressive neoplastic diseases might be treated through the use of GPX4 inhibitors10–12.
Inhibition of GPX4 with small molecules has been challenging due to the flat surface surrounding the active site and the lack of known regulatory sites13,14. To date, all known GPX4 inhibitors are assumed to be covalent inhibitors that react with the active site selenocysteine of GPX48,15–20. The most widely used and potent inhibitor, (1S, 3R)-RSL3 (hereinafter referred to as “RSL3” ), as well as an additional inhibitor named ML162, exhibit nanomolar potencies for inducing ferroptosis to cancer cells, but have poor drug-like properties in part due to their reactive chloroacetamide warheads8,15–17,21, preventing development for clinical use. Additionally, developing drug-like compounds that selectively bind to the flat active site of GPX4, but not to other selenocysteines of glutathione peroxidases in this enzyme family may be challenging. Although extensive medicinal chemistry efforts have been devoted to optimizing existing inhibitors, their potencies and selectivities have not been improved18–20. These observations indicate the necessity of obtaining a deeper understanding of these inhibitors and a search for alternative binding sites before implementing a structure-based approach to development of drug-like GPX4 inhibitors.
In addition to its peroxidase function, as a moonlighting protein under specific condition, GPX4 can polymerize into an enzymatically inactive, oxidatively cross-linked, insoluble structural element of the mitochondrial sheath of the midpiece of mature spermatozoa22,23. This unusual polymerization property may derive from GPX4’s lack of dependence on glutathione as a reducing substrate and the expression of multiple non-conserved surface cysteines, which are distinct features compared to other members of the GPX family23,24. The structural and mechanistic basis for this dual function and whether it is involved in the regulation of ferroptosis by GPX4 have not been evaluated. We hypothesized that further mechanistic study of this enzymatic-independent dual function might yield insight into how to discover potent, drug-like GPX4-specific inhibitors.
By analyzing the mechanism of known GPX4 inhibitors, we identified an unexpected allosteric binding site on GPX4 that allows for inactivation of GPX4 without targeting the active site selenocysteine. We found that this allosteric site is involved in the dual function of GPX4, based on which we developed an extended model for the normal GPX4 catalytic cycle. After identifying multiple compounds that covalently bind to this site, we screened a library of lead-optimized compounds and found a lead compound that covalently binds in the allosteric site to inhibit GPX4. This validates an allosteric approach to targeting GPX4 and provides a starting point for creating drug-like GPX4 inhibitors for aggressive cancers.
The region around Cysteine 66 is a covalent binding site for RSL3 and ML162 on GPX4
Aiming to decipher the mechanism of known GPX4 inhibitors, we determined the co-crystal structure of GPX4U46C with each of its covalent inhibitors, RSL3 and ML162; we used U46C, as it is more difficult to express large amounts of the native protein with selenocysteine-46 (U46) using bacterial expression systems, although recent efforts have enabled expression of some native selenoproteins in vitro. The structures that we solved revealed unexpectedly that RSL3 and ML162 both react with C66, as in an alternative binding site, rather than the expected active site selenocysteine/cysteine 46 (Figure 1A, 1B, S1A, and S1B).
Figure 1. Region around Cysteine 66 is a binding site of RSL3 and ML162 on GPX4.
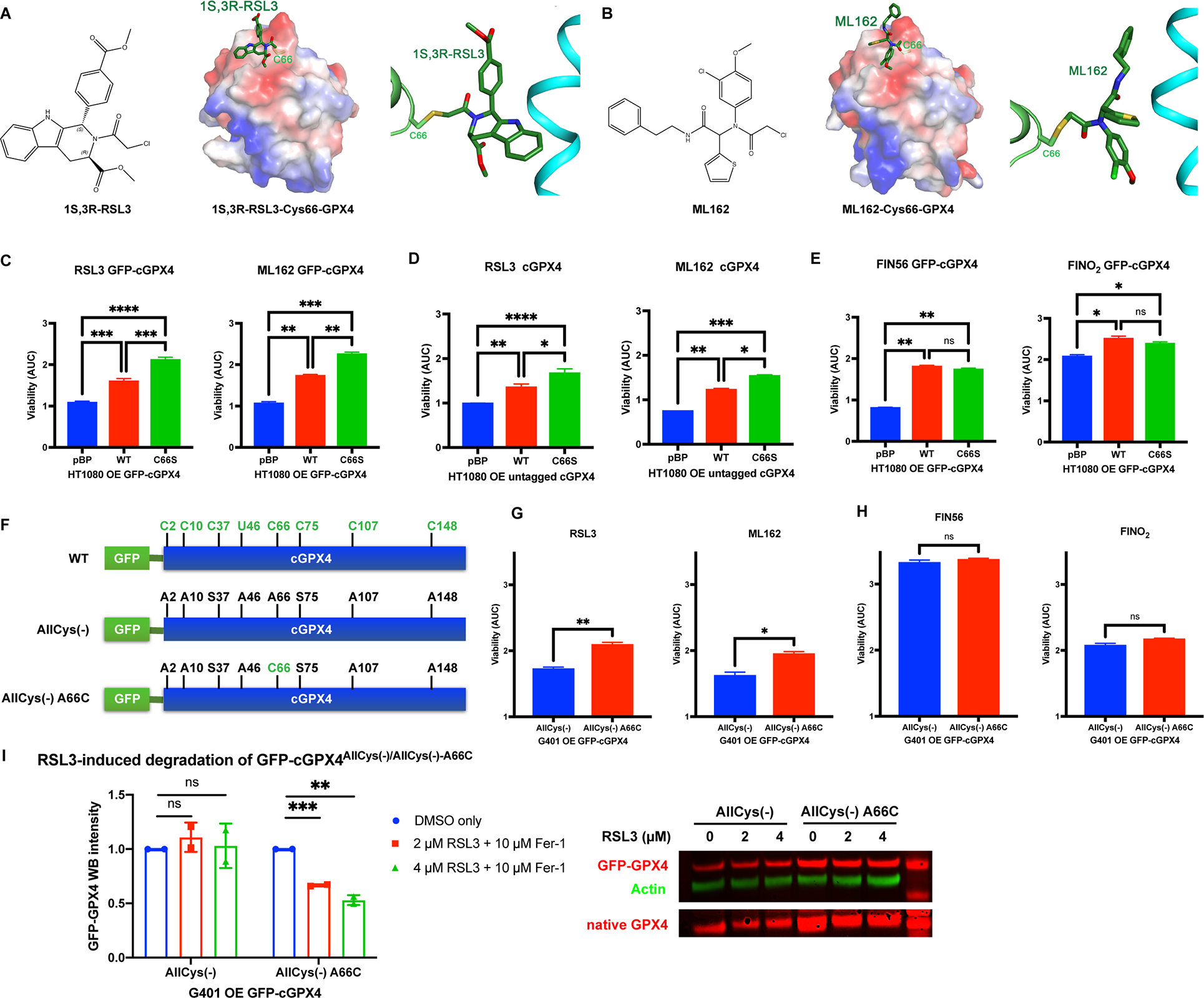
A, Crystal structure of GPX4U46C with RSL3. B, Crystal structure of GPX4U46C with ML162. C-E, HT-1080 overexpressing exogenous WT or C66S GFP-tagged or untagged GPX4 and a control line were tested for RSL3, ML162, FIN56, and FINO2 sensitivity (n=3). Based on the dose-response curve of viability, area under curve (AUC) values were calculated. One-way ANOVA followed by Tukey’s multiple comparisons test was performed: pns>0.05, p* < 0.05, p** < 0.01, p*** < 0.001, and p**** < 0.0001. F, Construction of GFP-tagged AllCys(−) and AllCys(−) A66C GPX4. G and H, G401 cells overexpressing GFP-GPX4 variants and a control line were tested for RSL3, ML162, FIN56, and FINO2 sensitivity (n=3). Unpaired t test was performed: pns>0.05, p* < 0.05, and p** < 0.01. I, The native GPX4 and GFP-GPX4 variants in G401 overexpressing exogenous GFP-GPX4 variants were tested for vulnerability to the degradation induced by RSL3 (n=2).
For C-E and G-I, data were presented as mean ± standard error (s.d.). See also Figure S1.
We initially supposed that the observed binding of the covalent inhibitors to C66 might be an in vitro artifact, due to the U46C mutation in the active site rendering the active site less reactive, and the possibility for adventitious reactions on other cysteines in vitro. However, a recent study on ML162 with wild-type U46 GPX4 also reported the detection of covalent binding of ML162 to C66, even in the presence of selenocysteine-4625, demonstrating that covalent binding to C66 occurs even with the wild-type protein. Thus, to evaluate the functional relevance of C66 on wild-type GPX4 in a cellular context, we stably overexpressed either GFP-tagged GPX4WT or GFP-tagged GPX4C66S, a binding-deficient mutant, in HT-1080 fibrosarcoma cells, where GPX4 functions to protect cells from ferroptosis8 (Figure S1C). Using HT-1080 cells transfected with empty vector (pBabe-puro) as a control, we found that overexpression of GFP-tagged-GPX4C66S, which lacks the C66 reactive residue, protected HT-1080 cells from ferroptosis induced by RSL3 and ML162 to a greater extent than overexpression of GFP-tagged-GPX4WT, suggesting that covalent binding of both RSL3 and ML162 to C66 on GPX4 normally causes inhibition of GPX4 in a cellular context (Figure 1C and S1D). Moreover, overexpression of tag-free GPX4C66S also protected HT-1080 cells from ferroptosis induced by RSL3 and ML162 to a greater extent than overexpression of tag-free GPX4WT, excluding the possibility of a GFP tag artifact (Figure 1D and S1E).
In addition to GPX4 inhibitors, we also examined whether the inhibitor-binding deficient mutant C66S affected other classes of ferroptosis inducers, namely FIN56, which depletes GPX4 protein and CoQ10, and FINO2, which oxidizes iron to drive lipid peroxidation and inactivation of GPX426–28. Consistent with these other compounds acting independently of the C66 site, overexpression of GFP-tagged-GPX4C66S provided equivalent protection to that conferred by GFP-tagged-GPX4WT against FIN56 and FINO2, confirming that the specific protection of GPX4C66S against RSL3 and ML162 is due to loss of the covalent binding at C66 specifically for these compounds (Figure 1E and S1F).
GPX4 contains a selenocysteine in the active site, and seven other cysteines, which are all potentially reactive with electrophiles (Figure 1F). In previous efforts attempting to locate the RSL3 binding site, we replaced all the electrophilic GPX4 residues with Ala or Ser (C2A, C10A, C37S, Sec46A, C66A, C75S, C107A, and C148A) and expressed the mutant GPX4 protein, termed allCys(−), in G401 renal carcinoma cells as a GFP fusion16 (Figure 1F). Each mutated residue on GFP-allCys(−)-GPX4 was then separately reverted to the original selenolcysteine or cysteine16. To verify the hypothesis that RSL3 binds to C66 in a cellular context, we examined the ferroptosis sensitivity of cells expressing allCys(−) or allCys(−) A66C GPX4, in which A66 in allCys(−) GPX4 was reverted to C66 (Figure 1F). We found that overexpression of this enzymatically inactive allCys(−) A66C GPX4 nonetheless provided significant protection against RSL3 and ML162 as compared to allCys(−) GPX4. This remarkable result suggested that C66 on the inactive GPX4 protein reacted with RSL3 and ML162 and therefore shielded endogenous enzymatically active GPX4 in G401 cells from the effects of these inhibitors (Figure 1G and S1G).
As expected, overexpression of enzymatically inactive allCys(−) A66C GPX4 exhibited no protective effects against FIN56 and FINO2 lethality as compared to allCys(−) GPX4, confirming specific protection of allCys(−) A66C GPX4 against RSL3 and ML162, due to the binding of these covalent inhibitors to C66 on allCys(−) A66C GPX4 (Figure 1H and S1H).
In our prior work, sepharose beads coupled with anti-fluorescein antibodies were not able to pull down a detectable amount of RSL3-fluorescein-treated allCys(−) A66C GPX4 from G401 cells16. We reasoned that this could be because RSL3 reacted with C66 on GPX4 and subsequently induced the degradation of allCys(−) A66C GPX4 in a cellular context26. In line with this hypothesis, we found that, while allCys(−) GPX4 could not be covalently modified by RSL3 and therefore was resistant to RSL3-induced degradation, which is observed with WT GPX4, the allCys(−) A66C GPX4 protein with the C66 reactive residue restored was vulnerable to the RSL3-induced degradation; this was determined using Western-blotting of G401 cells overexpressing allCys(−) or allCys(−) A66C GPX4 with or without treatment with RSL3 (Figure 1I). The dose-dependent degradation of allCys(−) A66C GPX4 induced by RSL3 suggested that covalent binding of RSL3 to C66 on GPX4 is sufficient to induce the degradation of GPX4 in a cellular context, indicating a role for C66 in the previously enigmatic mechanism of RSL3-induced GPX4 inhibition.
RSL3 and ML162 selectively react with selenocysteine-46 and C66 on GPX4
In addition to C66, we wondered whether other surface cysteines (e.g., C10, C107, or C148) on GPX4 might also be amenable to electrophilic attacks by RSL3 and ML162. Moreover, sec46 at the active site is expected to be more reactive towards electrophiles because of its lower pKa values compared to cysteine thiols16. To test whether sec46 or other surface cysteine residues on GPX4 also are compatible with RSL3 binding, we used G401 cell lines stably expressing the corresponding revertants of allCys(−) GPX416. Five such G401 cell lines (A10C, A46C, A46U, A107C, and A148C) were included in the test, along with G401 cells overexpressing WT, allCys(−), or allCys(−) A66C GPX4, for RSL3 and ML162 sensitivities (Figure 2A). A46C, in which the A46 in allCys(−) was replaced with cysteine, was also included to further evaluate the requirement of selenolcysteine at the active site of GPX4 for covalent binding with RSL3; in the co-crystal structure we didn’t observe binding of RSL3 to cys46 on the GPX4U46C protein.
Figure 2. RSL3 and ML162 specifically bind to Sec46 and Cys66 of GPX4.
