

Abstract
Regioselective functionalization of gem-difluoroalkenes enables convergent late-stage access to fluorinated functional groups, though most functionalization reactions proceed through defluorinative functionalization processes that deliver mono-fluorovinyl products. In contrast, fewer reactions undergo net hydrofunctionalization to generate difluorinated products. Herein, we report a photocatalytic hydrothiolation of gem-difluoroalkenes that enables access to a broad spectrum of α,α−difluoroalkylthioethers. Notably, the reaction successfully couples non-activated substrates, which expands the scope of accessible molecules relative to previously reported reactions involving organo- or photocatalytic strategies. Further, this reaction successfully couples biologically relevant molecules under aqueous conditions, highlighting potential applications in both late-stage and biorthogonal functionalizations.
Keywords: Hydrothiolation, gem-Difluoroalkene, Fluorination, Photochemistry, Aqueous
GRAPHICAL ABSTRACT

INTRODUCTION
Fluorinated functional groups are typically used by the medicinal and agricultural chemistry communities to influence the physicochemical, pharmacokinetic, and pharmacodynamic properties of small molecules.1–3 Thus, catalytic methods for preparing medicinally relevant fluorinated substructures have received substantial attention from the synthetic community.4–6 Since many of a molecule’s physicochemical properties are affected by heteroatom-containing functional groups, the introduction of one or more fluorine atoms in the vicinity of a heteroatom readily modulates these properties.7 For example, the introduction of two fluorine atoms alpha to the sulfur atom of a thioether reduces sulfur’s nucleophilicity and increases its oxidative stability relative to the non-fluorinated counterpart.8 Such changes in the physicochemical properties of sensitive thioethers encourages the development of a general and convergent catalytic strategy for accessing α,α−difluoroalkylthioethers.
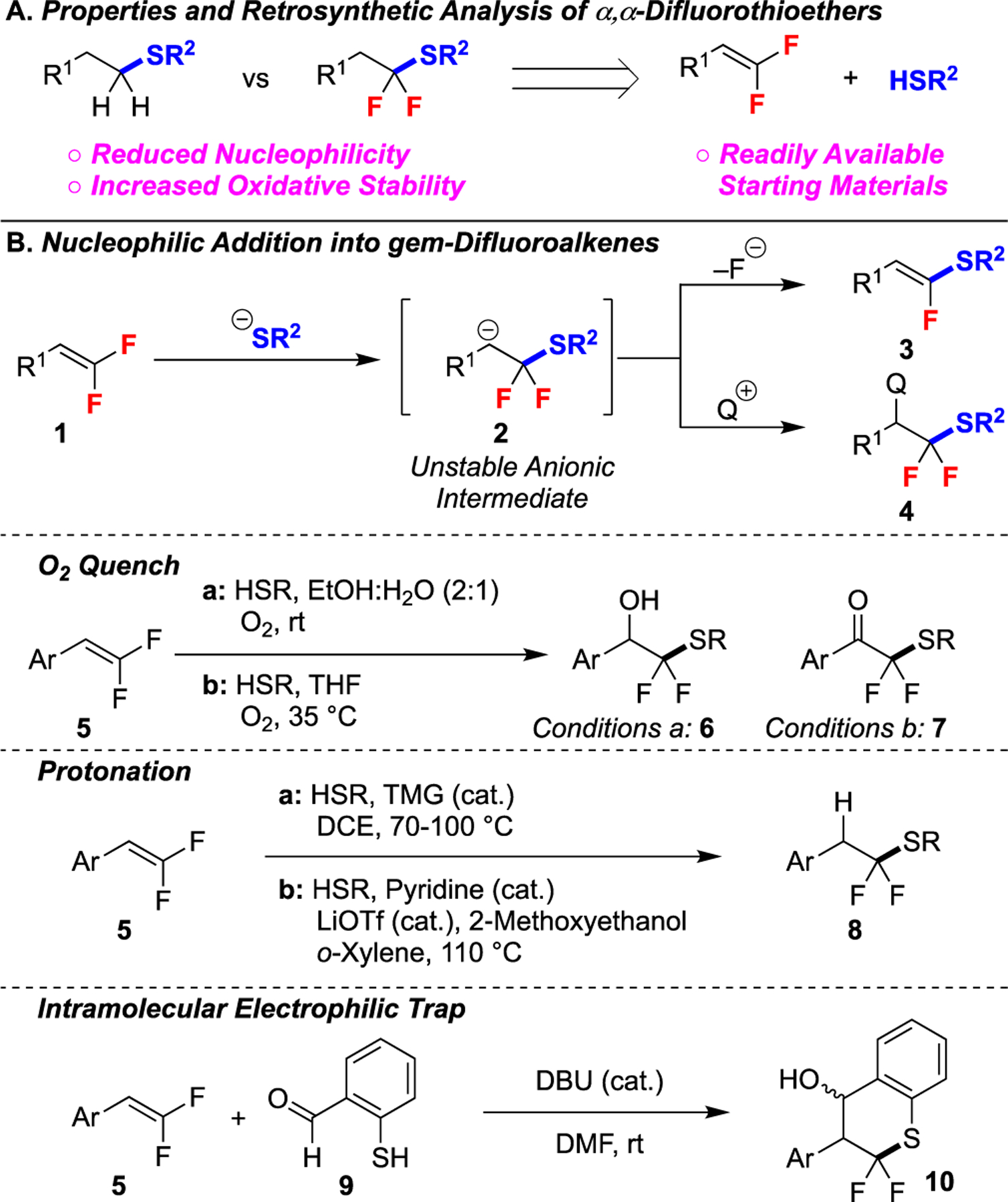
To access this substructure, an attractive retrosynthetic disconnection might involve the CF2–S bond, which would reveal a nucleophilic thiol and electrophilic difluorinated carbon atom (Figure 1A). In the forward reaction, the σ-withdrawing character of the fluorine atoms would activate the alkene for nucleophilic attack by the thiol.5,6,9–11 However, in practice, nucleophilic addition of a thiolate into gem-difluoroalkenes generates an anionic intermediate 2 that can readily undergo β-fluoride elimination.6,10 This elimination has been partially overcome using several different strategies (Figure 1B),5 such as kinetically quenching the anionic intermediate 2 with molecular oxygen,12 exploiting an acidic organocatalyst,8,13 or trapping the anion with an appended electrophile.14 However, these strategies only function on styrenyl gem-difluoroalkenes, which react through stabilized benzylic carbanion intermediates.
Figure 1: Hydrothiolation of gem-Difluoroalkenes.
In contrast, alternate strategies involving single electron processes can successfully couple thiols with both styrenyl and aliphatic gem-difluoroalkenes.15–17 These reactions involve the addition of a nucleophilic thiyl radical to the electrophilic difluorinated position of the alkene, which generates a β,β-difluororadical intermediate 11 (Figure 2A). This intermediate does not eject fluorine radicals, which allows for a hydrogen atom transfer (HAT) to quench the radical and deliver the difluorinated thioether product 12. However, existing single electron hydrothiolation methods still largely remain limited to styrenyl gem-difluoroalkenes,15,16,18,19 and radical hydrothiolation reactions of aliphatic gem-difluoroalkenes remain limited to three individual examples with thiophenols.15,17,20 The lack of reactions that couple aliphatic gem-difluoroalkenes with aliphatic thiols leaves a notable gap in the scope of accessible molecules. Herein, we report a general photocatalytic hydrothiolation of both aliphatic and styrenyl gem-difluoroalkenes with both thiols and thiophenols (Figure 2B).21 Notably, an aniline-derived co-catalyst enhances the yield of α,α−difluorothioethers, which contrasts related base-mediated photocatalytic reactions of thiols with gem-difluoroalkenes that undergo net C–F functionalization to deliver α-fluorovinylthioethers (Figure 2C).22 Further, this transformation is compatible with aqueous conditions, effectively coupling biologically relevant small molecules.
Figure 2: Radical Hydrothiolation of gem-Difluoroalkenes.

RESULTS AND DISCUSSION
Inspired by previous photochemical hydrothiolation reactions,16,18,19,23,24 we sought to develop a photocatalytic method to enable hydrothiolation of gem-difluoroalkenes (Figure 3D). In early scouting experiments, the reaction of gem-difluorostyrene 20 with octane-1-thiol occurred with trace yield and 36% conversion using 0.5 mol% [Ru(bpy)3]Cl2 in acetonitrile under irradiation with 34 W blue light (Figure 3A; S1). The addition of catalytic quantities of p-toluidine marginally improved both conversion (34%) and yield (10%, Figure 3B). In these reactions, the aniline might mediate the redox reactions and serve as a co-catalyst for a PCET event (See S1).25 While amines are frequently employed as PCET co-catalysts in photo- and electrochemical reactions, anilines remain underutilized in this role.26 Further screening of photocatalysts identified {Ir[dF(CF3)ppy]2(dtbpy)}PF6 as the optimal photocatalyst, which generated product 21 in 47% yield (Figure 3C; S2). Application of these conditions to a previously challenging substrate combination of an aliphatic gem-difluoroalkene with a thiophenol afforded the coupled product in 77% product (Figure 3C; S3). However, the reaction of alkyl difluoroalkenes afforded both linear and branched regioisomers, generally in a 10:1 ratio (Figure 3C). Moreover, these conditions suffered from a limited spectrum; specifically, electron-rich thiophenols reacted sluggishly. Further optimization involved the exploration of alternate anilines, and m-anisidine improved both the yield (23, 72%) and regioselectivity (14:1) of the hydrothiolation with electron rich thiols (see S4). However, the high yield observed with m-anisidine did not translate from 0.1 mmol to a 1.0 mmol scale. Additional screening of alternative anilines identified p-aminoacetophenone as a suitable additive that helped deliver the desired product 23 in >95% yield and excellent regioselectively (>20:1; Figure 3D, S4). Further routine optimization of reaction parameters provided general conditions of thiophenol (1.5 equiv.), {Ir[dF(CF3)ppy]2(dtbpy)}PF6 (0.25 mol%) and p-aminoacetophenone (10 mol%) in acetonitrile (0.5 M) under irradiation with a 40 W 427 nm LED lamp (See S5).
Figure 3: Optimization of Reaction for the Radical Hydrothiolation of gem-Difluoroalkenes.

Control experiments indicate that the aniline additives facilitate formation of the α,α−difluorinated thioether product (21, Figure 4A; S6), which contrasts previously reported conditions for defluorinative thiolation that utilized K2CO3 as an additive (Figure 2C).22 As a control, the reaction of gem-difluorostyrene 1 with octane-1-thiol proceeds in 45% yield and 19:1 selectivity in the absence of an additive (entry 1), while the addition of 10% p-aminoacetophenone increases the yield to 68% and selectivity beyond 25:1 (entry 2). In contrast, addition of K2CO3 (2.0 equiv.) increases the yield of undesired α-fluorovinylthioether 25 and decreases the selectivity of the reaction to 2:1 (entry 3), while the addition of both p-aminoacetophenone and K2CO3 provides yield and selectivity comparable to the control reaction that lacks both additives (entry 4 vs. 1). Of note, the difluorinated products are stable to the previously reported conditions (K2CO3, 30 °C, MeCN; Figure 4B; S11).22 Thus, difluorinated thioether 16 is not an intermediate en route to the monofluorovinylether product 3. At this point, we speculate that the aniline additive might mediate the redox events between the Ir-based catalyst and the thiol substrate, and also serve a role as an H• donor to quench radical intermediates (Figure 2B).25,26
Figure 4: Aniline Additives Improve Chemoselectivity of the Reaction.

aReactions were carried out with 20 (0.50 mmol), octane-1-thiol (0.75 mmol), additive(s) [p-aminoacetophenone (0.050 mmol) and/or K2CO3 (1.0 mmol)], and {Ir[dF(CF3)ppy]2(dtbpy)} PF6 (1.25 µmol) in MeCN (1.0 mL) irradiated with a 40 W 427 nm LED for 24 h under an atmosphere of N2. Conversion was determined with respect to starting material 20. All yields were determined by 19F NMR using α,α,α−trifluorotoluene as an internal standard. bReactions were carried out with 16 (0.10 mmol) and K2CO3 (0.20 mmol) in MeCN (0.20 mL) at 30 °C for 24 h under an atmosphere of N2. Conversion of 16 and formation of 3 were determined by 19F NMR using α,α,α−trifluorotoluene as an internal standard.
A range of aliphatic gem-difluoroalkenes successfully coupled with 3-mercaptophenol (26) to afford α,α-difluoroalkylthioethers in good to excellent yields (Figure 5). In general, fully substituted alkenes, an underrepresented substrate with only one prior example,18 underwent photocatalytic hydrothiolation to afford the desired products in high yields (27a, 90%; 27b, 90%). Moreover, many useful functional groups including boronate esters and secondary, Boc- and Cbz-protected amines which were not demonstrated with the previous systems were tolerated under the optimized conditions (27c – 27f, 83 – 94%).8,13 Further, the successful reaction of a functionalized carbohydrate 27g substrate demonstrated excellent compatibility with acetal, ketal, ester, and heterocycle groups.
Figure 5: Scope of gem-Difluoroalkenesa.

aUnless otherwise stated, all reactions were carried out with 1 (1.0 mmol), 3-mercaptophenol 26 (1.5 mmol), {Ir[dF(CF3)ppy]2(dtbpy)}PF6 (1.25 µmol), and p-aminoacetophenone (0.10 mmol) in MeCN (2.0 mL) irradiated with a 40 W 427 nm LED for 24 h under an atmosphere of N2. Isolated yields represent an average of two independent reactions. bReactions were carried out with 1 (0.50 mmol), 3-mercaptophenol 26 (0.75 mmol), {Ir[dF(CF3)ppy]2(dtbpy)}PF6 (1.25 µmol), and p-aminoacetophenone (0.05 mmol) in MeCN (1.0 mL) irradiated with a 40 W 427 nm LED for 24 h under an atmosphere of N2.
