

Abstract
The natural world is a prolific source of some of the most interesting, rare, and complex molecules known, harnessing sophisticated biosynthetic machinery evolved over billions of years for their production. Many of these natural products represent high-value targets of total synthesis, either for their desirable biological activities or for their beautiful structures outright; yet, the high sp3-character often present in nature’s molecules imparts significant topological complexity that pushes the limits of contemporary synthetic technology. Dearomatization is a foundational strategy for generating such intricacy from simple materials that has undergone considerable maturation in recent years. This review highlights the recent achievements in the field of dearomative methodology, with a focus on natural product total synthesis and retrosynthetic analysis. Disconnection guidelines and a three-phase dearomative logic are described, and a spotlight is given to nature’s use of dearomatization in the biosynthesis of various classes of natural products. Synthetic studies from 2011 to 2021 are reviewed, and 425 references are cited.
Graphical Abstract
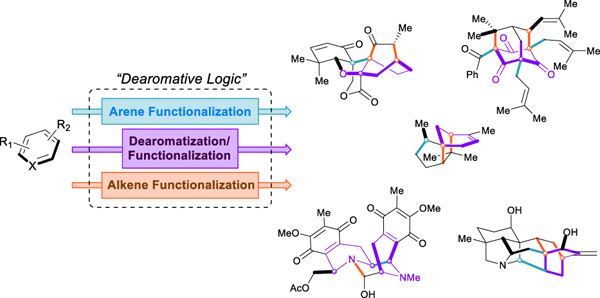
Introduction
Although once fixated on complex molecule structure elucidation and the exploration of synthetic feasibility, the objectives of natural product total synthesis have evolved recently under increased pressure for utility, efficiency, and novel reactivity.1–4 Simultaneously, organic synthesis has infiltrated other fields such as chemical biology, drug development, agriculture, food science, cosmetics and fragrances, alternative energy, microelectronics, and materials science, providing crucial support and molecular tools for interdisciplinary research with extraordinary societal impact.5 The need for practical and scalable approaches to complex molecules is apparent,6 both in support of these other areas as well as in its own right as the field of organic synthesis continues to mature.7
Questions of structure and reactivity notwithstanding, today’s synthetic chemists are primarily concerned with the assembly of molecules possessing desirable properties and functions,8 which, in the area of natural product total synthesis, lie almost exclusively within the vast realm of molecular recognition and its influence upon a biological system. The well-established correlation between structural complexity and biological activity demands synthetic chemists to construct highly saturated molecular architectures that encompass intricate systems of fused or caged rings, numerous and often contiguous stereogenic centers, and a diverse assortment of correctly placed functional groups.9,10 Although history has shown that synthetic chemists can access such intricate structures quite well, doing so in a practical and efficient manner remains a challenge. The continuous development of modern methods has fueled the emergence of distinct strategies that can address these limitations.11 The aim of the present review is to reiterate the value of disconnections-based retrosynthetic analysis12 through the widening lens of dearomative chemistry, a powerful, complexity-generating tactic that has gained traction in recent years due to significant advancements in methodology.13–18
Dearomative Logic in Retrosynthetic Analysis
Given the ubiquity of cyclic motifs within natural products, drugs, and other fine chemicals, the case for dearomative strategy in total synthesis can be stated quite clearly: it is often much easier and more efficient to acquire, manipulate, and then dearomatize arenes than it is to selectively functionalize cyclohexane derivatives or build them de novo (Figure 1). Simple aromatics are produced on hundred-million-ton scales annually and are some of the most inexpensive and abundant feedstock chemicals accessible; the robust methods available for their elaboration—including nucleophilic and electrophilic aromatic substitutions, condensations, directed ortho- metalation, aryne functionalizations, and cross-couplings—are considered to be so classical that they constitute the largest sections of organic chemistry textbooks as well as the bulk of synthetic research published to date.19 Arene functionalization is so well-developed as to make possible the synthesis of nearly every conceivable substitution pattern on a benzene ring.
Figure 1.
Dearomatization as a means to generate complex carbocycles.
Furthermore, the chemistry used to modify aromatic rings is often orthogonal to that used for saturated systems. Sufficiently advanced sp3 C–H activation methods may someday trivialize the construction and functionalization of saturated natural product scaffolds, and many admirable efforts have been made in this field,20–25 but until such technology reaches its pinnacle, synthetic chemists must rely upon the use of pre-functionalized building blocks to forge the requisite bonds of a particular natural product. By their very nature, arenes have embedded within them considerable synthetic potential; with benzene derivatives, for example, a chemist can perform up to three separate transformations and bond-forming events en route to a decorated cyclohexane ring—provided that the first encompass an olefin-like dearomatization. In asymmetric dearomative processes, aromatic rings provide an exponentially greater number of potential chiral compounds than any other class of prochiral substrates. Recently, tremendous advancements have been made in dearomative methodology, increasing the scope and sophistication of building blocks available from feedstock arenes. The rapid generation of complexity from simplicity through dearomative strategies allows us to postulate that this approach will find increased usage as chemists continue to place greater emphasis on efficiency and practicality in total synthesis.26
Disconnection Guidelines
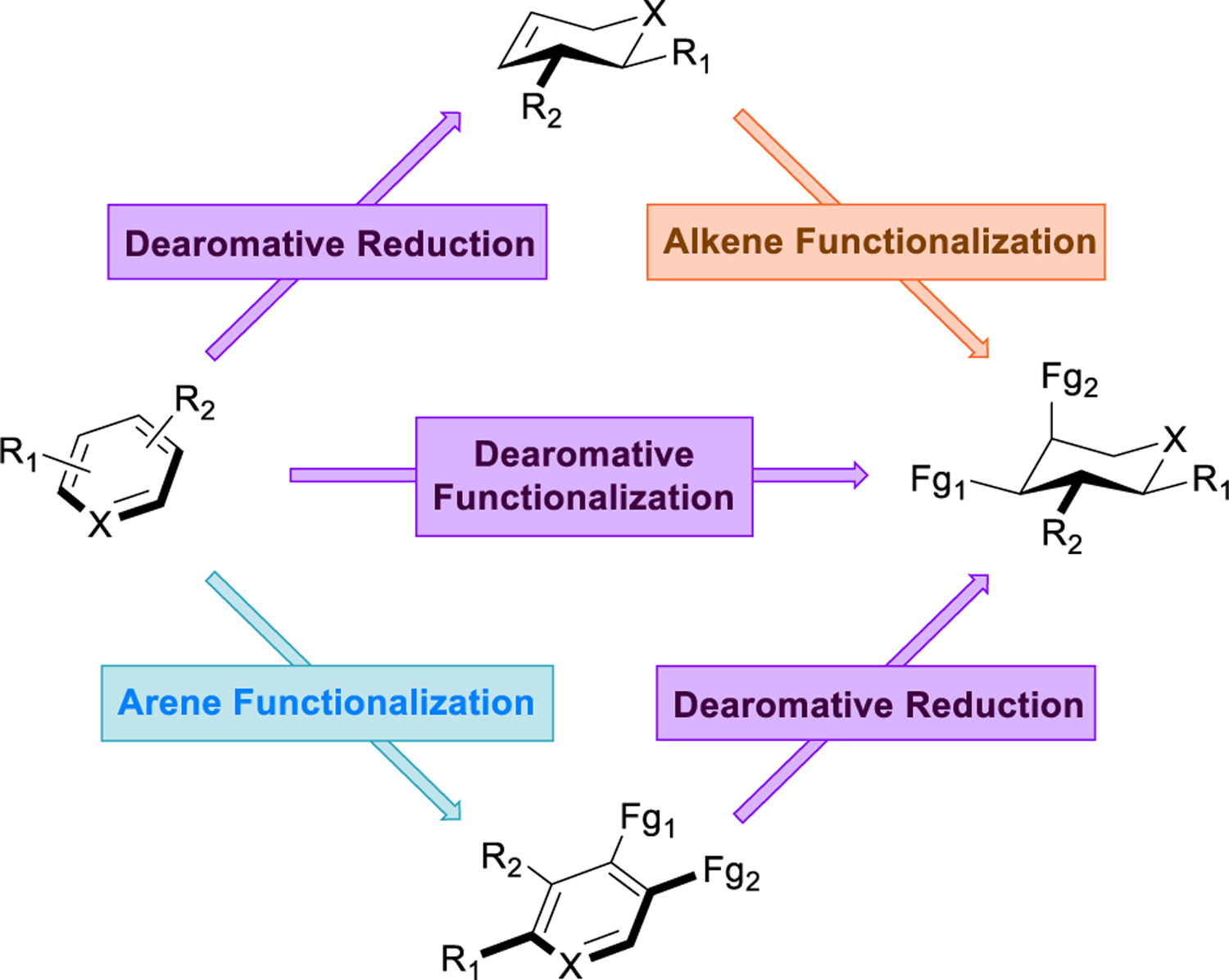
Dearomative logic can be used to complement most retrosynthetic strategies (Figure 2).12 Within transform-based approaches, dearomatization itself can function as powerfully simplifying transform when associated with concomitant C–C or C–X bond disconnection, exemplified by Wender’s use of the arene-alkene meta-photocycloaddition in his total synthesis of α–cedrene.27 Dearomatization can also serve as a means to obtain the precursors for other powerful complexity-generating transformations, such as cyclohexenones for conjugate addition or cyclohexa‑1,3‑dienes for subsequent [4+2] cycloaddition. Liao’s synthesis of penicillone A28 serves to illustrate nicely this tactic that has been used quite frequently in the construction of the bicyclo[2.2.2]octane scaffold. In structure-goal strategies, dearomative transforms can quickly simplify dense, three-dimensional structures into cheap and readily available aromatic compounds, desirable starting materials with the proper carbon count. In Sarlah’s synthesis of the blockbuster antidepressant sertraline, an asymmetric dearomative carboamination imprints all necessary functionality, with proper stereochemistry, directly onto the feedstock chemical naphthalene.29 Topological and network-guided analyses of intricate polycyclic and caged ring systems often dictate strategic bond disconnections that generate retrons for dearomative transforms, simplifying that particular ring even further.30 This approach is showcased in Sarpong’s syntheses of diterpenoid alkaloids like arcutinidine.31 Finally, dearomative transformations can be used to great advantage in stereochemical strategies, both to control relative stereochemistry, such as the use of arene hydrogenation to obtain an all-cis relationship for substituents within a cyclohexane ring, as well as to establish absolute stereochemistry (e.g., through asymmetric hydrogenation of heteroarenes32 or microbial arene oxidations,33 as in Lewis’s synthesis of zeylenol34).
Figure 2.
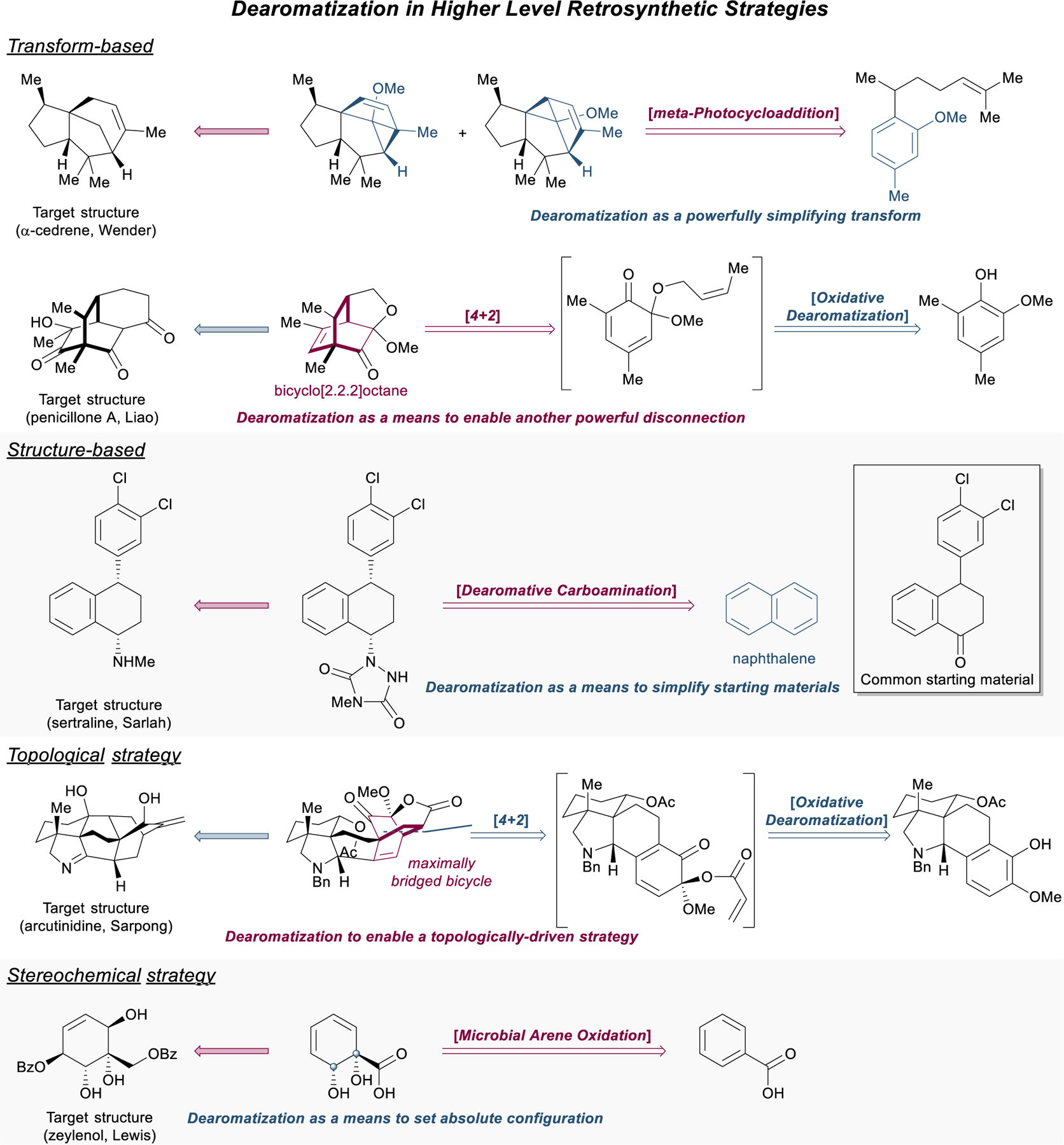
Applications of dearomative logic in high-level retrosynthetic strategies.
Within the framework of a dearomative retrosynthetic logic, there are three fundamental stages in which the decoupled chemistries of aromatics and aliphatics and the practicality of dearomatization can be put to good use (Figure 3). The first phase (denoted in orange) requires the application of any transform on a cyclic system that produces either a full or partial dearomative retron. In many cases this may simply entail a hydrogenation; when possible, however, it is highly strategic to disconnect C–C and C–X bonds that can be formed from the unsaturation left behind after the dearomative event. Typical examples of this include olefin functionalizations, β-functionalization of cyclohexenones through conjugate addition, and cycloadditions. When intramolecular, the latter two transformations can often be achieved concurrently with dearomatization, resulting in dramatic cascade reactions or one-pot processes.35 In this context, one may also consider conformational changes brought on through dearomatization that may facilitate other intramolecular processes (e.g., lactamization in Stoltz’s jorumycin synthesis, vide infra).
Figure 3.
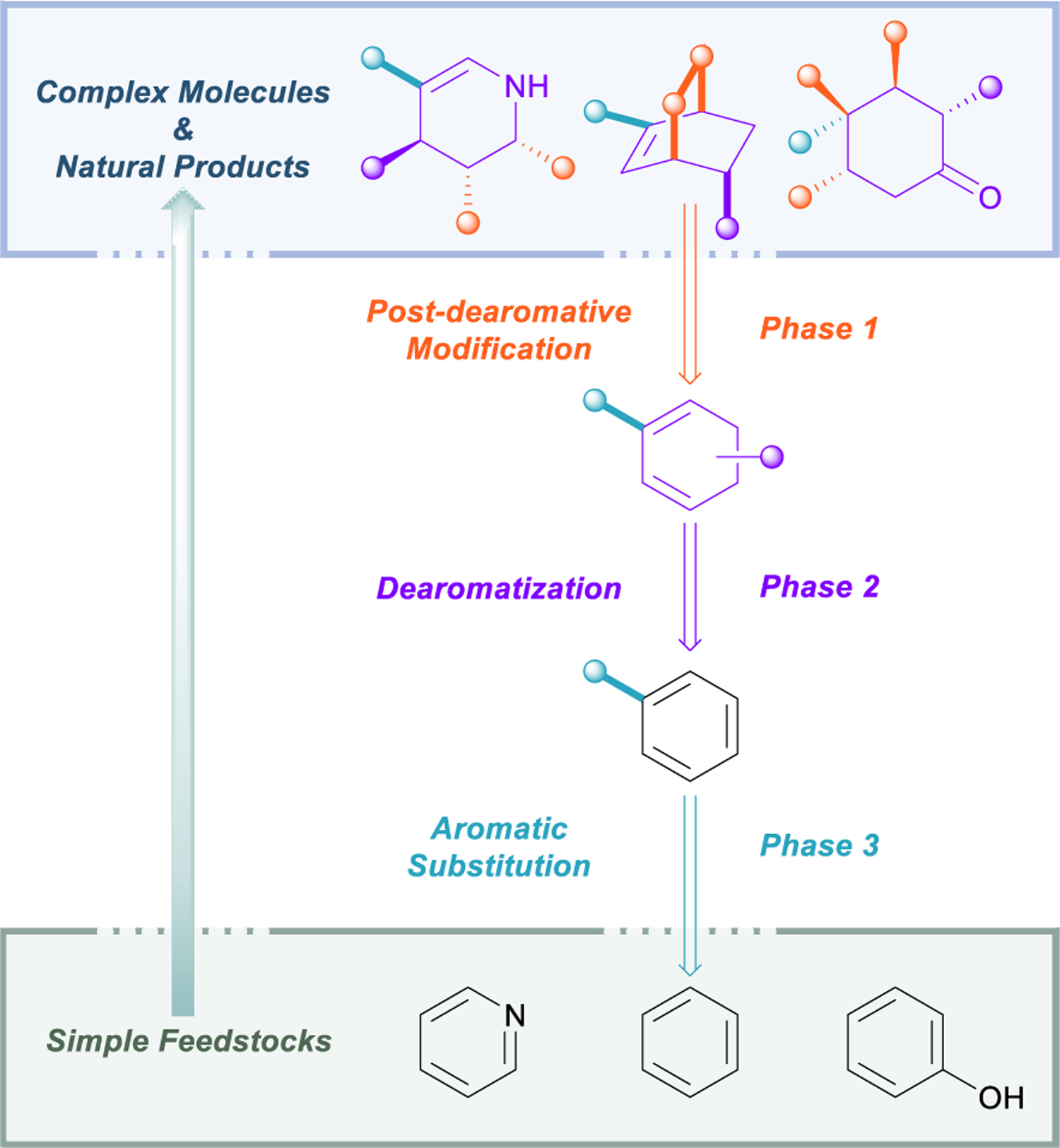
Three phases of dearomative retrosynthetic logic.
The second stage of dearomative analysis, depicted in purple, concerns the dearomative event itself. This of course depends entirely upon the retron generated during the first phase. There may arise situations in which several dearomative retrons could be generated from a single intermediate, and therefore several dearomative processes may be possible. In these cases, one must judge the dearomative and post-dearomative transformations and prioritize the route with the most structurally simplifying disconnection, whether this encompasses the formation of an important C–C bond or the establishment of a critical stereoenter.
Dearomative transforms bridge the gap between the orthogonal realms of arene chemistry and aliphatic/olefin chemistry. Thus, the third and final stage of dearomative logic (marked in light blue) regards those bonds that may be easily formed on an aromatic precursor, but which would be extremely challenging to install directly on an aliphatic system. This tactic is especially useful for constructing C–C and C–X bonds attached to sp3 carbons that are distal to other functional groups in the target structure. In this phase, it is important to keep in mind the availability and cost of variously substituted aromatic starting materials so that a balance may be struck between substituents that are more efficient to “buy” and those that are more practical to build through EAS or cross coupling.
The remainder of this review is devoted to the exhibition of outstanding applications of dearomative logic in the total synthesis of natural products. We have restrained ourselves to showcase examples primarily from the past decade, as an excellent review on this topic was published in 2011.36 Readers interested in earlier cases of dearomatization in total synthesis are highly encouraged to see this work, as well as others.37–40
Applications of Dearomative Logic in Natural Product Total Synthesis
Terpenes
ent-Kaurene diterpenoids:
Leaves and extracts from the Isodon (mint) family of plants were prized in ancient Chinese and Japanese folk medicine for their antibacterial, anticancer, anthelmintic, stomachic, and anti-inflammatory properties.41–43 Many of these herbal remedies are still in use today. Modern isolation studies revealed that many of these plants are prodigious sources of biologically active terpenoids; their ent-kaurene diterpenoids, in particular, possess excellent antibacterial and antitumor pharmacological profiles (Figure 4a). Notable examples include the potent, gram-positive antibiotic platensimycin (1), first reported by Merck in 2006, which inhibits type II bacterial fatty acid synthesis and shows no cross-resistance to any other strains of antibiotic-resistant bacteria,44–46 as well as oridonin (2), the major component of Rabdosia rubescens (donglingcao), a Chinese anti-inflammatory herbal remedy.47–49 Compound 2 has shown great promise for the treatment of a variety of cancers, and its analogue HAO472 (not shown) has recently entered a phase I clinical trial in China for acute myeloid leukemia.50 With their characteristic bicyclo[3.2.1]octene core embedded within a precisely oxidized hydrocarbon skeleton, the ent-kaurene diterpenoids pose a significant challenge for synthetic chemists, and at present only a handful of these natural products have succumbed to total synthesis.42,51,52 Nonetheless, the incipient medicinal properties of furnished ent-kaurene diterpenoids such as 2–4, and of their even further decorated seco-ent-kaurenoids like 1, 5, and 6, have in recent years renewed interest within the synthetic community to take on these formidable targets.
Figure 4.
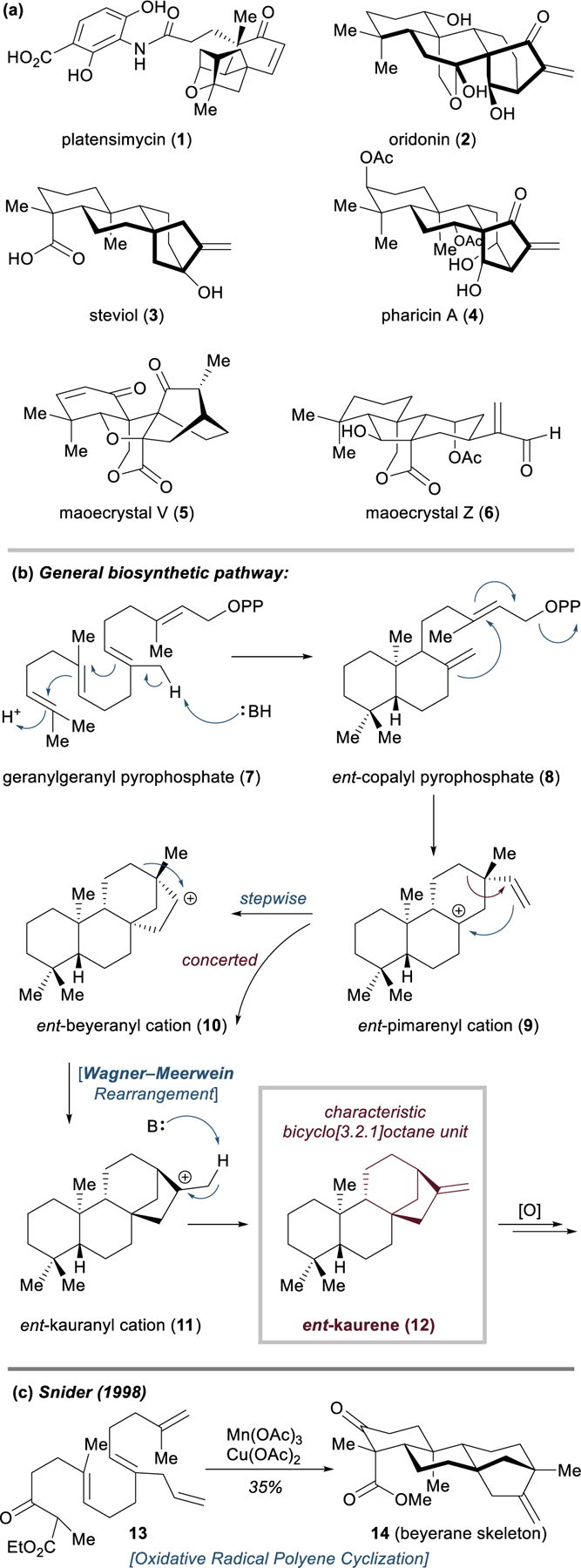
Structures, biosynthesis, and early synthetic work on ent-kaurenes.
ent-Kaurene biosynthesis proceeds first through protonation and conversion of geranylgeranyl pyrophosphate (7) to ent-copalyl pyrophosphate (8), followed by loss of the diphosphate group and 6-exo-trig cyclization (8 → 9), and finally a series of carbocation rearrangements (9 → 12) catalyzed by the ent‑kaurene synthase enzyme (Figure 4b).42,53 The intermediacy of secondary carbocations (e.g., 10) in this mechanism has been questioned in recent years, and a computational study from Tantillo and coworkers strongly supports a mechanism with direct conversion of the ent-pimarenyl cation (9) to the ent‑kaurenyl cation (11) through a concerted cyclization and alkyl shift.54 Without the use of an enzyme catalyst, this precisely orchestrated cascade of C–C bond formations and 1,2-rearrangements would seem an edifice beyond the reach of synthetic chemistry, and indeed such a polyene cyclization has never been reproduced in a laboratory setting. In their total synthesis of isosteviol (not shown), Snider and coworkers reported an impressive Mn(III)/Cu(II) oxidative radical polyene cyclization of 13 that could reliably deliver the isomeric beyerene carbon skeleton 14 (Figure 4c); however, such strategies are not easily adaptable towards the synthesis of the ent‑kaurenoids given the requisite alkyl shift.55 With the possibility of polyene cyclization precluded, multistep routes to the bicyclo[3.2.1]octane have continued to dominate synthetic efforts towards the ent-kaurenes.
Early work from Ireland and Ziegler established two classic dearomative strategies used to construct the C8 quaternary center and bridged ring system, both relying upon Birch reduction of fused phenyl ethers (Figure 5).56–61 Ireland’s synthesis of (±)-kaurene (12) proceeds through a Saucy–Marbet enol ether Claisen rearrangement, followed by intramolecular aldol reaction (15 → 16),56–59 a strategy later used by the Hong group in their total synthesis of cafestol (not shown).62 Pioneering efforts from Ziegler and Kloek established the [2+2] cycloaddition with allene (generating 17) as a powerful tool for the construction of the bicyclo[3.2.1]octane motif in steviol methyl ester (18).60,61 Despite the low yield of the ring expansion (17 → 18), the allene [2+2] has endured as a highly effective way to forge a C–C bond at C8; later, more efficient methods were utilized to trigger fragmentation of the methylenecyclobutane, such as ozonolysis, used in Corey’s synthesis of neotripterifordin (not shown).63 This allene [2+2] and ozonolysis sequence was also applied in Baran’s recent synthesis of steviol (3).64 It is worth noting that in all of these examples, the authors built the most complex and three-dimensional portion of the molecule from what had originally been a flat aromatic ring, leveraging its synthetic potential through dearomatization.
Figure 5.
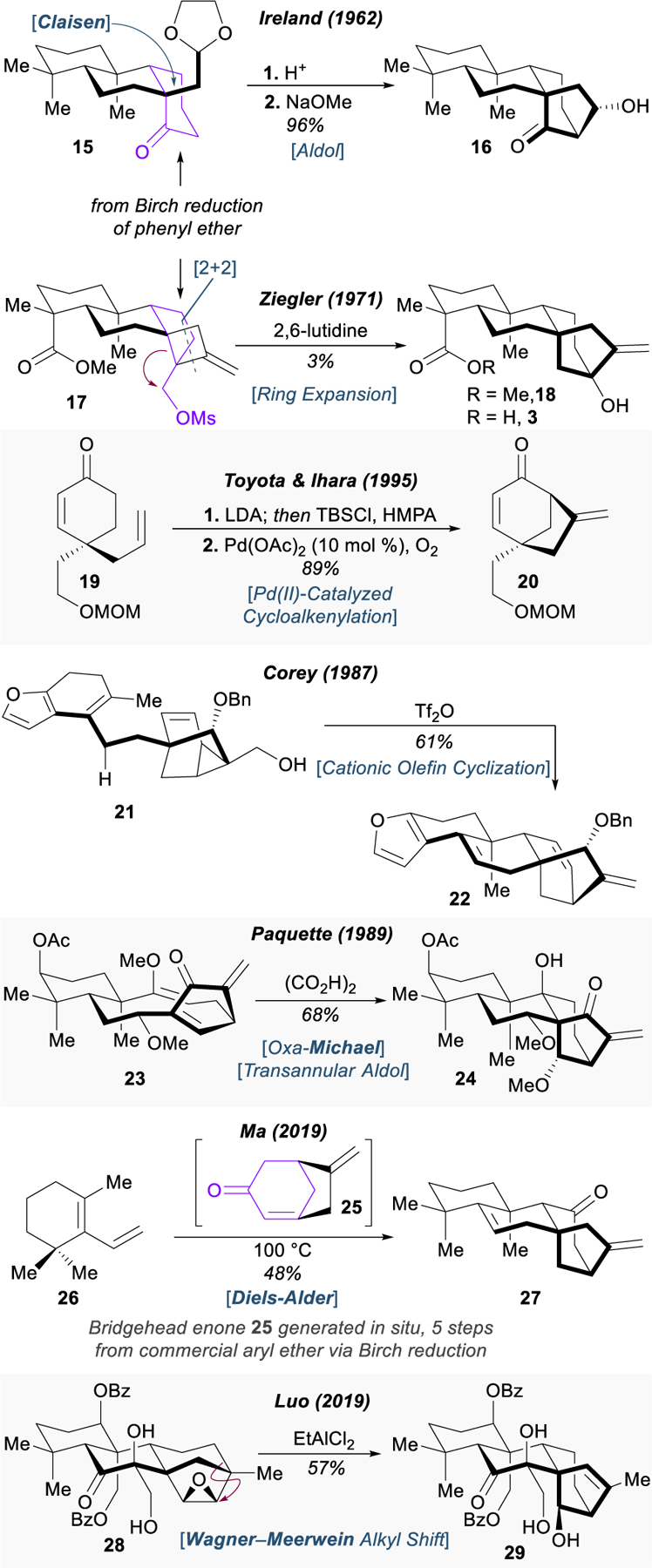
Selected approaches towards the bicyclo[3.2.1]octane motif of the ent-kaurenes.
One of the most widely-adopted methods for construction of the bicyclo[3.2.1]octane is the palladium-catalyzed cycloalkenylation (19 → 20) first reported by Toyota and Ihara in 199465,66 and later used in their total syntheses of the ent-kaurenoids (±)‑methyl atis-16-en-19-oate67 and (–)‑methyl kaurenoate,68 as well as for a number of other terpenoids.69 Toyota and Ihara’s oxidative cyclization reaction would later serve as a key step in Reisman’s syntheses of (–)-trichorabdal A and (–)-longikaurin E,70 and most recently has been used in Ma’s convergent asymmetric syntheses of lungshengenin D and 1⍺,6⍺‑diacteoxy‑ent‑kaura‑9(11),16‑dien‑12,15-dione.71
Beyond these canonical approaches, the architectural complexity of the ent-kaurene scaffold has inspired some interesting alternative strategies. During his synthesis of cafestol, Corey showcased the viability of the cyclopropyl methyl cation to trigger polyolefin cyclization, closing the final ring of the natural product (21 → 22).72 A dramatic cascade reaction incorporating enol ether hydrolysis, hemiacetal formation, oxa-Michael addition, and finally a transannular aldol reaction back into the original ketone served as the final step in Paquette’s synthesis of dimethylshikoccidin (23 → 24).73 Although requiring preinstallation of the bicyclo[3.2.1]octane motif (via radical cyclization), Ma has demonstrated that Diels–Alder cycloaddition with transiently generated bridgehead enone 25 is a powerful tactic to rapidly construct the ent-kaurene skeleton in a convergent fashion (26 → 27).74 This method was applied in the total syntheses of 11β-hydroxy-16-kaurene, 11α-hydroxy-16-kaurene, liangshanin G, and gesneroidin B. Biomimetic conversion of the beyerene skeleton to the kaurene skeleton through 1,2-alkyl shift (28 → 29) was recently achieved by Luo and coworkers, using Lewis acid-promoted epoxide opening to trigger the rearrangement.75 This synthesis is also notable for its application of an interesting interrupted Nazarov cyclization. While impressive, all of these approaches towards the tetracyclic, ent-kaurene scaffold either proceed from complex, advanced intermediates acquired through multistep syntheses, or suffer from a lack of generality.
A fundamentally new, dearomative approach towards the bicyclo[3.2.1]octane structure was reported by Ding and coworkers in 2017 (Figure 6).76 They found that, on mono-protected catechols with a pendent alkene side chain (e.g., 30), an oxidative dearomatization-induced [5+2] cycloaddition yielded cedrene-like cationic intermediates 31 with a proximal bridgehead methoxy group, which readily promoted a pinacol-type 1,2-acyl migration to reveal the bicyclo[3.2.1]octene framework of the ent-kaurenoids (32). A screen of solvents and oxidants was sufficient to optimize the yield of the desired product while minimizing the production of byproducts 33 and 34 from overoxidation or addition of solvent and [4+2] cycloaddition.
Figure 6.
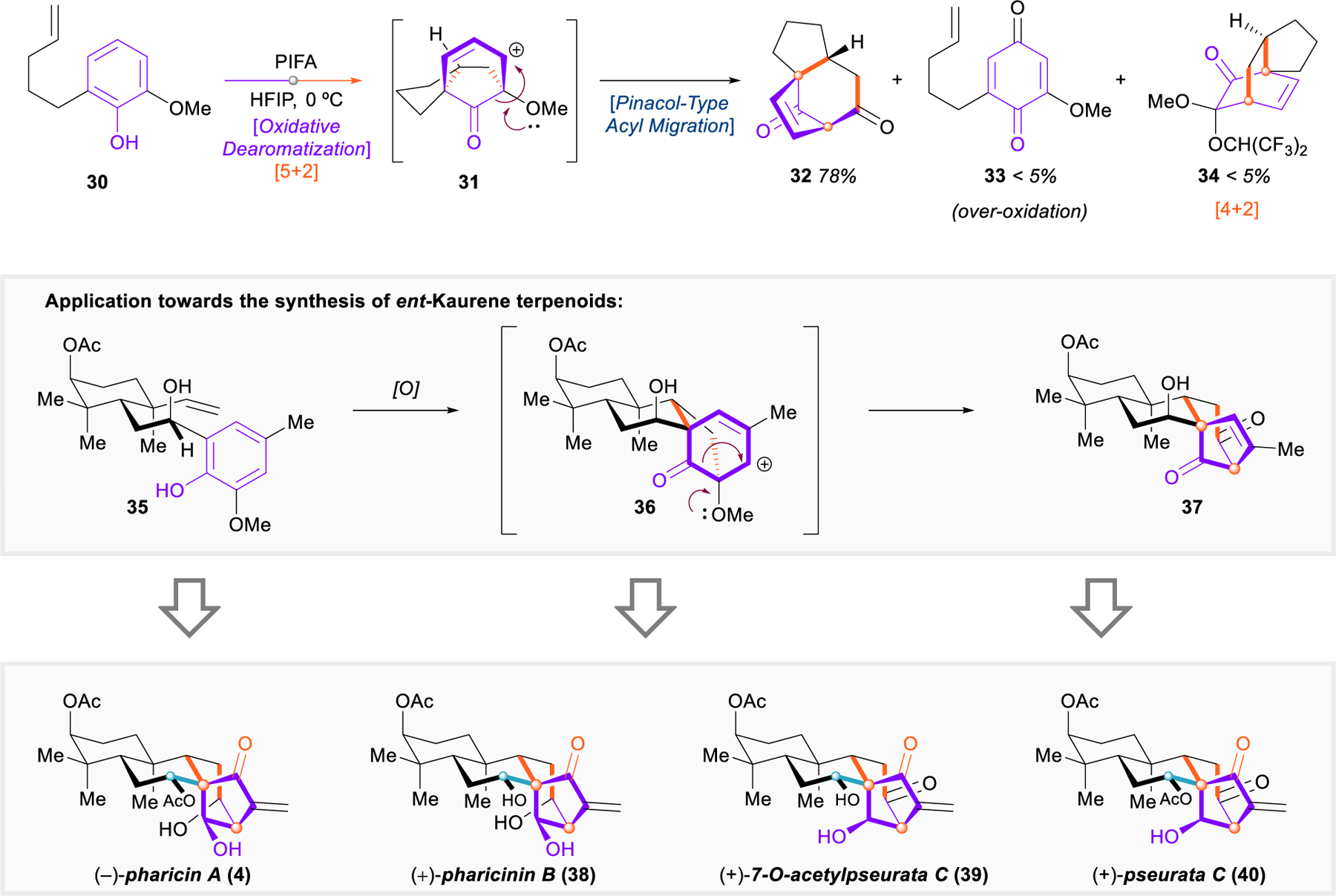
Ding’s oxidative dearomatization-induced [5+2] cycloaddition, and application towards ent-kaurenes.
The dearomative [5+2] cycloaddition/pinacol shift is an attractive strategy for the synthesis of ent‑kaurenoids (35 → 36 → 37). It generates considerable topological complexity within a single step, and it proceeds from relatively simple starting materials that can likely be derivatized and interchanged in a modular fashion, leaving open the possibility that this approach might be general. Ding and coworkers aptly demonstrated the utility of this method by completing the first asymmetric total syntheses of four ent-kaurenoids: (–)-pharicin A (4), (+)-pharicinin B (38), (+)-7-O-acetylpseurata C (39), and (+)-psuerata C (40) (Figure 7).76
Figure 7.
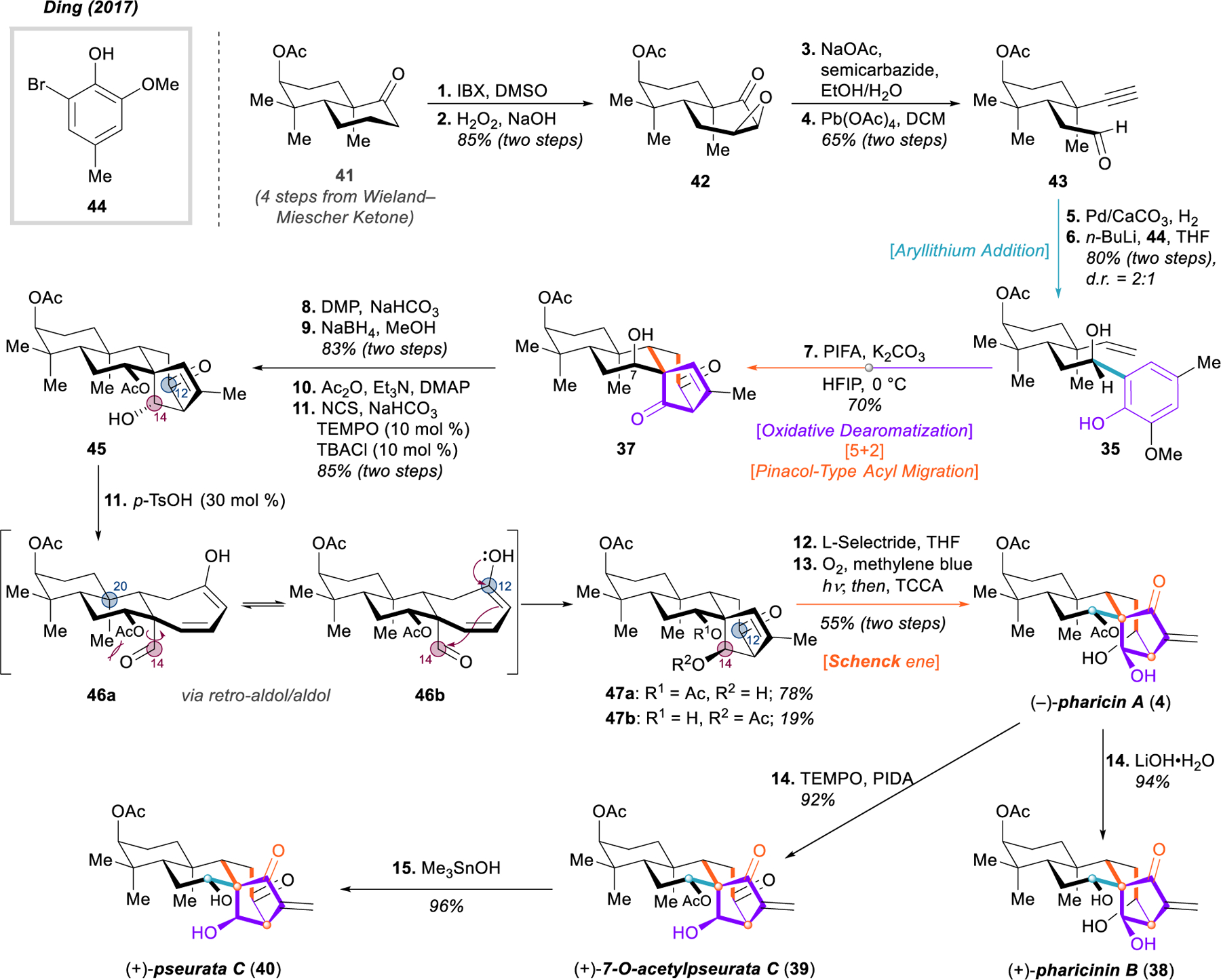
Ding’s total syntheses of (–)-pharicin A (4), (+)-pharicinin B (38), (+)-7-O-acetylpseurata C (39), and (+)-pseurata C (40).
Starting from known bicyclic ketone 41, made in four steps from commercially available Wieland–Miescher ketone, a Nicolaou oxidation followed by nucleophilic epoxidation delivered epoxy ketone 42 in 85% overall yield, which was then subjected to a modified Eschenmoser–Tanabe fragmentation (42 → 43). Likely due to the steric hindrance of this ketone, modified conditions for the fragmentation were required, using the more nucleophilic semicarbazide followed by lead tetraacetate oxidation and fragmentation.77 Lindlar reduction of the alkyne and nucleophilic addition of the aryllithium derived from aryl bromide 44 furnished 35, the requisite precursor for the dearomative cycloaddition. Treatment of 35 with PIFA in HFIP smoothly promoted the oxidative dearomatization-induced [5+2] cycloaddition, followed by pinacol-type 1,2-acyl shift, building two new rings and completely assembling the bicyclo[3.2.1]octene motif of 37 in a single step. Notably, the full tetracyclic ent-kaurene framework was completed in only seven steps.
The endgame of Ding’s total synthesis required selective reduction of both ketones, and inversion of the stereocenter at C7. The latter task was accomplished through DMP oxidation and subsequent reduction with sodium borohydride; however, during this sequence it became apparent that hydride reductions consistently reduced the ketone at C14 from the undesired face, despite a screen of reductants and attempts to use the free hydroxyl groups at C7 and C12 for assistance. Thus, an alternative strategy was envisioned involving a retro-aldol/aldol sequence, an approach Ding and coworkers had used in their recent synthesis of steenkrotin A.78 Preparation for this key manipulation required monoacetylation at C7 with acetic anhydride, and selective oxidation at C12 with catalytic TEMPO and NCS to give β-hydroxy ketone 45. The envisioned retro-aldol proceeded readily under acidic conditions to generate a transient, freely rotating aldehyde 46 at C14. Ding rationalizes the selectivity of the subsequent aldol reaction as the result of an unfavorable 1,3-diaxial interaction between this aldehyde and the C20 bridgehead methyl substituent, the aldehyde reacting preferably from rotamer 46b in which it points away from the ring, resulting in an apparent inversion of stereochemistry of the hydroxyl at C14. Partial acetyl transfer occurred during this process, generating 47a and 47b. Finally, the synthesis of (–)-pharicin A (4) was completed after reduction with L-Selectride and Shenck ene reaction with subsequent Kornblum–DeLaMare fragmentation. Simple protecting and functional group manipulations allowed Ding to elaborate (–)-pharicin A to three additional ent-kaurene terpenoids 38-40. Additionally, the authors later applied this transformation in a total synthesis of (–)-rhodomollanol A.79
Maoecrystal V:
Upon its structural elucidation in 2004,80 maoecrystal V (5) immediately captivated the imaginations of chemists around the globe and inspired over a decade of synthetic studies (Figure 8).81 Valiant efforts from the groups of Baran,82 Yang,83 Danishefsky,84,85 Singh,86 Trauner,87 Thomson,88,89 Zakarian,90 Nicolaou and Chen,91,92 Chisholm,93 May,94 Njardarson,95 Christie,96 and Sorenson97–99 brought to light the immense challenges awaiting those undertaking the synthesis of this unusual molecule. The crux of the synthetic problem imposed by the fascinating structure of maoecrystal V is the construction of two vicinal quaternary centers, adjacent to a tertiary alcohol, linking a cyclohexene subunit with a bicyclo[2.2.2]octan-2-one. A bridging δ‑valerolactone and strained oxolane ring amplify this problem considerably. Additionally, 5 was reported to have remarkably selective and potent antiproliferative effects against the HeLa cervical cancer cell line, thus presenting a highly attractive synthetic target despite—indeed because of—its difficult structure.
Figure 8.
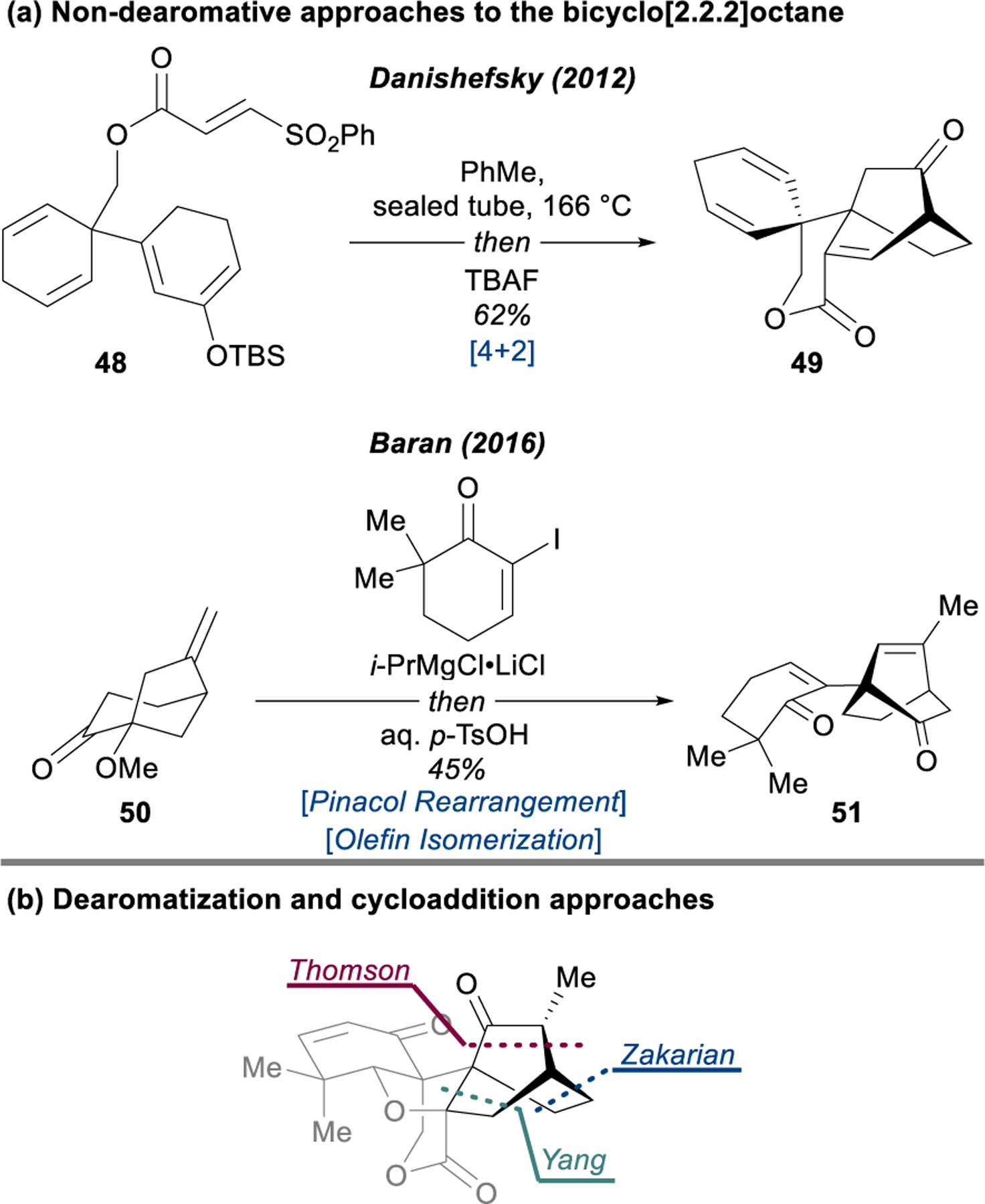
Key disconnections for dearomative and non-dearomative syntheses of maoecrystal V (5).
