

Abstract
Background & Aims
Aflatoxin exposure increases the risk for hepatocellular carcinoma (HCC) in hepatitis B virus (HBV)-infected individuals, particularly males. We investigated sex-based differences in the HCC genome and antitumor immunity.
Methods
Whole-genome, whole-exome, and RNA sequencing were performed on 101 HCC patient samples (47 males, 54 females) that resulted from HBV infection and aflatoxin exposure from Qidong. Androgen on the expression of aflatoxin metabolism-related genes and nonhomologous DNA end joining (NHEJ) factors were examined in HBV-positive HCC cell lines, and further tested in tumor-bearing syngeneic mice.
Results
Qidong HCC differed between males and females in genomic landscape and transcriptional dysfunction pathways. Compared with females, males expressed higher levels of aflatoxin metabolism-related genes, such as AHR and CYP1A1, and lower levels of NHEJ factors, such as XRCC4, LIG4, and MRE11, showed a signature of up-regulated type I interferon signaling/response and repressed antitumor immunity. Treatment with AFB1 in HBV-positive cells, the addition of 2 nmol/L testosterone to cultures significantly increased the expression of aflatoxin metabolism-related genes, but reduced NHEJ factors, resulting in more nuclear DNA leakage into cytosol to activate cGAS-STING. In syngeneic tumor-bearing mice that were administrated tamoxifen daily via oral gavage, favorable androgen signaling repressed NHEJ factor expression and activated cGAS-STING in tumors, increasing T-cell infiltration and improving anti–programmed cell death protein 1 treatment effect.
Conclusions
Androgen signaling in the context of genotoxic stress repressed DNA damage repair. The alteration caused more nuclear DNA leakage into cytosol to activate the cGAS-STING pathway, which increased T-cell infiltration into tumor mass and improved anti–programmed cell death protein 1 immunotherapy in HCCs.
Keywords: Sex Hormones, Aflatoxin, DNA Double-Strand Break, Immune Checkpoints, HBV
Abbreviations used in this paper: AFB1, aflatoxin B1; AHR, aryl hydrocarbon receptor; AR, androgen receptor; ARE, androgen responsive element; CC50, cytotoxicity concentration at 50%; cGAS, cyclic guanosine monophosphate–adenosine monophosphate synthase; CM, cell medium; CYP, cytochrome P450 isoenzyme; DEG, differentially expressed gene; DSB, double-strand break; ER, estrogen receptor; γH2AX, phosphorylated histone H2AX; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HNF4α, hepatocyte nuclear factor 4α; IFN, interferon; ITG, integration targeted gene; NHEJ, nonhomologous DNA end joining; PD-1, programmed cell death protein 1; QD, Qidong; RNA-seq, RNA sequencing; TCGA, The Cancer Genome Atlas; Tes, testosterone; TMB, tumor mutational burden; Veh, vehicle; WGS, whole-genome sequencing
Graphical abstract
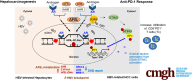
Summary.
Through repressing DNA double-strand break repair, androgen signaling enhanced aflatoxin B1 genotoxicity, potentially enhancing aflatoxin-related hepatocarcinogenesis in hepatitis B virus chronically infected males. Alternatively, favorable androgen signaling increased the tumor infiltration of CD8+ programmed cell death protein 1+ T cells to enhance the anti–programmed cell death protein 1 treatment effect, providing a new strategy against hepatocellular carcinoma.
Hepatocellular carcinoma (HCC) is common globally and is a leading cause of cancer-related death. Hepatitis B virus (HBV) infection remains the leading etiology underlying HCC development, and male sex has long been identified as an important HCC risk factor after HBV infection.1,2 Previous studies have indicated that androgen and estrogen have antagonistic behaviors in hepatocarcinogenesis after chronic HBV infection.2, 3, 4 Through direct binding to the cognate androgen responsive element (ARE) sites in enhancer I of the HBV genome, the ligand-stimulated androgen receptor (AR) enhances HBV transcription and replication,3 significantly increasing HCC risk. Estrogen represses HBV transcription by increasing the hepatic expression of estrogen receptor (ER)α, which interacts with hepatocyte nuclear factor (HNF)4α to prevent its binding to HBV, potentially reducing HCC risk.4 In cases of HBV integration in the promoter of telomerase reverse transcriptase (TERT), which is an early event in HCC development, the TERT transcription is enhanced by the AR pathway but repressed by the ERα pathway.5,6
Increasing evidence indicates that sex hormones and sex chromosomes have intrinsic roles in the control of cancer-initiating cell populations, tumor microenvironment, metabolism, and immune responses to cancer development.2 In HBV-infected individuals, HCC risk is synergistically enhanced by exposure to chemical carcinogens, including aflatoxin, owing to contamination of food crops.1,7,8 Through binding to the aryl hydrocarbon receptor (AHR) and activation by the cytochrome P450 (CYP450) oxidase family members, aflatoxin B1 (AFB1) becomes genotoxic via the formation of the AFB1-N7-guanine DNA (AFB1-DNA) adduct.9,10 Most HCC cases in China are related to HBV infection. However, in Qidong, a classic region of aflatoxin exposure, the incidence of liver cancer is higher and the male-to-female ratio is greater than the national level in China.7,8 Occult aflatoxin exposure resulting from food contamination is more common than previously thought.11 It is not well known how sex hormones and sex chromosomes affect aflatoxin carcinogenesis in HBV-infected individuals. In the most recent decade, cancer immunotherapy greatly improved the management of multiple types of cancer, including HCC patients. Nevertheless, the magnitude of cancer patient benefit from immune checkpoint inhibitors is sex-dependent.12 A better understanding of the differences in the genomic features of HCC based on sex could be of value for the prevention and treatment of HCC.
Results
Description of Patients
We obtained the tumor and matched nonneoplastic liver tissues from 101 HBV-related HCC patients (Qidong [QD]-HCCs, 47 males and 54 females), who received primary hepatectomy without systemic or radiation therapy and were followed up for 5 years by May 31, 2019. Aflatoxin exposure was confirmed by the record of aflatoxin M1 in their urine 3–18 years before HCC diagnosis. Tumor sizes greater than 3 cm in diameter were found in 40 males (85%) and 41 females (76%) of QD-HCC samples (Table 1). From The Cancer Genome Atlas (TCGA) database, we recognized 119 HBV-related HCCs (TCGA-HCCs) and recruited 113 cases, 73 males and 40 females, for analysis by excluding 6 cases that harbored mutational signatures of aflatoxin exposure.11 The median age of the male patients in QD-HCC and in TCGA-HCC was younger than that of female patients. No differences between males and females were observed either in QD-HCCs (Table 1) or in TCGA-HCCs (Table 2) regarding the distribution of serum levels of α-fetoprotein level, tumor sizes and numbers, Edmondson–Steiner grade, and the presence of liver cirrhosis.
Table 1.
Demographic and Clinical Characteristics of 101 Cases of QD-HCCs
| Variable | Male (N = 47) | Female (N = 54) | P valuea |
|---|---|---|---|
| Age, year | |||
| Median (IQR) | 50 (40.0–59.0) | 57 (48.8–64.3) | |
| AFM1 detection, years before HCC diagnosis | |||
| ≤10 | 18 (38.3) | 44 (81.5) | <.001 |
| >10 | 29 (61.7) | 10 (18.5) | |
| HBV infection status, n (%) | |||
| HBsAg (+) | 35 (74.5) | 40 (74.1) | 1.000 |
| HBsAg(-) and anti-HBc(+)b | 12 (25.5) | 14 (25.9) | |
| Serum AFP level, ng/mL, n (%) | |||
| <20 | 21 (44.7) | 20 (37.0) | .564 |
| ≥20 | 26 (55.3) | 34 (63.0) | |
| Cirrhosis, n (%) | |||
| No | 24 (51.1) | 30 (55.6) | .802 |
| Yes | 23 (48.9) | 24 (44.4) | |
| Tumor size, n (%) | |||
| ≤3 cm | 7 (14.9) | 13 (24.1) | .366 |
| >3 cm | 40 (85.1) | 41 (75.9) | |
| Tumor number, n (%) | |||
| 1 | 40 (85.1) | 51 (94.4) | .182 |
| >1 | 7 (14.9) | 3 (5.6) | |
| Edmondson–Steiner grade, n (%) | |||
| I | 0 (0.0) | 2 (3.7) | .247 |
| II–III | 47 (100.0) | 50 (92.6) | |
| Data missing | 0 (0.0) | 2 (3.7) | |
| Vital status,c n (%) | |||
| Alive | 18 (38.3) | 24 (44.4) | .101 |
| Dead | 25 (53.2) | 30 (55.6) | |
| Data missing | 4 (8.5) | 0 (0.0) | |
AFP, α-fetoprotein; anti-HBc, antibodies against the hepatitis B virus core antigen; HBsAg, hepatitis B surface antigen; IQR, interquartile range.
Chi-square test or the Fisher exact test.
The samples were positive for HBV-DNA test.
Followed up for 5 years by May 31, 2019.
Table 2.
Demographic and Clinical Characteristics of 113 HBV-Related TCGA-HCCs
| Variables | Male (N = 73) | Female (N = 40) | P valuea |
|---|---|---|---|
| Age, year | |||
| Median (IQR) | 58 (48.0–67.0) | 62.5 (54.0–72.0) | |
| Edmondson–Steiner grade, n (%) | |||
| I–II | 39 (53.4) | 23 (57.5) | .793 |
| III–IV | 30 (41.1) | 16 (40.0) | |
| Data missing | 4 (5.5) | 1 (2.5) | |
| HBV infection status, n (%) | |||
| HBsAg (+) | 34 (46.6) | 20 (50.0) | .102 |
| Anti-HBc (+) | 28 (38.4) | 19 (47.5) | |
| HBV DNA (+) | 11 (15.1) | 1 (2.5) | |
| Race, n (%) | |||
| Asian | 35 (47.9) | 12 (30.0) | .099 |
| Others | 38 (52.1) | 28 (70.0) | |
| Vital status,b n (%) | |||
| Alive | 43 (58.9) | 22 (55.0) | .840 |
| Dead | 30 (41.1) | 18 (45.0) |
anti-HBc, antibodies against the hepatitis B virus core antigen; HBsAg, hepatitis B surface antigen; IQR, interquartile range.
Chi-squared test or the Fisher exact test.
Downloaded on June 26, 2019.
Similarity of Mutation Patterns and Mutational Burden in Male and Female QD-HCCs
No significant differences between male and female QD-HCCs were found based on the numbers of single-base substitutions (Figure 1A) or the tumor mutational burden (TMB) (Figure 1B). The classic aflatoxin-associated HCC signature11 was present in both male and female QD-HCCs (Figure 1C). The mutation frequencies of 75 previously identified HCC driver genes13 were similar in both sexes including TP53, TERT promoter, AXIN1, KMT2D, and CTNNB1. The genes with mutation frequencies greater than 5% are listed in Figure 1D. TP53 was the most frequently mutated gene with a mutational hot spot at R249S. The mutation frequencies in the TERT promoter region of male QD-HCCs and female QD-HCCs were 34.04% and 37.04%, and in ADGRB1 were 12.77% and 14.81%, respectively (Figure 1E).
Figure 1.