A wide range of thiophenols were successfully coupled with Cbz-protected piperidine substrate 28 to deliver α,α–difluoroalkylthioethers (Figure 6). Substrates reacted smoothly to generate desired products bearing aryl halides (29a – 29c, 88 – 90%), an amide (29d, 68%), ethers (29e – 29f, 86 – 68%), and a benzyl alcohol (29g, 75%). Further, a sterically hindered thiophenol delivered product in good yield (29h; 96%), and a thiophenol bearing a boronic ester, which has not been demonstrated in the previous systems, reacted to generate the desired product and provide a functional handle for further synthetic elaboration (29i, 89%).
Figure 6: Scope of Hydrothiolation of gem-Difluoroalkenes with Thiophenolsa.

aUnless otherwise stated, all reactions were carried out with 28 (1.0 mmol), thiophenol 17 (1.5 mmol), {Ir[dF(CF3)ppy]2(dtbpy)}PF6 (1.25 µmol), and p-aminoacetophenone (0.10 mmol) in MeCN (2.0 mL) irradiated with a 40 W 427 nm LED for 24 h under an atmosphere of N2. Isolated yields represent an average of two independent reactions.
The optimized conditions successfully coupled alkylthiols with aliphatic gem-difluoroalkenes, which, relative to previous hydrothiolation reactions (Figure 7),8,15,16,18,19 represents a substantial expansion of previously accessible products. Similar to their reactions with thiophenols, fully substituted alkenes including cyclohexyl, Cbz-piperidine, and methyl substituted alkenes were coupled in excellent yields (30b – 30c, 95 – 98%). Of note, the reaction of octane-1-thiol with a cyclohexyl alkene generated a single diastereomer of product 30a (72%), which contrasts the reactions of thiophenols that generated 4:1 and 10:1 mixtures of diastereomeric products, respectively (Figure 5, 27a and 27d). The steric hindrance of the thiol did not greatly influence reactivity, with primary and secondary thiols reacting to give products in excellent yields (30d – 30h; 82 – 95%). Further, tertiary thiols, which have not been effectively coupled in other systems, successfully generated the product in moderate to excellent yields (30i, 87%; 30j, 39%). Interestingly, the reaction of a trisubstituted alkene provided product 30l in poor regioselectivity (branched:linear 8:5). Finally, the reaction of captopril proceeded to generate product 30k in high yield (86%), which demonstrated the compatibility of more complex reaction partners.
Figure 7: Scope of Hydrothiolation of gem-Difluoroalkenes with Alkyl Thiolsa.

aUnless otherwise stated, all reactions were carried out with 1 (1.0 mmol), thiol (1.5 mmol), {Ir[dF(CF3)ppy]2(dtbpy)}PF6 (1.25 µmol), and p-aminoacetophenone (0.10 mmol) in MeCN (2.0 mL) irradiated with a 40 W 427 nm LED for 24 h under an atmosphere of N2. Isolated yields represent an average of two independent reactions. bReactions were carried out with 1 (1.0 mmol), thiol (1.5 mmol), {Ir[dF(CF3)ppy]2(dtbpy)}PF6 (1.25 µmol), and p-fluoroaniline (0.10 mmol) in MeCN (1.0 mL) irradiated with a 40 W 427 nm LED for 24 h under an atmosphere of N2. cbranched:linear ratio of the crude reaction mixture was determined by 19F NMR.
In addition to successfully coupling aliphatic gem-difluoroalkenes, the optimized conditions also efficiently coupled thiols and thiophenols with gem-difluorostyrenes (Figure 8A), substrate combinations that have previously been reported using organocatalytic bases (e.g. TMG)13 and acids (pyridinium triflate)8. For reactions of thiophenols with gem-difluorosytrenes, the photoredox catalytic system generally tolerated a wide variety of useful functional groups, including hydroxyl, amine, ether, acetal, heterocycle, amide, carbamate, nitrile and nitro groups (Figure 8A). Further, in most cases, the photocatalytic system provided products in comparable or better yields than the previously reported systems (31a – 31f),8,13 though the nitro bearing gem-difluorostyrene delivered product 31g in an inferior yield relative to the pyridinium triflate system.8 In addition to reactions of gem-difluorostyrenes with thiophenols, reactions of alkyl thiols generated the desired products in improved yields relative to the acid catalyzed system (Figure 8B).8 Moreover, thiols that performed poorly or that were incompatible with the prior conditions delivered product in good yield (31h – 31l, 37 – 84%), thus demonstrating a clear expansion in the scope of accessible molecules.
Figure 8: Scope of Hydrothiolation of gem-Difluorostyrenes with Thiophenols and Alkyl Thiolsa.

aUnless otherwise stated, all reactions were carried out with gem-difluorostyrene (1.0 mmol), thiol (1.5 mmol), {Ir[dF(CF3)ppy]2(dtbpy)}PF6 (1.25 µmol), and p-aminoacetophenone (0.10 mmol) in MeCN (2.0 mL) irradiated with a 40 W 427 nm LED for 24 h under an atmosphere of N2. Isolated yields represent an average of two independent reactions. b5 (0.50 mmol), thiol (1.5 mmol), TMG (5 mol%) in DCE (2.0 mL) heated to 100 °C for 20 h under an atmosphere of N2. c5 (0.50 mmol), thiol (0.75 mmol), LiOTf (10 mol%), pyridine (20 mol%), and 2-methoxyethanol (1.0 mmol) in o-xylene (1.5 mL) heated at 110 °C for 24 h under an atmosphere of air. dReported selectivity refers to the corresponding mono-fluorovinyl product 3 (Figure 1B). e5 (1.0 mmol), thiol (1.5 mmol), and p-aminoacetophenone (0.10 mmol) in MeOH (2.0 mL) irradiated with a 40 W 427 nm LED for 24 h under an atmosphere of N2.
With slight modifications to the optimized reaction conditions, water-soluble substrates were successfully coupled with difluoroalkene 32 under aqueous conditions (Figure 9). Specifically, the reaction proceeded smoothly with N-protected, C-protected and unprotected cysteine to furnish the coupled products in good to excellent yields (33a – 33d, 75 − 92%). Moreover, coupling with L-glutathione delivered the desired product 33e in excellent yield (90%). These examples demonstrate the suitability of this general photocatalytic hydrothiolation method for the fluorination of biomolecules.
Figure 9: Hydrothiolation of gem-Difluoroalkenes Under Aqueous Conditions.
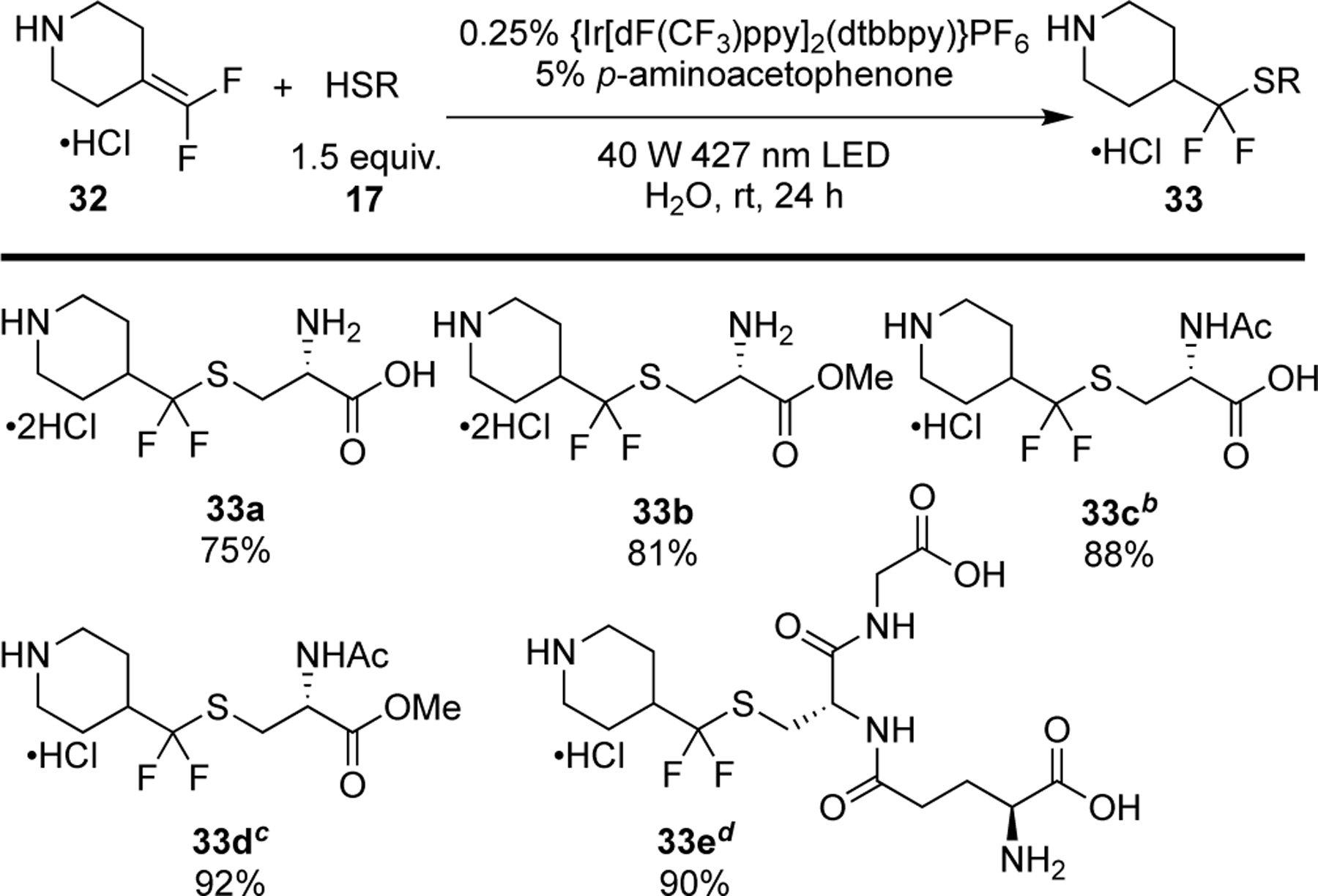
aUnless otherwise stated, all reactions were carried out with gem-difluoroalkene (0.50 mmol), thiol (0.65 mmol), {Ir[dF(CF3)ppy]2(dtbpy)}PF6 (1.25 µmol), and p-aminoacetophenone (0.025 mmol) in H2O (1.0 mL) irradiated with a 40 W 427 nm LED for 24 h under an atmosphere of N2. Isolated yields represent an average of two independent reactions. bgem-difluoroalkene (0.50 mmol), thiol (0.70 mmol), {Ir[dF(CF3)ppy]2(dtbpy)}PF6 (1.25 µmol), and p-aminoacetophenone (0.025 mmol) in H2O (1.0 mL) irradiated with a 40 W 427 nm LED for 24 h under an atmosphere of N2. cgem-difluoroalkene (0.50 mmol), thiol (0.60 mmol), {Ir[dF(CF3)ppy]2(dtbpy)}PF6 (1.25 µmol), and p-aminoacetophenone (0.025 mmol) in H2O (1.0 mL) irradiated with a 40 W 427 nm LED for 24 h under an atmosphere of N2. dgem-difluoroalkene (0.50 mmol), thiol (0.50 mmol), p-aminoacetophenone (0.025 mmol), and {Ir[dF(CF3)ppy]2(dtbbpy)}PF6 (2.5 µmol) in H2O (1.0 mL) irradiated with a 40 W 427 nm LED for 24 h under an atmosphere of N2.
In conclusion, we have developed a co-catalytic method for the hydrothiolation of gem-difluoroalkenes to generate α,α–difluoroalkylthioethers. This system substantially expands upon the existing scope of fluorine-retentive hydrothiolation of gem-difluoroalkenes, as access to α,α–difluoroalkylthioethers derived from each distinct pairing of aliphatic and aryl gem-difluoroalkenes and thiols are enabled by this method. Moreover, these mild conditions were applied to the late-stage functionalization of biologically relevant products in both organic solvents and water. Further, this reaction exploits an underutilized aniline-derived additive as a possible redox mediator and/or PCET co-catalyst, enhancing fluorine retentive hydrofunctionalization of gem-difluoroalkenes. The reported reaction contrasts related conditions that utilize K2CO3 that undergo a SET to generate anion 18 prior to β-fluoride elimination (Figure 2C).22 However, robust support for aniline’s role does not exist in the literature. Mechanistic studies of this iridium and aniline co-catalyzed reaction are underway and will be reported separately in due course.
EXPERIMENTAL SECTION
General Synthetic Information
Air- and moisture-sensitive reactions were carried out in oven-dried one-dram vials sealed with PTFE-lined septa or glassware sealed with rubber septa under an atmosphere of dry nitrogen. Plastic syringes equipped with stainless-steel needles were used to transfer air- and moisture-sensitive liquid reagents. Reactions were stirred using teflon-coated magnetic stir bars, and elevated temperatures were maintained using thermostat-controlled heating mantles. Light-promoted reactions were conducted using a EvoluChem PhotoRedOx Box™ photoreactor equipped with a Kessil PR160–427nm light with a peak intensity at 427 nm operating at 40 W. Organic solvents were removed using a rotary evaporator with a diaphragm vacuum pump. Thin-layer analytical chromatography was performed on silica gel UNIPLATE Silica Gel HLF UV254 plates, and spots were visualized by quenching of ultraviolet light (λ = 254 nm). Purification of products was accomplished by automated flash column chromatography on silica gel (VWR Common Silica Gel 60 Å, 40–60 μm). Unless otherwise noted, reagents were purchased from various commercial sources and used as received. NMR spectra were recorded on Bruker DRX 500 MHz (1H at 500 MHz, 13C{1H} at 126 MHz, and 19F at 470 MHz) or Bruker Avance III 800 with a QCI cryoprobe (1H at 800 and 13C{1H} at 201 MHz) nuclear magnetic resonance spectrometers. 1H NMR spectra were calibrated against the peak of the residual CHCl3 (7.26 ppm). 13C{1H} NMR spectra were calibrated against the peak of CDCl3 (77.2 ppm). NMR data are represented as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet), coupling constant in Hertz (Hz), integration. High-resolution mass determinations were obtained by electrospray ionization (ESI) or atmospheric-pressure chemical ionization (APCI) on a Waters LCT Premier™ mass spectrometer. Infrared spectra were measured on a Perkin Elmer Spectrum Two Fourier Transform Infrared Spectrometer by drying samples on a diamond ATR sample base plate. Uncorrected melting points were measured on a Chemglass Digital Melting Point apparatus.
