

Abstract
Proteolysis Targeting Chimeras (PROTACs) are attractive therapeutic modalities for degrading disease-causing proteins. While many PROTACs have been developed for numerous protein targets, current small-molecule PROTAC approaches cannot target undruggable proteins that do not have small-molecule binders. Here, we present a novel PROTAC approach, termed bridged PROTAC, which utilizes a small-molecule binder of the target protein’s binding partner to recruit the protein complex into close proximity with an E3 ubiquitin ligase to target undruggable proteins. Applying this bridged PROTAC strategy, we discovered MS28, the first-in-class degrader of cyclin D1, which lacks a small-molecule binder. MS28 effectively degrades cyclin D1, with faster degradation kinetics and superior degradation efficiency than CDK4/6, through recruiting the CDK4/6-cyclin D1 complex to the von Hippel–Lindau E3 ligase. MS28 also suppressed the proliferation of cancer cells more effectively than CDK4/6 inhibitors and degraders. Altogether, the bridged PROTAC strategy could provide a generalizable platform for targeting undruggable proteins.
Graphical Abstract

1. INTRODUCTION
Targeted protein degradation using proteolysis targeting chimeras (PROTACs) is a promising therapeutic strategy for the treatment of diseases such as cancer that are linked to aberrant high levels of disease-causing proteins. PROTACs are heterobifunctional small molecules with an E3 ubiquitin ligase binding moiety linked to a protein of interest (POI) binding moiety. The PROTAC-induced proximity leads to selective polyubiquitination of the POI and its subsequent degradation by the ubiquitin–proteasome system (UPS). Since PROTACs do not require high-affinity target binding nor binding to an active site, they have the potential to target disease-related proteins that are intractable to small-molecule inhibitors.1–3 Indeed, PROTACs have been successfully applied on difficult-to-target proteins, such as the STAT3 transcription factor.4 Additionally, alternative PROTAC strategies5–12 have been developed for proteins that lack small-molecule binders, such as TF-PROTACs,5–7 that target undruggable transcription factors (TFs) and RNA-PROTACs8 that target RNA-binding proteins. Despite these efforts, there still remains a large portion of the human proteome that is undruggable.13–15 In particular, although there are as many as 700 identified cancer-related protein targets, only approximately 40 of them have approved treatments,16 whereas the remaining targets, including ones commonly altered in cancer, do not.14
Cyclin D1 (encoded by CCND1) is an important cell cycle regulator that activates cyclin-dependent kinase 4/6 (CDK4/6).17,18 Upon activation, CDK4/6 phosphorylates the retinoblastoma (Rb) protein, thereby releasing its repression on the transcription factor E2F, which is then free to induce the expression of proteins involved in G1 to S phase transition.19 Due to its integral role in mitogen-dependent cell cycle control, cyclin D1 is frequently amplified and overexpressed in many cancer types, including esophageal, breast, and lung cancer (Figure S1). Cyclin D1 is essential in both tumor initiation and maintenance. Cyclin D1 ablation protected mice from developing breast cancer induced by the neu and ras oncogene,20 and inducible deletion of cyclin D1 in adult mice with ErbB2-driven breast cancer halted tumor growth.21 Despite the importance of cyclin D1 in cancer, it is largely dispensable for normal physiology as mice deficient in CCND1 were viable with minor and restricted developmental defects.21,22 Taken together, cyclin D1 is a promising therapeutic target for cancer. Indeed, the Cancer Dependency Map (DepMap) project ranked cyclin D1 as a top cancer drug target among 627 priority targets.14
However, cyclin D1 is undruggable, and there are no reported small-molecule cyclin D1 binders. Due to the lack of small-molecule binders, cyclin D1 cannot be targeted by the conventional PROTAC strategy. We hypothesized that cyclin D1 and other traditionally undruggable proteins could be targeted by a novel PROTAC approach, termed bridged PROTAC, which utilizes a small-molecule binder that recruits a bridge protein and its binding partner (POI), thus bringing the protein complex into proximity with an E3 ligase and inducing preferential degradation of the POI over the bridge protein (Figure 1A). While protein complex degraders have been reported23–29 such as EZH2 and EED PROTACs that also degraded other PRC2 (polycomb repressive complex 2) components23–26,29,30 and a hydrophobic tag-based CDK9 degrader that induced the degradation of both CDK9 and cyclin T1,31 none of these degraders preferentially degraded partner proteins over the protein that binds the degraders directly. In fact, the degradation of these partner proteins is likely the consequence of the initial degradation of the protein that directly binds the PROTAC and subsequent destabilization of the protein complex. On the other hand, we thought it is possible to preferentially degrade the partner protein (POI) over the bridge protein (Figure 1A) by utilizing appropriate linkers and/or E3 ligase ligands. To test this hypothesis, we selected cyclin D1 as the undruggable target because cyclin D1 forms a complex with CDK4/6,32 and highly potent and selective small-molecule inhibitors/binders of CDK4/6 have been developed.33–36
Figure 1.
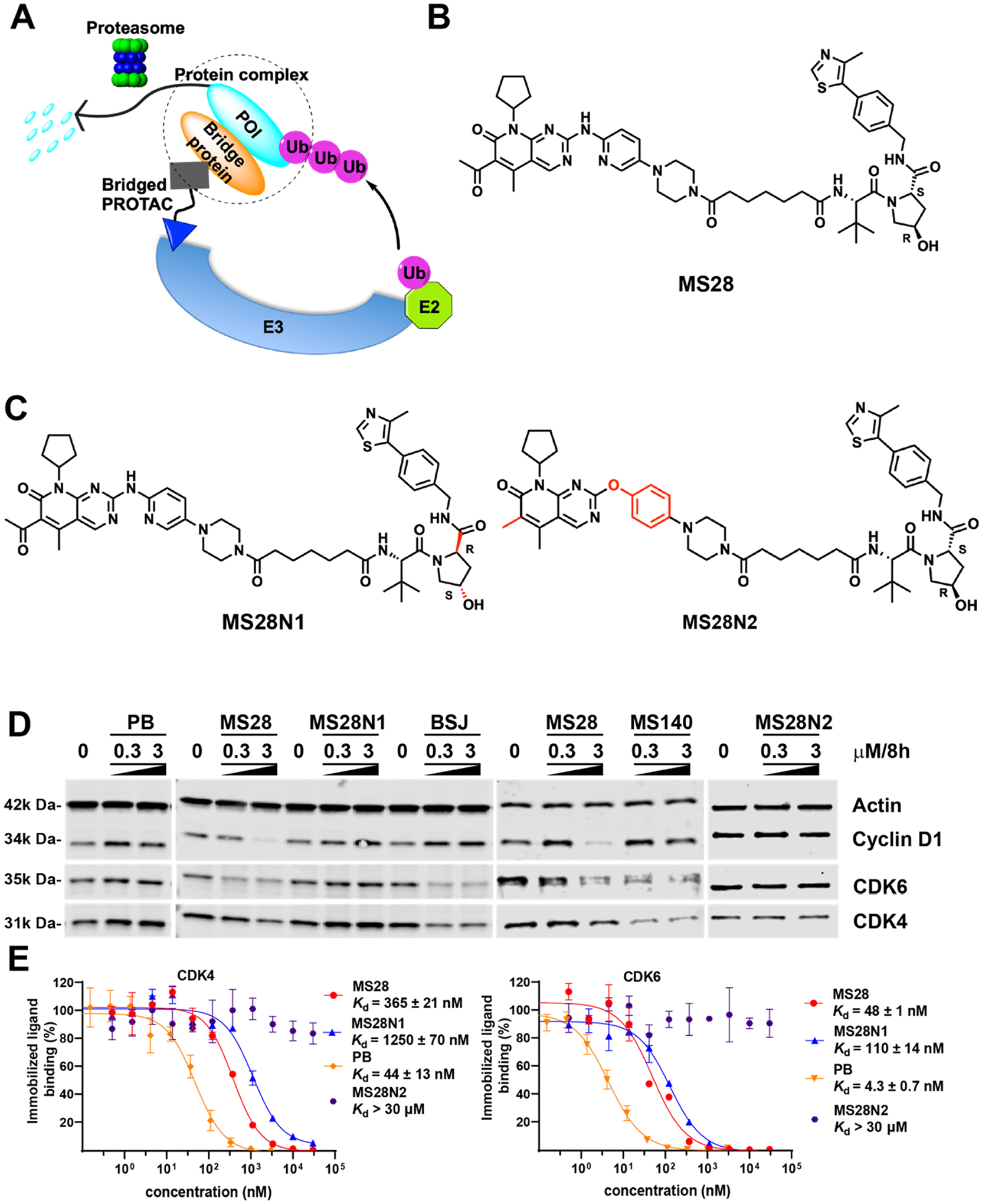
Discovery of MS28, the first cyclin D1 degrader. (A) Schematic representation of bridged PROTAC. The bridged PROTAC, which binds a bridge protein but not its binding partner (POI), recruits the protein complex into proximity with an E3 ligase, resulting in preferential polyubiquitination and subsequent degradation of the POI by the proteasome over the bridge protein. (B) Chemical structure of MS28. (C) Chemical structures of MS28N1 and MS28N2. (D) Western blot (WB) results of MS28, MS28N1, BSJ-03–204 (BSJ), MS140, and PB on degrading cyclin D1 and CDK4/6. Calu-1 cells were treated with the indicated compound at the indicated concentrations for 8 h. Results shown are representative of at least two independent experiments. (E) Binding affinities of MS28, MS28N1, MS28N2, and PB to CDK4 and CDK6 in biochemical assays. Results shown are the mean ± SD from duplicate experiments.