Five research groups have now completed the total synthesis of maoecrystal V: racemic approaches were reported by Yang in 2010,100 Danishefsky in 2012,101 and Zakarian in 2013.102 Asymmetric syntheses were disclosed from Thomson in 2014,103 a collaborative effort from Zakarian and Davies also in 2014,104 Yang in 2015,105 and most recently from Baran in 2017.106 Notably, the total synthesis completed in the Baran laboratory produced enough of the natural product to reproduce the biological assays, with the surprising result that maoecrystal V possessed no anticancer activity whatsoever.
In a retrosynthetic analysis of maoecrystal V, the utility of the Diels–Alder transform to disconnect the bicyclo[2.2.2]octane is apparent; indeed, ten of the synthetic studies and four out of the five completed total syntheses leverage this robust method (e.g., 48 → 49 in Danishefsky’s approach, Figure 8a). The lone alternative strategy, reported from the Baran laboratory, successfully applies a biomimetic pinacol-type shift of 50 → 51, inspired by the proposed conversion of epi-eriocalyxin A (52) to maoecrystal V (5) through a cationic intermediate such as 53 (Figure 9). When the cycloaddition tactic is applied in conjunction with dearomative disconnections, some attractive synthetic designs emerge (Figure 8b). The Yang, Zakarian/Davies, and Thomson syntheses all incorporate the oxidative dearomatization of a phenol and a [4+2] cycloaddition to assemble the bridging ring. Despite sharing this basic disconnection, the three approaches are quite distinct, each incidentally breaking the bicyclo[2.2.2]octane at a different location, with associated advantages and disadvantages.
Figure 9.
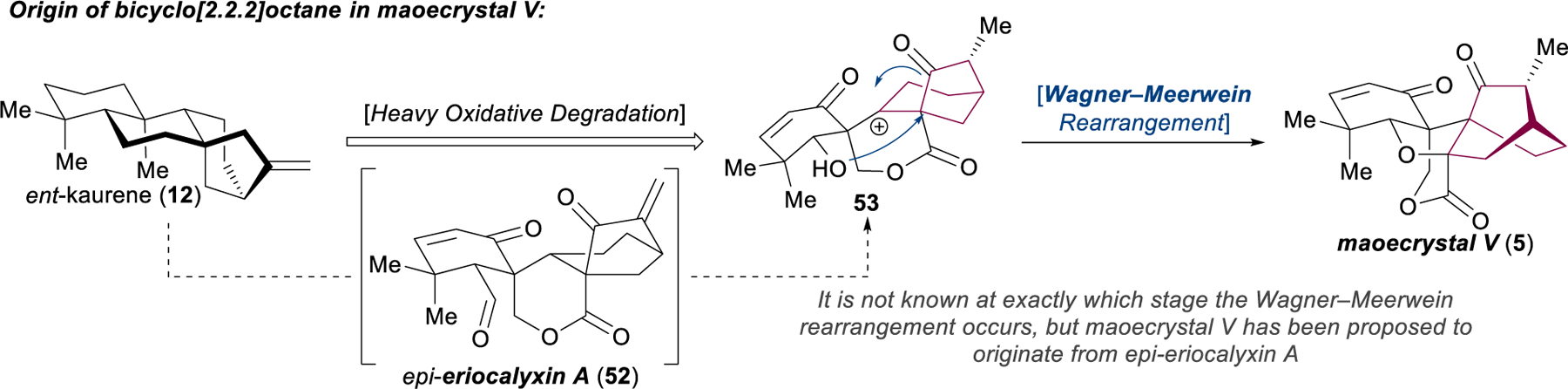
Proposed biosynthetic mechanism for the conversion of [3.2.1]-bicycle to [2.2.2].
Yang’s strategy adopts the most structurally simplifying of the three possible [4+2] cycloadditions—a powerful transformation—but one that necessarily proceeds from a fairly complex intermediate (54, Figure 10). The synthetic route addresses the problem of assembling one of the quaternary centers immediately, employing arylplumbane 56 in a high-yielding oxidative arylation of β-ketoester 57.107 The electron-rich phenol might have played an unexpected yet critical role in the following stage. Requiring diastereoselective ketone reduction from the more hindered face of the cyclohexenone to give alcohol 59a, Yang and coworkers found that many conventional hydride reducing agents were highly selective for the undesired isomer 59b. Some of the failed reductants include LiAlH4, DIBAL-H, NaBH4 with a Lewis acid additive, organoboranes, and hydrosilanes. Strikingly, the use of (Bu4N)BH4 effected a complete reversal in diasteroselectivity, producing alcohol 59a as a single isomer. The authors ascribe this unusual outcome to accelerating and directing effects that may result from a noncovalent cation-π interaction between the ammonium salt and the electron-rich phenol, to deliver the hydride syn to the arene. However, this rationale is speculative, and several examples of highly cis-selective reductions of related β-ketoesters lacking aromatic groups have been reported.108
Figure 10.
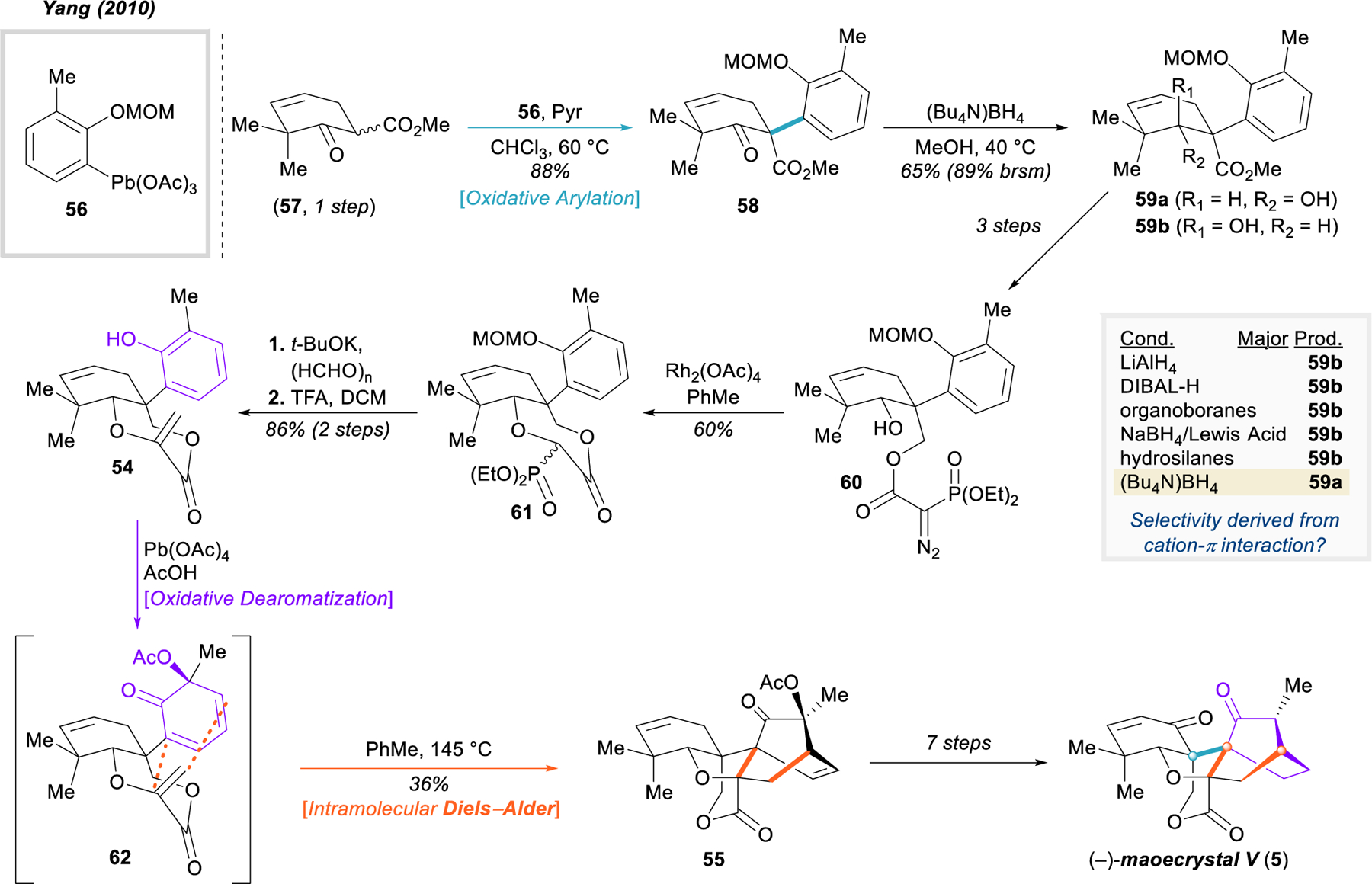
Yang’s synthesis of maoecrystal V (5).
After a three-step sequence of ester reduction, acylation, and diazo transfer (59a → 60), Yang’s team was able to quickly prepare a suitable substrate 54 for the oxidative dearomatization and [4+2] cycloaddition cascade. Rhodium-catalyzed intramolecular O–H insertion (60 → 61),109 deprotection of the phenol, and Wittig olefination furnished dioxepane 54. Notably, this process also created the precursors for the δ-lactone and oxolane rings; thus, upon Wessely oxidation and reflux in toluene, the envisioned dearomative cascade reaction occurred (via 62), simultaneously forging three rings as well as the second of the challenging vicinal quaternary centers. Although quite an impressive transformation, a drawback of this disconnection is the lack of diastereoselectivity, generating a mixture of three products and only 36% yield of the desired isomer 55. With the intricate skeletal framework completed, Yang and associates were able to complete the first total synthesis of maoecrystal V after seven additional redox maneuvers.100
Thomson’s strategy is unique among the Diels–Alder approaches in that its [4+2] cycloaddition is intermolecular and unrelated to the dearomative step, allowing instead for an oxidative phenol dearomatization to close the strained oxolane ring and establish the tertiary alcohol (63 → 64, Figure 11). Freeing the dienophile from a tether, the authors employed an elegantly streamlined sequence to swiftly assemble the complex framework from a simple intermediate, albeit at the cost of some late-stage functional group manipulations to access the optically pure precursor.
Figure 11.
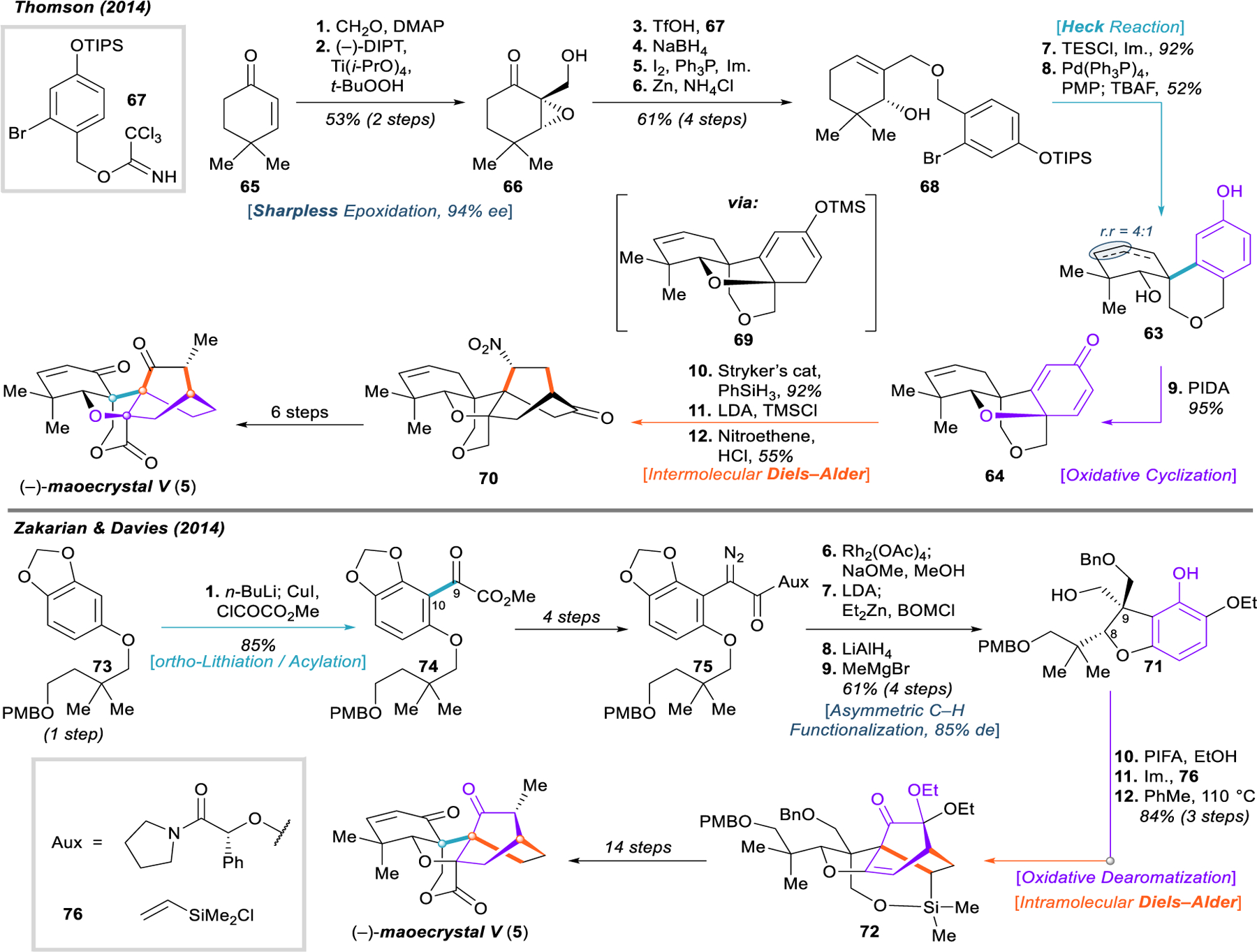
The Thomson and Zakarian/Davies syntheses of maoecrystal V (5).
The route to this precursor commences with a Baylis–Hilman reaction of enone 65 with formaldehyde. High optical purity (94% ee) was established with a Sharpless asymmetric epoxidation, and the remaining alcohol 66 was benzylated with trichloroimidate 67. Reductive fragmentation of the epoxide was achieved in a three-step sequence to reveal allylic alcohol 68, which was subsequently protected as the triethylsilyl ether.
In the first of a series of key steps, the C9 quaternary center was constructed with ease through an intramolecular Heck reaction with the arene, generating 63. The phenol was then immediately subjected to oxidative dearomatization with hypervalent iodine, closing the oxolane ring through intramolecular attack from the allylic alcohol. The resultant cyclohexadienone 64 was arranged for the impending Diels–Alder through conjugate reduction of the more exposed olefin and conversion to the unstable silyl dienol ether 69. Without purification, 69 was applied directly in a [4+2] cycloaddition with nitroethene as the active dienophile. Hydrolysis of the silyl enol ether was achieved upon acidic workup. Despite the missing and misplaced functionality of this complex intermediate 70, Thomson and coworkers were able to complete the synthesis of (–)-maoecrystal V in six additional steps.103
The [4+2] detachment that Zakarian and Davies chose (71 → 72)—arguably the most difficult of the three possibilities—may at first appear unorthodox, as it seems to require an ethylene equivalent as its dienophile. However, this disconnection highly advantageous because it unravels the complicated polycyclic system into simple benzofuran derivative 71. This judicious choice allowed the authors to address the construction of the strained oxolane ring early in the synthesis, the difficulty of which either presented significant problems in or arrested entirely numerous synthetic attempts requiring late-stage ring closure.81–85,88,89,93 The benzofuran intermediate 71 is also a strategic target for an asymmetric synthesis, as it could be generated through a catalytic, enantioselective C–H insertion of a rhodium carbenoid, a process previously reported by and heavily studied in the Davies laboratory.110 The solution the authors found for the Diels–Alder was to use a silicon-tethered olefin in an intramolecular reaction, which could be desilylated after the cycloaddition. The use of these two critical steps in concert to construct the oxolane and bicyclo[2.2.2]octane is tactful; however, the necessary intermediates are quite complex, and the strategy does little to accommodate the synthesis of the cyclohexenone, which required several functional- group transformations to assemble.
Aryl ether 73 was synthesized in one step through a Mitsunobu reaction of sesamol and the corresponding monoprotected, neopentylic diol. Regioselective ortho‑lithiation, transmetalation to zinc, and acylation with methyl chloroxoacetate furnished α-keto ester 74. At this early stage, classical arene chemistry was used to forge the incumbent C9–C10 bond, a vital connection that would be extremely difficult to make on the bicyclo[2.2.2]octane after dearomatization. Intermediate 75 was equipped for the enantioselective C–H insertion via a four-step sequence to install the diazo group and attach a mandelic acid-derived chiral auxiliary. The auxiliary was employed only after an exhaustive screen of chiral ligands and reaction conditions suffered from low diastereoselectivity at the C8 position. High enantioselectivity at C9 could be obtained with chiral ligands on rhodium; however, as C9 is immediately epimerized to provide the thermodynamically favored isomer, the poor diastereoselectivity of the C–H insertion effectively rendered the overall process racemic.
With the chiral auxiliary in place, rhodium-catalyzed C–H insertion proceeded in good yield and 84% diastereomeric excess. Cleavage of the auxiliary and epimerization at C9 to the trans-isomer was accomplished with sodium methoxide. This methyl ester was alkylated with benzyl chloromethyl ether and reduced with lithium aluminum hydride. Dioxolane cleavage with methylmagnesium bromide furnished benzofuran 71. The bicyclo[2.2.2]octane foundation 72 was assembled by a three-step sequence incorporating oxidative dearomatization with bis(trifluoroacetoxy)iodobenzene, silylation of the free alcohol with dimethylvinylchlorosilane (76), and reflux in toluene. With the construction of the A-ring cyclohexenone and the δ-lactone remaining, the authors achieved the total synthesis of (–)-maoecrystal V in fourteen steps from siloxacycle 72.104
Miscellaneous Terpenoids:
The oxidative dearomatization-induced [5+2] cycloaddition has been of great utility in a number of synthetic approaches to terpenes that feature bridged ring structures as well. A notable example was reported by Pettus and coworkers in their 2011 total synthesis of ⍺‑cedrene (77), sec-cedrenol, and ⍺‑pipitzol (Figure 12).111 Effective dearomative logic is used throughout the synthesis, beginning with the transformation of salicylaldehyde 78 into curcuphenol (79). Two equivalents of methyllithium facilitate 1,2-addition into the aldehyde and deprotonation of the phenol, followed by monocarbonylation of the dianion with di-tert-butyl dicarbonate (78 → 80). In the same pot, treatment with the homoprenylmagnesium bromide reagent 81 promotes carbonate elimination from the magnesium phenoxide and generates ortho-quinone methide 82, into which the magnesium reagent adds to furnish curcuphenol (79) in 84% yield overall. The authors report an asymmetric approach to this compound as well, via inverse electron demand hetero Diels–Alder reaction of ortho-quinone methide 82 with a vinyl ether bearing a chiral auxiliary and subsequent fragmentation of the resultant chroman ketal. The key oxidative dearomatization-induced [5+2] cycloaddition (79 → 83 → 84 → 85) failed with a variety of hypervalent iodine oxidants; however, the use of lead tetraacetate uniquely promoted clean formation of the cedrene scaffold 85 in 61% yield. From this intermediate, Pettus and coworkers were able to complete the syntheses of ⍺‑cedrene (77), sec-cedrenol, and ⍺‑pipitzol in a rapid fashion.
Figure 12.
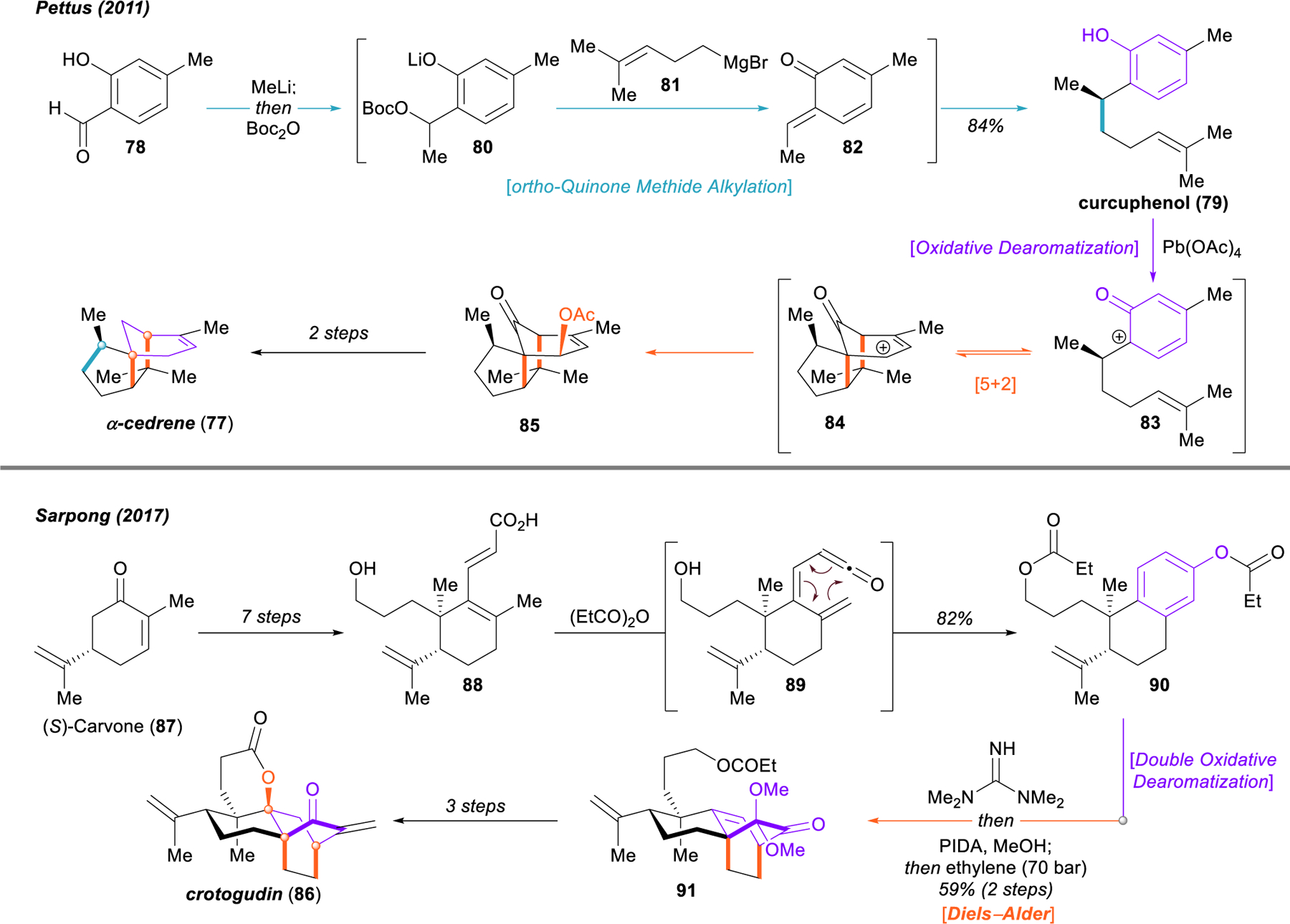
Pettus synthesis of cedrene (77) and Sarpong synthesis of crotogudin (86).
In 2017, the Sarpong group reported an interesting approach to crotogudin (86) through a benzannulation of (S)-carvone (87) and an unusual, regioselective double oxidative dearomatization and intramolecular [4+2] cycloaddition cascade.112 Seven steps were required to convert (S)-carvone into precursor 88. Heating this compound in solvent quantities of propionic anhydride resulted in the formation of ketene 89, and subsequent 6π-electrocyclization delivered the benzannulated derivative 90. Cleavage of the phenyl propionate with tetramethylguanidine, followed by regioselective iodine(III)-mediated oxidative dearomatization generated a dienone acetal that was carried through a high-pressure Diels–Alder reaction with ethylene to construct the bicyclo[2.2.2]octenone 91, bearing the full carbon skeleton of crotogudin (86). The authors were able to complete the synthesis of this natural product in an additional three steps.
Diels–Alder cycloadditions triggered by the oxidative dearomatization of phenols continue to find wide application in the synthesis of topologically complex, caged terpenoid structures. One natural product of particular interest, given its possible biosynthetic relationship113 to the enigmatic arcutane-type diterpenoid alkaloids (vide infra), is atropurpuran (92, Figure 13). Bearing a unique tetracyclo[5.3.3.0.4,9.04,12]tridecane framework with two adjacent bicyclo[2.2.2] octane ring systems, atropurpuran has been the subject of numerous syntheses114–117 since its initial isolation in 2009.118 Two successful campaigns, from the groups of Qin119 and Xu120 in 2016 and 2019, respectively, both make use of the dearomatization/cycloaddition tactic in the construction of either one or both of the [2.2.2] bicyclic motifs.
Figure 13.
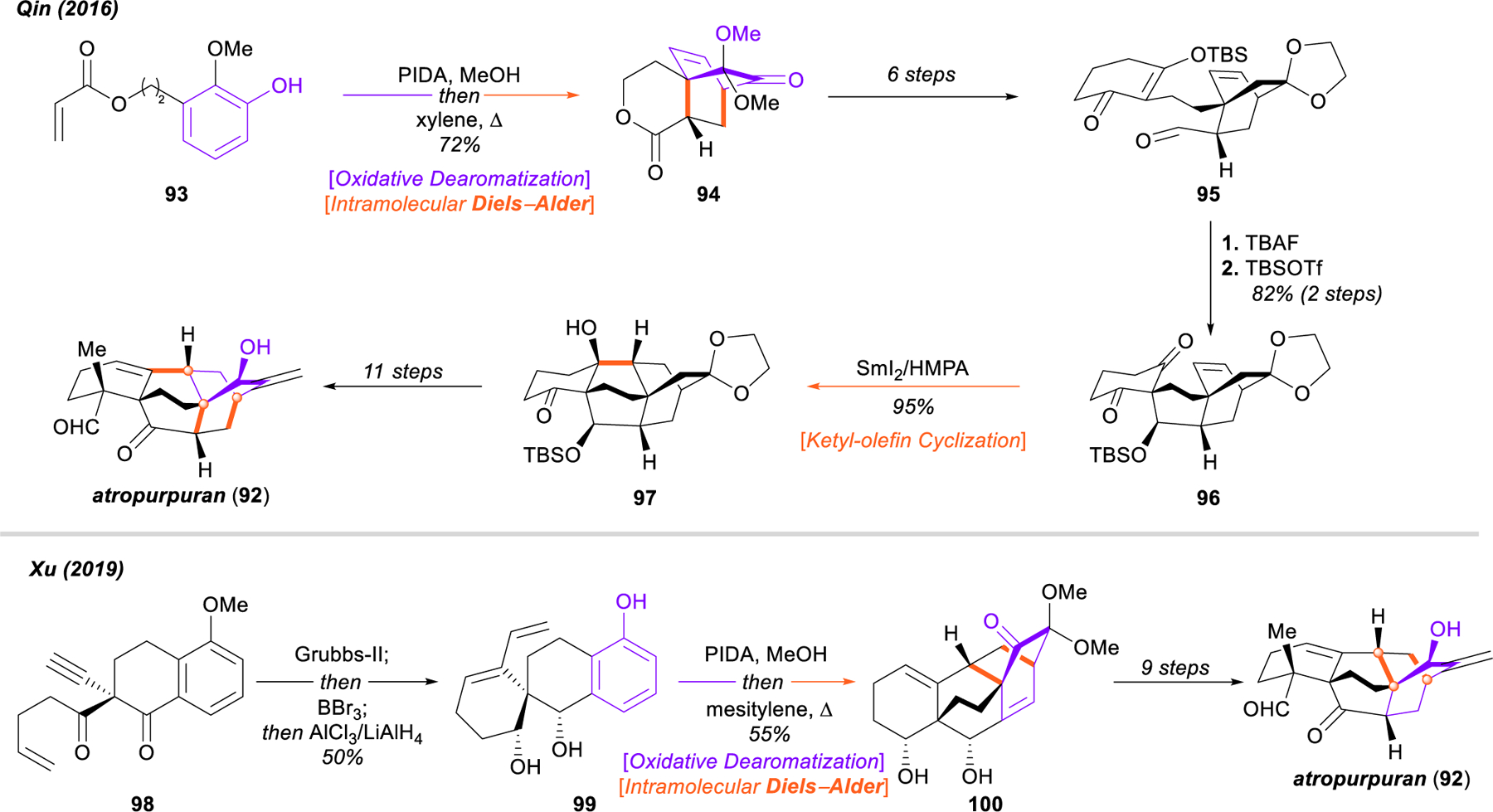
Qin and Xu syntheses of atropurpuran (92).
Qin’s approach carries out this operation at the outset, building tricyclic intermediate 94 from 93 in the first step under standard conditions. This was advanced to 95 in six steps, after which an intramolecular aldol reaction (95 → 96) followed by SmI2-promoted ketyl-olefin cyclization assembled the remaining bicyclo[2.2.2]octane system 97 and completed the synthesis of the complex carbon framework. Qin and coworkers were able to complete the first synthesis of atropurpuran (92) in an additional eleven steps.119
Xu’s 13-step 2019 approach began with the preparation of enyne 98 from commercially available 5‑methoxytetralone in two steps. Extremely rapid assembly of the full tetracyclo[5.3.3.0.4,9.04,12]tridecane scaffold was achieved through two subsequent C–C bond-forming events. Intramolecular ring-closing enyne metathesis with subsequent methyl ether deprotection and reduction of the 1,3-dicarbonyl delivered spirocycle 99, which was subject to a regioselective double oxidative dearomatization/intramolecular [4+2] cycloaddition cascade that notably builds both [2.2.2] bicycles in 100 in a single step. Following this impressive sequence, Xu and coworkers completed the total synthesis of atropurpuran (92) in nine additional steps.120
Diterpenoid Alkaloids:
Few molecular architectures have captivated the imaginations of synthetic organic chemists more than the diterpenoid alkaloids of the Aconitum, Delphinium, and Consolidum genera of plants (e.g., 101–110, Figure 14). These highly complex nitrogenous terpenoids are more appropriately classified as pseudo- or crypto-alkaloids with respect to their biosynthetic origins (vide infra) and are included in this section as such.121 As the main agents responsible for the toxic effects of Aconitum plants (monkshood/wolfsbane), some diterpenoid alkaloids have gained notoriety as poisons used in hunting, warfare, and a number of high-profile murders, and also feature prominently in the novels of Agatha Christie.122–127 Nonetheless, their strong affinity for voltage-gated Na+ and K+ ion channels lends these natural products powerful analgesic, antiarrhythmic, and other desirable medicinal properties, and herbal remedies containing aconite alkaloids continue to find use in traditional medicinal practices in China and Slovenia.128 Lappaconitine (not shown) is approved for the treatment of arrhythmia and pain in Russia and China, and the guan-fu base A (106) has been approved by the Chinese Food and Drug Administration for paroxysmol supraventricular tachycardia in 2005, and is currently in stage IV postclinical trials in China for similar indications.129–131,137 Several diterpenoid alkaloids have high channel subtype specificity and demonstrate potential therapeutic value in a variety of channelopathies.121,132–134
Figure 14.
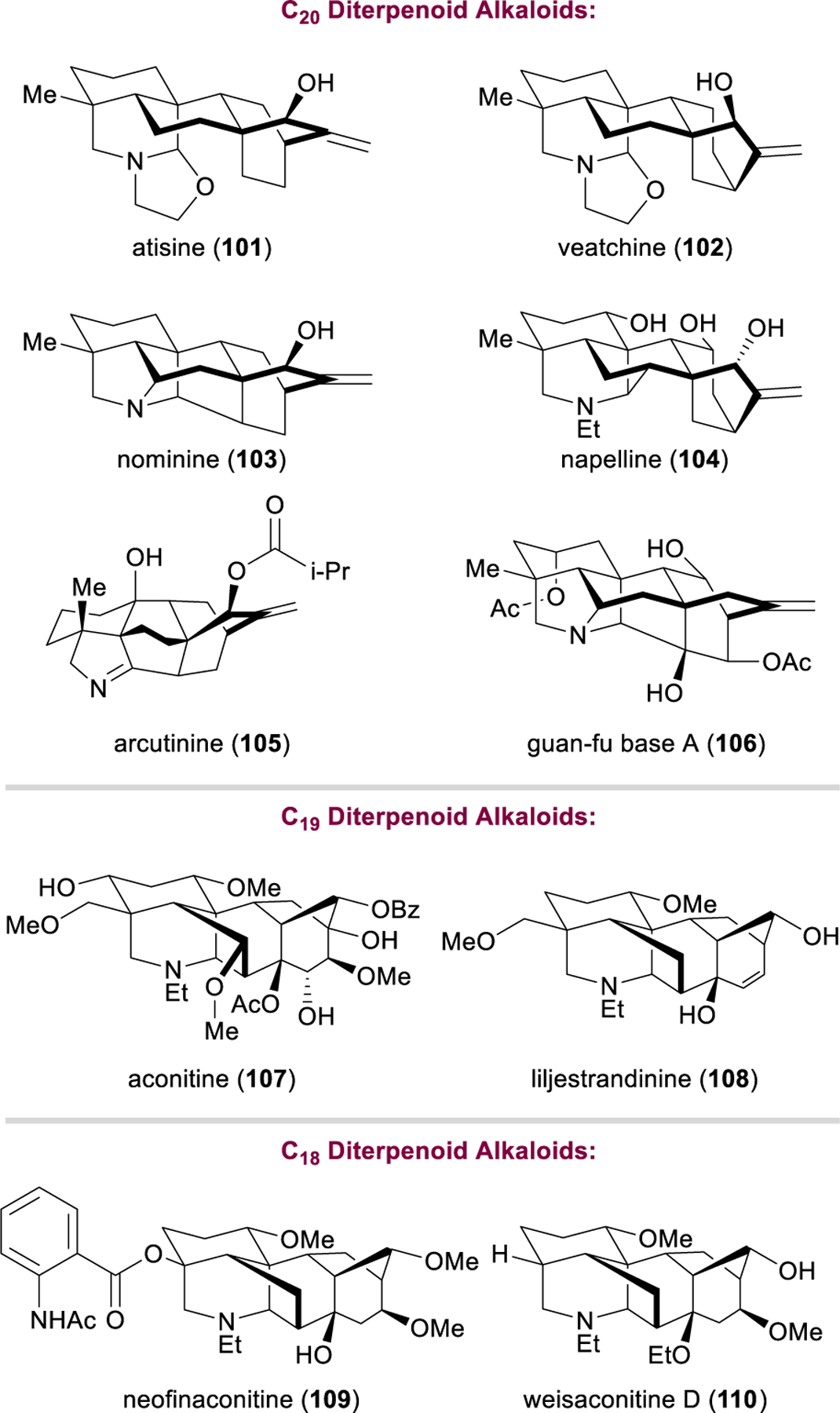
Structures of selected diterpenoid alkaloids.
The chemistry and biology of the diterpenoid alkaloids have been reviewed extensively, most notably by Wang and coworkers,121 who have segregated them by their carbon backbones into the C18,135 C19,136 and C20137 diterpenoid alkaloid families. The C20 diterpenoid alkaloids can be further classified into a variety of skeletal frameworks of increasing topological complexity. Unlike true amino acid-derived alkaloids, crypto-alkaloids are generated from the structurally related atisane and ent-kaurane diterpenoids via oxidation and subsequent amination with L-serine (Figure 15).137 A series of Wagner–Meerwein shifts and C–C bond forming events gives rise to a constellation of elaborate molecular architectures. Further rearrangement and excision of the exocyclic methylene group furnishes the C19 family of natural products,136 while cleavage of the remaining C4 methyl group delivers the C18 diterpenoid alkaloids.135 In addition to their daunting skeletal complexity, many diterpenoid alkaloids are heavily oxidized, providing the rich structural and functional diversity responsible for their myriad downstream biological effects.
Figure 15.
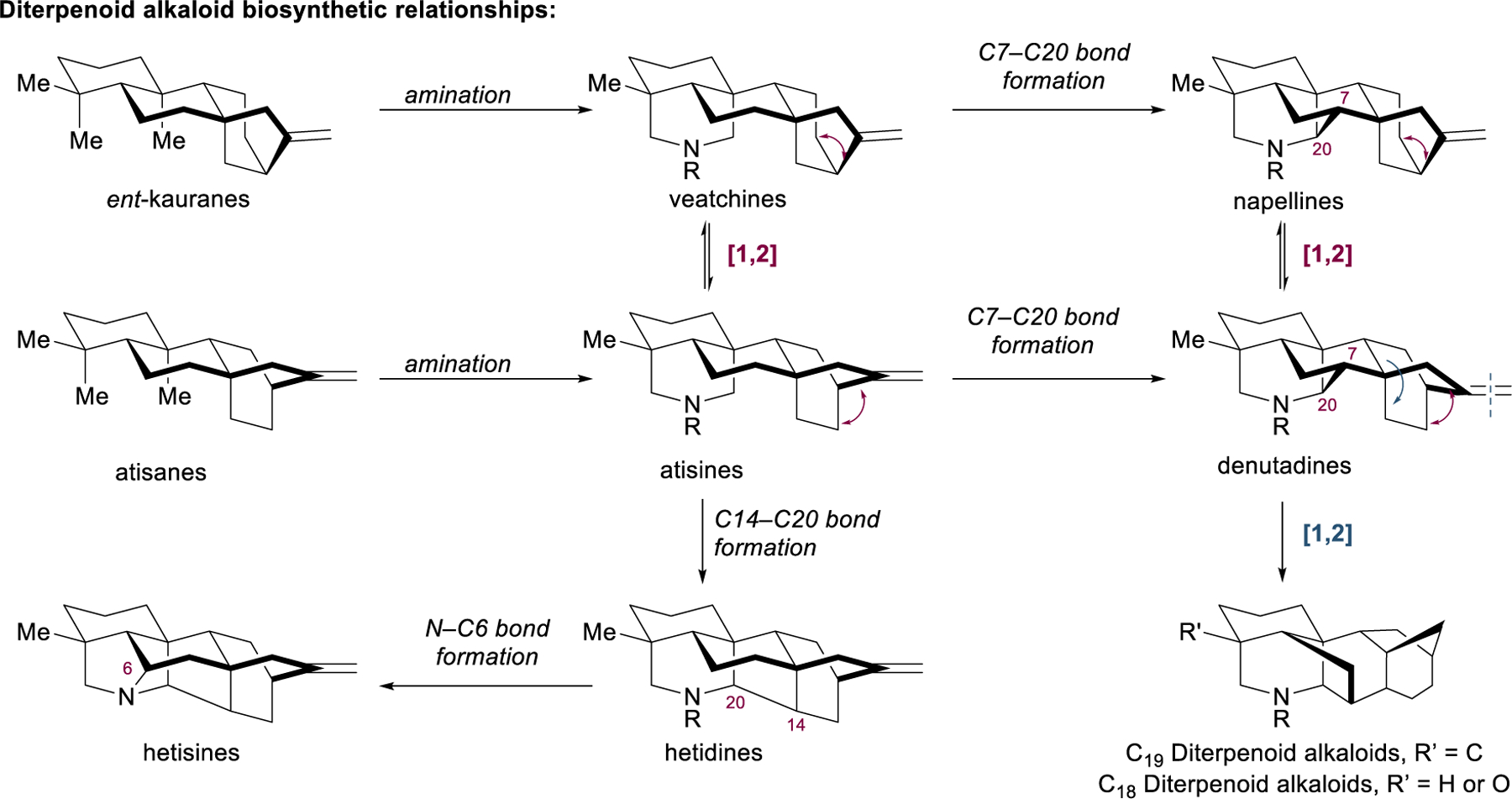
Biosynthetic relationships among the diterpenoids alkaloids as well as with other diterpenes.
Pioneering spectroscopic and degradative structural studies from Pelletier,138–140 Weisner and Büchi,141–143 and others144–146 revealed the alluring topologies and rich oxidative decor of the aconite alkaloids by the mid 1950s; however, such architectural features characteristic of these remarkable structures have proven so challenging to construct that efficient synthesis of diterpenoid alkaloids remains an active and difficult area of research even today, nearly seventy years later. In fact, despite landmark initial synthetic accomplishments from Nagata,147,148 Masamune,149–153 and Weisner154–160 in the 1960s and 1970s, perhaps only within the last decade has the state-of-the-art of diterpenoid alkaloid synthesis advanced significantly. This growing renaissance in aconite alkaloid synthesis can be attributed in large part to emerging strategies that place great emphasis on dearomative C–C bond-forming events that construct bridging rings within caged polycyclic frameworks.161 In these strategies, as was the case with the parent atisane and ent‑kaurene diterpenoid families, it is pertinent to note that the maximally bridged and most topologically complex portions of diterpenoid alkaloid scaffolds most often germinate from flat, aromatic building blocks through the use of dearomative chemistry.
This concept is not new—indeed, it lays the groundwork for nearly all successful diterpenoid alkaloid syntheses reported to date, including the seminal efforts from Nagata, Masamune, and Weisner (Figure 16). The two principal synthetic problems posed by all of the diterpenoid alkaloids is the installation of nitrogen during the construction of the azabicyclo[3.3.1]octane subunit, and the assembly of the highly caged polycyclic ring systems found in the eastern hemisphere of the molecules. Additional challenges are to be found on a case-by-case basis as more C–C and C–N bonds are made to form additional bridging rings.
Figure 16.
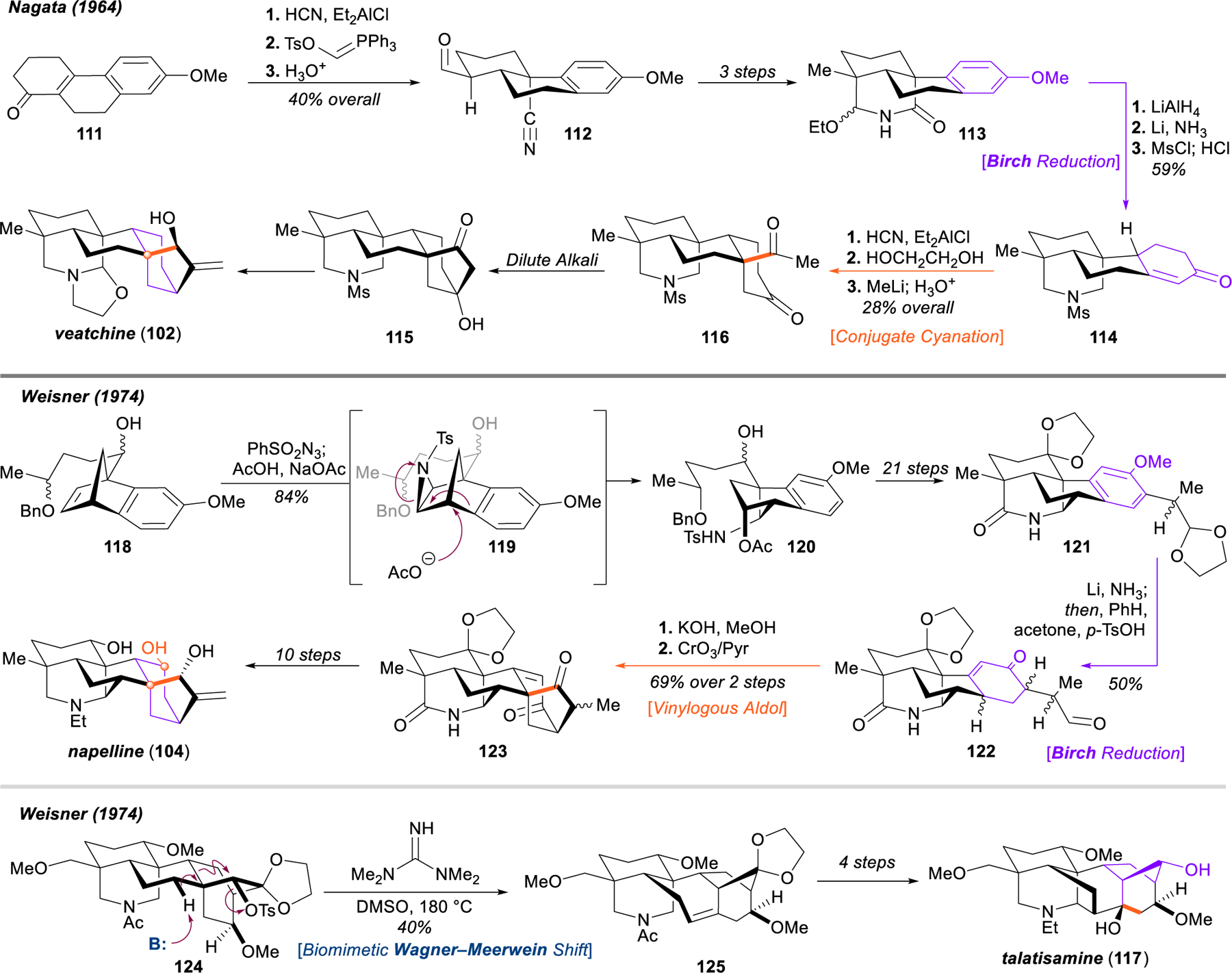
Landmark early syntheses of the diterpenoid alkaloids.
In Nagata’s classical approach to both atisine (101)147 and veatchine (102),148 a conjugate hydrocyanation of enone 111 served as a key step to affix the axial bridgehead methyl group as well as to bring in the nitrogen functionality preinstalled for subsequent closure of the azabicyclo[3.3.1]octane system. Wittig homologation delivered aldehyde 112, which successfully underwent ring closure after a sequence incorporating methylation and reduction of the nitrile (112 → 113). Treatment with lithium aluminum hydride and reductive dearomatization of the phenyl ether and resulting bis-hemiaminal under Birch conditions furnished enone 114, which could be used to construct the [3.2.1] bicycle 115 following another conjugate cyanation and generation of the methyl ketone (114 → 116), and intramolecular aldol reaction. This intermediate could be advanced to veatchine after several additional steps.148
Along with their groundbreaking structural studies141 and early total syntheses of atisine (101)156 and veatchine (102),155 the substantial synthetic accomplishments of Wiesner’s group included their successful pursuit of several members of more challenging diterpenoid alkaloid structural classes, including napelline (104)157–160 and the C19 norditerpenoid alkaloids talatisamine (117) and 13‑desoxydelphonine.162,163 Paramount to Wiesner’s approach to napelline (104) was the establishment of a C7–C20 bond during the assembly of the azabicyclo[3.3.1]octane system, which was achieved through Diels–Alder cycloaddition of benzeindene to construct benzonorbornene 118. Aziridination, acetolysis, and rearrangement (via 119) delivered sulfonamide 120, which could be advanced to the polycyclic scaffold 121 in 21 steps. Construction of the requisite bicyclo[3.2.1]octane motif 123 was accomplished through dearomative Birch reduction of the methyl phenyl ether and acetal deprotection (121 → 122), and vinylogous intramolecular aldol reaction. Diketone 123 was carried forward to the natural product napelline (104) in 10 steps.160 In another landmark effort from 1974, Wiesner and coworkers reported the first total synthesis of the C19 diterpenoid alkaloid talatisamine (117) through the use of a biomimetic Wagner–Meerwein rearrangement of the atisine-type [2.2.2] bicyclic system 124 to the bicyclo[3.2.1]heptane 125 characteristic of the aconitine alkaloids.162
Adoption of the powerful oxidative dearomatization/Diels–Alder cascade in aconite alkaloid synthesis can be traced to Wang’s 2012 formal synthesis of atisine (101) (Figure 17).164 This highly efficient approach served to inspire many subsequent synthetic efforts, and now constitutes one of the most fundamental strategies in the area of diterpenoid alkaloid synthesis.161 In Wang’s report, commercial β-ketoester 126 was transformed into azabicyclo[3.3.1]octane 127 in two steps through methylation and a double Mannich reaction with benzylamine. This intermediate was converted to cycloaddition precursor 128 in 13 steps. Oxidative dearomatization with PIDA and subsequent intramolecular [4+2] cycloaddition established the full atisine core in a very expedient fashion, and 129 could be advanced to atisine along Pelletier’s route,138 thus completing the formal synthesis.