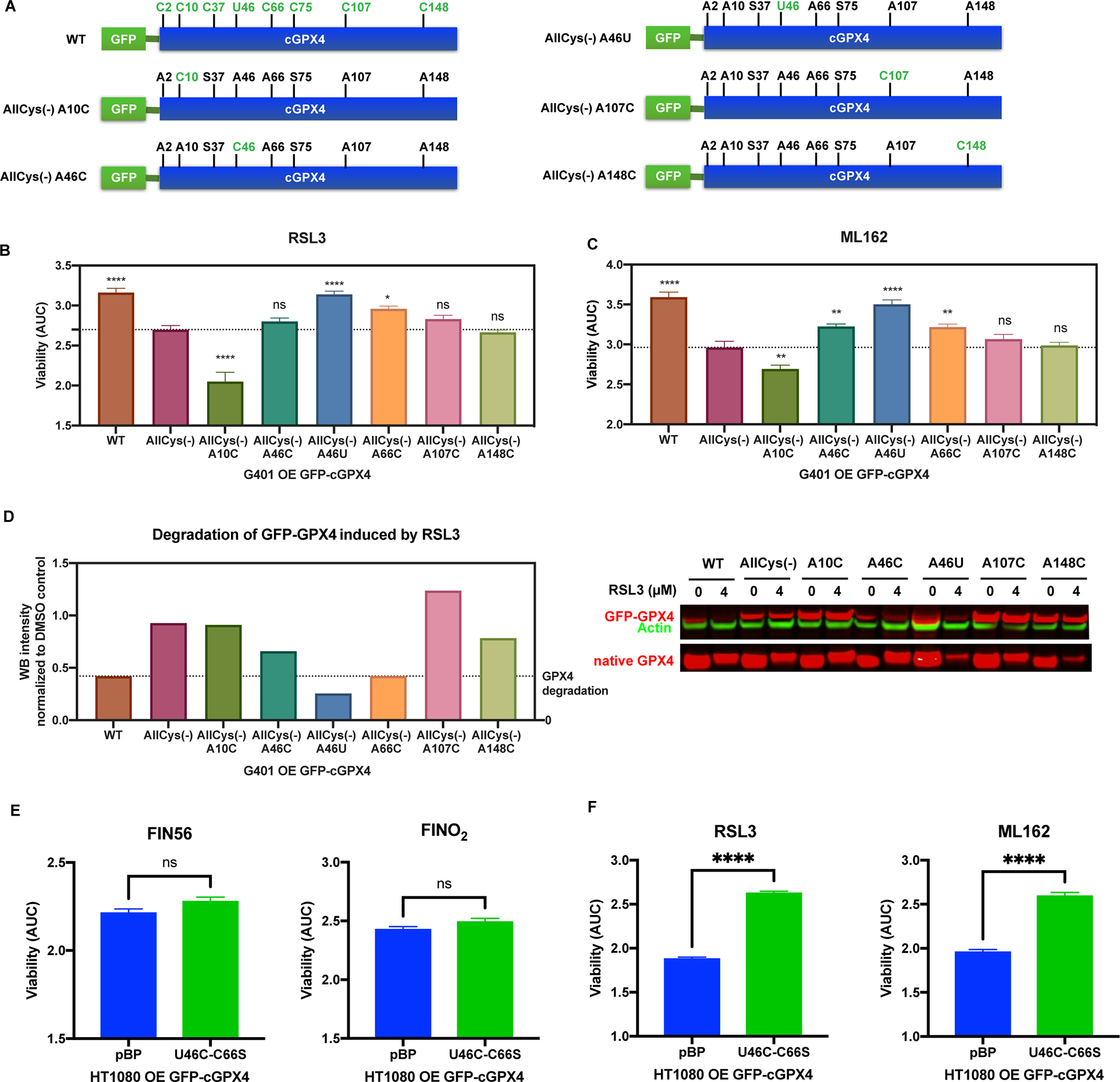
A, Construction of GFP-tagged AllCys(−) A10C, AllCys(−) A46C, AllCys(−) A46U, AllCys(−) A107C, and AllCys(−) A148C GPX4. B and C, G401 cells overexpressing exogenous WT or variants of GFP-GPX4 were tested for RSL3 and ML162 sensitivity (n=3). One-way ANOVA followed by Dunnett’s multiple comparisons test (compared to AllCys(−)) was performed: pns>0.05, p* < 0.05, p** < 0.01, and p**** < 0.0001. D, The native GPX4 along with WT and variants of GFP-GPX4 expressed in G401 was tested for vulnerability to the degradation induced by RSL3. E and F, HT-1080 overexpressing exogenous U46C-C66S GFP-GPX4 and a control line were tested for FIN56, FINO2, RSL3, and ML162 sensitivity (n=3). Unpaired t test was performed: pns>0.05 and p**** < 0.0001.
For B, C, E, and F, data are represented as mean ± s.d. See also Figure S2.
We found that overexpression of WT, allCys(−) A46U, or allCys(−) A66C GPX4 significantly protected G401 cells from both RSL3 and ML162 as compared with allCys(−), while overexpression of GPX4 containing other cysteines didn’t provide protection, suggesting that RSL3 and ML162 selectively bind to sec46 and C66 on GPX4 in a cellular context (Figure 2B, 2C, and S2A). Furthermore, in accordance with the viability results, we found that WT and allCys(−) A46U GPX4 were prone to RSL3-induced degradation, to the extent observed with allCys(−) A66C GPX4, while allCys(−) GPX4 with other cysteines were not susceptible to RSL3-induced degradation, confirming the selectivity of RSL3 towards selenocysteine 46 and cysteine 66 (Figure 2D).
It is noteworthy that overexpression of allCys(−) A46C GPX4 didn’t exhibit significant protection against RSL3 and only slight degradation was observed. This further confirmed the importance of selenocysteine for the binding of RSL3 to the GPX4 active site and explained the absence of this compound in the active site of the RSL3-GPX4U46C co-crystal structure16,29. Additionally, allCys(−) A46U GPX4 showed the most significant protection and degradation, which indicated a preference of inhibitor binding to sec46, potentially due to its higher activity against electrophiles, and explained why tagged RSL3 pulled down a larger amount of allCys(−) A46U than allCys(−) with any other reverted cysteines16,29.
To validate selenocysteine 46 and cysteine 66 as inhibitor binding sites on GPX4 in a cellular context, we stably overexpressed either GFP-tagged GPX4U46C_C66S, a double mutant which lacks both reactive residues, in HT-1080 cells (Figure S2B). Using HT-1080 cells transfected with empty vector (pBabe-puro) as a control, we found that GPX4U46C_C66S, which has much lower enzymatic activity than WT due to the U-to-C mutation at active site, exhibited no protective effects against treatment with FIN56 or FINO2 (Figure 2E and S2C). However, as expected, overexpression of GPX4U46C_C66S, which is less enzymatically active but completely devoid of both RSL3-reactive residues, significantly protected HT-1080 cells from ferroptosis induced by RSL3 and ML162 (Figure 2F and S2D). Together, these data suggest that RSL3 and ML162 specifically bind to both sec46 and C66 of GPX4 in a cellular context.
C66 and C10 modulate the activity of GPX4 under limited GSH conditions
Besides the three classes of ferroptosis inducers that we tested, imidazole ketone erastin (IKE), an inhibitor of cystine/glutamate antiporter system xc−, represents class 1 ferroptosis inducers28. IKE prevents cystine import, which leads to the depletion of the GPX4 cofactor GSH and a loss of GPX4 activity, causing ferroptosis30. When we tested G401 cells overexpressing WT, allCys(−), or individual revertants of allCys(−) GPX4 (A10C, A46C, A46U, A66C, A107C, and A148C) for IKE sensitivities, we found that overexpression of enzymatically inactive allCys(−) A66C GPX4 significantly protected G401 cells from IKE, the effect of which was comparable with the overexpression of enzymatically active WT GPX4 and AllCys(−) A46U GPX4 (Figure 3A and S3A). In contrast, overexpression of enzymatically inactive AllCys(−) A10C GPX4 sensitized G401 cells to ferroptosis induced by IKE. These observations suggested a potentially intrinsic role of cys66 and cys10 in modulating GPX4 activity under limited GSH conditions, which is independent of GPX4 inhibitor binding.
Figure 3. Cys66 and Cys10 are involved in modulating the dual function of GPX4.
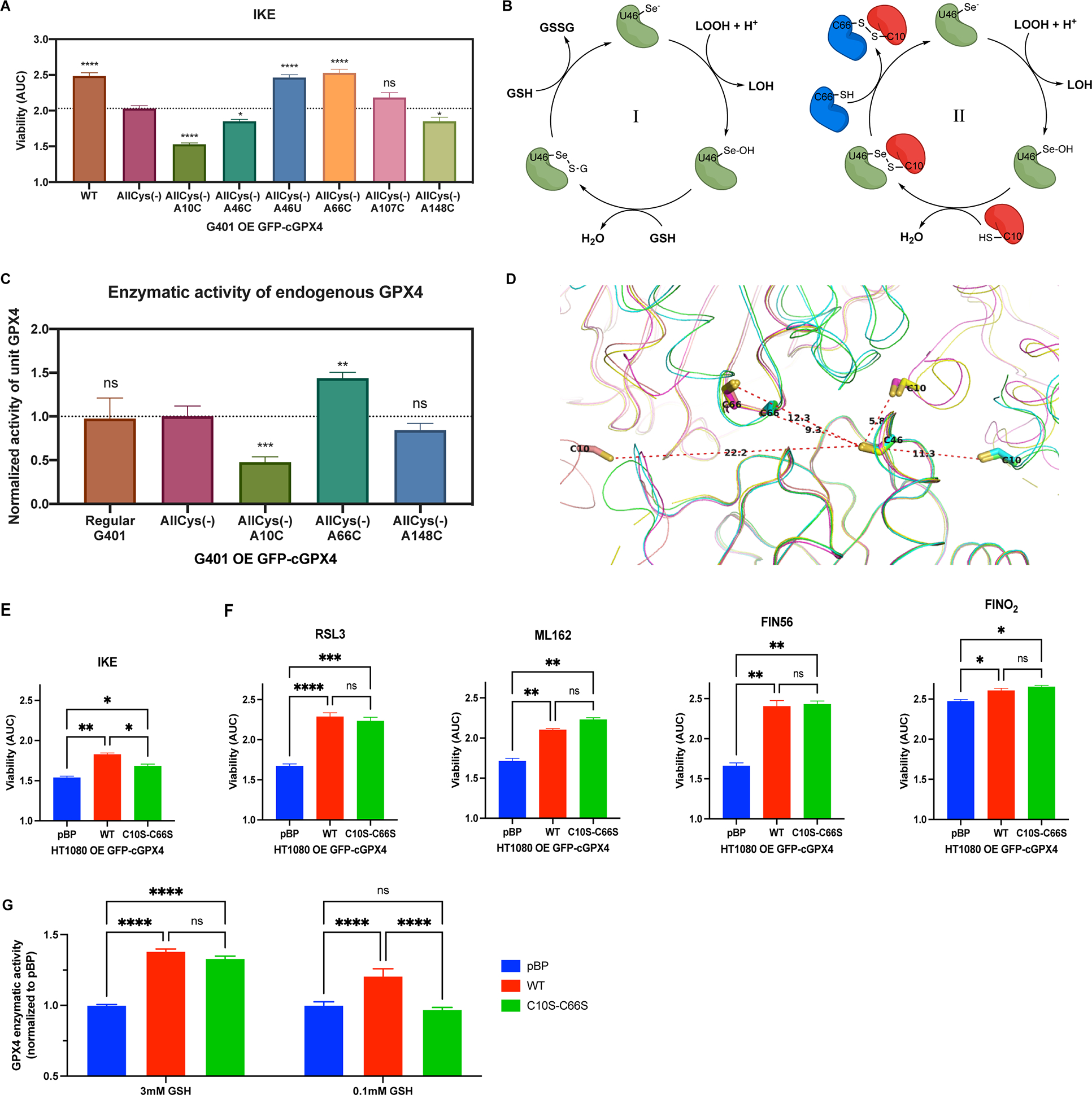
A, G401 cells overexpressing exogenous WT or variants of GFP-GPX4 were tested for IKE sensitivity (n=3). One-way ANOVA followed by Dunnett’s multiple comparisons test (compared to AllCys(−)) was performed: pns>0.05, p* < 0.05, and p**** < 0.0001. B, Canonical glutathione-dependent catalytic cycle of GPX4 (I), and proposed cysteine-thiol-dependent GPX4 catalytic cycle (II). C, Activity of the native GPX4 in regular G401 and G401 cells overexpressing exogenous GFP-GPX4 variants was acquired (n=20). Unpaired t test (compared to AllCys(−)) was performed: pns>0.05, p** < 0.01, and p*** < 0.001. D, The crystal packing of ligand-free GPX4U46C molecules crystallized in diverse space groups (P3121, P21, and P1). E and F, HT-1080 overexpressing exogenous WT or C10S-C66S GFP-GPX4 and a control line were tested for IKE, RSL3, ML162, FIN56, and FINO2 sensitivity (n=3). One-way ANOVA followed by Tukey’s multiple comparisons test was performed: pns>0.05, p* < 0.05, p** < 0.01, p*** < 0.001, and p**** < 0.0001. G, Total activity of GPX4 in HT-1080 overexpressing exogenous WT or C10S-C66S GFP-GPX4 and a control line were acquired at 3 mM or 0.1 mM GSH (n=11). Two-way ANOVA followed by Tukey’s multiple comparisons test was performed: pns>0.05 and p**** < 0.0001.
For A, C, E-G, data are represented as mean ± s.d. See also Figure S3.
Previous studies revealed that at low GSH concentrations, GPX4 acts as a protein thiol peroxidase to utilize specific protein thiols as the reductants in its catalytic cycle and structurally crosslink proteins, which is known as the dual function of GPX422,23. The reported low GSH conditions coincidentally matched the IKE-treated cellular conditions in our experiments22. We therefore hypothesized that, in addition to the canonical glutathione-dependent catalytic cycle of GPX4 (model I), GPX4 may utilize the thiol of C10 on a second GPX4 protein as a reductant to form a pseudo-dimer, which is then decomposed by the thiol of C66 on a third GPX4 protein molecule to complete the catalytic cycle (model II, Figure 3B). In this scenario, given that oxidatively cross-linked GPX4 was reported to be enzymatically inactive22, an overwhelming quantity of inactive GPX4 with C10 may lock the active selenocysteine-containing GPX4 into an inactive state if sufficient reductants (GSH or C66-SH) are not readily available. On the other hand, although also being enzymatically inactive, GPX4 with only C66 may accelerate the catalytic cycle via pushing oxidized GPX4 (GPX4-Se-S-G or GPX4-Se-S-cys10) into a regenerated active state31, effectively acting like GSH. A previous mutagenesis study, which showed a role of U46, C10, and C66 in GPX4 polymerization, also supports this model24.
In accordance with this model for GPX4 enzyme function, we found that overexpression of enzymatically inactive AllCys(−) A66C GPX4 in G401 cells boosted the enzymatic activity of endogenous WT GPX4, while AllCys(−) A10C GPX4 significantly suppressed the activity (Figure 3C and S3B). By comparison, overexpression of AllCys(−) or AllCys(−) A148C GPX4 exhibited no significant effects on the enzymatic activity of endogenous GPX4. Moreover, in the packing of multiple ligand-free GPX4U46C crystals that we solved in diverse space groups (P3121, P21, and P1), as well as previously reported GPX4 structures, C10, C46, and C66 are consistently in close proximity to each other, suggesting a structural foundation of this model24 (Figure 3D).
To further verify the potential role of C66 and C10 in modulating GPX4 activity under limited GSH conditions, we stably overexpressed GFP-tagged GPX4C10S_C66S, which is devoid of the C66 and C10 cross-linking sites, in HT-1080 cells and tested the sensitivity of these cells to IKE (Figure S3C). Using HT-1080 cells either overexpressing GFP-tagged GPX4WT or transfected with empty vector (pBabe-puro) as controls, we found that overexpression of GPX4C10S_C66S, which lacks the crosslinking sites, still protected HT-1080 cells from ferroptosis induced by IKE, but to a significantly lesser extent than the overexpression of GPX4WT, suggesting a role for both C10 and C66 in modulating GPX4 function under low GSH concentrations (Figure 3E and S3D). Additionally, as controls, overexpression of GPX4C10S_C66S protected HT-1080 cells from RSL3, ML162, FIN56, and FINO2 to an indistinguishable extent as overexpression of GPX4WT (Figure 3F and S3D). This confirmed a role for both C66 and C10 in modulating GPX4 activity specifically under limited GSH conditions (Figure S3E).
To further dissect the origin of the different IKE sensitivities observed, we tested the GPX4 activity of these three cell lines in vitro with different GSH concentrations. Although GPX4C10S_C66S and GPX4WT exhibited comparable activity at 3 mM GSH, GPX4WT was able to reduce more phospholipids hydroperoxides than GPX4C10S_C66S at 0.1 mM GSH (Figure 3G and S3F). Together, these observations support the role of C66 and C10 in modulating the activity of GPX4 under low GSH concentrations.
Region around C66 is an allosteric binding site of RSL3 on GPX4
These results demonstrated that RSL3, the primary proof-of-concept GPX4 inhibitor with nanomolar potency widely used for inactivating GPX4, not only binds to sec46 in the active site, but also specifically binds to C66, a non-conserved positive modulator of GPX4 activity, and subsequentially induces degradation of GPX4 in a cellular context. The potential advantage of targeting the binding site around C66 for the design of drugs targeting GPX4 is that this site may enable more selective compounds compared to targeting the active site, as C66 is not conserved across the GPX isoforms. To specifically examine the suitability of the binding site around C66 for drug design, in vitro purified GPX4U46C protein was used, as the above cellular data and co-crystal structure indicated that RSL3 solely bound to C66 on the GPX4U46C protein.