In this proof-of-concept study, we utilize this novel bridged PROTAC approach to degrade cyclin D1, an undruggable protein, by engaging the cyclin D1-CDK4/6 complex with the von Hippel–Lindau (VHL) E3 ligase through the use of a direct CDK4/6 small-molecule binder linked to a VHL ligand. The discovery and functional characterization of our lead bridged PROTAC, MS28, and its negative controls have demonstrated for the first time that, even without direct binding, PROTACs can effectively degrade an undruggable target by recruiting its partner protein, thereby potentially expanding the scope of the targetable human proteome.
2. RESULTS AND DISCUSSION
2.1. Discovery of the First Cyclin D1 Degrader MS28.
We first designed and synthesized a set of putative PROTACs based on the crystal structure (PDB ID: 5L2I)37 of CDK6 in complex with the FDA-approved CDK4/6 inhibitor Palbociclib33,34 (PB). We linked the solvent-exposed piperazine moiety (Figure S2A) to VHL-1, a well-known ligand of the E3 ligase VHL,38 via multiple linkers (Figure S2B). We then evaluated the effect of these compounds on reducing the cyclin D1 protein level in Calu-1 cells, a cyclin D1-dependent and -overexpressing non-small cell lung cancer (NSCLC) cell line (DepMap 22Q2). While most of these compounds did not degrade cyclin D1, several compounds, including MS28 (compound 5, Figure 1B), effectively degraded cyclin D1 in Calu-1 cells (Figure S2C,D). We next developed two negative controls for MS28, MS28N1, and MS28N2 (Figure 1C). MS28N1 contains the identical CDK4/6 binder and linker to MS28 to maintain a similar binding affinity to CDK4/6, but a diastereoisomer of VHL-1 that abrogates its binding to VHL.39 MS28N2 contains the identical VHL binder and linker to MS28 to maintain similar binding affinity to VHL, but a modified CDK4/6 binder that was designed to disrupt its binding to CDK4/6.
In addition to cyclin D1, MS28 selectively degraded CDK6 over CDK4 in Calu-1 cells (Figures 1D, S3). Specifically, MS28 at 0.3 μM reduced the CDK6 protein level close to 50%, while it had no effect on CDK4 (Figures 1D, S3), suggesting that MS28 may prefer the CDK6-cyclin D1 complex over CDK4-cyclin D1 complex. Interestingly, we found that two published cereblon (CRBN)-recruiting CDK4/6 PROTACs, BSJ-03–204 (BSJ)40 and MS140,41 did not degrade cyclin D1, even though they effectively degraded CDK4/6 (Figures 1D, S3). It was reported that CP-10, another CRBN-recruiting PROTAC that effectively degraded CDK4/6, also did not degrade cyclin D1.42 These results indicate that the cyclin D1 degradation by MS28 is not due to cyclin D1 destabilization from CDK4/6 degradation and dissociation of the cyclin D1-CDK4/6 complex. We next assessed the binding affinities of MS28, MS28N1, MS28N2, and PB to CDK4/6 in competitive binding assays. Compared with PB, MS28 and MS28N1 maintained sufficient binding affinities to CDK4/6, albeit they displayed 10- to 30-fold weaker affinities (Figure 1E). As expected, MS28N2 did not bind CDK4/6 at up to 30 μM. In addition, MS28 and MS28N1 displayed a higher binding affinity to CDK6 than CDK4 (Figure 1E), which could explain the selective degradation of CDK6 over CDK4 by MS28. Next, we evaluated the effect of MS28N1 and MS28N2 on degrading cyclin D1. As expected, none of the negative controls, nor PB, degraded cyclin D1 or CDK4/6 (Figure 1D), suggesting that the cyclin D1 degradation by MS28 is dependent on VHL and CDK4/6 binding.
2.2. MS28 Preferentially Degrades Cyclin D1 over CDK4/6.
We next assessed the time course of the cyclin D1 and CDK4/6 degradation induced by MS28. MS28 degraded cyclin D1 by approximately 50% as early as 1 h, whereas CDK4/6 degradation was not observed until 4 h of treatment (Figure 2A). At almost all treatment time points, MS28 reduced the protein level of cyclin D1 to a greater extent than that of CDK4/6. Together with the observation that BSJ and MS140 degraded CDK4/6 but not cyclin D1 (Figures 1D, S3), this time course study further supports that cyclin D1 degradation by MS28 is not a consequence of CDK4/6 degradation. Rather, since cyclin D1 degradation occurred before CDK4/6, MS28 likely induces a quaternary complex formation, leading to cyclin D1 being ubiquitinated and degraded first. MS28 also degraded cyclin D1 in a concentration-dependent manner in Calu-1 cells with a DC50 of 950 ± 66 nM and a Dmax of 90 ± 1% (Figure 2B,C) and displayed modest degradation effects on CDK4/6 with DC50 > 10 μM and Dmax < 50% at the 8 h time point (Figure S4). Since cyclin D1 is also a target of the downstream Rb-E2F pathway,43 we next determined the effect of MS28 on the CCND1 and CDK4/6 mRNA levels using RT-qPCR studies. MS28, as well as PB, did not change the mRNA levels of CCND1, CDK4, and CDK6 (Figure 2D). Thus, the reduction in cyclin D1 occurs at the protein level and is not due to changes in transcription.
Figure 2.
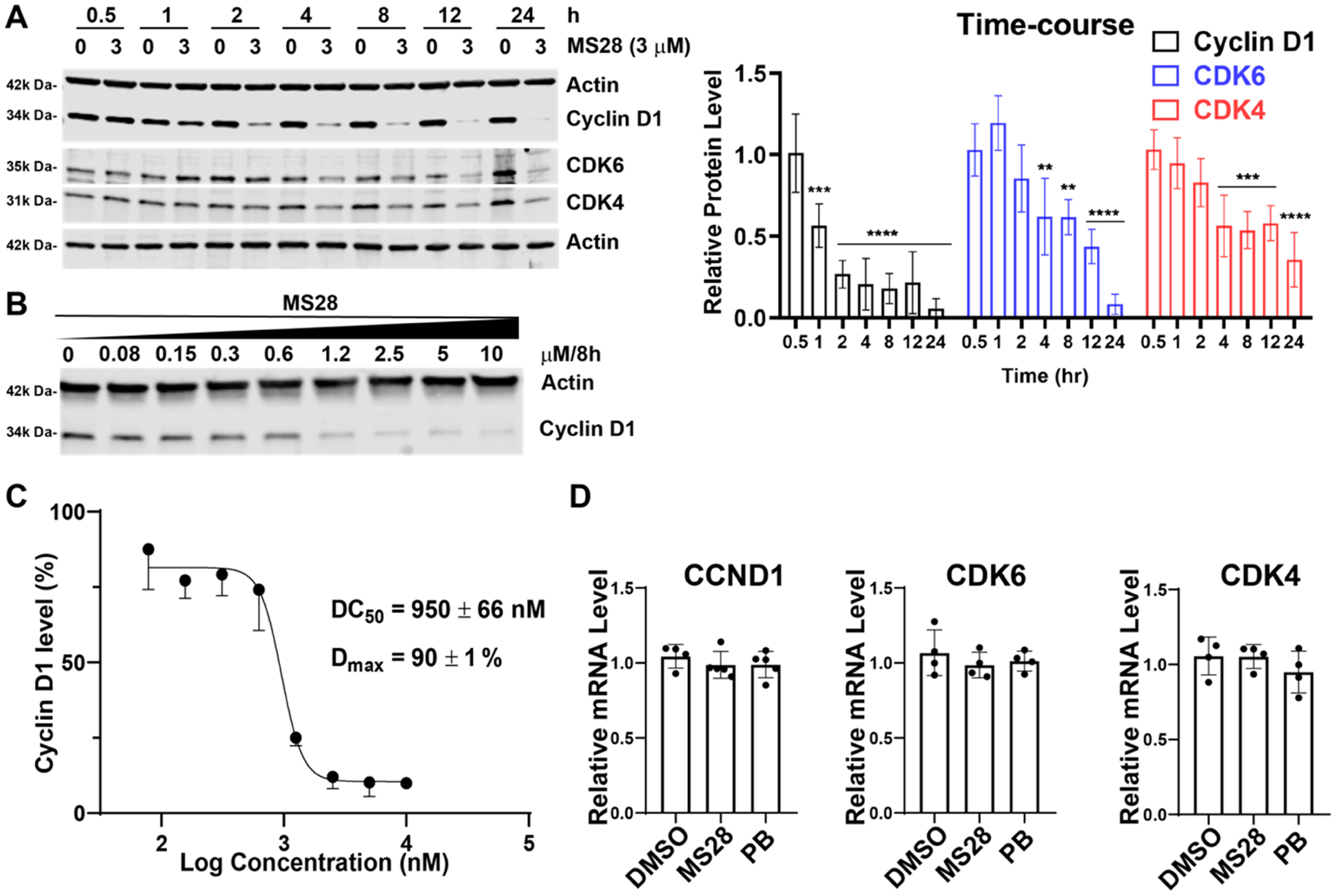
MS28 preferentially degrades cyclin D1 over CDK4/6 in Calu-1 cells. (A) Left, MS28 degrades cyclin D1 first and CDK4/6 subsequently. Calu-1 cells were treated with DMSO or 3 μM of MS28 for the indicated time. Results shown are representative of three independent experiments. Right, quantification of relative cyclin D1, CDK4, and CDK6 abundance at each time point, following 3 μM MS28 treatment (four technical repeats from three biological repeats). P-values were calculated for each protein relative to its abundance at 0.5 h. ****P < 0.0001, ***P < 0.001, and **P < 0.01. The line above the bars indicates that all of the encompassed bars share the same p-value above the line. (B) MS28 concentration-dependently degrades cyclin D1. Calu-1 cells were treated with MS28 at the indicated concentration for 8 h. Results shown are representative of two independent experiments. (C) DC50 and Dmax values of MS28 in Calu-1 cells (calculated from the WB data in panel B and biological repeat). (D) MS28 did not change the mRNA levels of CCND1, CDK4, and CDK6 in RT-qPCR studies. Calu-1 cells were treated with DMSO, 3 μM MS28, or 3 μM PB for 4 h. mRNA levels are relative to DMSO control. Results are representative of two biological repeats.