Mutation patterns and mutational burden in male and female QD-HCCs. (A) Mutation counts in the whole genome or exome of male and female QD-HCCs. (B) TMB of QD-HCCs, or TCGA-HCCs (73 males, 40 females), or Chinese HCC samples (159 males, 22 females) in the International Cancer Genome Consortium (ICGC) database (ICGC-HCCs). Data present means ± SEM. P values were calculated by the Mann–Whitney test. (C) Percentage of the 6 variant types in the exome of QD-HCCs and TCGA-HCCs. The P value was calculated by chi-squared test. (D) Reported HCC driver genes13 with more than 5% mutation frequencies in male and female QD-HCCs (P > .05, chi-squared test). (E) Mutations of TP53, TERT promoter, and ADGRB1 in male and female QD-HCCs. SBS, single-base substitution.
In TCGA-HCCs, the TMBs (Figure 1B) and the mutation patterns (Figure 1C) also showed no sex differences. We further retrieved 181 Chinese HCCs (159 males and 22 females) from the International Cancer Genome Consortium, and also detected no sex difference in the TMBs. In both TCGA-HCCs and International Cancer Genome Consortium HCCs, the TMBs were lower compared with QD-HCCs (Figure 1B). However, among the 75 previously identified HCC driver genes,13 the TP53 mutation frequency in male TCGA-HCCs was 30.14%, which was significantly higher than that in female TCGA-HCCs (10%) (Table 3).
Table 3.
Seven Genes With Different Mutation Frequencies Between Males and Females in HBV-Related TCGA-HCCs
| Gene | Males (N = 73) |
Females (N = 40) |
P valuea | ||
|---|---|---|---|---|---|
| n | Frequency | n | Frequency | ||
| TP53b | 22 | 30.14% | 4 | 10.00% | .028 |
| OBSCN | 11 | 15.07% | 0 | 0.00% | .007 |
| MUC17 | 8 | 10.96% | 0 | 0.00% | .049 |
| RYR3 | 8 | 10.96% | 0 | 0.00% | .049 |
| TACC2 | 0 | 0.00% | 3 | 7.50% | .042 |
| TRIP12 | 0 | 0.00% | 3 | 7.50% | .042 |
| FAM205A | 0 | 0.00% | 3 | 7.50% | .042 |
Chi-squared test or the Fisher exact test.
TP53 is one of the previously identified HCC driver genes.
Sex Differences in Mutation Profiles
In QD-HCC samples, we identified 71 genes with significantly different mutation frequencies between the 2 sexes (P < .05), 62 genes were associated more frequently with males, and 9 genes with females (Table 4). Figure 2A lists the 12 genes with significantly different mutation frequencies between the 2 sexes with P < .02. Although these genes have not been reported previously as HCC drivers,13 some have been found previously in other cancers. Mutations in PAK7 and CEP250 were reported in melanoma; RET in lung cancer, melanoma, and thyroid adenocarcinoma; and GTF2I in anal cancer and thymic carcinoma.13, 14, 15 Mutations in PAK7, ANP32E, CEP250, and ATXN2 detected in male QD-HCCs were associated significantly with poor survival of male patients but not female patients based on the 5-year follow-up results (Figure 3). The sex differences in mutation frequencies of specified genes also were observed in TCGA-HCCs (Table 3).
Table 4.
The 71 Identified Genes With Different Mutation Frequencies Between Male and Female QD-HCC Samples
| Genea | Males (N = 47) |
Females (N = 54) |
Pb | ||
|---|---|---|---|---|---|
| n | Frequency | n | Frequency | ||
| FLNCc | 11 | 23.40% | 3 | 5.56% | .021 |
| MEGF6d | 10 | 21.28% | 2 | 3.70% | .009 |
| APOB | 9 | 19.15% | 2 | 3.70% | .030 |
| SSPOd | 9 | 19.15% | 2 | 3.70% | .030 |
| DSTc,d | 9 | 19.15% | 0 | 0.00% | .001 |
| IGSF9B | 8 | 17.02% | 2 | 3.70% | .042 |
| STARD9c | 8 | 17.02% | 2 | 3.70% | .042 |
| CELSR3c | 8 | 17.02% | 1 | 1.85% | .011 |
| TEP1 | 7 | 14.89% | 1 | 1.85% | .024 |
| SPATA31D1d | 7 | 14.89% | 1 | 1.85% | .024 |
| RP1e | 6 | 12.77% | 1 | 1.85% | .048 |
| UNC13A | 6 | 12.77% | 1 | 1.85% | .048 |
| TSHZ1 | 6 | 12.77% | 1 | 1.85% | .048 |
| CEP250 | 6 | 12.77% | 1 | 1.85% | .048 |
| MYO10 | 6 | 12.77% | 1 | 1.85% | .048 |
| GPSM1 | 6 | 12.77% | 1 | 1.85% | .048 |
| IQSEC3 | 6 | 12.77% | 1 | 1.85% | .048 |
| XKR7 | 6 | 12.77% | 1 | 1.85% | .048 |
| ABCA2d,e | 6 | 12.77% | 1 | 1.85% | .048 |
| LRRK1 | 6 | 12.77% | 0 | 0.00% | .008 |
| HCN4d | 6 | 12.77% | 0 | 0.00% | .008 |
| CCDC129d | 5 | 10.64% | 0 | 0.00% | .019 |
| RAVER2 | 5 | 10.64% | 0 | 0.00% | .019 |
| FYCO1 | 5 | 10.64% | 0 | 0.00% | .019 |
| CITc | 5 | 10.64% | 0 | 0.00% | .019 |
| ATP2B1c | 5 | 10.64% | 0 | 0.00% | .019 |
| PHKA2 | 4 | 8.51% | 0 | 0.00% | .044 |
| CCDC136 | 4 | 8.51% | 0 | 0.00% | .044 |
| HK2 | 4 | 8.51% | 0 | 0.00% | .044 |
| RET | 4 | 8.51% | 0 | 0.00% | .044 |
| OR2L2 | 4 | 8.51% | 0 | 0.00% | .044 |
| BCR | 4 | 8.51% | 0 | 0.00% | .044 |
| ZNF429 | 4 | 8.51% | 0 | 0.00% | .044 |
| OPLAH | 4 | 8.51% | 0 | 0.00% | .044 |
| PKD1 | 4 | 8.51% | 0 | 0.00% | .044 |
| FER1L5 | 4 | 8.51% | 0 | 0.00% | .044 |
| PCF11c | 4 | 8.51% | 0 | 0.00% | .044 |
| SLC6A1 | 4 | 8.51% | 0 | 0.00% | .044 |
| SMAD6 | 4 | 8.51% | 0 | 0.00% | .044 |
| ITGA2d | 4 | 8.51% | 0 | 0.00% | .044 |
| DACH1c | 4 | 8.51% | 0 | 0.00% | .044 |
| DDX60Lc | 4 | 8.51% | 0 | 0.00% | .044 |
| MIB2 | 4 | 8.51% | 0 | 0.00% | .044 |
| CENPF | 4 | 8.51% | 0 | 0.00% | .044 |
| SLC9A3 | 4 | 8.51% | 0 | 0.00% | .044 |
| DNAJC6 | 4 | 8.51% | 0 | 0.00% | .044 |
| MEI1 | 4 | 8.51% | 0 | 0.00% | .044 |
| ERN2 | 4 | 8.51% | 0 | 0.00% | .044 |
| PDE10A | 4 | 8.51% | 0 | 0.00% | .044 |
| ESYT2c | 4 | 8.51% | 0 | 0.00% | .044 |
| ZNF318 | 4 | 8.51% | 0 | 0.00% | .044 |
| CCDC166 | 4 | 8.51% | 0 | 0.00% | .044 |
| IQGAP3 | 4 | 8.51% | 0 | 0.00% | .044 |
| PAK7 | 4 | 8.51% | 0 | 0.00% | .044 |
| SCN8A | 4 | 8.51% | 0 | 0.00% | .044 |
| ANO7 | 4 | 8.51% | 0 | 0.00% | .044 |
| ARSI | 4 | 8.51% | 0 | 0.00% | .044 |
| NKPD1 | 4 | 8.51% | 0 | 0.00% | .044 |
| ASPHD1 | 4 | 8.51% | 0 | 0.00% | .044 |
| LAMB1 | 4 | 8.51% | 0 | 0.00% | .044 |
| ANP32E | 4 | 8.51% | 0 | 0.00% | .044 |
| ATXN2 | 4 | 8.51% | 0 | 0.00% | .044 |
| COL6A3 | 3 | 6.38% | 10 | 18.52% | .044 |
| FLNA | 0 | 0.00% | 7 | 12.96% | .014 |
| GTF2Ic,e | 0 | 0.00% | 7 | 12.96% | .014 |
| ZNF628e | 0 | 0.00% | 6 | 11.11% | .029 |
| MYOFe | 0 | 0.00% | 6 | 11.11% | .029 |
| MACF1 | 0 | 0.00% | 6 | 11.11% | .029 |
| RUBCNL | 0 | 0.00% | 6 | 11.11% | .029 |
| MUCL3 | 0 | 0.00% | 6 | 11.11% | .029 |
| CROCC2 | 0 | 0.00% | 6 | 11.11% | .029 |
Reconfirmed by Integrated Genomics Viewer.
Chi-squared test or the Fisher exact test.
In the samples with no cirrhosis, mutation frequencies differed between males and females.
In the samples with cirrhosis, mutation frequencies differed between males and females.
In the samples from patients with serum α-fetoprotein level less than 20 ng/mL, mutation frequencies differed between males and females.
Figure 2.

Mutation profile and HBV integration in male and female QD-HCCs. (A) Twelve genes with significantly different mutation frequencies between the 2 sexes with P < .02 are listed, the others are provided in Table 4. P values were calculated by the chi-squared test or the Fisher exact test. Distribution of HBV breakpoints across the (B) HBV genome and the (C) human genome. P values were calculated by the chi-squared test. (D) Functional annotation of integration-targeted genes in male and female samples analyzed by DAVID. AFP, α-fetoprotein; bp, base pair; C, core gene; Del, deletion; Ins, insertion; P, polymerase gene; S, surface antigen genes; X, X gene.
Figure 3.

The association of specified gene mutations and survival of QD-HCC patients. The Kaplan–Meier method was used for survival analysis. Poor survival was observed in (A) male patients, but not in (B) female patients. No mutation of ANP32E and ATXN2 was detected in female QD-HCC samples.
HCC developed mainly in the cirrhotic liver, and CTNNB1 mutation has been implicated as an oncogenic driver in HCC and other cancers.13 In QD-HCCs with cirrhosis (23 males, 24 females), the CTNNB1 mutation frequency in males was 26.09%, which was significantly greater than that in females, which was 4.17%. HBV also can cause HCC without cirrhosis, but the mechanisms are unclear.16 In QD-HCCs without cirrhosis (24 males, 30 females), the males harbored more frequent mutations in FLNC (33.33%), and the females harbored more frequent mutations in GTF2I (20%) (Table 4). In the QD-HCC samples of patients with serum α-fetoprotein levels less than 20 ng/mL, the mutation frequencies of genes in males also differed from those in females (Table 4).
Sex Differences in HBV Integration
We performed whole-genome sequencing (WGS) on the samples from 88 QD-HCC patients (36 males, 52 females) and detected HBV integration in 37 (42.05%) of them, identifying 110 breakpoints. No difference in HBV breakpoint numbers was detected between males and females (Figure 4A). However, S-gene breakpoints were detected more often in male QD-HCCs, and X-gene breakpoints mapping to the direct repeat (DR)2–DR1 region were detected more often in female QD-HCCs. Only male QD-HCCs harbored HBV breakpoints that were reported to bind AR and HNF4α3,4 (Figure 2B). To validate this observation, we analyzed another cohort of HBV-related Chinese HCC samples with no record of aflatoxin exposure. Using sequence-capture probes that recognize 8 HBV genotypes,17 we detected 83 (81.4%) HCCs harboring HBV integration in 102 tumor tissues. The HBV breakpoints binding to AR and HNF4α were detected only in male HCC samples (Figure 4B), confirming previous reports that AR and HNF4α interact directly with HBV to promote HCC.3, 4, 5, 6
Figure 4.