Safety
Many thiols and thiophenols are acutely toxic and typically possess a pungent odor. Wear approved personal protective equipment (laboratory safety glasses, coat and gloves) to avoid contact with skin, eyes, and clothing. Wash hands immediately after handling materials.
Synthesis and Characterization
Preparation and Characterization of Substrates:
5-(2,2-Difluorovinyl)-1,2,3-trimethoxybenzene, 4-(2,2-difluorovinyl)dibenzo[b,d]thiophene, 2-(3-(2,2-difluorovinyl)phenyl)-5-(1,3-dioxolan-2-yl)pyridine, 1-(2,2-difluorovinyl)-3-nitrobenzene, 4-(2,2-difluorovinyl)benzonitrile, tert-butyl 4-(5-(2,2-difluorovinyl)thiazol-2-yl)piperazine-1-carboxylate, 4-(2,2-difluorovinyl)-1-phenyl-1H-pyrazole, 3-(2,2-difluorovinyl)benzonitrile, and ethyl (E)-3-(3-(2,2-difluorovinyl)phenyl)acrylate were prepared according to a previously reported procedure.13 (4,4-Difluorobut-3-en-1-yl)benzene was prepared according to a previously reported procedure.27
General Procedure A for the Preparation of gem-Difluoroalkenes:
An oven-dried 3-neck round-bottomed flask equipped with addition funnel and magnetic stir bar was charged with aldehyde (1.0 equiv.) and PPh3 (1.5 equiv.). The system was sealed with three rubber septa, and subsequently evacuated and backfilled with dry N2 three times. Dry DMF was added via syringe, and the system was charged to a heating mantle preheated to 90 °C. A solution of KO2CCF2Br (1.8 equiv.) in DMF was added dropwise using an addition funnel over 0.5 h, with the rate of addition controlling the evolution of CO2 gas. Once all of the KO2CCF2Br was added, the solution was allowed to stir for 0.5 h at 90 °C. Upon completion, the reaction was cooled to 0 °C and then quenched with H2O. Subsequently, Et2O was added to the mixture, and the organic layer was sequentially washed with H2O (three times), an aqueous solution of LiCl (10% in H2O; one time), then brine. The organic layer was dried over Na2SO4 and the solid was filtered away. Subsequently, MeI (1.5 equiv.) was added to the organic layer, and the mixture was stirred at room temperature for 30 min to methylate any residual PPh3. The crude material was eluted through a pad of silica gel with Et2O:pentane (1:1). The solution was concentrated, and the resulting residue was subjected to normal phase flash chromatography using EtOAc and hexanes.
General Procedure B for the Preparation of gem-Difluoroalkenes:
An oven-dried 3-neck round-bottomed flask equipped with magnetic stir bar and internal thermometer was charged with aldehyde (1.0 equiv.) and Ph3P+CF2CO2– (2.0 equiv.). The system was sealed with rubber septa, and subsequently evacuated and backfilled with dry N2 three times. Dry DMF was added via syringe, and the system was immersed in an oil bath preheated to 90 °C for 3 h. Upon completion, the reaction was cooled to 0 °C and then quenched with H2O. Et2O was added to the mixture, and the organic layer was sequentially washed with H2O (three times), an aqueous solution of LiCl (10% in H2O; one time), then brine. The organic layer was dried over Na2SO4 and the solid was filtered away. The solution was concentrated, and the resulting residue was subjected to normal phase flash chromatography using EtOAc and hexanes.
General Procedure C for the Preparation of gem-Difluoroalkenes:
An oven-dried 3-neck round-bottomed flask equipped with magnetic stir bar and internal thermometer was charged with ketone (1.0 equiv.) and (Me2N)3P+CF2CO2– (2.0 equiv.). The system was sealed with rubber septa, and subsequently evacuated and backfilled with dry N2 three times. A mixture of dry PhMe:DMA (3:1) was added via syringe, and the system was immersed in an oil bath preheated to 100 °C for 3 h. Upon completion, the reaction was cooled to 0 °C and then quenched with H2O. Subsequently, Et2O was added to the mixture, and the organic layer was sequentially washed with H2O (three times), an aqueous solution of LiCl (10% in H2O; one time), then brine. The organic layer was dried over Na2SO4 and the solid was filtered away. The solution was concentrated, and the resulting residue was subjected to normal phase flash chromatography using EtOAc and hexanes.
(4-(difluoromethylene)cyclohexyl)benzene:
Following general procedure C, (Me2N)3P+CF2CO2− (6.2 g, 24 mmol, 2.0 equiv.) and 4-phenylcyclohexan-1-one (2.1 g, 12 mmol) in PhMe:DMA (160 mL, 3:1, 0.075 M) were heated in a 100 °C oil bath and stirred for 3 hours. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:20) to furnish 1.8 g (73% yield) of desired product as a colorless crystalline solid. 1H NMR and 19F NMR spectra match a previous report.27
tert-butyl 4-(difluoromethylene)piperidine-1-carboxylate:
Following general procedure C, (Me2N)3P+CF2CO2− (6.2 g, 24 mmol, 2.0 equiv.) and tert-butyl 4-oxopiperidine-1-carboxylatein (2.4 g, 12 mmol) in PhMe:DMA (160 mL, 3:1, 0.075 M) were heated in a 100 °C oil bath and stirred for 3 hours. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:20) to furnish 2.1 g (75% yield) of desired product as a colorless crystalline solid. 1H NMR and 19F NMR spectra match a previous report.28
benzyl 4-(difluoromethylene)piperidine-1-carboxylate (28):
Following general procedure C, (Me2N)3P+CF2CO2− (5.0 g, 20 mmol, 2.0 equiv.) and benzyl 4-oxopiperidine-1-carboxylate (2.0 g, 8.5 mmol) in PhMe:DMA (110 mL, 3:1, 0.075 M) were heated in a 100 °C oil bath and stirred for 3 hours. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:4) to furnish 2.1 g (75% yield) of desired product as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.37 – 7.31 (m, 5H), 5.15 (s, 2H), 3.61 – 3.23 (m, 4H), 2.16 (s, 4H). 13C{1H} NMR (126 MHz, CDCl3) δ 152.5, 148.9 (t, J = 282.2 Hz), 134.2, 125.9, 125.5, 125.3, 82.9 (t, J = 20.3 Hz), 64.6, 41.4, 21.3 (d, J = 24.7 Hz). 19F NMR (470 MHz, CDCl3) δ −98.58. IR (Film) 2966, 2918, 2870, 1761, 1697, 1428, 1276, 1225, 1213, 1035, 697 cm–1. HRMS (APCI)+ m/z calc’d C14H16F2NO2 [M+H]+ 268.1149, found 268.1149.
benzyl 4-(difluoromethylene)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)piperidine-1-carboxylate:
Copper chloride (0.034 g, 0.25 mmol, 0.050 equiv.)), bis(pinacolato)diboron (1.9 g, 7.5 mmol, 1.5 equiv.), Xantphos (0.14 g, 0.25 mmol, 0.050 equiv.) and NaOMe (0.41 g, 7.5 mmol, 1.5 equiv.) were placed in a round-bottomed flask. The flask was connected to a nitrogen manifold and the flask was evacuated and backfilled with nitrogen. Dry THF (10.0 mL, 0.5 M) was added in the flask through the rubber septum using a syringe. After stirring for 30 min at 30 ºC, benzyl 4-(trifluoromethyl)-3,6-dihydropyridine-1(2H)-carboxylate (1.4 g, 5.0 mmol) was added to the mixture. After the reaction was complete, the reaction mixture was quenched by water and extracted with Et2O (three times). The combined organic layer was dried over MgSO4. After filtration through SiO2 (2 cm), the solvents were removed under a reduced pressure. Material was recrystallized from hot EtOAc/hexanes. Specifically, the material was dissolved in minimal EtOAc and heated to reflux. Hexanes were then slowly added while maintaining temperature until precipitate stated to form. The flask was then removed from heat and allowed to cool to rt for 5 hours. The material was then moved to 4 °C for 5 hours, then −20 °C for 15 h. The final product was obtained as a tan crystalline product in 75% yield (18.1 g). 1H NMR (500 MHz, CDCl3) δ 7.39 (s, 2H), 7.35 (s, 1H), 7.31 (s, 2H), 5.11 (s, 2H), 4.23 (s, 1H), 4.10 (s, 1H), 3.89 (s, 1H), 3.14 (s, 1H), 3.06 (s, 1H), 2.86 (s, 1H), 2.25 (d, J = 43.8 Hz, 1H), 1.19 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 152.52, 148.66 (t, J = 281.3 Hz), 134.15, 125.87, 125.35, 125.25, 83.66 (t, J = 20.2 Hz), 81.31, 64.45, 42.82, 41.39, 22.45, 22.12, 22.06, 20.70. 19F NMR (470 MHz, CDCl3) δ −97.47 (dd, J = 206.8, 59.6 Hz), −98.41 (dd, J = 87.7, 60.0 Hz). IR (Film) 2978, 2864, 1758, 1697, 1469, 1432, 1335, 1231, 1141, 1036, 738, 697 cm–1. M.P. 93 – 95 °C. HRMS (APCI)+ m/z calc’d C20H27BF2NO4 [M+H]+ 394.2001, found 394.2009.
4-(difluoromethylene)piperidin-1-ium chloride (32):
Following general procedure C, (Me2N)3P+CF2CO2− (5.0 g, 20 mmol, 2.0 equiv.) and tert-Butyl 4-(difluoromethylene)piperidine-1-carboxylate (2.4 g, 12 mmol) in PhMe:DMA (160 mL, 3:1, 0.075 M) were heated in a 100 °C oil bath and stirred for 3 hours. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:20) to furnish 2.1 g (75% yield) of desired product as a colorless crystalline solid. 1H NMR (500 MHz, CDCl3) δ 9.74 (s, 2H), 3.17 (s, 4H), 2.53 (s, 4H). 13C{1H} NMR (126 MHz, CDCl3) δ 151.5 (t, J = 283 Hz), 81.2 (t, J = 21 Hz), 43.6, 20.6. 19F NMR (470 MHz, CDCl3) δ −95.31. IR (Film) 2943, 2795, 2768, 2716, 2610, 2488, 1766, 1269, 1220, 1144, 1049, 725, 562 cm–1. M.P. 209 – 212 °C. HRMS (APCI)+ m/z calc’d C6H10F2N [M+H]+ 134.0776, found 134.0772.
((3aR,5S,6aR)-6-(difluoromethylene)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methyl benzoate carboxylate:
Following general procedure C, (Me2N)3P+CF2CO2− (5.0 g, 20 mmol, 2.4 equiv.) and ((3aR,5R,6aS)-2,2-dimethyl-6-oxotetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methyl benzoate (2.0 g, 8.5 mmol) in PhMe:DMA (110 mL, 3:1, 0.075 M) were heated in a 100 °C oil bath and stirred for 3 hours. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:1) to furnish 420 mg (26% yield) of desired product as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 7.4 Hz, 2H), 7.56 (t, J = 6.9 Hz, 1H), 7.43 (t, J = 7.7 Hz, 2H), 5.97 (d, J = 4.1 Hz, 2H), 4.64 (s, 1H), 4.56 (dd, J = 12.0, 2.4 Hz, 1H), 4.38 (dd, J = 12.0, 4.5 Hz, 1H), 1.52 (s, 3H), 1.40 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 165.9, 152.1 (t, J = 291 Hz), 133.1, 129.5, 128.3, 105.5, 90.0 (t, J = 21 Hz), 86.9, 78.5, 77.9, 75.2, 64.4, 27.3, 27.2. 19F NMR (470 MHz, CDCl3) δ −82.81 (d, J = 39.0 Hz), −84.13 (d, J = 39.0 Hz). IR (Film) 2993, 1731, 1699, 1429, 1270, 1226, 1115, 1071, 1025, 999.7 cm–1. HRMS (APCI)+ m/z calc’d C16H17F2O5 [M+H]+ 327.1044, found 327.1045.
(E)-4-(3,4,5-trimethoxyphenyl)but-3-en-2-one:
3,4,5-Trimethoxybenzaldehyde (19.6 g, 100 mmol) was added to a round-bottomed flask equipped with a magnetic stir bar. The system was sealed with a rubber septa, and subsequently evacuated and backfilled with dry N2 three times. Acetone (125 mL, 1.69 mol, 17 equiv.) and 1 M NaOH in H2O (100 mmol) was charged to the flask and stirred at room temperature for 1 h. The reaction was then monitored by TLC for the conversion of the aldehyde. Upon completion, the mixture was concentrated under reduced pressure. The resulting residue was diluted in H2O (300 mL) and washed with EtOAc (100 mL; three times). The organic fractions were then combined and washed with brine, dried over Na2SO4 and concentrated. The resulting residue was purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 7:3) to furnish 9.86 g (42% yield) of desired product as a pale-yellow solid. 1H NMR spectrum matches a previous report.29
4-(3,4,5-trimethoxyphenyl)butan-2-one :
(E)-4-(3,4,5-Trimethoxyphenyl)but-3-en-2-one (5.0 g, 21.2 mmol) and 5% Pd/C (450 mg, 0.21 mmol, 0.010 equiv.) were added to a round-bottomed flask equipped with a magnetic stir bar. The system was sealed with a rubber septa, and subsequently evacuated and backfilled with dry N2 three times. Dry THF:MeOH (100 mL, 9:1, 0.20 M) was added via syringe, and a balloon containing H2 was charged to the flask. The reaction was monitored for completion by TLC. Upon completion, the reaction mixture was filtered through a plug of celite using MeOH to remove Pd/C. Care was taken to maintain a layer of MeOH on the Pd/C to minimize the risk of ignition. The resultant filtrate was concentrated and purified by normal-phase flash chromatography using EtOAc and hexanes (1 − 7:3) to furnish 3.7 g (73% yield) of desired product as a pale-yellow oil. 1H NMR spectrum matches a previous report.30
5-(4,4-difluoro-3-methylbut-3-en-1-yl)-1,2,3-trimethoxybenzene:
Following general procedure D, (Me2N)3P+CF2CO2− (4.7 g, 18 mmol, 2.0 equiv.) and 4-(3,4,5-trimethoxyphenyl)butan-2-one (2.17 g, 9.1 mmol) in PhMe:DMA (120 mL, 3:1 0.075 M) were heated in a 100 °C oil bath and stirred for 3 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 − 1:2) to furnish 592 mg (24% yield) of desired product 3 as a pale-yellow oil. 1H NMR (500 MHz, CDCl3) δ 6.39 (s, 1H), 3.85 (s, 1H), 3.82 (s, 1H), 2.68 – 2.61 (m, 1H), 2.26 (ddt, J = 10.0, 8.2, 2.1 Hz, 1H), 1.59 (t, J = 3.1 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 153.0, 152.9 (t, J = 282.0 Hz), 136.9, 136.1, 105.0, 84.1 (t, J = 18.8 Hz), 60.8, 55.9, 33.8, 30.0, 11.9. 19F NMR (470 MHz, CDCl3) δ −97.5 (d, J = 57.6 Hz, 1F), −97.9 (d, J = 57.5 Hz, 1F). IR (Film) 2930, 2839, 1756, 1589, 1457, 1237, 1126, 1103, 1010, 826.4 cm–1. HRMS (APCI)+ m/z calc’d C14H18F2O3 [M+H]+ 273.1302, found 273.1308.