2.3. Cyclin D1 Degradation Induced by MS28 is Dependent on VHL, CDK6, and UPS.
For MS28 to be a bridged PROTAC, it must bind to CDK4/6 directly, thus recruiting the cyclin D1-CDK4/6 complex and not cyclin D1. As described above, we have already confirmed that MS28 bound CDK4/6 with high affinity (Figure 1E). We next assessed the binding of MS28 to cyclin D1 using isothermal titration calorimetry (ITC) and confirmed that MS28 did not bind cyclin D1, while the positive control, a 16-mer peptide based on the CDK4/6 binding sequence to cyclin D1,44 did bind cyclin D1 (Figure S5). Therefore, cyclin D1 degradation by MS28 is not due to direct interaction between cyclin D1 and MS28. Instead, it occurs through the binding of the CDK4/6-cyclin D1 complex.
To further demonstrate that the cyclin D1 degradation mediated by MS28 occurs through the recruitment of VHL and CDK6, we ablated VHL and CDK6, which are required for the formation of the cyclin D1-CDK6-MS28-VHL quaternary complex. Specifically, we generated VHL knockout (KO) via CRISPR-Cas9 (Figure 3A) and CDK6 knockdown (KD) via siRNA (Figure 3B) in Calu-1 cells. Upon VHL or CDK6 depletion, the ability of MS28 to degrade cyclin D1 was substantially weakened (cyclin D1 degradation was rescued by >50% in both cases), suggesting that MS28 degrades cyclin D1 by recruiting the CDK6-cyclin D1 complex to VHL. Since MS28 degraded CDK6 more effectively than CDK4 (Figure 2A) and displayed higher binding affinity to CDK6 than CDK4 (Figure 1E), it is not surprising that CDK6 KD alone effectively rescued MS28-induced cyclin D1 degradation. To demonstrate the formation of the cyclin D1-CDK6-MS28-VHL quaternary complex, an in vitro pull-down experiment was conducted using recombinant CDK6-cyclin D1 and VHL-ELOC-ELOB (VCB) protein complexes. Cyclin D1 and VHL were coeluted, along with CDK6, in the presence of MS28 (Figure 3C), thereby supporting that MS28 induces the interaction of the cyclin D1-CDK6 complex with VHL. Finally, to confirm that the cyclin D1 degradation by MS28 occurs through the UPS, we performed rescue studies by pretreating Calu-1 cells with the proteasome inhibitor MG13245 (Figure 3D) or the neddylation inhibitor MLN492446 (Figure 3E). Inhibition of the UPS successfully rescued MS28-induced cyclin D1 degradation. Taken together, these results indicate that MS28 degraded cyclin D1 in a VHL-, CDK6- and UPS-dependent manner.
Figure 3.

MS28 effectively degrades cyclin D1 in a VHL-, CDK6-, and UPS-dependent manner. (A) Left, VHL KO rescues the MS28-induced degradation of cyclin D1 and CDK4/6. Calu-1 cells were transduced with lentivirus containing VHL-targeting sgRNA or an empty vector. Antibiotic-selected cells were checked for VHL expression (Top: WB results confirm CRISPR-mediated VHL KO in Calu-1 cells), followed by treatment with MS28 for 8 h. Right, quantification of cyclin D1 and CDK4/6 abundance in the indicated experimental groups. P-values for each protein were calculated between the mock and VHL KO groups (indicated by bar) within each concentration. (B) Left, CDK6 KD via siRNA rescues the MS28-induced degradation of cyclin D1. Calu-1 cells were transfected with CDK6-targeting siRNA for 2 days, followed by treatment with MS28 for 8 h. Right, quantification of cyclin D1 abundance in the indicated experimental groups. P-values were calculated between the control and CDK6 KD group within each MS28 concentration. (C) VHL coelutes with cyclin D1-CDK6 in the presence of MS28. His-tagged CDK6 was immobilized on cobalt agarose resin and incubated overnight along with cyclin D1. The VCB complex was added the next day with either DMSO or MS28. Pretreatment with MG132 (D) or MLN4924 (E) rescues the MS28-induced degradation of cyclin D1 and CDK4/6. Quantification of cyclin D1 and CDK4/6 abundance are listed aside. For each protein, p-values were calculated between groups indicated by the lines above the bars. Calu-1 cells were pretreated with MG132 or MLN4924 for 1 h, followed by treatment with MS28 for 8 h. WB results shown in panels A–E are representative of at least two independent experiments. ****P < 0.0001, ***P < 0.001, **P < 0.01, and *P < 0.05.
2.4. Selectivity of MS28.
Next, using WB analysis, we confirmed that, in addition to cyclin D1, CDK6, and CDK4 (to a lesser extent), MS28 also degraded cyclin D3 and slightly reduced the cyclin A2 protein level (Figure 4A). This reduction in cyclin A2 is likely due to the downstream transcriptional effect of CDK4/6 inhibition (see below). We also confirmed that MS28 did not degrade cyclin D2 or other major cyclins and CDKs, such as cyclin B1 and CDK2. The negative control MS28N1 did not degrade any of these cyclins and CDKs in Calu-1 cells. We also evaluated the selectivity of MS28 against a panel of 58 kinases. MS28 at 1 μM significantly inhibited CDK6 but not the other 57 kinases (Figure 4B and Table S1). In an unbiased mass spectrometry (MS)-based global proteomic profiling study, we found that out of ~7500 proteins detected, cyclin D1 and CDK4/6 are among a very small portion of the proteins showing a significant decrease in protein levels in Calu-1 cells treated with MS28 versus DMSO or MS28N1 (Figure S6, Table S2). In addition, most of the other proteins shown to be significantly downregulated by MS28 have cell cycle-related functions, thus they are likely downregulated due to the degradation of cyclin D1 and CDK4/6, the key regulators of cell cycle progression. Consistent with our WB result (Figure 4A), cyclin A2, a downstream target of the cyclin D1-CDK4/6-RB-E2F pathway, was also downregulated in the proteomics study. Interestingly, cyclin D3 was also degraded, while cyclin D2 was not detected in the MS study due to its low expression level in Calu-1 cells. Collectively, these results support that MS28 is a selective cyclin D1/3 and CDK4/6 degrader.
Figure 4.