Figure 17.
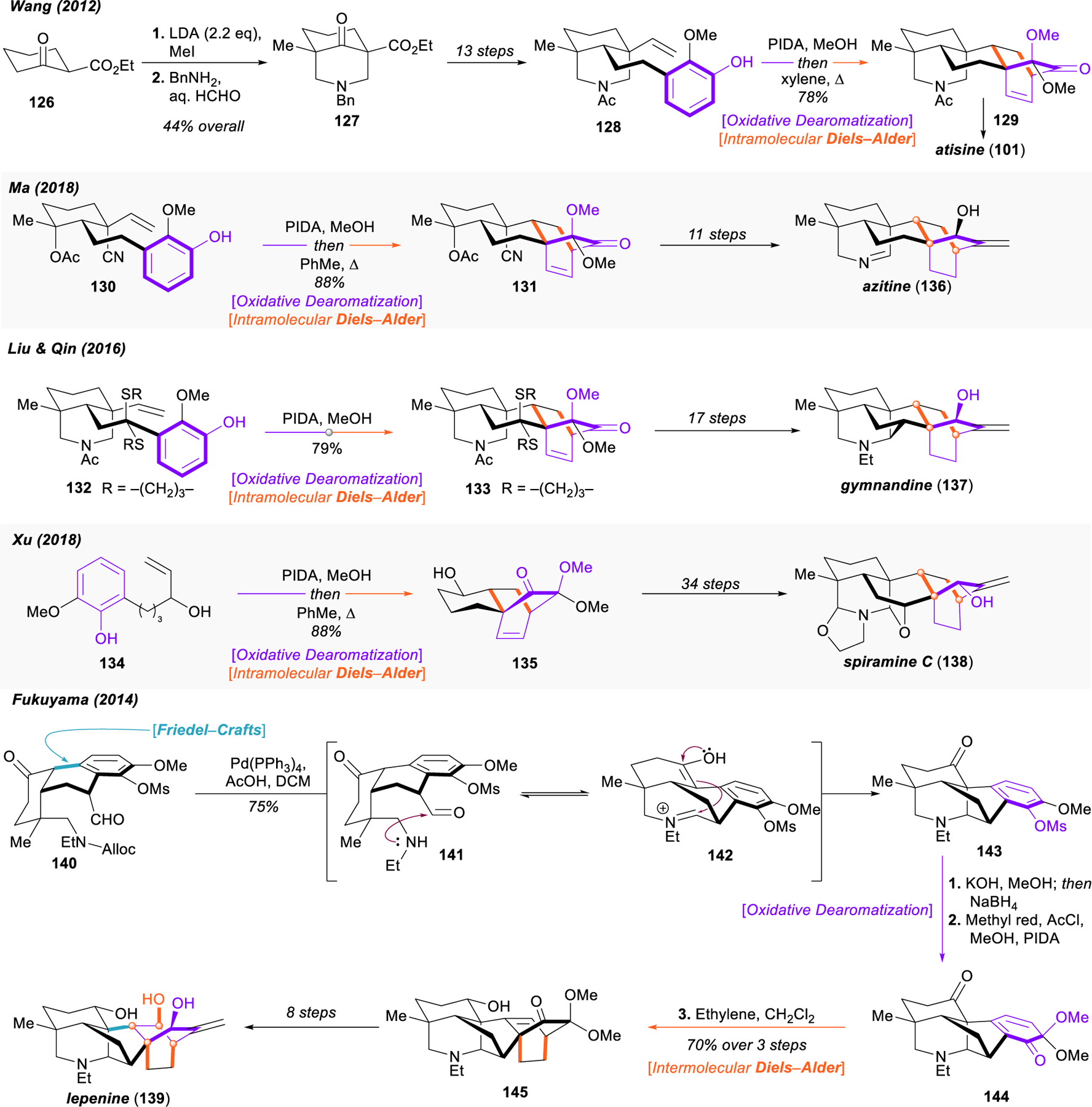
Selected total syntheses of diterpenoid alkaloids using the oxidative dearomatization/intramolecular Diels–Alder approach.
Similar oxidative dearomatization/intramolecular Diels–Alder sequences (e.g., 130 → 131, 132 → 133, and 134 → 135) have served as the backbone of numerous other synthetic approaches, such as Ma’s 2018 total synthesis of azitine (136),165 Liu and Qin’s synthesis of gymnandine (137),166 and Xu’s 2016 synthesis of spiramine C (138).167 An interesting variant on this strategy was reported by Fukuyama in 2014, using ethylene as the dienophile in an intermolecular cycloaddition to build the core of lepenine (139).168 Rapid construction of the caged polycyclic scaffold was achieved through back-to-back tandem reactions. Palladium-catalyzed deprotection of allyl carbamate 140 triggered intramolecular Mannich condensation to forge both challenging N–C20 and C10–C20 bonds in a single step (141 → 142 → 143). An intervening mesyl deprotection and hydride reduction then set the stage for oxidative dearomatization and high-pressure [4+2] cycloaddition (143 → 144 → 145) with ethylene to finish the synthesis of the denudatine core, which was advanced to the natural alkaloid lepenine (139) in eight steps.
Some of the most substantial recent contributions to diterpenoid alkaloid synthesis come from Sarpong, who has espoused the virtues of chemical network analysis in the deconstruction of topologically complex polycyclic scaffolds (Figure 18). In an early approach towards the hetidine family of natural products, Sarpong and coworkers utilized a gallium(III)-catalyzed cycloisomerization of enyne 146 to provide quick access to the key 6–7-6 tricyclic intermediate 147.169 Due to the lack of significant functionality, seven steps were required to convert 147 to benzylic alcohol 148. Treatment with thionyl chloride afforded piperidine ring closure, and the methyl phenyl ether was deprotected with sodium ethanethiolate under microwave irradiation, revealing 149. Hypervalent iodine-induced oxidative dearomatization para to the phenol occurred through intramolecular nucleophilic attack of the Boc-carbamate, installing requisite C14 oxygenation as well as generating a reactive enone to be used in the final ring-closure sequence. This process was achieved, after Johnson–Lemieux oxidation of the allyl group, via silica-mediated conjugate addition of the resultant aldehyde 150 into the enone, followed by rhodium/alumina-catalyzed hydrogenation and subsequent intramolecular aldol reaction to close the final ring of the hetidine skeleton. Compound 151 was advanced to the natural product dihydronavirine (152) in seven steps.
Figure 18.
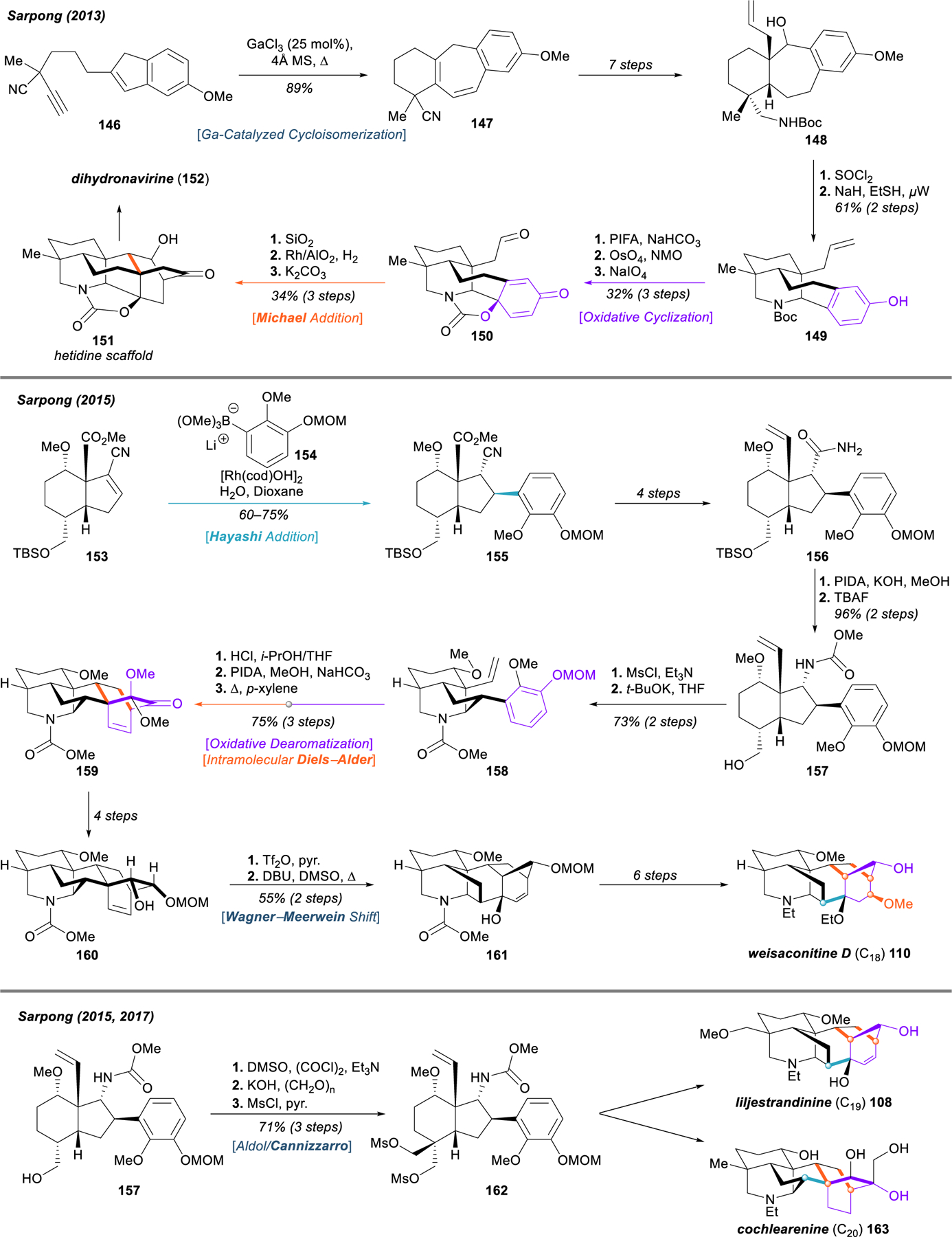
General approaches to C20, C19, and C18 diterpenoid alkaloids from the Sarpong group, using oxidative dearomatization and chemical network analysis.
In 2015, the Sarpong group reported an impressive hybrid application of dearomative logic and chemical network analysis that enabled the development of a general strategy towards C18, C19, and C20 diterpenoid alkaloids, and they demonstrated its efficacy by completing the first total syntheses of weisaconitine D (110) and liljestrandinine (108).170 Highly functionalized hydrindane 153 was obtained in four steps through Diels–Alder cycloaddition. Hayashi addition of lithium boronate 154 served as a key step to install the guaiacol motif that is later dearomatized. β-cyanoester 155 was converted in four steps to primary amide 156, which was then subject to a series of critical C–C and C–N bond-forming events that rapidly allow a denudatine-type core structure to take shape. PIDA-promoted Hofmann rearrangement of the amide and deprotection of the primary silyl ether with TBAF delivered methyl carbamate 157, which underwent ring-closure to form the crucial piperidine moiety of 158 upon treatment with methanesulfonyl chloride and potassium tert-butoxide. Cleavage of the MOM ester allowed for subsequent oxidative phenol dearomatization and intramolecular [4+2] cycloaddition to fabricate the remainder of a denudatine-like core framework 159, merely lacking the C4 methyl substituent. Four steps were required to prepare this scaffold for a biomimetic Wagner–Meerwein rearrangement (á la 124 → 125, Figure 16) to the C18 diterpenoid alkaloid core, which was achieved by triflation of the free hydroxyl group in 160 and heating in DMSO with concomitant addition of DBU. The resulting [3.2.1] bicyclic system 161 could be advanced to the natural product weisaconitine D (110) in eight additional steps.
In this and a follow up report in 2017,171 Sarpong and coworkers demonstrated an aldol/Cannizzaro sequence on an early intermediate from the weisaconitine D synthesis (157 → 162), enabling functionalization at the C4 position and extension of the synthetic strategy to both the C19 diterpenoid alkaloids as well as the denudatine-type C20 diterpenoid alkaloids. The generality of this approach was validated through the total synthesis of C19 diterpenoid alkaloids liljestrandinine (108),170 and later of three C20 diterpenoid alkaloids, including cochlearenine (163), paniculamine (not pictured), and N‑ethyl‑1⍺‑hydroxy‑17‑veratroyldictyzine (not pictured).171
In addition to their oxidatively dearomative strategies, the Sarpong group has recently reported an excellent joint application of reductively dearomative logic and chemical network analysis in a second-generation approach to the hetidine and hetisine scaffolds (Figure 19).172 Taking advantage of recent developments in aryne chemistry from Stoltz,173 Sarpong and coworkers were able to streamline their route to highly functionalized key 6–7-6 tricycle 164—reminiscent of intermediate 148 in their earlier synthesis of dihydronavirine (152)—through aryne insertion of 165 into β‑ketoester 166, obtained in four steps from the common hydrindane precursor 153 in their previous routes. Notably, the aryl bromide was required to achieve proper regioselectivity in the aryne insertion.174 A five-step sequence (164 → 167) encompassing the removal of the allyl ester, conversion of the silyl ether to a nitrile, and installation of the necessary C4 methyl group of the C20 diterpenoid alkaloids delivered 167, an important precursor compound for a series of highly productive C–C and C–N bond-forming events that swiftly build up the hetisine core scaffold in analogy to prior strategies from the Sarpong group. Chemoselective reduction of the nitrile with cobalt boride and borane tert-butylamine complex, followed by treatment with lithium aluminum hydride afforded ketone reduction, protodebromination, and direct cyclization to forge the N–C20 bond of 168. Photochemical hydroamination (168 → 169) formed the challenging N–C6 bond, and a Birch reduction/intramolecular Diels–Alder sequence (169 → 170 → 171 → 172) similar to that used by Gin in the synthesis of nominine (103)175 established the complete hetisine framework 172. With this advanced intermediate in hand, Sarpong and coworkers were able to complete the synthesis of cossonidine (173) in six steps.
Figure 19.
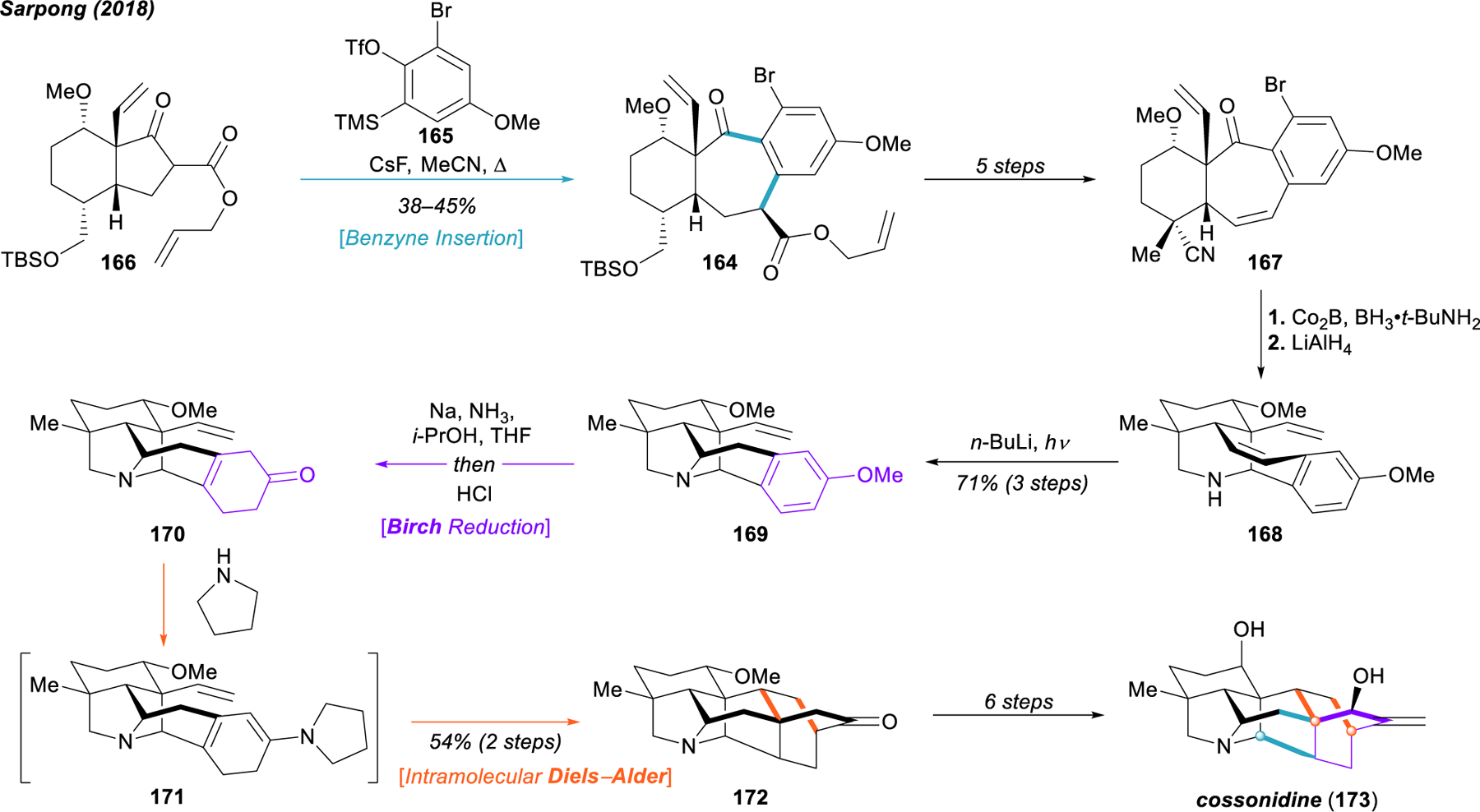
Sarpong’s synthesis of cossonidine (173) using reductive dearomatization and chemical network analysis.
Alkaloids
bis-Tetrahydroisoquinoline Alkaloids:
The remarkable story of the bis-tetrahydroisoquinoline (bis-THIQ) antitumor antibiotics begins in 1969 with the first reports of potent in vivo antitumor activity of crude extracts of the colonial sea squirt Ecteinascidia turbinate (e.g., 174–177, Figure 20).176 It would be another 21 years until the structure of ecteinascidin-743 (174), the agent responsible for this activity, was reported simultaneously by Rinehart177 and Wright,178 but the isolation of numerous other tetrahydroisoquinoline natural products captured the attention of researchers and catalyzed more than 40 years of study into their synthesis, structural elucidation, and chemical biology.179 Landmark efforts from Fukuyama,180–186 Kubo,187–201 Myers,202–204 Corey,205–209 Williams,210–217 and others218–224 have enabled synthetic access to many structurally complex bis-THIQ natural products, and have codified general strategies that build individual piperidine rings in a stepwise fashion via Pictet–Spengler, Bischler−Napieralski, and Pomeranz−Fritsch cyclizations179,225 Two classic approaches from Fukuyama181 and Myers202 towards saframycin A (178) are depicted in Figure 21, both of which serve to illustrate the critical role of electron-rich arenes in facilitating electrophilic aromatic substitution chemistry to construct each of the piperidine rings (e.g., 179 → 180, 181 → 182, 185 → 186, 187 → 188, 188 → 189).
Figure 20.
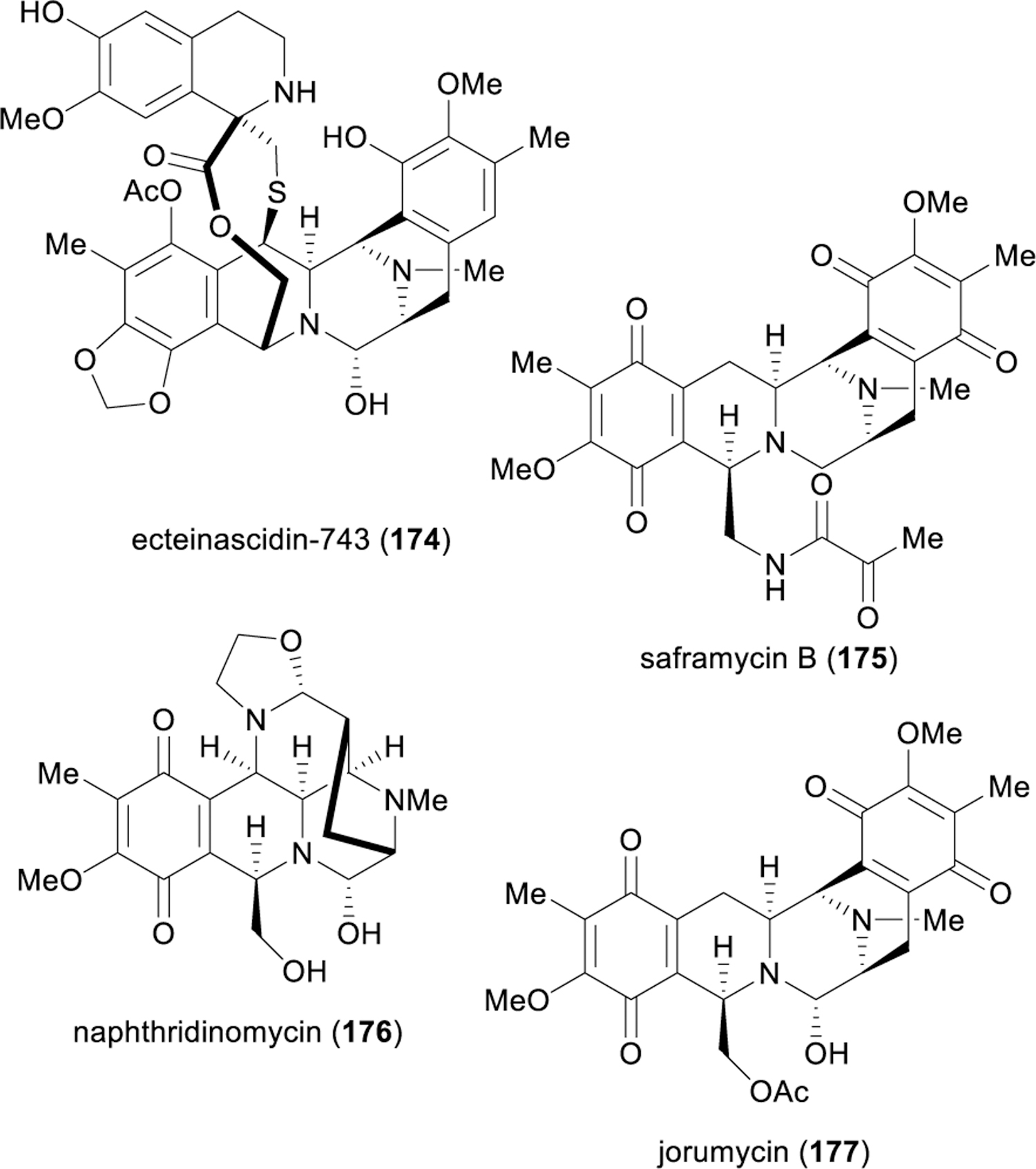
Structures of selected bis-tetrahydroisoquinoline alkaloids.
Figure 21.
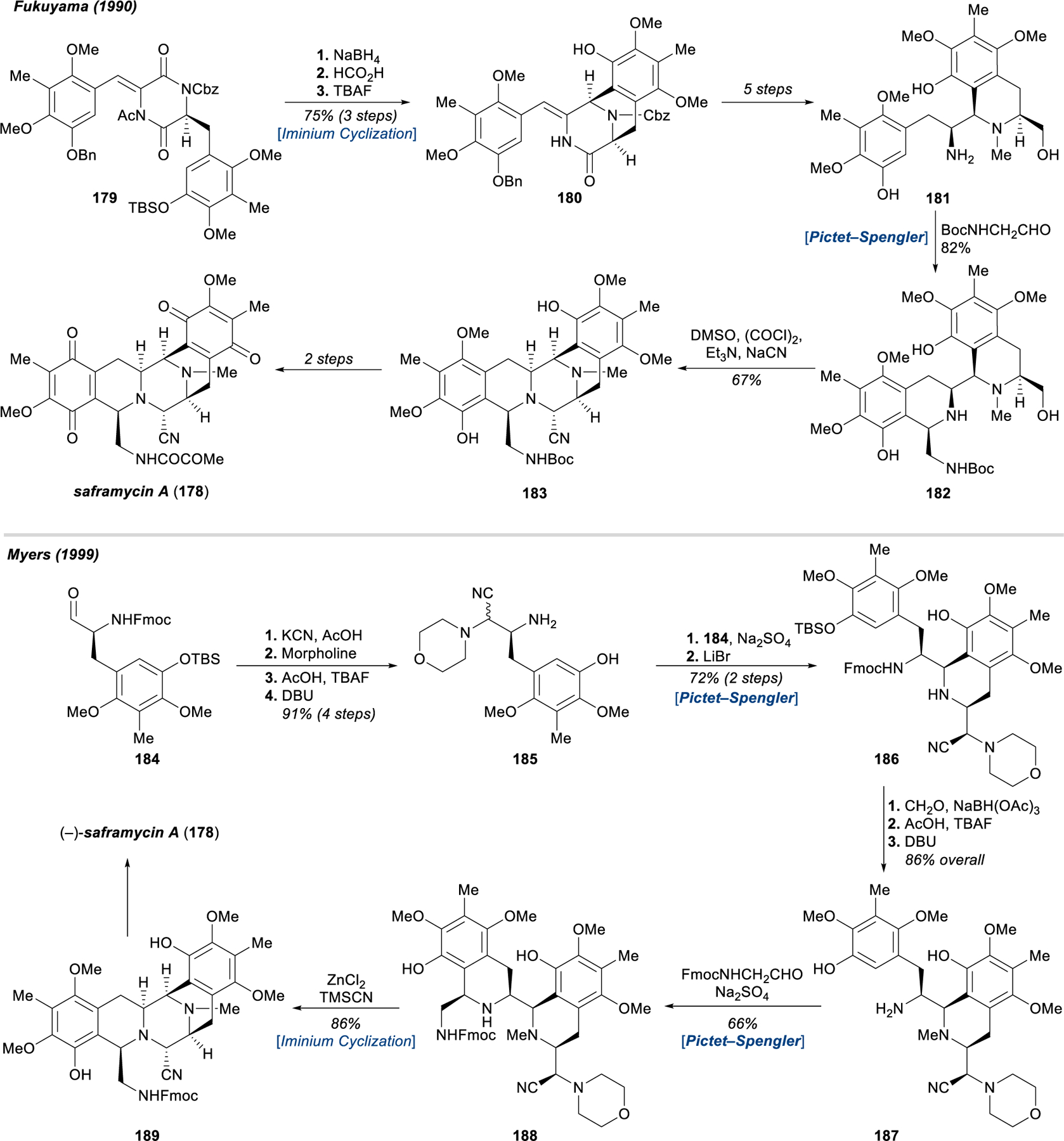
Fukuyama’s and Myers’s total syntheses of saframycin A (178), leveraging classical ring-forming reactions.
Ecteinascidin-743 (174) has achieved iconic status as the first marine natural product approved for the treatment of cancer (trabectin), and as one of the most structurally complex drugs produced through chemical synthesis to date.226 Although Corey’s total synthesis206 was able to supply enough material for clinical trials, industrial production required PharmaMar’s development of a scalable semisynthetic route from cyanosafracin B,222 averting the difficult construction of the functionalized bis-THIQ scaffold but also restricting the scope of analogues that could be generated for further medicinal chemistry endeavors. The biochemical profile of bis-THIQ natural products is very broad;179 thus, significant effort has been put towards the identification and synthesis of simpler bis-THIQ natural products or analogues that retain these desirable properties, but which are more synthetically tractable.204,207,209,212–214,216
One such compound is jorumycin (177), first isolated in 2000 from the Pacific nudibranch Jorunna funebris, an antitumor bis-THIQ of nearly equivalent potency as ecteinascidin-743 (174), but with a stripped-down core scaffold.227 An enantioselective synthesis of (–)-jorumycin was reported by Williams in 2005,228 followed by several others,229–231 all of which leverage biomimetic Pictet–Spengler reactions to construct both THIQ rings. One of the major synthetic challenges in the bis-THIQ natural products is the all-cis stereochemistry of sp3 C–C bonds relative to the central piperazine ring, which has plagued a significant number of bis-THIQ synthetic efforts and demanded subsequent epimerization after the Pictet–Spengler or similar cyclization.232
In a beautiful application of dearomative logic, the Stoltz group reported in 2019 the asymmetric synthesis of (–)-jorumycin (177) by a fundamentally different strategy, exploiting the all-cis stereochemical relationships through the use of a catalytic asymmetric hydrogenation of a simple bis-isoquinoline (190, Figure 22).233 The retrosynthetic application of late-stage quinone oxidation is the key transform that simplifies the structure of (–)-jorumycin to bis-THIQ 191, in which two retrons for the asymmetric hydrogenation of isoquinoline can now be readily identified. 191 can be traced back to 190 through hydrogenation and lactamization, and this bis-isoquinoline can easily be disconnected through arene coupling chemistry.
Figure 22.
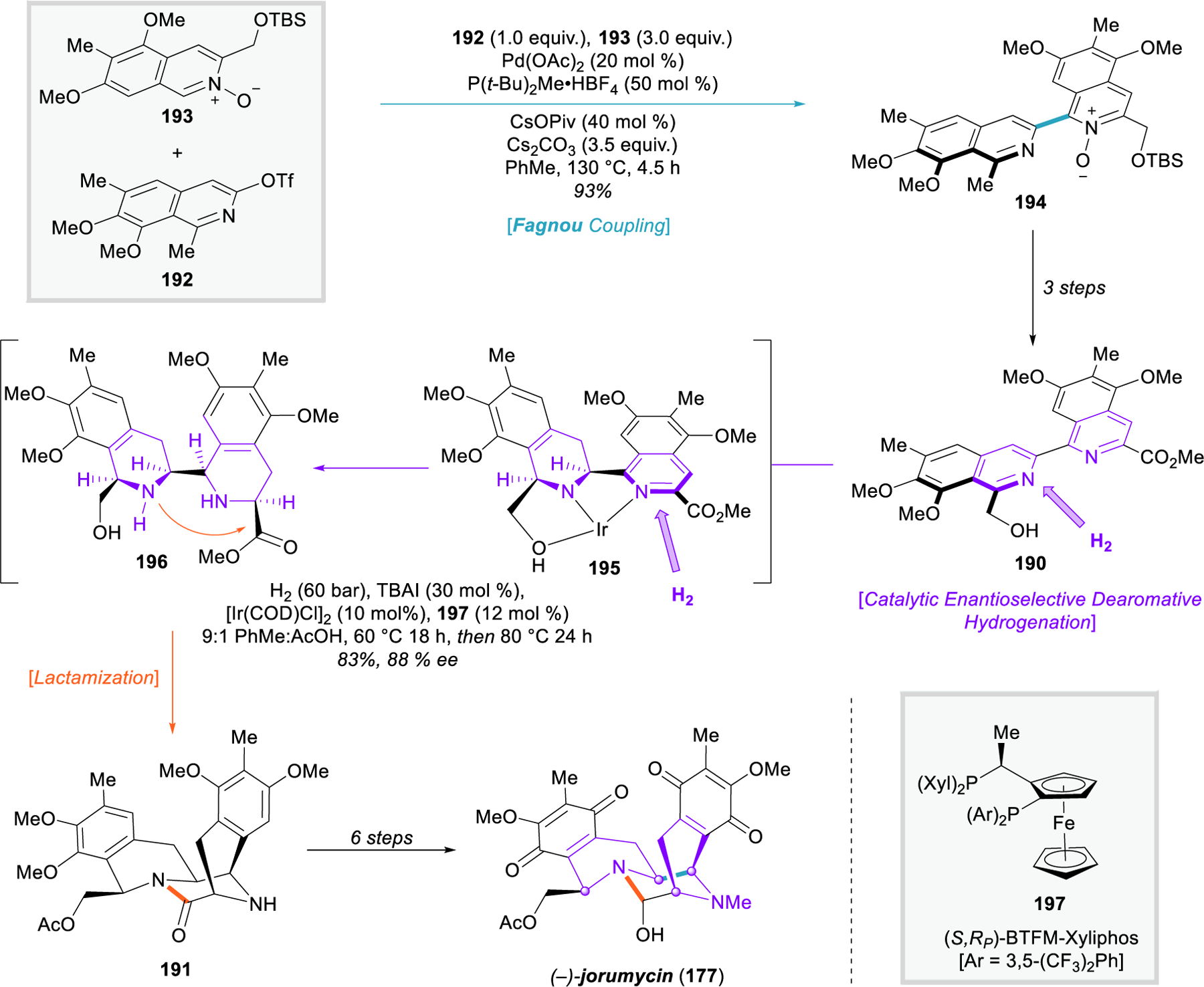
Stoltz’s synthesis of jorumycin (178), using enantioselective hydrogenation of isoquinolines.
This synthesis utilizes all three phases of dearomative logic to rapidly establish the core scaffold of the bis-THIQ natural products. Isoquinoline monomer 192 was synthesized in two steps using in-house aryne chemistry developed by the Stoltz group,234 while isoquinoline N-oxide 193 was synthesized in two steps through a silver(I)-catalyzed cyclization of an alkynylbenzaldoxime. Direct cross-coupling of these arenes under Fagnou’s conditions235 forged a critical bond in 194 as a pre-dearomative event. Several redox adjustments of 194 were necessary to deliver gram-quantities of the dearomative hydrogenation substrate 190. Notably, this cross-coupling strategy is unaffected by the electronics of the arenes, allowing for the synthesis of unnatural analogues with electron-withdrawing groups that would not be accessible through the conventional strategies based on electrophilic aromatic substitution.
Unlike most nitrogen-based heteroarenes, isoquinolines are very challenging substrates for enantioselective hydrogenation, and only a handful of examples have been reported.32 The Stoltz team planned to use the unprotected hydroxymethyl substituent in concert with the isoquinoline nitrogens to chelate a transition-metal catalyst and direct the hydrogenation to the B-ring first. They reasoned that the addition of two molar equivalents of hydrogen to 190 would force the second isoquinoline into a pseudoaxial conformation (195) on the newly-formed piperidine ring, providing substrate-reinforced diastereoselectivity for a second dearomative hydrogenation from the same face, and leaving compound 196 poised for rapid post-dearomative lactamization to construct the final ring of the pentacyclic core. After substantial screening, a catalytic system was developed that could achieve this remarkable transformation in 83% yield, >20:1 d.r., and 88% ee. This powerful dearomative event set four stereocenters and closed the fifth ring of the bis-THIQ in a single step. After lactamization, the Stoltz group was able to complete the synthesis of (–)-jorumycin (177) in six additional steps, intercepting the bis-THIQ natural product (–)-jorunnamycin en route. Using this non-biomimetic strategy, they prepared four unnatural analogues of 177 and investigated their cytotoxic effects through preliminary structure–activity relationship studies in collaboration with the Slamon group.
Amaryllidaceae Alkaloids:
The Amaryllidaceae alkaloids and isocarbostyril constituents (e.g., 198–205) have long served as benchmark compounds for synthetic chemists to test new methods and strategies but stand as important targets in their own right for their wide range of medicinal properties (Figure 23). Most notable are their potent antitumor and cholinesterase (AChE and BuChE) inhibitory activities, but these diverse natural products from the daffodil family have also demonstrated analgesic, antibacterial, antifungal, antimalarial, antiviral, anti-inflammatory, and even antidiabetic and anti-obesity effects.236 The alkaloid galanthamine (201) has been approved by the FDA for the treatment of mild to moderate dementia and Alzheimer’s disease due to its strong inhibition of acetylcholinesterase.237
Figure 23.
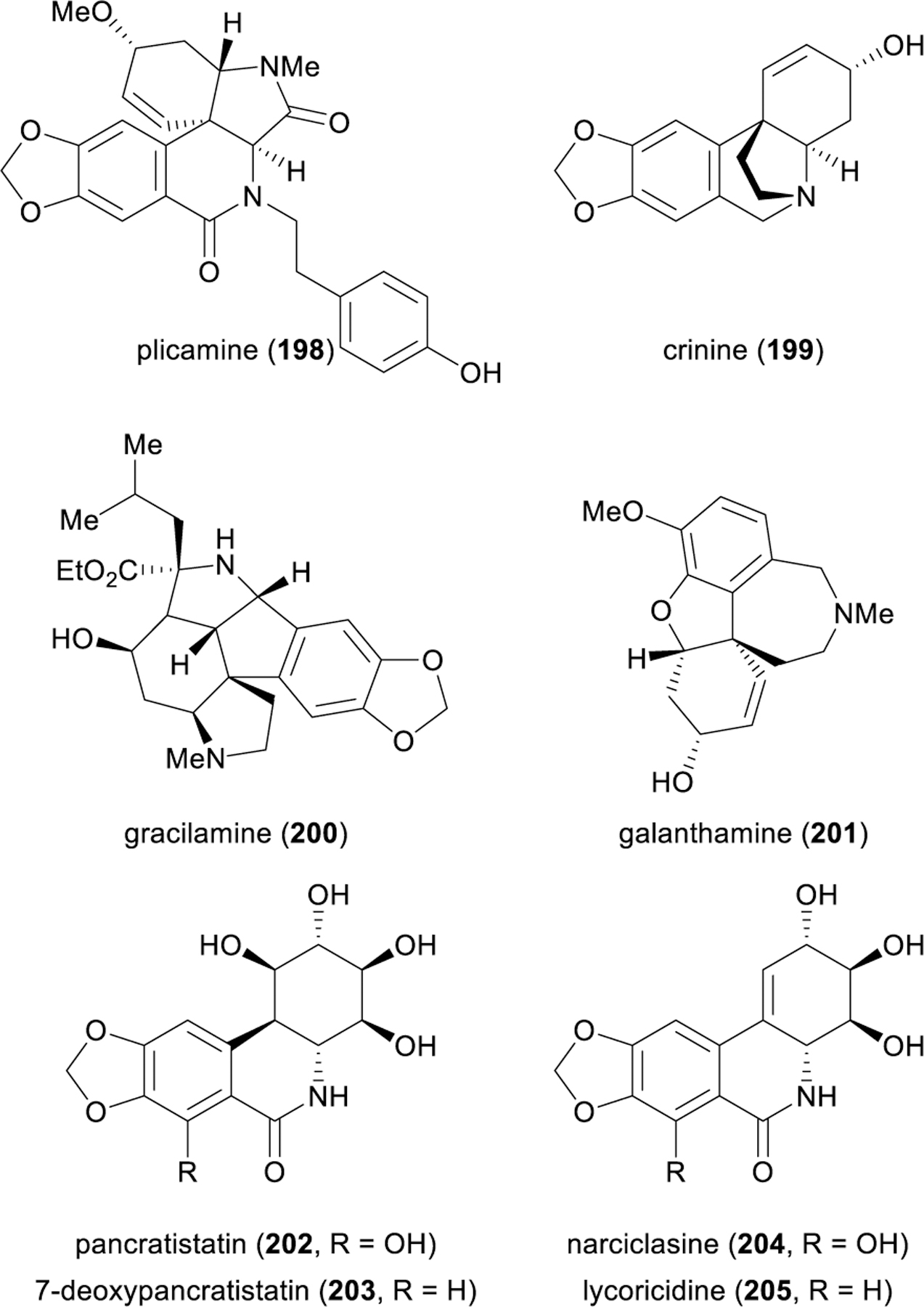
Structures of selected amaryllidaceae alkaloids.
Tyrosine-derived alkaloids such as 198–201 and isocarbostyril constituents 202–205 are particularly appropriate subjects for dearomative analysis because this is how nature makes them;238 different oxidative phenol couplings of 4ʹ-O-methylnorbelladine (206), generated from L-phenylalanine and L-tyrosine via reductive amination of 207 and 208, give rise to three primary alkaloid scaffolds (199, 201, 210), which in turn may convert to several other major alkaloid families through bond cleavage and oxidative transformations (Figure 24).236 Indeed, numerous early synthetic reports of dearomative routes to the lycorine (210),239,240 galanthamine (201),241–253 plicamine (198),254–256 and crinine (199)257–268 families of alkaloids have demonstrated the efficiency of this biomimetic strategy. Dearomative approaches towards the phenanthridine isocarbostyrils 202–205 through microbial arene oxidation have also been quite successful,269–271 with notably strong work in this area from Hudlicky and coworkers.272–280 Although non-biomimetic, these strategies showcase the power of dearomative logic in the construction of densely functionalized carbocyles.
Figure 24.
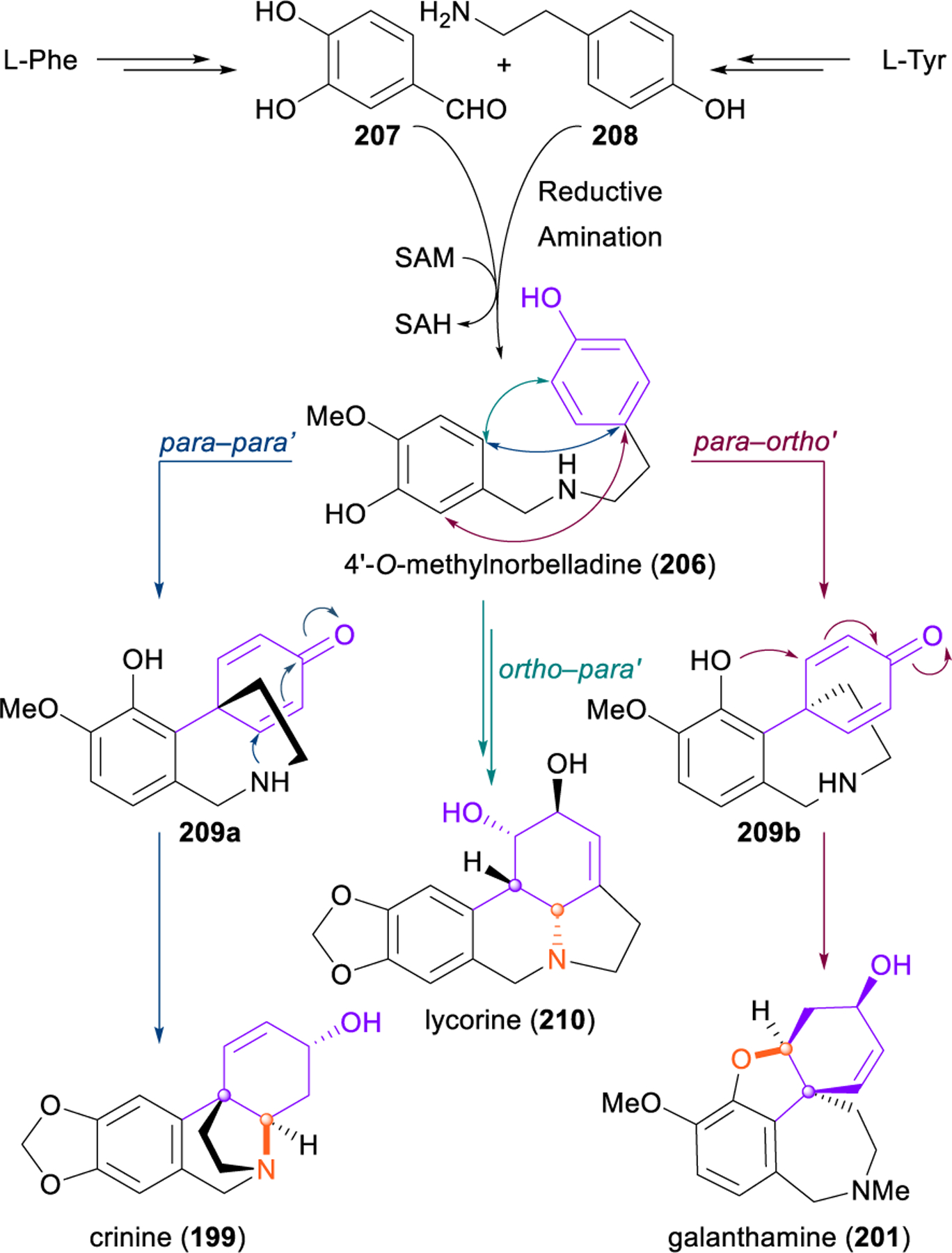
Different oxidative phenol couplings in Amaryllidaceae alkaloid biosynthesis generate diverse scaffolds.
The crinine scaffold in particular has been the subject of intense dearomative study. The requisite para–paraʹ oxidative coupling has been achieved photolytically,257 electrochemically,258 and with stoichiometric oxidants such as VOCl3,259 Tl(TFA)2,260–262 [Fe(DMF)3Cl2][FeCl4],263,264 VOF3–TFA/TFAA,265 and hypervalent iodine reagents.266–268 In addition to the use of a stoichiometric (and often toxic) oxidant, a major drawback of these methods is their lack of enantioselectivity, although a limited number of asymmetric syntheses have been reported using chiral controller groups that require subsequent removal.262,263
Building off strong preliminary work from Buchwald281–283 and You,284 Tang and coworkers reported a dramatic advance in this coupling methodology in 2015 (Figure 25).285 They found that, under palladium catalysis, dihydrostilbenoids containing a para-substituted phenol and an aryl bromide (e.g., 211) could engage in a dearomative intramolecular Heck-type coupling to build a quaternary center and assemble the phenanthrenone skeleton 212. Bulky chiral phosphine ligand 214 enables profound catalyst discrimination between the two enantiotopic faces of the phenol, and the product is generated in very high (up to 99%) enantiomeric excess. Although similar in appearance to previously reported oxidative phenol couplings, this method is actually isohypsic, thereby exchanging the use of stoichiometric oxidants for the requirement of an aryl bromide substrate.
Figure 25.
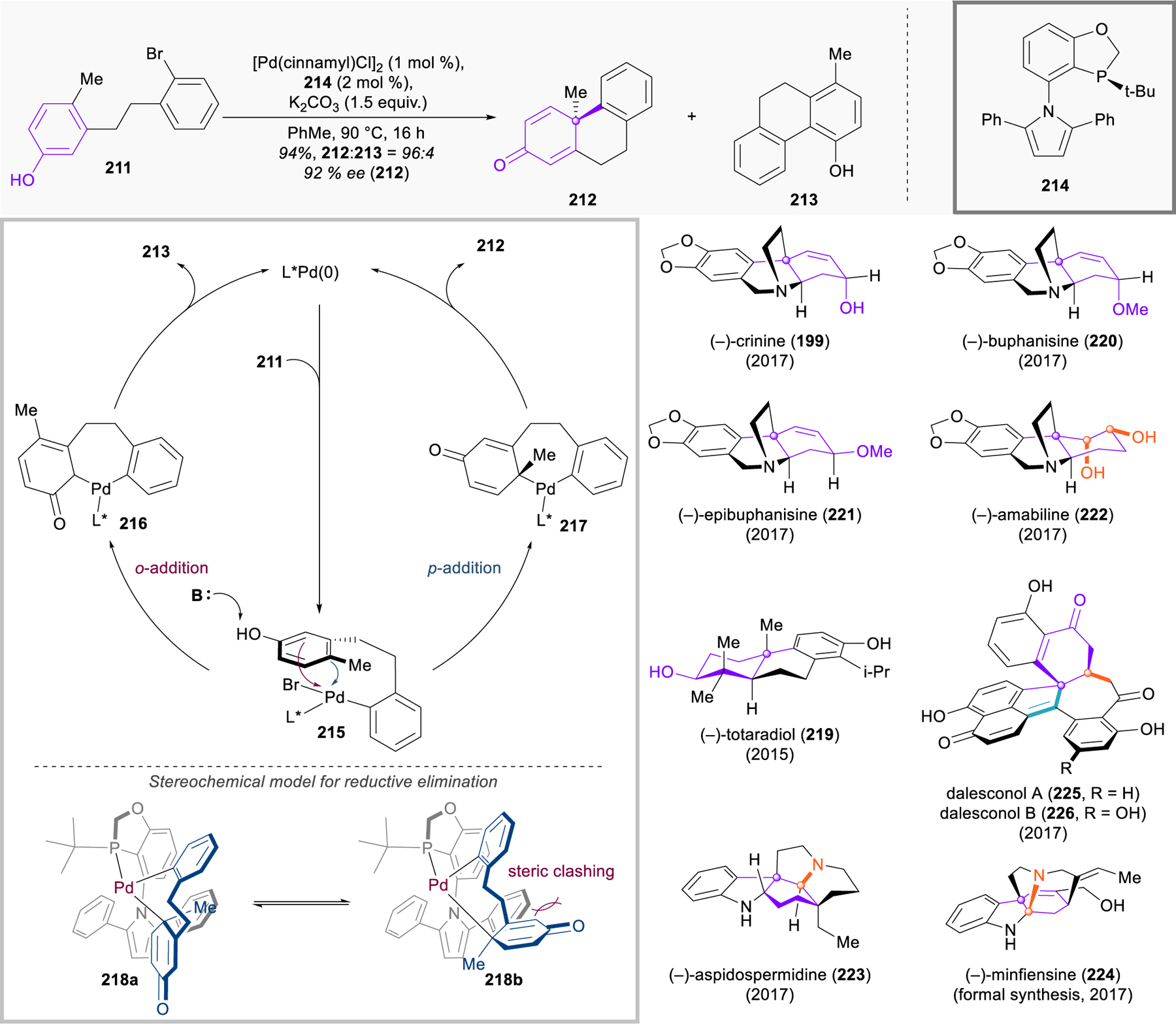
Palladium-catalyzed enantioselective dearomatization of phenols, and structures of natural products synthesized by the Tang group using this method.