To evaluate potentially selective binding to C66, we conducted intact protein MALDI MS analysis of GPX4U46C pre-incubated with excess RSL3, which showed 1:1 covalent binding of RSL3 to GPX4U46C protein (Figure 4A). To demonstrate the single covalent modification by RSL3 is not on cys46, we expressed and purified tag-free GPX4allCys(−) A46C protein, in which A46 in allCys(−) GPX4 was reverted to C46 (Figure 2A). While the control compound ML162 was able to modify C46 and shift the mass of GPX4allCys(−) A46C, which is consistent with the increased viability of G401 AllCys(−) A46C over G401 AllCys(−) when treated with ML162 (Figure 2C), we observed no mass shift induced by RSL3 in parallel, confirming no RSL3 modification on C46 (Figure S4A). In addition, we found that pre-incubation of GPX4U46C-C66S, which is devoid of the C66 reactive residue, with excess RSL3 resulted in no mass shift as well, suggesting the single covalent modification by RSL3 on GPX4U46C is on cys66 (Figure 4A). We further confirmed this by performing trypsin digestion and peptide analysis of GPX4U46C pre-incubated with excess RSL3, where selective modification of cys66 by RSL3 was observed (Figure S4B). With the co-crystal structure of GPX4U46C-RSL3 also being considered, we concluded that RSL3 solely reacted with C66 when selenocysteine 46 is mutated to cysteine in GPX4U46C (Figure 1A).
Figure 4. Surface region around C66 is an allosteric binding site of RSL3 on GPX4.
A, Intact protein MALDI MS of GPX4U46C or GPX4U46C-C66S preincubated with DMSO or RSL3. B, Enzymatic activity of GPX4U46C and GPX4U46C_C66S preincubated with DMSO, RSL3 (20 μM), or ML162 (20 μM) were acquired (n=3). Data are represented as mean ± s.d. One-way ANOVA followed by Dunnett’s multiple comparisons test was performed: pns>0.05 and p** < 0.01. C, Backbone assignments of 1H, 15N-HSQC-NMR spectrum for 15N-GPX4U46C. Horizontal red lines point out the peaks corresponding to Asparagine and Glutamine side chain NH2. D, Overlap of 1H, 15N-HSQC-NMR spectrum of 50 μM 15N-GPX4U46C alone and its mixture with 100 μM RSL3 (both in 5.0 % d6-DMSO), with zoom-in panels on selected peaks of prominent changes.
See also Figure S4.
To evaluate the impact on GPX4 of RSL3 binding to C66, we measured the enzymatic activity of GPX4U46C after incubation with RSL3 and observed effective inhibition (Figure 4B). Furthermore, we found that RSL3 exhibited no inhibition on the enzymatic activity of GPX4U46C-C66S, which is devoid of the C66 reactive residue, while the positive control ML162, which can covalently bind to both C46 and C66, consistently inhibited both GPX4U46C and GPX4U46C-C66S. This suggested that binding of RSL3 to C66 directly inhibited the enzymatic activity of GPX4. Loss of the positive activity modulation function of C66 after RSL3 binding likely contributes to the observed decrease in enzymatic activity.
Aiming to gain more structural insight into RSL3 inhibition in solution, we used our previously reported 1H, 15N- heteronuclear single quantum coherence (HSQC) NMR spectrum of GPX4 in the presence and absence of RSL3; this prior work was limited by the lack of HSQC resonance spectrum assignments to each GPX4 amino acid residue, and we could thus only probe binding without knowing the structural binding site on GPX427. To address the previous limitation and to benefit future studies of GPX4 inhibitors by the community, we now solved the backbone resonance assignments of 1H, 15N-HSQC-NMR spectrum for 15N isotope-labeled GPX4U46C, which enabled investigation of the specific binding modes of GPX4 inhibitors in solution (Figure 4C). With the spectrum assignments and the overlap of 1H, 15N-HSQC-NMR spectrum of 15N-GPX4U46C alone and in complex with RSL3, we found the peaks corresponding to residues (Y63, E65, G67, and R69) around C66, which directly reacted with RSL3 in the crystal structure, consistently exhibited the most outstanding changes in the HSQC-NMR spectrum (Figure 4D). Additionally, we observed prominent changes in peaks corresponding to another group of residues: K140, I143, K145, V150, and R152. This indicated a secondary effect of RSL3 binding on GPX4, which is not limited to the binding site. Since our recent study revealed that perturbation of R152 would destabilize a critical loop and disrupt the active site32, the observed impact of RSL3 on R152 may contribute to its inhibitory effect after binding to C66.
As we further examined the co-crystal structure of GPX4U46C with RSL3, unlike the non-selective control ML162, RSL3 exhibited well-defined electron density, in particular around the tricyclic fused rings and the attached methyl ester moiety (Figure S1A). The fused aromatic indole ring makes extensive hydrophobic interactions with the side chains of Y63, L166, P167, and F170. Accordingly, residues L166-F170 undergo a conformational change from a loop in our previously reported structure of GPX4 alone (PDB code: 7L8K) to a one-turned α-helical entity via 180° flipping of the P167 side chain (Figure S4C). As a result, a two-turned α-helix, as observed in the GPX4 alone structure, becomes a 3-turned α-helix extending to residue Y169. This is in line with the chemical shifts observed on the HSQC peaks corresponding to these residues (Figure 4D). As we found the carboxyl group of F170 residue formed a hydrogen bond either with C66 thiol of the same GPX4 (PDB code: 7L8K) or C46 thiol of neighboring GPX4 (PDB code: 2OBI) in the crystal structures of GPX4 alone (Figure S4D), F170 may facilitate the regeneration of GPX4 by activating the cysteine thiols, especially in enzymatic model II of GPX4 that we proposed in this study. Therefore, the observed structural interference of RSL3 with L166-F170 may contribute to its inhibitory effect after binding to C66. This is structurally evidenced by loss of hydrogen bond between F170 and C66 in GPX4-RSL3 structure (Figure S4C) and a change in pseudo-trimer packing pattern between GPX4-RSL3 and GPX4 alone (Figure S4E).
To further examine the allosteric binding site around C66, we prepared a derivative of RSL3 without the reactive warhead and termed it RSL3-minus-Cl, as we substituted the chlorine atom in the RSL3 chloroacetamide with a hydrogen atom to make it non-covalent (Figure S4F). We found that RSL3-minus-Cl, which is devoid of reactive warhead, was able to change the melting temperature of GPX4U46C in a thermal shift assay, suggesting reversible binding of RSL3-minus-Cl to GPX4 (Figure S4G). We then measured its binding affinity in a Surface Plasmon Resonance (SPR) assay and observed a KD value of 60 μM (Figure S4H). With the overlap of 1H, 15N-HSQC-NMR spectrum of 15N-GPX4U46C with and without RSL3-minus-Cl, we observed prominent changes in peaks corresponding to residues around C66 (Y63, G67, L166, H168, Y169, and F170; Figure S4I). Together, these data suggested molecular interactions of the RSL3 structure with the binding site around C66 beyond the chloroacetamide reactive warhead, though the affinity requires further improvement via modifications of RSL3 structures or development of alternative inhibitors.
Accordingly, we conclude that region around C66 is a genuine allosteric binding site of RSL3 on GPX4. Furthermore, we propose the application of 1H, 15N-HSQC-NMR in GPX4 inhibitor studies, especially along with the spectrum assignments, will enable a rapid investigation into the binding modes and potentially the allosteric effects of future GPX4 inhibitors.
Additional compounds bind to the C66 allosteric site
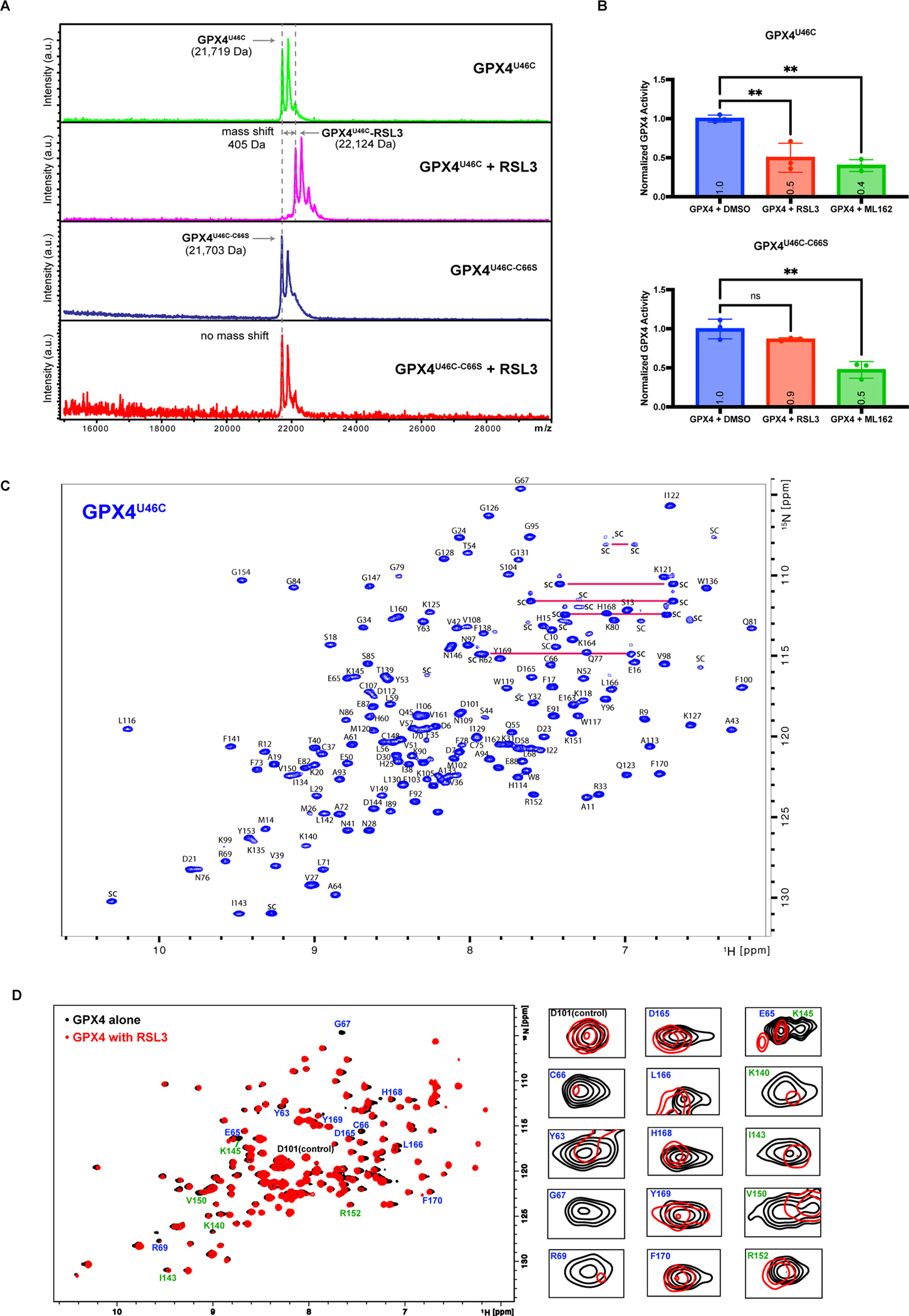
Since we demonstrated that the region around C66 is an allosteric binding site of RSL3 on GPX4, we initiated a search for additional compounds that also react with this site, to validate the druggability of the site for future discovery of therapeutics targeting GPX4. We started with two fragments of ML162 (CDS9 and TMT10) which share similar warheads and structures as RSL3 and ML162. Intact protein MALDI MS analysis of GPX4U46C preincubated with CDS9 and TMT10 demonstrated covalent binding (Figure S5A). We proceeded to solve the co-crystal structures of GPX4U46C with each of the compounds, which showed that both fragments bound to the C66 site (Figure 5A, 5B, S5B, and S5C). During our analysis, we found that another protein cysteine modifier, MAC-557633, was able to covalently modify GPX4 (Figure S5D). The co-crystal structure of GPX4 with MAC-5576 revealed that it also bound to the C66 allosteric site (Figure 5C and S5E).
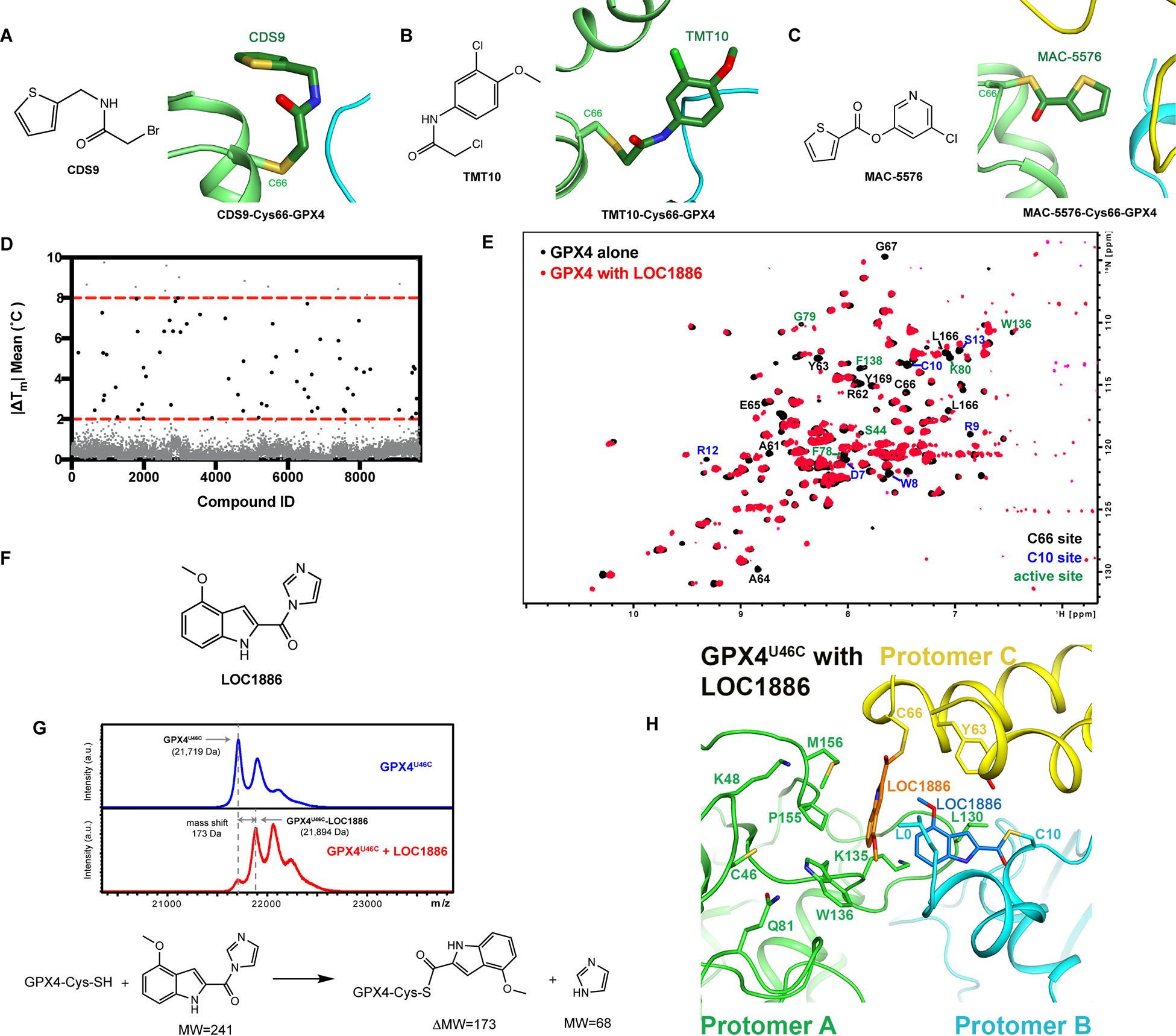
Figure 5. Screening of Lead-Optimized-Compound library identified lead compound binding to the Cys66 allosteric site.
A-C, Crystal structure of GPX4U46C with CDS9, TMT10, or MAC5576. D, Thermal shift assay was applied to screen 9,719 compounds in the Lead-Optimized-Compound library for in vitro binders of GPX4U46C. E, Overlap of 1H, 15N-HSQC-NMR spectrum of 50 μM 15N-GPX4U46C alone and its mixture with 800 μM LOC1886. F, Structure of LOC1886. G, Intact protein MALDI MS analysis of GPX4U46C preincubated with DMSO or LOC1886 and the proposed nucleophilic substitution reaction based on the observed mass shift. H, Co-crystal structure of GPX4U46C with LOC1886. The LOC1886 molecules bound to cysteine 66 (yellow) and cysteine 10 (cyan) of GPX4U46C are shown in orange and marine, respectively. The three protomers (A-C) are colored light-green, cyan, and yellow, and residues interacting with two LOC1886 molecules are shown as stick models and labeled.