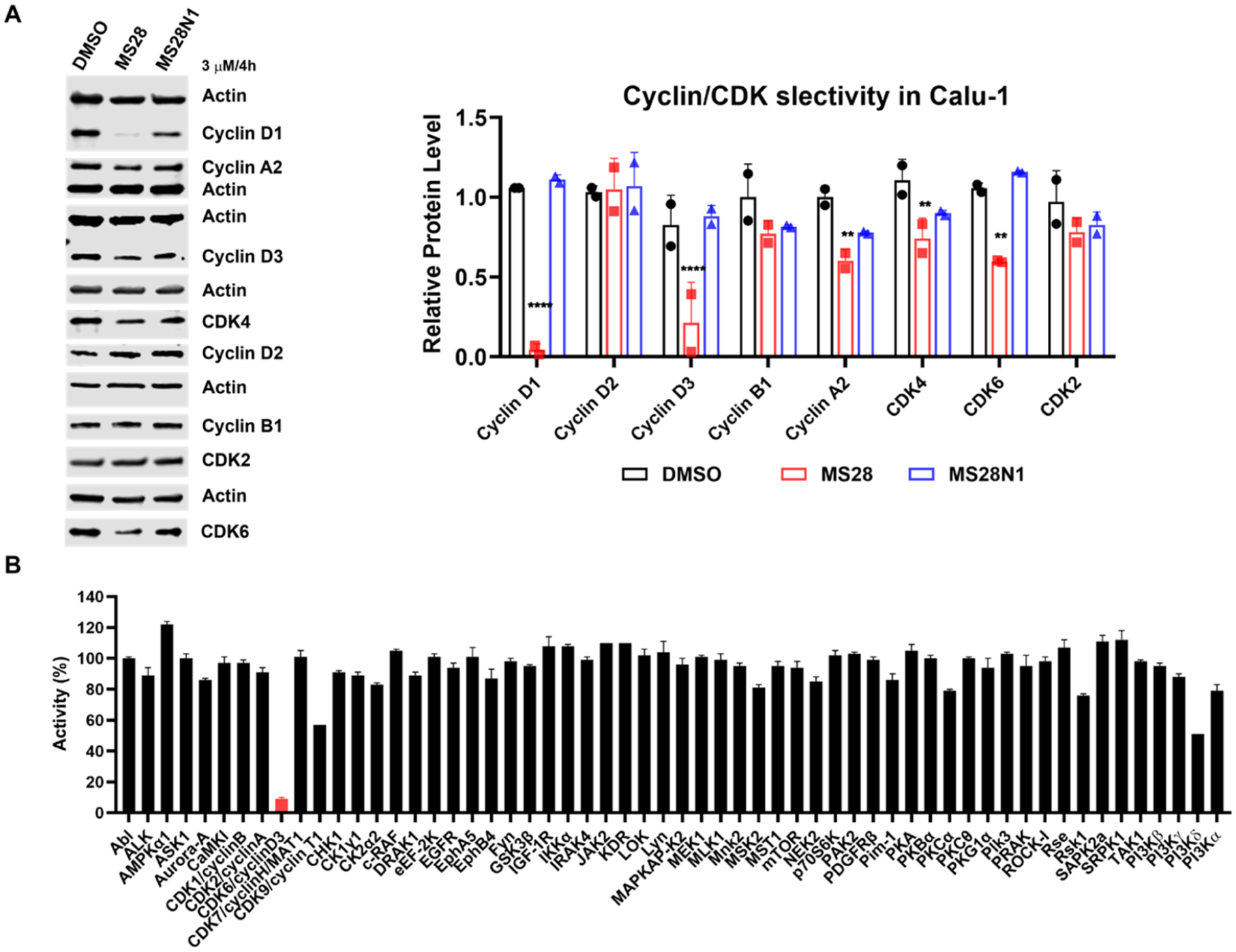
MS28 is selective for cyclin D1/D3 and CDK4/6. (A) Left, MS28, but not MS28N1, degrades cyclin D1/3 and CDK4/6, and reduces the cyclin A2 level, but not cyclin D2/B1 and CDK2 in Calu-1 cells (4 h-treatment at 3 μM). WB data shown are representative of two biological repeats. Right, quantification of the relative abundance of cyclins and CDKs analyzed in the WB upon DMSO, MS28, or MS28N1 treatment. P-values were calculated relative to the DMSO control for each protein. ****P < 0.0001, ***P < 0.001, and **P < 0.01. (B) MS28 is selective for CDK6 over a panel of 57 other kinases at 1 μM concentration. Data are the means ± SD from duplicate experiments.
2.5. MS28 Effectively Suppresses the Growth of NSCLC Cells.
Since cyclin D1 is well-known for its regulatory role on CDK4/6 and the Rb-E2F pathway, we next assessed the effect of MS28 on the downstream E2F target genes using RT-qPCR. MS28, as well as PB, significantly reduced the mRNA levels of E2F target genes at 8 h of treatment in Calu-1 cells (Figure 5A). This result also suggests that the decrease in the cyclin A2 protein level observed previously (Figure 4A) is a consequence of the CCNA2 transcription downregulation. Importantly, MS28 (GI50 = 1.0 ± 0.32 μM) also more effectively suppressed the proliferation of Calu-1 cells than PB and the CDK4/6 degrader, BSJ, which had negligible effects at up to 10 μM (Figure 5B). The effect of MS28 phenocopied that of CCND1 KO in Calu-1 cells (Figure S7). Furthermore, we evaluated the effect of MS28, MS28N1, BSJ, and PB on the clonogenicity of Calu-1 cells in a soft agar colony formation assay. Following 20 days of treatment with 0.3 μM of each compound, MS28 inhibited the clonogenicity of Calu-1 cells more profoundly than MS28N1, BSJ, or PB, as all of the colonies in the MS28 treatment condition were less than 100 μm in diameter (Figures 5C and S8).
Figure 5.
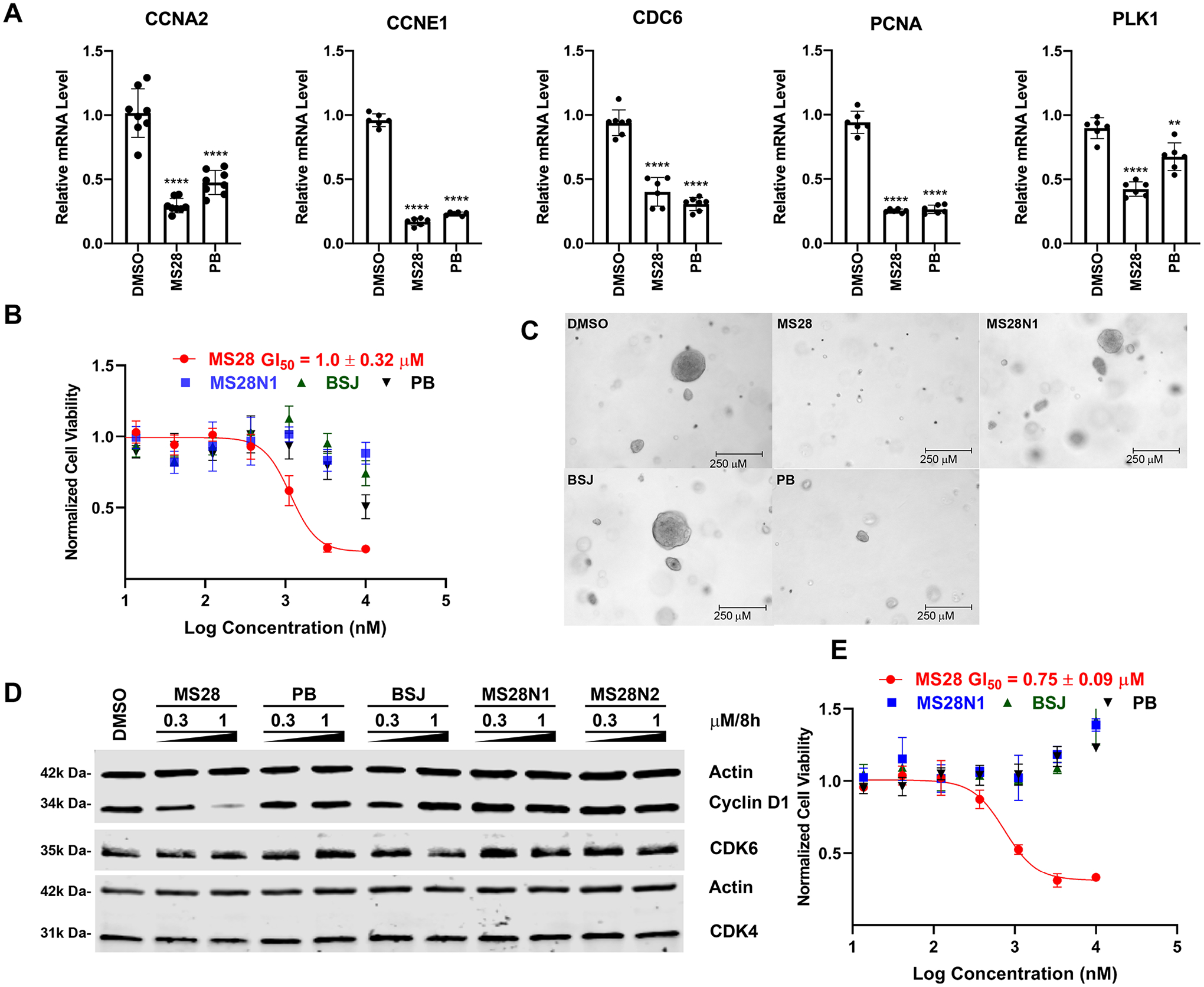
MS28 effectively suppresses the downstream Rb-E2F pathway, proliferation, and tumorigenesis in cancer cells. (A) MS28 and PB significantly reduced mRNA levels of E2F target genes (CCNA2, CCNE1, CDC6, PCNA, and PLK1) in RT-qPCR studies. Calu-1 cells were treated with DMSO, 3 μM of MS28, or PB for 8 h. P-values were calculated in comparison to DMSO from two biological repeats. ****P < 0.0001, ***P < 0.001, **P < 0.01, and *P < 0.05. (B) MS28 inhibits cell growth much more effectively than PB, BSJ, and MS28N1 in Calu-1 cells (treated with the indicated compound for 5 days). Data shown are the mean values ± SD from two biological repeats (each with three technical repeats). (C) MS28 suppresses clonogenicity of Calu-1 cells in a soft agar assay more effectively than PB, BSJ, and MS28N1. Images were taken at the end of the 20-day treatment. Each treatment group (at 0.3 μM) is representative of three independent experiments, each with at least five technical repeats. (D) MS28, not MS28N1, BSJ, or PB, effectively degrades cyclin D1 in NCI-H2110 cells (treated with the indicated compound at the indicated concentrations for 8 h). Results are representative of two biological repeats. (E) MS28, but not MS28N1, BSJ or PB, potently inhibits the growth of NCI-H2110 cells (5 days treatment, three biological repeats).