General procedure D, for the Photochemical Hydrothiolation of gem-Difluoroalkenes:
An oven-dried one-dram vial equipped with a magnetic stir bar was charged with gem-difluoroalkene (1.0 mmol, 1.0 equiv.), 1-(4-aminophenyl)ethan-1-one (0.10 mmol, 0.10 equiv.), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.5 µmol, 0.0025 equiv.). The system was sealed with PTFE lined septa, and subsequently evacuated and backfilled with dry N2 three times. Dry MeCN (0.50 M) and thiol (1.5 mmol, 1.5 equiv.) were added via syringe, and the vial was irradiated by a 40 W 427 nm LED lamp cooled by a fan (< 30 °C) for 24 h. Upon completion, the reaction was concentrated onto diatomaceous earth and purified by normal-phase flash chromatography using EtOAc and hexanes to provide the desired product in >95% purity.
General Procedure E for the Photochemical Hydrothiolation of gem-Difluoroalkene:
An oven-dried one-dram vial equipped with a magnetic stir bar was charged with gem-difluoroalkene (0.50 mmol, 1.0 equiv.), 1-(4-aminophenyl)ethan-1-one (0.025 mmol, 0.050 equiv.), (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (1.25 µmol, 0.0025 equiv.) and thiol (0.50 – 0.70 mmol, 1.0 – 1.4 equiv.). The system was sealed with PTFE lined septa, and subsequently evacuated and backfilled with dry N2 three times. Water (0.50 M) was added via syringe, and the vial was irradiated by a 40 W 427 nm LED lamp cooled by a fan (< 30 °C) for 24 h. Upon completion, the reaction was concentrated onto diatomaceous earth and purified by reverse-phase flash chromatography using MeCN and 1% AcOH in H2O to provide the desired product in >95% purity.
3-((difluoro(4-phenylcyclohexyl)methyl)thio)phenol (27a):
Following general procedure D, (4-(difluoromethylene)cyclohexyl)benzene (0.208 g, 1.00 mmol) was reacted with 3-mercaptophenol (0.153 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:5) to furnish 0.301 g (90% yield) of desired product 27a as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.5 Hz, 2H), 7.26 (t, J = 7.9 Hz, 2H), 7.25 – 7.18 (m, 4H), 7.12 (s, 1H), 6.90 (dd, J = 7.9, 2.4 Hz, 1H), 4.88 (s, 1H), 2.53 (dt, J = 11.4, 5.6 Hz, 1H), 2.16 (d, J = 11.8 Hz, 2H), 2.13 – 2.06 (m, 2H), 2.03 (d, J = 11.7 Hz, 2H), 1.51 (h, J = 12.9 Hz, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 152.9, 144.1, 127.4, 126.0, 125.9, 125.8 (t, J = 272 Hz), 124.2, 123.6, 120.2, 114.2, 43.4 (t, J = 21.5 Hz), 41.1, 30.5, 24.2. 19F NMR (470 MHz, CDCl3) δ −81.76 (d, J = 9.3 Hz). IR (Film) 3393, 2935, 2860, 1585, 1450, 1171, 1041, 943, 759, 781, 699 cm–1. M.P. 76 – 78 °C. HRMS (APCI)+ m/z calc’d C19H20FOS [M-F]+ 315.1219, found 315.1215.
3-((1,1-difluoro-2-methyl-4-(3,4,5-trimethoxyphenyl)butyl)thio)phenol (27b):
Following general procedure D, 5-(4,4-difluoro-3-methylbut-3-en-1-yl)-1,2,3-trimethoxybenzene (0.267 g, 1.00 mmol) was reacted with 3-mercaptophenol (0.153 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 7:3) to furnish 0.383 g (96% yield) of desired product 27b as a pale-yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.19 (t, J = 7.8 Hz, 1H), 7.13 (s, 1H), 7.11 (s, 1H), 6.95 – 6.88 (m, 2H), 6.43 (s, 2H), 3.87 (s, 3H), 3.85 (s, 6H), 2.83 – 2.65 (m, 1H), 2.60 – 2.45 (m, 1H), 2.40 – 1.97 (m, 2H), 1.64 (dtd, J = 14.3, 9.5, 9.0, 4.6 Hz, 1H), 1.21 (d, J = 6.9 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 156.2, 153.0, 137.7, 135.6, 132.5 (t, J = 282.0 Hz), 129.8, 128.0, 127.5, 123.0, 117.0, 105.3, 61.0, 56.1, 41.4 (t, J = 21.5 Hz), 33.5, 32.4, 13.9. 19F NMR (470 MHz, CDCl3) δ −77.75 – −78.31 (dd, J = 202.9, 13.7 Hz, 1F), −78.60 (dd, J = 202.9, 13.7 Hz, 1F). IR (Film) 3391, 2940, 2840, 1590, 1461, 1238, 1126, 996, 952, 886 cm–1. HRMS (APCI)+ m/z calc’d C20H24F2O4S [M+H]+ 399.1442, found 399.1434.
tert-butyl 4-(difluoro((3-hydroxyphenyl)thio)methyl)piperidine-1-carboxylate (27c):
Following general procedure D, tert-butyl 4-(difluoromethylene)piperidine-1-carboxylate (0.233 g, 1.00 mmol) was reacted with 3-mercaptophenol (0.153 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:10) to furnish 0.304 g (85% yield) of desired product 27c as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 7.23 (t, J = 7.6 Hz, 1H), 7.12 (s, 1H), 7.11 (d, J = 6.9 Hz, 1H), 6.95 – 6.89 (m, 1H), 6.48 (s, 1H), 4.22 (d, J = 11.4 Hz, 2H), 2.67 (t, J = 12.5 Hz, 2H), 2.14 (ddd, J = 15.2, 7.4, 3.3 Hz, 1H), 1.92 (d, J = 13.1 Hz, 2H), 1.53 (qd, J = 12.7, 4.3 Hz, 2H), 1.47 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 152.3, 135.0, 127.4, 125.4, 124.6, 120.2, 114.6, 42.1, 25.9, 23.3. 19F NMR (470 MHz, CDCl3) δ −82.74. IR (Film) 3258, 1661, 1589, 1475, 1432, 1370, 1247, 1166, 1138, 1035, 961, 784, 698, 685 cm–1. M.P. 129 – 131 °C. HRMS (ESI)− m/z calc’d C17H22F2NO3S [M-H]− 358.1289, found 358.1278.
benzyl 4-(difluoro((3-hydroxyphenyl)thio)methyl)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)piperidine-1-carboxylate (27d):
Following general procedure D, benzyl 4-(difluoromethylene)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)piperidine-1-carboxylate (0.394 g, 1.00 mmol) was reacted with 3-mercaptophenol (0.153 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:2) to furnish 0.437 g (84% yield) of desired product 28d as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 9.70 (s, 1H), 7.47 – 7.38 (m, 3H), 7.34 (dd, J = 8.2, 5.8 Hz, 1H), 7.29 (t, J = 7.9 Hz, 1H), 7.17 – 7.12 (m, 1H), 7.08 (t, J = 7.4 Hz, 1H), 6.98 (ddd, J = 8.2, 2.4, 0.8 Hz, 1H), 5.25 – 5.17 (m, 1H), 5.12 (s, 1H), 4.28 (t, J = 13.9 Hz, 1H), 4.21 – 4.06 (m, 21H), 3.24 (s, 1H), 3.12 – 2.96 (m, 1H), 2.46 (dt, J = 26.6, 12.0 Hz, 1H), 2.18 – 1.95 (m, 1H), 1.80 (d, J = 12.2 Hz, 1H), 1.25 (s, 3H), 1.21 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 158.4, 154.5, 137.6, 131.7, 130.0, 128.5, 127.8, 127.7, 127.3, 126.6, 122.8, 117.3, 83.2, 66.5, 47.5, 45.1, 43.3, 24.6, 24.2. 19F NMR (470 MHz, CDCl3) δ −76.14 – −78.25 (m). IR (Film) 3316, 2975, 2927, 2864, 1672, 1584, 1474, 1334, 1141, 1103, 968, 848, 761, 694 cm–1. M.P. 53 – 56 °C. HRMS (APCI)+ m/z calc’d C26H33BF2NO5S [M+H]+ 520.2141, found 520.2138.
4-(difluoro((3-hydroxyphenyl)thio)methyl)piperidin-1-ium chloride (27e):
Following general procedure D, 4-(difluoromethylene)piperidin-1-ium chloride (0.133 g, 1.00 mmol) was reacted with 3-mercaptophenol (0.153 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated by precipitation with Et2O and filtering to furnish 0.289 g (98% yield) of desired product 27e as a colorless solid. 1H NMR (500 MHz, DMSO-d6)) δ 8.01 (dd, J = 8.3, 1.3 Hz, 2H), 7.75 – 7.52 (m, 1H), 7.52 – 7.34 (m, 1H), 6.02 – 5.94 (m, 2H), 5.34 – 5.11 (m, 1H), 4.68 – 4.31 (m, 2H), 1.53 (s, 2H), 1.42 (s, 2H). 13C{1H} NMR (126 MHz, DMSO-d6) δ 152.2 (t, J = 294 Hz), 133.2, 129.5, 128.4, 113.3, 105.6, 78.6, 75.3, 64.5, 27.4 (d, J = 12.2 Hz). 19F NMR (470 MHz, DMSO-d6)) δ −83.74 (d, J = 10.6 Hz). IR (Film) 3211, 2958, 2805, 2727, 2486, 1583, 1474, 1437, 1039, 978, 887, 780, 692 cm–1. M.P. 179 – 181 °C. HRMS (APCI)+ m/z calc’d C12H16F2NOS [M+H]+ 260.0921, found 260.0915.
benzyl 4-(difluoro((3-hydroxyphenyl)thio)methyl)piperidine-1-carboxylate (27f):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with 3-mercaptophenol (0.153 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:2) to furnish 0.361 g (92% yield) of desired product 27f as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 7.28 – 7.22 (m, 5H), 7.12 (t, J = 7.9 Hz, 1H), 7.01 (d, J = 8.7 Hz, 1H), 6.80 (dd, J = 8.2, 2.3 Hz, 2H), 5.04 (s, 2H), 4.20 (s, 2H), 2.64 (s, 2H), 2.15 – 1.97 (m, 1H), 1.82 (s, 2H), 1.53 – 1.35 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 153.6, 152.8, 133.8, 127.4, 125.9, 125.6, 125.4, 124.5, 120.2, 114.6, 64.9, 42.0, 40.8, 23.2. 19F NMR (470 MHz, CDCl3) δ −82.74 (d, J = 36.2 Hz). IR (Film) 3315, 2954, 2927, 2885, 1699, 1468, 1430, 1228, 1027, 963, 764, 696 cm–1. M.P. 128 – 131 °C. HRMS (ESI)− m/z calc’d C20H20F2NO3S [M-H]− 392.1132, found 392.1125.
((3aR,5S,6aR)-6-(difluoro((3-hydroxyphenyl)thio)methyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methyl benzoate (27g):
Following general procedure D, ((3aR,5S,6aR)-6-(difluoromethylene)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methyl benzoate carboxylate (0.325 g, 1.00 mmol) was reacted with 3-mercaptophenol (0.153 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:2) to furnish 0.129 g (57% yield) of desired product 27g as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 8.01 (d, J = 8.3 Hz, 1H), 7.66 – 7.55 (m, 2H), 7.45 (t, J = 7.8 Hz, 2H), 5.99 (d, J = 4.2 Hz, 1H), 5.46 – 5.30 (m, 1H), 5.23 (s, 1H), 4.58 (dd, J = 12.0, 2.4 Hz, 1H), 4.40 (dd, J = 12.0, 4.5 Hz, 0H), 1.53 (s, 3H), 1.42 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 165.9, 152.1, 133.2, 129.9, 129.5, 128.4, 127.9, 122.6, 117.2, 113.2, 105.5, 104.4, 90.0, 79.5, 78.5, 75.2, 64.4, 63.3, 27.4, 27.3. 19F NMR (470 MHz, CDCl3) δ −67.63 (dd, J = 216.8, 8.8 Hz), −71.63 (dd, J = 216.8, 14.0 Hz). IR (Film) 3412, 2990, 2940, 1880, 1721, 1452, 1269, 1245, 1024, 866, 709, 687 cm–1. HRMS (ESI)− m/z calc’d C22H21F2O6S [M-H]− 451.1027, found 451.1020.