The Tang group proposes the following mechanism: Oxidative addition of the palladium(0) catalyst to the aryl bromide 211 yields the palladium(II) complex 215. The phenol may then undergo nucleophilic addition to the palladium(II) complex from either the ortho (215 → 216) or the para (215 → 217) positions, leading to products 213 and 212, respectively. Although 213 is the thermodynamically favored product, Tang proposes that palladacycle 217 is formed preferentially for kinetic reasons. Reductive elimination forges a new C–C bond, a quaternary stereocenter, and regenerates the palladium(0) catalyst.
This catalytic enantioselective dearomative cyclization holds enormous potential for application in the total synthesis of natural products, and the Tang group has done a fine job demonstrating this. In addition to disclosing the method, their initial publication included streamlined routes to a key chiral intermediate used in kaurene synthesis, the full skeleton of the anabolic steroid boldenone, and the terpenoid natural product (–)-totaradiol (219). This work was followed up in 2017 with highly efficient, biomimetic asymmetric total syntheses of several crinine-type alkaloids, including (–)-buphanisine (220), (–)-epibuphanisine (221), (–)-amabiline (222), and (–)-crinine (199) itself.286 The same report also described the enantioselective syntheses of the strychnos alkaloids (–)-aspidospermidine (223, vide infra) and (–)-minfiensine (224), and a later publication describes the enantioselective syntheses of the complex aromatic polyketides dalesconol A and B (225 and 226, vide infra).287
The plicamine isocarbostyrils, first isolated in 1999,288 have received considerably less attention than the crinine alkaloids but are in fact susceptible to the identical dearomative analysis, as they biosynthetically derive from crinine via ring cleavage and amination with tyramine. Thus, they contain the same dearomatized phenol and fused pyrrolidine, a fact that Ley and coworkers used to great advantage when they reported the first and only asymmetric total synthesis of (+)-plicamine (198) in 2002.254,255 In addition to its biomimetic oxidative phenol dearomatization, this 16-step sequence is notable for using almost entirely solid-supported reagents and scavengers, requiring no chromatography. This strategy was later applied to the total syntheses of (–)-obliquine and (+)-plicane as well.256
A very interesting adaptation of Ley’s route was reported by Miranda and Mijangos in 2016 (Figure 26).289 The authors cleverly noticed that Ley’s key intermediate 227 prior to dearomatization could essentially be synthesized in a single step through an Ugi four-component condensation of p‑hydroxybenzaldehyde (228), piperonylamine (229), formic acid (230), and methylisocyanide (231) (the only difference being a formate-protected amine vs. Ley’s trifluoroacetamide). This reaction was optimized and found to proceed best under microwave irradiation. The Ugi adducts 227 were immediately subject to hypervalent iodine-mediated oxidative dearomatization (227 → 232), and the authors were able to develop a one-pot sequence incorporating this dearomative cyclization with base-promoted aza-Michael addition (232 → 233) to fully assemble the plicamine scaffold in just three steps from very simple precursors. Given the modularity of the Ugi reaction, a library of seventeen plicamine-type analogues were readily synthesized by changing the carboxylic acid and isocyanide components. Finally, they demonstrated the applicability of this method by intercepting a late-stage intermediate (234) from Ley’s route, completing an 8-step formal synthesis of (±)-plicamine (198). Although the synthesis is racemic, Miranda’s use of the Ugi four- component reaction reduces the step count by half and provides nearly endless opportunities for analogue synthesis.
Figure 26.
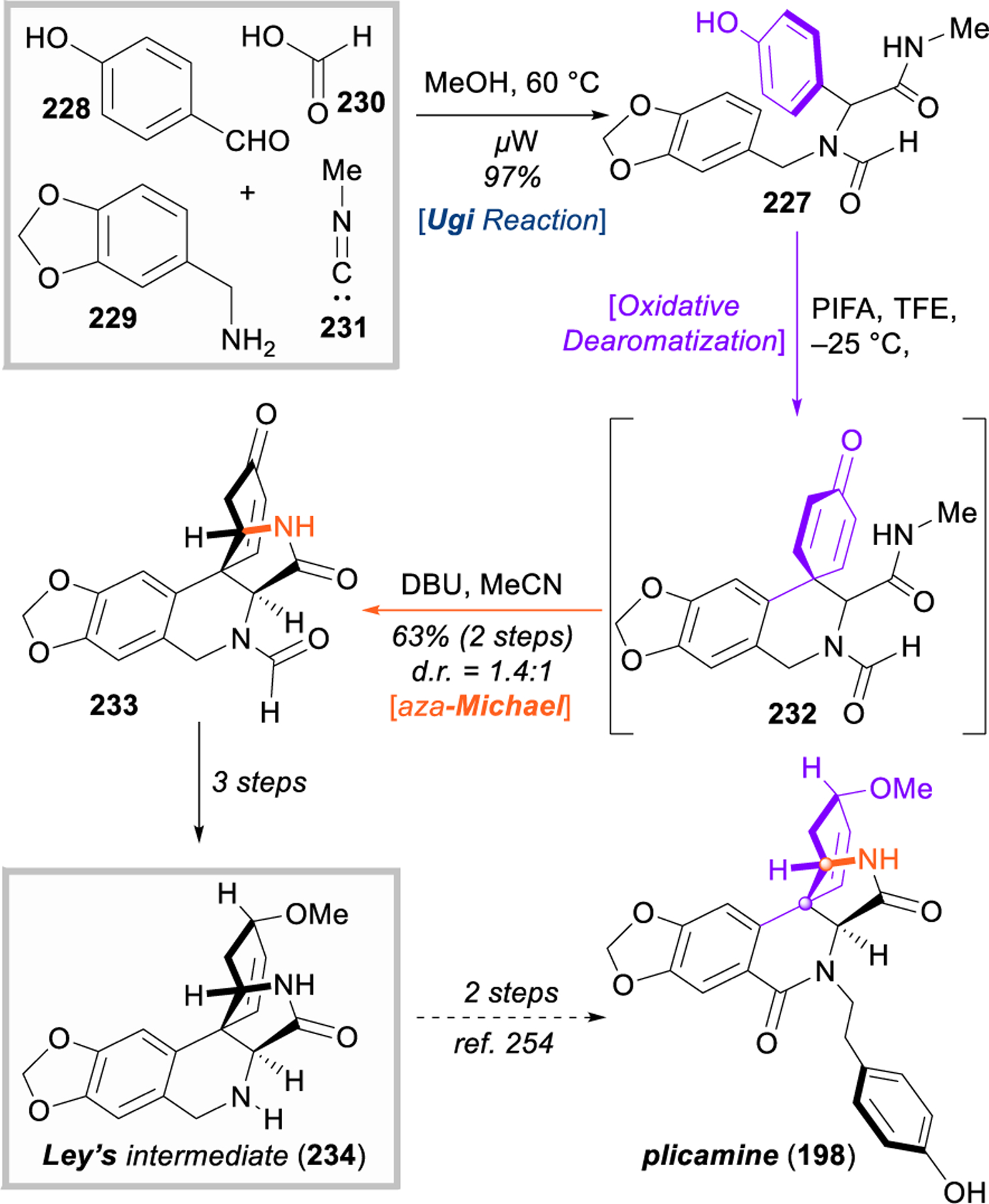
Miranda and Mijangos’s approach to plicamine, featuring a four-component Ugi reaction and oxidative dearomatization.
Although only a recent addition to the family of Amaryllidaceae alkaloids, the rare pentacyclic dinitrogenous base gracilamine (200)—isolated in 2005— has been the subject of no fewer than eight synthetic efforts within the past ten years.290–298 The intriguing structure of gracilamine is proposed to derive biosynthetically from (+)-epicrinine (235, Figure 27). Oxidative cleavage of the B-ring and condensation of aldehyde 236 with L-leucine generates imine 237. Intramolecular 1,3-dipolar cycloaddition completes the pentacyclic core and, followed by esterification, furnishes (+)-graciliamine (200).236 In addition to a complex ring system containing two basic amines, a major challenge in the synthesis of gracilamine is the construction of the highly congested all-carbon quaternary stereocenter embedded within the polycyclic scaffold at the junction of three rings. This quaternary center is established through oxidative dearomative cyclization in the biosynthesis of crinine, which suggests that this motif may be amenable to dearomative retrosynthetic logic; however, the majority of synthetic efforts towards gracilamine have avoided this approach.290
Figure 27.
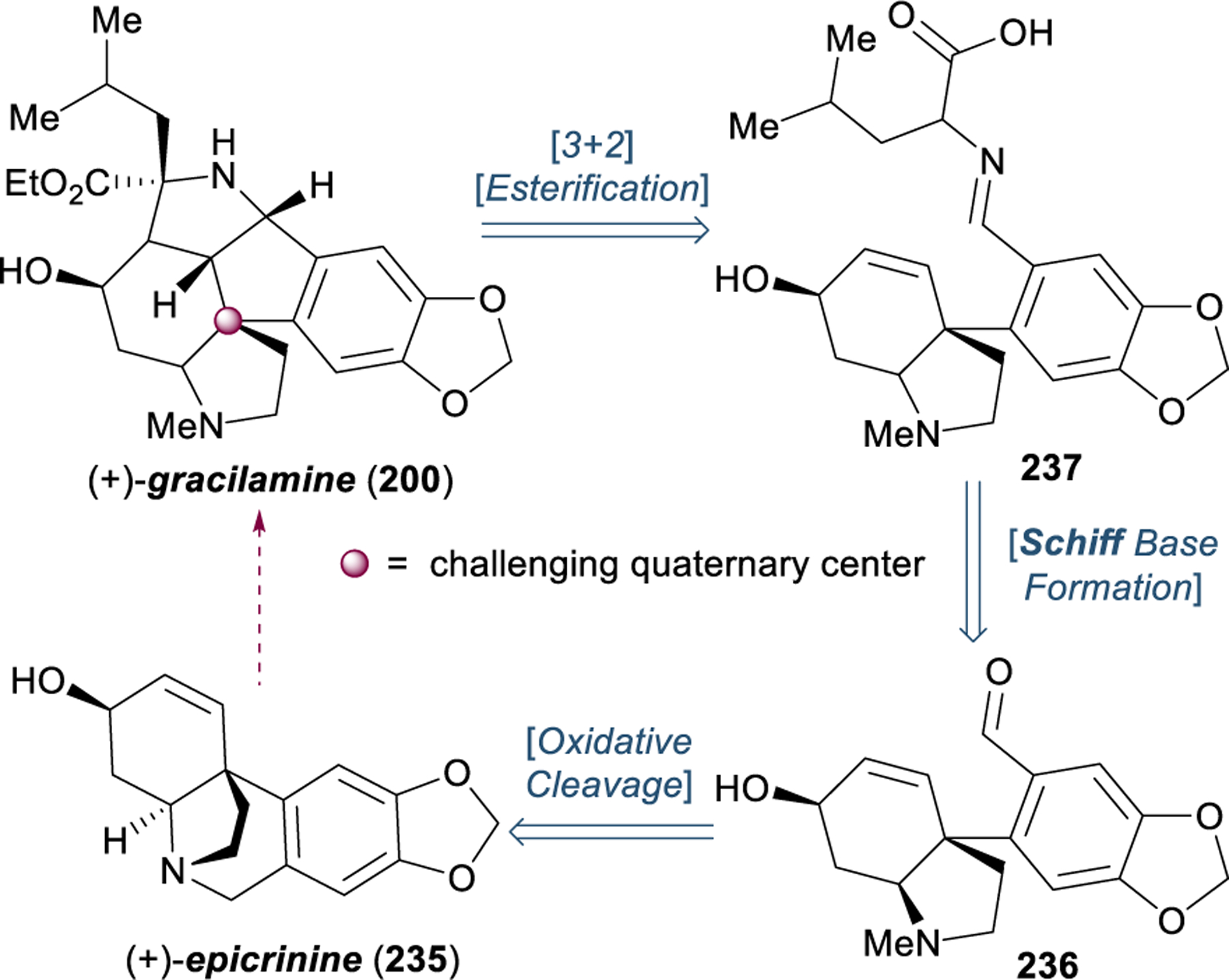
Proposed biosynthetic pathway from crinine to gracilamine.
The first total synthesis, reported by Ma in 2012, actually did construct the quaternary carbon via oxidative dearomatization, but carried symmetric achiral intermediates most of the way through the route, finally establishing it as a stereocenter with a biomimetic 1,3‑dipolar cycloaddition (238 → 239) in good d.r. (5.8:1, Figure 28).291 Gao’s 2014 synthesis forged this critical asymmetric carbon center through photo-Nazarov cyclization (240 → 241),292 while a 2016 formal synthesis from Snyder utilized an intramolecular [4+2]/retro-[4+2] cycloaddition of a pyrone (242 → 243).293 More exotic strategies from Yu (formal synthesis, 2016),294 Pandey (2017),295 and Banwell (2017)296 construct this stereocenter through Rh-catalyzed [3+2+1] cycloaddition (244 → 245), ring opening of an aza-norbornene derivative (246 → 247), and Pd-catalyzed intramolecular Alder-ene reaction (248 → 249), respectively. In 2017, Zhou and Xie published a notable semisynthesis of (+)‑gracilamine from a crinine derivative that proceeds through oxidative cleavage, Schiff-base formation with L‑leucine, and intramolecular 1,3-dipolar cycloaddition, lending strong support to the proposed biosynthesis.297
Figure 28.
Key steps forming the congested quaternary center in several gracilamine total syntheses.
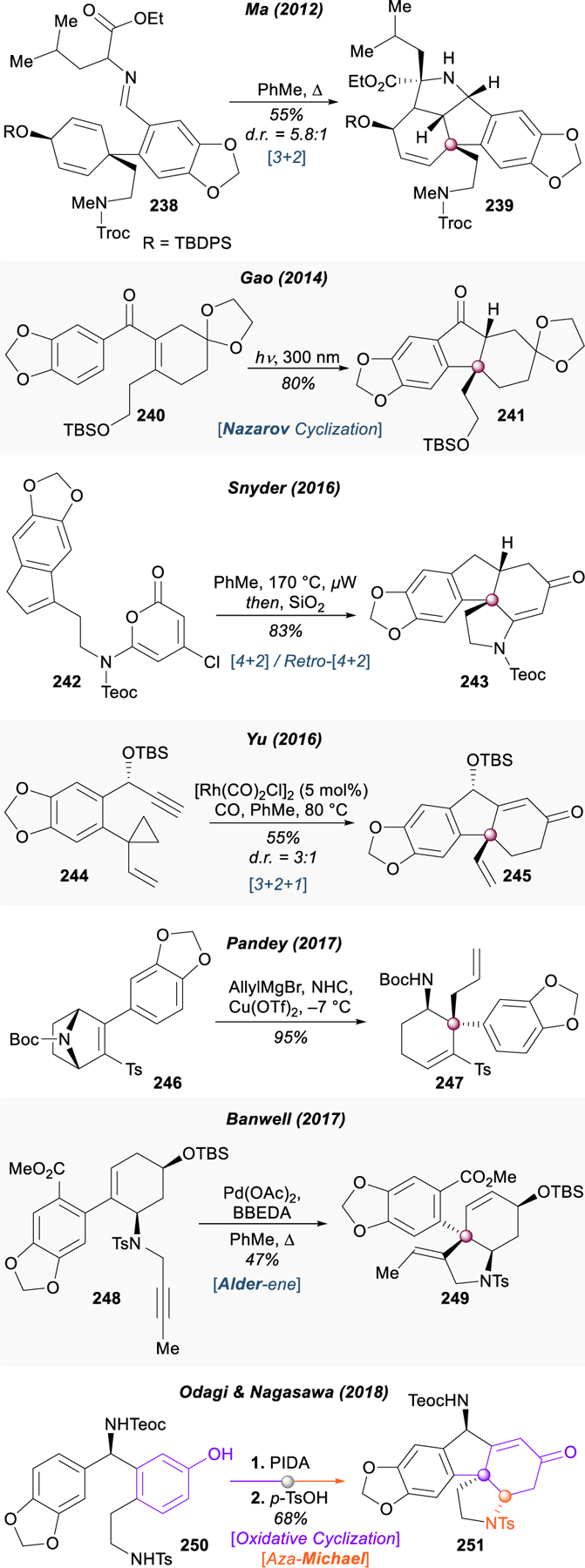
An elegant approach to (+)‑gracilamine (200) was reported by Odagi and Nagasawa in 2018, utilizing a non-biomimetic oxidative dearomative phenol coupling (250 → 251) to first assemble the somewhat strained 6–5-6 core ring system, after which the periphery rings could be installed stepwise (Figure 29).298 Eschewing the biomimetic 1,3-dipolar cycloaddition strategy used by Ma, Pandey, Zhou and Xie, and Banwell to build the E-ring, Odagi and Nagasawa opted to disconnect this motif through the retro-Mannich transformation first reported by Gao and used in the formal syntheses of Snyder and Yu. A logical aza-Michael transform unveils a catechol-bound derivative that could be disconnected in a number of different ways; in this context the authors chose to use an organocatalytic enantioselective aza-Friedel–Crafts reaction that had been previously developed in their laboratory.299
Figure 29.
Total synthesis of gracilamine from Odagi and Nagasawa.
All three phases of dearomative logic were applied during the construction of the quaternary stereocenter and the adjacent pyrrolidine ring. Benzaldimine 252 was synthesized from 3‑hydroxybenzoic acid in seven steps, using standard EAS to brominate para to the phenol. Subsequent cross coupling with allyltributylstannane installed the pendent allyl substituent, which would later be transformed to an amine. In the presence of chiral guanidine–bisthiourea bifunctional organocatalyst 253, the enantioselective aza-Friedel–Crafts reaction with sesamol (254) proceeded smoothly, delivering benzhydrylamine 255 in 94% yield and 91% ee. This compound was converted to the dearomative cyclization precursor through a six-step sequence (255 → 250) incorporating triflation of the phenol, reductive ozonolysis and nucleophilic azidation of the allyl group, and catalytic hydrogenation to cleave the triflate and reduce the azide to the amine. The amine was protected as a toluenesulfonylamide, and the Boc was exchanged for a Teoc group due to solubility issues.
Oxidative dearomatization of 250 with phenyliodine(III) diacetate closed the B-ring of gracilamine, forming the central quaternary center of 256 with good yield and as a single isomer. The authors propose that this excellent diastereoselectivity derives from the configuration of the carbamate set during the aza-Friedel–Crafts reaction, driving a conformational preference (257a → 257b) in the transition state to avoid steric interactions between the bulky protected amines and yielding exclusively the trans-isomer 256. The resulting cyclohexadienone was used immediately to build the pyrrolidine ring in 251 through an acid-catalyzed aza-Michael addition. Having essentially arrived at Gao’s intermediate,292 Odagi and Nagasawa were able to complete the total synthesis of (+)-gracilamine (200) in four steps encompassing hydrogenation of the enone, condensation of an α–ketoester with subsequent Mannich annulation, hydride reduction of the ketone, and removal of protecting groups. Notably, this enantioselective dearomative route enabled the authors to synthesize enough material to determine its absolute configuration, a characteristic that had remained a mystery for thirteen years.
Isocarbostyril Alkaloids:
The Amaryllidaceae constituents that have attracted by far the most interest from the synthetic community are the isocarbostyrils narciclasine (204), lycoricidine (205), pancratistatin (202), and 7‑deoxypancratistatin (203, Figure 23). The potent antitumor effects of crude daffodil extracts were known to ancient Greek physicians by the time of Hippocrates and have found applications in various medicinal practices throughout the world for over two thousand years.300 The identification of the isocarbostyrils 202–205 as the specific metabolites responsible for this activity stimulated intense research efforts aimed to develop these auspicious compounds into modern anticancer treatments through natural product total synthesis, analogue development, and structure–activity relationship studies.301
To address the difficulties encountered in their isolation, particularly regarding the low yield of the pancratistatins 202 and 203, dozens of total syntheses of the isocarbostyril alkaloids have been reported; however, the densely packed functionality decorating their compact aminocyclitol cores poses a daunting challenge for practical and efficient total synthesis, and despite the brevity and elegance of many approaches, until recently, only milligram quantities of the isocarbostyril alkaloids have been prepared. Several excellent reviews summarizing the state of Amaryllidaceae isocarbostyril alkaloid synthesis have been published300,302 and thus only one particularly strong pioneering example will be discussed here.
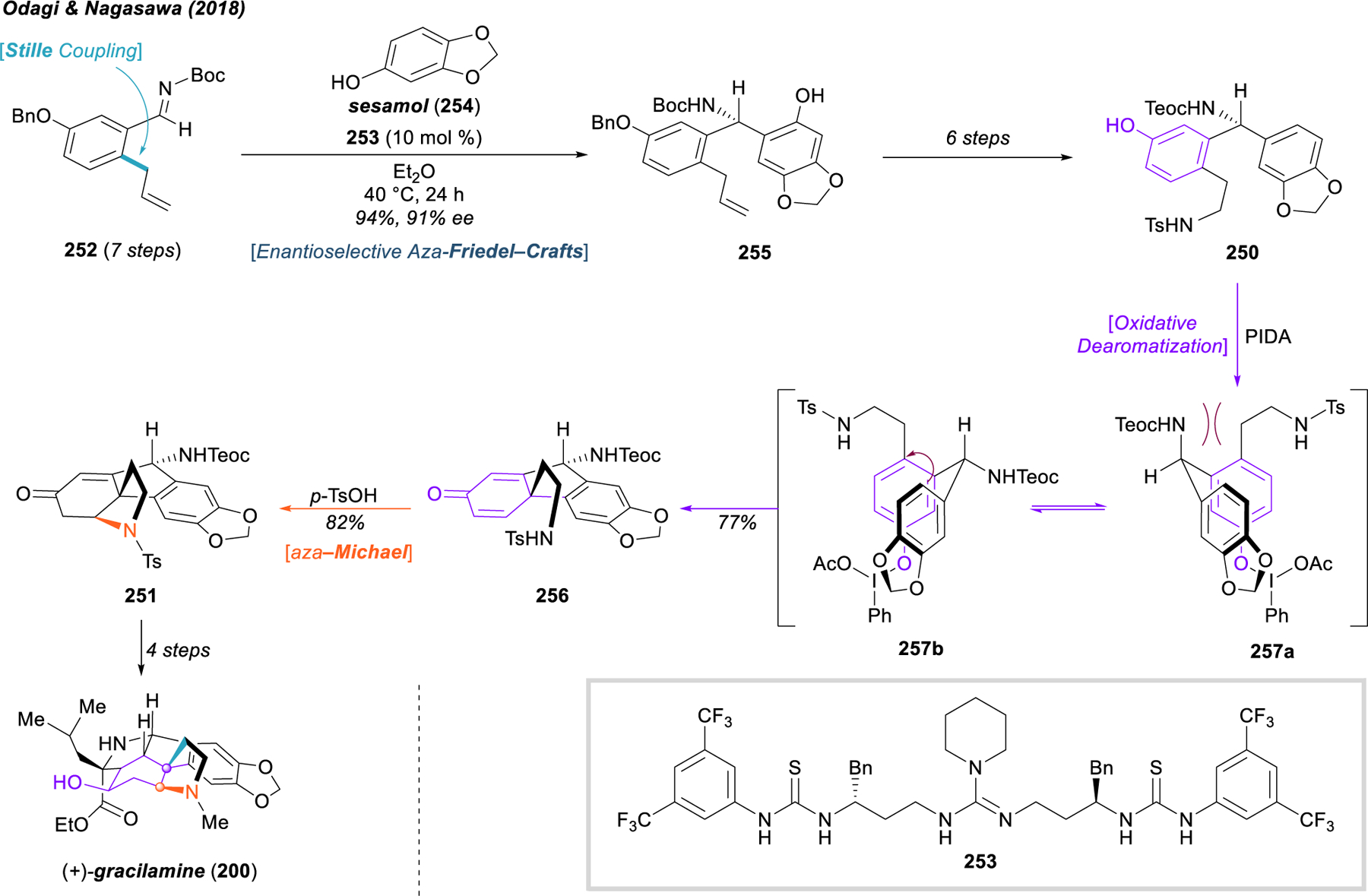
In 1995, Hudlicky and coworkers published the first asymmetric total synthesis of (+)-pancratistatin (202), leveraging the power of dearomative microbial oxidation of bromobenzene (258) to acquire optically pure, cis-dihydrodiol 259 (Figure 30).274 This highly strategic starting material, with the necessary asymmetry and vicinal syn-diol, possesses two sterically and electronically differentiated olefins suitable for further elaboration to the full aminocyclitol core through a series of olefin and functional-group manipulations. Acetonide protection of the diol was followed by aziridination to install a C–N bond, and the vinyl bromide—no longer necessary to maintain asymmetry—was removed via radical dehalogenation (259 → 260). Vinylaziridine ring opening with the higher-order aryl cuprate derived from arene 261 cleanly produced tosylamide 262 in 75% yield, bearing four requisite components of the hexafunctionalized aminocyclitol core. At this stage, seven steps were required to adjust the functionality and protecting groups from 262 to 263 to prepare for the final olefin functionalization and δ-lactam closure. The final trans-diol 264 was synthesized through a Sharpless directed epoxidation and benzoate-catalyzed epoxide opening, with concomitant cleavage of the Boc group and cyclization of the lactam. Although some debenzylation was observed in the previous step, the authors reported a more efficient deprotection via hydrogenolysis that delivered (+)‑pancratistatin (202) in 80% yield.
Figure 30.
Hudlicky’s total synthesis of pancratistatin.
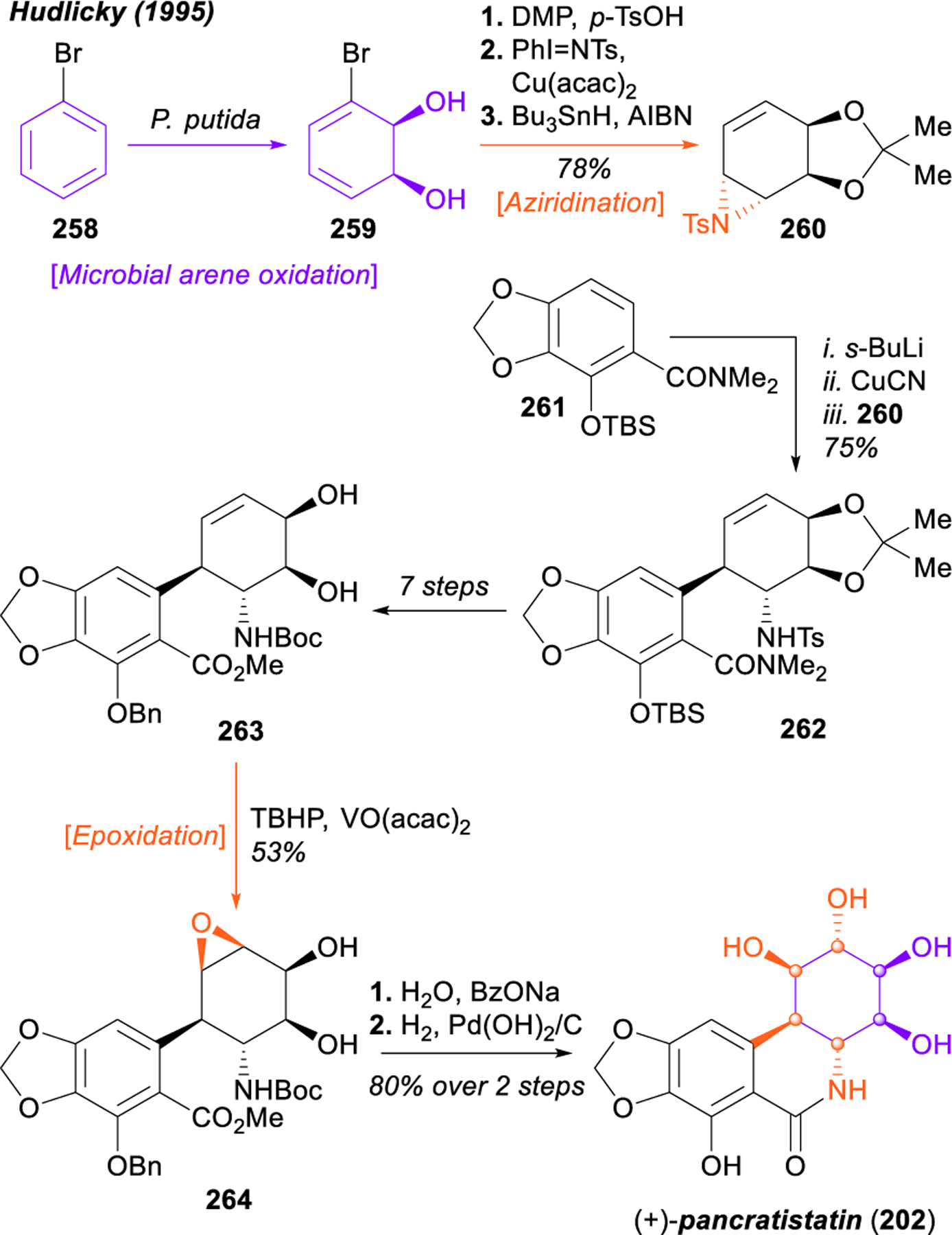
With modern developments in dearomative methods, Sarlah and coworkers reported rapid strategies towards the isocarbostyril alkaloids in 2017 and 2019 (Figure 31).303,304 Using the arenophile-mediated asymmetric dearomative anti-carboamination, a catalytic desymmetrization of benzene (265) with MTAD (266) was achieved in 98:2 er. The product, critical diene intermediate 267, could easily be elaborated to the full aminocyclitol core of the pancratistatins in just two steps, consisting of an anti- dihydroxylation followed by a syn-dihydroxylation (267 → 270 → 271). Notably, all six components of the aminocyclitol core were installed in just three steps, treating benzene as a surrogate for 1,3,5-cyclohexatriene subjected to three subsequent olefin difunctionalization reactions. This intermediate was carried forward to complete the synthesis of (+)-7-deoxypancratistatin (203) in just three steps. Alternatively, diene 267 underwent a bromohydrin reaction with concomitant aryl bromination with NBS in water. Upjohn dihydroxylation of 272 was followed by treatment with potassium carbonate to facilitate epoxide closure, and the diol was protected as the acetonide 273. This intermediate was readily advanced to the natural product (+)-lycoricidine (205) in three steps. Although the syntheses of the hydroxylated isocarbostyrils pancratistatin (202) and narciclasine (204) could be completed through analogous routes that incorporated an appropriately oxidized Grignard reagent in the dearomative carboamination, Sarlah’s strategy towards these natural products is unique in that it provides the first direct access from their deoxygenated congeners. After global silylation of the free hydroxyl moieties of 203 and 205 with HMDS, a novel, amide-directed arylcupration, developed by Uchiyama and coworkers,305 allowed for the direct oxidation with TBHP to complete the syntheses of both (+)-pancratistatin (202) and (+)-narciclasine (204) in a total of seven steps from benzene.
Figure 31.
Sarlah’s catalytic desymmetrization of benzene and application to the total synthesis of pancratistatin, 7-deoxypancaratistatin, narciclasine, and lycoricidine.
Morphinan and Hasuban Alkaloids:
Given their potent analgesic activity and therapeutic applications, morphinan (274)-based opioid narcotics such as morphine (275), codeine (276), hydrocodone (277), and oxycodone (278) have been the subject of extensive studies (Figure 32).306 Morphinan biosynthesis begins with the condensation of two tyrosine derivatives, dopamine (279) and 4-hydroxyphenylacetaldehyde (280). Multiple methyl transfers, hydroxylation of C4, and epimerization at C9 furnishes the key biosynthetic precursor (R)-reticuline (281). Salutaridinol (282), the first promorphinan alkaloid, is produced via cytochrome P450 (SalSyn)-catalyzed intramolecular phenol coupling. Ketone C7 reduction catalyzed by NADPH-dependent reductase (Sal) is followed by acetyl transfer to deliver intermediate 283. Spontaneous rearrangement occurs, yielding thebaine (284), the first pentacyclic morphinan alkaloid. Several additional biocatalytic steps are required for conversion of thebaine into various morphinans such as morphine (275) and codeine (276).
Figure 32.
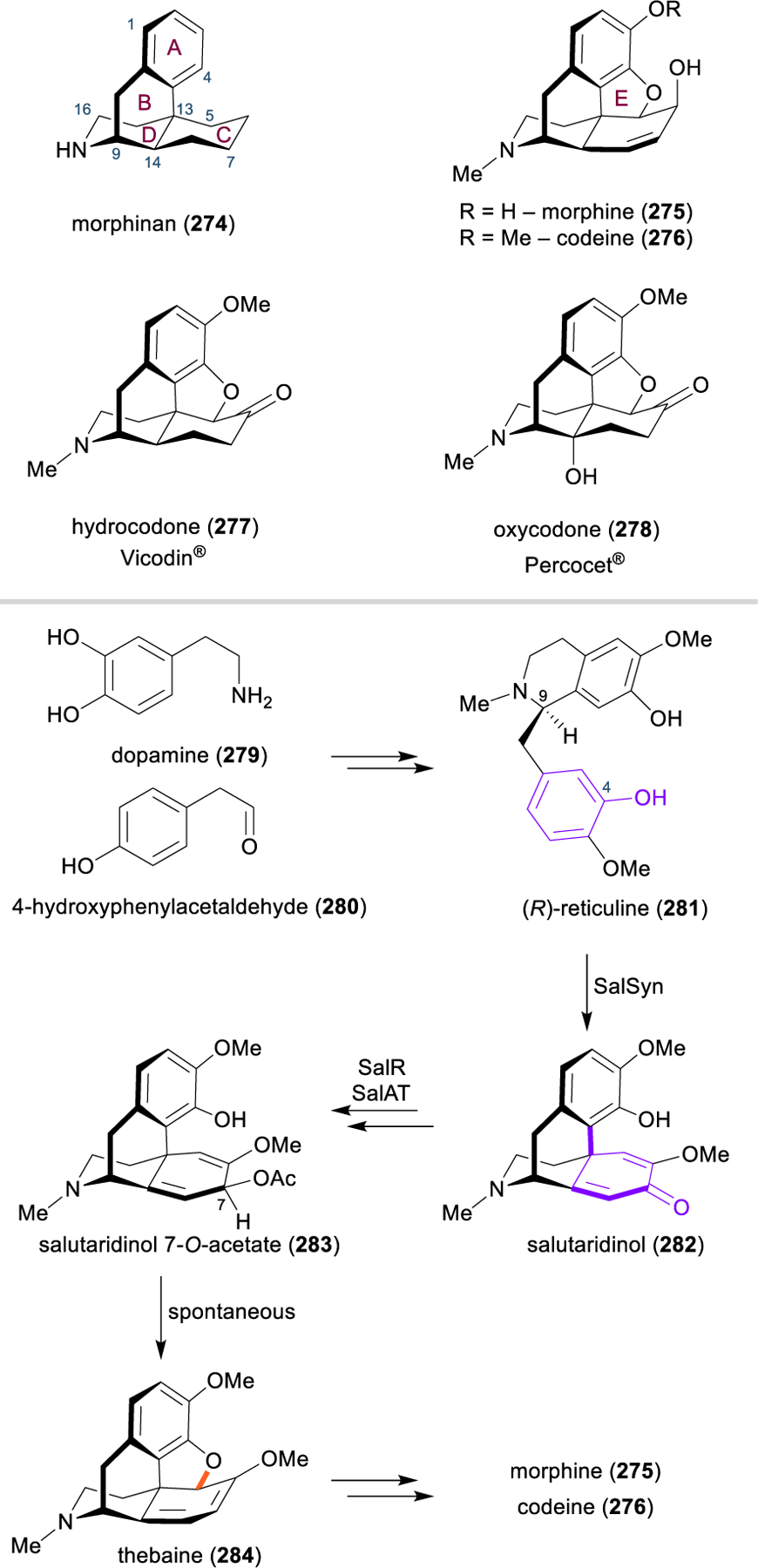
Structures of selected morphinan alkaloids and proposed biosynthetic pathway.
The exceptional medicinal importance of the morphinan alkaloids stimulated copious synthetic studies since the early 1950s.307,308 More than 30 total and formal syntheses have been reported to date (Figure 33). Construction of the quaternary stereocenter at C13, buried within the caged structure, might be considered to be the most significant obstacle en route to morphinans. Informed by the biosynthetic hypothesis, researchers have often incorporated dearomative cyclization as a key transform in their retrosynthesis. Indeed, dearomative logic in the context of morphinan synthesis has proven quite fruitful, as the corresponding reticuline-like aromatic precursors such as 285 can be easily synthesized from simple building blocks (e.g., via Bischler–Napieralski reaction of 286 and 287).
Figure 33.
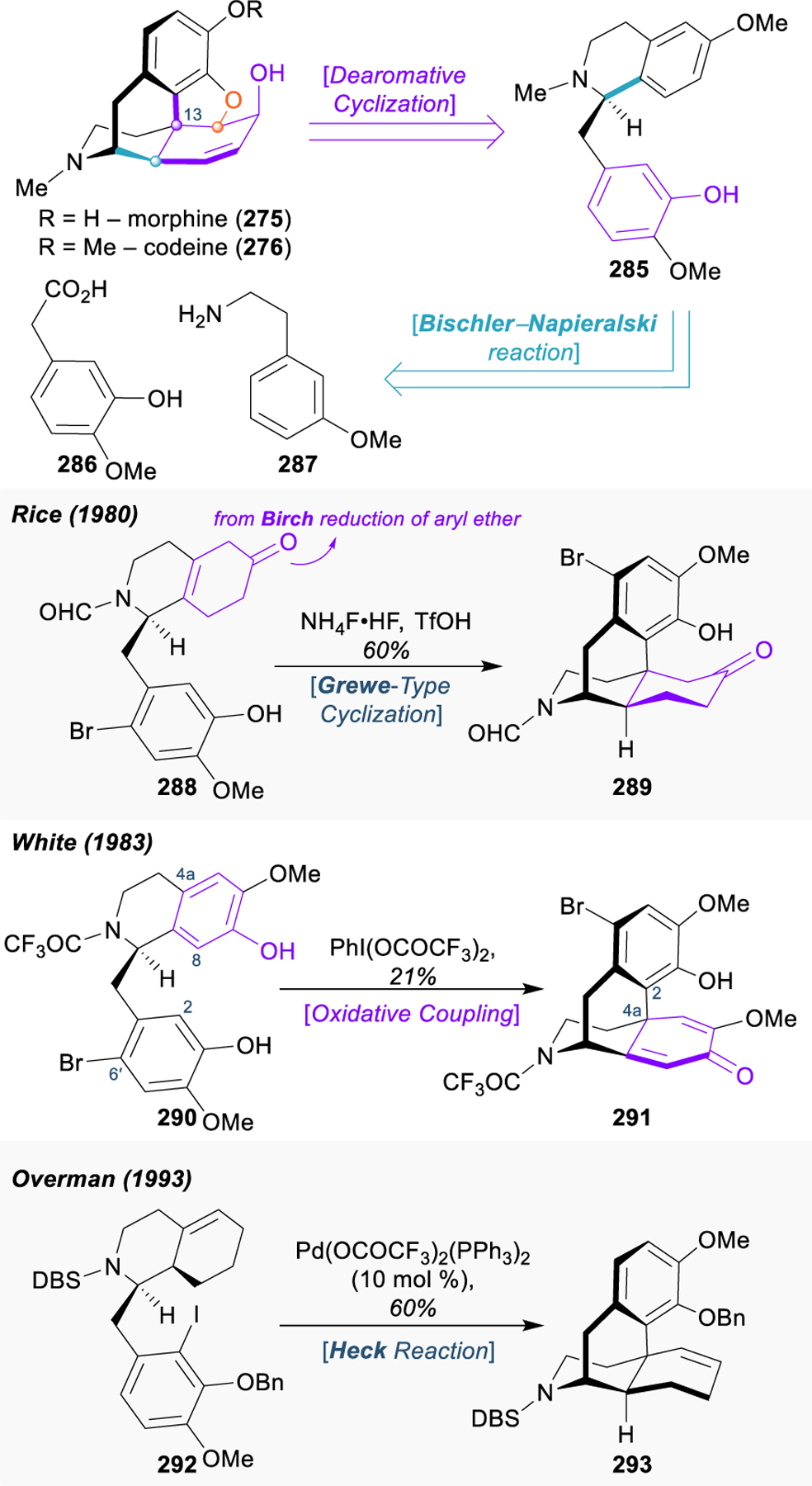
Classical approaches for the synthesis of morphinan alkaloids.
The first practical synthesis, reported by Rice in 1980, is a milestone in the field of morphinan alkaloids (Figure 33).309 The key C12/C13 bond formation is secured by Grewe-type cyclization under strong acidic conditions. The requisite precursor 288 was obtained via Birch reduction of the corresponding methoxy-substituted aromatic compound, and a bromine atom was used as a blocking group on the arene to achieve necessary site-selectivity during Grewe cyclization (288 → 289). Thus, in this case dearomative cyclization is separated into two distinct chemical operations. Compound 289 was further elaborated to (±)-dihydrothebaione, a precursor for morphine (275), codeine (276) and thebainone, in only six steps from commercial material with 37% overall yield. Another classical approach is a direct ortho-para phenolic coupling (290 → 291). Notably, this biomimetic transformation is challenging to reproduce in a flask, as four potential constitutional isomers can be expected. Significant efforts were needed to identify effective conditions, since inherent reaction selectivity lies towards undesired 8,6´- and 4a,6´-isomers.310 In 1983, White and coworkers published a synthesis of (–)-codeine employing a bromine substituent at the 6´-position to overcome this intrinsic bias and furnish the desired scaffold of the natural product in 21% yield.311 In 1993, Overman applied a Heck cyclization (292 → 293) in his enantioselective synthesis of dihydrocodeinone and morphine.312 This approach would become canonical in the field. Of note, even though dearomatization is not involved in the key transformation, substrates like 292 are often synthesized from corresponding aromatic counterparts. While other elegant strategies towards morphinans have emerged over the years,313–316 considering the tremendous success of dearomative methods, it is not surprising that, even after 35 years, these approaches still dominate the field.
During last decade, significant progress was made to increase the selectivity and efficiency of phenolic oxidative couplings (Figure 34). To avoid regioselectivity issues in his formal synthesis of (–)-morphine, Gaunt used substrate 294 for oxidative coupling, the structure of which significantly differs from biosynthetic precursor 281.317 The existing stereocenter at C16 serves as a controlling element in the following Michael addition (295 → 296). The overall process delivers the desired product in 48% yield and furnishes three new stereocenters with exclusive diastereoselectivity. Downstream intermediate 297 was subjected to a sequence comprising Luche reduction, treatment with acid, and subsequent reductive amination in order to rearrange the core into the morphinan scaffold (298 → 299 → 300).
Figure 34.
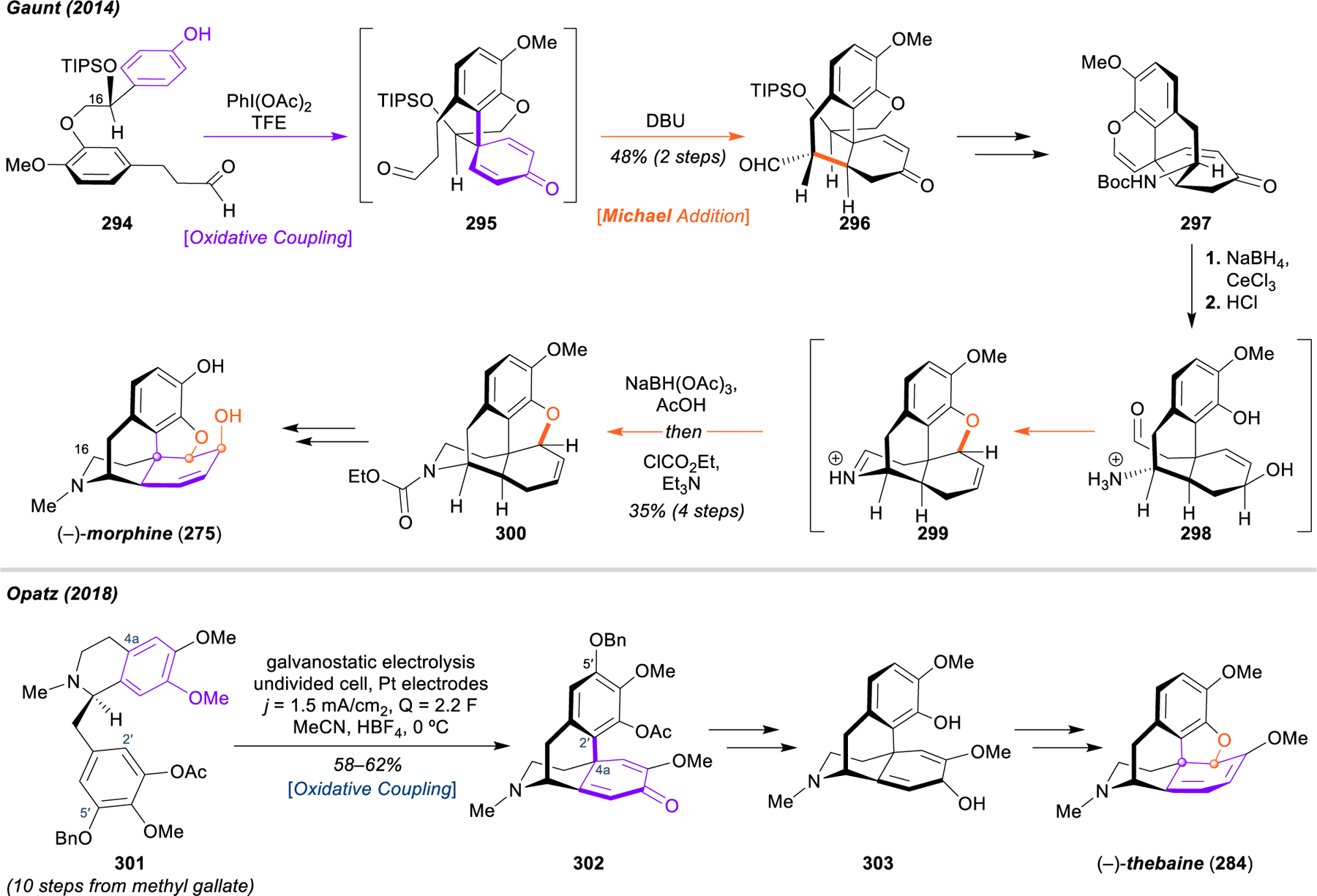
Modern syntheses of morphine and thebaine with improved oxidative coupling conditions.