We next explored the degradation effect of MS28 in another NSCLC cell line, NCI-H2110, which is also dependent on CCND1 (Figure S9). MS28 preferentially degraded cyclin D1, while BSJ had no effect on degrading cyclin D1 and CDK4/6 (Figures 5D, S10). As expected, PB, MS28N1 and MS28N2 did not degrade cyclin D1 and CDK4/6. Interestingly, in contrast to the potent degradation of cyclin D1, MS28 slightly reduced the CDK4 level at 1 μM and had no effect on the CDK6 level in NCI-H2110 cells treated for 8 h, suggesting a different degradation kinetic in this cell line. Furthermore, different cellular contexts, such as the abundance of cyclin D1, CDK4/6, and the cyclin D1-CDK4 or cyclin D1-CDK6 binary complexes, may lead to different degradation profiles of MS28. In addition, consistent with results in other tested cell lines, the cyclin D3 protein level can also be effectively reduced by MS28 in NCI-H2110 cells (Figure S11). We also tested the effect of MS28, PB, and BSJ on cell viability in NCI-H2110 cells. Similar to Calu-1 cells, MS28 displayed a superior anti-proliferative effect (GI50 = 750 ± 90 nM) than PB and BSJ, which were ineffective in suppressing NCI-H2110 proliferation even at 10 μM (Figure 5E). We also evaluated MS28 in another cancer type, the BT-549 breast cancer cell line. Similarly, MS28 preferentially degraded cyclin D1 and cyclin D3 over cyclin D2 and CDK2/4/6 (Figure S12). Next, we evaluated the cytotoxicity of MS28 in normal human PNT2 cells. Although MS28 concentration-dependently reduced the cyclin D1 and CDK4/6 protein levels, it did not show significant antiproliferation effect in PNT2 cells (Figure S13), suggesting that degradation of cyclin D1 and CDK4/6 are nontoxic to normal cells. Finally, we evaluated mouse PK properties of MS28. Following a single intraperitoneal (i.p.) injection of MS28 at 30 mg/kg dose, compound concentrations in plasma were maintained above 1 μM up to 12 h (Figure S14). Together, these results indicate that MS28 is more effective than CDK4/6 inhibitors and degraders in suppressing the proliferation and tumorigenesis of cancer cells, safe in normal tissue, and suitable for in vivo efficacy studies.
3. CONCLUSIONS
While PROTACs have been applied successfully in targeting numerous proteins, the current small-molecule-based PROTAC approaches require a small-molecule binder of the POI, which means that undruggable proteins, which lack small-molecule binders, cannot be targeted by these traditional PROTAC approaches. In this study, we present a novel PROTAC strategy, termed bridged PROTAC, to target currently undruggable proteins using a small-molecule binder of the POI’s binding partner. We demonstrated the applicability of this bridged PROTAC strategy to degrade cyclin D1, an undruggable protein, by engaging the cyclin D1-CDK4/6 complex with the VHL E3 ligase through the use of a direct CDK4/6 binder. Our lead cyclin D1 bridged PROTAC, MS28, preferentially degrades cyclin D1 over CDK4/6 in a time-, concentration-, VHL-, CDK6- and UPS-dependent manner. MS28 induces the formation of a cyclin D1-CDK6-MS28-VHL quaternary complex. MS28 also inhibited the proliferation and tumorigenesis in NSCLC cells much more effectively than CDK4/6 inhibitors and degraders, which did not degrade cyclin D1. Overall, the bridged PROTAC strategy has the potential to target currently undruggable proteins such as TFs that lack small-molecule binders but interact with other proteins with well-characterized small-molecule ligands. This potentially generalizable platform could expand the targetable human proteome.
4. EXPERIMENTAL METHODS
4.1. Compound Synthesis.
Synthesis and characterization of MS28, MS28N1, MS28N2, and compounds 1–4, 6–9, as well as the intermediates, are detailed in the Supporting Information.
4.2. Cell Lines and Tissue Culture.
Human NSCLC cell line, Calu-1 (American Tissue Culture Collection [ATCC], HTB-54), was cultured in DMEM base medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% Penicillin/Streptomycin. The NSCLC cell line, NCI-H2110 (ATCC, CRL-5924), the BT-549 (ATCC, HTB-122) breast cancer cell line, and the PNT2 normal prostate cell line (Sigma-Aldrich, cat# 95012613) were cultured in RPMI 1640 base medium supplemented with 10% heat-inactivated FBS and 1% Penicillin/Streptomycin. CCND1 and/or VHL knock-outs (KOs) were generated in Calu-1 and NCI-H2110 cell lines using CRISPR/Cas9-based strategies detailed below, and corresponding KO cell lines were maintained in the same culture condition as their parental cell lines. CDK6 knockdown (KD) Calu-1 cells were generated using Silencer Select siRNA from Thermo Fisher Scientific and were maintained in the same culture condition as the Calu-1 parental cell line during cellular assays.
4.3. Antibodies and Immunoblotting.
Total cell lysate was used for western blots, as previously described.47 The following primary antibodies were used in the study: β-Actin (Cell Signaling Technology [CST], 3700), VHL (CST, 68547), cyclin D1 (CST, 55506), CDK4 (CST, 12790), CDK6 (CST, 13331), CDK2 (CST, 2546), cyclin B1 (CST, 4138), cyclin A2 (CST, 4656T), vinculin (CST, 13901S), cyclin D3 (CST, 2936S), and cyclin D2 (Santa Cruz, sc-56305). Blots were imaged using fluorescence-labeled secondary antibodies on LI-COR Odyssey CLx Imaging Systems.
4.4. CDK4/6 Binding Assays.
CDK4/6 binding affinities were determined with the KINOMEscan assay performed by Eurofins, as described previously.48 Test compounds were prepared as 111× stocks in 100% DMSO. Kd values were determined using an 11concentration 3-fold compound dilution series (top concentration = 30,000 nM for MS28, MS28N1, and MS28N2; 3000 nM for PB), with three DMSO control points in duplicates.
4.5. Isothermal Titration Calorimetry (ITC).
Binding of PSTAIRE peptide (16-mer peptide) (Sigma-Aldrich) to cyclin D1 as a positive control and binding of MS28 to cyclin D1 were analyzed using a MicroCal iTC200 (Malvern, UK) in 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 5% glycerol, 1% DMSO. After an initial 0.4 μL injection, 13 injections from the syringe solution (200 μM of 16-mer peptide or MS28) were titrated into 300 μL of the protein solution (20 μM of Cyclin D1) in the cell, which was stirred at 750 rpm. The data were fitted by single-binding site model using Microcal Origin 7.0 (Malvern). The reported values represent the mean ± SD from two independent measurements.