See also Figure S5.
To fully exploit the versatility of the C66 site to accommodate structurally diverse compounds, we applied a thermal shift assay to screen 9,719 compounds from a Lead-Optimized-Compound (LOC) library34 for in vitro binders of GPX4U46C, which would shift the melting temperature of GPX4U46C (|ΔTm| > 2 °C, Figure 5D). The LOC library was assembled via stringently filtering a database of 3,372,615 commercially available small molecules for structurally diverse compounds with desired drug-like properties and suitability for lead development34. Top hits from this screen were then tested by the 1H, 15N-HSQC-NMR assay that we developed to examine their individual binding modes. Accordingly, we found that LOC1886, a thermal shift screening hit compound (|ΔTm|=3°C), strongly interacted with the binding site around C66, based on the significant chemical shifts of the residues around the site (Figure 5E, 5F, and S5F). Intact protein MALDI MS analysis of GPX4U46C pre-incubated with LOC1886 revealed that it covalently bound to GPX4U46C, which resembled RSL3 and other proof-of-concept GPX4 inhibitors (Figure 5G). The observed mass shift suggested that the cysteine thiol of GPX4 substituted the imidazole ring in LOC1886 and formed a thioester via a nucleophilic substitution reaction, which represented an alternative warhead for the design of GPX4 inhibitors. The subsequent trypsin digestion and peptide analysis demonstrated that LOC1886 covalently bound to C66 of GPX4 (Figure S5G).
We then determined the co-crystal structure of GPX4U46C with LOC1886, which confirmed that LOC1886 bound to C66 via the proposed nucleophilic substitution reaction (Figure 5H and S5H). Similar to the GPX4 crystal structures that we solved with the other five GPX4 binders (RSL3, ML162, CDS9, TMT10, and MAC-5576), residues L166-F170 undergo a conformational change from a loop into a helix, suggesting a shared GPX4 conformational change upon small molecule binding to C66 site. Naturally, the hydrogen bond involving the F170 carboxyl was lost in all six inhibitor-bound structures. In addition, LOC1886 also modified C10 in the crystal structure, which is consistent with the outstanding chemical shifts of the residues around the C10 site in 1H, 15N-HSQC-NMR spectrum of GPX4U46C with LOC1886 (Figure 5E). Such modifications would be expected to strongly interfere with the modulation by C66 and C10 of the active site in GPX4 catalysis. This is evidenced by a unique pseudo-trimer packing pattern observed on GPX4-LOC1886, which is different from those of GPX4-RSL3 and apo GPX4 (Figure S5I).
Indeed, LOC1886 inhibited the ability of cellular GPX4 in HT-1080 cell lysates to reduce phospholipid hydroperoxides, as well as the enzymatic activity of purified GPX4 (Figure 6A and S5J). In addition, we observed degradation of GPX4 in HT-1080 cells treated with LOC1886 (Figure 6B and S5K). Using the fluorescent probe C11-BODIPY as a lipid ROS indicator and RSL3 as control, we found that LOC1886 induced a significant increase in lipid peroxidation in HT-1080 cells, which resembled the effect of RSL3 and was rescued by treatment with ferrostatin-1, a ferroptosis-specific inhibitor9 (Figure 6C). Furthermore, LOC1886 was able to induce ferroptosis to HT-1080 fibrosarcoma cells, which could be rescued by ferrostatin-1 and by the lipophilic antioxidant alpha-tocopherol, but not rescued by apoptosis inhibitor Z-VAD-FMK and necroptosis inhibitor Nec-1s, indicating activity as ferroptosis-inducing agent (Figure 6D).
Figure 6. LOC1886 inhibits and degrades GPX4 to induce ferroptosis.
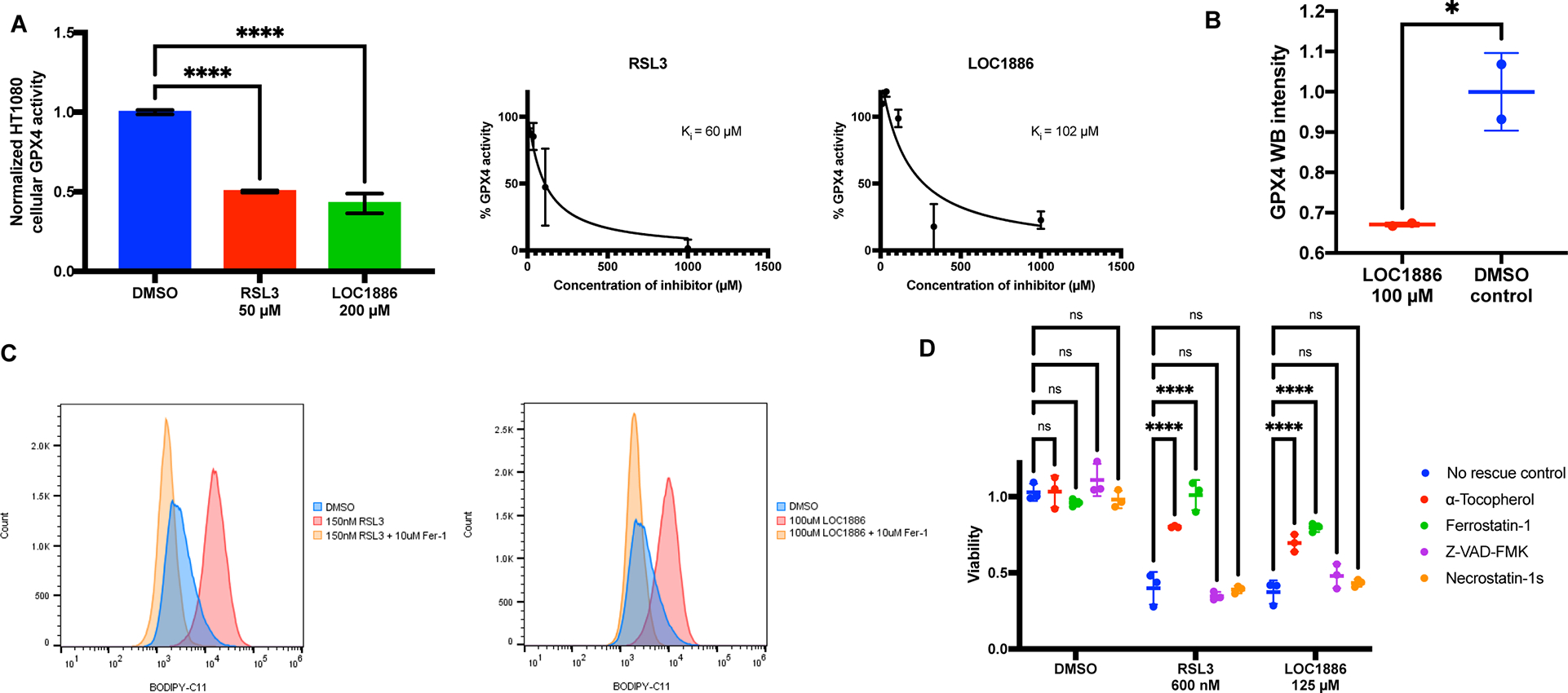
A, Left: Enzymatic activity of GPX4 in HT1080 cell lysates preincubated with DMSO, 50 μM RSL3, or 200 μM LOC1886 was acquired (n=10). One-way ANOVA followed by Dunnett’s multiple comparisons test was performed: p**** < 0.0001. Right: Measurement of inhibition constants (Ki) of RSL3 and LOC1886 on purified GPX4U46C protein (n=2). B, Native GPX4 in HT-1080 cells were tested for vulnerability to degradation induced by 100 μM LOC1886 (n=2). Unpaired t test was performed: p* < 0.05. C, Lipid peroxidation in HT-1080 cells treated with DMSO, 200 nM RSL3, or 100 μM LOC1886, with or without fer-1 rescue, was assayed by flow cytometry using C11-BODIPY. D, Viability of HT1080 cells treated with DMSO, 600 nM RSL3, or 125 μM LOC1886, with or without a-Tocopherol (100 μM), Ferrostatin-1 (100 μM), Z-VAD-FMK (20 μM), and Necrostatin-1s (20 μM) rescue (n=3). Two-way ANOVA followed by Dunnett’s multiple comparisons test was performed: pns>0.05 and p**** < 0.0001.
For A, B, and D, data are represented as mean ± s.d. See also Figure S5.
We plotted the surface potential around C66 in six co-crystal structures (Figure S6A–F) and performed a structural overlay of six GPX4 binders onto the GPX4-RSL3 structure in which relevant residues were highlighted (Figure S6G). First, on the left side of C66, there exists a hydrophobic pocket formed by residues Y63, L166, P167, F170, R62, and E163, the last two of which form a bidentate salt bridge (Figure S6G). The pocket and the position of residues forming it stay mostly the same in all six structures. In contrast, the side chain of E65, which resides near C66, adopts different conformations in six structures, suggesting this residue should be properly engaged for drug design. While the fused ring moiety of RSL3 fits well onto the hydrophobic cavity, the upward-projecting 4-(methoxycarbonyl)phenyl group of RSL3 does not make energetically-favored interactions with its surrounding negatively charged surface around E65, which suggested opportunities for modifying the RSL3 structure with targeted incorporation of hydrogen-bond donors (Figure S6G). Likewise, the thiophene and 3-chloro-4-methoxyphenyl group of ML162 interact well with the hydrophobic cavity, whereas the upward-projecting hydrophobic 2-phenylethyl group cannot engage the negatively charged side chain of E65 and is mostly disordered in the co-crystal structure (Figure S1B). The co-crystals of GPX4 with CDS9 and TMT10 both showed that the hydrophobic cavity on the left side of C66 was occupied by the side chain of residues I129 and L130 from neighboring GPX4 molecule, suggesting that a hydrophobic moiety is most likely suited for the cavity (Figure S4C and S4D). However, the thiophene moiety of CDS9 and 3-chloro-4-methoxyphenyl group of TMT10 were not recognized properly by the cavity, resulting in poor electron density for both (Figure S5B and S5C). In contrast, MAC5576 and LOC1886 both have well-defined electron density in the co-crystals, as we observed that the aromatic ring of each inhibitor was interacting with the aforementioned hydrophobic cavity (Figure S5E and S5H). Together, the consistent observations on all six co-crystal structures provide a convergent clue as how to specifically design potent binders for the C66 allosteric site.
Discussion
Our investigation into the binding modes of RSL3 and ML162 revealed that they not only interact with the active site selenocysteine, but also selectively bind to cysteine 66 of GPX4. With the U46C and AllCys(−)-A66C GPX4 constructs, which excluded binding of inhibitors to the active site, we found that binding of inhibitors to the C66 site contributed to the inactivation of GPX4. In particular, using RSL3, we found that its binding to C66 caused conformational change, compromised GPX4 regeneration via the pseudo-trimer mechanism, inhibition of activity, and a subsequent degradation in the cellular context. The additional proof-of-concept reactive fragments binding to this allosteric site further provide insight into the design of potent GPX4 inhibitors featuring specific interactions with the C66 site of GPX4. Moreover, since C66 is not conserved across the GPX isoforms and, unlike selenocysteine, is unique for GPX4, we expect inhibitors designed for C66 allosteric sites to be selective towards GPX424. In addition, since this study revealed an intrinsic role of C66 in modulating GPX4 activity under limited GSH conditions, we envision that inhibitors designed for C66 would synergize with system xc− inhibitors, such as IKE, which can deplete cellular GSH and slow tumor growth in vivo, to maximize therapeutic effects30.
Limitations of the study
LOC1886, along with other GPX4 reactive fragments that we identified for the C66 site, exhibited lower potencies than RSL3 and ML162. Although they may provide insight into the design of potent GPX4 inhibitors featuring specific interactions with C66 site, extensive optimization would be required to develop these compounds into therapeutic agents. Since the surface region around C66 of GPX4 is not a deep, well-defined binding pocket, the druggability of the C66 site needs to be further validated by the optimized binders.
Significance
As cancers of diverse lineages evolve into aggressive and drug-resistant forms, they uniformly acquire an exquisite sensitivity to GPX4 inhibition, which indicates a tantalizing possibility that aggressive neoplastic diseases might be treated through the use of GPX4 inhibitors. However, to date, no significant improvements over the original proof-of-concept GPX4 inhibitors have been achieved with medicinal chemistry efforts. Here, by deciphering the mechanism of known GPX4 inhibitors, we discovered an unexpected shared allosteric inhibitor binding site. We found that covalent binding of compounds to this allosteric site caused conformational change, inhibition of enzymatic activity, and also a subsequent degradation of GPX4 in the cellular context. Furthermore, we found that this non-conserved site is involved in the characteristic dual function of GPX4, based on which we propose an extended model of GPX4 catalytic cycle, where enzymatic function of GPX4 is modulated by this allosteric site. After identification of additional proof-of-concept reactive fragments binding to this allosteric site, we screened a library of lead-optimized compounds and found a compound that covalently binds in the allosteric site to inhibit and degrade GPX4. Our work not only proposes a therapeutic strategy for targeting GPX4, but also provides insightful guidance for inhibitor design with co-crystal structures of six distinct compounds binding to the allosteric site.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents should be directed to, and will be fulfilled by the Lead Contact, Brent R. Stockwell (bstockwell@columbia.edu).
Materials availability
All of the materials support the conclusions relevant to this manuscript are available upon reasonable request from the Lead Contact without restriction.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. Structural data for the GPX4 protein in complex with RSL3, ML162, CDS9, TMT10, MAC-5576, and LOC1886 were deposited in the Protein Data Bank (PDB). Backbone NMR chemical shifts assignment of GPX4 were deposited in the Biological Magnetic Resonance Data Bank (BMRB, entry 51659). Proteomics data of GPX4 treated with inhibitors were deposited in Mass Spectrometry Interactive Virtual Environment (MassIVE, MSV000090526). All deposited data are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
Key resources table.