Another demonstration of the unique ability of dearomative approaches to convert easily accessible arenes into three-dimensional complex structures is formal synthesis of (–)-thebaine (284) completed by Opatz and coworkers.310 In the quest to overcome the undesired regioselectivity of the oxidative coupling, substrate 301 with a symmetrically oxygenated aromatic B-ring was employed. Anodic oxidation (301 → 302) in an undivided cell reliably affords the cyclized product in good yield. A six-step sequence allows for orthogonal deprotection, deoxygenation of C5´ and E-ring formation. Ultimately, the phenolic coupling strategy can be rightfully considered to be one of the most powerful for the synthesis of morphinan alkaloids. However, further improvements are desirable, such as exclusion of excessive substitution in aromatic precursors, which inevitably hampers efficiency of the synthesis.
Tandem dearomatization/Heck cyclization is a remarkably effective tactic for construction of the morphinan scaffold (Figure 35). For that reason, this approach has been widely implemented in key retrosynthetic disconnections. Chen and coworkers applied oxidative dearomatization of the arene 304 with singlet oxygen.318 Desymmetrization (305 → 306), catalyzed by chiral phosphoric acid 307, followed by hydrogenation and fragmentation, afforded intermediate 308. Pd-catalyzed cyclization yielded key ABC-ring system 309 in 69% yield. Finally, Ueno–Stork cyclization served for construction of the C13 stereocenter within tetracyclic fragment 310. Seven additional steps were required to complete total synthesis of ent-oxycodone (278).
Figure 35.
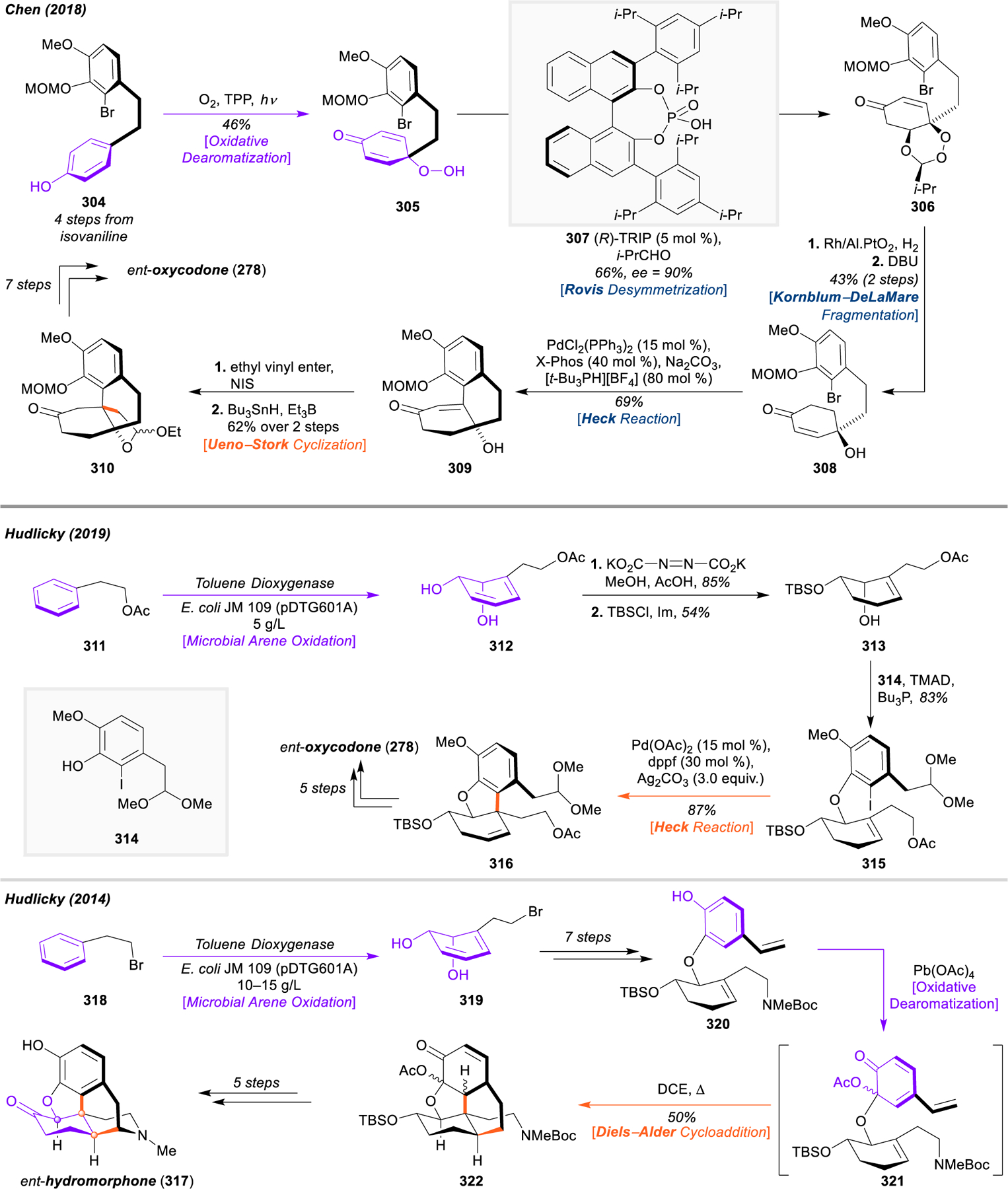
Approaches to morphinan alkaloids using dearomatization/Heck sequences.
The use of enzymatic arene oxidation in the context of the dearomatization/Heck-based strategy towards morphinans was explored by Hudlicky and coworkers.319 Phenethylacetate 311 was subjected to microbial dihydroxylation in whole-cell fermentation with E.coli JM109(pDTG601A) to furnish 312 in 5 g/L yield. Semireduction of the diene followed by protection (312 → 313) and Mitsunobu substitution with phenol 314 afforded intermediate 315. Thus, only four steps including the key dearomatization of a simple arene were necessary to bring significant complexity that would otherwise be hard to achieve. Heck cyclization (315 → 316) forged the AEC-ring system of oxycodone with concomitant formation of the stereocenter at C13 in 89% yield. Four distinct approaches were developed by the group to complete the total synthesis of ent-oxycodone with the most efficient comprising only five steps from 316.320
Besides evolution of the classical approaches towards morphinans, new tactics have emerged over the years. For example, Hudlicky reported the total synthesis of ent-hydromorphone (317) using a double dearomatization/cycloaddition strategy.321 Commencing with an analogous microbial dearomatization of phenethylbromide (318 → 319), compound 320 was attained through a nine-step synthetic sequence. Lead-mediated oxidative dearomatization (320 → 321) of the phenol followed by in situ Diels–Alder cycloaddition (321 → 322) proceeded in 50% yield. The simultaneous formation of the ABCE-ring system of morphine along with quaternary and tertiary stereocenters in the course of a single operation demonstrates a good suitability of such dearomative logic. ent-Hydromorphone (317) was obtained after five additional steps. Notably, only the unnatural series of morphinans can be synthesized using the chemoenzymatic approaches shown in Figure 35 due to the inability to switch the sense of enantioinduction during microbial oxidation.
The hasubanan (323) alkaloids are closely related natural products to morphinans but with a rearranged [4.4.3]-propellane core (Figure 36).322 They are mostly produced by plants of genus Stephania. Comparatively little is known regarding their biosynthesis as well as medicinal potential. In 2011, the Herzon323 and Reisman324 laboratories published enantioselective total syntheses of the hasubanan alkaloids (–)-runanine (324) and (–)-8-demethoxyrunanine (325) employing very similar strategies (Figure 37): the sequence of 1,2-addition to a quinone derivative followed by Friedel–Crafts cyclization was utilized in order to furnish the desired scaffold. Interestingly, in both instances easily accessible arenes were used as starting materials, following a structure-based retrosynthetic strategy. Of note, even though dearomatization is not involved in the key bond-forming events, the assortment of functional groups available after the dearomative process and their orthogonal reactivities are crucial for expedient elaboration of the synthetic precursors.
Figure 36.
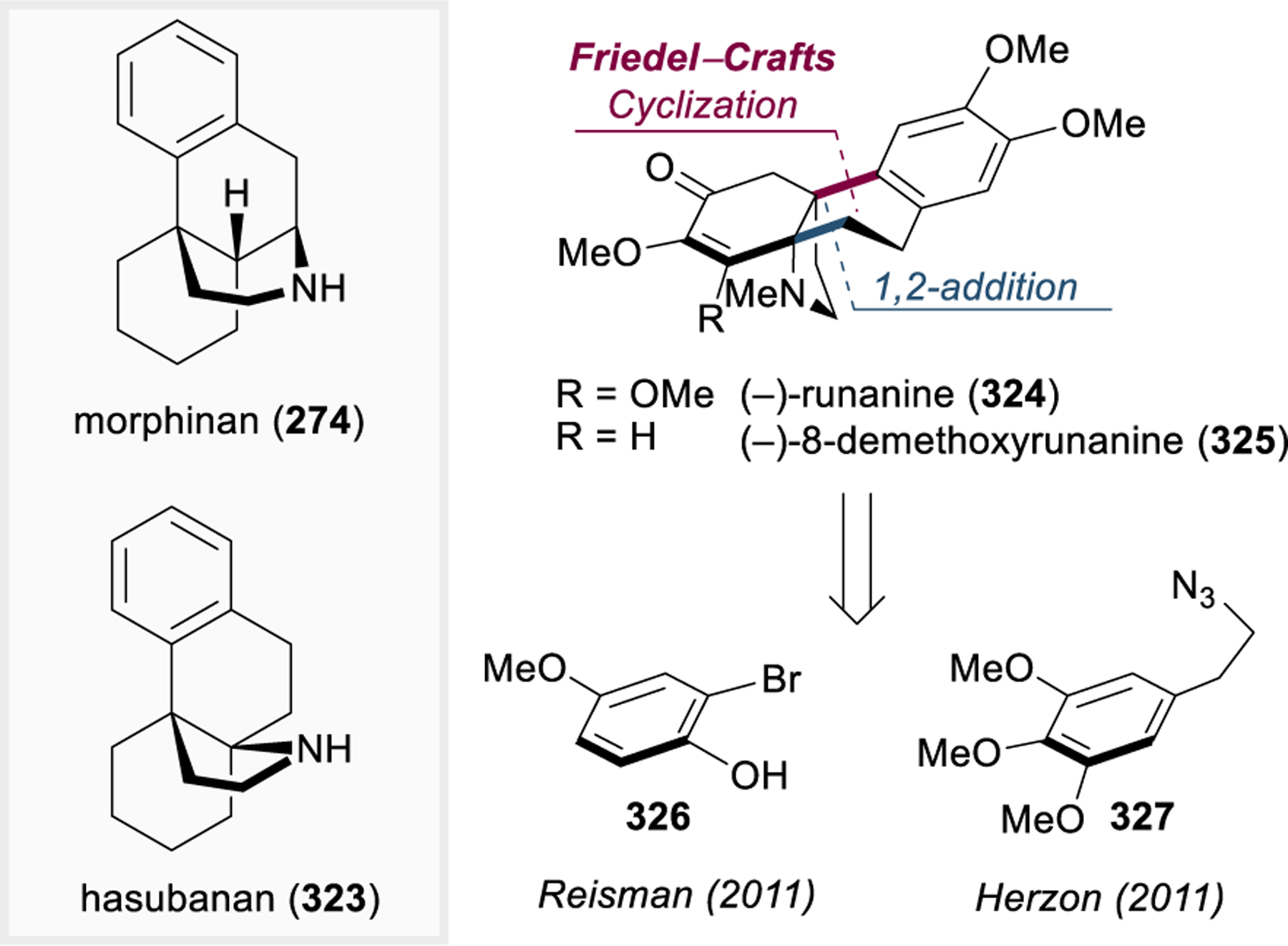
Comparison of morphinan and hasubanan alkaloid scaffolds, and summary of Herzon’s and Reisman’s synthetic approaches.
Figure 37.
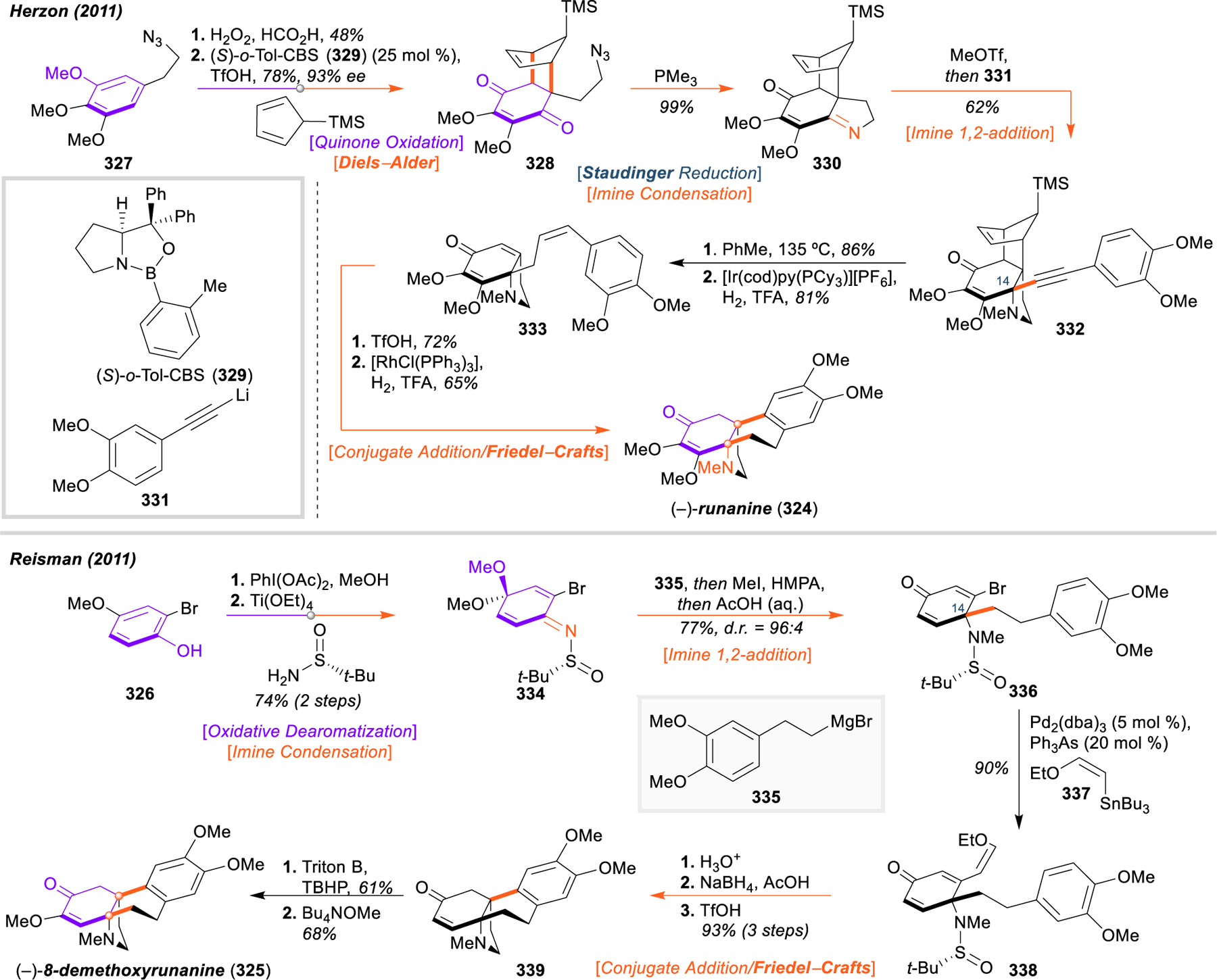
Herzon’s and Reisman’s total syntheses of hasubanan alkaloids.
In Herzon’s synthesis, substituted pyrogallol 327 was oxidized to the corresponding quinone.323 Regio- and enantioselective cycloaddition with 5-trimethylsilylcyclopentadiene was exploited to mask the quinone motif, prevent tautomerization in the following steps, and render the synthesis enantioselective. Compound 328 was obtained in 78% yield and 93% ee. Staudinger reduction, methylation of the resulting imine, and addition of alkynyl lithium reagent 331 into the iminium cation produced 332 in 62% yield. Once the new C–C bond was formed at C14, the masked olefin was revealed via thermal retro-cycloaddition. Semireduction of the alkyne followed by triflic acid-mediated intramolecular Friedel–Crafts alkylation and the final reduction of the cis-olefin furnished natural product (–)-runanine (324) in eight steps and 7.6% overall yield.
Analogously, in Reisman’s synthesis, substituted phenol 326 was oxidized to the dimethyl acetal of quinone.324 Chiral imine 334 was formed upon condensation with Ellman’s auxiliary. Diastereoselective addition of Grignard reagent 335, followed by methylation, furnished intermediate 336 with a properly installed stereocenter at C14. In order to form the D-ring, two-carbon building block 337 was merged with vinyl bromide 336 via Stille cross-coupling. Treatment of 338 with acid promotes cleavage of the sulfonamide with concomitant condensation to form the indolone motif. Chemoselective reduction followed by similar Friedel–Crafts cyclization yielded key intermediate 339. Finally, sequential epoxidation, nucleophilic opening, and elimination afforded the natural product (–)-8-demethoxyrunanine (325) in nine steps and 19% overall yield.
Akuammiline Alkaloids:
Akuammiline alkaloids are plant-derived monoterpenoid indole natural products (e.g., 340–344, Figure 38).325–330 Their namesake compound, akuammilline (340), was isolated in 1932 from seeds of Picralima klaineana.331 However, some of its other members have been known over 145 years. Biosynthetically, these alkaloids derive from cyclization of geissoschizine (341), forming a bond between C7 and C16.332,333 Some members, including akuammiline itself or aspidophylline A (342), possess N4-C3 connectivity, whereas others such as vincorine 343 or echitamine 344 feature N4-C2 connectivity instead. Further oxidative modifications bring wide diversity to the akuammiline alkaloid family with more than 100 members being isolated to date. Importantly, while some hypotheses regarding their biosynthetic origin have been offered, the exact mechanism is yet to be determined. A broad range of biological properties have been reported for the akuammiline alkaloids, such as anticancer, antimicrobial, anti-inflammatory, and analgesic activities.334–339 Aspidophylline A has demonstrated the ability to reverse drug resistance in KB cells.340 Picraline has displayed selective activity towards renal cortex membraine protein SGLT2, which regulates glucose reabsorption and represents a potential target for type-II diabetes intervention.341,342 Of note, due to minute isolation yields, most of these alkaloids have not been studied in depth and their full medicinal potential remains underexplored.
Figure 38.
Structures of selected akuammiline alkaloids and proposed biosynthetic synthons.
Early synthetic studies towards akuammiline alkaloids suggested the densely substituted 6-membered ring with several quaternary stereocenters, including one at C7, to be the most problematic structural feature of these natural products.343 Given the substitution pattern and oxidation state of the core (345), it became evident that dearomatization of the indole motif—perhaps through intramolecular alkylation of synthons 346 or 347—would be the most productive maneuver for the assembly of the caged scaffold. Consequentially, this dearomative logic has been incorporated in a variety of elegant total syntheses and approaches. Even though the first isolation report of an akuammiline alkaloid is dated to 1875, only during the last decade have these molecules capitulated to total synthesis campaigns. This surge can be attributed to the development of new technologies suitable for dearomative construction of the C7 stereocenter, such as transition-metal catalyzed cyclizations and oxidative coupling. Of note, dearomative construction of the C7 quaternary stereocenter is typically performed at the final stage of the synthesis, demanding a robust and highly effective process.
The first total synthesis of an akuammiline alkaloid was reported by Qin in 2009 (Figure 39).344 The key complexity-generating event was a tandem transformation consisting of intramolecular dearomative cyclopropanation (348 → 349) and fragmentation of the donor-acceptor cyclopropane to iminium 350, followed by aminocyclization to form pyrrolidine 351. Thus, nearly the entire framework of (±)-vincorine (343) was assembled from a simple starting material in a single step. This precedent set the stage for application of dearomative methods for future syntheses of akuammiline natural products. MacMillan345 and Yang346 developed unique approaches employing catalytic enantioselective methods for dearomatization of the indole moiety based on sequential Diels–Alder cycloaddition/amination (352 → 353) and Ir-catalyzed allylic substitution reaction (355 → 356), respectively. Both syntheses are discussed in greater details in a previous review.347
Figure 39.
Qin, MacMillan, and Yang syntheses of akuammiline alkaloids.
C6–C7 formation and assembly of the intricate skeleton of akuammiline alkaloids based on dearomative alkylation (formally through 346) was one of the first successful strategies applied (Figure 40). It has been shown that not all substrates are suitable for this cyclization due to high strain energy associated with the corresponding products,343 and harsh conditions (high temperature, strong base, etc.) are often necessary. Nevertheless, this approach proved to be quite effective. For instance, Smith and coworkers completed first total synthesis of (+)-scholarisine A (358) in 2012 using this strategy.348 Readily available bicycle 359 was subjected to heterogeneous nitrile reduction followed by spontaneous nucleophilic opening of epoxide. Protection of the newly revealed secondary amine and oxidation of the alcohol delivers substrate 360 for Fischer indole synthesis. The use of 1-benzyl-1-phenylhydrazine (361) proved to be critical for successful annulation. The resultant indole 362 possesses the majority of the structural features of the natural product. Redox adjustments, protecting group manipulations, and incorporation of a one-carbon synthon at C5 were required to furnish precursor 363 for the final key step. With the aid of the strong base BTPP (364), dearomative alkylation resulted in the formation of the final ring of 365 and a quaternary stereocenter in good yield. Only deprotection and oxidation of the secondary amine were required to complete total synthesis of (+)-scholarisine A (358).
Figure 40.
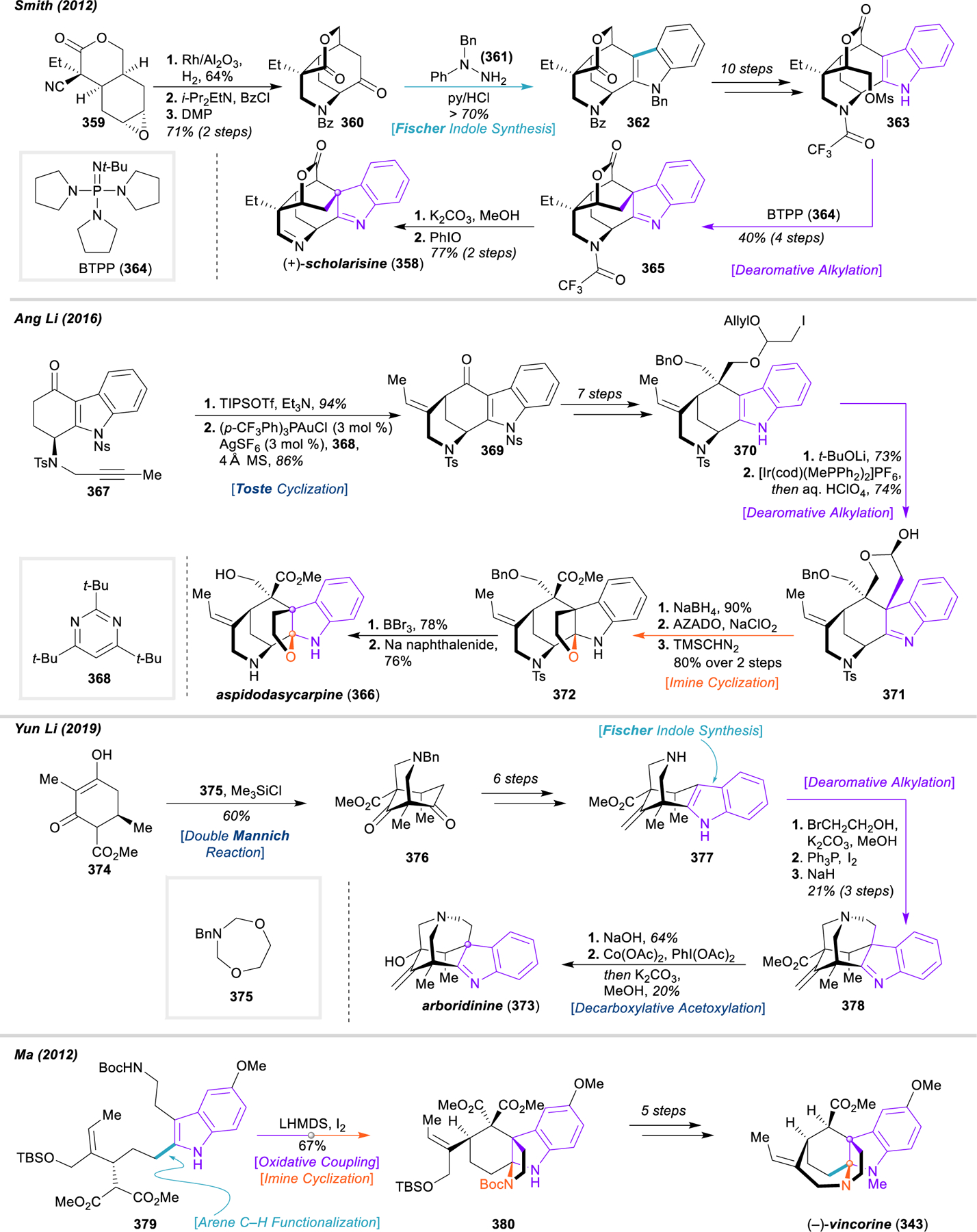
Smith’s, Ang Li’s, Yun Li’s, and Ma’s syntheses of akuammiline alkaloids.
Ang Li and coworkers employed a Toste cyclization and dearomative alkylation as key C–C bond forming reaction in the first and asymmetric total syntheses of aspidodasycarpine (366) and lonicerine.349 Towards this end, tricyclic ketone 367 was converted to the silyl enol ether, which was subjected to a gold-catalyzed cycloisomerization reaction (367 → 369). After several functional group manipulations, tetracyclic intermediate 369 was converted to 370, the precursor for a dearomative cyclization. Reaction with lithium tert-butoxide furnished the quaternary stereocenter in 73% yield with exclusive diastereoselectivity, and subsequent deprotection afforded key intermediate 371. Only five additional steps were required for adjustment of oxidation states (371 → 372) and global deprotection (372 → 366) to deliver the natural product. In 2019, this approach was applied by the same group in the syntheses of seven other alkaloids including akuammiline (340), echitamine (344), and rhazicine.350
Arboridinine (373) was isolated in 2015 from the bark of the Malayan Kopsia arborea tree.351 While this alkaloid bears the features of akuammiline alkaloids, it has an unusual connectivity pattern. Yun Li and coworkers reported an elegant enantioselective total synthesis of (+)-arboridinine in 2019.352 Their first key complexity-generating transformation was a double Mannich cyclization (374 → 376) under Brimble conditions. Bridged bicycle 376 was produced in 60% yield with three of the requisite stereocenters. The indole motif was incorporated via Fischer reaction, and a few additional chemical operations led to the key intermediate 377. An ethylene linchpin between N4 and C7 was introduced over three steps to form the final ring and quaternary stereocenter of 378 in 21% overall yield. Hydrolysis of the ester, followed by an ambitious decarboxylative acetoxylation, delivered the natural product. These selected examples demonstrate the utility of dearomative alkylation as a reliable and straightforward method for the synthesis of various arduously caged alkaloids.
A conceptually distinct approach was disclosed by the Ma group in their total synthesis of (–)-vincorene (343).353 Instead of the C7–C6 disconnection used in the dearomative alkylation strategy, the key quaternary stereocenter and basic framework of the natural product was assembled via formation of the C7–C16 bond (synthon 347). This disconnection was realized by intramolecular oxidative coupling between indole and malonate motifs. The key precursor 379, with a single stereocenter, was prepared from 5-methoxytryptamine. Sequential oxidative coupling/aminal formation (379 → 380) were carried out under basic conditions in presence of iodine as an oxidant with 67% yield and exclusive diastereoselectivity. Thus, a single dearomative operation allowed for formation of the complex polycyclic structure and two quaternary stereocenters, enabling rapid access to the akuammiline alkaloid. Only five additional steps were required to complete the synthesis. Notably, this design has been extended further to the total synthesis of (±)-aspidophylline (342) reported in 2014.354
Alternatively, C7–C16 bond formation can be accomplished via 6-endo-dig cyclization between the indole and an alkyne. Pioneering studies by Wang and coworkers (not shown) demonstrated the feasibility of this dearomative approach towards akuammiline alkaloid scaffold using gold catalysis.355 Inspired by these initial results, the Snyder group elaborated upon this strategy and completed total syntheses of strictamine (402, Figure 41),356 arboridinine (373),357 and arborisidine (381).358
Figure 41.
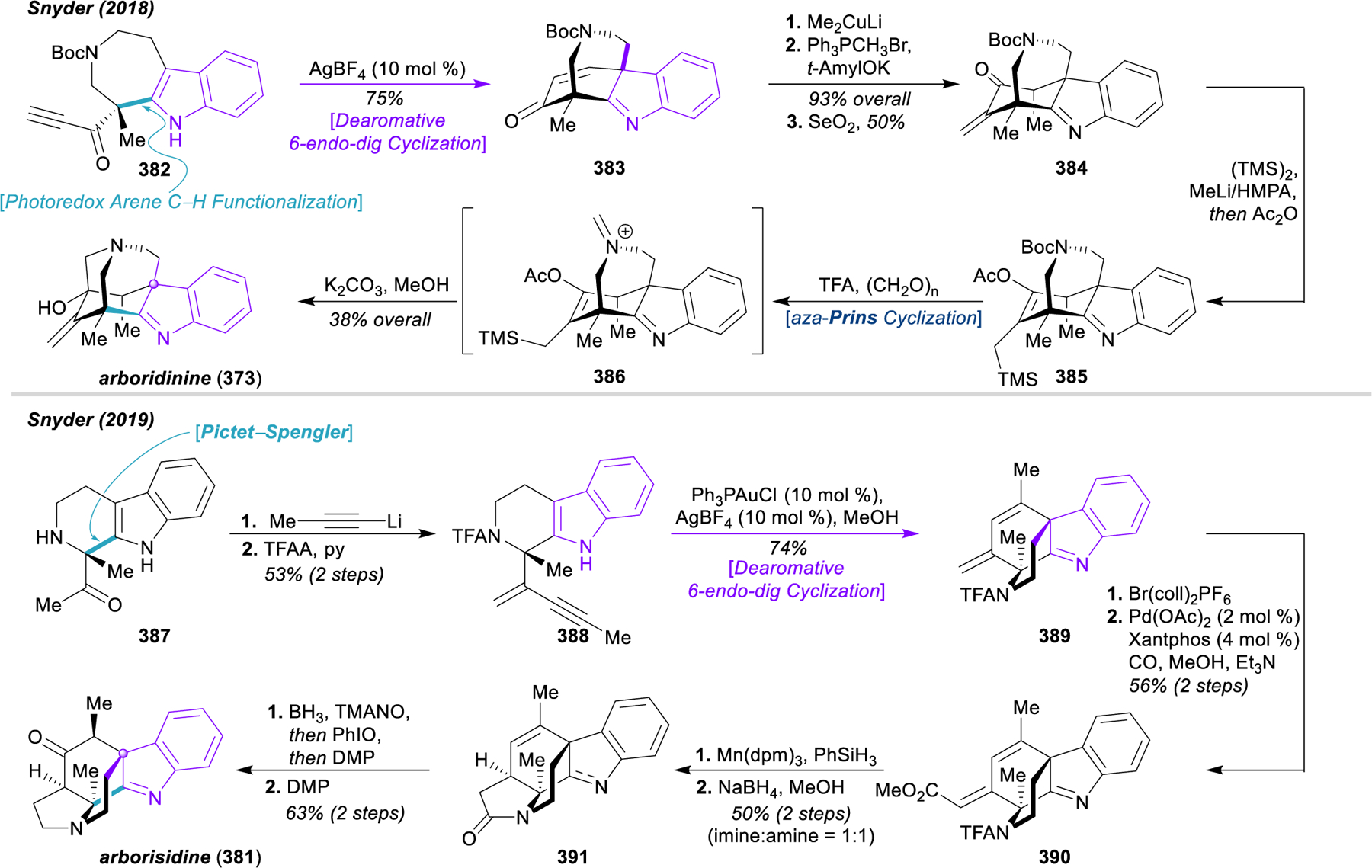
Snyder’s total syntheses of arboridine and arborisidine.
The first total synthesis of aboridinine leveraged a 6-endo-dig cyclization of the compound 382, accessible from tryptamine.359 The authors discovered that a silver catalyst is sufficient for high conversion, affording key intermediate 383 in 75% yield. Addition of Gilman’s reagent followed by methylenation and allylic oxidation delivered compound 384 in 46% yield over three steps. This α,β–unsaturated enone was converted into allylsilane 385. Under acidic conditions in presence of paraformaldehyde, the Boc-protected amine was converted to iminium cation 386, which underwent aza-Prins cyclization to close the final ring and set a quaternary stereocenter at C15. In situ hydrolysis of the acetate furnished arboridinine (373) in 38% yield from 385.357
An analogous dearomative cyclization was employed in the first total synthesis of (+)-arborisidine (381) (Figure 41).358 First, the acetyl group of readily synthesized compound 387 was converted into enyne 388. A rapid increase of complexity was attained via gold-catalyzed cycloisomerization (388 → 389) in 74% yield. Only formation of the pyrrolidine ring and adjustment of oxidation states were required to complete the synthesis. Towards this end, diene 389 was converted to the dienyl bromide, followed by palladium-catalyzed carboxylation. Sequential HAT-type reduction of α,β,γ,δ–unsaturated ester 390 and deprotection of secondary amine resulted in formation of pyrrolidinone 391 in 50% yield. Partial undesired but inconsequential reduction of the imine was observed during this process. Reduction of the lactam, formation of the ketone at C15, and reestablishment of the imine functionality was achieved in two steps, affording (+)-arborisidine (381) in 63% from 391.
It is important to acknowledge that not only dearomative strategies have been successful in the past. Construction of the crucial structural element—the C7 quaternary stereocenter—was realized through a C7–C8 disconnection as well (Figure 42). A series of alkaloids have been prepared by Garg and coworkers, exploiting the interrupted Fischer indolization methodology developed in their laboratory.359–364 Azabicyclo[3.3.1]-nonane precursor 393 was quickly accessed via Heck cyclization of 392. Closure of the δ–lactone during a multistep sequence from 393 to 395 provided additional rigidity to the system. 395 was treated with phenylhydrazine in presence of TFA, and the generated intermediate 396 was trapped with a hydroxyl unit released in situ via basic hydrolysis. Thus, compound 397 with a furoindole core and the C7 stereocenter was synthesized in 70% yield. Functional group swap at N4 delivered racemic aspidophylline A (342).359 The Garg group applied a similar strategy for the first synthesis of picrinine (398) in 2014.361 In this case 5-membered protected diol 399 was incorporated to induce necessary rigidity, which was oxidatively cleaved to aldehyde 400 directly after indolization (399 → 401). Zhu and coworkers completed a total synthesis of strictamine (402) in 2016, utilizing sequential functionalization of a 1,3-cyclohexanedione to assemble the C7 stereocenter of 403.365 Reduction of the nitro group in the elaborated synthetic intermediate 404 was followed by simultaneous condensation to afford indole 405 in 74% yield, which was advanced to strictamine (402) in three steps.
Figure 42.
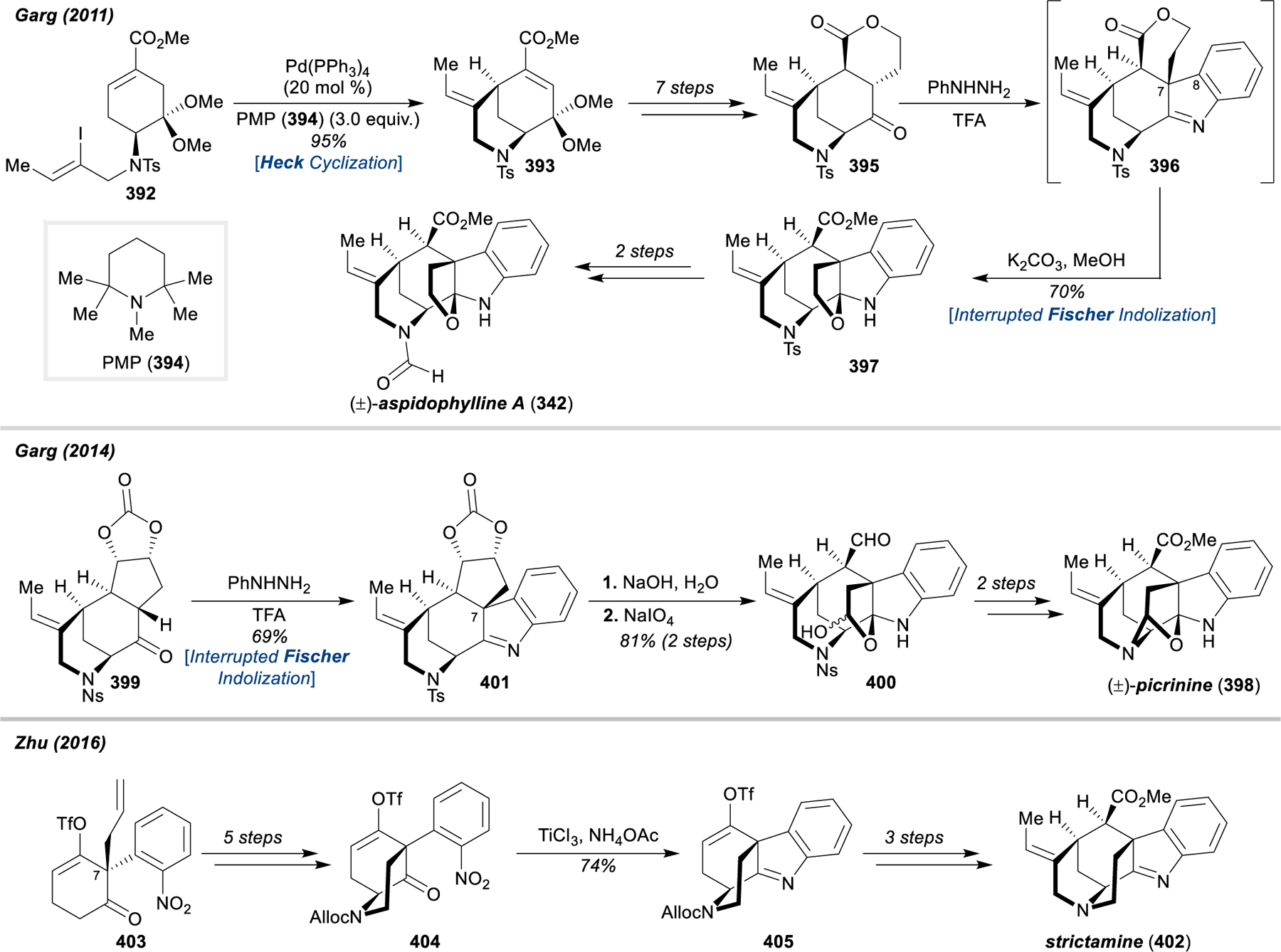
Non-dearomative approaches to akuammiline alkaloids from Garg and Zhu.
Miscellaneous Alkaloids:
The total synthesis of Strychnos and Aspidosperma alkaloids using indole dearomatization is a rich field of study, and numerous examples have been reported within the last decade; however, they will not be discussed here and instead the reader is referred to an excellent review that has been recently published on this topic.366 Although the dearomatization of tryptamine derivatives has been the most widely adopted approach to these indole alkaloid natural products, several alternative and efficient dearomative strategies have been disclosed that deviate from this paradigm.
A noteworthy example that proceeds through phenol dearomatization is Tang’s 2017 total synthesis of aspidospermidine (223) (Figure 43),286 utilizing their previously developed catalytic asymmetric dearomative cyclization methodology (vide supra).285 Effective use of dearomative logic enabled swift assembly of a tetracyclic scaffold bearing appropriate functionality for further elaboration, using the natural reactivity of the intermediates. Buchwald–Hartwig coupling of ortho‑dibromobenzene and aniline 406, prepared in three steps from a commercially available nitrated phenethylamine, forged a critical Csp2–N bond. Protection of nitrogen as the phosphoramide 407 set the stage for a key, palladium-catalyzed dearomative asymmetric cyclization (407 → 408) to establish the indoline system and an all- carbon quaternary center. An additional benefit of this transformation was the generation of a reactive cyclohexadienone perfectly set up for an aza-Michael addition to close the pyrrolidine ring of 410, following deprotection of the benzylamine. From this intermediate, the authors completed the total synthesis of (–)-aspidospermidine (223) in six steps. Additionally, intermediate 408 could be taken forward in seven steps to intercept Overman’s intermediate 411,367 thus completing a formal asymmetric synthesis of (–)-minfiensine (224) as well.
Figure 43.
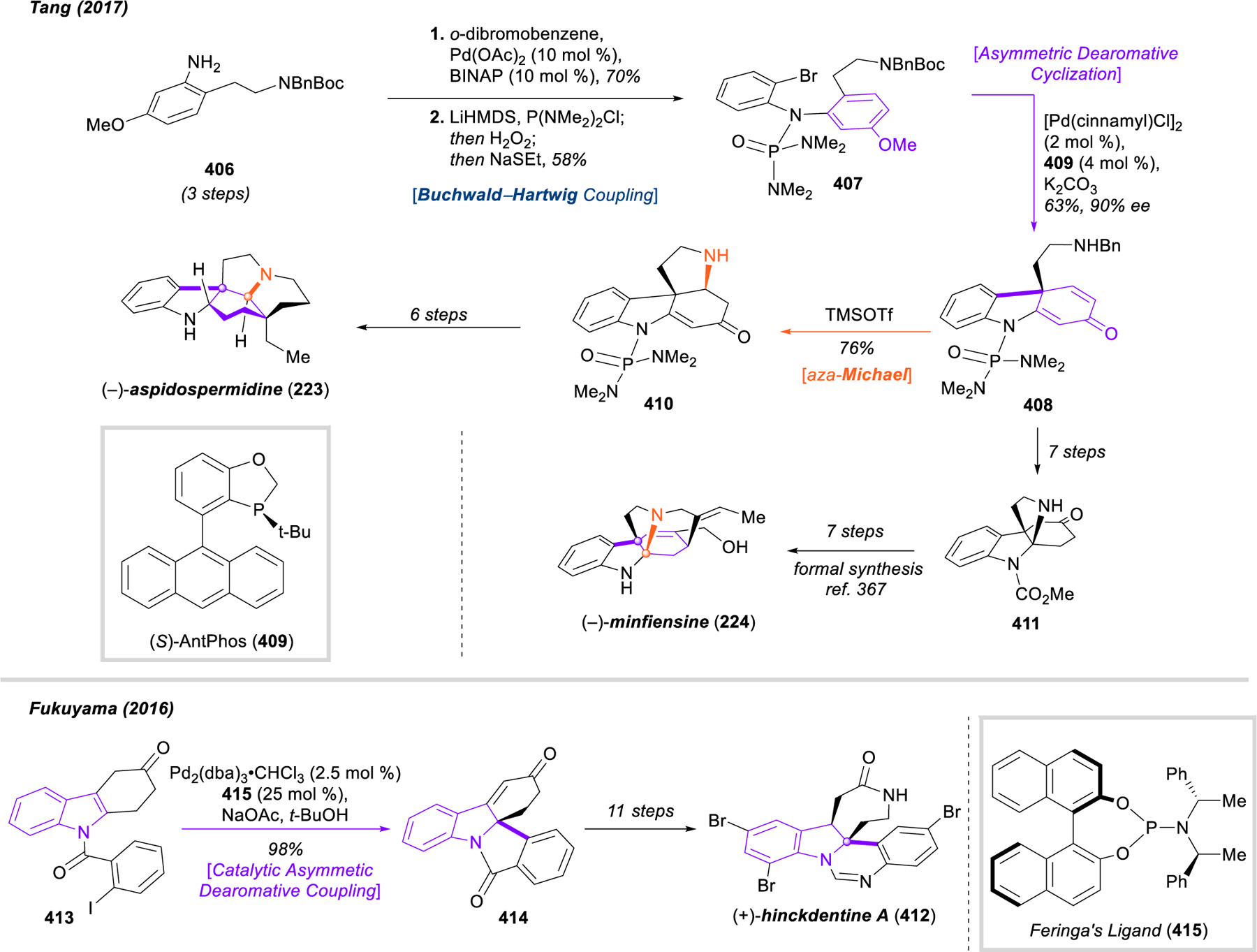
Tang’s synthesis of aspidospermidine and formal synthesis of minfiensine, using enantioselective palladium-catalyzed dearomatization of phenols. Fukuyama’s synthesis of hinckdentine A using a catalytic asymmetric dearomative coupling.
Another powerful catalytic asymmetric dearomatization was reported by Fukuyama in 2016, during the total synthesis of the unusual indole alkaloid (+)-hinckdentine A (412).368 Tetrahydrocarbazole 413, with a pendent aryl iodide, proved a competent substrate for dearomative Heck-type cyclization (413 → 414) to establish an all-carbon quaternary center at C2 of the indole. This process was rendered asymmetric with the use of Feringa’s ligand 415, and the Fukuyama team was able to complete the total synthesis of (+)-hinckdentine A (412) after eleven steps.
Related catalytic asymmetric dearomatization (CADA) reactions of indoles, such as those pioneered by MacMillan,369–374 You,375–380 Reisman,381–383 and Knowles384 have been utilized with great success in the synthesis of numerous other indole alkaloids during the past decade.366 For more examples and an in-depth discussion of CADA reactions of indoles in total synthesis, the reader is referred to a recent review from You on the subject.347
Polyketides and Meroterpenoids
Dalesconols:
The Snyder group has completed a large and impressive body of work relating to the synthesis of oligomeric, resveratrol-based natural products, leveraging unique and divergent reactivity modes of unnatural biaryl alcohol building blocks similar to 416 (Figure 44).385–387 In 2010, they reported an extension of this methodology towards the synthesis of the potent immunosuppressant polyphenolic natural products dalesconol A (225) and dalesconol B (226),388 featuring a powerful dearomative cascade sequence in which carbon 19 served as both a nucleophile and electrophile to forge the quaternary center embedded in the center of the molecules. Assembly of 416 from phosphonate ester 417 was achieved in two steps via Horner–Wadsworth–Emmons olefination with 418, followed by lithium-halogen exchange and aryllithium addition into naphthaldehyde derivative 419. Debenzylation and hydrogenation of the stilbene delivered biaryl alcohol 420, which underwent immediate Friedel–Crafts cyclization (420 → 421) upon treatment with TFA, and oxidative dearomatization and cyclization with PIDA (421 → 422) to furnish the entire heptacyclic scaffold of the dalesconols in a single operation. Snyder and coworkers were able to complete the synthesis of dalesconol B (226) in another seven steps, as well as a synthesis of dalesconol A (225) in nine steps LLS following an analogous strategy.
Figure 44.