HBV integration in the 2 sexes. (A) Number of HBV breakpoints in male and female QD-HCC samples. Data represent means ± SEM. The P value was calculated by the Mann–Whitney test. (B) Distribution of HBV breakpoints across the HBV genome in 83 HBV-related HCCs with no record of aflatoxin exposure. bp, base pair; C, core gene; DR, direct repeat ; P, polymerase gene; S, surface antigen genes; X, X gene.
Across the human genome, HBV breakpoints in QD-HCC samples were detected mostly on chromosome 5 and no sex difference was observed (P = .36). But the samples from males harbored more breakpoints on chromosomes 17 (P = .06) and 19 (P = .02) than those from females; the samples from females harbored more breakpoints on chromosome 3 than those from males (P = .03) (Figure 2C). HBV breakpoints were detected mostly within 100 kb of the transcription start sites of the integration targeted genes (ITGs), and the TERT promoter region was altered most frequently in both sexes. However, many ITGs in male QD-HCCs differed from those in female QD-HCCs (Figure 2D). HBV integration in MLL4, previously reported as a recurrent hotspot for HBV integration,6 was detected mainly in male samples. MLL4 performs intrinsic histone lysine-specific methylation, acting as the tumor protein p53 (TP53) coactivator.18 In female samples, the ITGs included PRDM7 and SMYD3, which perform histone lysine-specific methylation.19,20 In summary, QD-HCCs differed between 2 sexes on the basis of somatic mutation profiles and HBV integration.
Sex Differences in Multiple Biological Capabilities of HCC
We performed RNA sequencing (RNA-seq) on the samples from 87 QD-HCC patients, 43 males and 44 females, identifying 3070 significantly differentially expressed genes (DEGs) between males and females with a fold change greater than 2 and a false discovery rate of less than 0.05. In the samples from males, 2352 DEGs were up-regulated and 718 DEGs were down-regulated compared with those from females (Figure 5A). The transcription levels of estrogen receptor (ESR) 1 and 2 were similar between male and female QD-HCCs. However, male QD-HCCs expressed higher levels of AR and the genes defining early response to estrogen (Figure 5B). In TCGA-HCCs, enhanced AR expression also was detected in males (Figure 6A).
Figure 5.

Expression patterns of male and female QD-HCCs. (A) DEGs between 43 male QD-HCC and 44 female QD-HCCs were identified with fold change greater than 2 and false discovery rate (FDR) less than 0.05. Gene expression in tumor tissues was normalized by matched non-neoplastic liver samples. (B) Expression levels of AR, ESR1, ESR2, and the genes related to the estrogen response early pathway in male and female QD-HCCs. (C) Expression of the genes related to multiple biological capabilities in male and female QD-HCCs. (D) Sex differences in gene expression related to inflammation and adaptive immune responses in male and female QD-HCCs. CCR5, C-C motif chemokine receptor 5; CREB, cAMP responsive element binding protein; DAI, DNA-dependent activator of IFN-regulatory factors; FCER1, fc ε receptor 1; FGFR4, fibroblast growth factor receptor 4; FPKM, fragments per kilobase of transcript per million fragments mapped; IFNG, interferon γ; IgFC, log2 fold change; IL, interleukin; logFC, log2 fold change; MET, mesenchymal-epithelial transition; MHC, major histocompatibility complex; NES, normalized enrichment score; NFκB, nuclear factor-κB; RIP, receptor interacting protein.
Figure 6.

Sex differences in gene expression of HBV-related TCGA-HCCs. (A) The expression levels of AR and ESR1, ESR2. Each dot represents 1 case. (B) The expression of gene sets involved in specified pathways in QD-HCCs and in TCGA-HCCs. The yellow boxes with positive numbers indicate the pathways were up-regulated, the green boxes with negative numbers indicate the pathways were down-regulated in male HCCs compared with female HCCs. AHSP, α-hemoglobin stabilizing protein; AKAP13, a-kinase anchoring protein 13; BMP, bone morphogenetic protein; CDK5, cyclin-dependent kinase-5; CREB, cAMP responsive element binding protein; FCER1, fc ε receptor 1; FGFR4, fibroblast growth factor receptor 4; FPKM, fragments per kilobase of transcript per million fragments mapped; IFNG, interferon γ; logFC, log2 fold change; MHC, major histocompatibility complex; MYC, myelocytomatosis oncogene; NFκB, nuclear factor-κB; PAR1, protease-activated receptor-1; PCAF, p300/CBP-associating factor; pol III, RNA polymerase III; V2, version 2.
We then conducted gene set variation analysis to understand HCC transcriptional dysfunction in males and females. QD-HCC samples of male patients showed different biological capabilities from those of females. Several signaling pathways related to HCC development and progression were up-regulated in males, including fibroblast growth factor receptor (FGFR4) ligand binding and activation, Wnt/β-catenin signaling, and Notch signaling. Some signaling pathways in males were down-regulated, such as DNA double-strand break (DSB) repair (Figure 5C). Distinct gene signatures of immune responses and inflammation were observed between males and females. In males, QD-HCCs showed repression of specific antitumor immunity, including the antigen processing/presentation, regulation of interferon γ (IFNG) signaling, and CD28 costimulation. However, the inflammation-related gene expression was augmented in male QD-HCCs; for example, nuclear factor-B activation levels were enhanced (Figure 5D). In TCGA-HCCs, differences in gene signatures between males and females also were detected (Figure 6B).
Sex Differences in Gene Expression Related to Aflatoxin Metabolism
QD-HCC samples were from the patients with aflatoxin exposure subsequent to HBV infection (Table 1).7,8 AFB1 induces HCC through binding AHR9 and bioactivation by CYP450 oxidases, including CYP1A1, CYP1A2, and CYP3A4. Glutathione-S-transferase M1 can detoxicate AFB1 genotoxicity.10 We, therefore, analyzed the genes associated with aflatoxin metabolism. Male QD-HCC samples expressed greater levels of AHR and CYP1A1, but lower levels of GSTM1 genes compared with female samples (Figure 7A). The enhanced protein expression of AHR and CYP1A1 in QD-HCC samples from males was confirmed by immunohistochemistry staining (Figure 7B). Sex-based differences of these genes also were observed in TCGA-HCCs (Figure 7A).
Figure 7.

Sex hormones on gene expression related to aflatoxin metabolism. (A) Expression levels of the genes in QD-HCCs and TCGA-HCCs. (B) Expression of AHR and CYP1A1 determined by immunohistochemistry in QD-HCCs. Shown are the representatives from 10 male samples and 10 female samples. Scale bar: 100 μm. (C) Bar graphs show AHR and CYP1A1 expression levels in HepG2.2.15 (left) and PLC/PRF/5 (right) in the presence of different concentrations of testosterone after AFB1 treatment, determined by real-time quantitative polymerase chain reaction. Glyceraldehyde-3-phosphate dehydrogenase was used as control. The curves show AFB1 toxicity on HepG2.2.15 (left) and PLC/PRF/5 (right) in the presence of 2 nmol/L testosterone (blue lines, AFB1+Tes) or AFB1 only (red lines, AFB1). (D) Images show the AFB1–DNA adducts (brown) in HepG2.2.15 (left) and PLC/PRF/5 (right) detected by immunohistochemistry, after AFB1 treatment only (AFB1+vehicle) or in the presence of 2 nmol/L testosterone (AFB1+Tes). Bar graphs indicate the average AFB1–DNA positive numbers every 10 cells in 5 fields of 3 independent experiments. F, female; FPKM, fragments per kilobase of transcript per million fragments mapped; M, male.
We then used HBV-positive HepG2.2.15 cells and PLC/PRF/5 cells to test sex hormones in the regulation of AHR and CYP1A1, impacting AFB1 cytotoxicity. The expression of androgen receptors and estrogen receptors was confirmed in the 2 cell lines (Figure 8A). After AFB1 treatment, the addition of testosterone to the cultures significantly enhanced the transcription levels of AHR and CYP1A1, depending on the concentration of testosterone and AFB1. The cytotoxicity concentration of AFB1 at 50% (CC50) of AFB1 was 0.9 μg/mL in HepG2.2.15 cells, and 3.0 μg/mL in PLC/PRF/5 cells. When 2 nmol/L testosterone was added, the CC50 of AFB1 was reduced to 0.5 μg/mL in HepG2.2.15 cells, and to 1.4 μg/mL in PLC/PRF/5 cells. The dose of AFB1 causing cell death was reduced by 50% in the presence of testosterone (Figure 7C). In addition, more AFB1-DNA adducts were detected in these cells treated with AFB1 plus 2 nmol/L testosterone than the cells treated with the same concentration of AFB1 alone (Figure 7D).
Figure 8.

Expression of AR and ERα incell lines by immunoblot. Images show 1 representative of 2 independent experiments in (A) human HCC cell lines and (B) mice hepatoma cell lines, with actin as the loading control.
Sex Differences in DNA Damage Responses
The generation of AFB1-DNA adducts results in DNA damage, activating various DNA repair mechanisms. We further analyzed different pathways in response to DNA damage in QD-HCCs. The most down-regulated pathway in male QD-HCCs was nonhomologous end joining (NHEJ), the predominant DSB repair in mammalian cells.21 QD-HCCs in males expressed significantly lower levels of NHEJ factors than in females, including XRCC4, MRE11, ATM, XRCC5, and NBN (Figure 9A). To validate this observation, we examined the gene expression levels in some cancer types in the TCGA database. TCGA-HCCs in males expressed significantly lower levels of LIG4, MRE11, and ATM than in females. Colon adenocarcinoma in TCGA from males expressed significantly lower levels of ATM, XRCC5, and NBN than those from females (Figure 10).
Figure 9.

Sex hormones on expression of the genes related to DNA damage responses and cGAS-STING activation. (A) The NHEJ pathway genes differentially expressed in male and female QD-HCCs that were confirmed with HBV integration. (B) Bar graphs show transcriptional levels of some NHEJ genes in HepG2.2.15 cells and PLC/PRF/5 cells that were treated with AFB1 in the presence of different concentrations of Tes, or 17β-estradiol (E2), determined by real-time quantitative polymerase chain reaction with glyceraldehyde-3-phosphate dehydrogenase as control. Images show the γH2AX expression in total cellular proteins analyzed by immunoblot of 3 independent experiments. (C) The schematic effects of AFB1 plus androgen on HCC cells. Graphs show the results of QD-HCC samples. The augmented genes related to innate immune responses analyzed by Gene Ontology are shown in the bubble graph. Bubble sizes indicate variated numbers of related genes in the specified pathway, the biggest includes 25 genes and the smallest includes 4 genes. Scatter plots show the specified genes expressed in male and female QD-HCCs. (D and E) HepG2.2.15 cells and PLC/PRF/5 cells were treated for 72 hours with AFB1 (CC50 concentration) plus 2 nmol/L testosterone (AFB1/Tes) or with the same concentration of AFB1 alone (AFB1/Veh). (D) Representative images (×400) show the cytosolic double-strand DNA analyzed by fluorescent microscopy. Scale bars: 10 μm. (E) Analysis of cGAS-STING pathway activation in differently treated HepG2.2.15 cells and PLC/PRF/5 cells by immunoblot. Images show 1 representative of 3 independent experiments, with actin as the loading control. Bar graphs show the band density of the specific genes. (F) The transcriptional levels of interferon β (IFNB1) in the cells of HepG2.2.15, PLC/PRF/5, and HepG2 transfected with 1.3 × HBV plasmid (HepG2-HBV) or empty-vector (HepG2-pcDNA3.1), described in PMID: 36058909. All cells were treated as described in panel E, with glyceraldehyde-3-phosphate dehydrogenase as control. Data represent means ± SEM. Each dot represents 1 independent experiment. P values were calculated by an unpaired Student t test. logFC, log2 fold change.
Figure 10.