(CELL-CHEMICAL-BIOLOGY-D-21-00395)
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-GPX4 | R&D systems | Cat# MAB5457, RRID:AB_2232542 |
| Rabbit monoclonal anti-actin | Cell Signaling | Cat# 8456, RRID:AB_10998774 |
| Rabbit polyclonal anti-GAPDH | Abcam | Cat# ab9485, RRID:AB_307275 |
| Bacterial and virus strains | ||
| E. coli BL21-Gold (DE3) competent cells | Agilent | Cat# 230132 |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| His-tagged-c-GPX4U46C | This paper | N/A |
| His-tagged-c-GPX4U46C_R152H | This paper | N/A |
| His-tagged-c-GPX4U46C_C66S | This paper | N/A |
| His-tagged-c-GPX4AllCys(-)-A46C | This paper | N/A |
| 15N-labeled His-tagged-c-GPX4U46C | This paper | N/A |
| 13C, 15N-labeled His-tagged-c-GPX4U46C | This paper | N/A |
| (1S,3R)-RSL3 | Yang et al.16 | N/A |
| (1S,3R)-RSL3-minus-Cl | This paper | N/A |
| ML162 | Aobious | Cat# AOB1514 |
| FIN56 | Gaschler et al.27 | N/A |
| FINO2 | Gaschler et al.27 | N/A |
| Fer-1 | Gaschler et al.27 | N/A |
| IKE | Zhang et al.30 | N/A |
| CDS9 | Sigma-Aldrich | Cat# CDS006509 |
| TMT10 | Millipore Sigma | Cat# TMT00610 |
| MAC-5576 | Maybridge | Cat# MAC-5576 |
| LOC1886 | This paper | N/A |
| Critical commercial assays | ||
| Site-directed mutagenesis kit QuickChange II | Agilent | Cat# 200521 |
| CellTiter-Glo luminescent cell viability | Promega | Cat# G7573 |
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat# 23225 |
| Deposited data | ||
| GPX4-U46C crystal structure | Liu et al.32 | PDB ID: 7L8K |
| GPX4-U46C-R152H crystal structure | Liu et al.32 | PDB ID: 7L8L |
| GPX4-RSL3 crystal structure | This paper | PDB ID: 7U4N |
| GPX4-ML162 crystal structure | This paper | PDB ID: 7U4K |
| GPX4-CDS9 crystal structure | This paper | PDB ID: 7U4I |
| GPX4-TMT10 crystal structure | This paper | PDB ID: 7U4J |
| GPX4-MAC-5576 crystal structure | This paper | PDB ID: 7U4L |
| GPX4-LOC1886 crystal structure | This paper | PDB ID: 7U4M |
| Backbone NMR Chemical Shift Assignments of GPX4 | This paper | BMRB entry 51659 |
| Proteomics data of GPX4 treated with inhibitors | This paper | MassIVE code: MSV000090526 |
| Experimental models: Cell lines | ||
| HT-1080 | ATCC | Cat# CCL-121 |
| HT-1080 OE GFP-tagged-cyto-GPX4WT | This paper | N/A |
| HT-1080 OE GFP-tagged-cyto-GPX4C66S | This paper | N/A |
| HT-1080 OE GFP-tagged-cyto-GPX4C10S-C66S | This paper | N/A |
| HT-1080 OE GFP-tagged-cyto-GPX4U46C | This paper | N/A |
| HT-1080 OE GFP-tagged-cyto-GPX4U46C-C66S | This paper | N/A |
| HT-1080 OE tag-free-cyto-GPX4WT | This paper | N/A |
| HT-1080 OE tag-free-cyto-GPX4C66S | This paper | N/A |
| G401 | ATCC | Cat# CRL-1441 |
| G401 OE GFP-tagged-GPX4WT | Yang et al.16 | N/A |
| G401 OE GFP-tagged-GPX4allCys(-)-A10C | Yang et al.16 | N/A |
| G401 OE GFP-tagged-GPX4allCys(-)-A46C | Yang et al.16 | N/A |
| G401 OE GFP-tagged-GPX4allCys(-)-A46U | Yang et al.16 | N/A |
| G401 OE GFP-tagged-GPX4allCys(-)-A66C | Yang et al.16 | N/A |
| G401 OE GFP-tagged-GPX4allCys(-)-A107C | Yang et al.16 | N/A |
| G401 OE GFP-tagged-GPX4allCys(-)-A148C | Yang et al.16 | N/A |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| R152H mutagenesis primers Foward: CTGCGTGGTGAAGCACTACGGACCCATGG | This paper | N/A |
| R152H mutagenesis primers Reverse: CCATGGGTCCGTAGTGCTTCACCACGCAG | This paper | N/A |
| C66S mutagenesis primers Foward: CCCGATACGCTGAGAGTGGTTTGCGGATC | This paper | N/A |
| C66S mutagenesis primers Reverse: GATCCGCAAACCACTCTCAGCGTATCGGG | This paper | N/A |
| C10S mutagenesis primers Foward: GGAGCGCGCACTGCGCCAGTCG | This paper | N/A |
| C10S mutagenesis primers Reverse: GGAGCGCGCACTGCGCCAGTCG | This paper | N/A |
| Recombinant DNA | ||
| pET-15b-His-tagged-c-GPX4U46C | Yang et al.16 | N/A |
| pET-15b-His-tagged-c-GPX4U46C−R152H | This paper | N/A |
| pET-15b-His-tagged-c-GPX4U46C_C66S | This paper | N/A |
| pET-15b-His-tagged-c-GPX4AllCys(-)-A46C | Yang et al.16 | N/A |
| pBabe-puro GFP-tagged-cyto-GPX4WT | Yang et al.8 | N/A |
| pBabe-puro tag-free-cyto-GPX4WT | Yang et al.8 | N/A |
| pBabe-puro GFP-tagged-cyto-GPX4U46C | Yang et al.8 | N/A |
| pBabe-puro GFP-tagged-cyto-GPX4C66S | This paper | N/A |
| pBabe-puro tag-free-cyto-GPX4C66S | This paper | N/A |
| pBabe-puro GFP-tagged-cyto-GPX4U46C-C66S | This paper | N/A |
| pBabe-puro GFP-tagged-cyto-GPX4C10S-C66S | This paper | N/A |
| Software and algorithms | ||
| Maestro, v2020-3 | Schrodinger | https://www.schrodinger.com/products/maestro |
| PyMOL, v2.5.0 | Schrodinger | https://pymol.org/2/ |
| GraphPad Prism 9 | GraphPad Software | https://www.graphpad.com/ |
| MOLREP, v11.0 | Vagin and Teplyakov37 | https://www.ccp4.ac.uk/html/molrep.html |
| XtalView 4.0 | McRee38 | https://www.sdsc.edu/CCMS/Packages/XTALVIEW/xtalview.html |
| Coot, 0.9.1 | Emsley et al.39 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix, 1.14-3260 | Adams et al.40 | https://phenix-online.org/download |
| ImageJ, v1.51 | Schneider et al. 42 | https://imagej.nih.gov/ij/download.html |
| Protein Thermal Shift, v1.4 | Thermo Fisher | https://www.thermofisher.com/order/catalog/product/4466038 |
| CcpNmr, v3 | Skinner et al.47 | https://ccpn.ac.uk/software/downloads/ |
| Thermo Xcalibur, v4.5.445.18 | Thermo Fisher | https://www.thermofisher.com/order/catalog/product/OPTON-30965 |
| Other | ||
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cells and reagents
HT-1080 (deposited as a fibrosarcoma cell line derived from a 35-year-old white male) cells were obtained from ATCC and grown in DMEM with glutamine and sodium pyruvate (Corning, Cat# 10–013) supplemented with 10% FBS (Gibco), 1% non-essential amino acids (Invitrogen) and 1% penicillin-streptomycin mix (Invitrogen). G401 (deposited as a Rhabdoid Tumor cell line derived from a 3 month old white male) cells were obtained from ATCC and grown in McCoy’s 5A medium (Thermo Fisher, Cat# 16600108) supplemented with 10% FBS and 1% penicillin-streptomycin mix. The pBabe-puro vectors incorporated with the cDNA of GFP-tagged-cyto-GPX4WT, tag-free-cyto-GPX4WT, or GFP-tagged-cyto-GPX4U46C were prepared in previous work8. With the vectors as template, the following mutagenesis primers were designed using the Agilent QuikChange Primer Design application: C10S (5’- GGA GCG CGC ACT GCG CCA GTC G -3’, 5’- ACG ACT GGC GCA GTG CGC GCT C -3’) and C66S (5’- CCC GAT ACG CTG AGA GTG GTT TGC GGA TC -3’, 5’- GAT CCG CAA ACC ACT CTC AGC GTA TCG GG -3’). Primers were purchased from Integrated DNA Technologies. Site-directed mutagenesis kit (QuickChange II, Agilent Cat# 200521) was then used to acquire pBP-GFP-cGPX4C66S, pBP-tag-free-cGPX4C66S, pBP-GFP-cGPX4U46C_C66S, and pBP-GFP-cGPX4C10S_C66S. All mutations and the resulted plasmids were confirmed by sequencing at GENEWIZ. HT-1080 cells were seeded into a 6-well dish at a density of 300,000 cells/well the night before lipofection. 2.5 μg DNA (empty pBabe-puro vector and the above GPX4 expressing pBabe-puro vectors, separately), 7.5 μL Lipofectamine 3000 (Invitrogen, Cat# L3000015), and 250 μL Opti-MEM were incubated for 5 min at room temperature before adding to the HT-1080 cells. Following transfection, cells were passaged several times in HT-1080 media supplemented with 1.5 mg/mL puromycin and grown in this media for all experiments performed. Expression of the exogenous GFP-tagged-GPX4 was confirmed with fluorescence microscope and Western Blot with both GFP and GPX4 antibodies. HT1080 cells expressing GFP-tagged-cyto-GPX4 were further selected with cell sorting for high GFP expression cells. The G401 cells overexpressing WT, allCys(−), allCys(−) A10C, allCys(−) A46C, allCys(−) A46U, allCys(−) A66C, allCys(−) A107C, or allCys(−) A148C GPX4 were reported in the previous work16. The G401 cells overexpressing exogenous GPX4 were cultured in G401 media supplemented with 1.5 mg/mL puromycin.
METHOD DETAILS
Expression and purification of GPX4 protein
Bacterial expression vectors pET-15b-His-tagged-c-GPX4U46C and pET-15b-His-tagged-c-GPX4AllCys(−)-A46C were prepared in the previous work16. In a separate study32, we found GPX4 protein with the R152H mutation crystalized at an accelerated rate and yielded high-resolution crystal structures. Therefore, we used both GPX4U46C and GPX4R152H-U46C protein in the co-crystallization of GPX4 with small molecule inhibitors. Practically, with the pET-15b-His-tagged-c-GPX4U46C vector as template, R152H mutagenesis primers were designed using the Agilent QuikChange Primer Design application (F: 5’- CTG CGT GGT GAA GCA CTA CGG ACC CAT GG -3’, R: 5’- CCA TGG GTC CGT AGT GCT TCA CCA CGC AG -3’). To demonstrate region around C66 as binding site, C66S mutagenesis primers were also designed accordingly (F: 5’- CCC GAT ACG CTG AGA GTG GTT TGC GGA TC -3’, R: 5’- GAT CCG CAA ACC ACT CTC AGC GTA TCG GG -3’). Primers were purchased from Integrated DNA Technologies. Site-directed mutagenesis kit (QuickChange II, Agilent Cat# 200521) was then used to acquire pET-15b-His-tagged-c-GPX4U46C_R152H and pET-15b-His-tagged-c-GPX4U46C_C66S, which were confirmed by sequencing at GENEWIZ. All four variants of GPX4 protein were separately expressed in E. coli and purified according to a published protocol with minor modifications24, with experimental details described below.
Isolated colonies of BL21-Gold (DE3) competent cells (Agilent, Cat# 230132) with each plasmid were separately transferred to 8 mL of LB medium with 100 μg/mL ampicillin, and the inoculated culture was incubated while being shaken (225 rpm) at 37 °C for 16 h. 3 mL of the starter culture was added to 1 L of fresh LB medium with 100 μg/mL ampicillin. The culture was incubated while being shaken at 37 °C and 225 rpm until the OD600 reached 0.9. The temperature was then decreased to 15 °C. Cells were incubated with 1 mM isopropyl β-D-1- thiogalactopyranoside (IPTG) while being shaken at 15 °C and 225 rpm overnight. The next day, the bacteria were harvested by centrifugation at 4000g for 20 min at 4 °C and the pellet obtained was ready for purification or stored at −20 °C. The pellet was resuspended in 25 mL of chilled lysis buffer (100 mM Tris pH 8.0, 300 mM NaCl, 20 mM imidazole, 3 mM TCEP, and protease inhibitor cocktail (Roche-Sigma, Cat# 11836170001)). The bacteria were lysed by sonication on ice for 6 min, and the lysate was centrifuged at 10000 rpm for 20 min at 4 °C to remove cell debris. The clarified lysate was incubated with Ni Sepharose 6 Fast Flow beads (GE Life Sciences, via Cytiva Cat# 17-5318-01) on a rotator at 4 °C for at least 1 h. The beads were washed with wash buffer (100 mM Tris pH 8.0, 300 mM NaCl, 50 mM imidazole, and 3 mM TCEP) to remove nonspecific binding. The protein was eluted with 100 mM Tris pH 8.0, 300 mM NaCl, 100 mM imidazole, and 3 mM TCEP. The protein was further purified using a gel filtration Superdex 200 column in FPLC buffer containing 100 mM Tris pH 8.0, 300 mM NaCl, and 3 mM TCEP. The fractions containing GPX4 were pooled together and analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE).
Intact protein MALDI MS analysis
GPX4U46C, GPX4U46C-C66S, or GPX4AllCys(−)-A46C protein was pre-incubated with DMSO control or the inhibitor to be tested prior to MALDI MS analysis: 50 μM GPX4 protein was incubated with 500 μM inhibitors in FPLC buffer (100 mM Tris pH 8.0, 300 mM NaCl, and 3 mM TCEP) with 5% DMSO at RT for 1 hour before transferring to 4°C overnight.
1 μl of the ligand-free protein (pre-incubated with DMSO) or protein-inhibitor complex (pre-incubated with the inhibitor to be tested) was mixed with 9 μl of 10 mg/ml sinapinic acid in the matrix solution (70:30 water/acetonitrile, with 0.1% TFA). 1.0 μl of the final mix was deposited onto the target carrier and allowed to air dry. MALDI spectrum was recorded using Bruker ultrafleXtreme MALDI-TOF instrument. The range of m/z detection and suppression was adjusted to accommodate the molecular weight of target protein. 2000 Hz and 50% intensity was applied for the laser setting. For each sample, five cumulative spectra were collected and the sum was recorded for analysis. All MALDI spectra of protein-inhibitor complex were compared with ligand-free protein to determine the mass shift. Mass shifts were aligned with the mass of potential staying group of each inhibitor to conclude covalent binding.
Protein crystallography
In a separate study32, we found GPX4 protein with the R152H mutation crystalized at an accelerated rate and yielded high-resolution crystal structures. Therefore, we used both GPX4U46C and GPX4R152H-U46C protein in the co-crystallization of GPX4 with small molecule inhibitors. While we attempted to obtain crystal structures of inhibitors in complex with GPX4U46C, we succeeded in crystallizing complexed structures for three inhibitors RSL3, MAC5576, and LOC1886. We obtained the remaining complexes of ML162, CDS9, and TMT10 in presence of the R152H mutant of GPX4U46C. The corresponding GPX4 protein was pre-incubated with the covalent inhibitors before crystallization using the following specifically optimized condition. RSL3 and ML162 condition: 50 μM GPX4 incubated with 150 μM RSL3 or ML162 in the reaction buffer (20 mM Tris pH 9.0, 100 mM NaCl, 3 mM TCEP, 1.5% DMSO) at 15 °C for 1 hour before transferring to 4°C overnight. CDS9 condition: 25 μM GPX4 incubated with 2 mM CDS9 in the reaction buffer (100 mM Tris pH 9.0, 300 mM NaCl, 1mM TCEP, 2% DMSO) at 37°C for 2 hours before transferring to 4°C overnight. TMT10 condition: 25 μM GPX4 incubated with 2 mM TMT10 in the reaction buffer (100 mM Tris pH 9.0, 300 mM NaCl, 2% glycerol, 2% DMSO) at 37°C for 4 hours before transferring to 4°C overnight. MAC-5576 condition: 50 μM GPX4 protein was incubated with 500 μM MAC-5576 in the reaction buffer (100 mM Tris pH 9.0, 300 mM NaCl, and 3 mM TCEP, 5% DMSO) at RT for 1 hour before transferring to 4°C overnight. LOC1886 condition: 40 μM GPX4 protein was incubated with 375 μM LOC1886 in the reaction buffer (20 mM Tris pH 9.0, 100 mM NaCl, and 2 mM TCEP, 1.5% DMSO) at 37°C for 4 hours before transferring to 4°C overnight.
After confirmation of covalent binding using intact protein MALDI MS analysis, the protein-inhibitor complex were exchanged into crystallization buffer (20 mM Tris pH 8.0, 300 mM NaCl, 3 mM TCEP) and concentrated to 5mg/ml before setting up for crystallization.
Rod-shaped crystals of GPX4U46C in complex with RSL3 were grown in a COY anaerobic glove box, with O2 level <2 ppm, at 23°C using a crystallization reagent comprising 0.1 M HEPES, pH 7.5, 0.1 M magnesium nitrate hexahydrate, and 20% (w/v) PEG 8000 with protein to crystallization reagent ratio of 2 μL :1 μL. All crystals were subsequently transferred into a similar crystallization reagent that was supplemented by 20% (v/v) ethylene glycol and flash-frozen in liquid nitrogen in the glove box. A native dataset was collected on a crystal of GPX4U46C with RSL3 at beamline NSLS-II of the Brookhaven National Laboratory, NY.
Protein samples of GPX4U46C-R152H in complex with each of inhibitors: ML162, CDS9, and TMT10, and GPX4U46C with LOC1886 were initially screened at the High-Throughput Crystallization Screening Center 35 of the Hauptman-Woodward Medical Research Institute (HWI) (https://hwi.buffalo.edu/high-throughput-crystallization-center/). The most promising crystal hits were reproduced using under oil micro batch method at 23°C.