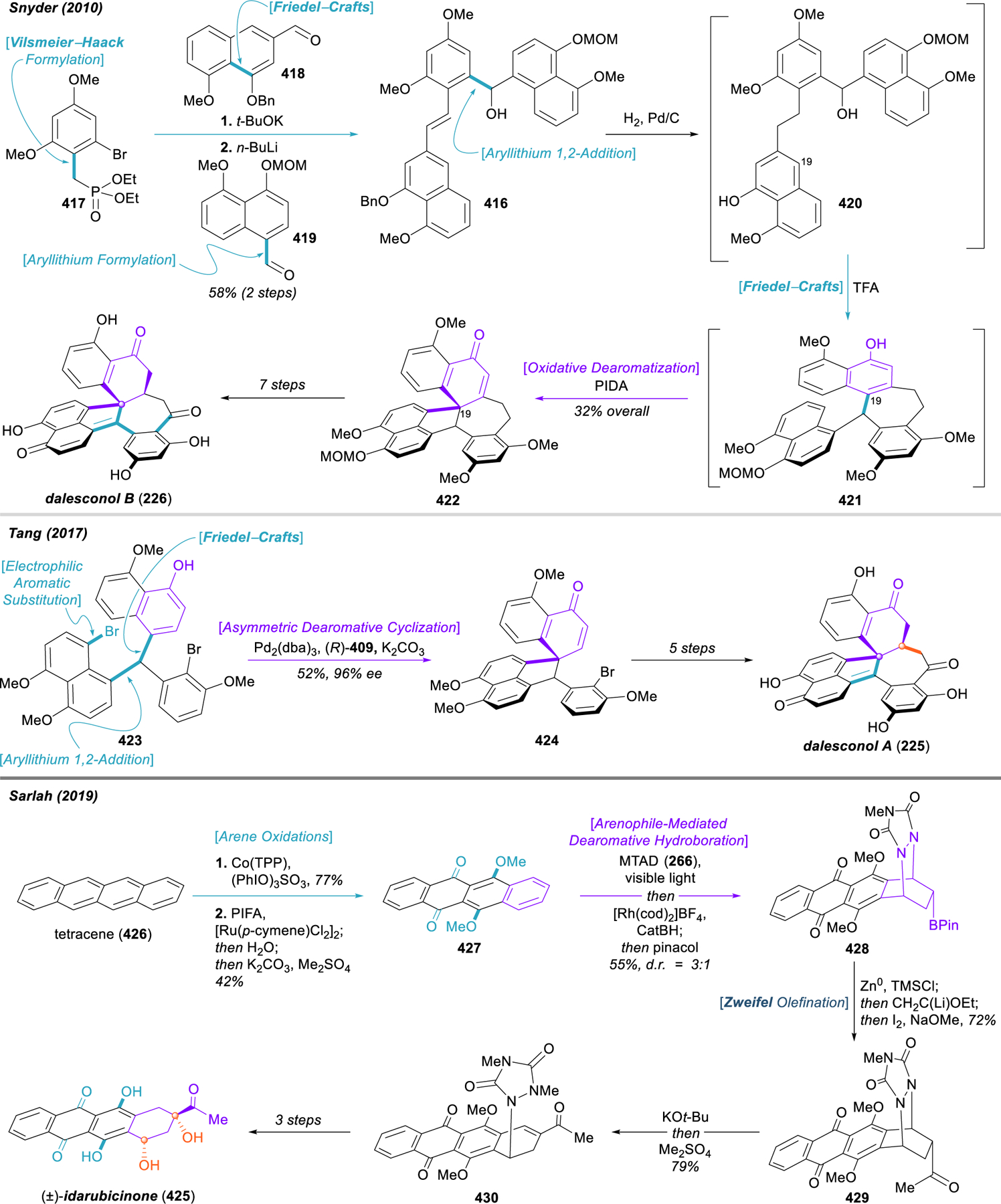
Snyder and Tang’s syntheses of dalesconals, and Sarlah’s synthesis of idarubicinone from tetracene.
In 2017, the Tang group reported an alternative approach towards the dalesconol natural products, utilizing their palladium-catalyzed dearomative cyclization (423 → 424) to construct the central quaternary center. This method allowed them to complete asymmetric syntheses of both dalesconols in nine steps LLS.287
Anthracyclines:
The anthracycline polyketides have provided some of the most effective chemotherapeutics and antibiotics used in the clinic for over 50 years.389 These drugs possess well-characterized, cumulative dose-dependent cardiotoxicity,390 the severity of which has fueled decades of research into the synthesis and identification of suitable analogues that maintain their desirable potency but with reduced harmful side effects.391 The vast canon of synthetic approaches consists entirely of convergent strategies that utilize annulation processes to generate at least one of the four carbocyclic rings.
Sarlah and coworkers disclosed a fundamentally different, non-annulative strategy towards the idarubicin aglycone (425) in 2019, via global functionalization of a polynuclear arene already in possession of the full tetracyclic scaffold (Figure 44).392 Starting from tetracene (426), two sequential, selective C–H oxidation events delivered protected tetracenequinone 427. An arenophile-mediated, dearomative hydroboration, developed specifically for this synthesis, furnished boronic ester 428, which was subject to Zweifel olefination (428 → 429) to forge the remaining C–C bond to the pendent acetyl group. β-elimination and methylation of the urazole (429 → 430) set the stage for the completion of the synthesis, which required three steps for the installation of two hydroxyl groups and deprotection of the hydroquinone. Notably, this novel, “functionality-imprinting” approach resulted in the shortest synthesis of idarubicinone (425) to date.
Azaphilones:
So named for their characteristic affinity for ammonia, the azaphilones comprise a small family of fungal pigments bearing a dearomatized pyranoquinone bicyclic core with a chiral tertiary alcohol (Figure 45).393 Their propensity to alkylate nitrogenous biomolecules via substitution reactions that form vinylogous γ–pyridones lends them numerous desirable properties including antifungal, antimicrobial, antiviral, and cytotoxic activities.394 As attractive targets for total synthesis, several successful approaches have emerged, most commonly employing oxidative dearomatization of appropriately functionalized resorcinol derivatives.393 However, as both (R)- and (S)-configurations of the C7 quaternary center have been observed in nature, the enantioselective synthesis of azaphilones has been hampered by the relative dearth of methods available for asymmetric oxidative dearomatization.
Figure 45.
Porco’s enantiodivergent oxidative dearomatization of resorcinols, applied within the total syntheses of mitorubrin and sclerotioin, and Narayan’s enantiodivergent chemoenzymatic syntheses of trichoflectins.
A major breakthrough in this area was disclosed by Porco in 2006, during the total synthesis of (–)-mitorubrin (431, Figure 45).395 Using copper-oxo (–)-sparteine complex 432, the asymmetric oxidative dearomatization of alkynylbenzaldehyde 433 was carried out to establish the (R)-configured quaternary center in 98% ee. A copper(I)-catalyzed cycloisomerization then furnished the remainder of the pyranoquinone framework (435), which was advanced to (–)‑mitorubrin (431) in two additional steps. With the (+)‑sparteine enantiomer not readily available, this method was initially unsuitable to be used for the synthesis of the (S)-series of azaphilones; however, Porco and coworkers screened a variety of surrogates that might be used for this transformation, and they reported in 2011 the successful application of a copper-oxo complex with O’Brian’s N-ethyl-(+)-sparteine mimic (436) to promote the (S)-selective oxidative dearomatization (434 → 437) of alkynylbenzaldehydes.396 The Porco group then used this modified approach to access a number of (S)-configured azaphilone natural products including (+)-sclerotiorin (438) and (+)-8-O-methylsclerotiorinamine.
With the exception of microbial arene oxidations providing access to cis-dihydrodiols,397,398 the use of biocatalysis in dearomative synthesis remains an emerging field. In a rare example of enantiodivergent dearomative biocatalysis, Narayan and coworkers reported in 2019 the chemoenzymatic total synthesis of both (R)- and (S)-trichoflectin [(S)-439 & (R)-439], alongside a series of epimeric azaphilones including deflectin-1a and lunatoic acid A.399,400 Their syntheses hinged on the curious observation that both C7 epimers of certain azaphilones occur in nature, but are produced in different fungal species (i.e., not as a racemate). At the time, all flavin-dependent monooxygenases, such as AzaH, known to perform the requisite asymmetric oxidative dearomatization only produced the (R) enantiomer. Since azaphilones with the (S)-configuration were known, the authors postulated that enzymes capable of assuring this stereochemical outcome might be discovered via bioinformatic analysis. A sequence similarity network of flavin-dependent monooxygenases was assembled, from which several distinct clusters of related proteins emerged. Representative sequences from these clusters were selected for their proximity in the network to previously reported monooxygenases that carry out oxidative dearomatization of resorcinols; these were expressed in E. coli and screened for this desired reactivity, revealing a set of enzymes indeed able to catalyze the oxidative dearomatization of resorcinols with (S)-configuration. With these powerful tools in hand, Narayan and coworkers embarked upon the total synthesis of (S)-trichoflectin [(S)-439]. Enone 440 was obtained in a 5-step sequence from methyl atratate. Oxidative dearomatization [440 → (R)-435] with AzaH proceeded in 96% yield and >99% ee. Esterification with the acylketene of 441 and in situ Knoevenagel condensation quickly delivered the natural product, whose optical rotation was of the opposite sign to that reported in the literature. Chemoenzymatic synthesis of (R)-trichoflectin [(R)-439)], exploiting the newly discovered (S)-selective oxidative dearomatization with AfoD, confirmed the misassignment of absolute configuration in the original isolation report.
Bisorbicillinoids:
Gulder’s 2017 synthesis of bisorbicillinoid natural products stands as another exceptional case of biocatalytic, asymmetric oxidative dearomatization with flavin-dependent monooxygenases (Figure 46).401 Many sorbicillinoids, including bisorbicillinol (442), are effective radical scavengers with powerful antioxidant properties. Several of these fungal secondary metabolites also possess admirable anticancer [e.g., trichodimerol (443) and bisorbicilinbutenolide (444)], antiviral, and antimicrobial [e.g., bisorbicillinol (442)] activities as well. Their complex, cage-like architectures arise biosynthetically through spontaneous homodimerization of the cyclohexadienone sorbicillinol (445), itself derived from the namesake aromatic compound sorbicillin (446) via precise, enzymatic oxidative dearomatization of its resorcinol motif.402 The brevity and simplifying power of the biosynthetic approach presents an attractive strategy for total synthesis; indeed, several biomimetic total syntheses of bisorbicillinoids have been reported from Corey,403 Nicolaou,404,405 Pettus,406 and Deng,407 exploiting the spontaneous homodimerization of sorbicillinol derivatives. While Deng’s route successfully constructs the monomeric building block in enantioselective fashion through a nine-step sequence, Corey’s, Nicolaou’s, and Pettus’s expedient approaches proceed with poor or no stereoselectivity during oxidative dearomatization, greatly diminishing the yield and efficiency of this process.
Figure 46.
Gulder’s chemoenzymatic synthesis of bisorbicillinoids.
Gulder and coworkers reported an elegant solution to this problem using biocatalytic oxidative dearomatization with SorbC, the native enzyme responsible for this key step in the biosynthesis of bisorbicillinoids.401 Like AzaH and AfoD, SorbC is a flavin-dependent oxidoreductase able to catalyze the oxidative dearomatization of resorcinols, but it does so with complementary site-selectivity. A convenient in vitro protocol with NADH in pH 8 phosphate buffer and acetone was developed, facilitating efficient conversion of sorbicillin (446) to sorbicillinol (445) in >99.5% ee, the structure of which was confirmed after trapping with acetyl chloride. Perhaps counterintuitively, 445 was quite stable in the aqueous system, but the authors were able to leverage some remarkable solvent effects to deliberatively promote three different dimerization mechanisms and assemble diverse bisorbicillinoid scaffolds. Whereas quenching the biocatalytic reaction with DCM resulted in smooth core/core Diels–Alder reaction, furnishing bisorbicillinol (442) in 27% yield, the use of DMF as cosolvent was sufficient to alter the prevailing mechanism to double Michael addition/ketalization, such that trichodimerol (443) became the major product in 27% yield, with an additional 20% of bisorbicillinol. Furthermore, the authors found that heating the extract of the DMF reaction with pyridine allowed for the addition of sorbiquinol (447) to the product distribution, the result of core/side-chain [4+2] cycloaddition. Simple conversions of bisorbincillinol (442) to bisorbibutenolide (444) and bisorbicillinolide (448) have previously been reported by Nicolaou405 and Deng,407 respectively; therefore, Gulder’s biocatalytic approach to bisorbicillinol constitutes formal, chemoenzymatic syntheses of these natural products as well. Overall, the enzymatic oxidative dearomatization of sorbicillin (446) enables the first truly enantioselective, biomimetic syntheses of numerous polyketides within the bisorbicillinoid family of natural products.
PPAPs:
The polycyclic polyprenylated acylphloroglucinols (PPAPs) comprise a large family of natural products primarily isolated from the plants of the Hypericum and Garcinia genera (Figure 47).408 More than 500 members of this family are known, most of which can be separated into two major types: type A features a characteristic bicycle[3.3.1]nonane core with an α-acyl-β-hydroxyenone motif, while the type B PPAPs bear that acyl group in the bridgehead position instead. Beyond their intriguing structures, these natural products also possess important biological properties. For example, although not isolated or characterized until 1971, hyperforin (449) has been used since the time of ancient Greece as the major component of St. John’s wort, a well-known plant with antidepressant properties. In the modern era, hyperforin has become a popular treatment for anxiety and mild depression. It has been suggested that PPAP natural products may activate cation channels such as TRPC6,409 leading to an influx of Na+ and Ca2+ and diminished membrane electrochemical gradient, thus indirectly inhibiting synaptosomal reuptake of various neurotransmitters. Nemorosone (450) and clusianone (451) display antimicrobial and anti-HIV activities, along with chemopreventive properties.410 Garsubellin A (452) was identified as a potent inducer of choline acyltransferase, which is a key enzyme in the biosynthesis of the essential neurotransmitter acetylcholine.411 Low concentrations of this neurotransmitter are associated with the development of neuorodegenerative diseases such as Alzheimer’s disease.
Figure 47.
Structures of selected PPAPs and proposed biosynthetic pathways to type A and type B scaffolds.
The biosynthesis of PPAPs begins with condensation of one molecule of acetyl-CoA (453) and three molecules of malonyl-CoA (454), as was elucidated by labeling and enzymology studies.408 The resulting tetraketide 455 undergoes Dieckman condensation followed by dearomative alkylation of phloroglucinol moiety (455 → 456). One of the geminal prenyl groups in intermediate 456 is further alkylated by another molecule of prenyl pyrophosphate (457), furnishing tertiary carbocation 458. From this divergent point, two pathways for additional cyclization are possible: cyclization at C1 leads to the type A PPAPs (459), whereas type B PPAPs (460) are constructed after cyclization at C5. Additional cyclizations may occur, which endow this family of natural products with great diversity [e.g., garsubellin A (452)]. Accordingly, several attempts have been made to implement such a powerful transformation into synthetic approaches towards PPAPs in a laboratory setting. Although modified substrates successfully engaged in biomimetic cationic cyclizations, the fully decorated core was not susceptible to the desired transformation. This phenomenon was ascribed to the substantial buildup of strain in the transition-state as well as steric interactions associated with formation of two vicinal quaternary stereocenters. Thus, the most significant synthetic challenge posed by these molecules is the assembly of the bicycle[3.3.1]nonane core structure with quaternary stereocenters at the bridgehead positions.
The intricate architectures of the PPAPs coupled with their important biological properties have galvanized significant synthetic endeavors, and a variety of approaches and completed total syntheses have been published to date. Due to specific features of the basic scaffold (461), several common themes and disconnections can be unearthed within these synthetic studies. In 2006, Simpkins and coworkers disclosed the total synthesis of (±)-clusianone (451, Figure 48).412 The key feature of their synthesis was a regioselective and highly efficient lithiation of the bicycle[3.3.1]nonane core (462) to install a requisite quaternary stereocenter. Thus, compound 463 was obtained in 91% yield by direct alkylation of 462 at the C5 bridgehead position with prenyl bromide (464). Acylation of C1 and hydrolysis then furnished the natural product (461) in a concise manner. This approach, where substituents R3 and R4 (Figure 46) are installed via directed metalation, has been widely adopted and this tactic constitutes the endgame in the majority of PPAP syntheses. Another quite widespread strategy for construction of the bicycle[3.3.1]nonane core relies on sequential α,α’-functionalization of the C6 carbonyl motif of cyclohexanones like 465 with an appropriate substitution pattern, followed by a ring-closing event. For example, Plietker employed sequential double alkylation (465 → 466) and Dieckman condensation (466 → 467) to forge desired core 467 with both quaternary stereocenters in place.413 The modularity and robust nature of this approach allowed for the synthesis of four different type B PPAPs in only 7 steps and good overall yield. Several alternative strategies for accessing the bicyclic core of PPAPs have emerged recently. Maimone and coworkers completed the enantioselective total synthesis of hyperforin (449), employing several unorthodox transformations.414 First, a newly developed annulation method with diketene (468) was performed on ketone 469 to generate a bicyclic 1,3-diketone, after which methylation gave β–methoxyenone 470. This compound was subjected to oxidative fragmentation with PIDA (470 → 471 → 472), leading to a 1,2-alkyl shift in high yield. From bicyclo[3.3.1]nonane 472, Maimone and coworkers were able to complete the synthesis of hyperforin (449) after four additional steps. As noted above, functionalization of C1 and C3 still relies on the Simpkins direct metalation strategy.
Figure 48.
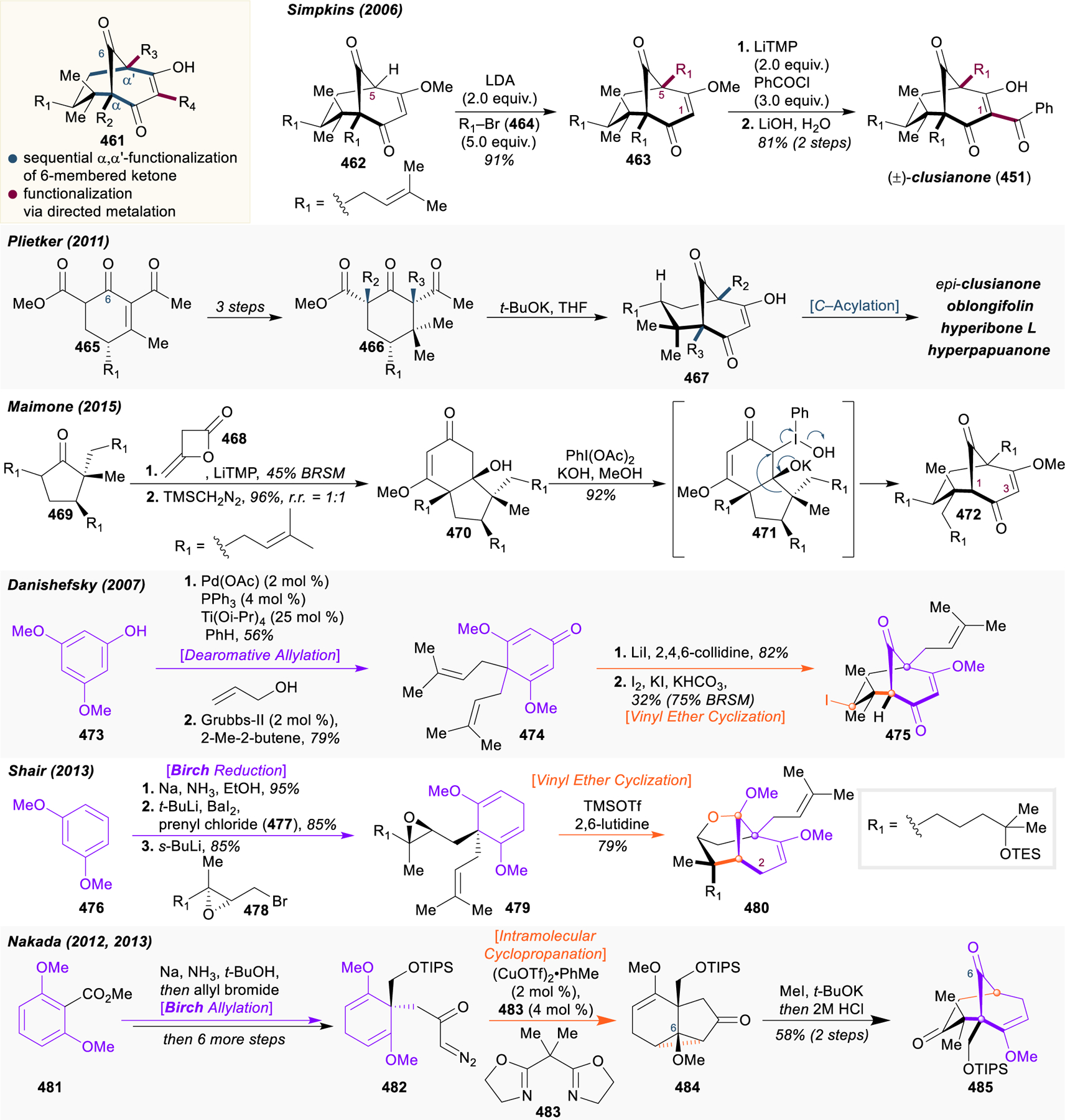
Synthetic considerations and summary of non-dearomative and dearomative approaches to type B PPAPs.
The arrangement of three carbonyl groups within one 6-membered ring of the bicycle[3.3.1]nonane core suggests that dearomatization of phloroglucinol derivatives would be a powerful disconnection, in line with the biosynthetic hypothesis; indeed, this biomimetic strategy was recognized and resulted in total syntheses of various members of PPAP family. The earliest example of such approach can be found in Danishefsky’s total synthesis of garsubellin A (452), which has been extensively covered elsewhere. An analogous approach was used for the synthesis of nemorosone (450) and clusianone (451) by the same group.415 Towards this end, Pd-catalyzed dearomative alkylation of phloroglucinol derivative 473 with allyl alcohol, followed by cross-metathesis with 2-methyl-2-butene, led to bisprenylated precursor 474. Of note, conversion of the allyl to prenyl group via cross metathesis is a standard way to circumvent an otherwise rather challenging α–selective prenylation. Demethylation followed by cyclization of electron-rich vinyl ether onto the appended olefin activated via formation of iodonium ion (474 → 475) led to the formation of the bicyclic core in moderate yield. In 2013, Shair and coworkers described an enantioselective total synthesis of hyperforin (449) using a similar disconnection between the vinyl ether as post-dearomative handle and an electrophilic site on the appended side-chain.416 Reductive dearomatization of dimethyl resorcinol (476) followed by double alkylation with prenyl chloride (477) and bromoepoxide 478 afforded the precursor 479 for the ring-forming event. TMSOTf was employed as a Lewis acid to facilitate ring-opening of the trisubstituted epoxide by the vinyl ether, resulting in a highly diastereoselective cyclization. Product 480 was formed in 79% yield and contains the desired scaffold with the quaternary stereocenter incorporated at the bridgehead position. The residual carbonyl functionality at C2 was installed via allylic C–H oxidation. Nakada and coworkers completed the total synthesis of nemorosone (450) in 2012,417 and garsubellin A (452) in the following year,418 using intramolecular cyclopropanation as a key step. To prepare the precursor, an alkylative Birch reduction of 481 was conducted, followed by several functional group interconversions to reach compound 482. Cu-catalyzed cyclopropanation reaction between the diazo motif and the aforementioned vinyl ether as post-dearomative handle in compound 482 yielded the overbred intermediate 484. Subsequent dimethylation of C6 and cyclopropane fragmentation delivered the core of PPAPs 485 in 58% yield, which was elaborated further into corresponding natural products 450 and 452.
Natural products such as plukenetione A (486) and hyperibon K (487), representing type A and type B PPAPs, respectively, feature an unusual adamantane core (Figure 49). This structure derives from an additional cyclization within the more common bicyclo[3.3.1]nonane (bond formation between C10 and C3/C1). Porco and coworkers rationalized that application of a synthon such as 488, that could form three bonds and the entire framework from simple aromatic compound 489 in a single operation, would be highly beneficial for synthetic efficiency. The authors identified enals of type 490 as likely synthetic equivalents of 488. Based on their previous work,419 an enantioselective, alkylative dearomatization/annulation process (489 → 491) was realized through phase-transfer catalysis.420 During this process, clusiaphenone B (489, accessible in one step from acylphloglucinol via direct aromatic prenylation) undergoes sequential Michael addition/elimination with 490, followed by Michael addition/aldol reaction. Judicious optimization of catalyst structure (492) allowed for the transformation to proceed in high yield, enantioselectivity, and exclusive chemo- and diastereoselectivity, delivering the core of the type B PPAPs (491). A subsequent retro-aldol and 1,2-addition process with vinylmagnesium bromide 493 delivered a mixture of isomeric allylic alcohols 494, which upon treatment with Lewis acid in nitromethane as a cation-stabilizing solvent, ionized and cyclized to form hyperibon K (487) in 50% overall yield.
Figure 49.
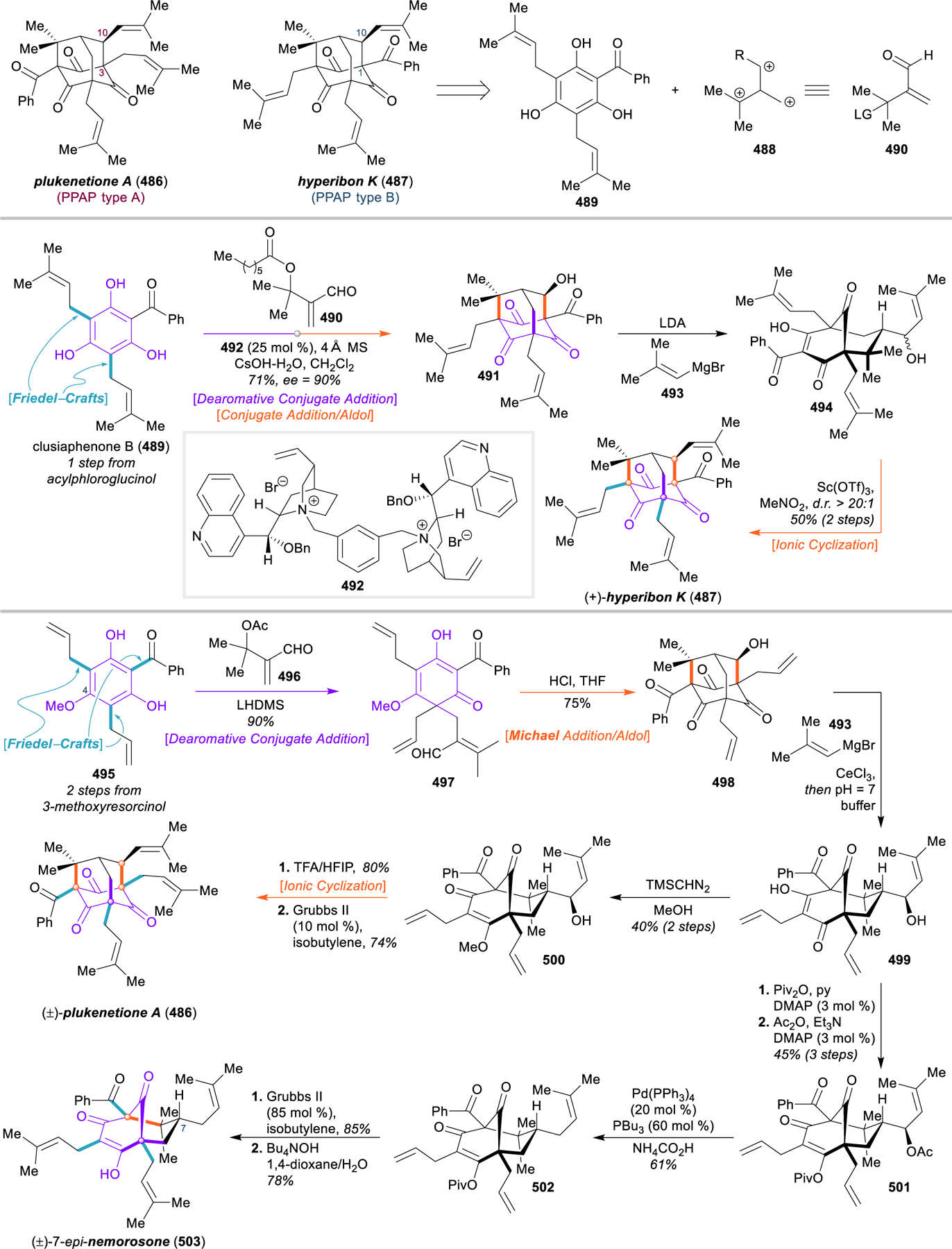
Porco’s synthetic approaches to type A and B PPAPs.
In order to swing the chemoselectivity of the annulation process towards the core of type A PPAPs, a blocking strategy was applied.421 The C4 phenolic group of 495 was protected as a methyl ether and the previously developed conditions were applied (495 → 497). In contrast to the cascade reaction observed with free acylphloroglucinol 498, the Michael reaction of 496 with methyl-protected 495 forged a single C–C bond, perhaps the result of rapid reversibility during the second Michael addition. Acid-mediated demethylation/cyclization of 497 delivered adamantane core 498. A retro-aldol/alkylation (498 → 499), analogous to that used in the synthesis of hyperibon K (487), was followed by methylation to afford compound 500. The moderate yield of the sequence is ascribed to the nonselective methylation of the unsymmetrical 1,3-diketone 499. Ionization/cyclization and subsequent cross metathesis leads to (±)-plukenetione A (486). The same strategy was applied to the total synthesis of 7-epi-nemorosone (503).422 Intermediate 499 was used as a point of divergency. Its 1,3-dicarbonyl motif was protected as pivaloyl ester and the allylic alcohol was acylated (499 → 501). Pd-catalyzed reduction (501 → 502), followed by cross metathesis and hydrolysis (502 → 487), furnished the natural product. Overall, the alkylative dearomatization/annulation strategy has proven to be a powerful tool for the synthesis of bicyclo[3.3.1]nonane-containing natural products. However, introduction of the stereocenter at C8 remains rather challenging.
Inspired by the biosynthesis of PPAPs, several years later Porco and coworkers developed a second- generation strategy realizing the biomimetic cationic cyclization (Figure 50).423 The previously exploited persubstituted compound 495 was competent for dearomative alkylation (495 → 504) with enantiopure triflate 505 in 72% yield but with little diastereoselectivity. After extensive screening for an optimal promoter, Porco’s team discovered that formic acid induces formation of a tertiary carbocation from the 1,1-disubstituted olefin of 504. The following electrophilic cyclization delivers the core of type A PPAPs with high chemoselectivity in 72% yield. Ordinary cross-metathesis yields (–)-clusianone (451). The brevity of this approach allows for expedient synthesis of the natural product in only six steps from commercial methoxyresorcinol. In order to manipulate the relative reactivity of C1 vs. C3 and as a result render this biomimetic strategy divergent, an alternative mode of cyclization was investigated. It was found that upon irradiation with purple LEDs, intermediate 504 underwent excited-state intramolecular proton transfer (ESIPT) to form zwitterion 506.424 Since, under these conditions, C1 is more activated for cyclization than C3, the core of type B PPAPs is selectively assembled to give 507. Cross metathesis followed by deprotection delivers (–)-nemorosone (450) in seven steps from commercial material, which represents the shortest synthesis of this natural product reported to date. Ultimately, this biomimetic strategy may provide access to a large variety of biologically important PPAP natural products, and stereoisomers thereof, in a concise manner from readily available commercial materials. However, more studies are needed to address yet unresolved challenges in this field. For example, a diastereoselective dearomative alkylation, which could avoid significant loss in reaction output and tedious separation of stereoisomers, would be highly beneficial for such an approach.
Figure 50.
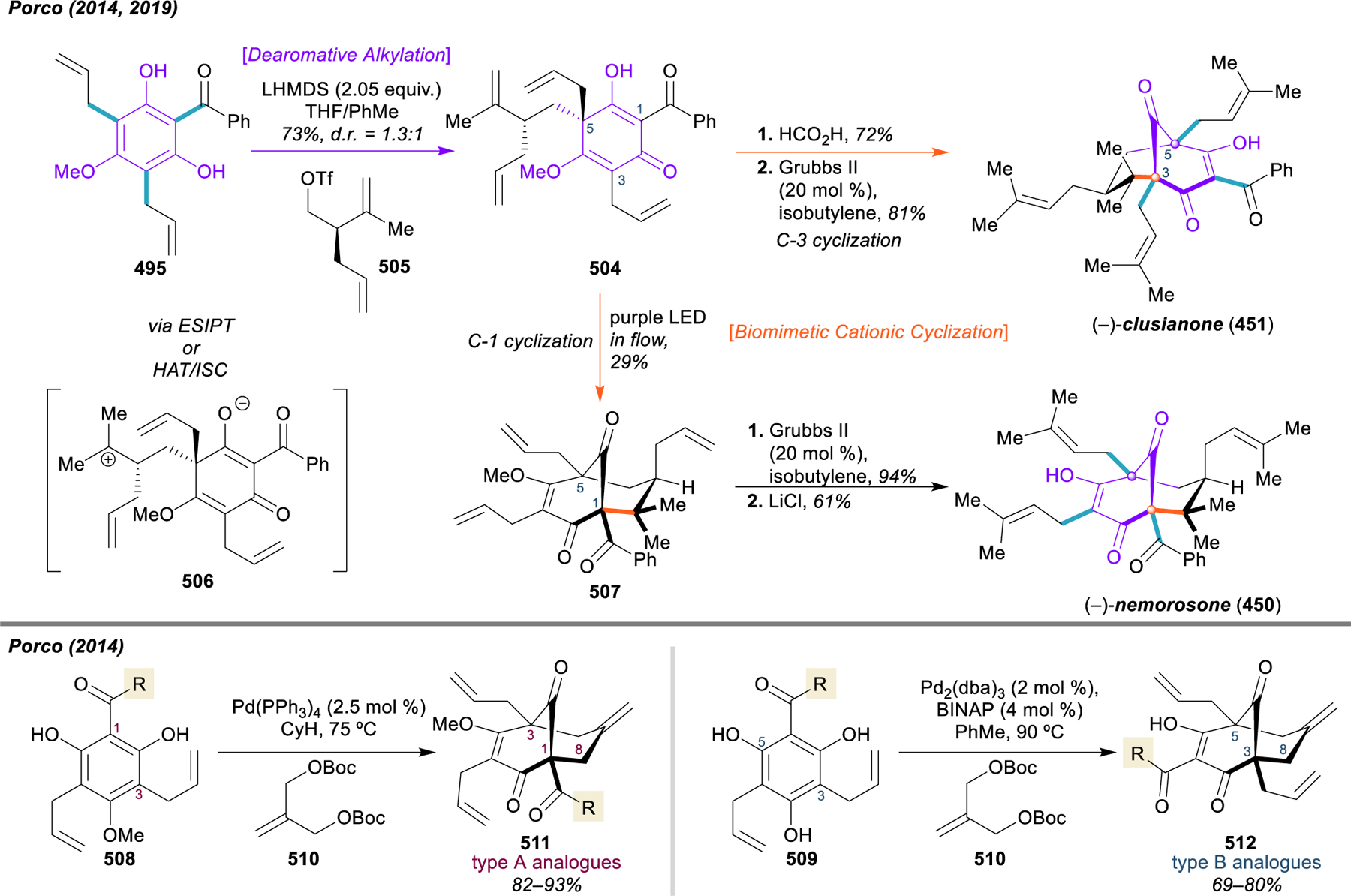
Porco’s second-generation strategy towards the PPAPs, using a biomimetic cationic cyclization.
Finally, it is noteworthy to mention another dearomative strategy towards bicyclo[3.3.1]nonane scaffold developed in Porco’s laboratory.425 Pd-catalyzed conjunctive allylic annulation of substrates like 508 and 509 with TMM precursor 510 can yield bicyclic structures like 511 and 512, with both quaternary stereocenters at bridgehead position in place. Using their previous results,421 a similar blocking strategy was utilized to achieve divergent access to the frameworks of both families of natural products. Thus, depending on the substitution pattern of the aromatic precursor (508 vs. 509), both type A and type B cores are accessible in high yields. Although this approach cannot be used for synthesis of PPAP natural products, due to the missing substitution at C8 that comes from TMM-precursor 510, it provides expedient access to valuable analogues for medicinal chemistry studies.
Summary and Conclusions
The complex, saturated architectures underlying natural products continue to challenge chemists and defy existing strategies and tactics within the synthetic canon. Dearomatization offers a strategic means of egress from the established chemistry of aromatic feedstocks into natural product scaffolds. Recent progress in dearomative methodology has initiated a paradigm shift in the way chemists disconnect molecules and has enabled groundbreaking achievements in the construction of such molecules. A three-phase dearomative logic encompassing arene substitution, dearomative reduction or functionalization, and alkene transformation provides myriad opportunities to decorate intermediates in demanding positions en route to ornate natural products. The presently insurmountable challenges posed by many natural products have stimulated intense study within the synthetic community, and novel developments in dearomative chemistry continue to nourish the blossoming field of natural product total synthesis with increased practicality and efficiency. This review presents powerful advances in dearomative methods from the past decade that have streamlined the synthesis of complex natural products, as well as modern approaches showcasing classical transformations in a new light. As nascent dearomative technology continues to mature and more chemists are exposed to the advantages of dearomative logic, we expect these approaches to see increased adoption among chemists seeking pragmatic solutions to complex synthetic problems. We hope the present review will serve to advance this movement.
Acknowledgements
This work was supported by the University of Illinois, the National Science Foundation (CHE-1654110), and the NIH/National Institute of General Medical Sciences (GM122891). C.J.H. acknowledges support from a National Science Foundation Graduate Research Fellowship as well as a Robert C. and Carolyn J. Springborn Fellowship. Y. D. B. acknowledges support from the University of Illinois (Seemon H. Pines graduate fellowship) and Bristol-Myers Squibb (predoctoral fellowship). Finally, we would also like to thank David Ryffel and Peter Ryffel for helpful discussions and careful proofreading of the manuscript.
Footnotes
Conflicts of interest
There are no conflicts of interest to declare.