Transcription levels of specified NHEJ genes of some types of cancers in TCGA database. The specified NHEJ gene expression in (A) 73 male and 40 female HBV-related HCC samples in (B) 247 male and 224 female colon adenocarcinoma samples. Data represent means ± SEM. P values were calculated by an unpaired Student t test or the Mann–Whitney test. FPKM, fragments per kilobase of transcript per million fragments mapped.
The genes encoded by sex chromosomes can confer sex-distinct cancer vulnerabilities.2 However, the analysis of sex on gene expression across human tissues from 838 adult individuals indicated that sex differences reflect the sum of distinct sex hormone–related transcriptional regulation plus tissue cell–specific regulation.22 We used the web-based MEME Suite (http://meme-suite.org)23 to predict whether AHR, CYP1A1, XRCC4, LIG4, and MRE11 are the AR target genes. Results indicated that AREs were presented in the promoter areas of these specified genes (Figure 11A). To test the effects of androgen on the regulation of NHEJ factors, we treated HBV-positive HepG2.2.15 cells and PLC/PRF/5 cells with AFB1 plus testosterone. As a control, these cell lines were treated with the same concentration of AFB1 alone. The transcriptional levels of XRCC4 and LIG4 in HepG2.2.15 cells and XRCC4, LIG4, and MRE11 in PLC/PRF/5 cells that were treated with AFB1 plus testosterone were reduced significantly compared with those treated with AFB1 alone. The reduction depended on the concentrations of AFB1 and testosterone in the culture medium. Notably, the addition of 17β-estradiol estrogen partially reversed the reduction of XRCC4 and MRE11 expression (Figure 9B). The half-lives of XRCC4 and MRE11 in both cell lines treated with AFB1 plus testosterone were shortened significantly compared with those treated with AFB1 alone after new RNA synthesis was blocked with actinomycin D (Figure 11B). Without AFB1, the alteration of these NHEJ factors by testosterone alone was insignificant (Figure 12). We then measured the expression levels of phosphorylated histone H2AX (γH2AX), which quantitatively reflects the extent of DNA damage. Both HepG2.2.15 and PLC/PRF/5 cells treated with AFB1 plus testosterone expressed more γH2AX than the cells treated with AFB1 alone (Figure 9B), reflecting the synergetic signaling effects mediated by AFB1 and androgen. We also transiently transfected HepG2 cells with an HBV plasmid containing 1.3 × HBV genomes (HepG2-HBV) to test the combined effects of AFB1 plus testosterone on these specified genes. The alterations of the gene transcription in HepG2-HBV cells were similar to that in the HepG2.2.15 cells. Without HBV, the syngeneic effects of AFB1 plus testosterone on the transcription of NHEJ factors in HepG2 cells were much weaker (Figure 13).
Figure 11.

Predicted AREs in the promoters of specified genes and the half-lives of these genes in HCC cell lines after different treatment. (A) The predicted AREs in the promoter areas of AHR, CYP1A1, XRCC4, LIG4, and MRE11 analyzed with web-based MEME Suite (http://meme-suite.org). (B) Based on the determined AFB1 CC50, the HepG2.2.15 cells were treated with 0.5 μg/mL AFB1, PLC/PRF/5 with 1.4 μg/mL AFB1 plus 2 nmol/L testosterone (AFB1/Tes), or the same concentration of AFB1 alone (AFB1/Veh) for 72 hours. The cells with medium only (No) was used as control. Each treatment was performed in triplicate. Actinomycin D at 5 μg/mL then was added for the indicated period (0, 4, 8, 12, and 24 hours). Transcriptional levels of XRCC4, LIG4, and MRE11 were determined using real-time quantitative polymerase chain reaction, with glyceraldehyde-3-phosphate dehydrogenase as control. One of 2 independent experiments is shown. Chr, chromosome.
Figure 12.

Transcription levels of some aflatoxin metabolism-related genes and NHEJ factors in differently treated HepG2.2.15 and PLC/PRF/5 cells. The cells were treated with the indicated concentration of testosterone for 72 hours and gene expression was determined by real-time quantitative polymerase chain reaction with glyceraldehyde-3-phosphate dehydrogenase as control. Data represent means ± SEM. P values were calculated by 1-way analysis of variance.
Figure 13.

Transcription levels of the specified genes in transiently HBV-transfected HepG2 cells. HepG2 cells were transfected with a plasmid containing 1.3 × HBV genome (HepG2-HBV) or with an empty-vector (HepG2-pcDNA3.1), followed by treatment with the indicated concentration of AFB1 and testosterone for 72 hours. The gene expression was determined by real-time quantitative polymerase chain reaction with glyceraldehyde-3-phosphate dehydrogenase as control. Data represent means ± SEM. P values were calculated by 1-way analysis of variance. The information for 2 plasmids is described in PMID: 36058909.
It has been shown that chronic genotoxic stress induces nuclear DNA leakage into the cytosol, where double-strand DNA intrinsically is detected by cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS) to activate STING-dependent cytokine production,24 depending on the severity of the DNA damage.25,26 QD-HCCs in males showed different gene signatures of immune responses and inflammation from that in females (Figure 5D). To identify the pathways related to tumor immune responses in QD-HCCs, we performed Gene Ontology analysis on the DEGs that showed statistical significance between the 2 sexes. The genes related to interferon (IFN)-I signaling and response were up-regulated significantly in males, but not in females. Notably, significantly higher levels of STING and IRF3 were detected in male QD-HCC samples (Figure 9C).
To validate this observation, we treated HepG2.2.15 cells and PLC/PRF/5 cells with AFB1 plus testosterone, or with the same concentration of AFB1 alone. In the HepG2.2.15 cells and PLC/PRF/5 cells treated with AFB1 plus testosterone, more double-strand DNA was detected in the cytoplasm compared with the cells treated with the same concentration of AFB1 alone (Figure 9D). We collected the cells and examined activation of the cGAS-STING pathway. Significantly higher levels of cGAS, phosphorylated STING, and phosphorylated interferon regulatory factor 3 (IRF3) were detected in the cells treated with AFB1 plus testosterone than in the cells treated with AFB1 alone (Figure 9E). Greater transcription levels of IFNB1 also were detected in HepG2.2.15 cells and in HepG2-HBV cells treated with AFB1 plus testosterone compared with those treated with AFB1 alone (Figure 9F).
Sex Differences in Antitumor Immunity Against HCC
QD-HCC samples from males showed repression of antigen-specific antitumor immunity (Figure 5D). We then analyzed the infiltration of CD8+ T cells and the expression of immune checkpoints that affect antitumor immunity. Significantly increased CD8+ T-cell infiltration was detected in tumor tissues of QD-HCC samples from males, but not from females (Figure 14A). Compared with adjacent non-neoplastic tissues, tumor tissues from male QD-HCC patients but not from female patients showed significantly higher transcriptional levels of PD-1 and CTLA-4, 2 immune checkpoint receptors on activated T cells. The tumor tissues from males also showed significantly greater levels of B7-H2 and B7-H3, which provide negative signals, and less CD80 (B7.1) and CD86 (B7.2), which provide positive signals in T-cell activation (Figure 14B). The enhanced expression of programmed cell death protein 1 (PD-1) and B7-H2 in the tumor tissues of male QD-HCCs was confirmed by immunohistochemistry staining (Figure 14C).
Figure 14.

Infiltration of CD8+T cells and expression levels of different immune checkpoint molecules in male and female QD-HCCs. (A) Images show the representative staining of CD8+ T cells (brown) of 5 male QD-HCCs, and 5 female QD-HCCs, with the average of CD8+ T-cell density presented in the bar graph. Scale bar: 10 μm. Scatter plots indicate the tumor expression levels of CD8α related to matched non-neoplastic tissues in 43 male QD-HCCs and 44 female QD-HCCs, determined by RNA-seq. Each dot represents 1 case. (B) Expression levels of the specified molecules in QD-HCCs. Each dot represents 1 case. (C) Representative images of PD-1 (left panel) and B7-H2 (right panel) detected by immunohistochemistry staining in tumor and adjacent non-neoplastic tissues of 5 male QD-HCCs and 5 female QD-HCCs, with the average scores of PD-1 and B7-H2 presented in the bar graphs. Scale bar: 100 μm. (D) Surface expression of B7-H2 and B7-H3 in HepG2.2.15 cells treated for 72 hours with 2 nmol/L testosterone only (Tes, yellow lines), 5 μg/mL of AFB1 alone (AFB1/Veh, red lines), and 5 μg/mL of AFB1 plus 2 nmol/L testosterone (AFB1/Tes, blue lines). Grey lines are the staining of cells treated with vehicle. The profiles of B7-H2 and B7-H3 in PLC/PRF/5 are shown in Figure 15B. (E) Contoured plots show the staining of T cells migrated into the lower chambers of the Transwell insert (5-μm pore size) in a chemotaxis assay using the conditioned medium collected from differently treated HepG2.2.15 cells. The results of differently treated PLC/PRF/5 cells are provided in Figure 15C. Bar graphs indicate the average of migrated CD8+ T cells calculated from 3 independent experiments. ∗P ≤ .05, ∗∗∗P ≤ .001. FPKM, fragments per kilobase of transcript per million fragments mapped; N, non-neoplastic tissue; Tu, tumor tissue.
To validate the sex-distinct antitumor immunity observed in QD-HCC samples, we treated HBV-positive HepG2.2.15 cells and PLC/PRF/5 cells for 72 hours with a CC50 dose of AFB1 plus 2 nmol/L testosterone (AFB1/Tes) or the same concentration of AFB1 alone (AFB1/vehicle [Veh]) (Figure 15A). The expression levels of B7-H2 and B7-H3 in the 2 HCC cell lines were increased significantly after AFB1/Tes treatment compared with AFB1/Veh treatment. Without AFB1, testosterone alone did not alter the expression of B7-H2 or B7-H3 in both HepG2.2.15 cells (Figure 14D) and PLC/PRF/5 cells (Figure 15B). We collected HCC cell medium (CM) from the different treatments and conducted a chemotaxis assay to examine the effect of soluble factors on immune cells, mimicking the infiltration of immune cells into tumor tissues. In response to the CM from either AFB1/Tes-treated HepG2.2.15 cells (Figure 14E), or PLC/PRF/5 cells (Figure 15C), more CD8+PD-1+ T cells migrated into the lower chambers than the cells in response to the CM from AFB1/Veh-treated or only testosterone-treated HCC cells. The addition of AFB1 or testosterone in the medium directly increased the chemotaxis of T cells, especially PD-1+ T cells. However, compared with the AFB1/Tes-treated HCC cell medium, the direct effect of these chemicals on PD-1+ T-cell chemotaxis was much weaker (Figure 15D).
Figure 15.

Determination of the effects of differently treated HepG2.2.15 and PLC/PRF/5 cells on T-cell infiltration. (A) The schematic diagram of experiments. HBV-positive HepG2.2.15 cells or PLC/PRF/5 cells were differently treated for 72 hours as shown. (B) Shown is the surface expression of B7-H2 and B7-H3 in PLC/PRF/5 cells treated with AFB1 plus testosterone (AFB1/Tes) or with the same concentration of AFB1 alone, or with the testosterone alone (Tes). The controls were the cells treated with 0.1% dimethyl sulfoxide plus 0.1% ethanol (Veh, shade profile). (C) The T cells that had migrated into the lower chambers of the Transwell inserts (5-μm pore size) in response to the CM derived from differently treated PLC/PRF/5 cells. Contoured plots show the staining of the T cells in 1 of 3 chemotaxis assays using 3 healthy males. Bar graphs indicate the average of migrated CD8+ T cells calculated from 3 independent experiments. (D) The chemotaxis effect of chemical AFB1 or testosterone or their combination on T cells. The assay was conducted in parallel with the chemicals and differently treated HepG2.2.15 cell–derived CM using the same males. Flow cytometry profiles show 1 of 3 representative independent male donors and the bar graph shows the average of them. ∗P ≤ .05, ∗∗∗P ≤ .001.
Androgen Signaling Improved the Anti–PD-1 Effect in Murine Models
The earlier-described data indicated that androgen signaling in established HBV-related HCCs contribute to the development of an immunosuppressive microenvironment, which could render the tumor sensitive to anti–PD-1 immunotherapy. We then examined the impact of a favorable androgen pathway on anti–PD-1 treatment effects against hepatoma using syngeneic tumor-bearing mouse models. H22 cells were injected subcutaneously into the left flanks of Balb/c mice, and Hepa1–6 cells were injected into the left flanks or the left lobe of the liver of C57BL/6 mice. The expression of AR, ERα, and ERβ of these cells was confirmed (Figure 8B). To favor the androgen signaling, we administered tamoxifen to the mice to block ER signaling (Figure 16A). In both male and female mice, tamoxifen administration synergistically enhanced the anti–PD-1 effects to eradicate the established tumor quickly in C57BL/6 (Figure 16B) and Balb/c mice (Figure 16C). To mimic human HCC, we injected Hepa1–6 cells into the left lobe of C57BL/6 mouse livers. The synergistic effect of tamoxifen administration on anti–PD-1 was confirmed in both male and female mice (Figure 16D). To further validate the effects of androgen signaling on antitumor immunity, we administered flutamide to tumor-bearing C57BL/6 mice to block the androgen pathway. No significant difference in tumor growth was observed between the female mice that were given flutamide or peanut oil. However, in male mice, tumors grew faster in the flutamide-treated mice than in the vehicle-treated mice (Figure 16E).
Figure 16.