Benzyl 4-(((4-fluorophenyl)thio)difluoromethyl)piperidine-1-carboxylate (29a):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with 4-fluorobenzenethiol (192 mg, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 2:5) to furnish 0.357 g (90% yield) of desired product 29a as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 7.58 (d, J = 5.3 Hz, 2H), 7.39 – 7.35 (m, 4H), 7.36 – 7.29 (m, 1H), 7.08 (t, J = 8.6 Hz, 2H), 5.14 (s, 2H), 4.32 (d, J = 21.6 Hz, 2H), 2.75 (s, 2H), 2.17 (tdd, J = 11.7, 10.0, 5.8 Hz, 1H), 1.94 (s, 2H), 1.58 (d, J = 21.8 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.9 (t, J = 250.6 Hz), 155.0, 138.6 (d, J = 8.6 Hz), 136.6, 128.4, 128.0, 127.8, 121.2, 116.3, 116.1, 67.1, 44.5 (t, J = 22.5 Hz), 43.2, 25.7. 19F NMR (470 MHz, CDCl3) δ −82.90 (t, J = 15.6 Hz). IR (Film) 2980, 2962, 2864, 1696, 1588, 1490, 1227, 1130, 961, 832, 697 cm–1. HRMS (APCI)+ m/z calc’d C20H21F3NO2S [M+H]+ 396.1245, found 396.1239.
Benzyl 4-(((4-chlorophenyl)thio)difluoromethyl)piperidine-1-carboxylate (29b):
Following general procedure A, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with 4-chlorobenzenethiol (217 mg, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 2:5) to furnish 0.384 g (93% yield) of desired product 29b as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 8.4 Hz, 2H), 7.41 – 7.30 (m, 7H), 5.15 (s, 2H), 4.32 (d, J = 33.9 Hz, 2H), 2.75 (s, 2H), 2.25 – 2.11 (m, 1H), 1.93 (s, 2H), 1.56 (s, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 155.2, 137.7, 136.8, 136.6, 130.7 (t, J = 281.5 Hz), 129.4, 128.6, 128.2, 128.0, 124.7, 67.3, 44.8 (t, J = 22.4 Hz), 43.4, 25.9. 19F NMR (470 MHz, CDCl3) δ −82.55 (dd, J = 55.6, 11.8 Hz, 2F). IR (Film) 2954, 2934, 2862, 1695, 1472, 1429, 1309, 1228, 1130, 960, 763, 696 cm–1. M.P. 72 – 74 °C. HRMS (APCI)+ m/z calc’d C20H21ClF2NO2S [M+H]+ 412.0950, found 412.0942.
Benzyl 4-(((4-bromophenyl)thio)difluoromethyl)piperidine-1-carboxylate (29c):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with 4-bromobenzenethiol (284 mg, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 2:5) to furnish 0.434 g (95% yield) of desired product 29c as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 4.2 Hz, 3H), 7.33 (dt, J = 9.1, 4.6 Hz, 1H), 5.15 (s, 2H), 4.32 (d, J = 24.9 Hz, 2H), 2.75 (s, 2H), 2.18 (pt, J = 11.9, 3.6 Hz, 1H), 1.93 (s, 2H), 1.56 (s, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 155.2, 137.9, 136.7, 132.4, 130.6 (t, J = 281.6 Hz), 128.6, 128.2, 128.0, 125.3, 124.8, 67.3, 44.8 (t, J = 22.3 Hz), 43.3, 25.9. 19F NMR (470 MHz, CDCl3) δ −82.45 (dd, J = 56.4, 11.5 Hz, 2F). IR (Film) 3066, 3031, 2941, 2863 1696, 1475, 1429, 1228, 1130, 961, 928, 747, 697 cm–1. M.P. 53 – 54 °C. HRMS (APCI)+ m/z calc’d C20H21BrF2NO2S [M+H]+ 456.0444, found 456.0442.
benzyl 4-(((4-acetamidophenyl)thio)difluoromethyl)piperidine-1-carboxylate (29d):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with N-(4-mercaptophenyl)acetamide (251 mg, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 The material was isolated according to the general procedure and purified by reversed-phase flash chromatography using MeCN and 0.1% AcOH in H2O (5 – 100%) to furnish 0.323 g (74% yield) of desired product 29d as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 7.73 (s, 1H), 7.53 (q, J = 8.9 Hz, 3H), 7.41 – 7.29 (m, 5H), 5.14 (s, 2H), 4.30 (s, 2H), 2.74 (s, 2H), 2.17 (s, 4H), 1.92 (s, 2H), 1.53 (dd, J = 13.1, 3.6 Hz, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.5, 155.1, 139.6, 137.3, 136.5, 130.6 (t, J = 287.7 Hz), 128.4, 128.0, 127.8, 120.5, 119.8, 67.1, 44.4 (t, J = 22.5 Hz), 43.2, 25.7, 24.6. 19F NMR (470 MHz, CDCl3) δ −80.74 (dd, J = 42.7, 12.2 Hz, 2F). IR (Film) 3314, 1675, 1591, 1528, 1473, 1434, 1396, 1367, 1310, 1232, 1178, 1132, 1036, 962, 834, 697 cm–1. M.P. 134 – 136 °C. HRMS (APCI)+ m/z calc’d C22H24F2N2O3S [M+H]+ 435.1553, found 435.1560.
benzyl 4-(difluoro((3-methoxyphenyl)thio)methyl)piperidine-1-carboxylate (29e):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with 3-methoxybenzenethiol (186 µL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 1:9) to furnish 0.372 g (91% yield) of desired product 29e as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.39 – 7.36 (m, 4H), 7.35 – 7.31 (m, 1H), 7.29 (t, J = 8.0 Hz, 1H), 7.18 (d, J = 7.7 Hz, 1H), 7.15 – 7.13 (m, 1H), 6.97 (dd, J = 7.8, 3.1 Hz, 1H), 5.14 (s, 2H), 4.31 (d, J = 26.7 Hz, 2H), 3.82 (s, 3H), 2.74 (s, 2H), 2.23 – 2.11 (m, 1H), 1.95 (s, 2H), 1.57 (s, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 159.6, 155.0, 136.6, 130.7 (t, J = 281.2 Hz), 129.7, 128.4, 128.3, 128.0, 127.8, 127.1, 121.2, 115.7, 67.1, 55.3, 44.6 (t, J = 22.7 Hz), 43.2, 25.7. 19F NMR (470 MHz, CDCl3) δ −80.21 (dd, J = 39.7, 12.2 Hz, 2F). IR (Film) 2956, 1698, 1591, 1576, 1476, 1365, 1310, 1282, 1231, 1177, 1038, 962, 693 cm–1. HRMS (APCI)+ m/z calc’d C21H23F2NO3S [M+H]+ 408.1444, found 408.1449.
benzyl 4-(difluoro((4-methoxyphenyl)thio)methyl)piperidine-1-carboxylate (29f):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with 4-methoxybenzenethiol (210 mg, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 1:9) to furnish 0.296 g (73% yield) of desired product 29f as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 8.9 Hz, 2H), 7.43 – 7.29 (m, 5H), 6.90 (d, J = 8.9 Hz, 2H), 5.14 (s, 2H), 4.30 (d, J = 24.2 Hz, 2H), 3.82 (s, 3H), 2.74 (s, 2H), 2.22 – 2.06 (m, 1H), 1.94 (s, 2H), 1.54 (s, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 161.0, 155.0, 138.2, 136.6, 130.5 (t, J = 284.6 Hz), 128.4, 128.0, 127.8, 116.5, 114.6, 67.1, 55.3, 44.3 (t, J = 22.7 Hz), 43.2, 25.7. 19F NMR (470 MHz, CDCl3) δ −81.31 (dd, J = 51.9, 12.2 Hz, 2F). IR (Film) 2955, 1698, 1591, 1495, 1432, 1310, 1250, 1230, 1174, 1131, 1029, 962, 829, 698 cm–1. HRMS (APCI)+ m/z calc’d C21H23F2NO3S [M+H]+ 408.1444, found 408.1455.
benzyl 4-(difluoro((2-(hydroxymethyl)phenyl)thio)methyl)piperidine-1-carboxylate (29g):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with (2-mercaptophenyl)methanol (210 mg, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 2:5) to furnish 0.343 g (84% yield) of desired product 29g as a colorless solid. 1H NMR (800 MHz, CDCl3) δ 7.62 (dd, J = 22.9, 7.7 Hz, 2H), 7.46 (t, J = 7.5 Hz, 1H), 7.38 (d, J = 6.3 Hz, 2H), 7.36 – 7.33 (m, 1H), 7.31 (t, J = 7.5 Hz, 1H), 5.14 (s, 2H), 4.88 (s, 2H), 4.31 (d, J = 11.0 Hz, 1H), 2.82 – 2.62 (m, 2H), 2.33 – 2.16 (m, 2H), 1.95 (s, 2H), 1.57 (d, J = 10.3 Hz, 2H). 13C{1H} NMR (201 MHz, CDCl3) δ 155.1, 146.1, 138.5, 136.6, 130.8 (t, J = 281.6 Hz), 130.7, 128.5, 128.1, 127.9, 123.7, 67.2, 63.3, 44.9 (t, J = 22.2 Hz), 43.2, 25.8. 19F NMR (470 MHz, CDCl3) δ −81.74. IR (Film) 3426, 3063, 2954, 2934, 2864, 1696, 1678, 1435, 1230, 1131, 959, 756, 697 cm–1. HRMS (APCI)+ m/z calc’d C21H24F2NO3S [M+H]+ 408.1441, found 408.1441.
benzyl 4-(((2,6-dimethylphenyl)thio)difluoromethyl)piperidine-1-carboxylate (29h):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with 2,6-dimethylbenzenethiol (207 mg, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 2:5) to furnish 0.391 g (96% yield) of desired product 29h as a colorless solid. 1H NMR (800 MHz, CDCl3) δ 7.38 (d, J = 4.5 Hz, 4H), 7.33 (dt, J = 8.6, 4.4 Hz, 1H), 7.22 (t, J = 7.5 Hz, 1H), 7.15 (d, J = 7.5 Hz, 2H), 5.16 (s, 2H), 4.34 (d, J = 47.7 Hz, 2H), 2.79 (s, 2H), 2.52 (s, 6H), 2.32 – 2.17 (m, 2H), 2.01 (s, 1H), 1.63 (s, 2H). 13C{1H} NMR (201 MHz, CDCl3) δ 155.1, 145.5, 136.7, 131.9 (t, J = 281.9 Hz), 130.0, 128.5, 128.2, 128.0, 127.9, 124.9, 67.2, 45.4, 43.3, 25.9, 22.4. 19F NMR (470 MHz, CDCl3) δ −78.85 (d, J = 9.6 Hz). IR (Film) 3064, 3033, 2955, 2924, 2863, 1697, 1429, 1308, 1227, 1129, 1027, 958, 766, 696 cm–1. M.P. 99 – 101 °C. HRMS (APCI)+ m/z calc’d C22H26F2NO2S [M+H]+ 406.1652, found 406.1637.
benzyl 4-(difluoro((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)thio)methyl)piperidine-1-carboxylate (29i):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzenethiol (354 mg, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 2:5) to furnish 0.450 g (89% yield) of desired product 29i as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 7.9 Hz, 2H), 7.59 (d, J = 7.9 Hz, 2H), 7.37 (d, J = 3.5 Hz, 4H), 7.34 – 7.30 (m, 1H), 5.14 (s, 2H), 4.28 (s, 2H), 2.73 (s, 2H), 2.14 (dd, J = 22.7, 11.0 Hz, 2H), 1.93 (s, 2H), 1.55 (d, J = 9.8 Hz, 2H), 1.35 (s, 12H). 13C{1H} NMR (126 MHz, CDCl3) δ 155.0, 136.6, 135.2, 135.1, 130.1 (t, J = 142.1 Hz), 128.4, 128.0, 127.8, 84.0, 67.1, 44.6 (t, J = 22.5 Hz), 43.2, 25.7, 24.8. 19F NMR (470 MHz, CDCl3) δ −80.04 (dd, J = 39.4, 10.2 Hz). IR (Film) 3056, 2978, 2934, 2870, 1697, 1358, 1229, 1141, 734, 697 cm–1. M.P. 114 – 116 °C. HRMS (APCI)+ m/z calc’d C26H32BF2NO4S [M+H]+ 504.2191, found 504.2186.
(difluoro(4-phenylcyclohexyl)methyl)(octyl)sulfane (30a):
Following general procedure D, (4-(difluoromethylene)cyclohexyl)benzene (0.208 g, 1.00 mmol) was reacted with octane-1-thiol (0.260 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:5) to furnish 0.327 g (92% yield) of desired product 30a as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 7.35 – 7.29 (m, 1H), 7.23 (s, 1H), 7.22 – 7.20 (m, 1H), 2.97 – 2.80 (m, 1H), 2.54 (dt, J = 18.1, 9.1 Hz, 1H), 2.11 (d, J = 8.9 Hz, 2H), 2.02 (d, J = 8.0 Hz, 2H), 1.80 – 1.67 (m, 1H), 1.61 (dd, J = 14.1, 6.8 Hz, 1H), 1.54 – 1.46 (m, 3H), 1.44 – 1.39 (m, 2H), 1.33 (dd, J = 14.0, 5.2 Hz, 11H), 0.97 – 0.80 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 146.6, 138.2 (t, J = 277.9 Hz), 128.3, 126.6, 126.0, 46.2 (t, J = 21.8 Hz), 43.6, 33.0, 31.7, 29.9, 29.1, 29.0, 28.8, 27.3, 26.7, 22.5, 14.0. 19F NMR (470 MHz, CDCl3) δ −80.30 (d, J = 10.2 Hz). IR (Film) 2925, 2855, 1493, 1451, 1169, 1024, 974, 956, 756, 698 cm–1. HRMS (APCI)+ m/z calc’d C21H32F1S [M-F]+ 335.2209, found 335.2202.
benzyl 4-(difluoro(octylthio)methyl)piperidine-1-carboxylate (30b):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with octane-1-thiol (0.260 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:2) to furnish 0.406 g (98% yield) of desired product 30b as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.36 – 7.26 (m, 5H), 5.13 (s, 1H), 4.29 (d, J = 30.0 Hz, 2H), 2.94 – 2.78 (m, 1H), 2.73 (s, 2H), 2.23 – 2.05 (m, 1H), 1.87 (s, 2H), 1.65 (p, J = 7.4 Hz, 2H), 1.50 (s, 1H), 1.43 – 1.35 (m, 2H), 1.30 (dd, J = 14.6, 6.1 Hz, 11H), 0.89 (t, J = 6.9 Hz, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 152.5, 134.2, 129.9 (t, J = 278.1 Hz), 125.9, 125.5, 125.3, 64.6, 42.6 (t, J = 22.8 Hz), 40.7, 29.2, 27.4, 26.6, 26.3, 24.9, 23.3, 20.1, 11.6. 19F NMR (470 MHz, CDCl3) δ −83.43 (t, J = 11.1 Hz). IR (Film) 2954, 2927, 2855, 1699, 1430, 1309, 1228, 1130, 1027, 963, 764, 696 cm–1. HRMS (APCI)+ m/z calc’d C22H34F2NO2S [M+H]+ 414.2278, found 414.2271.