References
- 1.Seeman JI, On the Relationship between Classical Structure Determination and Retrosynthetic Analysis/Total Synthesis, Isr. J. Chem, 2018, 58, 28–44. [Google Scholar]
- 2.Nicolaou KC, Hale CRH, The endeavor of total synthesis and its impact on chemistry, biology and medicine, Natl. Sci. Rev, 2014, 1, 233–252. [Google Scholar]
- 3.Gaich T, Baran PS, Aiming for the ideal synthesis, J. Org. Chem, 2010, 75, 4657–4673. [DOI] [PubMed] [Google Scholar]
- 4.Armaly AM, Deporre YC, Groso EJ, Riehl PS, Schindler CS, Discovery of Novel Synthetic Methodologies and Reagents during Natural Product Synthesis in the Post-Palytoxin Era, Chem. Rev, 2015, 115, 9232–9276. [DOI] [PubMed] [Google Scholar]
- 5.Nicolaou KC, Organic synthesis: the art and science of replicating the molecules of living nature and creating others like them in the laboratory, Proc. R. Soc. A Math. Phys. Eng. Sci, 2014, 470: 20130690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuttruff CA, Eastgate MD, Baran PS, Natural product synthesis in the age of scalability, Nat. Prod. Rep, 2014, 31, 403–606. [DOI] [PubMed] [Google Scholar]
- 7.Nicolaou KC, The Emergence and Evolution of Organic Synthesis and Why It is Important to Sustain It as an Advancing Art and Science for Its Own Sake, Isr. J. Chem, 2018, 58, 104–113. [Google Scholar]
- 8.Heathcock CH, Panel Discussion I As We Head Into the 21st Century, is There Still Value in Total Synthesis of Natural Products as a Research Endeavor?, in Chemical synthesis: gnosis to prognosis, eds. Chatgilialoglu C and Snieckus V, Kluwer Academic, Dordrecht, 1996, pp. 223–244. [Google Scholar]
- 9.Lovering F, Bikker J, Humblet C, Escape from flatland: Increasing saturation as an approach to improving clinical success, J. Med. Chem, 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- 10.Lovering F, Escape from Flatland 2: complexity and promiscuity, Med. Chem. Commun 2013, 4, 515–519. [Google Scholar]
- 11.Eastgate MD, Schmidt MA, Fandrick KR, On the design of complex drug candidate syntheses in the pharmaceutical industry, Nat. Rev. Chem, 2017, 1, 1–16. [Google Scholar]
- 12.Corey EJ, Cheng X-M, The Logic of Chemical Synthesis, John Wiley & Sons, Inc., New York. 1995. [Google Scholar]
- 13.Huck CJ, Sarlah D, Shaping Molecular Landscapes: Recent Advances, Opportunities, and Challenges in Dearomatization, Chem, 2020, 6, 1589–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wertjes WC, Southgate EH, Sarlah D, Recent advances in chemical dearomatization of nonactivated arenes, Chem. Soc. Rev, 2018, 48, 7996–8017. [DOI] [PubMed] [Google Scholar]
- 15.Wiesenfeldt MP, Nairoukh Z, Dalton T, Glorius F, Selective Arene Hydrogenation for Direct Access to Saturated Carbo- and Heterocycles, Angew. Chem. Int. Ed, 2019, 58, 10460–10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liebov BK, Harman WD, Group 6 Dihapto-Coordinate Dearomatization Agents for Organic Synthesis, Chem. Rev, 2017, 117, 13721–13755. [DOI] [PubMed] [Google Scholar]
- 17.Pape AR, Kaliappan KP, Kündig EP, Transition-Metal-Mediated Dearomatization Reactions, Chem. Rev, 2000, 100, 2917–2940. [DOI] [PubMed] [Google Scholar]
- 18.Ortiz FL, Iglesias MJ, Fernández I, Sánchez CMA, Gómez GR, Nucleophilic dearomatizing (DNAr) reactions of aromatic C,H-Systems. A mature paradigm in organic synthesis, Chem. Rev, 2007, 107, 1580–1691. [DOI] [PubMed] [Google Scholar]
- 19.Snieckus V, Directed Aromatic Metalation: A Continuing Education in Flatland Chemistry, in Chemical synthesis: gnosis to prognosis, eds. Chatgilialoglu C and Snieckus V, Kluwer Academic, Dordrecht, 1996, pp. 191–222. [Google Scholar]
- 20.Gutekunst WR, Baran PS, C–H functionalization logic in total synthesis, Chem. Soc. Rev, 2011, 40, 1976–1991. [DOI] [PubMed] [Google Scholar]
- 21.Hartwig JF, Larsen MA, Undirected, homogeneous C-H bond functionalization: Challenges and opportunities, ACS Cent. Sci, 2016, 2, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White MC, Zhao J, Aliphatic C-H Oxidations for Late-Stage Functionalization, J. Am. Chem. Soc, 2018, 140, 13988–14009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartwig JF, Catalyst-controlled site-selective bond activation, Acc. Chem. Res, 2017, 50, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neufeldt SR, Sanford MS, Controlling site selectivity in palladium-catalyzed C-H bond functionalization, Acc. Chem. Res, 2012, 45, 936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He J, Wasa M, Chan KSL, Shao Q, Yu JQ, Palladium-Catalyzed Transformations of Alkyl C-H Bonds, Chem. Rev, 2017, 117, 8754–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baran PS, Natural Product Total Synthesis: As Exciting as Ever and Here To Stay, J. Am. Chem. Soc, 2018, 140, 4751–4755. [DOI] [PubMed] [Google Scholar]
- 27.Wender PA, Howbert JJ, Synthetic studies on arene-olefin cycloadditions: total synthesis of (±)-α-cedrene, J. Am. Chem. Soc, 1981, 103, 688–690. [Google Scholar]
- 28.Hsu D-S, Liao C-C, First Total Syntheses of (±)-Penicillones A and B, Org. Lett, 2007, 9, 4563–4565. [DOI] [PubMed] [Google Scholar]
- 29.Tang C, Okumura M, Zhu Y, Hooper AR, Zhou Y, Lee YH, Sarlah D, Palladium-Catalyzed Dearomative syn-1,4-Carboamination with Grignard Reagents. Angew. Chem. Int. Ed, 2019, 58, 10245–10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corey EJ, Howe WJ, Orf HW, Pensak DA, Petersson G, General methods of synthetic analysis. Strategic bond disconnections for bridged polycyclic structures, J. Am. Chem. Soc, 1975, 97, 6116–6124. [Google Scholar]
- 31.Owens KR, McCowen SV, Blackford KA, Ueno S, Hirooka Y, Weber M, Sarpong R, Total Synthesis of the Diterpenoid Alkaloid Arcutinidine Using a Strategy Inspired by Chemical Network Analysis, J. Am. Chem. Soc, 2019, 141, 13713–13717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang DS, Chen QA, Lu SM, Zhou YG, Asymmetric Hydrogenation of Heteroarenes and Arenes, Chem. Rev, 2011, 112, 2557–2590. [DOI] [PubMed] [Google Scholar]
- 33.Lewis SE, Applications of biocatalytic arene ipso,ortho cis-dihydroxylation in synthesis, Chem. Commun, 2014, 50, 2821–2830. [DOI] [PubMed] [Google Scholar]
- 34.Palframan MJ, Kociok-Köhn G, Lewis SE, Photooxygenation of a microbial arene oxidation product and regioselective Kornblum-DeLamare rearrangement: Total synthesis of zeylenols and zeylenones, Chem. Eur. J, 2012, 18, 4766–4774. [DOI] [PubMed] [Google Scholar]
- 35.Nicolaou KC, Edmonds DJ, Bulger PG, Cascade reactions in total synthesis. Angew. Chem. Int. Ed, 2006, 45, 7134–7186. [DOI] [PubMed] [Google Scholar]
- 36.Roche SP, Porco JA, Dearomatization strategies in the synthesis of complex natural products, Angew. Chem. Int. Ed, 2011, 50, 4068–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mander LN, Exploitation of Aryl Synthons in the Synthesis of Polycyclic Natural Products, Synlett, 1991, 134–144. [Google Scholar]
- 38.Pouységu L, Deffieux D, Quideau S, Hypervalent iodine-mediated phenol dearomatization in natural product synthesis, Tetrahedron, 2010, 66, 2235–2261. [Google Scholar]
- 39.Heravi M, Fard M, Faghihi Z, Recent Applications of Birch Reduction in Total Synthesis of Natural Products, Curr. Org. Chem, 2015, 19, 1491–1525. [Google Scholar]
- 40.George JH, Biomimetic Dearomatization Strategies in the Total Synthesis of Meroterpenoid Natural Products, Acc. Chem. Res, 2021, 54, 1843–1855. [DOI] [PubMed] [Google Scholar]
- 41.Sun H-D, Huang S-X, Han Q-B, Diterpenoids from Isodon species and their biological activities. Nat. Prod. Rep, 2006, 23, 673–698. [DOI] [PubMed] [Google Scholar]
- 42.Ding Y, Chen H, Zhou J, Chemistry and Bioactivity of Ent-Kaurene Diterpenoids. Studies in Natural Product Chemistry, 2017, 54, 141–197. [Google Scholar]
- 43.Liu M, Wang W-G, Sun H-D and Pu J-X, Diterpenoids from Isodon species: an update. Nat. Prod. Rep, 2017, 34, 1090–1140. [DOI] [PubMed] [Google Scholar]
- 44.Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C, Hernandez L, Allocco J, Basilio ngela, Tormo JR, Genilloud O, Vicente F, Pelaez F, Colwell L, Ho Lee S, Michael B, Felcetto T, Gill C, Silver LL, Hermes JD, Bartizal K, Barrett J, Schmatz D, Becker JW, Cully D, Singh SB, Platensimycin is a selective FabF inhibitor with potent antibiotic properties, Nature, 2006, 441, 358–361. [DOI] [PubMed] [Google Scholar]
- 45.Martens E, Demain AL, Platensimycin and platencin: Promising antibiotics for future application in human medicine. J. Antibiot. (Tokyo), 2011, 64, 705–710. [DOI] [PubMed] [Google Scholar]
- 46.Shang R, Liang J, Yi Y, Liu Y, Wang J, Review of Platensimycin and Platencin: Inhibitors of β-Ketoacyl-acyl Carrier Protein (ACP) Synthase III (FabH). Molecules, 2015, 20, 16127–16141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Z, Chen Y, Oridonin, a Promising Antitumor Natural Product in the Chemotherapy of Hematological Malignancies. Curr. Pharm. Biotechnol, 2014, 15, 1083–1092. [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Xu J, Zhou J, Shen Q, Oridonin and its derivatives for cancer treatment and overcoming therapeutic resistance. Genes & Diseases, 2021, 8, 448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu J, Wold E, Ding Y, Shen Q, Zhou J, Therapeutic Potential of Oridonin and Its Analogs: From Anticancer and Antiinflammation to Neuroprotection. Molecules, 2018, 23, 474–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.CTR20150246; http://www.chinadrugtrials.org.cn
- 51.Riehl PS, DePorre YC, Armaly AM, Groso EJ and Schindler CS, New avenues for the synthesis of ent-kaurene diterpenoids, Tetrahedron, 2015, 71, 6629–6650. [Google Scholar]
- 52.Lazarski KE, Moritz BJ and Thomson RJ, Angew. Chem., Int. Ed, 2014, 53, 10588–10599. [DOI] [PubMed] [Google Scholar]
- 53.Coates RM, Cavender PL, Stereochemistry of the Enzymatic Cyclization of Copalyl Pyrophosphate to Kaurene in Enzyme Preparations from Marah macrocarpus, J. Am. Chem. Soc, 1980, 102, 6358–6359. [Google Scholar]
- 54.Hong YJ, Tantillo DJ, Formation of beyerene, kaurene, trachylobane, and atiserene diterpenes by rearrangements that avoid secondary carbocations, J. Am. Chem. Soc, 2010, 132, 5375–5386. [DOI] [PubMed] [Google Scholar]
- 55.Snider BB, Kiselgof JY, Foxman BM, Total Syntheses of (±)-Isosteviol and (±)-Beyer-15-ene-3β,19-diol by Manganese(III)-Based Oxidative Quadruple Free-Radical Cyclization, J. Org. Chem, 1998, 63, 7945–7952. [Google Scholar]
- 56.Church RF, Ireland RE, Marshall JA, Experiments Directed toward the Total Synthesis of Terpenes. VII. The Synthesis of (±)-8β-Carbomethoxy-13-oxopodocarpanone, a Degradation Product of Phyllocladene, J. Org. Chem, 1966, 31, 2526–2530. [DOI] [PubMed] [Google Scholar]
- 57.Bell RA, Ireland RE, Partyka RA, Experiments Directed toward the Total Synthesis of Terpenes. VIII. The Total Synthesis of (±)-Kaurene and (±)-Atisirene, J. Org. Chem, 1966, 31, 2530–2536. [DOI] [PubMed] [Google Scholar]
- 58.Church RF, Ireland RE, Marshall JA, The stereospecific total synthesis of d1–8β-carbomethoxy-13-oxopodocarpane, a degradation product of phyllocladane, Tetrahedron Lett, 1960, 1, 1–4. [Google Scholar]
- 59.Bell RA, Ireland RE, Partyka RA, The Total Synthesis of dl-Kaurene. J. Org. Chem, 1962, 27, 3741–3744. [DOI] [PubMed] [Google Scholar]
- 60.Ziegler FE, Kloek JA, 1-Hydroxy-7-methylene bicyclo[3.2.1]octane: a gibbane-steviol c/d ring model. Tetrahedron Lett, 1971, 12, 2201–2203. [Google Scholar]
- 61.Ziegler FE, Kloek JA, The stereocontrolled photoaddition of allene to cyclopent-1-ene-1-carboxaldehydes. A total synthesis of (±) steviol methyl ester and isosteviol methyl ester. Tetrahedron, 1977, 33, 373–380. [Google Scholar]
- 62.Zhu L, Luo J, Hong R, Total synthesis of (±)-cafestol: A late-stage construction of the furan ring inspired by a biosynthesis strategy. Org. Lett, 2014, 16, 2162–2165. [DOI] [PubMed] [Google Scholar]
- 63.Corey EJ, Liu K, Enantioselective total synthesis of the potent Anti-HIV agent neotripterifordin. Reassignment of stereochemistry at C(16). J. Am. Chem. Soc, 1997, 119, 9929–9930. [Google Scholar]
- 64.Cherney EC, Green JC, Baran PS, Synthesis of ent -Kaurane and Beyerane Diterpenoids by Controlled Fragmentations of Overbred Intermediates. Angew. Chemie Int. Ed, 2013, 52, 9019–9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Toyota M, Wada T, Nishikawa Y, Yanai K, Fukumoto K, Pd2+ -Promoted Cyclization in Gibberellin Synthesis - A New Strategy for C20 Gibberellin Synthesis. Synlett, 1994, 597–598. [Google Scholar]
- 66.Toyota M, Wada T, Nishikawa Y, Yanai K, Fukumoto K, Kabuto C, Simple design for the construction of complex gibberellin framework-Stereoselective synthesis of a possible key intermediate to GA12 via Pd2+-promoted cycloalkenylation reaction. Tetrahedron, 1995, 51, 6927–6940. [Google Scholar]
- 67.Toyota M, Wada T, Ihara M, Total syntheses of (–)-methyl atis-16-en-19-oate, (–)-methyl kaur-16-en-19-oate, and (–)-methyl trachyloban-19-oate by a combination of palladium-catalyzed cycloalkenylation and homoallyl-homoallyl radical rearrangement. J. Org. Chem, 2000, 65, 4565–4570. [DOI] [PubMed] [Google Scholar]
- 68.Toyota M, Wada T, Fukumoto K, Ihara M, Total synthesis of (±)-methyl atis-16-en-19-oate via homoallyl- homoallyl radical rearrangement. J. Am. Chem. Soc, 1998, 120, 4916–4925. [DOI] [PubMed] [Google Scholar]
- 69.Toyota M, Wada T, Fukumoto K, INOC reaction in gibberellin synthesis-a practical synthesis of the key intermediate for gibberellin A12, Heterocycles, 1995, 41, 1135–1138. [Google Scholar]
- 70.Yeoman JTS, Mak VW, Reisman SE, A unified strategy to ent -kauranoid natural products: Total syntheses of (–)-trichorabdal A and (–)-longikaurin e. J. Am. Chem. Soc, 2013, 135, 11764–11767. [DOI] [PubMed] [Google Scholar]
- 71.Zhao X, Li W, Wang J, Ma D, Convergent Route to ent-Kaurane Diterpenoids: Total Synthesis of Lungshengenin D and 1α,6α-Diacetoxy-ent-kaura-9(11),16-dien12,15-dione. J. Am. Chem. Soc, 2017, 139, 2932–2935. [DOI] [PubMed] [Google Scholar]
- 72.Corey EJ, Wess G, Bin Xiang Y, Singh AK, Stereospecific Total Synthesis of (±)-cafestol. J. Am. Chem. Soc, 1987, 109, 4717–4718. [Google Scholar]
- 73.Backhaus D, Paquette LA, Synthetic entry into the ent-kaurene framework. Application of an unprecedented transannular cyclization for forming the central bond common to the B and C rings. Tetrahedron Lett, 1997, 38, 29–32. [Google Scholar]
- 74.Wang J, Ma D, 6-Methylenebicyclo[3.2.1]oct-1-en-3-one: A Twisted Olefin as Diels–Alder Dienophile for Expedited Syntheses of Four Kaurane Diterpenoids. Angew. Chemie Int. Ed, 2019, 58, 15731–15735. [DOI] [PubMed] [Google Scholar]
- 75.Kong L, Su F, Yu H, Jiang Z, Lu Y, Luo T, Total Synthesis of (−)-Oridonin: An Interrupted Nazarov Approach, J. Am. Chem. Soc, 2019, 141, 20048–20052. [DOI] [PubMed] [Google Scholar]
- 76.He C, Hu J, Wu Y, Ding H, Total Syntheses of Highly Oxidized ent-Kaurenoids Pharicin A, Pharicinin B, 7-O-Acetylpseurata C, and Pseurata C: A [5+2] Cascade Approach, J. Am. Chem. Soc, 2017, 139, 6098–6101. [DOI] [PubMed] [Google Scholar]
- 77.Macalpine GA, Warkentin J, Thermolysis of Δ3–1,3,4-oxadiazolin-2-ones and 2-phenylimino- Δ3–1,3,4-oxadiazolines derived from α,β-epoxyketones. An alternative method for the conversion of a α,β-epoxyketones to alkynones and alkynals, Can. J. Chem, 1978, 56, 308–315. [Google Scholar]
- 78.Pan S, Xuan J, Gao B, Zhu A, Ding H, Total Synthesis of Diterpenoid Steenkrotin A. Angew. Chemie Int. Ed, 2015, 54, 6905–6908. [DOI] [PubMed] [Google Scholar]
- 79.Gao J, Rao P, Xu K, Wang S, Wu Y, He C, Ding H, Total Synthesis of (−)-Rhodomollanol A. J. Am. Chem. Soc, 2020, 142, 4592–4597. [DOI] [PubMed] [Google Scholar]
- 80.Li S-H, Wang J, Niu X-M, Shen Y-H, Zhang H-J, Sun H-D, Li M-L, Tian Q-E, Lu Y, Cao P, Zheng Q-T, Maoecrystal V, Cytotoxic Diterpenoid with a Novel C19 Skeleton from Isodon eriocalyx (Dunn.) Hara. Org. Lett, 2004, 6, 4327–4330. [DOI] [PubMed] [Google Scholar]
- 81.Smith BR, Njardarson JT, Review of synthetic approaches toward maoecrystal V. Org. Biomol. Chem, 2018, 16, 4210–4222. [DOI] [PubMed] [Google Scholar]
- 82.Krawczuk PJ, Schone N and Baran PS, A synthesis of the carbon skeleton of maoecrystal V, Org. Lett, 2009, 11, 4774–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gong J, Lin G, Li C and Yang Z, Synthetic study toward the total synthesis of maoecrystal V, Org. Lett, 2009, 11, 4770–4773. [DOI] [PubMed] [Google Scholar]
- 84.Peng F, Yu M and Danishefsky SJ, Synthetic Studies towards Maoecrystal V, Tetrahedron Lett, 2009, 50, 6586–6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Peng F and Danishefsky SJ, Toward the total synthesis of maoecrystal V: an intramolecular Diels-Alder route to the maoecrystal V pentacyclic core with the appropriate relative stereochemistry, Tetrahedron Lett, 2011, 52, 2104–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Singh V, Bhalerao P and Movin SM, A tandem oxidative dearomatization/intramolecular Diels–Alder reaction: a short and efficient entry into tricyclic system of maoecrystal V, Tetrahedron Lett, 2010, 51, 3337–3339. [Google Scholar]
- 87.Baitinger I, Mayer P and Trauner D, Toward the total synthesis of maoecrystal V: establishment of contiguous quaternary stereocenters, Org. Lett, 2010, 12, 5656–5659. [DOI] [PubMed] [Google Scholar]
- 88.Lazarski KE, Hu DX, Stern CL and Thomson RJ, A synthesis of the carbocyclic core of maoecrystal V, Org. Lett, 2010, 12, 3010–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lazarski KE, Akpinar B and Thomson RJ, Evaluation of ‘east-to-west’ ether-forming strategies for the total synthesis of maoecrystal V, Tetrahedron Lett, 2013, 54, 635–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gu Z and Zakarian A, Studies toward the synthesis of maoecrystal V, Org. Lett, 2011, 13, 1080–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nicolaou KC, Dong L, Deng L, Talbot AC and Chen DY-K, Synthesis of functionalized maoecrystal V core structures, Chem. Commun, 2010, 46, 70–72. [DOI] [PubMed] [Google Scholar]
- 92.Dong L, Deng L, Lim Y-H, Leung GYC and Chen DY-K, Synthesis of an advanced maoecrystal V core structure, Chem. – Eur. J, 2011, 17, 5778–5781. [DOI] [PubMed] [Google Scholar]
- 93.Carberry P, Viernes DR, Choi LB, Fegley MW and Chisholm JD, An unusual intramolecular Diels–Alder approach toward maoecrystal V, Tetrahedron Lett, 2013, 54, 1734–1737. [Google Scholar]
- 94.Jansone-Popova S and May JA, Stereoelectronic factors in bridgehead C–H bond insertion: studies toward the total synthesis of maoecrystal V, Tetrahedron, 2016, 72, 3734–3747. [Google Scholar]
- 95.Smith BR and Njardarson JT, Double-Diels-Alder Approach to Maoecrystal V. Unexpected C-C Bond-Forming Fragmentations of the [2.2.2]-Bicyclic Core, Org. Lett, 2017, 19, 5316–5319. [DOI] [PubMed] [Google Scholar]
- 96.Chang TM, PhD Dissertation, University of Arizona, 2012. [Google Scholar]
- 97.McLeod DD, PhD Dissertation, Princeton University, 2010. [Google Scholar]
- 98.Smith MJ, PhD Dissertation, Princeton University, 2013. [Google Scholar]
- 99.Naylor MR, PhD Dissertation, Princeton University, 2014. [Google Scholar]
- 100.Gong J, Lin G, Sun W, Li C-C and Yang Z, Total synthesis of (±) maoecrystal V, J. Am. Chem. Soc, 2010, 132, 16745–16746. [DOI] [PubMed] [Google Scholar]
- 101.Peng F and Danishefsky SJ, Total synthesis of (±)-maoecrystal V, J. Am. Chem. Soc, 2012, 134, 18860–18867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lu P, Gu Z and Zakarian A, Total synthesis of maoecrystal V: early-stage C-H functionalization and lactone assembly by radical cyclization, J. Am. Chem. Soc, 2013, 135, 14552–14555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zheng C, Dubovyk I, Lazarski KE and Thomson RJ, Enantioselective total synthesis of (–)-maoecrystal V, J. Am. Chem. Soc, 2014, 136, 17750–17756. [DOI] [PubMed] [Google Scholar]
- 104.Lu P, Mailyan A, Gu Z, Guptill DM, Wang H, Davies HML and Zakarian A, Enantioselective synthesis of (–)-maoecrystal V by enantiodetermining C-H functionalization, J. Am. Chem. Soc, 2014, 136, 17738–17749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang W-B, Shao W-B, Li F-Z, Gong J-X and Yang Z, Asymmetric Total Synthesis of (–)-Maoecrystal V, Chem. – Asian J, 2015, 10, 1874–1880. [DOI] [PubMed] [Google Scholar]
- 106.Cernijenko A, Risgaard R and Baran PS, 11-Step Total Synthesis of (–)-Maoecrystal V, J. Am. Chem. Soc, 2016, 138, 9425–9428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pinhey JT and Rowe BA, The chemistry of aryllead(IV) tricarboxylates. Reaction with β-keto esters: a convenient route to α-arylated ketones, Aust. J. Chem, 1980, 33, 113–120. [Google Scholar]
- 108.Fraga CAM, Teixeira LHP, Menezes C. M. de S., Sant’Anna CMR, Ramos M. da C. K. V., de Aquino Neto FR, Barreiro EJ, Studies on diastereoselective reduction of cyclic β-ketoesters with boron hydrides. Part 4: The reductive profile of functionalized cyclohexanone derivatives, Tetrahedron, 2004, 60, 2745–2755. [Google Scholar]
- 109.Paulissen R, Reimlinger H, Hayez E, Hubert AJ, Ph. Teyssié, Transition metal catalysed reactions of diazocompounds - II insertion in the hydroxylic bond, Tetrahedron Lett, 1973, 14, 2233–2236. [Google Scholar]
- 110.Davies HML, Grazini MVA, and Aouad E, Asymmetric Intramolecular C−H Insertions of Aryldiazoacetates, Org. Lett 2001, 3, 1475–1477. [DOI] [PubMed] [Google Scholar]
- 111.Green JC, Pettus TRR, An oxidative dearomatization-induced [5 + 2] cascade enabling the syntheses of α-cedrene, α-pipitzol, and sec -cedrenol. J. Am. Chem. Soc, 2011, 133, 1603–1608. [DOI] [PubMed] [Google Scholar]
- 112.Finkbeiner P, Murai K, Röpke M, Sarpong R, Total Synthesis of Terpenoids Employing a “Benzannulation of Carvone” Strategy: Synthesis of (−)-Crotogoudin. J. Am. Chem. Soc, 2017, 139, 11349–11352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weber M, Owens K, Sarpong R, Atropurpuran—missing biosynthetic link leading to the hetidine and arcutine C20-diterpenoid alkaloids or an oxidative degradation product? Tetrahedron Lett, 2015, 56, 3600–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Suzuki T, Sasaki A, Egashira N, & Kobayashi S, S. A synthetic study of atropurpuran: construction of a pentacyclic framework by an intramolecular reverse-electron-demand Diels–Alder reaction. Angew. Chem. Int. Ed, 2011, 50, 9177–9179. [DOI] [PubMed] [Google Scholar]
- 115.Hayashi R, Ma ZX, & Hsung RP, A tandem 1,3-H-shift–6pi- electrocyclization–cyclic 2-amido-diene intramolecular Diels–Alder cycloaddition approach to BCD-ring of atropurpuran. Org. Lett, 2012, 14, 252–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen H, Zhang D, Xue F, & Qin Y, Synthesis of the atropurpuran A-ring via an organocatalytic asymmetric intramolecular Michael addition. Tetrahedron, 2013, 69, 3141–3148. [Google Scholar]
- 117.Chen H, Li X-H, Gong J, Song H, Liu X-Y, & Qin Y, Synthetic approach to the functionalized tricyclic core of atropurpuran. Tetrahedron, 2016, 72, 347–353. [Google Scholar]
- 118.Tang P, Chen QH, Wang FP, Atropurpuran, a novel diterpene with an unprecedented pentacyclic cage skeleton, from Aconitum hemsleyanum var. atropurpureum, Tetrahedron Lett, 2009, 50, 460–462. [Google Scholar]
- 119.Gong J, Chen H, Liu X-Y, Wang Z-X, Nie W, Qin Y, Total synthesis of atropurpuran. Nat. Commun, 2016, 7, 12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xie S, Chen G, Yan H, Hou J, He Y, Zhao T, Xu J, 13-Step Total Synthesis of Atropurpuran. J. Am. Chem. Soc, 2019, 141, 3435–3439. [DOI] [PubMed] [Google Scholar]
- 121.Wang F-P, Chen QH, Liu X-Y, Diterpenoid alkaloids. Nat. Prod. Rep, 2010, 27, 529–570. [DOI] [PubMed] [Google Scholar]
- 122.Wada K, Yamashita H, Cytotoxic Effects of Diterpenoid Alkaloids Against Human Cancer Cells. Molecules, 2019, 24, 2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ohno Y, The Experimental Approach to the Murder Case of Aconite Poisoning. J. Tox.: Tox. Rev, 2009, 17, 1–11. [Google Scholar]
- 124.Van Landeghem AA, De Letter EA, Lambert WE, Van Peteghem CH, Piette MHA Aconitine involvement in an unusual homicide case. Int. J. Legal Med,. 2007, 121, 214–219. [DOI] [PubMed] [Google Scholar]
- 125.Colomboa ML, Bugattib C, Davanzoc F, Persicob A, Ballabiob C, Restanib P, Analytical Aspects of Diterpene Alkaloid Poisoning with Monkshood. Nat. Prod. Comm, 2009, 4, 1551–1552. [PubMed] [Google Scholar]
- 126.Miyaguchi H, Sekine H, Homicide involving Aconitum tuberous root: LC-MS-MS analysis of Aconitum alkaloids and their hydrolysates in formalin-fixed tissues. Forensic. Toxicol, 2010, 28, 47–51. [Google Scholar]
- 127.Bonnici KB, Stanworth D, Simmonds MSJ, Mukherjee E, Ferner RE, Flowers of evil. The Lancet, 2010, 376, 1616. [DOI] [PubMed] [Google Scholar]
- 128.Povšnar M, Koželj G, Kreft S, Lumpert M, Rare tradition of the folk medicinal use of Aconitum spp. is kept alive in Solčavsko, Slovenia. J. Enthobiol. Enthomed, 2017, 13, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Huang X, Yang Y, Zhu J, Dai Y, Pu J, The Effects of a Novel Anti-arrhythmic Drug, Acehytisine Hydrochloride, on the Human Ether-a-go-go Related Gene K+ Channel and Its Trafficking. Basic Clin. Pharmacol. Toxicol, 2008, 104, 145–154. [DOI] [PubMed] [Google Scholar]
- 130.Gao X, Zhu J, Yang YM, Li JD, Yang ZM, Liu JH, Acehytisine Hydrochloride Injection Phase II Clinic Trial Group Investigators, Efficacy of intravenous Acehytisine Hydrochloride versus propafenone on terminating paroxysmal supraventricular tachycardia: a double-blinded, randomized multi-center study. Zhonghua Xin Xue Guan Bing Za Zhi, 2007, 35, 151–154. In Chinese. [PubMed] [Google Scholar]
- 131.Yang YM, Zhu J, Gao X, Yin YL, Hao YX, Liu JH, Guanfu Base A Hydrochloride Injection Phase II Clinic Trial Group Investigators, Effect of Guanfu Base A in patients with ventricular arrhythmias. Zhonghua Xin Xue Guan Bing Za Zhi, 2006, 34, 329–332. In Chinese. [PubMed] [Google Scholar]
- 132.Stevens M, Peigneur S, Tytgat J, Neurotoxins and their binding areas on voltage-gated sodium channels. Front Pharmacol, 2011, 2, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Anger T, Madge DJ, Mulla M, Riddall D, Medicinal Chemistry of Neuronal Voltage-Gated Sodium Channel Blockers. J. Med. Chem, 2001, 44, 115–137. [DOI] [PubMed] [Google Scholar]
- 134.Wang S-Y, Wang GK, Voltage-gated sodium channels as primary targets of diverse lipid-soluble neurotoxins. Cell. Signal, 2003, 15, 151–159. [DOI] [PubMed] [Google Scholar]
- 135.Wang F-P, Chen QH, Liang X-T, The C18-diterpenoid alkaloids. Alkaloids Chem Biol, 2009, 67, 1–78. [DOI] [PubMed] [Google Scholar]
- 136.Wang F-P, Chen QH, The C19-diterpenoid alkaloids. Alkaloids Chem. Biol, 2010, 69, 1–577. [DOI] [PubMed] [Google Scholar]
- 137.Wang FP, Liang X-T, C20 diterpenoid alkaloids, Alkaloids Chem. Biol 2002, 59, 1–280. [DOI] [PubMed] [Google Scholar]
- 138.Pelletier SW, Parthasarathy PC, The Diterpene Alkaloids: A Partial Synthesis of Atisine. Tetrahedron Lett, 1963, 4, 205–208. [Google Scholar]
- 139.Pelletier SW, Jacobs WA, The aconite alkaloids. XXVII. The structure of atisine, J. Am. Chem. Soc, 1954, 76, 4496–4497. [Google Scholar]
- 140.Mody NV, Pelletier SW 13C nuclear magnetic resonance spectroscopy of atisine and veatchine-type C20-diterpenoid alkaloids from aconitum and garrya species. Tetrahedron, 1978, 34, 2421–2431. [Google Scholar]
- 141.Wiesner K, Edwards JA, The basicity and steric configuration of the diterpene alkaloids veatchine and atisine, Experientia, 1955, 11, 255–259. [DOI] [PubMed] [Google Scholar]
- 142.Wiesner K, Bickelhaupt F, Babin DR, Gotz M, The structure of delphinine, Tetrahedron Lett, 1959, 1, 3, 11–14. [Google Scholar]
- 143.Wiesner K, Götz M, Simmons DL, Fowler LR, Bachelor FW, Brown RFC, Büchi G, The structure of aconitine, Tetrahedron Lett, 1959, 1, 2, 15–24. [Google Scholar]
- 144.Edwards OE, Singh T, Atsine: The heterocyclic ring and functional groups, Can. J. Chem, 1954, 32, 465–473. [Google Scholar]
- 145.Edwards OE, Singh T, Atsine: The functional groups, Can. J. Chem, 1955, 33, 448–451. [Google Scholar]
- 146.Jacobs WA, The Aconite Alkaloids. XXIV. The Degradation of Atisine and Isoatisine, J. Org. Chem, 1951, 16, 1593–1602. [Google Scholar]
- 147.Nagata W, Sugasawa T, Narisada M, Wakabayashi T, Hayase Y, Stereospecific Total Synthesis of dl-Atisine. J. Am. Chem. Soc, 1963, 85, 2342–2343. [Google Scholar]
- 148.Nagata W, Narisada M, Wakabayashi T, Sugasawa T, Total Synthesis of dl-garryine and dl-veatchine. J. Am. Chem. Soc, 1964, 85, 929–930. [Google Scholar]
- 149.Masamune S, Synthesis of 4a,6-ethano-5.6 7,8-tetrahydro- 2(4a)-naphthalenone. J. Am. Chem. Soc, 1961, 83, 1009–1010. [Google Scholar]
- 150.Masamune S, Total Syntheses of Diterpenes and Diterpene Alkaloids. II. A Tetracyclic Common Intermediate. J. Am. Chem. Soc, 1964, 86, 288–289. [Google Scholar]
- 151.Masamune S, Total Syntheses of Diterpenes and Diterpene Alkaloids. III. Kaurene. J. Am. Chem. Soc, 1964, 86, 289–290. [Google Scholar]
- 152.Masamune S, Total Syntheses of Diterpenes and Diterpene Alkaloids. IV. Garryine. J. Am. Chem. Soc, 1964, 86, 290–291. [Google Scholar]
- 153.Masamune S, Total Syntheses of Diterpenes and Diterpene Alkaloids. V. Atisine. J. Am. Chem. Soc, 1964, 86, 291–292. [Google Scholar]
- 154.Valenta Z, Wiesner K, Wang CH, Synthesis in the diterpene alkaloid series - II. A total synthesis of the garrya alkaloids. Tetrahedron Lett, 1964, 5, 2437–2442. [Google Scholar]
- 155.Guthrie RW, Henry WA, Immer H, Wong CM, Valenta Z, K, Wiesner, The total synthesis of the Garrya veatchii alkaloids. Collect. Czech. Chem. Commun, 1966, 31, 602–621. [Google Scholar]
- 156.Guthrie RW, Valenta Z, Wiesner K, Synthesis in the series of diterpene alkaloids VI. A simple synthesis of atisine. Tetrahedron Lett, 1966, 7, 4645–4654. [DOI] [PubMed] [Google Scholar]
- 157.Wiesner K, Ho P-T, Chang D, Lam YK, Pan CSJ, Ren WY, The Synthesis of Songorine: A Simplified Synthesis of the Aromatic Intermediate. Can. J. Chem, 1973, 51, 3978–3988. [Google Scholar]
- 158.Wiesner K, Ho P, Tsai C. S. J. (Pan), The Total Synthesis of a Hexacyclic Relay for the Alkaloid Napelline. Can. J. Chem, 1974, 52, 2353–2355. [Google Scholar]
- 159.Wiesner K, Ho P, Tsai C. S. J. (Pan), Lam YK, The Total Synthesis of Racemic Napelline. Can. J. Chem, 1974, 52, 2355–2357. [Google Scholar]
- 160.Sethi SP, Atwal KS, Marini-Bettolo RM, Tsai TYR, Wiesner K, A stereospecific synthesis of napelline. Can. J. Chem, 1980, 58, 1889–1891. [Google Scholar]
- 161.Liu X-Y, Qin Y, Enabling syntheses of diterpenoid alkaloids and related diterpenes by an oxidative dearomatization/Diels–Alder cycloaddition strategy. Nat. Prod. Rep, 2017, 34, 1044–1050. [DOI] [PubMed] [Google Scholar]
- 162.Weisner K, Tsai TYR, Huber K, Bolton SE, Vlahov R, Total synthesis of talatisamine, a delphinine type alkaloid, J. Am. Chem. Soc, 1974, 96, 4990–4992. [Google Scholar]
- 163.Wiesner K, Tsai TYR, Nambiar KP, A new stereospecific total synthesis of chasmanine and 13-desoxydelphonine, Can. J. Chem, 2011, 56, 1451–1454. [Google Scholar]
- 164.Liu X-Y, Cheng H, Li X-H, Chen Q-H, Xu L, Wang F-P, Oxidative dearomatization/intramolecular Diels-Alder cycloaddition cascade for the syntheses of (±)-atisine and (±)-isoazitine, Org. Biomol. Chem, 2012, 10, 1411–1417. [DOI] [PubMed] [Google Scholar]
- 165.Liu J, Ma D, A Unified Approach for the Assembly of Atisine- and Hetidine-type Diterpenoid Alkaloids: Total Syntheses of Azitine and the Proposed Structure of Navirine C, Angew. Chem. Int. Ed, 2018, 57, 6676–6680. [DOI] [PubMed] [Google Scholar]
- 166.Li X-H, Zhu M, Wang Z-X, Liu X-Y, Song H, Zhang D, Wang F-P, Qin Y, Synthesis of Atisine, Ajaconine, Denudatine, and Hetidine Diterpenoid Alkaloids by a Bioinspired Approach, Angew. Chem. Int. Ed, 2016, 55, 15667–15671. [DOI] [PubMed] [Google Scholar]
- 167.Cheng H, Zeng F-H, Yang X, Meng Y-J, Xu L, Wang F-P, Collective Total Syntheses of Atisane-Type Diterpenes and Atisine-Type Diterpenoid Alkaloids: (±)-Spiramilactone B, (±)-Spiraminol, (±)-Dihydroajaconine, and (±)-Spiramines C and D, Angew. Chem. Int. Ed, 2016, 55, 392–396. [DOI] [PubMed] [Google Scholar]
- 168.Nishiyama Y, Han-Ya Y, Yokoshima S, Fukuyama T, Total Synthesis of (−)-Lepenine, J. Am. Chem. Soc, 2014, 136, 6598–6601. [DOI] [PubMed] [Google Scholar]
- 169.Hamlin AM, de Jesus Cortez F, Lapointe D, Sarpong R, Gallium(III)-Catalyzed Cycloisomerization Approach to the Diterpenoid Alkaloids: Construction of the Core Structure for the Hetidines and Hetisines, Angew. Chem. Int. Ed, 2013, 52, 4854–4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Marth CJ, Gallego GM, Lee JC, Lebold TP, Kulyk S, Kou KGM, Qin J, Lilien R, Sarpong R, Network-analysis-guided synthesis of weisaconitine D and liljestrandinine, Nature, 2015, 528, 493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kou KGM, Kulyk S, Marth CJ, Lee JC, Doering NA, Li BX, Gallego GM, Lebold TP, Sarpong R, A Unifying Synthesis Approach to the C 18-, C 19-, and C 20-Diterpenoid Alkaloids, J. Am. Chem. Soc, 2017, 139, 13882–13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kou KGM, Pflueger JJ, Kiho T, Morrill LC, Fisher EL, Clagg K, Lebold TP, Kisunzu JK, Sarpong R, A Benzyne Insertion Approach to Hetisine-Type Diterpenoid Alkaloids: Synthesis of Cossonidine (Davisine), J. Am. Chem. Soc, 2018, 140, 8105–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tambar UK, Stoltz BM, The direct acyl-alkylation of arynes, J. Am. Chem. Soc, 2005, 127, 5340–5341. [DOI] [PubMed] [Google Scholar]
- 174.Medina JM, MacKey JL, Garg NK, Houk KN, The role of aryne distortions, steric effects, and charges in regioselectivities of aryne reactions, J. Am. Chem. Soc, 2014, 136, 15798–15805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Peese KM, Gin DY, Efficient Synthetic Access to the Hetisine C20-Diterpenoid Alkaloids. A Concise Synthesis of Nominine via Oxidoisoquinolinium-1,3-Dipolar and Dienamine-Diels-Alder Cycloadditions, J. Am. Chem. Soc, 2006, 128, 8734–8735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sigel MM, Wellham LL, Lichter W, Dudeck LE, Gargus JL, and Lucas LH, “Anticellular and antitumor activity of extracts from tropical marine invertebrates.” Food-Drugs From the Sea: Proceedings, 1969; ed. Youngken HW Jr., Marine Technology Society, Washington DC, 1970, 281–294. [Google Scholar]
- 177.Rinehart KL, Holt TG, Fregeau NL, Stroh JG, Keifer PA, Sun F, Li LH and Martin DG, Ecteinascidins 729, 743, 745, 759A, 759B, and 770: potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinate. J. Org. Chem, 1990, 55, 4512–4515; [Google Scholar]; Rinehart KL, Holt TG, Fregeau NL, Stroh JG, Keifer PA, Sun F, Li LH and Martin DG, Ecteinascidins 729, 743, 745, 759A, 759B, and 770: potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata [Erratum to document cited in CA113(9):75189d]. J. Org. Chem, 1991, 56, 1676. [Google Scholar]
- 178.Wright AE, Forleo DA, Gunawardana PG, Gunasekera SP, Koehn FE and McConnell OJ, Antitumor tetrahydroisoquinoline alkaloids from the colonial ascidian Ecteinascidia turbinate. J. Org. Chem, 1990, 55, 4508–4512. [Google Scholar]
- 179.Scott JD, Williams RD, Chemistry and Biology of the Tetrahydroisoquinoline Antitumor Antibiotics. Chem. Rev 2002, 102, 1669–1730. [DOI] [PubMed] [Google Scholar]
- 180.Fukuyama T, Sachleben RA, Stereocontrolled Total Synthesis of (±)-saframycin B. J. Am. Chem. Soc, 1982, 104, 4957–4958. [Google Scholar]
- 181.Fukuyama T, Yang L, Ajeck KL, Sachleben RA, Total Synthesis of (±)-saframycin A. J. Am. Chem. Soc, 1990, 112, 3712–3713. [Google Scholar]
- 182.Fukuyama T, Linton SD, Tun MM, A stereocontrolled total synthesis of (±)-renieramycin A. Tetrahedron Lett, 1990, 31, 5989–5992. [Google Scholar]
- 183.Endo A, Kann T, Fukuyama T, Synthetic Study on Ecteinascidin 743 Starting from d-Glucose. Synlett, 1999, 7, 1103–1105. [Google Scholar]
- 184.Fukuyama T, Laird AA, Synthetic approaches toward naphthyridinomycin. I. Stereoselective synthesis of a tetracyclic intermediate. Tetrahedron Lett, 1986, 27, 6173–6176. [Google Scholar]
- 185.Fukuyama T, Li L, Laird AA, Frank RK, Stereocontrolled total synthesis of (±)-cyanocycline A. J. Am. Chem. Soc, 1987, 109, 1587–1589. [Google Scholar]
- 186.Fukuyama T, Nunes JJ, Stereocontrolled total synthesis of (±)-quinocarcin. J. Am. Chem. Soc, 1988, 110, 5196–5198. [Google Scholar]
- 187.Kubo A, Saito N, Yamauchi R, Sakai S, Synthesis of Saframycins. I Total Synthesis of (±)-Saframycin B and its Congeners. Chem. Pharm. Bull, 1987, 35, 2158–2161. [DOI] [PubMed] [Google Scholar]
- 188.Kubo A, Saito N, Yamato H, Kawakami Y, Preparations and Reactions of (Z)-3-Arylidene-6-arylmethyl-2,5-piperazinediones Having Highly Oxygenated Benzene Rings. Chem. Pharm. Bull, 1987, 35, 2525–2532. [Google Scholar]
- 189.Kubo A, Saito N, Nakamura M, Ogata K, Sakai S, A Promising Cyclization of the 3-Arylidene-6-arylmethyl-2,5-piperazinedione to Construct Tricyclic Lactam as an Intermediate to Saframycin Synthesis. Heterocycles, 1987, 26, 1765–1770. [Google Scholar]
- 190.Kubo A, Saito N, Yamato H, Yamauchi R, Hiruma K, Inoue S, Synthesis of Saframycins. II.: Preparations and Reactions of N-Methyl-2,5-piperazinediones. Chem. Pharm. Bull, 1988, 36, 2607–2614. [DOI] [PubMed] [Google Scholar]
- 191.Kubo A, Saito N, Yamato H, Masubuchi K, Nakamura M, Stereoselective Total Synthesis of (±)-saframycin B. J. Org. Chem, 1988, 53, 4295–4310. [Google Scholar]
- 192.Saito N, Ohira Y, Kubo A, Synthesis of Saframycins. IV.: Selenium Oxide Oxidation of 4-oxo-hexahydro-1,5-imino-3-benzazocin-7,10-dione; Promising Method to Construct Saframycins C and D from Saframycin B. Chem. Pharm. Bull, 1990, 38, 821–823. [Google Scholar]
- 193.Saito N, Ohira Y, Wada N, Kubo A, A. Synthesis of saframycins. V. Selenium oxide oxidation of hexahydro-1,5-imino-3-benzazocin-7,10-dione; a useful method for constructing saframycins C and D from saframycin B. Tetrahedron, 1990, 46, 7711–7728. [Google Scholar]
- 194.Saito N, Harada S, Nishida M, Inouye I, Kubo A, Synthesis of Saframycins. X. Transformation of (–)-Saframycin A to (−)-Saframycin Mx Type Compound with the Structure Proposed for Saframycin E. Chem. Pharm. Bull, 1995, 43, 777–782. [DOI] [PubMed] [Google Scholar]
- 195.Saito N, Nishida M, Kubo A, Synthesis of Saframycins. VI. The Useful Transformation of (–)-saframycin A to (–)-saframycin Mx Type Compound. Chem. Pharm. Bull, 1991, 39, 1343–1345. [DOI] [PubMed] [Google Scholar]
- 196.Saito N, Harada S, Inouye I, Yamaguchi K, Kubo A, Synthesis of saframycins. XII. 1 total synthesis of (–)-N-acetylsaframycin Mx 2 and Its epi-(+)-enantiomer. Tetrahedron, 1995, 51, 8231–8246. [Google Scholar]
- 197.Saito N, Tanitsu M, Betsui T, Suzuki R, Kubo A, Synthesis of Novel Octahydro-1,5-imino-3-benzazocin-4,7,10-trione Derivatives Having a Methyl Group at the C-2 Position as ABC Ring Models of Saframycins. Chem. Pharm. Bull, 1997, 45, 1120–1129. [Google Scholar]
- 198.Saito N, Yamauchi R, Kubo A, Synthesis of Saframycins. VII. The Synthesis of Novel Renieramycin Congeners. Heterocycles, 1991, 32, 1203–1214. [Google Scholar]
- 199.Saito N, Tachi M, Seki R, Kamayachi H, Kubo A, A Practical Synthesis of the ABC Ring Model of Ecteinascidins. Chem. Pharm. Bull, 2000, 48, 1549–1557. [DOI] [PubMed] [Google Scholar]
- 200.Saito N, Kamayachi H, Tachi M, Kubo A, An Improved Synthesis of the ABC Ring Model of Ecteinascidins. Heterocycles, 1999, 51, 9–12. [DOI] [PubMed] [Google Scholar]
- 201.Saito N, Tashiro K, Maru Y, Yamaguchi K, Kubo A, Synthetic approaches toward ecteinascidins. Part 1. Preparation of an (E)-2-arylidene-3-benzyl-1,5-imino-3-benzazocin-4-one having a protected phenol in the E-ring. J. Chem. Soc., Perkin Trans 1, 1997, 53–70. [Google Scholar]
- 202.Myers AG, Kung DW, A Concise, Stereocontrolled Synthesis of (−)-Saframycin A by the Directed Condensation of α-Amino Aldehyde Precursors. J. Am. Chem. Soc, 1999, 121, 10828–10829. [Google Scholar]
- 203.Myers AG, Kung DW, One-Step Construction of the Pentacyclic Skeleton of Saframycin A from a “Trimer” of α-Amino Aldehydes. Org. Lett, 2000, 2, 3019–3022. [DOI] [PubMed] [Google Scholar]
- 204.Myers AG, Plowright AT, Synthesis and Evaluation of Bishydroquinone Derivatives of (−)-Saframycin A: Identification of a Versatile Molecular Template Imparting Potent Antiproliferative Activity. J. Am. Chem. Soc, 2001, 123, 5114–5115. [DOI] [PubMed] [Google Scholar]
- 205.Martinez EJ, Corey EJ, Enantioselective Synthesis of Saframycin A and Evaluation of Antitumor Activity Relative to Ecteinascidin/Saframycin Hybrids. Org. Lett, 1999, 1, 75–78. [DOI] [PubMed] [Google Scholar]
- 206.Corey EJ, Gin DY, Kania RS, Enantioselective Total Synthesis of Ecteinascidin 743. J. Am. Chem. Soc, 1996, 118, 9202–9203. [Google Scholar]
- 207.Martinez EJ, Corey EJ, A New, More Efficient, and Effective Process for the Synthesis of a Key Pentacyclic Intermediate for Production of Ecteinascidin and Phthalascidin Antitumor Agents. Org. Lett, 2000, 2, 993–996. [DOI] [PubMed] [Google Scholar]
- 208.Corey EJ, Gin DY, A convergent enantioselective synthesis of the tetrahydroisoquinoline unit in the spiro ring of ecteinascidin 743. Tetrahedron Lett, 1996, 37, 7163–7166. [Google Scholar]
- 209.Martinez EJ, Owa T, Schreiber SL, Corey EJ, Phthalascidin, a synthetic antitumor agent with potency and mode of action comparable to ecteinascidin 743. Proc. Natl. Acad. Sci, 1999, 96, 3496–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Herberich B, Kinugawa M, Vazquez A, Williams RM, Sequential Staudinger/Pictet–Spengler cyclization strategy for the construction of tetrahydroisoquinolines of the bioxalomycin and ecteinascidin family of alkaloids. Tetrahedron Lett, 2001, 42, 543–546. [Google Scholar]
- 211.Flanagan ME, Williams RM, Synthetic Studies on Quinocarcin: Total Synthesis of (±)-Quinocarcinamide via Dipole Cycloaddition of an Azomethine Ylide Generated by NBS Oxidation. J. Org. Chem, 1995, 60, 6791–6797. [Google Scholar]
- 212.Williams RM, Glinka T, Gallegos R, Ehrlich PP, Flanagan ME, Coffman H, Park G, Synthesis, conformation, crystal structures and DNA cleavage abilities of tetracyclic analogs of quinocarcin. Tetrahedron, 1991, 47, 2629–2642. [Google Scholar]
- 213.Flanagan ME, Rollins SB, Williams RM, Netropsin and spermine conjugates of a water-soluble quinocarcin analog: analysis of sequence-specific DNA interactions. Chem. Biol, 1995, 2, 147–146. [DOI] [PubMed] [Google Scholar]
- 214.Herberich B, Scott JD, Williams RM, Synthesis of a netropsin conjugate of a water-soluble epi-quinocarcin analogue: the importance of stereochemistry at nitrogen. Bioorg. Med. Chem, 2000, 8, 523–532. [DOI] [PubMed] [Google Scholar]
- 215.Scott JD, Williams RM, Total Synthesis of (−)-Tetrazomine and Determination of Its Stereochemistry. Angew. Chem 2001, 40, 1463–1465. [DOI] [PubMed] [Google Scholar]
- 216.Scott JD, Williams RM, Total Synthesis of (−)-Tetrazomine. Determination of the Stereochemistry of Tetrazomine and the Synthesis and Biological Activity of Tetrazomine Analogues. J. Am. Chem. Soc, 2002, 124, 2951–2956. [DOI] [PubMed] [Google Scholar]
- 217.Scott JD, Williams RM, Synthetic studies on tetrazomine: lipase PS resolution of racemic cis-β-hydroxypipecolic acid. Tetrahedron Lett, 2000, 41, 8413–8416. [Google Scholar]