Favorable androgen pathway enhanced anti–PD-1 effects to eradicate established tumors. (A) Experimental scheme. In each C57BL/6 mouse, 5 × 106 Hepa1–6 cells were inoculated subcutaneously, 2 × 106 cells into the left lobe of the liver. In each Balb/c mouse, 5 × 104 H22 cells were inoculated subcutaneously. A palpable tumor mass formed 3 days after cell inoculation, the mice in the tamoxifen group (Tam) received tamoxifen (30 mg/kg) dissolved in peanut oil, the vehicle group (Veh) received the same amounts of peanut oil daily by oral gavage until scarification. Anti–PD-1 (100 μg/mouse) was injected intraperitoneally 3 days after tamoxifen or peanut oil. Control (Ctrl) mice were administered peanut oil and rat IgG at the same time points. C57BL/6 mice that were injected subcutaneously with tumor cells also received flutamide (300 mg/kg, Flu) dissolved in peanut oil without administration of anti–PD-1. (B and C) Shown are 1 of 2 independent experiments. Left: Tumor volumes at different time points of variated groups. Middle and Right: Tumor growth in an individual mouse in the tamoxifen group (Tam + anti–PD-1) and the vehicle group (anti–PD-1 only), respectively. Images are the removed tumors in the mice that were killed on day 20. Scatter plot shows the tumor weights (means ± SEM), each dot represents 1 mouse. P values were calculated by 1-way analysis of variance. (D) Shown are the representative liver images of C57BL/6 mice that were inoculated with Hepa1–6 cells and were killed 20 days after tumor injection. Graphs show the average liver tumor volume (means ± SEM). Each dot represents 1 mouse, n = 5 per group. (E) Hepa1–6 cells were inoculated subcutaneously into C57BL/6 mice that were administered flutamide on day 3. Graphs show the tumor volumes (means ± SEM) at different time points of variated groups, n = 5 per group. Images are the removed tumor in the mice killed on day 15. The handwritten numbers in images indicate mice identification. ∗P ≤ .05, ∗∗P ≤ .01. N, non-neoplastic tissue; Tu, tumor tissue.
Favorable Androgen Signaling Increased T-Cell Infiltration Into the Tumor Mass
We sampled mouse tumors and examined the tumor T-cell infiltration by tamoxifen administration to favor androgen signaling, and to repress androgen signaling by flutamide administration. Without injection of anti–PD-1 antibody (control), male mice showed greater tumor T-cell infiltration than females, especially C57BL/6 mice. With 1 dose of anti–PD-1 injection, favorable androgen signaling significantly increased tumor T-cell infiltration regardless of the mouse strain and sex (Figure 17A). However, when androgen signaling was blocked through flutamide administration, tumor T-cell infiltration was reduced significantly (Figure 17B).
Figure 17.