4.6. Quantitative Reverse Transcriptase Polymerase Chain Reaction (RT-qPCR).
Calu-1 cells were seeded in a 60 mm dish at a density of 1.5 million cells. The next day, cells were treated with DMSO, 3 μM MS28, or 3 μM PB for 4 h or 8 h. Cells were collected at the end of treatment, and RNA was extracted using the Monarch Total RNA Miniprep Kit (New England BioLabs Inc., T2010S) according to the manufacturer’s protocol. Using 1 μg of total RNA, cDNA was generated using the SuperScript III First-Strand Synthesis System kit (Thermo Fisher Scientific, 18080051) according to the manufacturer’s protocol. Gene expression was then measured using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, A25742) on the ViiA 7 Real-Time PCR machine. The mRNA expression for each target gene was first normalized to internal GAPDH and then calculated relative to the DMSO control. Experiments were performed twice in quadruplicate. The human primer sequences used in the RT-qPCR studies are listed in Table 1.
Table 1.
Human Primer Sequences Used in RT-qPCR Studies
| target gene | forward primer | reverse primer |
|---|---|---|
| CCND1 | GCTGCGAAGTGGAAACCATC | CCTCCTTCTGCACACATTTGAA |
| CDK4 | ATGGCTACCTCTCGATATGAGC | CATTGGGGACTCTCACACTCT |
| CDK6 | TGCACAGTGTCACGAACAGA | ACCTCGGAGAAGCTGAAACA |
| CCNA2 | TGGAAAGCAAACAGTAAACAGCC | GGGCATCTTCACGCTCTATTT |
| CDC6 | ACCTATGCAACACTCCCCATT | TGGCTAGTTCTCTTTTGCTAGGA |
| PLK1 | ACTGGCAACCAAAGTCGAATA | CTCGAAACTGTGCCCTTTCT |
| CCNE1 | AAGGAGCGGGACACCATGA | ACGGTCACGTTTGCCTTCC |
| PCNA | GCGTGAACCTCACCAGTATGT | TCTTCGGCCCTTAGTGTAATGAT |
| GAPDH | CTGGGCTACACTGAGCACC | AAGTGGTCGTTGAGGGCAATG |
4.7. CRISPR/Cas9-Mediated Knockout (KO) (VHL + Cyclin D1).
The sgRNA sequences used in CRISPR/Cas9-mediated KO studies are listed in Table 2.
Table 2.
sgRNA Sequences Used in CRISPR/Cas9-Mediated KO Studies
| ID | target gene | sequence |
|---|---|---|
| VHL sgRNA | VHL | 5′-GCCGTCGAAGTTGAGCCATA-3′ |
| CCND1 sgRNA 1 | CCND1 | 5′-TTTTCACGGGCTCCAGCGAC-3′ |
| CCND1 sgRNA 2 | CCND1 | 5′-ATGCCAACCTCCTCAACGAC-3′ |
The sgRNA sequence targeting human VHL gene was chosen from the CRISPR sgRNA database from GenScript (https://www.genscript.com/gRNA-database.html). The sgRNA sequences targeting the human CCND1 gene were selected from predesigned Alt-R CRISPR-Cas9 guide RNAs from Integrated DNA Technologies (IDT; https://www.idtdna.com/site/order/designtool/index/CRISPR_PREDESIGN). All sgRNA sequences cloned into the pLentiCRISPR v2 vector (Addgene #52961) were purchased from GenScript. sgRNA-containing pLentiCRISPR v2 plasmid or the empty pLentiCRISPR v2 plasmid was transfected into HEK293T cells along with the lentiviral packaging plasmid Pax2 (Addgene #12260) and the envelope plasmid VSVG (Addgene #8454) using Lipofectamine 3000 (Thermo Fisher Scientific). Media were collected on day 2 and day 3 and filtered using Corning 0.45 μm filter tube. The lentivirus from the filtered media was condensed using Takara Lenti-X Concentrator (Takara, Cat. # 631232) and titered using Lenti-X GoStix Plus (Takara, Cat. # 631280). Target cell lines, Calu-1 and NCI-H2110, were transduced with lentiviruses in the presence of Polybrene for 4 days and selected using 2 μg/mL of puromycin. Once all cells in the control well (not containing lentiviruses) died of puromycin treatment, lentiviral-transduced cells were collected to check for cyclin D1 and VHL levels. CCND1 KO and VHL KO were examined by immunoblotting with anti-Cyclin D1 antibody (Cell Signaling Technology, #55506) and anti-VHL antibody (Cell Signaling Technology, #68547), respectively. Once cells showed efficient target gene KO, the cells were used in the following experiments.
4.8. siRNA-Mediated CDK6 Knockdown (KD).
Validated Silencer Select predesigned siRNA targeting CDK6 and a Silencer Cy3-labeled negative control siRNA (Thermo Fisher Scientific, AM4621) were purchased from Thermo Fisher Scientific. Calu-1 cells were seeded at a density of 500,000 cells/well in a 6-well plate and transfected with siRNA targeting CDK6 using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific) for 2 days, according to the manufacturer’s recommendations. The KD efficiency was analyzed using Western blot analysis. Once target gene KD achieved more than 80% on the protein level compared to nontransfected parental cells, cells were collected, split into 3 fractions, seeded onto 12-well plates, and treated with DMSO or MS28 (1 or 3 μM) for 8 h.
4.9. Recombinant Proteins.
Recombinant human CCND1 protein, full length (1–295aa), containing an N-terminal GST-tag, and recombinant human CDK6 protein, full length (1–326aa), containing an N-terminal His-tag were purchased from BPS Bioscience (San Diego, CA). Untagged recombinant human ELOB/ELOC/VHL (VCB) complex was purchased from Bio-Techne R&D Systems (Minneapolis, MN). These proteins were used in the pull-down studies. For the ITC experiment, recombinant human CCND1 protein (1–295aa) fused to N-terminal His-tag was purchased from Creative Biomart.
4.10. Recombinant His-CDK6 Pull-Down Experiments.
Recombinant His-tagged CDK6 and GST-tagged cyclin D1 complex were purchased from BPS Bioscience (cat. # 40097), and recombinant human VCB complex was purchased from R&D Systems (cat. # E3–600). The Pierce His Protein Interaction Pull-Down Kit (Thermo Fisher Scientific, cat. # 21277) was used for all pull-down experiments. Briefly, 0.5 μg of His-CDK6/GST-cyclin D1 complex was incubated with cobalt agarose resin overnight at 4 °C in Pierce Pull-down Lysis Buffer (25 mM Tris-HCl pH 7.4, 150 mM NaCl) for immobilization. His-CDK6-coupled cobalt beads were washed with diluted Pierce Pull-down Lysis Buffer (wash buffer) and incubated with 0.5 μg of the VCB complex in the presence of DMSO or 3 μM MS28 for at least 4 h at 4 °C. Beads were then washed three times with wash buffer, eluted with 290 mM Imidazole Elution Buffer, boiled for 10 min, and loaded onto polyacrylamide gel for Western blot analysis.
4.11. Proteomics Studies.
Proteomics studies were conducted following the previously reported method using TMT-based proteomics global profiling samples.49
4.12. Selectivity Assays.
Selectivity of MS28 against a panel of 58 human kinases was carried out in radiometric kinase enzymatic inhibition assays by Eurofins Cerep. The assays were performed in duplicate, and MS28 was tested at 1 μM.
4.13. Cell Proliferation Assay for Evaluating MS28 in Calu-1, NCI-H2110, and PNT2 Cells.
Cell proliferation assays were performed for MS28 (and PB, BSJ, MS28N1) in Calu-1 and NCI-H2110 cells. Cells were seeded (4000 cells/well for Calu-1, 10,000 cells/well for NCI-H2110, and 5000 cells/well for PNT2) in white, cell culture-treated, flat and clear-bottom 96-well plates in triplicate and incubated at 37 °C overnight. After 16 h, cells were treated with 0.01% DMSO or MS28 at 7 concentrations, with 10 μM as the highest concentration and 13 nM as the lowest concentration (3-fold dilution). Following a 5-day incubation with MS28 at 37 °C and 5% CO2, cell viability was assessed by CCK-8 (Cell Counting Kit-8, WST-8). Briefly, cells were treated with 1× solution of CCK-8 (Dojindo, CK04) and re-incubated at 37 °C for 2–4 h until the absorbance at 450 nm reads 1.0 or greater for the DMSO control group. Absorbance of 690 nm was used as a reference using an Infinite F PLEX plate reader (TECAN). GI50 values were calculated using GraphPad Prism 8 using the nonlinear regression analysis. Error bars represent ± SD for three biological independent experiments.