(1,1-difluoro-2-methyl-4-(3,4,5-trimethoxyphenyl)butyl)(octyl)sulfane (30c):
Following general procedure D, 5-(4,4-difluoro-3-methylbut-3-en-1-yl)-1,2,3-trimethoxybenzene (0.267 g, 1.00 mmol) was reacted with octane-1-thiol (0.260 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 7:10) to furnish 0.351 g (84% yield) of desired product 30c as a pale-yellow oil. 1H NMR (500 MHz, CDCl3) δ 6.40 (s, 2H), 3.85 (s, 6H), 3.82 (s, 3H), 2.82 (t, J = 7.5 Hz, 2H), 2.71 (ddd, J = 14.7, 10.2, 5.1 Hz, 1H), 2.51 (ddd, J = 13.8, 10.0, 6.8 Hz, 1H), 2.19 – 2.11 (m, 1H), 2.06 (dddd, J = 13.9, 10.5, 6.8, 3.7 Hz, 1H), 1.64 (p, J = 7.3 Hz, 2H), 1.56 (dtd, J = 14.9, 9.8, 5.2 Hz, 1H), 1.41 – 1.33 (m, 2H), 1.31 – 1.22 (m, 8H), 1.14 (d, J = 6.9 Hz, 3H), 0.87 (t, J = 6.6 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 153.3, 137.4, 136.3, 134.2 (t, J = 279.3 Hz), 105.3, 61.0, 56.2, 42.1 (t, J = 21.8 Hz), 33.7, 32.6, 31.9, 30.1, 29.3, 29.2, 29.0, 27.5, 22.8, 14.2. 19F NMR (470 MHz, CDCl3) δ −81.58 (d, J = 12.2 Hz, 2F). IR (Film) 2927, 2855, 1589, 1456, 1420, 1340, 1238, 1127, 1009, 954.6, cm–1. HRMS (APCI)+ m/z calc’d C22H36F2O3S [M+H]+ 418.2431, found 419.2422.
Benzyl 4-(((3-butoxy-3-oxopropyl)thio)difluoromethyl)piperidine-1-carboxylate (30d):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with butyl 3-mercaptopropanoate (0.244 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 1:2) to furnish 0.420 g (98% yield) of desired product 30d as a pale-yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.35 (s, 2H), 7.34 – 7.28 (m, 3H), 5.12 (s, 2H), 4.27 (d, J = 28.9 Hz, 2H), 4.10 (t, J = 6.7 Hz, 2H), 3.07 (t, J = 7.2 Hz, 2H), 2.70 (t, J = 7.2 Hz, 4H), 2.13 (p, J = 11.6 Hz, 1H), 1.85 (s, 2H), 1.60 (p, J = 6.6 Hz 2H), 1.47 (s, 2H), 1.37 (h, J = 7.5 Hz, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 171.6, 155.1, 136.7, 132.5 (t, J = 278.8 Hz), 128.5, 128.1, 127.9, 67.2, 64.7, 45.1 (t, J = 22.5 Hz), 43.3, 35.6, 30.6, 25.8, 22.6, 19.1, 13.7. 19F NMR (470 MHz, CDCl3) δ −83.45 (s, 2F). IR (Film) 2958, 2870, 2931, 1733, 1697, 1428, 1228, 1129, 963, 928, 764, 697 cm–1. HRMS (APCI)+ m/z calc’d C21H30F2NO4S [M+H]+ 430.1864, found 430.1854.
benzyl 4-((benzylthio)difluoromethyl)piperidine-1-carboxylate (30e):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with phenylmethanethiol (0.176 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:2) to furnish 0.385 g (98% yield) of desired product 30e as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 7.38 – 7.35 (m, 4H), 7.35 – 7.31 (m, 5H), 7.29 – 7.26 (m, 1H), 5.13 (s, 2H), 4.28 (s, 2H), 4.08 (s, 2H), 2.72 (s, 2H), 2.13 (ddd, J = 15.1, 7.5, 3.3 Hz, 1H), 1.87 (s, 2H), 1.49 (d, J = 10.5 Hz, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 155.0, 136.7, 136.6, 132.0 (t, J = 279.1 Hz), 129.0, 128.6, 128.5, 128.0, 127.9, 127.4, 67.1, 44.9 (t, J = 22.5 Hz), 43.2, 31.8, 25.8.19F NMR (470 MHz, CDCl3) δ −80.70. IR (Film) 2954, 2937, 2859, 1695, 1429, 1228, 1129, 962, 928, 763, 695, 603 cm–1. M.P. 55 – 57 °C. HRMS (APCI)+ m/z calc’d C21H24F2NO2S [M+H]+ 392.1496, found 392.1487.
benzyl 4-((cyclohexylthio)difluoromethyl)piperidine-1-carboxylate (30f):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with cyclohexanethiol (184 µL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 1:4) to furnish 0.353 g (92% yield) of desired product 30f as a pale-yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.45 – 7.27 (m, 5H), 5.13 (s, 2H), 4.63 – 4.16 (m, 2H), 3.40 – 3.20 (m, 1H), 2.86 – 2.62 (m, 2H), 2.16 – 2.07 (m, 1H), 2.06 – 1.99 (m, 2H), 1.93 – 1.82 (m, 2H), 1.78 – 1.69 (m, 2H), 1.62 – 1.56 (m, 1H), 1.53 – 1.32 (m, 6H), 1.30 – 1.23 (m, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 155.0, 136.6, 132.7, 128.4, 127.9, 127.8, 67.0, 45.4 (t, J = 23.1 Hz), 45.2, 43.2, 41.5, 34.6, 25.8, 25.3. 19F NMR (470 MHz, CDCl3) δ −79.07 (s, 2F). IR (Film) 2933, 1699, 1428, 1308, 1227, 1128, 1026, 962, 738, 696 cm–1. HRMS (APCI)+ m/z calc’d C20H28F2NO2S [M+H]+ 384.1808, found 384.1818.
benzyl 4-(difluoro((1-hydroxyhexan-3-yl)thio)methyl)piperidine-1-carboxylate (30g):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with 3-mercaptohexan-1-ol (207 µL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 2:3) to furnish 0.388 g (96% yield) of desired product 30g as a pale-yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.41 – 7.28 (m, 5H), 5.12 (s, 2H), 4.27 (s, 2H), 3.87 – 3.62 (m, 2H), 3.45 – 3.28 (m, 1H), 2.72 (s, 2H), 2.24 – 2.00 (m, 2H), 1.99 – 1.80 (m, 3H), 1.80 – 1.70 (m, 1H), 1.70 – 1.54 (m, 2H), 1.54 – 1.35 (m, 4H), 0.91 (t, J = 7.3 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 155.0, 136.5, 132.6 (t, J = 278.1 Hz), 128.4, 127.9, 127.8, 67.1, 60.0, 45.3 (t, J = 22.9 Hz), 43.2, 40.3, 38.7, 38.4, 25.8, 19.6, 13.7. 19F NMR (470 MHz, CDCl3) δ −78.08 (d, J = 205.2 Hz, 1F), −79.14 (d, J = 205.8 Hz, 1F). IR (Film) 3442, 2958, 1698, 1435, 1310, 1231, 1132, 1030, 765, 697 cm–1. HRMS (APCI)+ m/z calc’d C20H29F2NO3S [M+H]+ 402.1914, found 402.1922.
benzyl 4-(((1-ethoxy-1-oxopropan-2-yl)thio)difluoromethyl)piperidine-1-carboxylate (32h):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with ethyl 2-mercaptopropanoate (198 µL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 1:4) to furnish 0.369 g (92% yield) of desired product 32h as a pale-yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.45 – 7.23 (m, 5H), 5.10 (s, 2H), 4.46 – 4.07 (m, 4H), 3.94 (q, J = 7.2 Hz, 1H), 2.70 (s, 2H), 2.26 – 2.02 (m, 1H), 1.83 (s, 2H), 1.52 (d, J = 7.3 Hz, 3H), 1.45 (d, J = 7.8 Hz, 2H), 1.24 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 172.4, 154.9, 136.6, 131.7 (t, J = 280.4 Hz), 128.4, 127.9, 127.8, 67.0, 61.5, 44.8 (t, J = 22.3 Hz), 43.1, 39.2, 25.6, 18.8, 13.9. 19F NMR (470 MHz, CDCl3) δ −79.30 (dd, J = 204.1, 10.1 Hz, 1F), −80.44 (dd, J = 204.2, 110.5 Hz, 1F). IR (Film) 2937, 1736, 1699, 1431, 1311, 1230, 1177, 1027, 764, 698 cm–1. HRMS (APCI)+ m/z calc’d C19H25F2NO4S [M+H]+ 402.1550, found 402.1561.
benzyl 4-((adamantan-1-ylthio)difluoromethyl)piperidine-1-carboxylate (30i):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with 1-adamantanethiol (200 mg, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:9) to furnish 0.373 g (86% yield) of desired product 30i as a pale-yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.52 – 7.28 (m, 5H), 5.14 (d, J = 7.7 Hz, 2H), 4.28 (d, J = 24.9 Hz, 2H), 3.62 – 3.39 (m, 1H), 2.72 (s, 2H), 2.10 (s, 7H), 2.05 (s, 3H), 1.87 (s, 2H), 1.71 (s, 5H), 1.59 – 1.39 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 155.0, 136.6, 134.2 (t, J = 279.5), 128.4, 127.9, 127.8, 67.1, 67.0, 50.9, 46.2 (t, J = 23.0 Hz), 44.4, 43.9, 43.2, 36.0, 29.9, 25.7. 19F NMR (470 MHz, CDCl3) δ −75.16 (d, J = 10.4 Hz, 2F). IR (Film) 2907, 1701, 1430, 1302, 1278, 1228, 1128, 1026, 958, 696 cm–1. HRMS (APCI)+ m/z calc’d C24H31F2NO2S [M+H]+ 436.2121, found 436.2108.
benzyl 4-(difluoro((2-(4-methyl-2-oxocyclohexyl)propan-2-yl)thio)methyl)piperidine-1-carboxylate (30j):
With slight modification to general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with 2-(2-mercaptopropan-2-yl)-5-methylcyclohexan-1-one (279 µL, 1.50 mmol) in the presence of 4-fluoroaniline (9.5 µL, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (1 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by reverse-phase flash chromatography using MeCN and 0.1% AcOH in H2O (5 – 100%) to furnish 0.182 g (40% yield) of desired product 30j as a pale-yellow oil. The d.r. of the product mixture was determined by the ratio of 1H NMR peak integrations at δ 1.01 (major) and δ 0.93 (minor). The NMR data are reported as a 4.4:1 mixture of diastereomers in CDCl3. 1H NMR for the major diastereomer (500 MHz, CDCl3) δ 7.38 – 7.32 (m, 4H), 7.33 – 7.28 (m, 1H), 5.11 (s, 2H), 4.26 (d, J = 32.1 Hz, 2H), 2.84 (dd, J = 13.0, 4.5 Hz, 1H), 2.70 (s, 2H), 2.59 – 2.39 (m, 1H), 2.32 – 2.22 (m, 1H), 2.13 – 1.98 (m, 2H), 1.97 – 1.78 (m, 4H), 1.69 (s, 3H), 1.59 – 1.19 (m, 7H), 1.01 (d, J = 6.3 Hz, 3H). 1H NMR for the minor diastereomer (500 MHz, CDCl3) δ 7.38 – 7.32 (m, 4H), 7.33 – 7.28 (m, 1H), 5.11 (s, 2H), 4.26 (d, J = 32.1 Hz, 2H), 2.70 (s, 2H), 2.59 – 2.39 (m, 1H), 2.45 – 2.38 (m, 1H), 2.38 – 2.31 (m, 1H), 2.13 – 1.98 (m, 2H), 1.97 – 1.78 (m, 4H), 1.69 (s, 3H), 1.59 – 1.19 (m, 7H), 0.93 (d, J = 7.1 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 210.7, 210.4, 155.1, 136.8, 134.0 (t, J = 280.2 Hz), 128.6, 128.1, 128.0, 67.2, 59.1, 53.5, 52.3, 50.3, 46.3 (t, J = 22.9 Hz), 43.4, 36.8, 34.5, 32.6, 31.4, 29.9, 29.0, 28.8, 25.9, 25.8, 25.7, 22.3, 18.7. 19F NMR (470 MHz, CDCl3) δ −75.33 – −80.62 (m, 2F). IR (Film) 2955, 2869, 1698, 1430, 1307, 1228, 1127, 1089, 1026, 961 cm–1. HRMS (APCI)+ m/z calc’d C24H33F2NO3S [M+H]+ 454.2227, found 454.2235.