Favorable androgen signaling enhanced the infiltration of T cells and activation of cGAS-STING in established tumor. (A) Representative images of CD8+ T cells (brown) detected in the removed tumor tissues of the tamoxifen group (Tam), vehicle group (Veh), and control-group (Ctrl). Scale bar: 100 μm. The graphs showed average scores of CD8+ T cells calculated from the group of mice (n = 5). Data present means ± SEM. P values were calculated by 1-way analysis of variance. (B) Representative images of CD8+ T cells (brown) detected in the removed tumor tissues from male C57BL/6 mice received flutamide (Flu), and peanut oil only (Ctrl). Graph shows the CD8+ T-cell scores (means ± SEM) calculated from the group of mice (n = 5). P values were calculated by an unpaired Student t test. (C and D) Shown are the expression levels of genes in the NHEJ and cGAS-STING pathways calculated from 5 mice from a specified group determined by real-time quantitative polymerase chain reaction. Data present means ± SEM. P values were calculated by an unpaired Student t test. IFNB, interferon β.
We further processed the tumor tissues and analyzed the expression levels of the genes involved in NHEJ and cGAS-STING activation. The expression levels of XRCC4, LIG4, and MRE11 in tumors were reduced significantly, while cGAS, STING, and IFNβ were augmented in both male and female mice that received tamoxifen compared with those mice without tamoxifen (Figure 17C). However, the tumors of flutamide-treated male mice showed enhanced expression of XRCC4, LIG4, and MRE11; and decreased expression of cGAS, STING, and IFNβ compared with those of the control male mice (Figure 17D).
Discussion
We examined 101 QD-HCC patient samples that resulted from aflatoxin exposure subsequent to HBV infection.7,8 QD-HCC samples of males showed different features in genome and antitumor immunity from those of females based on mutation frequencies in different genes, dissimilar HBV integration, and varied transcriptional dysfunction pathways. Compared with female QD-HCCs, male QD-HCCs expressed significantly higher levels of AR and aflatoxin metabolism-related genes that could enhance AFB1 genotoxicity,9,10 and lower levels of the genes involved in NHEJ, which is the predominant DSB repair pathway in mammalian cells.21 A signature of repressed antigen-specific antitumor immunity with more CD8+PD1+ T-cell infiltration was detected in male QD-HCCs, related to activation of the cGAS-STING pathway in tumors. Sex chromosomes can encode genes that are associated with distinct cancer vulnerabilities between males and females.2 However, these differences reflected the sum of distinct sex hormone–related transcriptional regulation plus tissue cell–specific regulation based on the results from the Genotype-Tissue Expression project.22 Our analysis of the cultures of HBV-positive HCC cell lines indicated that androgen stimulation increased AHR and CYP1A1, reduced NHEJ factors, and enhanced DNA damage after AFB1 treatment. These alterations potentially promote AFB1 hepatocarcinogenesis. Genotoxic stress causes nucleosome leakage into the cytosol to activate the cGAS–STING pathway, depending on the severity of the DNA damage.24, 25, 26 We confirmed the significant up-regulation of genes related to cGAS-STING activation in HBV-positive HCC cell lines treated with AFB1 plus testosterone compared with those treated with AFB1 alone. In 2 strains of syngeneic tumor-bearing mice, favorable androgen signaling significantly improved the anti–PD-1 effect, probably owing to activation of the cGAS-STING pathway in tumors and increasing T-cell infiltration. HBV DNA integration occurs early in infection, preferentially at the site of DSBs in the cell genome, resulting in genomic and chromosomal instability to promote hepatocarcinogenesis.27 On the other hand, the genomic and chromosomal instability with defects in DNA damage repair render tumors sensitive to immunotherapy, which is associated with the cGAS-STING activation of tumors.26 Ealy studies in hepatic AR knockout mice have shown that AR signaling in hepatocytes increased the cellular oxidative stress and DNA damage to promote hepatocarcinogenesis.28 However, once a hepatoma formed, hepatic AR signaling enhanced anoikis and suppressed migration of HCC cells.29 Androgen signaling in hepatocytes and HCC cells appears to be a double-edged sword.
HBV integration is the most important hepatocarcinogenesis mechanism that alters host gene function and expression.3,6,17,27,30,31 HBV integration was detected in 76.9% of tumor tissues of 426 pathologically characterized HBV-associated HCCs by using sequence-capture HBV probes,17 93.1% of 101 HCC samples by using the capture-next-generation-sequencing platform,6 and in 59.0% of 61 HBV-positive liver cancer specimens based on WGS data.32 HBV integration was detected in 42% of 88 QD-HCC samples and higher TMB. As a genotoxic agent, AFB1 binds to AHR and is bioactivated by CYP450 oxidase family members to form AFB1-DNA adducts, directly inducing various types of DNA damage in hepatocarcinogenesis.9,10 Exposure to AFB1 after HBV infection intensifies somatic mutation to increase HCC risk.7,11 The unique features of QD-HCCs in genetic alteration and transcription dysfunction reflected the combination effects of carcinogenic AFB1 and HBV infection. In addition, we identified 71 genes in QD-HCCs with significantly different mutation frequencies between the 2 sexes. Although none was reported as the HCC driver,13 some were found previously in the other cancers. The roles of these genes in HCC development require further illustration.
Compared with QD-HCC of female samples, AHR and CYP1A1 were increased significantly in the male samples. Meanwhile, lower levels of the NHEJ genes were detected in the male samples than in the female samples. The sex differences of these genes also were observed in the HBV-related TCGA-HCCs. In the cultures of 2 HBV-positive HCC cell lines, the expression levels of AHR and CYP1A1 were increased significantly, NHEJ factors were repressed significantly, and more γH2AX was generated in the cells treated with AFB1 plus testosterone than in those treated with AFB1 alone, depending on the concentration of testosterone and AFB1. Our current results indicate that androgen signaling in hepatic cells after HBV infection could repress the DSB repair in the context of genotoxic stress, which is probably another mechanism of increased HCC risk in HBV-infected males. AR is a ligand-dependent transcription factor that regulates target gene expression through binding to AREs in the presence of androgens. Indeed, analysis of web-based MEME Suite23 indicated that the AHR, CYP1A1, and some NHEJ factors, including XRCC4, LIG4, and MRE11, harbored AREs in their promotor areas, suggesting that AR might regulate these genes directly. AFB1 was found to trigger the nuclear translocation by directly binding to the N-terminus of AHR, which is a well-known ligand-activated transcription factor mediating the genotoxicity and tumor-promoting properties of the carcinogen 2,3,7,8-tetra-chlorodibenzo-p-dioxin, an industrial component.9 Crosstalk between AHR and androgen has been confirmed.33,34 One study using HepG2 transfected with a plasmid that encodes luciferase under the control of consensus dioxin response element showed that androgen treatment produces dioxin-like effects through AHR to increase the expression of CYP1A1 both in transcriptional and translational levels, but no significantly affected cell viability.33 One study on rat granulosa cells reported that testosterone stimulated the expression of AHR and its interaction with AR, promoting the recruitment of the AR/AHR complex to the promoter of the liver receptor homolog 1,34 which is recognized to coordinate a multitude of hepatic metabolic processes.35 Analysis of AR chromatin immunoprecipitation sequencing has recognized the DNA repair genes as AR target genes.36 RNA-seq data from multiple prostate cancer cell lines found that androgen signaling represses MRE11 transcription.37 The more severe DNA damage after AFB1 exposure in the presence of androgen might be owing to the dual-regulation by AR and AHR. However, the molecular regulation by androgen in the context of genotoxic reagents needs to be clarified further.
Based on a meta-analysis that included 20 randomized controlled trials, a significant sex difference in responses to immune checkpoint inhibitors was observed between 7646 male and 3705 female advanced/metastatic cancer patients, with melanoma and non–small-cell lung cancer as the most common types.12 The current clinical trials on advanced HCC patients did not report the sex difference in the response to immunotherapy,38,39 probably owing to the small number of female HCC patients who received the immunotherapy. HCCs, resulting from different etiologies, differ in the genetic alterations and transcriptomic dysregulation that shape tumor development and microenvironment formation, and influence antitumor responses.40 Nivolumab (antibody against PD-1) for advanced HCCs results in the objective response rate of 15% (21 of 139) in male patients and 12% (5 of 43) in females, regardless of the etiology. However, in the Asian cohort, in which HCC was related mainly to HBV infection, the objective response rate was 17% (11 of 65) in male patients and was 10% (2 of 20) in female patients.39 We observed the unique genetic alterations and transcriptomic dysfunction pathways in HBV-related HCCs exposed to AFB1. The synergistic genotoxic effects of androgen and AFB1 on HBV-positive cells were validated in in vitro cultures. We used syngeneic graft mouse models in which inoculated tumor cells were derived from mice with the same genetic background. The tumor-bearing inbred mice are identical except for the sex chromosomes and different levels of sex hormones. The authentic effects of androgen-receptor signaling on established tumors and their sensitivity to anti–PD-1 immunotherapy were confirmed by blocking the estrogen signaling via tamoxifen administration to favor the androgen pathway. Our results indicated that androgen signaling repressed NHEJ factors when exposed to DNA damage reagents. The improved anti–PD-1 effects are related to activation of the cGAS-STING pathway in HCC cells by increased nuclear DNA leakage into the cytosol. In addition, to induce IRF3 phosphorylation, which drives the expression of a plethora of antiviral genes (including IFNB), activated STING also performs additional functions that can be mechanistically and functionally separated from IRF3 activation.41 Based on the cell culture results, IFN-I–related soluble factors generated from the HBV-positive HCC cell lines that are triggered by cGAS-STING signaling might be one class of the mediators. However, the specified products need to be recognized in the future.
Previous randomized studies have shown that tamoxifen imparts no antitumoral effect and no survival benefit in HCC patients.42 One study observed a significantly higher risk of HCC-related death treated with a high dose of tamoxifen,43 indicating that tamoxifen has a limited antitumor effect against HCCs. A previous study of treatment of STING-/- mice and wild-type mice with 7,12-dimethylbenz(a)anthracene showed that chronic exposure to chemical carcinogens induces DNA adduct formation and breakage, causing nucleosome leakage into the cytoplasm to activate the cytosolic cGAS-STING.25 We observed repressed antitumor immunity in the hepatectomy samples from patients with aflatoxin exposure subsequent to HBV infection and these events were confirmed in HBV-positive HCC cell lines. Some drugs used in cancer chemotherapy, such as cisplatin, cause the generation of DNA lesions that may exert some of their antitumor activity by triggering STING-dependent innate immune activity. One study in tumor-bearing mice has shown that cisplatin treatment facilitates tumor clearance by activation of the STING pathway to enhance the effect of immune checkpoint inhibitors.44 It is tempting to increase the nuclear DNA leakage into the cytosol to activate cGAS-STING by favorable androgen signaling for stimulating anti-tumor–adaptive immune responses. Indeed, previous clinical studies have shown the benefit of radiation therapy in the treatment of male HCC patients, but not female patients.45 Thus, therapeutics that favor androgen signaling and/or blocking estrogen signaling may provide a new strategy to improve the efficacy of immune checkpoint inhibitors against HCC in combination with radiotherapy or chemotherapy that induced DNA damage. The adjuvant effects of tamoxifen for favorable androgen signaling to boost the anti–PD-1 effect in HCC patients needs future study in a prospective HCC cohort.
Materials and Methods
Ethics Statement
Study protocols for using human samples were approved by the Institutional Review Board of the National Cancer Center, Cancer Hospital, protocols involving mice were approved by the Institutional Animal Care and Use Committee at the National Cancer Center, Cancer Hospital, Chinese Academy of Medical Sciences (Beijing, China). All authors had access to the study data and reviewed and approved the final manuscript.
Patients and Samples
We obtained the tumor and matched non-neoplastic liver tissues from 101 QD-HCC patients (47 males, 54 females) who received primary hepatectomy without further systemic or radiation therapy. The patients were all Qidong local residents, and were followed up for 5 years by May 31, 2019. Aflatoxin exposure was confirmed by recording aflatoxin M1 in their urine 3–18 years before the HCC diagnosis (Table 1). From TCGA database, 119 HBV-related HCCs (TCGA-HCCs) were recognized. Six cases were excluded (IDs: A3M9, A3MB, AA8I, A8HT, A7XP, and A8HU) that harbored mutational signatures of aflatoxin exposure,11 therefore we recruited the 113 cases of TCGA-HCCs (73 males, 40 females) for analysis (Table 2).