4.14. Soft Agar Assay.
For the 1% agarose base layer, prewarmed DMEM medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% Penicillin/Streptomycin was mixed in a 1:1 ratio with autoclaved 2% agarose (Research Products International [RPI], A20070–50.0) and transferred into a 24-well plate, which was then incubated at 4 °C for 10 min for solidifying. Subsequently, Calu-1 cells were collected via trypsinization and counted. Enough cells required for a seeding density of 10,000 cells per well were resuspended in a complete DMEM medium and passed through a 70-micron filter to obtain single cells. Cells were then mixed with the 2% agarose in a 1:3 ratio to make the 0.5% agarose layer containing cells, which were pipetted on top of the base layer. Once the top agarose cell layer solidifies, a medium containing complete DMEM with compounds was added, and the plate was cultured under standard cell culture conditions. Medium with compounds was changed every 3 days. The colonies were visualized and measured around 3 weeks of culturing using normal light microscopy.
4.15. Statistics and Reproducibility.
Experimental data are presented as the mean ± SD or SEM of three independent experiments unless otherwise noted. Statistical analysis was performed using an unpaired two-sided Student’s t test for comparing two sets of data with assumed normal distribution. The results for immunoblotting are representative of at least three biologically independent experiments unless otherwise noted. All statistical analyses and visualizations were performed using GraphPad (Prism v8.4.2) or Excel.
Supplementary Material
ACKNOWLEDGMENTS
This work utilized the NMR Spectrometer Systems at Mount Sinai acquired with funding from National Institutes of Health SIG Grants 1S10OD025132 and 1S10OD028504. J.J. acknowledges the support by the Grants R01CA218600, R01CA230854, R01CA260666, R01CA268384, and R01CA268519 from the U.S. National Institutes of Health. Y.Z. acknowledges the support by the National Institute of General Medical Sciences (NIGMS)-funded Integrated Pharmacological Sciences Training Program T32GM062754.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.2c09255
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c09255.
Experimental procedures and characterization data of new compounds, 1H NMR, 13C NMR, and LC-MS spectra of MS28, MS28N1, and MS28N2 (Figures S1–S14 and Tables S1 and S2) (PDF)
The authors declare the following competing financial interest(s): The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, Inc., Cullgen, Inc. and Cullinan Oncology, Inc. J.J. is a cofounder and equity shareholder in Cullgen, Inc. and a consultant for Cullgen, Inc., EpiCypher, Inc., and Accent Therapeutics, Inc.
Contributor Information
Yan Xiong, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Yue Zhong, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Hyerin Yim, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Xiaobao Yang, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Kwang-Su Park, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Ling Xie, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Poulikos I. Poulikakos, Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States
Xiaoran Han, Cullgen Inc., San Diego, California 92130, United States.
Yue Xiong, Cullgen Inc., San Diego, California 92130, United States.
Xian Chen, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Jing Liu, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Jian Jin, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
REFERENCES
- (1).Békés M; Langley DR; Crews CM PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discovery 2022, 21, 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Dale B; Cheng M; Park KS; Kaniskan HÜ; Xiong Y; Jin J Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 21, 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Schapira M; Calabrese MF; Bullock AN; Crews CM Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discovery 2019, 18, 949–963. [DOI] [PubMed] [Google Scholar]
- (4).Bai L; Zhou H; Xu R; Zhao Y; Chinnaswamy K; McEachern D; Chen J; Yang C-Y; Liu Z; Wang M; Liu L; Jiang H; Wen B; Kumar P; Meagher JL; Sun D; Stuckey JA; Wang S A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell 2019, 36, 498–511.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Liu J; Chen H; Kaniskan HÜ; Xie L; Chen X; Jin J; Wei W TF-PROTACs enable targeted degradation of transcription factors. J. Am. Chem. Soc 2021, 143, 8902–8910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Samarasinghe KTG; Jaime-Figueroa S; Burgess M; Nalawansha DA; Dai K; Hu Z; Bebenek A; Holley SA; Crews CM Targeted degradation of transcription factors by TRAFTACs: TRAnscription Factor TArgeting Chimeras. Cell Chem. Biol 2021, 28, 648–661.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Shao J; Yan Y; Ding D; Wang D; He Y; Pan Y; Yan W; Kharbanda A; Li H.-y.; Huang H Destruction of DNA-binding proteins by programmable oligonucleotide PROTAC (O’PROTAC): Effective targeting of LEF1 and ERG. Adv. Sci 2021, 8, No. 2102555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ghidini A; Cléry A; Halloy F; Allain FHT; Hall J RNA-PROTACs: Degraders of RNA-binding proteins. Angew. Chem 2021, 133, 3200–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Costales MG; Matsumoto Y; Velagapudi SP; Disney MD Small molecule targeted recruitment of a nuclease to RNA. J. Am. Chem. Soc 2018, 140, 6741–6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Disney MD Targeting RNA with small molecules to capture opportunities at the intersection of chemistry, biology, and medicine. J. Am. Chem. Soc 2019, 141, 6776–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Childs-Disney JL; Yang X; Gibaut QMR; Tong Y; Batey RT; Disney MD Targeting RNA structures with small molecules. Nat. Rev. Drug Discovery 2022, 21, 736–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Naganuma M; Ohoka N; Tsuji G; Tsujimura H; Matsuno K; Inoue T; Naito M; Demizu Y Development of chimeric molecules that degrade the estrogen receptor using decoy oligonucleotide ligands. ACS Med. Chem. Lett 2022, 13, 134–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Neklesa TK; Winkler JD; Crews CM Targeted protein degradation by PROTACs. Pharmacol. Ther 2017, 174, 138–144. [DOI] [PubMed] [Google Scholar]
- (14).Behan FM; Iorio F; Picco G; Gonçalves E; Beaver CM; Migliardi G; Santos R; Rao Y; Sassi F; Pinnelli M; Ansari R; Harper S; Jackson DA; McRae R; Pooley R; Wilkinson P; van der Meer D; Dow D; Buser-Doepner C; Bertotti A; Trusolino L; Stronach EA; Saez-Rodriguez J; Yusa K; Garnett MJ Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 2019, 568, 511–516. [DOI] [PubMed] [Google Scholar]
- (15).Schneider M; Radoux CJ; Hercules A; Ochoa D; Dunham I; Zalmas L-P; Hessler G; Ruf S; Shanmugasundaram V; Hann MM; Thomas PJ; Queisser MA; Benowitz AB; Brown K; Leach AR The PROTACtable genome. Nat. Rev. Drug Discovery 2021, 20, 789–797. [DOI] [PubMed] [Google Scholar]
- (16).Duffy MJ; Crown J Drugging “undruggable” genes for cancer treatment: Are we making progress? Int. J. Cancer 2021, 148, 8–17. [DOI] [PubMed] [Google Scholar]
- (17).Xiong Y; Connolly T; Futcher B; Beach D Human D-type cyclin. Cell 1991, 65, 691–699. [DOI] [PubMed] [Google Scholar]
- (18).Matsushime H; Roussel MF; Ashmun RA; Sherr CJ Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell 1991, 65, 701–713. [DOI] [PubMed] [Google Scholar]
- (19).Xiong Y; Zhang H; Beach D D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell 1992, 71, 505–514. [DOI] [PubMed] [Google Scholar]
- (20).Yu Q; Geng Y; Sicinski P Specific protection against breast cancers by cyclin D ablation. Nature 2001, 411, 1017–1021. [DOI] [PubMed] [Google Scholar]
- (21).Choi YJ; Li X; Hydbring P; Sanda T; Stefano J; Christie AL; Signoretti S; Look AT; Kung AL; von Boehmer H; Sicinski P The requirement for cyclin D function in tumor maintenance. Cancer Cell 2012, 22, 438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Fantl V; Stamp G; Andrews A; Rosewell I; Dickson C Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev 1995, 9, 2364–2372. [DOI] [PubMed] [Google Scholar]