((S)-3-(((1-((benzyloxy)carbonyl)piperidin-4-yl)difluoromethyl)thio)-2-methylpropanoyl)-L-proline (30k):
Following general procedure D, benzyl 4-(difluoromethylene)piperidine-1-carboxylate (0.267 g, 1.00 mmol) was reacted with captopril (326 mg, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by reversed-phase flash chromatography using MeCN and 1% AcOH in H2O (5 – 100%) to furnish 0.415 g (86% yield) of desired product 30k as a pale-yellow oil. 1H NMR (500 MHz, CDCl3) δ 10.83 (s, 1H), 7.43 – 7.27 (m, 5H), 5.11 (s, 2H), 4.64 – 4.50 (m, 1H), 4.26 (d, J = 23.8 Hz, 2H), 3.68 – 3.46 (m, 2H), 3.16 – 3.03 (m, 1H), 3.04 – 2.92 (m, 1H), 2.93 – 2.84 (m, 1H), 2.83 – 2.62 (m, 2H), 2.23 (s, 1H), 2.17 – 1.96 (m, 4H), 1.84 (s, 2H), 1.44 (s, 2H), 1.22 (dd, J = 6.7, 2.8 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 174.9, 174.4, 155.0, 136.5, 132.4 (t, J = 279.1 Hz), 128.4, 128.0, 127.8, 67.2, 59.1, 47.2, 44.9 (t, J = 22.5 Hz), 43.1, 39.5, 30.4, 28.2, 25.6, 24.6, 16.9. 19F NMR (470 MHz, CDCl3) δ −79.58 (dd, J = 204.1, 83.5 Hz, 1F), −81.76 (dd, J = 204.1, 80.0 Hz, 1F). IR (Film) 2956, 1698, 1644, 1435, 1312, 1231, 1176, 1029, 753, 699 cm–1. HRMS (APCI)+ m/z calc’d C23H30F2N2O5S [M+H]+ 485.1921, found 485.1934.
(1,1-difluoro-4-phenylbutan-2-yl)(octyl)sulfane (30l):
Following general procedure D, (4,4-difluorobut-3-en-1-yl)benzene (0.168 g, 1.00 mmol) was reacted with octane-1-thiol (0.260 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal phase preparative thin layer chromatography using 100% pentane to furnish 0.128 g (41% yield) of desired product 30l as a colorless oil. The branched:linear regioselectivity (1.6:1) of the crude reaction mixture was determined by the ratio of 19F NMR peak integrations at δ −118.7, −120.9 (branched) and δ −74.4 (linear). 1H NMR (500 MHz, CDCl3) δ 7.37 – 7.28 (m, 2H), 7.23 (d, J = 7.4 Hz, 3H), 5.80 (td, J = 56.8, 4.3 Hz, 1H), 2.98 (ddd, J = 14.0, 9.1, 5.0 Hz, 1H), 2.87 – 2.68 (m, 2H), 2.61 (t, J = 7.4 Hz, 2H), 2.19 – 2.01 (m, 1H), 1.84 – 1.69 (m, 1H), 1.58 (dtd, J = 15.0, 7.4, 4.6 Hz, 2H), 1.43 – 1.35 (m, 2H), 1.35 – 1.20 (m, 8H), 0.90 (t, J = 6.8 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 141.0, 128.6, 126.3, 118.2 (t, J = 246.1 Hz), 47.1 (t, J = 20.8 Hz), 32.5, 32.0, 31.8, 30.5, 29.8, 29.3, 29.2, 29.0, 22.8, 14.3. 19F NMR (470 MHz, CDCl3) δ −118.21 (ddd, J = 275.2, 56.8, 11.9 Hz, 1F), −120.41 (ddd, J = 274.6, 57.1, 14.8 Hz, 1F). IR (Film) 2955, 2926, 2856, 1497, 1455, 1380, 1130, 1031, 750.1, 698.7 cm–1. HRMS (APCI)+ m/z calc’d C18H28F2S [M+H]+ 315.1958, found 315.1956.
3-((1,1-difluoro-2-(3,4,5-trimethoxyphenyl)ethyl)thio)phenol (31a):
Following general procedure D, 5-(2,2-difluorovinyl)-1,2,3-trimethoxybenzene (0.230 g, 1.00 mmol) was reacted with 3-mercaptophenol (0.153 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:5) to furnish 0.314 g (88% yield) of desired product 31a as a colorless oil. 1H NMR and 19F NMR spectra match a previous report.13
3-((1,1-difluoro-2-(3,4,5-trimethoxyphenyl)ethyl)thio)aniline (31b):
Following general procedure D, 5-(2,2-difluorovinyl)-1,2,3-trimethoxybenzene (0.246 g, 1.00 mmol) was reacted with 3-aminobenzenethiol (0.159 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using MeOH and CH2Cl2 (1:20) to furnish 0.299 g (84% yield) of desired product 31b as a colorless oil. 1H NMR and 19F NMR spectra match a previous report.13
2-(3-(2,2-difluoro-2-(phenylthio)ethyl)phenyl)-5-(1,3-dioxolan-2-yl)pyridine (31c):
Following general procedure D, 2-(3-(2,2-difluorovinyl)phenyl)-5-(1,3-dioxolan-2-yl)pyridine (0.289 g, 1.00 mmol) was reacted with benzenethiol (0.153 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:5) to furnish 0.304 g (76% yield) of desired product 31c as a colorless solid. 1H NMR and 19F NMR spectra match a previous report.13
4-(2,2-difluoro-2-(phenylthio)ethyl)dibenzo[b,d]thiophene (31d):
Following general procedure D, 4-(2,2-difluorovinyl)dibenzo[b,d]thiophene (0.246 g, 1.00 mmol) was reacted with benzenethiol (0.153 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:10) to furnish 0.335 g (94% yield) of desired product 31d as a colorless solid. 1H NMR and 19F NMR spectra match a previous report.13
tert-butyl 4-(5-(2,2-difluoro-2-(phenylthio)ethyl)thiazol-2-yl)piperazine-1-carboxylate (31e):
Following general procedure D, tert-butyl 4-(5-(2,2-difluorovinyl)thiazol-2-yl)piperazine-1-carboxylate (0.331 g, 1.00 mmol) was reacted with benzenethiol (0.153 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 7:3) to furnish 0.314 g (71% yield) of desired product 31e as a colorless solid. 1H NMR and 19F NMR spectra match a previous report.13
4-(2,2-difluoro-2-(phenylthio)ethyl)benzonitrile (31f):
Following general procedure D, 4-(2,2-difluorovinyl)benzonitrile (0.165 g, 1.00 mmol) was reacted with benzenethiol (0.153 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (0 – 1:5) to furnish 0.226 g (82% yield) of desired product 31f as a colorless solid. 1H NMR and 19F NMR spectra match a previous report.13
(1,1-difluoro-2-(3-nitrophenyl)ethyl)(phenyl)sulfane (31g):
Following general procedure D, 1-(2,2-difluorovinyl)-3-nitrobenzene (0.230 g, 1.00 mmol) was reacted with benzenethiol (0.153 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:10) to furnish 0.159 g (54% yield) of desired product 31g as a colorless oil. 1H NMR and 19F NMR spectra match a previous report.13
4-(2,2-difluoro-2-(octylthio)ethyl)-1-phenyl-1H-pyrazole (31h):
Following general procedure D, 4-(2,2-difluorovinyl)-1-phenyl-1H-pyrazole (0.206 g, 1.00 mmol) was reacted with octane-1-thiol (0.260 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:10) to furnish 0.303 g (86% yield) of desired product 31h as a colorless solid. 1H NMR and 19F NMR spectra match a previous report.8
3-(2-(benzylthio)-2,2-difluoroethyl)benzonitrile (31i):
Following general procedure D, 3-(2,2-difluorovinyl)benzonitrile (0.246 g, 1.00 mmol) was reacted with phenylmethanethiol (0.176 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:10) to furnish 0.204 g (70% yield) of desired product 31i as a colorless oil. 1H NMR and 19F NMR spectra match a previous report.8
3-(2,2-difluoro-2-(phenethylthio)ethyl)benzonitrile (31j):
Following general procedure D, 3-(2,2-difluorovinyl)benzonitrile (0.246 g, 1.00 mmol) was reacted with 2-phenylethane-1-thiol (0.176 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:10) to furnish 0.258 g (85% yield) of desired product 31j as a colorless oil. 1H NMR and 19F NMR spectra match a previous report.8
3-((1,1-difluoro-2-(1-phenyl-1H-pyrazol-4-yl)ethyl)thio)-1-methoxy-1-oxopropan-2-aminium chloride (31k):
With slight modification to general procedure D, 4-(2,2-difluorovinyl)-1-phenyl-1H-pyrazole (0.206 g, 1.00 mmol) was reacted with L-cysteine methyl ester hydrochloride (0.257 g, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeOH (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by reversed-phase flash chromatography using MeCN and 0.0005% HCl in H2O (5 – 100%) to furnish 0.145 g (38% yield) of desired product 31k as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.80 (s, 3H), 8.48 (s, 1H), 7.85 – 7.77 (m, 2H), 7.69 (s, 1H), 7.54 – 7.46 (m, 2H), 7.31 (td, J = 7.4, 1.1 Hz, 1H), 4.32 (t, J = 5.9 Hz, 1H), 3.73 (s, 3H), 3.51 (t, J = 15.1 Hz, 2H), 3.43 – 3.33 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.4, 142.1, 139.7, 129.9, 128.2, 126.6, 118.4, 113.5, 53.3, 52.2, 34.2 (t, J = 25.3 Hz), 27.6. 19F NMR (470 MHz, DMSO-d6) δ −72.72 (dt, J = 201.4, 16.0 Hz), −73.38 (dt, J = 201.4, 16.0 Hz). IR (Film) 3392, 2957, 2856, 2720, 2638, 1747, 1598, 1502, 1440, 1403, 1382, 1333, 1242, 1160, 1031, 1008, 983, 954, 868, 760, 691, 670 cm–1. M.P. 161 − 164 ºC. HRMS (APCI)+ m/z calc’d C15H18F2N3O2S [M+H]+ 342.1087, found 342.1091.
3-(2,2-difluoro-2-(phenethylthio)ethyl)benzonitrile (31l):
Following general procedure D, 3-(2,2-difluorovinyl)benzonitrile (0.246 g, 1.00 mmol) was reacted with 3-mercaptopropanoic acid (0.131 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using MeOH and CH2Cl2 (1:10) to furnish 0.226 g (83% yield) of desired product 31l as a colorless oil. 1H NMR and 19F NMR spectra match a previous report.8
3-(2,2-difluoro-2-((6-hydroxyhexyl)thio)ethyl)benzonitrile (31m):
Following general procedure D, 3-(2,2-difluorovinyl)benzonitrile (0.165 g, 1.00 mmol) was reacted with 6-mercaptohexan-1-ol (0.205 mL, 1.50 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (0.014 g, 0.10 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in MeCN (2.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by normal-phase flash chromatography using EtOAc and hexanes (1:5) to furnish 0.198 g (66% yield) of desired product 31m as a colorless oil. 1H NMR and 19F NMR spectra match a previous report.8
S-(difluoro(piperidin-4-yl)methyl)-L-cysteine dihydrochloride (33a):
Following general procedure E, 4-(difluoromethylene)piperidin-1-ium chloride ( 84.8 mg, 0.500 mmol) was reacted with L-cysteine hydrochloride (0.102 g, 0. 650 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (3.4 mg, 0.025 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (1.4 mg, 1.25 µmol) in H2O (1.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by reversed-phase flash chromatography using MeCN and 1% AcOH in H2O (5 – 100%) to furnish 0.128 g (75% yield) of desired product 33a as a colorless solid. 1H NMR (500 MHz, D2O) δ 4.14 – 4.08 (m, 1H), 3.57 – 3.47 (m, 3H), 3.40 – 3.30 (m, 1H), 3.09 – 2.96 (m, 2H), 2.62 – 2.49 (m, 1H), 2.15 (d, J = 11.5 Hz, 2H), 1.80 – 1.68 (m, 2H). 13C{1H} NMR (126 MHz, D2O) δ 171.2, 131.2 (t, J = 279.5 Hz), 53.6, 42.9, 41.4 (t, J = 22.7 Hz), 27.4, 22.3. 19F NMR (470 MHz, D2O) δ −79.93 (dd, J = 206.0, 12.2 Hz, 1F), −81.52 (dd, J = 206.0, 12.2 Hz, 1F). IR (Film) 3378, 2943, 2822, 1945, 1738, 1599, 1508, 1456, 1426, 1389 cm–1. M.P. 178 − 180 ºC. HRMS (APCI)+ m/z calc’d C9H16F2N2O2S [M+H]+ 255.0973, found 255.0969.
methyl S-(difluoro(piperidin-4-yl)methyl)-L-cysteinate dihydrochloride (33b):
Following general procedure E, 4-(difluoromethylene)piperidin-1-ium chloride (84.8 mg, 0.500 mmol) was reacted with L-cysteine methyl ester hydrochloride (0.112 g, 0.650 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (3.4 mg, 0.025 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (1.4 mg, 1.25 µmol) in H2O (1.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by reversed-phase flash chromatography using MeCN and 1% AcOH in H2O (5 – 100%) to furnish 0.132 g (81% yield) of desired product 33b as a colorless solid. 1H NMR (500 MHz, D2O) δ 4.59 – 4.42 (m, 1H), 3.84 (s, 3H), 3.61 – 3.43 (m, 4H), 3.01 (t, J = 11.7 Hz, 2H), 2.66 – 2.43 (m, 1H), 2.15 (d, J = 12.7 Hz, 2H), 1.80 – 1.66 (m, 2H). 13C{1H} NMR (126 MHz, D2O) δ 168.4, 131.0 (t, J = 280.1 Hz), 53.8, 52.7, 42.9, 41.3 (t, J = 22.5 Hz), 26.7, 22.3. 19F NMR (470 MHz, CDCl3) δ −80.43 (dd, J = 206.0, 12.2 Hz, 1F), −81.33 (dd, J = 206.0, 12.2 Hz, 1F). IR (Film) 3399, 2955, 2826, 1750, 1598, 1518, 1441, 1332, 1245, 1181 cm–1. M.P. 186 − 188 ºC. HRMS (APCI)+ m/z calc’d C10H18F2N2O2S [M+H]+ 269.1135, found 269.1136.