Crystals of GPX4U46C-R152H in complex with ML162 were grown using crystallization reagent comprising 8% Tacsimate, pH 6, and 20% (w/v) PEG 3350. The crystals were subsequently transferred into a similar crystallization reagent that was supplemented by 20% (v/v) ethylene glycol and flash-frozen in liquid nitrogen. Crystals of GPX4U46C-R152H with CDS9 and TMT10 both were grown using crystallization condition comprising 0.2 M potassium thiocyanate, pH 7, and 20% (w/v) PEG 3350, and flash-frozen in liquid nitrogen after supplementing the crystallization reagent with 20% (v/v) ethylene glycol. Crystals of GPX4U46C in complex with MAC-5576 were grown using crystallization condition similar to that used for GPX4U46C-R152H with ML162. In contrast, crystals of GPX4U46C with LOC1886 were grown using crystallization reagent comprising 2 M sodium chloride and 0.1 M sodium acetate trihydrate, pH 4.6. Crystals of GPX4U46C with MAC-5576 and LOC1886 were subsequently transferred to their respective crystallization reagent that was respectively supplemented by 20% (v/v) glycerol and 20% (v/v) ethylene glycol and flash-frozen in liquid nitrogen.
Crystals of GPX4U46C with RSL3 diffracted X-ray at the beamline NSLSII to resolution 1.60 Å, while GPX4U46C-R152H with ML162, GPX4U46C-R152H with CDS9, GPX4U46C with TMT10, GPX4U46C with MAC-5576, and GPX4U46C with LOC1886 diffracted X-ray at the beamline NE-CAT24-ID-C to resolution 1.69 Å, 1.91 Å, 1.73 Å, 2.25 Å, and 1.93 Å, respectively. The images were respectively processed and scaled in space group P21, C2, C2, C2, P21, and C2221, using XDS36. The structure of each protein was determined by molecular replacement method using MOLREP37 program and the previously determined32 crystal structures of GPX4U46C (PDB id: 7L8K) and GPX4U46C-R152H (PDB id: 7L8L) both were used as search models for structure determination and validation of six crystal structures reported here. The geometry of each crystal structure was fixed using programs XtalView38 and COOT39, and refined by Phenix40. The number of GPX4 protomers in the asymmetric unit (ASU) of each crystal varies from one to four. ASU of the crystal with GPX4 in complex with either ML162, TMT10, or LOC1886 has one protomer, while those in complex with RSL3 and CDS9 have two protomers. The ASU of GPX4U46C with MAC-5576 has four protomers. The crystallographic statistics is shown in Supplementary Table 1.
All figures depicting crystal structures and surface potential were produced using PyMOL (https://pymol.org/2/) with the APBS plug-in41.
Cellular viability assay
1000 cells of specified HT-1080 or G401 cell line were plated 36 μL per well of a 384-well plate on day 1. The remaining cells were immediately tested for Western Blot (to monitor GPX4 protein overexpression level). For dose response curves, compounds were dissolved in DMSO and a 12-point, twofold dilution series was prepared, unless otherwise specified in the corresponding figure legends. The compounds were then diluted 1:50 in media and 4 μL was added to each well of the plates on day 2. Final concentrations of the compounds on the 384-well plate started from 2 μM for RSL3/ML162 and 20 μM for IKE/FIN56/FINO2, unless otherwise specified in the corresponding figure legends. For single-point ferroptosis test, regular HT-1080 cells were treated either with DMSO, 600 nM RSL3, or 125 μM LOC1886, with or without supplementation of 20 μM a-Tocopherol, Ferrostatin-1, Z-VAD-FMK, or Necrostatin-1s. Specific concentration of RSL3 was determined by pilot experiments to approximately induce half-maximum effect, while concentration of LOC1886 is consistent with degradation experiment presented in Figure 6B. After 48 h of treatment, the viability of cells was measured using 1:1 dilution of the CellTiter-Glo luminescent reagent (Promega, Cat# G7573) with media, which was read on a Victor 5 plate reader after 10 min of shaking at room temperature on day 4. The intensity of luminescence was normalized to that of DMSO control. Results were quantified using GraphPad Prism 9. Based on the dose-response curve of viability, area under curve (AUC) values were calculated, with a uniform AUC calculation method consistently applied across all data sets. Practically, GraphPad Prism interprets a curve as a series of connected XY points, defines Y=0 as the baseline, and then computes the area under the curve using the trapezoid rule, without smoothing or extrapolation. Therefore AUC reported a cumulative measurement of drug effect in all concentrations being tested. AUC values were then reported in bar graph formats with standard errors using GraphPad Prism 9.
Western blot assay
For GPX4 degradation study with RSL3, specified G-401 cells were seeded at 800,000 per well in a 60-mm plate and allowed to adhere overnight. Cells were then treated with 10 μM Fer-1 and 0 (vehicle), 2, or 4 μM RSL3 for 10 h. For GPX4 degradation study with LOC1886, regular HT-1080 cells were seeded at 4 million per well in 100-mm plates and allowed 8 hours to adhere. Cells were then treated with 100 μM α-Tocopherol and 0 (vehicle) or 100 μM LOC1886 for 24 h. For both degradation studies, cells were harvested with trypsin (Invitrogen, Cat# 25200-114), pelleted, washed with PBS, and lysed with RIPA buffer (supplemented with cOmplete™ protease inhibitor cocktail).
For the quantification of GPX4 protein level as a part of cellular viability and GPX4-specific activity assay, each cell line subject to the cellular viability and GPX4-specific activity was tested by Western Blot in technical duplicates. In particular, cells were harvested with trypsin (Invitrogen, Cat# 25200-114), pelleted, and lysed by LCW lysis buffer (0.5 % TritonX-100, 0.5 % sodium deoxycholate salt, 150 mM NaCl, 20 mM Tris-HCl pH 7.5, 10 mM EDTA, 30 mM Na-pyrophosphate, and cOmplete™ protease inhibitor cocktail). While part of the cell lysates was blotted for protein quantification, the other part of lysates was used for the GPX4-specific activity assay.
For all Western Blot experiments, cell lysates were blotted and imaged as previously described8: The concentration of protein in the lysate was determined using BCA assay kit (Thermo Fisher, Cat# 23225) using BSA as standards. Samples were mixed with 5X SDS loading buffer and separated by SDS-polyacrylamide gel electrophoresis. Western transfer was performed using the iBlot system (Invitrogen). Membranes were blocked for 1 hour in Phosphate-buffered saline (pH 7.4) with 0.1% Tween-20 (PBS-T) with 50% Intercept blocking buffer (LI-COR, Cat# 927-70001) and incubated in primary antibody overnight at 4°C. Following 5 min washes in PBS-T for three times, the membrane was incubated with secondary antibodies for 1 hr. The membrane was washed again in PBS-T for 5 min for three times prior to visualization. Antibodies used were actin (Cell Signaling, Cat# D18C11, 1:3,000 dilution), GPX4 (R&D systems, Cat# MAB5457, 0.5 μg/mL), and GAPDH (Abcam, Cat# ab9485, 1:2000 dilution). Results were quantified using a LI-COR Odyssey CLx IR scanner, ImageJ42, and GraphPad Prism 9.
Determination of cellular GPX4-specific activity
We applied a NADPH-coupled cellular GPX4 enzymatic activity assay as previously reported with minor modifications43,44. Oxidized glutathione, generated by GPX4 during reducing its specific phospholipid hydroperoxides substrate, was reduced by Glutathione Reductase at the expense of NADPH, the decrease in the characteristic absorbance of which at 340 nm was monitored and quantified as GPX4 activity. The GPX4-specific substrate PCOOH was prepared by enzymatic hydroperoxidation of phosphatidylcholine by soybean lipoxidase type IV: 22 mL of 0.2 M Tris-HCl, pH 8.8, containing 3 mM sodium deoxycholate and 0.3 mM phosphatidylcholine was incubated at room temperature, under continuous stirring, for 30 min with 0.7 mg of soybean lipoxidase type IV. The mixture was loaded on a Sep-Pak C18 cartridge (Waters-Millipore) washed with methanol and equilibrated with water. After washing with 10 volumes of water, phosphatidylcholine hydroperoxides were eluted in 2 mL of methanol. 50 millions of particular G401 or HT-1080 cells were harvested and lysed by LCW lysis buffer (0.5 % TritonX-100, 0.5 % sodium deoxycholate salt, 150 mM NaCl, 20 mM Tris-HCl pH 7.5, 10 mM EDTA, 30 mM Na-pyrophosphate, and cOmplete protease inhibitor cocktail). The concentration of protein in the lysate was determined using BCA assay kit (Thermo Fisher, Cat# 23225) using BSA as standards. Then, on a 96-well plate, 250μL 1.5 μg/μL cell lysate was incubated in the GPX4 activity assay buffer (0.1 % Triton X-100, 100 mM Tris-HCl pH 7.4, 10mM NaN3, 5 mM EDTA, 0.6 IU/mL Glutathione reductase, 0.5 mM NADPH, and 3 mM GSH unless otherwise noted) at 37°C for 10 min. For evaluation of GPX4 inhibitors, DMSO, 50 μM RSL3, or 200μM LOC1886 was added to the 250μL cell lysate in GPX4 activity assay buffer before the 10 min incubation. Specific concentrations of compounds were determined by pilot experiments to approximately induce half-maximum effect. PCOOH was then added to the mixture to initiate GPX4 reaction. Absorbance of NADPH at 340 nm was determined kinetically at 1 min interval over the 20 min time. Experiments using lysis buffer instead of cell lysate and controls without addition of PCOOH were also done to measure the particular activity of GPX4 to reduce phospholipid hydroperoxides. Total GPX4 activity of each sample were normalized to their specific GPX4 protein level based on Western Blot for unit GPX4 enzymatic activity. For comparison, GPX4 activity was then normalized to the control of each experiment (AllCys(−), pBP, or DMSO-treated) before presentation in the figure. Results were quantified using GraphPad Prism 9.
Inhibition of the activity of purified GPX4U46C protein
Similar to the determination of cellular GPX4-specific activity, by coupling the oxidation of NADPH to NADP+ by oxidized glutathione produced by GPX4 in the presence of glutathione reductase, GPX4U46C and GPX4U46C-C66S activity was assessed by measuring the decrease in NADPH (absorbance at 340 nm). GPX4 reaction buffer was prepared by adding 0.05 U/mL glutathione reductase, 210 μM GSH, 250 μM NADPH into 50 mM Tris HCl, pH 8.0, 0.5 mM EDTA. For evaluation of GPX4 inhibitors, DMSO, 50 μM RSL3, or 50 μM ML162 was added to 100μL 15μM GPX4U46C or GPX4U46C-C66S in GPX4 reaction buffer and then incubated at 37 °C for 10 min. 50 μM Cumene-OOH was then added to the mixture to initiate GPX4 reaction. Absorbance of NADPH at 340 nm was determined kinetically over the 20 min time. Experiments using GPX4 stock buffer instead of GPX4 protein and controls without addition of Cumene-OOH were also done to measure the particular activity of GPX4 to reduce Cumene-OOH. GPX4 activity was then normalized to the DMSO control. Results were quantified using GraphPad Prism 9.
The inhibition constants (Ki) of GPX4 protease inhibitors were measured as previously described 45. First, for the quantitative determination of Km of GPX4U46C on cumene-OOH, the Cumene-OOH substrate ranging from 33.3 to 200 μM was applied to GPX4 reactions with 15 μM GPX4U46C. Reactions were monitored kinetically. The initial velocity of the proteolytic activity was monitored. The initial velocity was plotted against the substrate concentration with the classic Michaelis–Menten equation in Prism software (double reciprocal plot) to determine Km of GPX4U46C.
Sequentially, serial dilutions of the test compound were added to 100μL 15μM GPX4U46C in GPX4 reaction buffer and then incubated at 37 °C for 10 min. 50 μM cumene-OOH was then added to the mixture to initiate GPX4 reaction. Absorbance of NADPH at 340 nm was determined kinetically over the 20 min time. Experiments using GPX4 stock buffer instead of GPX4 protein and controls without addition of Cumene-OOH were also done to measure the particular activity of GPX4 to reduce Cumene-OOH. Ki values were fit to the Morrison equation with the enzyme concentration parameter fixed to 15 μM, the Km parameter fixed to 180 μM, and the substrate concentration parameter fixed to 50 μM using Prism software.
Thermal shift assay
Since the binding of small molecules may alter the thermostability of protein, we applied thermal shift assay to screen the LOC library, which determined binding of ligand from the change of the unfolding transition temperature (ΔTm) obtained in the presence of ligands relative to that obtained in the absence of ligands34,46. For higher throughput purpose, we adapted the assay on 384-well PCR plate. With the assistance of Biomek Automated Liquid Handler (Beckman), 2 μL 10 mM stock solution (in 100% DMSO) of each compound was transferred from 384-well library plates to 38 μL FPLC buffer (100mM Tris pH 8.0, 300 mM NaCl, 3mM TCEP) and mixed in each well of a 384-well mother plate. The compound solution was then dispensed, 12 μL per well, into three 384-well PCR plates. For the determination of binding of a specific compound (RSL3-minus-Cl) to GPX4, the compound was manually serially diluted as in 12 μL per well in triplicates. GPX4U46C protein and Sypro orange were then manually added to the plate, with the final concentrations of 5 μM and 5x, respectively, to make a volume of 20 μL per well right before thermal shift analysis of the specific plate. The thermal shift assay was performed on the ViiA 7 Real-Time PCR system (Thermo Fisher) with the thermal protocol: 25° for 15s, Increase temp to 99° at a rate of 0.05°/sec, 99° for 15 sec. The fluorescence was recorded and analyzed by Protein Thermal Shift™ software. The mean |ΔTm| value for the biological triplicates were calculated and used for hit identification.
SPR binding assay
The binding affinity of RSL3-minus-Cl for GPX4 was assayed using the SPR-based Biacore X100 instrument (Cytiva). His-tagged GPX4U46C was immobilized on a CM5 sensor chip by using His Capture Kit (Cytiva, Cat# 28995056) under standard condition at 25 °C with running buffer HBS-EP+ (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, and 0.05% v/v surfactant P-20). A reference flow cell was activated and blocked in the absence of GPX4. The GPX4 immobilization level was fixed at 1,000 response units (RU), and then different concentrations of RSL3-minus-Cl were serially injected into the channel to evaluate binding affinity. Between injections of RSL3-minus-Cl samples of different concentrations, regeneration of sensor chip was performed by washing with the regeneration buffer (20 mM Glycine, pH 1.5) provided by the same His Capture Kit (Cytiva, Cat# 28995056). The equilibrium dissociation constant (KD) of the RSL3-minus-Cl was obtained by fitting binding response units to the Hill equation.
1H, 15N-HSQC-NMR spectrum for 15N isotope-labeled GPX4U46C
Uniformly 15N-labeled GPX4U46C protein with N-terminal His6 tag was prepared. The GPX4U46C construct was expressed in E. coli BL21-Gold (DE3) cells (Stratagene) growing at 37°C in M9 minimal medium supplemented with 100 mg/mL ampicillin, 2 mM MgSO4, 100 mM CaCl2, 1X trace metals, 1X RPMI 1640 vitamin stock (Sigma-Aldrich, Cat# R7256), 10 mg/mL biotin, 10 mg/mL thiamine hydrochloride, 4 g/L glucose, and 3 g/L 15NH4Cl as the sole nitrogen source. The following induction, lysis, and protein purification was the same as for the non-isotope-labeled His-tagged GPX4U46C described above, except the N-terminal His6 tag was removed by adding 5 U/mg thrombin before FPLC purification and that HSQC buffer was used as FPLC buffer. The thrombin reaction was performed after a buffer exchange to remove imidazole, which otherwise would inhibit thrombin, and was allowed to proceed overnight at 4°C.
For HSQC spectrum of GPX4 used in backbone peak assignment, 50 μM 15N-labeled GPX4 (final concentration) was prepared in HSQC buffer (100 mM MES, 5 mM TCEP, pH 6.5, 10% D2O added for the field frequency lock).