- (23).Hsu JH-R; Rasmusson T; Robinson J; Pachl F; Read J; Kawatkar S; O’ Donovan DH; Bagal S; Code E; Rawlins P; Argyrou A; Tomlinson R; Gao N; Zhu X; Chiarparin E; Jacques K; Shen M; Woods H; Bednarski E; Wilson DM; Drew L; Castaldi MP; Fawell S; Bloecher A EED-targeted PROTACs degrade EED, EZH2, and SUZ12 in the PRC2 complex. Cell Chem. Biol 2020, 27, 41–46.e17. [DOI] [PubMed] [Google Scholar]
- (24).Potjewyd F; Turner A-MW; Beri J; Rectenwald JM; Norris-Drouin JL; Cholensky SH; Margolis DM; Pearce KH; Herring LE; James LI Degradation of polycomb repressive complex 2 with an EED-targeted bivalent chemical degrader. Cell Chem. Biol 2020, 27, 47–56.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Liu Z; Hu X; Wang Q; Wu X; Zhang Q; Wei W; Su X; He H; Zhou S; Hu R; Ye T; Zhu Y; Wang N; Yu L Design and synthesis of EZH2-based PROTACs to degrade the PRC2 complex for targeting the noncatalytic activity of EZH2. J. Med. Chem 2021, 64, 2829–2848. [DOI] [PubMed] [Google Scholar]
- (26).Tu Y; Sun Y; Qiao S; Luo Y; Liu P; Jiang Z-X; Hu Y; Wang Z; Huang P; Wen S Design, synthesis, and evaluation of VHL-based EZH2 degraders to enhance therapeutic activity against lymphoma. J. Med. Chem 2021, 64, 10167–10184. [DOI] [PubMed] [Google Scholar]
- (27).Shen Y; Gao G; Yu X; Kim H; Wang L; Xie L; Schwarz M; Chen X; Guccione E; Liu J; Bedford MT; Jin J Discovery of first-in-class protein arginine methyltransferase 5 (PRMT5) degraders. J. Med. Chem 2020, 63, 9977–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yu X; Li D; Kottur J; Shen Y; Kim HS; Park KS; Tsai YH; Gong W; Wang J; Suzuki K; Parker J; Herring L; Kaniskan H; Cai L; Jain R; Liu J; Aggarwal AK; Wang GG; Jin J A selective WDR5 degrader inhibits acute myeloid leukemia in patient-derived mouse models. Sci. Transl. Med 2021, 13, No. eabj1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wang J; Yu X; Gong W; Liu X; Park K-S; Ma A; Tsai Y-H; Shen Y; Onikubo T; Pi W-C; Allison DF; Liu J; Chen W-Y; Cai L; Roeder RG; Jin J; Wang GG EZH2 noncanonically binds cMyc and p300 through a cryptic transactivation domain to mediate gene activation and promote onco-genesis. Nat. Cell Biol 2022, 24, 384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Dale B; Anderson C; Park K-S; Kaniskan HÜ; Ma A; Shen Y; Zhang C; Xie L; Chen X; Yu X; Jin J Targeting triple-negative breast cancer by a novel proteolysis targeting chimera degrader of enhancer of zeste homolog 2. ACS Pharmacol. Transl. Sci 2022, 5, 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Li J; Liu T; Song Y; Wang M; Liu L; Zhu H; Li Q; Lin J; Jiang H; Chen K; Zhao K; Wang M; Zhou H; Lin H; Luo C Discovery of small-molecule degraders of the CDK9-cyclin T1 complex for targeting transcriptional addiction in prostate cancer. J. Med. Chem 2022, 65, 11034–11057. [DOI] [PubMed] [Google Scholar]
- (32).Guiley KZ; Stevenson JW; Lou K; Barkovich KJ; Kumarasamy V; Wijeratne TU; Bunch KL; Tripathi S; Knudsen ES; Witkiewicz AK; Shokat KM; Rubin SM p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 2019, 366, No. eaaw2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Toogood PL; Harvey PJ; Repine JT; Sheehan DJ; Vanderwel SN; Zhou H; Keller PR; McNamara DJ; Sherry D; Zhu T; Brodfuehrer J; Choi C; Barvian MR; Fry DW Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem 2005, 48, 2388–2406. [DOI] [PubMed] [Google Scholar]
- (34).Fry DW; Harvey PJ; Keller PR; Elliott WL; Meade M; Trachet E; Albassam M; Zheng X; Leopold WR; Pryer NK; Toogood PL Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther 2004, 3, 1427–1438. [PubMed] [Google Scholar]
- (35).Kim S; Loo A; Chopra R; Caponigro G; Huang A; Vora S; Parasuraman S; Howard S; Keen N; Sellers W; Brain C Abstract PR02: LEE011: An orally bioavailable, selective small molecule inhibitor of CDK4/6– reactivating Rb in cancer. Mol. Cancer Ther 2013, 12, PR02. [Google Scholar]
- (36).Álvarez-Fernández M; Malumbres M Mechanisms of sensitivity and resistance to CDK4/6 Inhibition. Cancer Cell 2020, 37, 514–529. [DOI] [PubMed] [Google Scholar]
- (37).Chen P; Lee NV; Hu W; Xu M; Ferre RA; Lam H; Bergqvist S; Solowiej J; Diehl W; He Y-A; Yu X; Nagata A; Vanarsdale T; Murray BW Spectrum and degree of CDK drug interactions predicts clinical performance. Mol. Cancer Ther 2016, 15, 2273–2281. [DOI] [PubMed] [Google Scholar]
- (38).Bondeson DP; Mares A; Smith IED; Ko E; Campos S; Miah AH; Mulholland KE; Routly N; Buckley DL; Gustafson JL; Zinn N; Grandi P; Shimamura S; Bergamini G; Faelth-Savitski M; Bantscheff M; Cox C; Gordon DA; Willard RR; Flanagan JJ; Casillas LN; Votta BJ; Den Besten W; Famm K; Kruidenier L; Carter PS; Harling JD; Churcher I; Crews CM Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol 2015, 11, 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Raina K; Lu J; Qian Y; Altieri M; Gordon D; Rossi AMK; Wang J; Chen X; Dong H; Siu K; Winkler JD; Crew AP; Crews CM; Coleman KG PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U.S.A 2016, 113, 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Jiang B; Wang ES; Donovan KA; Liang Y; Fischer ES; Zhang T; Gray NS Development of dual and selective degraders of cyclin-dependent kinases 4 and 6. Angew. Chem., Int. Ed 2019, 58, 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wu X; Yang X; Xiong Y; Li R; Ito T; Ahmed TA; Karoulia Z; Adamopoulos C; Wang H; Wang L; Xie L; Liu J; Ueberheide B; Aaronson SA; Chen X; Buchanan SG; Sellers WR; Jin J; Poulikakos PI Distinct CDK6 complexes determine tumor cell response to CDK4/6 inhibitors and degraders. Nat. Cancer 2021, 2, 429–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Su S; Yang Z; Gao H; Yang H; Zhu S; An Z; Wang J; Li Q; Chandarlapaty S; Deng H; Wu W; Rao Y Potent and preferential degradation of CDK6 via proteolysis targeting chimera degraders. J. Med. Chem 2019, 62, 7575–7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ezhevsky SA; Ho A; Becker-Hapak M; Davis PK; Dowdy SF Differential regulation of retinoblastoma tumor suppressor protein by G 1 cyclin-dependent kinase complexes in vivo. Mol. Cell. Biol 2001, 21, 4773–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Pines J Cyclins and cyclin-dependent kinases: a biochemical view. Biochem. J 1995, 308, 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lee DH; Goldberg AL Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in saccharomyces cerevisiae. J. Biol. Chem 1996, 271, 27280–27284. [DOI] [PubMed] [Google Scholar]
- (46).Soucy TA; Smith PG; Milhollen MA; Berger AJ; Gavin JM; Adhikari S; Brownell JE; Burke KE; Cardin DP; Critchley S; Cullis CA; Doucette A; Garnsey JJ; Gaulin JL; Gershman RE; Lublinsky AR; McDonald A; Mizutani H; Narayanan U; Olhava EJ; Peluso S; Rezaei M; Sintchak MD; Talreja T; Thomas MP; Traore T; Vyskocil S; Weatherhead GS; Yu J; Zhang J; Dick LR; Claiborne CF; Rolfe M; Bolen JB; Langston SP An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [DOI] [PubMed] [Google Scholar]
- (47).Cai L; Rothbart SB; Lu R; Xu B; Chen WY; Tripathy A; Rockowitz S; Zheng D; Patel DJ; Allis CD; Strahl BD; Song J; Wang GG An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol. Cell 2013, 49, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Fabian MA; Biggs WH; Treiber DK; Atteridge CE; Azimioara MD; Benedetti MG; Carter TA; Ciceri P; Edeen PT; Floyd M; Ford JM; Galvin M; Gerlach JL; Grotzfeld RM; Herrgard S; Insko DE; Insko MA; Lai AG; Lélias J-M; Mehta SA; Milanov ZV; Velasco AM; Wodicka LM; Patel HK; Zarrinkar PP; Lockhart DJ A small molecule–kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol 2005, 23, 329–336. [DOI] [PubMed] [Google Scholar]
- (49).Wei J; Meng F; Park K-S; Yim H; Velez J; Kumar P; Wang L; Xie L; Chen H; Shen Y; Teichman E; D L; Wang GG; Chen X; Kaniskan HÜ; Jin J Harnessing the E3 ligase KEAP1 for targeted protein degradation. J. Am. Chem. Soc 2021, 143, 15073–15083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.