Sequencing and Data Analysis
Whole genome sequencing was conducted in 88 QD-HCC samples, and whole exome sequencing in 13 samples. Somatic mutations were analyzed as we described previously.11 Common single-nucleotide polymorphisms and single-nucleotide polymorphism frequencies of greater than 1% of 1000 genomes were filtered from the somatic mutation list. To avoid false-positive calls, the quality of mutations with significantly different mutation frequencies between male HCCs and female HCCs was reviewed manually with Integrated Genomics Viewer (Cambridge, MA). HBV integration was analyzed in 88 cases with WGS data, also following the same procedures as we described previously.11 Integration sites were mapped to human genome 19 and the genes with their transcription start sites at a distance of 1 Mb or less were considered as the ITGs.20 The Database for Annotation, Visualization, and Integrated Discovery (DAVID) was used for gene functional annotation analysis, choosing the categories of Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathways as background databases. The results were chosen with a Benjamini adjusted P value of less than .05.
RNA-seq was performed on 87 QD-HCC samples. Illumina (San Diego, CA) Next-seq 500 was used to assess the library quality, followed by data acquisition on the Illumina Hi-seq 4000. Transcription levels of the genes were analyzed with Hisat, StringTie, and Ballgown (Baltimore, MD). Gene expression in tumor tissues was normalized to that in matched non-neoplastic liver samples. Gene set variation analysis was used to estimate the variation of pathway activities.46
Immunohistochemistry
Immunohistochemistry was conducted in paraffin-embedded tissues with standard laboratory protocols. The stained sections were scanned and analyzed using Aperio ScanScope software (Aperio Technologies, Vista, CA). A positive score of AHR, CYP1A1, and B7-H2 in HCC samples was quantified based on the percentage area and intensity of positive staining as reported.47 The CD8+ T-cell score and the PD-1 score was expressed as the average of positive cell numbers in each section from 5 independent fields under 400×.48
Cell Cultures
Human HCC cell line PLC/PRF/5, which contains HBV fragments and secretes envelope proteins, and murine hepatoma cell line H22 were purchased from the National Infrastructure of Cell Line Resource (Beijing, China). The murine hepatoma Hepa1–6 cell line was purchased from American Type Culture Collection (Manassas, VA). The HepG2.2.15 cell line is modified HepG2 with an integrated HBV genome secreting the HBV envelope protein that was kindly provided by Professor Yong Liao from Chongqing Medical University (Chongqing, China). The cells were cultured following the manufacturer’s instructions. Expression of AR and ER in these cell lines was determined by quantitative real-time polymerase chain reaction and immunoblotting. The CC50 and gene expression in cultured cells were determined after addition of different concentrations of AFB1, testosterone, or 17β-estradiol following the protocol of our previous report.9 All of the reagents were purchased from Sigma-Aldrich (St. Louis, MO). The primers, antibodies, and reagents are listed in Tables 5 and 6.
Table 5.
List of Primers Used in Quantitative Real-Time Polymerase Chain Reaction
| Gene | Orientation | Sequence, 5' to 3' |
|---|---|---|
| Human | ||
| AHR | Forward | ACATCACCTACGCCAGTCG |
| Reverse | CGCTTGGAAGGATTTGACTTGA | |
| CYP1A1 | Forward | TTCCGACACTCTTCCTTCGT |
| Reverse | ATGGTTAGCCCATAGATGGG | |
| XRCC4 | Forward | ATGTTGGTGAACTGAGAAAAGCA |
| Reverse | GCAATGGTGTCCAAGCAATAAC | |
| LIG4 | Forward | AGCAAAAGTGGCTTATACGGATG |
| Reverse | TGAGTCCTACAGAAGGATCATGC | |
| MRE11 | Forward | ATCGGCCTGTCCAGTTTGAAA |
| Reverse | TGCCATCTTGATAGTTCACCCAT | |
| STING | Forward | CCAGAGCACACTCTCCGGTA |
| Reverse | CGCATTTGGGAGGGAGTAGTA | |
| IRF3 | Forward | AGAGGCTCGTGATGGTCAAG |
| Reverse | AGGTCCACAGTATTCTCCAGG | |
| IFNB1 | Forward | ATGACCAACAAGTGTCTCCTCC |
| Reverse | GGAATCCAAGCAAGTTGTAGCTC | |
| B7-H2 | Forward | GCAGCCTTCGAGCTGATACTC |
| Reverse | GTTTTCGACTCACTGGTTTGC | |
| B7-H3 | Forward | CTCTGCCTTCTCACCTCTTTG |
| Reverse | CCTTGAGGGAGGAACTTTATC | |
| AR | Forward | CCAGGGACCATGTTTTGCC |
| Reverse | CGAAGACGACAAGATGGACAA | |
| ESR1 | Forward | CCCACTCAACAGCGTGTCTC |
| Reverse | CGTCGATTATCTGAATTTGGCCT | |
| ESR2 | Forward | AGCACGGCTCCATATACATACC |
| Reverse | TGGACCACTAAAGGAGAAAGGT | |
| GAPDH | Forward | GGAGCGAGATCCCTCCAAAAT |
| Reverse | GGCTGTTGTCATACTTCTCATGG | |
| Mice | ||
| AR | Forward | TCCAAGACCTATCGAGGAGCG |
| Reverse | GTGGGCTTGAGGAGAACCAT | |
| ESR1 | Forward | CCCGCCTTCTACAGGTCTAAT |
| Reverse | CTTTCTCGTTACTGCTGGACAG | |
| ESR2 | Forward | CTGTGATGAACTACAGTGTTCCC |
| Reverse | CACATTTGGGCTTGCAGTCTG | |
| XRCC4 | Forward | CTGGAGGAGAGTACCAAACCT |
| Reverse | CTGGGGTAGTGAAGAGGCAAG | |
| LIG4 | Forward | ATGGCTTCCTCACAAACTTCAC |
| Reverse | TTTCTGCACGGTCTTTACCTTT | |
| MRE11A | Forward | TCCTGGTTGCCACTGATATTCA |
| Reverse | CCATCCTGGTAGTTCACCCA | |
| STING | Forward | TCGCACGAACTTGGACTACTG |
| Reverse | CCAACTGAGGTATATGTCAGCAG | |
| CGAS | Forward | GAGGCGCGGAAAGTCGTAA |
| Reverse | TTGTCCGGTTCCTTCCTGGA | |
| IFNB1 | Forward | CAGCTCCAAGAAAGGACGAAC |
| Reverse | GGCAGTGTAACTCTTCTGCAT | |
| GAPDH | Forward | CTCCCACTCTTCCACCTTCG |
| Reverse | TAGGGCCTCTCTTGCTCAGT | |
Table 6.
List of Antibodies and Reagents
| Antibodies or reagents | Application | Source | Catalog number | Dilution |
|---|---|---|---|---|
| Antigen-specific antibody | ||||
| Human AHR | IHC | Proteintech (Rosemont, IL) | 17840-1-AP | 1:200 |
| Human CYP1A1 | IHC | Proteintech (Rosemont, IL) | 13241-1-AP | 1:200 |
| AFB1-DNA adduct | IHC | Novus (Novus Littleton, CO) | NB600-443 | 1:100 |
| Human CD8 | IHC | ZSGB-BIO (Beijing, CN) | ZA-0508 | 1:100 |
| Human PD-1 | IHC | Proteintech (Rosemont, IL) | 18106-1-AP | 1:200 |
| Human B7-H2 | IHC | Proteintech (Rosemont, IL) | 14922-1-AP | 1:200 |
| Mouse CD8 | IHC | Santa Cruz (Dallas, TX) | sc-53063 | 1:100 |
| dsDNA | IF | Abcam (Cambridge, UK) | Ab273137 | 1:500 |
| Human γH2AX | WB | BioLegend (San Diego, CA) | 613401 | 1:1000 |
| Human STING | WB | Cell Signaling Technology (Danvers, MA) | 13647T | 1:1000 |
| Human pSTING(S366) | WB | Cell Signaling Technology (Danvers, MA) | 50907T | 1:1000 |
| Human cGAS | WB | Cell Signaling Technology (Danvers, MA) | 15102T | 1:1000 |
| Human IRF3 | WB | Cell Signaling Technology (Danvers, MA) | 11904T | 1:1000 |
| Human pIRF3(S396) | WB | Cell Signaling Technology (Danvers, MA) | 29047T | 1:1000 |
| Human/mouse AR | WB | Abcam (Cambridge, UK) | Ab108341 | 1:1000 |
| Human/mouse ERα | WB | Cell Signaling Technology (Danvers, MA) | 8644T | 1:1000 |
| Human/mouse actin | WB | Proteintech (Rosemont, IL) | 66009-1-lg | 1:2000 |
| Human B7-H2 | FC | BioLegend (San Diego, CA) | 309407 | |
| Human B7-H3 | FC | BioLegend (San Diego, CA) | 351009 | |
| Human CD80 | FC | BioLegend (San Diego, CA) | 305219 | |
| Human CD86 | FC | BioLegend (San Diego, CA) | 305419 | |
| Human PD-1 | FC | BioLegend (San Diego, CA) | 329908 | |
| Human CD8 | FC | BioLegend (San Diego, CA) | 300906 | |
| Mouse PD-1 | Animal | BioXcell (West Lebanon, NH) | BE0273 | |
| Reagents | ||||
| Aflatoxin B1 | Cell culture | Sigma (Burlington, MA) | A6636 | |
| Testosterone | Cell culture | Sigma (Burlington, MA) | A8380 | |
| 17β-estradiol | Cell culture | Sigma (Burlington, MA) | E8875 | |
| Tamoxifen | Animal | MedChemExpress (Monmouth Junction, NJ) | HY-13757A | |
| Flutamide | Animal | MedChemExpress (Monmouth Junction, NJ) | HY-B0022 | |
dsDNA, double-strand DNA; FC, flow cytometry; IF, immunofluorescence; IHC, immunohistochemistry; WB, Western blot.
Immunofluorescence, Chemotaxis, and Flow Cytometry
Immunofluorescence microscopy was performed as reported25 to analyze nuclear DNA leakage into the cytosol of differently treated human HCC cells. Chemotaxis was conducted to examine the T cells in response to the soluble factors secreted from differently treated HCC cells to mimic the tumor infiltration of immune cells.49 Briefly, peripheral blood mononuclear cells from healthy males were isolated. Following the manufacturer’s instructions, Dynabeads Human T-Activator CD3/CD28 (Gibco, Fisher Scientific, Waltham, MA) was added to the cell suspension for 24 hours. The conditioned medium from differently treated HepG2.2.15 cells or PLC/PRF/5 cells was mixed with same volume of completed RPMI-1640, and 600 μL was added to each well. In the upper chamber of a Transwell insert (5-μm pore size), 1 × 106 activated cells in 100 μL were added, and cultured at 37°C for 2 hours. The cells that had migrated into the lower chambers were collected, counted, and analyzed by flow cytometry as we previously reported.49 To determine the expression of B7 molecules on HCC cells, single-cell suspension at a concentration of 1 × 106 cells/mL was prepared from differently treated HCC cells, and fluorescein-conjugated antibodies were added. Data were acquired in a LSR-II (BD, San Diego, CA) and analyzed using FlowJo software (Tree Star, Inc, Ashland, OR).
Animal Experiments
We used syngeneic graft mouse models in which inoculated tumor cells were derived from mice with the same genetic background. The tumor-bearing mice were an inbred stain that is identical except for the sex chromosomes and different levels of sex hormone. C57BL/6 mice and Balb/c mice were purchased from Huafukang Biological Technology (Beijing, China) and used at 10 weeks of age. To observe tumor growth at different time points, 5 × 106 Hepa1–6 cells were injected subcutaneously into the left flank of syngeneic C57BL/6 mice, and 5 × 104 H22 cells into Balb/c mice. When tumor mass formed, the androgen signaling was favored by administering 30 mg/kg tamoxifen50 or blocked by 300 mg/kg flutamide dissolved in peanut oil to each mouse via oral gavage daily. In the vehicle group, mice were given the same amount of peanut oil. After the administration of tamoxifen or peanut oil for 3 days, each mouse in the tamoxifen group and vehicle group was injected intraperitoneally with 1 dose of anti–PD-1 antibody (100 μg/mouse). In the control group, mice received the same amount of peanut oil and 100 μg/mouse of rat IgG at the same time points. The tumor was measured every 2–3 days, and the volume was calculated with the following formula: (length × width2)/2. To observe tumor development in liver mimicking human HCCs, 2 × 106 Hepa1–6 cells were injected into the left lobe of C57BL/6 mice livers. The mice were treated in the same way as they were injected subcutaneously with tumor cells. To examine the treatment effects, tumor tissues from each mouse were divided into 2 parts. One part was embedded in paraffin for regular histology and for analysis of CD8+ T-cell infiltration after immunohistochemistry staining. Another part was processed for quantification of the gene expression by real-time quantitative polymerase chain reaction using the primers listed in Table 5.
Statistical Analysis
Statistical analyses were performed using R version 3.4.2 (Vienna, Austria). The chi-squared test and the Fisher exact test were used to compare categoric variables. Continuous variables were compared with an unpaired Student t test between 2 groups, and by 1-way analysis of variance between more than 2 groups. The differences of discrete variables were compared using the Mann–Whitney test. A P value less than .05 (2-tailed) was considered statistically significant.
CRediT Authorship Contributions
Chungui Xu, PhD (Data curation: Equal; Investigation: Equal; Methodology: Equal; Validation: Equal; Writing – original draft: Equal)
Shaoyan Cheng, PhD (Data curation: Equal; Investigation: Equal; Methodology: Equal; Validation: Equal; Writing – original draft: Equal)
Kun Chen, PhD (Data curation: Equal; Methodology: Lead; Validation: Equal; Writing – original draft: Equal)
Qianqian Song, PhD (Data curation: Equal; Validation: Supporting)
Chang Liu, MD (Data curation: Supporting; Methodology: Supporting; Validation: Supporting)
Chunsun Fan, MD; PhD (Data curation: Supporting; Resources: Equal; Validation: Supporting)
Ruochan Zhang, PhD candidate (Methodology: Supporting; Validation: Supporting)
Qing Zhu, PhD (Methodology: Supporting; Validation: Supporting)
Zhiyuan Wu, BSci (Investigation: Supporting; Methodology: Supporting)
Yuting Wang, PhD (Data curation: Supporting; Investigation: Supporting; Methodology: Supporting; Resources: Supporting; Validation: Supporting)
Jian Fan, MD (Resources: Equal; Validation: Supporting)
Hongwei Zheng, BSci (Resources: Supporting; Validation: Supporting)
Lingling Lu, MSci (Resources: Supporting; Validation: Supporting)
Taoyang Chen, MD (Resources: Supporting; Validation: Supporting)
Hong Zhao, MD (Resources: Lead; Writing – original draft: Supporting; Writing – review & editing: Supporting)
Yuchen Jiao, MD; PhD (Conceptualization: Equal; Data curation: Equal; Formal analysis: Lead; Investigation: Equal; Methodology: Equal; Supervision: Equal; Writing – original draft: Equal; Writing – review & editing: Equal)
Chunfeng Qu, MD, PhD (Conceptualization: Lead; Data curation: Equal; Formal analysis: Equal; Funding acquisition: Lead; Investigation: Lead; Supervision: Lead; Writing – original draft: Lead; Writing – review & editing: Lead)
Footnotes
Conflicts of interest The authors disclose no conflicts.
Funding Supported by the National Natural Science Foundation Fund of China (81972628), Innovation Fund for Medical Sciences of Chinese Academy of Medical Sciences (2021-I2M-1-066), State Key Project for Infectious Diseases (2017ZX10201201006), and the PhD supporting program of Peking Union Medical College (2019-1001-01). The sponsors of the study had no role in the study design, data collection, data analysis, data interpretation, or writing of the manuscript.
Contributor Information
Hong Zhao, Email: zhaohong@cicams.ac.cn.
Yuchen Jiao, Email: jiaoyuchen@cicams.ac.cn.