- 218.Zhou B, Edmondson S, Padron J, Danishefsky SJ, Synthetic explorations in the saframycin-ecteinascidin series: construction of major chiral subunits through catalytic asymmetric induction. Tetrahedron Lett, 2000, 41, 2039–2042. [Google Scholar]
- 219.Zhou B, Guo J, Danishefsky SJ, A novel face specific Mannich closure providing access to the saframycin-ecteinascidin series of piperazine based alkaloids. Tetrahedron Lett, 2000, 41, 2043–2046. [Google Scholar]
- 220.Danishefsky SJ, Harrison PJ, Webb RR II, O’Neil BT, Total synthesis of quinocarcinol methyl ester. J. Am. Chem. Soc, 1985, 107, 1421–1423. [Google Scholar]
- 221.Shawe TT, Liebeskind LS, Saframycin synthetic studies. Tetrahedron, 1991, 47, 5643–5666. [Google Scholar]
- 222.Cuevas C, Pérez M, Martín MJ, Chicharro JL, Fernádez-Rivas C, Flores M, Francesch A, Gallego P, Zarzuelo M, de la Calle F, García J, Polanco C, Rodríguez I, Manzanares I, Synthesis of Ecteinascidin ET-743 and Phthalascidin Pt-650 from Cyanosafracin B. Org. Lett, 2000, 2, 2545–2548. [DOI] [PubMed] [Google Scholar]
- 223.Evans DA, Biller SA, The total synthesis of (±)-naphthyridinomycin. I. Preparation of a key tricyclic lactam intermediate. Tetrahedron Lett, 1985, 26, 1907–1910. [Google Scholar]
- 224.Evans DA, Biller SA, The total synthesis of (±)-naphthyridinomycin. II. Construction of the pentacyclic carbon skeleton. Tetrahedron Lett, 1985, 26, 1911–1914. [Google Scholar]
- 225.Chrzanowska M, Grajewska A, Rozwadowska MD, Asymmetric Synthesis of Isoquinoline Alkaloids: 2004−2015. Chem. Rev, 2016, 116, 12369–12465. [DOI] [PubMed] [Google Scholar]
- 226.Newman DJ, Cragg GM, Drugs and Drug Candidates from Marine Sources: An Assessment of the Current “State of Play.” Planta Med, 2016, 82, 775–789. [DOI] [PubMed] [Google Scholar]
- 227.Fontana A, Cavaliere P, Wahidulla S, Naik CG, Cimino G, A New Antitumor Isoquinoline Alkaloid from the Marine Nudibranch Jorunna funebris. Tetrahedron, 2000, 56, 7305–7308. [Google Scholar]
- 228.Lane JW, Chen Y, Williams RM, Asymmetric Total Syntheses of (–)-Jorumycin, (–)-Renieramycin G, 3-epi-Jorumycin, and 3-epi-Renieramycin G. J. Am. Chem. Soc, 2005, 127, 12684–12690. [DOI] [PubMed] [Google Scholar]
- 229.Wu Y-C, Zhu J, Asymmetric Total Syntheses of (–)-Renieramycin M and G and (–)-Jorumycin Using Aziridine as a Lynchpin. Org Lett, 2009, 11, 5558–5561. [DOI] [PubMed] [Google Scholar]
- 230.Liu W, Liao X, Dong W, Yan Z, Wang N, Liu Z, Total synthesis and cytotoxicity of (–)-jorumycin and its analogues. Tetrahedron, 2012, 68, 2759–2764. [Google Scholar]
- 231.Chen R, Liu H, Chen X, Asymmetric Total Synthesis of (−)-Jorunnamycins A and C and (−)-Jorumycin from L‑Tyrosine. J. Nat. Prod, 2013, 76, 1789–1795. [DOI] [PubMed] [Google Scholar]
- 232.Ong CW; Lee HC A Rapid Synthesis of 2,3,11,11a-Tetrahydro-6H-pyrazino[1,2-b]isoquinoline-1,4-diones Through an Amido Iminium Ion Cyclization. Aust. J. Chem, 1990, 43, 773–775. [Google Scholar]
- 233.Welin ER, Ngamnithiporn A, Klatte M, Lapointe G, Pototschnig GM, McDermott MSJ, Conklin D, Gilmore CD, Tadross PM, Haley CK, Negoro K, Glibstrup E, Grünanger CU, Allan KM, Virgil SC, Slamon DJ, Stoltz BM, Concise total syntheses of (–)-jorunnamycin A and (–)-jorumycin enabled by asymmetric catalysis. Science, 2019, 363, 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Allan KM, Hong BD, Stoltz BM, Expedient synthesis of 3-hydroxyisoquinolines and 2-hydroxy-1,4-naphthoquinones via one-pot aryne acyl-alkylation/condensation. Org. Biomol. Chem, 2009, 7, 4960–4964. [DOI] [PubMed] [Google Scholar]
- 235.Campeau L-C, Schipper DJ, Fagnou K, Site-Selective sp2 and Benzylic sp3 Palladium-Catalyzed Direct Arylation. J. Am. Chem. Soc, 2008, 130, 3266–3267. [DOI] [PubMed] [Google Scholar]
- 236.Jin Z, Yao G, Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep 2019, 36, 1462–1488. [DOI] [PubMed] [Google Scholar]
- 237.Scott LJ, Goa KL, Galantamine: a review of its use in Alzheimer’s disease. Drugs, 2000, 60, 1095–1122. [DOI] [PubMed] [Google Scholar]
- 238.Dewick PM, in Medicinal Natural Products: A Biosynthetic Approach, John Wiley & Sons Ltd, West Sussex, 2nd edn, 2002, ch. 6, pp. 291–404. [Google Scholar]
- 239.Schultz AG, Holoboski MA, Smyth MS, The first asymmetric synthesis of a lycorine alkaloid. Total synthesis of (+)-1-deoxylycorine. J. Am. Chem. Soc, 1993, 115, 7904–7905. [Google Scholar]
- 240.Schultz AG, Holoboski MA, Smyth MS, The First Asymmetric Total Syntheses of (+)-Lycorine and (+)-1-Deoxylycorine. J. Am. Chem. Soc, 1996, 118, 6210–6219. [Google Scholar]
- 241.Kametani T, Shishido K, Hayashi E, Seino C, Kohno T, Shibuya S, Fukumoto K, Synthesis of heterocyclic compounds CCCXCVI. Alternative total synthesis of (±)-galanthamine. J. Org. Chem, 1971, 36, 1295–1297. [Google Scholar]
- 242.Kametani T, Yarnaki K, Terui T, Shibuya S, Fukumoto K, Studies on the syntheses of heterocyclic compounds. Part CDLXVI. Synthesis of narwedine-type enones by photochemical cyclisation. J. Chem. Soc. Perkin Trans 1, 1972, 1513–1516. [Google Scholar]
- 243.Kametani T, Yamaki K, Terui T, Studies on the syntheses of heterocyclic compounds. Part DVII. A synthesis of (±)-N-norgalanthamine. J. Heterocyclic Chem, 1973, 10, 35–37. [Google Scholar]
- 244.Shimizu K, Tomioka K, Yamada S-I, Koga K, A Biogenetic-type Asymmetric Synthesis of Optically Active Amaryllidaceae Alkaloids: (+)- and (−)-Galanthamine from L-Tyrosine. Heterocycles, 1977, 8, 277–282. [Google Scholar]
- 245.Shimizu K, Tomioka K, Yamada S-I, Koga K, Stereochemical Studies. LIV. A Biogenetic-type Asymmetric Synthesis of optically Active Galanthamine from L-Tyrosine. Chem. Pharm. Bull, 1978, 26, 3765–3771. [Google Scholar]
- 246.Szewczyk J, Lewin AH, Carroll FI, An improved synthesis of galanthamine. J. Heterocyclic Chem, 1988, 25, 1809–1811. [Google Scholar]
- 247.Szewczyk J, Wilson JW, Lewin AH, Carroll FI, Facile synthesis of (±)-, (+)-, and (–)-galanthamine. J. Heterocyclic Chem, 1995, 32, 195–199. [Google Scholar]
- 248.Holton RA, Sibi MP, Murphy WS, Palladium-mediated biomimetic synthesis of narwedine. J. Am. Chem. Soc, 1988, 110, 314–366. [Google Scholar]
- 249.Malachowski WP, Paul T, Phounsavath S, The Enantioselective Synthesis of (−)-Lycoramine with the Birch−Cope Sequence. J. Org. Chem, 2007, 72, 6792–6796. [DOI] [PubMed] [Google Scholar]
- 250.Czollner L, Treu M, Froehlich J, Kueenburg B, Jordis U, Synthesis of galanthamine. ARKIVOC, 2001, 2, 191–200. [Google Scholar]
- 251.Lewin AH, Szewczyk J, Wilson JW, Carroll FI, Galanthamine analogs: 6H-benzofuro[3a,3,2,-e,f][1]benzazepine and 6H-benzofuro[3a,3,2-e,f][3]benzazepine. Tetrahedron, 2005, 61, 7144–7152. [Google Scholar]
- 252.Node M, Kodama S, Hamashima Y, Baba T, Hamamichi N, An Efficient Synthesis of (±)-Narwedine and (±)-Galanthamine by an Improved Phenolic Oxidative Coupling. K. Nishide, Angew Chem., Int. Ed 2001, 40, 3060–3062. [DOI] [PubMed] [Google Scholar]
- 253.Kodama S, Hamashima Y, Nishide K, Node M, Total Synthesis of (–)-Galanthamine by Remote Asymmetric Induction. Angew. Chem., Int. Ed 2004, 43, 2659–2661. [DOI] [PubMed] [Google Scholar]
- 254.Baxendale IR, Ley SV, Piutti C, Total Synthesis of the Amaryllidaceae Alkaloid (+)-Plicamine and Its Unnatural Enantiomer by Using Solid-Supported Reagents and Scavengers in a Multistep Sequence of Reactions. Angew. Chem., Int. Ed 2002, 41, 2194–2197. [DOI] [PubMed] [Google Scholar]
- 255.Baxendale IR, Ley SV, Nessi M, Piutti C, Total synthesis of the amaryllidaceae alkaloid (+)-plicamine using solid-supported reagents. Tetrahedron, 2002, 58, 6285–6304. [DOI] [PubMed] [Google Scholar]
- 256.Baxendale IR and Ley SV, Synthesis of the Alkaloid Natural Products (+)-Plicane and (−)-Obliquine, Using Polymer-Supported Reagents and Scavengers. Ind. Eng. Chem. Res, 2005, 44, 8588–8592. [Google Scholar]
- 257.Kametani T, Kohno T, Shibuya S, Fukumoto K, Studies on the syntheses of heterocyclic compounds-CDXXXVI: Photolytic synthesis of the crinine ring system-formal total synthesis of (±)-maritidine. Tetrahedron, 1971, 27, 5441–5444. [Google Scholar]
- 258.Kotani E, Takeuchi N, Tobinaga S, Total synthesis of the alkloids (±)-oxocrinine and (±)-oxomaritidine via anodic oxidation. J. Chem. Soc. Chem. Commun, 1973, 550–551. [Google Scholar]
- 259.Schwartz MA, Holton RA, Intramolecular oxidative phenol coupling. II. Biogenetic-type synthesis of (±)-maritidine. J. Am. Chem. Soc, 1970, 92, 1090–1092. [Google Scholar]
- 260.Schwartz MA, Rose BF, Vishnuvajjala B, Intramolecular oxidative phenol coupling. III. Two-electron oxidation with thallium(III) trifluoroacetate. J. Am. Chem. Soc, 1973, 95, 612–613. [Google Scholar]
- 261.Schwartz MA, Rose BF, Holton RA, Scott SW, Vishnuvajjala B, Intramolecular oxidative coupling of diphenolic, monophenolic, and nonphenolic substrates. J. Am. Chem. Soc, 1977, 99, 2571–2578. [Google Scholar]
- 262.Tomioka K, Koga K, Yamada S-I, Stereochemical Studies. XLIX. A Biogenetic-type Total Synthesis of Natural (+)-Maritidine from L-Tyrosine using highly Specific Asymmetric Cyclization. Chem. Pharm. Bull, 1977, 25, 2681–2688. [Google Scholar]
- 263.Yamada S-I, Tomioka K, Koga K, A biogenetic-type asymmetric synthesis of natural (+)-maritidine from L-tyrosine. Tetrahedron Lett, 1976, 17, 57–60. [Google Scholar]
- 264.Kotani E, Takeuchi N, Tobinaga S, Biogenetic-type synthesis of (±)-oxomaritidine by the catalytic oxidation with [Fe(dmf)3Cl2][FeCl4]. Tetrahedron Lett, 1973, 2735–2736. [Google Scholar]
- 265.Kupchan SM, Dhingra OP, Kim C-K, Efficient intramolecular monophenol oxidative coupling. J. Org. Chem, 1978, 43, 4076–4081. [Google Scholar]
- 266.White JD, Chong WKM, Thirring K, Phenolic oxidative coupling with hypervalent iodine. A synthesis of 6a-epipretazettine. J. Org. Chem, 1983, 48, 2300–2302. [Google Scholar]
- 267.Kita Y, Takada T, Gyoten M, Tohma H, Zenk MH, Eichhorn J, An Oxidative Intramolecular Phenolic Coupling Reaction for the Synthesis of Amaryllidaceae Alkaloids Using a Hypervalent Iodine(III) Reagent. J. Org. Chem, 1991, 61, 5857–5864. [Google Scholar]
- 268.Kodama S, Takita H, Kajimoto T, Nishide K, Node M, Synthesis of Amaryllidaceae alkaloids, siculine, oxocrinine, epicrinine, and buflavine. Tetrahedron, 2004, 60, 4901–4907. [Google Scholar]
- 269.Elango S, Yan T-H, A Short Synthesis of (+)-Lycoricidine. Tetrahedron, 2002, 58, 7335–7338. [Google Scholar]
- 270.Elango S, Yan T-H, A Short Synthesis of (+)-Narciclasine via a Strategy Derived from Stereocontrolled Epoxide Formation and SnCl4-Catalyzed Arene-Epoxide Coupling. J. Org. Chem, 2002, 67, 6954–6959. [DOI] [PubMed] [Google Scholar]
- 271.Phung AN, Zannetti MT, Whited G, Fessner W-D, Stereospecific Biocatalytic Synthesis of Pancratistatin Analogues. Angew. Chem. Int. Ed, 2003, 42, 4821–4824. [DOI] [PubMed] [Google Scholar]
- 272.Hudlicky T, Olivo HF, A short synthesis of (+)-lycoricidine. J. Am. Chem. Soc, 1992, 114, 9694–9696. [Google Scholar]
- 273.Tian X, Maruya R, Konigsberger K, Hudlicky T, Asymmetric Total Synthesis of (+)-7-Deoxypancratistatin. Synlett, 1995, 1125–1126. [Google Scholar]
- 274.Tian X, Hudlicky T, Konigsberger K, First Total Synthesis of (+)-Pancratistatin: An Unusual Set of Problems. J. Am. Chem. Soc, 1995, 117, 3643–3644. [Google Scholar]
- 275.Hudlicky T, Tian X, Konigsberger K, Maurya R, Rouden J, Fan B, Toluene Dioxygenase-Mediated cis-Dihydroxylation of Aromatics in Enantioselective Synthesis. Asymmetric Total Syntheses of Pancratistatin and 7-Deoxypancratistatin, Promising Antitumor Agents. J. Am. Chem. Soc, 1996, 118, 10752–10765. [Google Scholar]
- 276.Hudlicky T, Rinner U, Gonzalez D, Akgun H, Schilling S, Siengalewicz P, Martinot TA, Pettit GR, Total Synthesis and Biological Evaluation of Amaryllidaceae Alkaloids: Narciclasine, ent-7-Deoxypancratistatin, Regioisomer of 7-Deoxypancratistatin, 10b-epi-Deoxypancratistatin, and Truncated Derivatives. J. Org. Chem, 2002, 67, 8726–8743. [DOI] [PubMed] [Google Scholar]
- 277.Akgün H, Hudlicky T, Total syntheses of ent-conduramine A and ent-7-deoxypancratistatin. Tetrahedron Lett, 1999, 40, 3081–3084. [Google Scholar]
- 278.Rinner U, Siengalwicz P, Hudlicky T, Total Synthesis of epi-7-Deoxypancratistatin via Aza-Payne Rearrangement and Intramolecular Cyclization. Org. Lett, 2001, 4, 115–117. [DOI] [PubMed] [Google Scholar]
- 279.Rinner U, Hillebrenner H, Adams DR, Hudlicky T, Pettit GR, Synthesis and biological activity of some structural modifications of pancratistatin. Bioorg. Med. Chem. Lett, 2004, 14, 2911–2915. [DOI] [PubMed] [Google Scholar]
- 280.Rinner U, Hudlicky T, Gordon H, Pettit GR, A β-Carboline-1-one Mimic of the Anticancer Amaryllidaceae Constituent Pancratistatin: Synthesis and Biological Evaluation. Angew. Chem. Int. Ed, 2004, 43, 5342–5346. [DOI] [PubMed] [Google Scholar]
- 281.García-Fortanet J, Kessler F, Buchwald SL, Palladium-catalyzed asymmetric dearomatization of naphthalene derivatives, J. Am. Chem. Soc, 2009, 131, 6676–6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Rousseaux S, García-Fortanet J, Del Aguila Sanchez MA, Buchwald SL, Palladium(0)-catalyzed arylative dearomatization of phenols, J. Am. Chem. Soc, 2011, 133, 9282–9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Milner PJ, Maimone TJ, Su M, Chen J, Müller P, Buchwald SL, Investigating the dearomative rearrangement of biaryl phosphine-ligated Pd(II) complexes, J. Am. Chem. Soc, 2012, 134, 19922–19934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Xu RQ, Gu Q, Wu WT, Zhao ZA, You SL, Construction of erythrinane skeleton via pd(0)-catalyzed intramolecular dearomatization of para -aminophenols, J. Am. Chem. Soc, 2014, 136, 15469–15472. [DOI] [PubMed] [Google Scholar]
- 285.Du K, Guo P, Chen Y, Cao Z, Wang Z, Tang W, Enantioselective Palladium-Catalyzed Dearomative Cyclization for the Efficient Synthesis of Terpenes and Steroids. Angew. Chem. Int. Ed, 2015, 54, 3033–3037. [DOI] [PubMed] [Google Scholar]
- 286.Du K, Yang H, Guo P, Feng L, Xu G, Zhou Q, Lung B, Chung W, Tang W, Efficient syntheses of (–)-crinine and (–)-aspidospermidine, and the formal synthesis of (–)-minfiensine by enantioselective intramolecular dearomative cyclization, Chem. Sci, 2017, 8, 6247–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Zhao G, Xu G, Qian C, Tang W Efficient Enantioselective Syntheses of (+)-Dalesconol A and B. J. Am. Chem. Soc, 2017, 139, 3360–3363. [DOI] [PubMed] [Google Scholar]
- 288.Ünver N, Gözler T, Walch N, Gözler B, Hesse M, Two novel dinitrogenous alkaloids from Galanthus plicatus subsp. byzantinus (Amaryllidaceae), Phytochemistry, 1999, 50, 1255–1261. [Google Scholar]
- 289.Mijangos MV, Miranda LD, Multicomponent access to indolo[3,3a-c]-isoquinolin-3,6-diones: formal synthesis of (±)-plicamine, Org. Biomol. Chem, 2016, 14, 3677. [DOI] [PubMed] [Google Scholar]
- 290.Shi Y, He H, Gao S, Recent advances in the total synthesis of gracilamine, Chem. Commun, 2018, 54, 12905–12913. [DOI] [PubMed] [Google Scholar]
- 291.Tian S, Zi W, Ma D, Potentially Biomimetic Total Synthesis and Relative Stereochemical Assignment of (±)-Gracilamine, Angew. Chem. Int. Ed, 2012, 51, 10141–10144. [DOI] [PubMed] [Google Scholar]
- 292.Shi Y, Yang B, Cai S, Gao S, Total Synthesis of Gracilamine, Angew. Chem. Int. Ed, 2014, 53, 9539–9543. [DOI] [PubMed] [Google Scholar]
- 293.Gan P, Smith MW, Braffman NR, Snyder SA, Pyrone Diels-Alder Routes to Indolines and Hydroindolines: Syntheses of Gracilamine, Mesembrine, and Δ7 -Mesembrenone, Angew. Chem. Int. Ed, 2016, 55, 3625–3630. [DOI] [PubMed] [Google Scholar]
- 294.Bose S, Yang J, Yu Z-X, Formal Synthesis of Gracilamine Using Rh(I)-Catalyzed [3 + 2 + 1] Cycloaddition of 1-Yne−Vinylcyclopropanes and CO, J. Org. Chem, 2016, 81, 6757–6765. [DOI] [PubMed] [Google Scholar]
- 295.Chandra A, Verma P, Negel A, Pandey G, Asymmetric Total Synthesis of (–)-Gracilamine Using a Bioinspired Approach, Eur. J. Org. Chem, 2017, 6788–6792. [Google Scholar]
- 296.Gao N, Banwell MG, Willis AC, Biomimetic Total Synthesis of the Pentacyclic Amaryllidaceae Alkaloid Derivative Gracilamine, Org. Lett, 2017, 19, 162–165. [DOI] [PubMed] [Google Scholar]
- 297.Zuo X, Guo S, Yang R, Xie J, Zhou Q, Asymmetric Total Synthesis of Gracilamine and Determination of Its Absolute Configuration, Org. Lett, 2017, 19, 5240–5243. [DOI] [PubMed] [Google Scholar]
- 298.Odagi M, Yamamoto Y, Nagasawa K, Total Synthesis of (+)-Gracilamine Based on an Oxidative Phenolic Coupling Reaction and Determination of Its Absolute Configuration, Angew. Chem. Int. Ed, 2018, 57, 2229–2232. [DOI] [PubMed] [Google Scholar]
- 299.Kato M, Hirao S, Nakano K, Sato M, Yamanaka M, Sohtome Y, Nagasawa K, Entropy-Driven 1,2-Type Friedel–Crafts Reaction of Phenols with N-tert-Butoxycarbonyl Aldimines, Chem. Eur. J, 2015, 21, 18606–18612. [DOI] [PubMed] [Google Scholar]
- 300.Kornienko A, Evidente A, Chemistry, Biology, and Medicinal Potential of Narciclasine and its Congeners, Chem. Rev, 2008, 108, 1982–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Ingrassia L, Lefranc F, Mathieu V, Darro F, Kiss R, Amaryllidaceae Isocarbostyril Alkaloids and Their Derivatives as Promising Antitumor Agents, Transl. Oncol, 2008, 1, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Van Otterlo WAL, Green IR, A Review on Recent Syntheses of Amaryllidaceae Alkaloids and Isocarbostyrils (Time period mid-2016 to 2017), Nat. Prod. Commun, 2018, 13, 255–277. [Google Scholar]
- 303.Hernandez LW, Pospech J, Klöckner U, Bingham TW, Sarlah D, Synthesis of (+)-Pancratistatins via Catalytic Desymmetrization of Benzene, J. Am. Chem. Soc, 2017, 139, 15656–15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Bingham TW, Hernandez LW, Olson DG, Svec RL, Hergenrother PJ, Sarlah D, Enantioselective Synthesis of Isocarbostyril Alkaloids and Analogs Using Catalytic Dearomative Functionalization of Benzene, J. Am. Chem. Soc, 2019, 141, 657–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Tezuka N, Shimojo K, Hirano K, Komagawa S, Yoshida K, Wang C, Miyamoto K, Saito T, Takita R, Uchiyama M, Direct Hydroxylation and Amination of Arenes via Deprotonative Cupration, J. Am. Chem. Soc, 2016, 138, 9166–9171. [DOI] [PubMed] [Google Scholar]
- 306.Onoyovwe A, Hagel JM, Chen X, Khan MF, Schriemer DC, Facchini PJ, Morphine Biosynthesis in Opium Poppy Involves Two Cell Types: Sieve Elements and Laticifers, Plant Cell, 2013, 25, 4110–4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Gates M, Tschudi G, The synthesis of morphine, J. Am. Chem. Soc, 1952, 74, 1109–1110. [Google Scholar]
- 308.Gates M, Tschudi G, The synthesis of morphine, J. Am. Chem. Soc, 1956, 78, 1380–1393. [Google Scholar]
- 309.Rice KC, Synthetic opium alkaloids and derivatives, A short total synthesis of (±)-dihydrothebainone, (±)-dihydrocodeinone, and (±)-nordihydrocodeinone as an approach to a practical synthesis of morphine, codeine, and congeners, J. Org. Chem 1980, 45, 3135–3137. [Google Scholar]
- 310.Lipp A, Ferenc D, Gütz C, Geffe M, Vierengel N, Schollmeyer D, Schäfer HJ, Waldvogel SR, T. Opatz A Regio- and Diastereoselective Anodic Aryl–Aryl Coupling in the Biomimetic Total Synthesis of (–)-Thebaine, Angew. Chem. Int. Ed 2018, 57, 11055–11059. [DOI] [PubMed] [Google Scholar]
- 311.White JD, Caravatti G, Kline TB, Edstrom E, Rice KC, Brossi A, Biomimetic total synthesis of (−)-codeine, Tetrahedron, 1983, 39, 2393–2397. [Google Scholar]
- 312.Hong CY, Kado N, Overman LE, Asymmetric synthesis of either enantiomer of opium alkaloids and morphinans, Total synthesis of (−)- and (+)-dihydrocodeinone and (−)- and (+)-morphine, J. Am. Chem. Soc, 1993, 115, 11028–11029. [Google Scholar]
- 313.Brousseau J, Xolin A, Barriault L, A Nine-Step Formal Synthesis of (±)-Morphine, Org. Lett, 2019, 21, 1347–1349. [DOI] [PubMed] [Google Scholar]
- 314.Chu S, Münster N, Balan T, Smith MD, A Cascade Strategy Enables a Total Synthesis of (±)-Morphine, Angew. Chem. Int. Ed, 2016, 55, 14306–14309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Umihara H, Yokoshima S, Inoue M, Fukuyama T, Total Synthesis of (–)-Morphine, Chem. Eur. J, 2017, 23, 6993–6995. [DOI] [PubMed] [Google Scholar]
- 316.Trost BM, Tang W, Enantioselective Synthesis of (–)-Codeine and (–)-Morphine, J. Am. Chem. Soc, 2002, 124, 14542–14543. [DOI] [PubMed] [Google Scholar]
- 317.Tissot M, Phipps RJ, Lucas C, Leon RM, Pace RDM, Ngouansavanh T, Gaunt MJ, Gram-Scale Enantioselective Formal Synthesis of Morphine through an Ortho–Para Oxidative Phenolic Coupling Strategy., Angew. Chem. Int. Ed, 2014, 53, 13498–13501. [DOI] [PubMed] [Google Scholar]
- 318.Park KH, Chen DY-K, A Desymmetrization-Based Approach to Morphinans: Application in the Total Synthesis of Oxycodone, Chem. Commun, 2018, 54, 13018–13021. [DOI] [PubMed] [Google Scholar]
- 319.Reed JW, Hudlicky T, The Quest for a Practical Synthesis of Morphine Alkaloids and Their Derivatives by Chemoenzymatic Methods, Acc. Chem. Res, 2015, 48, 674–687. [DOI] [PubMed] [Google Scholar]
- 320.Makarova M, Endoma-Arias MAA, Dela Paz HE, Simionescu R, Hudlicky T, Chemoenzymatic Total Synthesis of Ent-Oxycodone: Second-, Third-, and Fourth-Generation Strategies, J. Am. Chem. Soc, 2019, 141, 10883–10904. [DOI] [PubMed] [Google Scholar]
- 321.Varghese V, Hudlicky T, Short Chemoenzymatic Total Synthesis of Ent-Hydromorphone: An Oxidative Dearomatization/Intramolecular [4+2] Cycloaddition/Amination Sequence, Angew. Chem. Int. Ed, 2014, 53, 4355–4358. [DOI] [PubMed] [Google Scholar]
- 322.King SM, Herzon SB, Chapter Three – The Hasubanan and Acutumine Alkaloids, The Alkaloids: Chemistry and Biology, 2014, 73, 161–222. [DOI] [PubMed] [Google Scholar]
- 323.Herzon SB, Calandra NA, King SM, Efficient Entry to the Hasubanan Alkaloids: First Enantioselective Total Syntheses of (−)-Hasubanonine, (−)-Runanine, (−)-Delavayine, and (+)-Periglaucine B, Angew. Chem. Int. Ed, 2011, 50, 8863–8866. [DOI] [PubMed] [Google Scholar]
- 324.Chuang KV, Navarro R, Reisman SE, Short, Enantioselective Total Syntheses of (−)-8-Demethoxyrunanine and (−)-Cepharatines A, C, and D, Angew. Chem. Int. Ed, 2011, 50, 9447–9451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Ramirez A, Garcia-Rubio S, Current Progress in the Chemistry and Pharmacology of Akuammiline Alkaloids, Curr. Med. Chem, 2003, 10, 1891–1915. [DOI] [PubMed] [Google Scholar]
- 326.Kam TS, Alkaloids: Chem. Biol. Perspect., 1999, 14, 285. [Google Scholar]
- 327.Arai H, Hirasawa Y, Rahman A, Kusumawati I, Zaini NC, Sato S, Aoyama C, Takeo J, Morita H, Alstiphyllanines E–H, picraline and ajmaline-type alkaloids from Alstonia macrophylla inhibiting sodium glucose cotransporter, Bioorg. Med. Chem, 2010, 18, 2152–2158. [DOI] [PubMed] [Google Scholar]
- 328.Pearce HL, in The Alkaloids, Vol. 37 (Eds.: Brossi A, Suffness M), Academic Press, San Diego, 1990, p. 145. [Google Scholar]
- 329.Eckermann R, Gaich T, The Akuammiline Alkaloids; Origin and Synthesis, Synthesis, 2013, 45, 2813–2823. [Google Scholar]
- 330.Smith JM, Moreno J, Boal BW, Garg NK, Cascade Reactions: A Driving Force in Akuammiline Alkaloid Total Synthesis, Angew. Chem. Int. Ed, 2015, 54, 400–412. [DOI] [PubMed] [Google Scholar]
- 331.Goodson JA, Echitamine, J. Chem. Soc, 1932, 2759–2768. [Google Scholar]
- 332.Smith GF, Echitamine: hypothetical biogenetic scheme, Chem. Ind, 1961, 29, 1120. [Google Scholar]
- 333.Wenkert E, Wickberg BJ, General methods of synthesis of indole alkaloids. IV. A synthesis of dl-eburnamonine, J. Am. Chem. Soc, 1965, 87, 1580–1589. [DOI] [PubMed] [Google Scholar]
- 334.Leewanich P, Tohda M, Matsumoto K, Subhadhirasakul S, Takayama H, Watanabe H, Behavioral Studies on Alkaloids Extracted from the Leaves of Hunteria zeylanica, Biol. Pharm. Bull, 1996, 19, 394–399. [DOI] [PubMed] [Google Scholar]
- 335.Saraswathi V, Subramanian S, Ramamoorthy N, Mathuram V, Govindasamy S, In vitro cytotoxicity of echitamine chloride and adriamycin on Ehrlich ascites carcinoma cell cultures, Med. Sci. Res, 1997, 25, 167–170. [Google Scholar]
- 336.Maier A, Maul C, Zerlin SM Grabley, R. Thiericke, A Novel Screening Approach for the Discovery of Biologically Active Secondary Metabolites, J. Antibiot, 1999, 52, 952–959. [DOI] [PubMed] [Google Scholar]
- 337.Baliga MS, Jagetia GC, Ulloor JN, Baliga MP, Venkatesh P, Reddy R, Rao KVNM, Baliga BS, Devi S, Raju SK, Veeresh V, Reddy TK, Bairy KL, The evaluation of the acute toxicity and long term safety of hydroalcoholic extract of Sapthaparna (Alstonia scholaris) in mice and rats, Toxicol. Lett, 2004, 151, 317–326. [DOI] [PubMed] [Google Scholar]
- 338.Kamarajan P, Sekar N, Mathuram V, Govindasamy S, Antitumor effect of echitamine chloride on methylcholonthrene induced fibrosarcoma in rats, Biochem. Int, 1991, 25, 491–498. [PubMed] [Google Scholar]
- 339.Duwiejua M, Woode E, Obiri DD, Pseudo-akuammigine, an alkaloid from Picralima nitida seeds, has anti-inflammatory and analgesic actions in rats, J. Ethnopharmacol, 2002, 81, 73–79. [DOI] [PubMed] [Google Scholar]
- 340.Subramaniam G, Hiraku O, Hayashi M, Koyano T, Komiyama K, Kam T-S, Biologically Active Aspidofractinine, Rhazinilam, Akuammiline, and Vincorine Alkaloids from Kopsia, J. Nat. Prod, 2007, 70, 1783–1789. [DOI] [PubMed] [Google Scholar]
- 341.Arai H, Hirasawa Y, Rahman A, Kusumawati I, Zaini NC, Sato S, Aoyama C, Takeo J, Morita H, Alstiphyllanines E–H, picraline and ajmaline-type alkaloids from Alstonia macrophylla inhibiting sodium glucose cotransporter, Bioorg. Med. Chem, 2010, 18, 2152–2158. [DOI] [PubMed] [Google Scholar]
- 342.Meng W, Ellsworth BA, Nirschl AA, McCann PJ, Patel M, Girotra R, Wu G, Sher PM, Morrison EP, Biller SA, Zahler R, Deshpande PP, Pullockaran A, Hagan DL, Morgan N, Taylor JR, Obermeier MT, Humphreys WG, Khanna A, Discenza L, Robertson JG, Wang A, Han S, Wetterau JR, Janovitz EB, Flint OP, Whaley JM, Washburn WN, Discovery of Dapagliflozin: A Potent, Selective Renal Sodium-Dependent Glucose Cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes, J. Med. Chem, 2008, 51, 1145–1149. [DOI] [PubMed] [Google Scholar]
- 343.Adams G, Smith AB III, The Chemistry of the Akuammiline Alkaloids. in The Alkaloids; Knolker H-J, Ed.; Elsevier: New York, 2016; Vol. 76, pp 171–257. [DOI] [PubMed] [Google Scholar]
- 344.Zhang M, Huang X, Shen L, Qin Y, Total Synthesis of the Akuammiline Alkaloid (±)-Vincorine, J. Am. Chem. Soc, 2009, 131, 6013–6020. [DOI] [PubMed] [Google Scholar]
- 345.MacMillan DWC, Nine-Step Enantioselective Total Synthesis of (−)-Vincorine, J. Am. Chem. Soc, 2013, 135, 6442–6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Jiang S-Z, Zeng X-Y, Liang X, Lei T, Wei K, Yang Y-R, Iridium-Catalyzed Enantioselective Indole Cyclization: Application to the Total Synthesis and Absolute Stereochemical Assignment of (–)-Aspidophylline A, Angew. Chem. Int. Ed, 2016, 55, 4044–4048. [DOI] [PubMed] [Google Scholar]
- 347.Zheng C, You S-L, Catalytic asymmetric dearomatization (CADA) reaction-enabled total synthesis of indole-based natural products, Nat. Prod. Rep, 2019, 36, 1589–1605. [DOI] [PubMed] [Google Scholar]
- 348.Adams GL, Carroll PJ, Smith AB, Total Synthesis of (+)-Scholarisine A, J. Am. Chem. Soc, 2012, 134, 4037–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Li Y, Zhu S, Li J, Li A, Asymmetric Total Syntheses of Aspidodasycarpine, Lonicerine, and the Proposed Structure of Lanciferine, J. Am. Chem. Soc, 2016, 138, 3982–3985. [DOI] [PubMed] [Google Scholar]
- 350.Zhang X, Kakde BN, Guo R, Yadav S, Gu Y, Li A, Total Syntheses of Echitamine, Akuammiline, Rhazicine, and Pseudoakuammigine, Angew. Chem. Int. Ed, 2019, 58, 6053–6058. [DOI] [PubMed] [Google Scholar]
- 351.Wong SP, Gan CY, Lim KH, Ting KN, Low YY, Kam TS, Arboridinine, a Pentacyclic Indole Alkaloid with a New Cage Carbon-Nitrogen Skeleton Derived from a Pericine Precursor, Org. Lett, 2015, 17, 3628–3631. [DOI] [PubMed] [Google Scholar]
- 352.Zhang Z, Xie S, Cheng B, Zhai H, Li Y, Enantioselective Total Synthesis of (+)-Arboridinine, J. Am. Chem. Soc, 2019, 141, 7147–7154. [DOI] [PubMed] [Google Scholar]
- 353.Zi W, Xie W, Ma D, Total Synthesis of Akuammiline Alkaloid (−)-Vincorine via Intramolecular Oxidative Coupling, J. Am. Chem. Soc, 2012, 134, 9126–9129. [DOI] [PubMed] [Google Scholar]
- 354.Teng M, Zi W, Ma D, Total Synthesis of the Monoterpenoid Indole Alkaloid (±)-Aspidophylline A, Angew. Chem. Int. Ed, 2014, 53, 1814–1817. [DOI] [PubMed] [Google Scholar]
- 355.Xu W, Wang W, Wang X, Gold-Catalyzed Cyclization Leads to a Bridged Tetracyclic Indolenine that Represses β-Lactam Resistance, Angew. Chem. Int. Ed, 2015, 54, 9546–9549. [DOI] [PubMed] [Google Scholar]
- 356.Smith MW, Zhou Z, Gao AX, Shimbayashi T, Snyder SA, A 7-Step Formal Asymmetric Total Synthesis of Strictamine via an Asymmetric Propargylation and Metal-Mediated Cyclization, Org. Lett, 2017, 19, 1004–1007. [DOI] [PubMed] [Google Scholar]
- 357.Gan P, Pitzen J, Qu P, Snyder SA, Total Synthesis of the Caged Indole Alkaloid Arboridinine Enabled by aza-Prins and Metal-Mediated Cyclizations, J. Am. Chem. Soc, 2018, 140, 919–925. [DOI] [PubMed] [Google Scholar]
- 358.Zhou Z, Gao AX, Snyder SA, Total Synthesis of (+)-Arborisidine, J. Am. Chem. Soc, 2019, 141, 7715–7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Zu L, Boal BW, Garg NK, Total Synthesis of (±)-Aspidophylline A, J. Am. Chem. Soc, 2011, 133, 8877–8879. [DOI] [PubMed] [Google Scholar]
- 360.Simmons BJ, Hoffmann M, Champagne PA, Picazo E, Yamakawa K, Morrill LA, Houk KN, Garg NK, Understanding and Interrupting the Fischer Azaindolization Reaction, J. Am. Chem. Soc, 2017, 139, 14833–14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Smith JM, Moreno J, Boal BW, Garg NK, Total Synthesis of the Akuammiline Alkaloid Picrinine, J. Am. Chem. Soc, 2014, 136, 4504–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Smith JM, Moreno J, Boal BW, Garg NK, Fischer Indolizations as a Strategic Platform for the Total Synthesis of Picrinine, J. Org. Chem, 2015, 80, 8954–8967. [DOI] [PubMed] [Google Scholar]
- 363.Moreno J, Picazo E, Morrill LA, Smith JM, Garg NK, Enantioselective Total Syntheses of Akuammiline Alkaloids (+)-Strictamine, (−)-2(S)-Cathafoline, and (−)-Aspidophylline A, J. Am. Chem. Soc, 2016, 138, 1162–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Picazo E, Morrill LA, Susick RB, Moreno J, Smith JM, Garg NK, Enantioselective Total Syntheses of Methanoquinolizidine-Containing Akuammiline Alkaloids and Related Studies, J. Am. Chem. Soc, 2018, 140, 6483–6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Ren W, Wang Q, Zhu J, Total Synthesis of (±)-Strictamine, Angew. Chem. Int. Ed, 2016, 55, 3500–3503. [DOI] [PubMed] [Google Scholar]
- 366.Saya JM, Ruijter E, Orru RVA, Total Synthesis of Aspidosperma and Strychnos Alkaloids through Indole Dearomatization, Chem. Eur. J, 2019, 25, 8916–8935. [DOI] [PubMed] [Google Scholar]
- 367.Dounay AB, Humphreys PG, Overman LE and Wrobleski AD, Total Synthesis of the Strychnos Alkaloid (+)-Minfiensine: Tandem Enantioselective Intramolecular Heck−Iminium Ion Cyclization, J. Am. Chem. Soc, 2008, 130, 5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Douki K, Ono H, Taniguchi T, Shimokawa J, Kitamura M, and Fukuyama T, Enantioselective Total Synthesis of (+)-Hinckdentine A via a Catalytic Dearomatization Approach, J. Am. Chem. Soc, 2016, 138, 14578–14581. [DOI] [PubMed] [Google Scholar]
- 369.Austin JF, Kim SG, Sinz CJ, Xiao WJ, MacMillan DWC, Enantioselective organocatalytic construction of pyrroloindolines by a cascade addition-cyclization strategy: Synthesis of (−)-flustramine B, Proc. Natl. Acad. Sci. U. S. A, 2004, 101, 5482–5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Jones SB, Simmons B, MacMillan DWC, Nine-step enantioselective total synthesis of (+)-minfiensine, J. Am. Chem. Soc, 2009, 131, 13606–13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Jones SB, Simmons B, Mastracchio A, MacMillan DWC, Collective synthesis of natural products by means of organocascade catalysis, Nature, 2011, 475, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Knowles RR, Carpenter J, Blakey SB, Kayano A, Mangion IK, Sinz CJ, MacMillan DWC, Total synthesis of diazonamide A, Chem. Sci, 2011, 2, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Zhu S, MacMillan DWC, Enantioselective copper-catalyzed construction of aryl pyrroloindolines via an arylation-cyclization cascade, J. Am. Chem. Soc, 2012, 134, 10815–10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Jiang R, Ding L, Zheng C, You SL, Iridium-catalyzed Z-retentive asymmetric allylic substitution reactions, Science, 2021, 371, 380–386. [DOI] [PubMed] [Google Scholar]
- 375.Wu QF, He H, Liu WB, You S-L, Enantioselective construction of spiroindolenines by Ir-catalyzed allylic alkylation reactions, J. Am. Chem. Soc, 2010, 132, 11418–11419. [DOI] [PubMed] [Google Scholar]
- 376.Liu C, Zhang W, Dai LX, You SL, Copper(I)-catalyzed cascade dearomatization of 2-substituted tryptophols with arylidonium salts, Org. Lett, 2012, 14, 4525–4527. [DOI] [PubMed] [Google Scholar]
- 377.Wu Q-F, Zheng C, You SL, Enantioselective Synthesis of Spiro Cyclopentane-1,3′-indoles and 2,3,4,9-Tetrahydro-1H-carbazoles by Iridium-Catalyzed Allylic Dearomatization and Stereospecific Migration, Angew. Chem. Int. Ed, 2012, 51, 1680–1683. [DOI] [PubMed] [Google Scholar]
- 378.Liu C, Yi JC, Zheng ZB, Tang Y, Dai LX, You SL, Enantioselective Synthesis of 3a-Amino-Pyrroloindolines by Copper-Catalyzed Direct Asymmetric Dearomative Amination of Tryptamines, Angew. Chem. Int. Ed, 2016, 55, 751–754. [DOI] [PubMed] [Google Scholar]
- 379.Tu HF, Zhang X, Zheng C, Zhu M, You SL, Enantioselective dearomative prenylation of indole derivatives. Nat. Catal, 2018, 1, 601–608. [Google Scholar]
- 380.Liu XJ, Zheng C, Yang YH, Jin S, You SL, Iridium-Catalyzed Asymmetric Allylic Aromatization Reaction, Angew. Chem. Int. Ed, 2019, 58, 10493–10499. [DOI] [PubMed] [Google Scholar]
- 381.Kieffer ME, Chuang KV, Reisman SE, A copper-catalyzed arylation of tryptamines for the direct synthesis of aryl pyrroloindolines, Chem. Sci, 2012, 3, 3170–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Kieffer ME, Chuang KV, Reisman SE, Copper-catalyzed diastereoselective arylation of tryptophan derivatives: Total synthesis of (+)-naseseazines A and B, J. Am. Chem. Soc, 2013, 135, 5557–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Wang H, Reisman SE, Enantioselective Total Synthesis of (−)-Lansai B and (+)-Nocardioazines A and B, Angew. Chem. Int. Ed, 2014, 53, 6206–6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Gentry EC, Rono LJ, Hale ME, Matsuura R, Knowles RR, Enantioselective Synthesis of Pyrroloindolines via Noncovalent Stabilization of Indole Radical Cations and Applications to the Synthesis of Alkaloid Natural Products, J. Am. Chem. Soc, 2018, 140, 3394–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Snyder SA, Zografos AL, Lin Y Total Synthesis of Resveratrol-Based Natural Products: A Chemoselective Solution. Angew. Chem. Int. Ed, 2007, 46, 8186–8191. [DOI] [PubMed] [Google Scholar]
- 386.Snyder SA, Gollner A, Chiriac MI Regioselective Reactions for Programmable Resveratrol Oligomer Synthesis, Nature, 2011, 474, 461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Snyder SA, Breazzano SP, Ross AG, Lin Y, Zografos AL Total Synthesis of Diverse Carbogenic Complexity within the Resveratrol Class from a Common Building Block, J. Am. Chem. Soc, 2009, 131, 1753–1765. [DOI] [PubMed] [Google Scholar]
- 388.Snyder SA, Sherwood TC, Ross AG Total Syntheses of Dalesconol A and B, Angew. Chem. Int. Ed, 2010, 49, 5146–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Hulst MB, Grocholski T, Neefjes JJC, van Wezel GP, Metsä-Ketalä M, Anthracyclines: biosynthesis, engineering and clinical applications, Nat. Prod. Rep, 2022, 39, 814–841. [DOI] [PubMed] [Google Scholar]
- 390.Zucchi R, Danesi R Cardiac Toxicity of Antineoplastic Anthracyclines, Curr. Med. Chem.: Anti-Cancer Agents, 2003, 3, 151–171. [DOI] [PubMed] [Google Scholar]
- 391.Binaschi M, Bigioni M, Cipollone A, Rossi C, Goso C, Maggi CA, Capranico G, Animati F Anthracyclines: Selected New Developments, Curr. Med. Chem.: Anti-Cancer Agents, 2001, 1, 113–130. [DOI] [PubMed] [Google Scholar]
- 392.Dennis DG, Okumura M, Sarlah D Synthesis of (±)-Idarubicinone via Global Functionalization of Tetracene, J. Am. Chem. Soc, 2019, 141, 10193–10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Gao J-M, Yang S-X, Qin J-C Azaphilones: Chemistry and Biology, Chem. Rev, 2013, 113, 4755–4811. [DOI] [PubMed] [Google Scholar]
- 394.Osmanova N, Schultze W, Ayoub N Azaphilones: a class of fungal metabolites with diverse biological activities, Phytochem. Rev, 2010, 9, 315–342. [Google Scholar]
- 395.Zhu J, Porco JA Asymmetric Syntheses of (−)-Mitorubrin and Related Azaphilone Natural Products, Org. Lett, 2006, 8, 5169–5171. [DOI] [PubMed] [Google Scholar]
- 396.Germain AR, Bruggemeyer DM, Zhu J, Genet C, O’Brien P, Porco JA, Synthesis of the Azaphilones (+)-Sclerotiorin and (+)-8- O -Methylsclerotiorinamine Utilizing (+)-Sparteine Surrogates in Copper-Mediated Oxidative Dearomatization, J. Org. Chem, 2011, 76, 2577–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Johnson RA Microbial Arene Oxidations. In Organic Reactions; American Cancer Society, 2004; pp 118–264. [Google Scholar]
- 398.Lewis SE Asymmetric Dearomatization Under Dearomative Conditions. In Asymmetric Dearomatization Reactions; Wiley-VCH Verlag GmbH & Co. KGaA., 2016; pp 279–346. [Google Scholar]
- 399.Pyser JB, Baker Dockrey SA, Benítez AR, Joyce LA, Wiscons RA, Smith JL, Narayan ARH, Stereodivergent, Chemoenzymatic Synthesis of Azaphilone Natural Products, J. Am. Chem. Soc, 2019, 141, 18551–18559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Chakrabarty S, Romero EO, Pyser JB, Yazarians JA, Narayan ARH, Chemoenzymatic Total Synthesis of Natural Products. Acc. Chem. Res, 2021, 54, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Sib A, Gulder TAM, Stereoselective Total Synthesis of Bisorbicillinoid Natural Products by Enzymatic Oxidative Dearomatization/Dimerization, Angew. Chemie Int. Ed, 2017, 56, 12888–12891. [DOI] [PubMed] [Google Scholar]
- 402.Kahlert L, Bassiony EF, Cox RJ, Skellam EJ, Diels–Alder Reactions During the Biosynthesis of Sorbicillinoids., Angew. Chemie Int. Ed, 2020, 59, 5816–5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Barnes-Seeman D, Corey EJ, A Two-Step Total Synthesis of the Natural Pentacycle Trichodimerol, a Novel Inhibitor of TNF-α Production, Org. Lett, 1999, 1, 1503–1504. [DOI] [PubMed] [Google Scholar]
- 404.Nicolaou KC, Jautelat R, Vassilikogiannakis G, Baran PS, Simonsen KB, Studies towards Trichodimerol: Novel Cascade Reactions and Polycyclic Frameworks, Chem. Eur. J, 1999, 5, 3651–3665. [Google Scholar]
- 405.Nicolaou KC, Vassilikogiannakis G, Simonsen KB, Baran PS, Zhong YL, Vidali VP, Pitsinos EN, Couladouros EA, Biomimetic Total Synthesis of Bisorbicillinol, Bisorbibutenolide, Trichodimerol, and Designed Analogues of the Bisorbicillinoids, J. Am. Chem. Soc, 2000, 122, 3071–3079. [Google Scholar]
- 406.Pettus LH, Van De Water RW, Pettus TRR, Synthesis of (±)-Epoxysorbicillinol Using a Novel Cyclohexa-2,5-dienone with Synthetic Applications to Other Sorbicillin Derivatives, Org. Lett, 2001, 3, 905–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Hong R, Chen Y, Deng L, Catalytic Enantioselective Total Syntheses of Bisorbicillinolide, Bisorbicillinol, and Bisorbibutenolide, Angew. Chem. Int. Ed, 2005, 44, 3478–3481. [DOI] [PubMed] [Google Scholar]
- 408.Ciochina R, Grossman RB, Polycyclic Polyprenylated Acylphloroglucinols, Chem. Rev, 2006, 106, 3963–3986. [DOI] [PubMed] [Google Scholar]
- 409.Leuner K, Kazanksi V, Müller M, Essin K, Henke B, Gollasch M, Harteneck C, W. E. Müller, Hyperforin—a key constituent of St. John’s wort specifically activates TRPC6 channels, FASEB J, 2007, 21, 4101–4111. [DOI] [PubMed] [Google Scholar]
- 410.Cuesta-Rubio O, Velez-Castro H, Frontana-Uribe BA, Cárdenas J, Nemorosone, the major constituent of floral resins of Clusia rosea, Phytochemistry, 2001, 57, 279–283. [DOI] [PubMed] [Google Scholar]
- 411.Fukuyama Y, Minami H, Kuwayama A, Garsubellins, polyisoprenylated phloroglucinol derivatives from Garcinia subelliptica, Phytochemistry, 1998, 49, 853–857. [Google Scholar]
- 412.Rodeschini V, Ahmad NA, Simpkins NS, Synthesis of (±)-Clusianone: High-Yielding Bridgehead and Diketone Substitutions by Regioselective Lithiation of Enol Ether Derivatives of Bicyclo[3.3.1]nonane-2,4,9-triones, Org. Lett, 2006, 8, 5283–5285. [DOI] [PubMed] [Google Scholar]
- 413.Biber N, Möws K, Plitker B, The total synthesis of hyperpapuanone, hyperibone L, epi-clusianone and oblongifolin A, Nat. Chem, 2011, 3, 938–942. [DOI] [PubMed] [Google Scholar]
- 414.Ting CP, Maimone TJ, Total Synthesis of Hyperforin, J. Am. Chem. Soc, 2015, 137, 10516–10519. [DOI] [PubMed] [Google Scholar]
- 415.Tsukano C, Siegel DR, Danishefsky SJ, Differentiation of Nonconventional “Carbanions”—The Total Synthesis of Nemorosone and Clusianone, Angew. Chem. Int. Ed, 2007, 46, 8840–8844. [DOI] [PubMed] [Google Scholar]
- 416.Sparling BA, Moebius DC, Shair MD, Enantioselective Total Synthesis of Hyperforin, J. Am. Chem. Soc, 2013, 135, 644–647. [DOI] [PubMed] [Google Scholar]
- 417.Uwamori M, Saito A, Nakada M, Stereoselective Total Synthesis of Nemorosone, J. Org. Chem, 2012, 77, 5098–5107. [DOI] [PubMed] [Google Scholar]
- 418.Uwamori M, Nakada M, Stereoselective total synthesis of garsubellin A, J. Antibiot, 2013, 66, 141–145. [DOI] [PubMed] [Google Scholar]
- 419.Qi J, Porco JA Jr., Rapid Access to Polyprenylated Phloroglucinols via Alkylative Dearomatization−Annulation: Total Synthesis of (±)-Clusianone, J. Am. Chem. Soc, 2007, 129, 12682–12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Qi J, Beeler AB, Zhang Q, Porco JA Jr., Catalytic Enantioselective Alkylative Dearomatization−Annulation: Total Synthesis and Absolute Configuration Assignment of Hyperibone K, J. Am. Chem. Soc, 2010, 132, 13642–13644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Zhang Q, Mitasev B, Qi J, Porco JA Jr., Total Synthesis of Plukenetione A, J. Am. Chem. Soc, 2010, 132, 14212–14215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Zhang Q, Porco JA Jr., Total Synthesis of (±)-7-epi-Nemorosone, Org. Lett, 2012, 14, 1796–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Boyce JB, Porco JA Jr., Asymmetric, Stereodivergent Synthesis of (−)-Clusianone Utilizing a Biomimetic Cationic Cyclization, Angew. Chem. Int. Ed, 2014, 53, 7832–7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Wen S, Boyce JH, Kandappa SK, Sivaguru J, Porco JA Jr., Regiodivergent Photocyclization of Dearomatized Acylphloroglucinols: Asymmetric Syntheses of (−)-Nemorosone and (−)-6-epi-Garcimultiflorone A, J. Am. Chem. Soc, 2019, 141, 11315–11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Grenning AJ, Boyce JH, Porco JA Jr., Rapid Synthesis of Polyprenylated Acylphloroglucinol Analogs via Dearomative Conjunctive Allylic Annulation, J. Am. Chem. Soc, 2014, 136, 11799–11804. [DOI] [PMC free article] [PubMed] [Google Scholar]