For HSQC analysis of GPX4 with RSL3, since solubility of RSL3 in aqueous solution is low, 5.0 % d6-DMSO was added to the buffer to increase the solubility of RSL3. 5.0 % d6-DMSO was also added to the negative control GPX4 alone sample to prepare samples with and without RSL3 but with the same final DMSO concentration. Practically, to achieve this, RSL3 was first dissolved in 100% d6-DMSO at 2 mM. 2 mM RSL3 or 100% d6-DMSO vehicle was then diluted into GPX4 sample to make 5.0 % final concentration of DMSO. 50 μM 15N-labeled GPX4 was preincubated with 100 μM RSL3 (5.0 % DMSO) or 5.0 % DMSO vehicle in HSQC buffer (100 mM MES, 5 mM TCEP, pH 6.5, 10% D2O) for 12 h at 4 °C before recording NMR spectrum. Here, aiming to record the same spectrum with and without RSL3 but with the same protein concentration, same volume of GPX4 from the same stock batch was used in these samples.
For HSQC analysis of GPX4 with RSL3-minus-Cl, since solubility of RSL3-minus-Cl in aqueous solution is low, 10.0 % d6-DMSO was added to the buffer to increase the solubility of RSL3-minus-Cl. 10.0 % d6-DMSO was also added to the negative control GPX4 alone sample to prepare samples with and without RSL3-minus-Cl but with the same final DMSO concentration. Practically, to achieve this, RSL3-mius-Cl was first dissolved in 100% d6-DMSO at 10 mM. 10 mM RSL3-minus-Cl or 100% d6-DMSO vehicle was then diluted into GPX4 sample to make 10.0 % final concentration of DMSO. 10 μM 15N-labeled GPX4 was mixed with 1 mM μM RSL3-minus-Cl (10.0 % DMSO) or 10.0 % DMSO vehicle in HSQC buffer (100 mM MES, 5 mM TCEP, pH 6.5, 10% D2O) immediately before recording NMR spectrum. Here, aiming to record the same spectrum with and without RSL3-minus-Cl but with the same protein concentration, same volume of GPX4 from the same stock batch was used in these samples. Additionally, since presence of DMSO may shift the HSQC peaks, we also prepared GPX4 alone sample with various DMSO concentrations and recorded HSQC spectra, aiming to confirm the spectrum assignment performed at 0% DMSO still works for HSQC spectrum collected with 10% DMSO.
For HSQC analysis of GPX4 with LOC1886, samples were prepared similarly as above, with slight modifications to account for the lower potency of LOC1886 as compared to RSL3. 50 μM 15N-labeled GPX4 was preincubated with 800 μM inhibitor or DMSO vehicle to be tested for 6 h at room temperature in HSQC buffer (100 mM MES, 5 mM TCEP, pH 6.5, 10% D2O) before recording NMR spectrum.
The 1H-15N HSQC spectra were collected on Bruker Avance III 500 Ascend (500 MHz) spectrometers (Columbia University) at 298K. The 1H carrier frequency was positioned at the water resonance. The 15N carrier frequency was positioned at 115 ppm. The spectral width in the 1H dimension was 7,500 Hz and the width in the 15N dimension was 1,824.6 Hz. Suppression of water signal was accomplished using the WATERGATE sequence. Heteronuclear decoupling was accomplished using GARP decoupling scheme.
3D protein NMR experiments for 13C, 15N isotope-labeled GPX4U46C
Uniformly 13C, 15N-labeled GPX4U46C protein with N-terminal His6 tag was prepared. The GPX4U46C construct was expressed in E. coli BL21-Gold (DE3) cells (Stratagene) growing at 37°C in M9 minimal medium supplemented with 100 mg/mL ampicillin, 2 mM MgSO4, 100 mM CaCl2, 1X trace metals, 1X RPMI 1640 vitamin stock (Sigma-Aldrich, #R7256), 10 mg/mL biotin, 10 mg/mL thiamine hydrochloride, 4 g/L U-13C6-glucose as the sole carbon source, and 3 g/L 15NH4Cl as the sole nitrogen source. The following induction, lysis, protein purification, and tag removal were the same as for uniformly 15N-labeled GPX4U46C protein.
HNCACB and CBCA(CO)NH NMR spectrum of 50 μM uniformly 13C, 15N-labeled GPX4U46C protein in 100 mM MES pH 6.5, with 5mM TCEP and 5% D2O were collected on 700 MHz Avance III/TS 3.5.6 at NYSBC and applied for HSQC backbone resonance assignments. We performed the assignment based on standard triple resonance backbone assignment (Cα and Cβ) in the practical guide on protein NMR (https://www.protein-nmr.org.uk) and used CcpNmr Analysis software47. To verify the assignment, we also collected HNCO, HN(CA)CO, HNCA, and HN(CO)CA NMR spectrum, and performed Cα and C’ (carbonyl caron) based assignments in parallel as a cross-check. Of the expected 168 amino acid residues of our GPX4U46C construct (175 total residues excluding 7 prolines which has no H attached to its N and therefore does not give signal in HSQC), backbone resonances of 147 residues were assigned. The unassigned residues included a 10-residue N-terminal linker sequence, which was also not visible in the crystal structure of GPX4 (PDB: 2OBI)24. In the sequence of our construct which is corresponding to D6-F170 of human GPX4 (NCBI Reference Sequence: NP_001354761.1), DDWRC ARSMHEFSAK DIDGHMVNLD KYRGFVCIVT NVASQCGKTE VNYTQLVDLH ARYAECGLRI LAFPCNQFGK QEPGSNEEIK EFAAGYNVKF DMFSKICVNG DDAHPLWKWM KIQPKGKGIL GNAIKWNFTK FLIDKNGCVV KRYGPMEEPL VIEKDLPHYF, the assigned residues were underlined (154/165). The inability to detect and/or assign the remaining backbone peaks may reflect exchange broadening owing to conformational dynamics and mobility inherent to GPX4 in solution, which notably included cysteine 46 at the active site and other residues on the loop surrounding it 48.
Re-synthesis of LOC1886
LOC1886 was initially purchased from LabNetwork, Cat# STOCK6S-48996. After screening, we resynthesized and characterized LOC1886 for validation. An oven-dried 25 mL round-bottom flask equipped with magnetic stir bar was charged with 4-methoxy-indole-2-carboxylic acid (191 mg, 1 mmol) in anhydrous THF (10 mL). 1,1’-Carbonyldiimidazole (178 mg, 1.1 mmol) was added to the solution and the reaction was stirred at room temperature overnight. The reaction mixture was concentrated in vacuum and the residue was purified by column chromatography (silica gel, hexanes : ethyl acetate = 1 : 4 ) to afford the product ( 214 mg, 89% yield ) as white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 8.51 (d, J = 1.3 Hz, 1H), 7.90 (d, J = 1.4 Hz, 1H), 7.33 (s, 1H), 7.30 (t, J = 8.1 Hz, 1H), 7.19 (d, J = 1.0 Hz, 1H), 7.13 – 7.05 (d, 1H), 6.61 (d, J = 7.6 Hz, 1H), 3.92 (s, 3H). HRMS (ESI) m/z: [M + H]+ calculated for C13H12N3O2, 242.0930; found, 242.0930.
Synthesis of RSL3-minus-Cl
Methyl (1S,3R)-2-acetyl-1-(4-(methoxycarbonyl) phenyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b] indole-3-carboxylate (1S,3R-RSL3-Cl). An oven-dried 25 mL round-bottom flask equipped with a magnetic stirring bar was charged with (1S,3R)-1-(4-(methoxycarbonyl) phenyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b] indole-3-carboxylate (50.0 mg, 0.14 mmol, 1.0 equiv) in dry DCM (3 mL) and Et3N (48.8 μL, 0.35 mmol, 2.5 equiv). The mixture was cooled in an ice bath to 0 °C and acetyl chloride (12.9 μL, 0.18 mmol, 1.3 equiv) was added. The resulting solution was warmed to room temperature and stirred overnight. The reaction mixture was concentrated under reduced pressure and the crude residue was purified by flash column chromatography on silica gel (0–50% EtOAc/hexanes) to afford the title compound (33 mg, 58% yield) as an off-white amorphous solid. (Peaks in 1H-NMR spectrum split as a ~ 1:1.1 mixture due to the presence of rotamers. Both rotamers are described.) 1H NMR (400 MHz, CDCl3); δ 7.98 (d, J = 35.2 Hz, 2H), 7.70 (d, J = 33.0 Hz, 1H), 7.53 (d, J = 7.5 Hz, 1H), 7.43 (d, J = 17.5 Hz, 2H), 7.24 – 7.04 (m, 3H), 6.12 (s, 1H), 5.41 (s, 0.47H), 5.07 (s, 0.5H), 3.90 (s, 3H), 3.63 (s, 3.5H), 3.61 – 3.41 (m, 1H), 3.28 (m, 0.5H), 2.16 (s, 1.7H), 2.01 (s, 1.3H) ppm. HRMS (ESI): m/z: [M + H]+ calculated for C23H23N2O5, 407.1607; found, 407.1622. The analytical data were in good accordance with those reported in the literature49.
Identification of LOC1886 and RSL3 binding site on GPX4 by LC-MS/MS
40 μM purified GPX4U46C protein was incubated with 375 μM LOC1886 or DMSO vehicle in the reaction buffer (20 mM Tris pH 9.0, 100 mM NaCl, 2 mM TCEP, 1.5% DMSO) at 37°C for 2 hours before transferring to 4°C overnight. 50 μM purified GPX4U46C protein was incubated with 150 μM RSL3 in the same reaction buffer at 15 °C for 1 hour before transferring to 4°C overnight. After confirmation of covalent binding using intact protein MALDI MS analysis, the protein-inhibitor complex and vehicle protein were denatured in denaturation buffer50 (1% SDC, 100 mM TrisHCl pH 8.5,) and heat for 10 min at 60°C. Protein reduction and alkylation of cysteines were performed with 10 mM TCEP and 40 mM CAA at 45°C for 10 min followed by sonication in a water bath at room temperature. Protein digestion was conducted overnight by adding trypsin in a 1:50 ratio (μg of enzyme to μg of protein) at 37° C. Peptides were acidified by adding 1% TFA, vortexed, and subjected to StageTip clean-up via SDB-RPS. Peptides were loaded on one 14-gauge StageTip plugs. Peptides were washed two times with 200 μL 1% TFA 99% ethyl acetate followed by 200 μL 0.2% TFA/5%ACN in centrifuge at 3000 rpm. The samples were then eluted with 60 μL of 1% Ammonia, 50% ACN into Eppendorf tubes and dried at 45°C in a SpeedVac centrifuge. Samples were resuspended in 10 μL of LC buffer (3% ACN/0.1% FA). Peptide concentrations were determined using NanoDrop and 500 ng of each sample was used for DDA analysis on Orbitrap Fusion Tribrid.
Desalted peptides were injected in an EASY-Spray™ PepMap™ RSLC C18 50 cm × 75 cm ID column (Thermo Scientific) connected to an Orbitrap Fusion™ Tribrid™ (Thermo Scientific). Peptide elution and separation were achieved at a non-linear flow rate of 250 nl/min using a gradient of 5%−30% of buffer B (0.1% (v/v) formic acid, 100% acetonitrile) for 120 minutes with a temperature of the column maintained at 50 °C during the entire experiment. The Thermo Scientific Orbitrap Fusion Tribrid mass spectrometer was used for peptide tandem mass spectroscopy (MS/MS). Survey scans of peptide precursors are performed from 200 to 800 m/z at 120 K full width at half maximum (FWHM) resolution (at 200 m/z) with a 2 × 105 ion count target and a maximum injection time of 50 ms. The instrument was set to run in top speed mode with 3-second cycles for the survey and the MS/MS scans. After a survey scan, MS/MS was performed on the most abundant precursors, i.e., those exhibiting a charge state from 2 to 6 of greater than 5 × 103 intensity, by isolating them in the quadrupole at 1.6 Th. We used HCD with 30% collision energy and detected the resulting fragments with the auto scan mode in the orbitrap. The automatic gain control (AGC) target for MS/MS was set to 100 % and the maximum injection time was limited to 30 ms. The dynamic exclusion was set to 45 s with a 10 ppm mass tolerance around the precursor and its isotopes. Monoisotopic precursor selection was enabled. Raw mass spectrometric data were analyzed using Qual Browser of Thermo Xcalibur (version: 4.5.445.18, Thermo Fisher Scientific). YAECRSL3GLR and YAECLOC1886GLR peptide identities were determined by precursor ions mass and selected fragment ions at MS/MS level.
Detection of Lipid ROS
Lipid reactive oxygen species (ROS) were detected using flow cytometry. 2.5 × 105 HT-1080 cells were seeded per well in a 6-well plate. The next day, medium was replaced with 2 mL medium containing drug (150 nM RSL3 or 100 μM LOC1886, with or without 10μM Fer-1 as indicated) or vehicle alone and cells were incubated at 37°C for two hours. Specific concentration of RSL3 was determined by pilot experiments to sufficiently induce the lipid peroxidation effects, while concentration of LOC1886 is consistent with degradation experiment presented in Figure 6B. Subsequently, cells were stained with 1.5 μM C11-BODIPY (BODIPY 581/591 C11, a lipid ROS indicator, Thermo Fisher Scientific, Cat# D3861) for 20 minutes at 37 °C. Cells were then harvested with 500 μL Trypsin-EDTA, pelleted by centrifugation at 300g for 5 minutes, washed with HBSS, and resuspended in 500 μL HBSS. The cell suspension was passed through nylon mesh (40um, cell strainer) to remove cell aggregates, then analyzed on a Beckman Coulter CytoFLEX flow cytometer. Fluorescence intensity was measured on the FL1 channel with gating to record live single cells only (gate constructed from DMSO treatment group). A minimum of 50,000 cells were analyzed per condition.
Quantification and Statistical Analysis
All replicate experiments, unless otherwise indicated, are biological replicates based on distinct samples. All statistical analyses were performed using GraphPad Prism. Tukey’s test or Dunnett’s test was performed as a post-hoc test after ordinary one-way ANOVA, comparing all pairwise datasets, with alpha = 0.05. Tukey’s test or Dunnett’s test was performed as a post-hoc test after ordinary two-way ANOVA for grouped data, comparing pairwise datasets in each group. Only relevant pairwise comparisons are highlighted in the figures. Unpaired Student’s t-test was performed when indicated in the manuscript, for comparing two experimental conditions. All t-tests were two tailed and set with significance thresholds of pns > 0.05, p* < 0.05, p** < 0.01, p*** < 0.001, and p**** < 0.0001.
Supplementary Material
Highlights.
The region around Cysteine 66 (C66) is an allosteric binding site of RSL3 on GPX4.
C66 modulates the enzymatic activity of GPX4.
Covalent binding of compounds to C66 inhibits and degrades GPX4.
Co-crystal structures of six distinct compounds covalently binding to C66 are reported.