N-acetyl-S-(difluoro(piperidin-4-yl)methyl)-L-cysteine hydrochloride (33c):
Following general procedure E, 4-(difluoromethylene)piperidin-1-ium chloride (84.8 mg, 0.500 mmol) was reacted with N-acetyl-L-cysteine (0.120 g, 0.700 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (3.4 mg, 0.025 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (1.4 mg, 1.25 µmol ) in H2O (1.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by reversed-phase flash chromatography using MeCN and 1% AcOH in H2O (5 – 100%) to furnish 0.148 g (88% yield) of desired product 33c as a colorless oil. 1H NMR (500 MHz, D2O) δ 4.65 – 4.59 (m, 1H), 3.48 (d, J = 13.9 Hz, 2H), 3.44 – 3.38 (m, 1H), 3.25 – 3.11 (m, 1H), 2.98 (t, J = 11.7 Hz, 2H), 2.55 – 2.40 (m, 1H), 2.11 (d, J = 13.8 Hz, 2H), 2.00 (s, 3H), 1.76 – 1.63 (m, 2H). 13C{1H} NMR (126 MHz, D2O) δ 174.0, 173.2, 131.3 (t, J = 278.8 Hz), 52.8, 42.9, 41.5 (t, J = 23.1 Hz), 28.3, 22.4, 21.6. 19F NMR (470 MHz, D2O) δ −81.14 (s, 2F). IR (Film) 3391, 3265, 3033, 2939, 2812, 2730, 2498, 1725, 1647, 1548 cm–1. HRMS (APCI)+ m/z calc’d C11H18F2N2O3S [M+H]+ 297.1084, found 297.1088.
methyl N-acetyl-S-(difluoro(piperidin-4-yl)methyl)-L-cysteinate hydrochloride (33d):
Following general procedure E, 4-(difluoromethylene)piperidin-1-ium chloride (84.8 mg, 0.500 mmol) was reacted with methyl N-acetyl-L-cysteinate (0.106 g, 0.600 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (3.4 mg, 0.025 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (1.4 mg, 1.25 µmol ) in H2O (1.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure and purified by reversed-phase flash chromatography using MeCN and 1% AcOH in H2O (5 – 100%) to furnish 0.160 g (92% yield) of desired product 33d as a colorless oil. 1H NMR (500 MHz, D2O) δ 4.70 – 4.64 (m, 1H), 3.73 (s, 3H), 3.49 (d, J = 13.3 Hz, 2H), 3.44 – 3.36 (m, 1H), 3.26 – 3.15 (m, 1H), 2.99 (t, J = 13.2 Hz, 2H), 2.58 – 2.41 (m, 1H), 2.11 (d, J = 15.0 Hz, 2H), 2.00 (s, 3H), 1.78 – 1.61 (m, 2H). 13C{1H} NMR (126 MHz, D2O) δ 174.0, 171.9, 131.3 (t, J = 279.7 Hz), 53.2, 52.8, 43.0, 41.5 (t, J = 22.7 Hz), 28.3, 22.4, 21.6. 19F NMR (470 MHz, D2O) δ −81.14 – −81.16 (m, 2F). IR (Film) 3367, 2503, 1732, 1640, 1441, 1227, 1037, 971, 673, 660 cm–1. HRMS (APCI)+ m/z calc’d C12H20F2N2O3S [M+H]+ 311.1235, found 311.1243.
N5-((S)-1-((Carboxymethyl)amino)-3-((difluoro(piperidin-4-yl)methyl)thio)-1-oxopropan-2-yl)-L-glutamine (33e):
With slight modification to general procedure E, 4-(difluoromethylene)piperidin-1-ium chloride (0.0848 g, 0.500 mmol) was reacted with L-glutathione (0.154 g, 0.500 mmol) in the presence of 1-(4-aminophenyl)ethan-1-one (3.4 mg, 0.025 mmol), and (4,4’-di-tert-butyl-2,2’-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-KN)phenyl-KC]iridium(III) PF6 (2.8 mg, 2.5 µmol) in H2O (1.0 mL) using 40 W 427 nm LED cooled by a fan for 24 h. The material was isolated according to the general procedure B and purified by reversed-phase flash chromatography using MeCN and 1% AcOH in H2O (5 – 100%) to furnish 0.201 g (90% yield) of desired product 33e as a colorless solid. 1H NMR (500 MHz, D2O) δ 4.67 (dd, J = 8.5, 5.2 Hz, 1H), 3.92 (s, 2H), 3.78 (t, J = 6.4 Hz, 1H), 3.48 (dt, J = 13.2, 3.1 Hz, 2H), 3.39 (dd, J = 14.5, 5.2 Hz, 1H), 3.15 (dd, J = 14.5, 8.5 Hz, 1H), 2.99 (td, J = 13.2, 3.0 Hz, 2H), 2.50 (h, J = 8.0 Hz, 3H), 2.25 – 2.02 (m, 4H), 1.70 (qd, J = 12.9, 4.2 Hz, 2H). 13C{1H} NMR (126 MHz, D2O) δ 174.7, 173.6, 173.4, 171.9, 132.5 (t, J = 279.6 Hz), 53.7, 53.3, 42.9, 41.9 – 41.4 (m), 41.3, 31.2, 28.4, 26.0, 22.4. 19F NMR (470 MHz, D2O) δ −81.29 (s, 2F). IR (Film) 3391, 1648, 1448, 1317, 1189, 1086, 1033, 917, 832, 674 cm–1. M.P. >250 ºC. HRMS (APCI)+ m/z calc’d C16H26F2N4O6S [M+H]+ 441.1613, found 441.1601.
Supplementary Material
ACKNOWLEDGMENTS
Generous financial support from the National Institute of General Medical Sciences of the National Institutes of Health (R35 GM124661) is gratefully acknowledged. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Mohammed K. Abd El-Gaber and Ahmed Z. Abdelazem are thankful to the Egyptian Ministry of Higher Education for Overseas Visiting Postdoctoral Fellowships (SAB 2339, SAB 2346) and to Assiut University and Beni-Suef University for the sabbatical leaves. Ankit Kumar is thankful to the Indian Science and Engineering Research Board for an Overseas Visiting Doctoral Fellowship.
Footnotes
Supporting Information
The Supporting Information is available free of charge at:
Reaction optimization and copies of 1H, 13C{1H}, and 19F NMR spectra for synthesized compounds
The authors declare no competing financial interests.
Data availability Statement
The data underlying this study are available in the published article and its online supplementary material.
REFERENCES
- (1).Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of Fluorine in Medicinal Chemistry. J. Med. Chem 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- (2).Meanwell NA Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem 2018, 61, 5822–5880. 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- (3).Johnson BM; Shu Y; Zhuo X; Meanwell NA Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. J. Med. Chem 2020, 63, 6315–6386. 10.1021/acs.jmedchem.9b01877. [DOI] [PubMed] [Google Scholar]
- (4).Britton R; Gouverneur V; Lin J-H; Meanwell M; Ni C; Pupo G; Xiao J-C; Hu J Contemporary Synthetic Strategies in Organofluorine Chemistry. Nat Rev Methods Prim 2021, 1 (47). 10.1038/s43586-021-00042-1. [DOI] [Google Scholar]
- (5).Sorrentino JP; Altman RA Fluorine-Retentive Strategies for the Functionalization of Gem-Difluoroalkenes. Synthesis (Stuttg) 2021, 53, 3935–3950. 10.1055/a-1547-9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhang X; Cao S Recent Advances in the Synthesis and C-F Functionalization of Gem-Difluoroalkenes. Tetrahedron Lett 2017, 58 (5), 375–392. 10.1016/j.tetlet.2016.12.054. [DOI] [Google Scholar]
- (7).Hagan DO Understanding Organofluorine Chemistry. An Introduction to the C – F Bond. Chem. Soc. Rev 2008, 37, 308–319. 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]
- (8).Sorrentino JP; Orsi DL; Altman RA Acid-Catalyzed Hydrothiolation of Gem-Difluorostyrenes to Access α,α-Difluoroalkylthioethers. J. Org. Chem 2020, 86, 2297–2311. 10.1021/acs.joc.0c02440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Orsi DL; Altman RA Exploiting the Unusual Effects of Fluorine in Methodology. Chem. Comm 2017, 53, 7168–7181. 10.1039/c7cc02341c. [DOI] [PubMed] [Google Scholar]
- (10).Liu C; Zeng H; Zhu C; Jiang H Recent Advances in Three-Component Difunctionalization of Gem-Difluoroalkenes. Chem. Comm 2020, 56, 10442–10452. 10.1039/d0cc04318d. [DOI] [PubMed] [Google Scholar]
- (11).Koley S; Altman RA Recent Advances in Transition Metal-Catalyzed Functionalization of Gem-Difluoroalkenes. Isr. J. Chem 2020, 60, 313–339. 10.1002/ijch.201900173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Liu C; Zhu C; Cai Y; Jiang H Solvent-Switched Oxidation Selectivities with O2: Controlled Synthesis of α-Difluoro(Thio)Methylated Alcohols and Ketones. Angew. Chem 2021, 133, 2–10. 10.1002/ange.202017271. [DOI] [PubMed] [Google Scholar]
- (13).Orsi DL; Easley BJ; Lick AM; Altman RA Base Catalysis Enables Access to α,α -Difluoroalkylthioethers. Org. Lett 2017, 19 (7), 1570. 10.1021/acs.orglett.7b00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Li J; Xu C; Wei N; Wang M Synthesis of 2,2-Difluorinated 4-Isoflavanols/4-Thioisoflavanols via a Base-Catalyzed [4 + 2] Annulation Reaction of Gem-Difluoroolefins. J. Org. Chem 2017, 82 (21), 11348–11357. 10.1021/acs.joc.7b01635. [DOI] [PubMed] [Google Scholar]
- (15).Suda M Radical Addition Reactions on 1,1-Difluoro-1-Olefins. Tetrahedron Lett 1981, 22 (25), 2395–2396. [Google Scholar]
- (16).Ashirbaev SS; Levin VV; Struchkova MI; Dilman AD Addition of Thiols to Gem-Difluoroalkenes under Photoactivation Conditions. Fluor. Notes 2017, 115, 1–2. [Google Scholar]
- (17).Levin VV; Dilman AD Visible-Light-Mediated Organocatalyzed Thiol−Ene Reaction Initiated by a Proton-Coupled Electron Transfer. J. Org. Chem 2019, 84, 8337–8343. 10.1021/acs.joc.9b01331. [DOI] [PubMed] [Google Scholar]
- (18).Zubkov MO; Kosobokov MD; Levin VV; Kokorekin VA; Korlyukov AA; Hu J; Dilman AD A Novel Photoredox-Active Group for the Generation of Fluorinated Radicals from Difluorostyrenes. Chem. Sci 2020, 11, 737–741. 10.1039/c9sc04643g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Levin VVV; Dilman ADD Visible-Light-Mediated Organocatalyzed Thiol–Ene Reaction Initiated by a Proton-Coupled Electron Transfer. J. Org. Chem 2019, 84 (12), 8337–8343. 10.1021/acs.joc.9b01331. [DOI] [PubMed] [Google Scholar]
- (20).Dilman A Addition of Thiols to Gem-Difluoroalkenes under Photoactivation Conditions. Fluor. Notes 2017, 115 (6), 1–1. 10.17677/fn20714807.2017.06.01. [DOI] [Google Scholar]
- (21).Sorrentino J; Herrick R; Abd El-Gaber M; Abdelazem A; Kumar A; Altman RA A General Co-Catalytic Hydrothiolation of Gem-Difluoroalkenes. ChemRxiv 2022. 10.26434/chemrxiv-2022-txxbr. [DOI] [PMC free article] [PubMed]
- (22).Wang J; Huang B; Yang C; Xia W Visible-Light-Mediated Defluorinative Cross-Coupling of Gem-Difluoroalkenes with Thiols. Chem. Commun 2019, 55, 11103–11106. 10.1039/c9cc05293c. [DOI] [PubMed] [Google Scholar]
- (23).Kade MJ; Burke DJ; Hawker CJ The Power of Thiol-Ene Chemistry. J. Polym. Sci. Part A Polym. Chem 2010, 48 (4), 743–750. 10.1002/pola.23824. [DOI] [Google Scholar]
- (24).Sinha AK; Equbal D Thiol−Ene Reaction: Synthetic Aspects and Mechanistic Studies of an Anti-Markovnikov-Selective Hydrothiolation of Olefins. Asian J. Org.Chem 2019, 8, 32–47. [Google Scholar]
- (25).Hoffmann N Proton‐Coupled Electron Transfer in Photoredox Catalytic Reactions. Eur. J. Org. Chem 2017, 1982–1992.
- (26).Tyson EL; Niemeyer ZL; Yoon TP Redox Mediators in Visible Light Photocatalysis: Photocatalytic Radical Thiol–Ene Additions. J. Org. Chem 2014, 79 (3), 1427–1436. 10.1021/jo500031g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yuan K; Feoktistova T; Cheong PH-YY; Altman RA Arylation of Gem-Difluoroalkenes Using a Pd/Cu Co-Catalytic System That Avoids β-Fluoride Elimination. Chem. Sci 2021, 12 (4), 1363–1367. 10.1039/D0SC05192F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhao Y; Huang W; Zhu L; Hu J Difluoromethyl 2-Pyridyl Sulfone: A New Gem-Difluoroolefination Reagent for Aldehydes and Ketones. Org. Lett. 2010, 12, 1444–1447. 10.1021/ol100090r. [DOI] [PubMed] [Google Scholar]
- (29).Banks MR; Cadogan JIG; Gosney I; Gould RO; Hodgson PKG; Mcdougall D TETRAHEDRON Pergamon Tetrahedron 54 (1998) 9765–9784. Tetrahedron 1998, 54, 9765–9784. [Google Scholar]
- (30).Naikwadi DR; Naikwadi DR; Ravi K; Ravi K; Singh AS; Singh AS; Advani JH; Biradar AV; Biradar AV Gram-Scale Synthesis of Flavoring Ketones in One Pot via Alkylation-Decarboxylation on Benzylic Carbon Using a Commercial Solid Acid Catalyst. ACS Omega 2020, 5 (24), 14291–14296. 10.1021/acsomega.0c00416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its online supplementary material.