Acknowledgements
This study was supported by P01CA87497 (BRS), R35CA209896 (BRS), and R61NS109407 (BRS), as well as by SMP Oncology. We thank the staff of the High-Throughput Crystallization Screening Center of the Hauptman-Woodward Medical Research Institute for screening of crystallization conditions. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of Interests
Brent R. Stockwell is an inventor on patents and patent applications involving small molecule drug discovery, ferroptosis, and GPX4, and co-founded and serves as a consultant to Inzen Therapeutics, Exarta Therapeutics, and ProJenX Inc, and serves as a consultant to Weatherwax Biotechnologies Corporation and Akin Gump Strauss Hauer & Feld LLP, and receives sponsored research support from Sumitomo Dainippon Pharma Oncology.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference:
- 1.Dillekas H, Rogers MS, and Straume O (2019). Are 90% of deaths from cancer caused by metastases? Cancer Med 8, 5574–5576. 10.1002/cam4.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Zhang H, and Chen X (2019). Drug resistance and combating drug resistance in cancer. Cancer Drug Resistance 2, 141–160. 10.20517/cdr.2019.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al. (2015). Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476. 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agmon E, and Stockwell BR (2017). Lipid homeostasis and regulated cell death. Curr Opin Chem Biol 39, 83–89. 10.1016/j.cbpa.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas JP, Geiger PG, Maiorino M, Ursini F, and Girotti AW (1990). Enzymatic reduction of phospholipid and cholesterol hydroperoxides in artificial bilayers and lipoproteins. Biochim Biophys Acta 1045, 252–260. 10.1016/0005-2760(90)90128-k. [DOI] [PubMed] [Google Scholar]
- 6.Kuhn H, and Borchert A (2002). Regulation of enzymatic lipid peroxidation: the interplay of peroxidizing and peroxide reducing enzymes. Free Radic Biol Med 33, 154–172. 10.1016/s0891-5849(02)00855-9. [DOI] [PubMed] [Google Scholar]
- 7.Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, Paradkar S, Boehnke N, Deik AA, Reinhardt F, et al. (2020). Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 585, 603–608. 10.1038/s41586-020-2732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. 10.1038/nature24297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, Tseng YY, Deasy R, Kost-Alimova M, Dancik V, et al. (2019). A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun 10, 1617. 10.1038/s41467-019-09277-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakamoto K, Sogabe S, Kamada Y, Matsumoto SI, Kadotani A, Sakamoto JI, and Tani A (2017). Discovery of GPX4 inhibitory peptides from random peptide T7 phage display and subsequent structural analysis. Biochem Biophys Res Commun 482, 195–201. 10.1016/j.bbrc.2016.11.035. [DOI] [PubMed] [Google Scholar]
- 14.Borchert A, Kalms J, Roth SR, Rademacher M, Schmidt A, Holzhutter HG, Kuhn H, and Scheerer P (2018). Crystal structure and functional characterization of selenocysteine-containing glutathione peroxidase 4 suggests an alternative mechanism of peroxide reduction. Biochim Biophys Acta Mol Cell Biol Lipids 1863, 1095–1107. 10.1016/j.bbalip.2018.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Weiwer M, Bittker JA, Lewis TA, Shimada K, Yang WS, MacPherson L, Dandapani S, Palmer M, Stockwell BR, Schreiber SL, and Munoz B (2012). Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg Med Chem Lett 22, 1822–1826. 10.1016/j.bmcl.2011.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, and Stockwell BR (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113, E4966–4975. 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang WS, and Stockwell BR (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 15, 234–245. 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eaton JK, Ruberto RA, Kramm A, Viswanathan VS, and Schreiber SL (2019). Diacylfuroxans Are Masked Nitrile Oxides That Inhibit GPX4 Covalently. J Am Chem Soc 141, 20407–20415. 10.1021/jacs.9b10769. [DOI] [PubMed] [Google Scholar]
- 19.Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ, Zimmermann K, Cai LL, Niehues M, Badock V, et al. (2020). Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol 16, 497–506. 10.1038/s41589-020-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eaton JK, Furst L, Cai LL, Viswanathan VS, and Schreiber SL (2020). Structure-activity relationships of GPX4 inhibitor warheads. Bioorg Med Chem Lett 30, 127538. 10.1016/j.bmcl.2020.127538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allimuthu D, and Adams DJ (2017). 2-Chloropropionamide As a Low-Reactivity Electrophile for Irreversible Small-Molecule Probe Identification. ACS Chem Biol 12, 2124–2131. 10.1021/acschembio.7b00424. [DOI] [PubMed] [Google Scholar]
- 22.Ursini F, Heim S, Kiess M, Maiorino M, Roveri A, Wissing J, and Flohe L (1999). Dual function of the selenoprotein PHGPx during sperm maturation. Science 285, 1393–1396. 10.1126/science.285.5432.1393. [DOI] [PubMed] [Google Scholar]
- 23.Conrad M, Moreno SG, Sinowatz F, Ursini F, Kolle S, Roveri A, Brielmeier M, Wurst W, Maiorino M, and Bornkamm GW (2005). The nuclear form of phospholipid hydroperoxide glutathione peroxidase is a protein thiol peroxidase contributing to sperm chromatin stability. Mol Cell Biol 25, 7637–7644. 10.1128/MCB.25.17.7637-7644.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheerer P, Borchert A, Krauss N, Wessner H, Gerth C, Hohne W, and Kuhn H (2007). Structural basis for catalytic activity and enzyme polymerization of phospholipid hydroperoxide glutathione peroxidase-4 (GPx4). Biochemistry 46, 9041–9049. 10.1021/bi700840d. [DOI] [PubMed] [Google Scholar]
- 25.Moosmayer D, Hilpmann A, Hoffmann J, Schnirch L, Zimmermann K, Badock V, Furst L, Eaton JK, Viswanathan VS, Schreiber SL, et al. (2021). Crystal structures of the selenoprotein glutathione peroxidase 4 in its apo form and in complex with the covalently bound inhibitor ML162. Acta Crystallographica Section D 77, 237–248. doi: 10.1107/S2059798320016125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, and Stockwell BR (2016). Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol 12, 497–503. 10.1038/nchembio.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, Heindel DW, Zuckerman DS, Bos PH, Reznik E, et al. (2018). FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol 14, 507–515. 10.1038/s41589-018-0031-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng H, and Stockwell BR (2018). Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol 16, e2006203. 10.1371/journal.pbio.2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao J, Yang F, Che J, Han Y, Wang Y, Chen N, Bak DW, Lai S, Xie X, Weerapana E, and Wang C (2018). Selenium-Encoded Isotopic Signature Targeted Profiling. ACS Cent Sci 4, 960–970. 10.1021/acscentsci.8b00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, Uchida K, O’Connor OA, and Stockwell BR (2019). Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol 26, 623–633 e629. 10.1016/j.chembiol.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T, et al. (2018). Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 172, 409–422 e421. 10.1016/j.cell.2017.11.048. [DOI] [PubMed] [Google Scholar]
- 32.Liu H, Forouhar F, Seibt T, Saneto R, Wigby K, Friedman J, Xia X, Shchepinov MS, Ramesh SK, Conrad M, and Stockwell BR (2022). Characterization of a patient-derived variant of GPX4 for precision therapy. Nature Chemical Biology 18, 91–100. 10.1038/s41589-021-00915-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blanchard JE, Elowe NH, Huitema C, Fortin PD, Cechetto JD, Eltis LD, and Brown ED (2004). High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem Biol 11, 1445–1453. 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaplan A, Gaschler MM, Dunn DE, Colligan R, Brown LM, Palmer AG 3rd, Lo DC, and Stockwell BR (2015). Small molecule-induced oxidation of protein disulfide isomerase is neuroprotective. Proc Natl Acad Sci U S A 112, E2245–2252. 10.1073/pnas.1500439112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luft JR, Collins RJ, Fehrman NA, Lauricella AM, Veatch CK, and DeTitta GT (2003). A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J Struct Biol 142, 170–179. 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 36.Kabsch W (2010). Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr 66, 133–144. 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vagin A, and Teplyakov A (2010). Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 66, 22–25. 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 38.McRee DE (1999). XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol 125, 156–165. 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baker NA, Sept D, Joseph S, Holst MJ, and McCammon JA (2001). Electrostatics of nanosystems: Application to microtubules and the ribosome. Proceedings of the National Academy of Sciences 98, 10037. 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roveri A, Maiorino M, and Ursini F (1994). Enzymatic and immunological measurements of soluble and membrane-bound phospholipid-hydroperoxide glutathione peroxidase. Methods Enzymol 233, 202–212. 10.1016/s0076-6879(94)33023-9. [DOI] [PubMed] [Google Scholar]
- 44.Stolwijk JM, Falls-Hubert KC, Searby CC, Wagner BA, and Buettner GR (2020). Simultaneous detection of the enzyme activities of GPx1 and GPx4 guide optimization of selenium in cell biological experiments. Redox Biology 32, 101518. 10.1016/j.redox.2020.101518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoffman RL, Kania RS, Brothers MA, Davies JF, Ferre RA, Gajiwala KS, He M, Hogan RJ, Kozminski K, Li LY, et al. (2020). Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. Journal of Medicinal Chemistry 63, 12725–12747. 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, and Ellestad G (2004). Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem 332, 153–159. 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 47.Skinner SP, Fogh RH, Boucher W, Ragan TJ, Mureddu LG, and Vuister GW (2016). CcpNmr AnalysisAssign: a flexible platform for integrated NMR analysis. Journal of Biomolecular NMR 66, 111–124. 10.1007/s10858-016-0060-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vadrevu R, Falzone CJ, and Matthews CR (2003). Partial NMR assignments and secondary structure mapping of the isolated alpha subunit of Escherichia coli tryptophan synthase, a 29-kD TIM barrel protein. Protein Sci 12, 185–191. 10.1110/ps.0221103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jelinek A, Heyder L, Daude M, Plessner M, Krippner S, Grosse R, Diederich WE, and Culmsee C (2018). Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radical Biol. Med. 117, 45–57. 10.1016/j.freeradbiomed.2018.01.019. [DOI] [PubMed] [Google Scholar]
- 50.Kulak NA, Pichler G, Paron I, Nagaraj N, and Mann M (2014). Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nature Methods 11, 319–324. 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. Structural data for the GPX4 protein in complex with RSL3, ML162, CDS9, TMT10, MAC-5576, and LOC1886 were deposited in the Protein Data Bank (PDB). Backbone NMR chemical shifts assignment of GPX4 were deposited in the Biological Magnetic Resonance Data Bank (BMRB, entry 51659). Proteomics data of GPX4 treated with inhibitors were deposited in Mass Spectrometry Interactive Virtual Environment (MassIVE, MSV000090526). All deposited data are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
Key resources table.
(CELL-CHEMICAL-BIOLOGY-D-21-00395)
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-GPX4 | R&D systems | Cat# MAB5457, RRID:AB_2232542 |
| Rabbit monoclonal anti-actin | Cell Signaling | Cat# 8456, RRID:AB_10998774 |
| Rabbit polyclonal anti-GAPDH | Abcam | Cat# ab9485, RRID:AB_307275 |
| Bacterial and virus strains | ||
| E. coli BL21-Gold (DE3) competent cells | Agilent | Cat# 230132 |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| His-tagged-c-GPX4U46C | This paper | N/A |
| His-tagged-c-GPX4U46C_R152H | This paper | N/A |
| His-tagged-c-GPX4U46C_C66S | This paper | N/A |
| His-tagged-c-GPX4AllCys(-)-A46C | This paper | N/A |
| 15N-labeled His-tagged-c-GPX4U46C | This paper | N/A |
| 13C, 15N-labeled His-tagged-c-GPX4U46C | This paper | N/A |
| (1S,3R)-RSL3 | Yang et al.16 | N/A |
| (1S,3R)-RSL3-minus-Cl | This paper | N/A |
| ML162 | Aobious | Cat# AOB1514 |
| FIN56 | Gaschler et al.27 | N/A |
| FINO2 | Gaschler et al.27 | N/A |
| Fer-1 | Gaschler et al.27 | N/A |
| IKE | Zhang et al.30 | N/A |
| CDS9 | Sigma-Aldrich | Cat# CDS006509 |
| TMT10 | Millipore Sigma | Cat# TMT00610 |
| MAC-5576 | Maybridge | Cat# MAC-5576 |
| LOC1886 | This paper | N/A |
| Critical commercial assays | ||
| Site-directed mutagenesis kit QuickChange II | Agilent | Cat# 200521 |
| CellTiter-Glo luminescent cell viability | Promega | Cat# G7573 |
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat# 23225 |
| Deposited data | ||
| GPX4-U46C crystal structure | Liu et al.32 | PDB ID: 7L8K |
| GPX4-U46C-R152H crystal structure | Liu et al.32 | PDB ID: 7L8L |
| GPX4-RSL3 crystal structure | This paper | PDB ID: 7U4N |
| GPX4-ML162 crystal structure | This paper | PDB ID: 7U4K |
| GPX4-CDS9 crystal structure | This paper | PDB ID: 7U4I |
| GPX4-TMT10 crystal structure | This paper | PDB ID: 7U4J |
| GPX4-MAC-5576 crystal structure | This paper | PDB ID: 7U4L |
| GPX4-LOC1886 crystal structure | This paper | PDB ID: 7U4M |
| Backbone NMR Chemical Shift Assignments of GPX4 | This paper | BMRB entry 51659 |
| Proteomics data of GPX4 treated with inhibitors | This paper | MassIVE code: MSV000090526 |
| Experimental models: Cell lines | ||
| HT-1080 | ATCC | Cat# CCL-121 |
| HT-1080 OE GFP-tagged-cyto-GPX4WT | This paper | N/A |
| HT-1080 OE GFP-tagged-cyto-GPX4C66S | This paper | N/A |
| HT-1080 OE GFP-tagged-cyto-GPX4C10S-C66S | This paper | N/A |
| HT-1080 OE GFP-tagged-cyto-GPX4U46C | This paper | N/A |
| HT-1080 OE GFP-tagged-cyto-GPX4U46C-C66S | This paper | N/A |
| HT-1080 OE tag-free-cyto-GPX4WT | This paper | N/A |
| HT-1080 OE tag-free-cyto-GPX4C66S | This paper | N/A |
| G401 | ATCC | Cat# CRL-1441 |
| G401 OE GFP-tagged-GPX4WT | Yang et al.16 | N/A |
| G401 OE GFP-tagged-GPX4allCys(-)-A10C | Yang et al.16 | N/A |
| G401 OE GFP-tagged-GPX4allCys(-)-A46C | Yang et al.16 | N/A |
| G401 OE GFP-tagged-GPX4allCys(-)-A46U | Yang et al.16 | N/A |
| G401 OE GFP-tagged-GPX4allCys(-)-A66C | Yang et al.16 | N/A |
| G401 OE GFP-tagged-GPX4allCys(-)-A107C | Yang et al.16 | N/A |
| G401 OE GFP-tagged-GPX4allCys(-)-A148C | Yang et al.16 | N/A |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| R152H mutagenesis primers Foward: CTGCGTGGTGAAGCACTACGGACCCATGG | This paper | N/A |
| R152H mutagenesis primers Reverse: CCATGGGTCCGTAGTGCTTCACCACGCAG | This paper | N/A |
| C66S mutagenesis primers Foward: CCCGATACGCTGAGAGTGGTTTGCGGATC | This paper | N/A |
| C66S mutagenesis primers Reverse: GATCCGCAAACCACTCTCAGCGTATCGGG | This paper | N/A |
| C10S mutagenesis primers Foward: GGAGCGCGCACTGCGCCAGTCG | This paper | N/A |
| C10S mutagenesis primers Reverse: GGAGCGCGCACTGCGCCAGTCG | This paper | N/A |
| Recombinant DNA | ||
| pET-15b-His-tagged-c-GPX4U46C | Yang et al.16 | N/A |
| pET-15b-His-tagged-c-GPX4U46C−R152H | This paper | N/A |
| pET-15b-His-tagged-c-GPX4U46C_C66S | This paper | N/A |
| pET-15b-His-tagged-c-GPX4AllCys(-)-A46C | Yang et al.16 | N/A |
| pBabe-puro GFP-tagged-cyto-GPX4WT | Yang et al.8 | N/A |
| pBabe-puro tag-free-cyto-GPX4WT | Yang et al.8 | N/A |
| pBabe-puro GFP-tagged-cyto-GPX4U46C | Yang et al.8 | N/A |
| pBabe-puro GFP-tagged-cyto-GPX4C66S | This paper | N/A |
| pBabe-puro tag-free-cyto-GPX4C66S | This paper | N/A |
| pBabe-puro GFP-tagged-cyto-GPX4U46C-C66S | This paper | N/A |
| pBabe-puro GFP-tagged-cyto-GPX4C10S-C66S | This paper | N/A |
| Software and algorithms | ||
| Maestro, v2020-3 | Schrodinger | https://www.schrodinger.com/products/maestro |
| PyMOL, v2.5.0 | Schrodinger | https://pymol.org/2/ |
| GraphPad Prism 9 | GraphPad Software | https://www.graphpad.com/ |
| MOLREP, v11.0 | Vagin and Teplyakov37 | https://www.ccp4.ac.uk/html/molrep.html |
| XtalView 4.0 | McRee38 | https://www.sdsc.edu/CCMS/Packages/XTALVIEW/xtalview.html |
| Coot, 0.9.1 | Emsley et al.39 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix, 1.14-3260 | Adams et al.40 | https://phenix-online.org/download |
| ImageJ, v1.51 | Schneider et al. 42 | https://imagej.nih.gov/ij/download.html |
| Protein Thermal Shift, v1.4 | Thermo Fisher | https://www.thermofisher.com/order/catalog/product/4466038 |
| CcpNmr, v3 | Skinner et al.47 | https://ccpn.ac.uk/software/downloads/ |
| Thermo Xcalibur, v4.5.445.18 | Thermo Fisher | https://www.thermofisher.com/order/catalog/product/OPTON-30965 |
| Other | ||