Chunfeng Qu, Email: quchf@cicams.ac.cn.
References
- 1.Kulik L., El-Serag H.B. Epidemiology and management of hepatocellular carcinoma. Gastroenterology. 2019;156:477–491. doi: 10.1053/j.gastro.2018.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haupt S., Caramia F., Klein S.L., Rubin J.B., Haupt Y. Sex disparities matter in cancer development and therapy. Nat Rev Cancer. 2021;21:393–407. doi: 10.1038/s41568-021-00348-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu M.H., Ma W.L., Hsu C.L., Chen Y.L., Ou J.H., Ryan C.K., Hung Y.C., Yeh S., Chang C. Androgen receptor promotes hepatitis B virus-induced hepatocarcinogenesis through modulation of hepatitis B virus RNA transcription. Sci Transl Med. 2010;2:32ra5. doi: 10.1126/scitranslmed.3001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang S.H., Yeh S.H., Lin W.H., Yeh K.H., Yuan Q., Xia N.S., Chen D.S., Chen P.J. Estrogen receptor alpha represses transcription of HBV genes via interaction with hepatocyte nuclear factor 4alpha. Gastroenterology. 2012;142:989–998 e4. doi: 10.1053/j.gastro.2011.12.045. [DOI] [PubMed] [Google Scholar]
- 5.Nault J.C., Martin Y., Caruso S., Hirsch T.Z., Bayard Q., Calderaro J., Charpy C., Copie-Bergman C., Ziol M., Bioulac-Sage P., Couchy G., Blanc J.F., Nahon P., Amaddeo G., Ganne-Carrie N., Morcrette G., Chiche L., Duvoux C., Faivre S., Laurent A., Imbeaud S., Rebouissou S., Llovet J.M., Seror O., Letouze E., Zucman-Rossi J. Clinical impact of genomic diversity from early to advanced hepatocellular carcinoma. Hepatology. 2020;71:164–182. doi: 10.1002/hep.30811. [DOI] [PubMed] [Google Scholar]
- 6.Jeschke U., Zhang X., Kuhn C., Jalaguier S., Colinge J., Pfender K., Mayr D., Ditsch N., Harbeck N., Mahner S., Sixou S., Cavaillès V. The prognostic impact of the aryl hydrocarbon receptor (AhR) in primary breast cancer depends on the lymph node status. Int J Mol Sci. 2019;20:1016. doi: 10.3390/ijms20051016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ming L., Thorgeirsson S.S., Gail M.H., Lu P., Harris C.C., Wang N., Shao Y., Wu Z., Liu G., Wang X., Sun Z. Dominant role of hepatitis B virus and cofactor role of aflatoxin in hepatocarcinogenesis in Qidong, China. Hepatology. 2002;36:1214–1220. doi: 10.1053/jhep.2002.36366. [DOI] [PubMed] [Google Scholar]
- 8.Kensler T.W., Qian G.S., Chen J.G., Groopman J.D. Translational strategies for cancer prevention in liver. Nat Rev Cancer. 2003;3:321–329. doi: 10.1038/nrc1076. [DOI] [PubMed] [Google Scholar]
- 9.Zhu Q., Ma Y.R., Liang J.B., Wei Z.W., Li M., Zhang Y., Liu M., He H., Qu C., Cai J., Wang X., Zeng Y.X., Jiao Y. AHR mediates the aflatoxin B1 toxicity associated with hepatocellular carcinoma. Signal Transduct Target Ther. 2021;6:299. doi: 10.1038/s41392-021-00713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng J., Zhao L., Zhang N.Y., Karrow N.A., Krumm C.S., Qi D.S., Sun L.H. Aflatoxin B(1) metabolism: regulation by phase I and II metabolizing enzymes and chemoprotective agents. Mutat Res. 2018;778:79–89. doi: 10.1016/j.mrrev.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Zhang W., He H., Zang M., Wu Q., Zhao H., Lu L.L., Ma P., Zheng H., Wang N., Zhang Y., He S., Chen X., Wu Z., Wang X., Cai J., Liu Z., Sun Z., Zeng Y.X., Qu C., Jiao Y. Genetic features of aflatoxin-associated hepatocellular carcinoma. Gastroenterology. 2017;153:249–262. doi: 10.1053/j.gastro.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 12.Conforti F., Pala L., Bagnardi V., De Pas T., Martinetti M., Viale G., Gelber R.D., Goldhirsch A. Cancer immunotherapy efficacy and patients' sex: a systematic review and meta-analysis. Lancet Oncol. 2018;19:737–746. doi: 10.1016/S1470-2045(18)30261-4. [DOI] [PubMed] [Google Scholar]
- 13.Martínez-Jiménez F., Muiños F., Sentís I., Deu-Pons J., Reyes-Salazar I., Arnedo-Pac C., Mularoni L., Pich O., Bonet J., Kranas H., Gonzalez-Perez A., Lopez-Bigas N. A compendium of mutational cancer driver genes. Nat Rev Cancer. 2020;20:555–572. doi: 10.1038/s41568-020-0290-x. [DOI] [PubMed] [Google Scholar]
- 14.Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6 doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E., Antipin Y., Reva B., Goldberg A.P., Sander C., Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chayanupatkul M., Omino R., Mittal S., Kramer J.R., Richardson P., Thrift A.P., El-Serag H.B., Kanwal F. Hepatocellular carcinoma in the absence of cirrhosis in patients with chronic hepatitis B virus infection. J Hepatol. 2017;66:355–362. doi: 10.1016/j.jhep.2016.09.013. [DOI] [PubMed] [Google Scholar]
- 17.Zhao L.H., Liu X., Yan H.X., Li W.Y., Zeng X., Yang Y., Zhao J., Liu S.P., Zhuang X.H., Lin C., Qin C.J., Zhao Y., Pan Z.Y., Huang G., Liu H., Zhang J., Wang R.Y., Yang Y., Wen W., Lv G.S., Zhang H.L., Wu H., Huang S., Wang M.D., Tang L., Cao H.Z., Wang L., Lee T.L., Jiang H., Tan Y.X., Yuan S.X., Hou G.J., Tao Q.F., Xu Q.G., Zhang X.Q., Wu M.C., Xu X., Wang J., Yang H.M., Zhou W.P., Wang H.Y. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat Commun. 2016;7 doi: 10.1038/ncomms12992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee J., Kim D.H., Lee S., Yang Q.H., Lee D.K., Lee S.K., Roeder R.G., Lee J.W. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc Natl Acad Sci U S A. 2009;106:8513–8518. doi: 10.1073/pnas.0902873106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blazer L.L., Lima-Fernandes E., Gibson E., Eram M.S., Loppnau P., Arrowsmith C.H., Schapira M., Vedadi M.P.R. Domain-containing protein 7 (PRDM7) is a histone 3 lysine 4 trimethyltransferase. J Biol Chem. 2016;291:13509–13519. doi: 10.1074/jbc.M116.721472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazur P.K., Reynoird N., Khatri P., Jansen P.W., Wilkinson A.W., Liu S., Barbash O., Van Aller G.S., Huddleston M., Dhanak D., Tummino P.J., Kruger R.G., Garcia B.A., Butte A.J., Vermeulen M., Sage J., Gozani O. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature. 2014;510:283–287. doi: 10.1038/nature13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scully R., Panday A., Elango R., Willis N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 2019;20:698–714. doi: 10.1038/s41580-019-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oliva M., Munoz-Aguirre M., Kim-Hellmuth S., Wucher V., Gewirtz A.D.H., Cotter D.J., Parsana P., Kasela S., Balliu B., Vinuela A., Castel S.E., Mohammadi P., Aguet F., Zou Y., Khramtsova E.A., Skol A.D., Garrido-Martin D., Reverter F., Brown A., Evans P., Gamazon E.R., Payne A., Bonazzola R., Barbeira A.N., Hamel A.R., Martinez-Perez A., Soria J.M., Consortium G.T., Pierce B.L., Stephens M., Eskin E., Dermitzakis E.T., Segre A.V., Im H.K., Engelhardt B.E., Ardlie K.G., Montgomery S.B., Battle A.J., Lappalainen T., Guigo R., Stranger B.E. The impact of sex on gene expression across human tissues. Science. 2020;369:6509. doi: 10.1126/science.aba3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bailey T.L., Johnson J., Grant C.E., Noble W.S. The MEME suite. Nucleic Acids Res. 2015;43:W39–W49. doi: 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun L., Wu J., Du F., Chen X., Chen Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahn J., Xia T., Konno H., Konno K., Ruiz P., Barber G.N. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun. 2014;5:5166. doi: 10.1038/ncomms6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li T., Chen Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215:1287–1299. doi: 10.1084/jem.20180139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tu T., Budzinska M.A., Shackel N.A., Urban S. HBV DNA integration: molecular mechanisms and clinical implications. Viruses. 2017;9:75. doi: 10.3390/v9040075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma W.L., Hsu C.L., Wu M.H., Wu C.T., Wu C.C., Lai J.J., Jou Y.S., Chen C.W., Yeh S., Chang C. Androgen receptor is a new potential therapeutic target for the treatment of hepatocellular carcinoma. Gastroenterology. 2008;135:947–955. doi: 10.1053/j.gastro.2008.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma W.L., Hsu C.L., Yeh C.C., Wu M.H., Huang C.K., Jeng L.B., Hung Y.C., Lin T.Y., Yeh S., Chang C. Hepatic androgen receptor suppresses hepatocellular carcinoma metastasis through modulation of cell migration and anoikis. Hepatology. 2012;56:176–185. doi: 10.1002/hep.25644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiu Y.T., Wong J.K., Choi S.W., Sze K.M., Ho D.W., Chan L.K., Lee J.M., Man K., Cherny S., Yang W.L., Wong C.M., Sham P.C., Ng I.O. Novel pre-mRNA splicing of intronically integrated HBV generates oncogenic chimera in hepatocellular carcinoma. J Hepatol. 2016;64:1256–1264. doi: 10.1016/j.jhep.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 31.Li X., Zhang J., Yang Z., Kang J., Jiang S., Zhang T., Chen T., Li M., Lv Q., Chen X., McCrae M.A., Zhuang H., Lu F. The function of targeted host genes determines the oncogenicity of HBV integration in hepatocellular carcinoma. J Hepatol. 2014;60:975–984. doi: 10.1016/j.jhep.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 32.Zapatka M., Borozan I., Brewer D.S., Iskar M., Grundhoff A., Alawi M., Desai N., Sultmann H., Moch H., Pathogens P., Cooper C.S., Eils R., Ferretti V., Lichter P., Consortium P. The landscape of viral associations in human cancers. Nat Genet. 2020;52:320–330. doi: 10.1038/s41588-019-0558-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moon H.Y., Kim S.H., Ryu S.H., Suh P.G. The androgenic anabolic steroid tetrahydrogestrinone produces dioxin-like effects via the aryl hydrocarbon receptor. Toxicol In Vitro. 2012;26:1129–1133. doi: 10.1016/j.tiv.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 34.Wu Y., Baumgarten S.C., Zhou P., Stocco C. Testosterone-dependent interaction between androgen receptor and aryl hydrocarbon receptor induces liver receptor homolog 1 expression in rat granulosa cells. Mol Cell Biol. 2013;33:2817–2828. doi: 10.1128/MCB.00011-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Y., Demagny H., Schoonjans K. Emerging functions of the nuclear receptor LRH-1 in liver physiology and pathology. Biochim Biophys Acta Mol Basis Dis. 2021;1867 doi: 10.1016/j.bbadis.2021.166145. [DOI] [PubMed] [Google Scholar]
- 36.Polkinghorn W.R., Parker J.S., Lee M.X., Kass E.M., Spratt D.E., Iaquinta P.J., Arora V.K., Yen W.F., Cai L., Zheng D., Carver B.S., Chen Y., Watson P.A., Shah N.P., Fujisawa S., Goglia A.G., Gopalan A., Hieronymus H., Wongvipat J., Scardino P.T., Zelefsky M.J., Jasin M., Chaudhuri J., Powell S.N., Sawyers C.L. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013;3:1245–1253. doi: 10.1158/2159-8290.CD-13-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jividen K., Kedzierska K.Z., Yang C.S., Szlachta K., Ratan A., Paschal B.M. Genomic analysis of DNA repair genes and androgen signaling in prostate cancer. BMC Cancer. 2018;18:960. doi: 10.1186/s12885-018-4848-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu A.X., Finn R.S., Edeline J., Cattan S., Ogasawara S., Palmer D., Verslype C., Zagonel V., Fartoux L., Vogel A., Sarker D., Verset G., Chan S.L., Knox J., Daniele B., Webber A.L., Ebbinghaus S.W., Ma J., Siegel A.B., Cheng A.L., Kudo M. KEYNOTE-224 Investigators. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19:940–952. doi: 10.1016/S1470-2045(18)30351-6. [DOI] [PubMed] [Google Scholar]
- 39.Yau T., Hsu C., Kim T.Y., Choo S.P., Kang Y.K., Hou M.M., Numata K., Yeo W., Chopra A., Ikeda M., Kuromatsu R., Moriguchi M., Chao Y., Zhao H., Anderson J., Cruz C.D., Kudo M. Nivolumab in advanced hepatocellular carcinoma: sorafenib-experienced Asian cohort analysis. J Hepatol. 2019;71:543–552. doi: 10.1016/j.jhep.2019.05.014. [DOI] [PubMed] [Google Scholar]
- 40.Rebouissou S., Nault J.C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J Hepatol. 2020;72:215–229. doi: 10.1016/j.jhep.2019.08.017. [DOI] [PubMed] [Google Scholar]
- 41.Hopfner K.P., Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol. 2020;21:501–521. doi: 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- 42.Llovet J.M., Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: chemoembolization improves survival. Hepatology. 2003;37:429–442. doi: 10.1053/jhep.2003.50047. [DOI] [PubMed] [Google Scholar]
- 43.Chow P.K., Tai B.C., Tan C.K., Machin D., Win K.M., Johnson P.J., Soo K.C. Asian-Pacific Hepatocellular Carcinoma Trials Group. High-dose tamoxifen in the treatment of inoperable hepatocellular carcinoma: a multicenter randomized controlled trial. Hepatology. 2002;36:1221–1226. doi: 10.1053/jhep.2002.36824. [DOI] [PubMed] [Google Scholar]
- 44.Lee S.E., Jang G.Y., Lee J.W., Park S.H., Han H.D., Park Y.M., Kang T.H. Improvement of STING-mediated cancer immunotherapy using immune checkpoint inhibitors as a game-changer. Cancer Immunol Immunother. 2022;71:3029–3042. doi: 10.1007/s00262-022-03220-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salem R., Lewandowski R.J., Mulcahy M.F., Riaz A., Ryu R.K., Ibrahim S., Atassi B., Baker T., Gates V., Miller F.H., Sato K.T., Wang E., Gupta R., Benson A.B., Newman S.B., Omary R.A., Abecassis M., Kulik L. Radioembolization for hepatocellular carcinoma using Yttrium-90 microspheres: a comprehensive report of long-term outcomes. Gastroenterology. 2010;138:52–64. doi: 10.1053/j.gastro.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 46.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:1471–2105. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qu C., Zheng D., Li S., Liu Y., Lidofsky A., Holmes J.A., Chen J., He L., Wei L., Liao Y., Yuan H., Jin Q., Lin Z., Hu Q., Jiang Y., Tu M., Chen X., Li W., Lin W., Fuchs B.C., Chung R.T., Hong J. Tyrosine kinase SYK is a potential therapeutic target for liver fibrosis. Hepatology. 2018;68:1125–1139. doi: 10.1002/hep.29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang C.Y., Wang H., Liao W., Han F., Li Y.Q., Chen S.W., Lao X.M. Transforming growth factor beta is a poor prognostic factor and inhibits the favorable prognostic value of CD8+ CTL in human hepatocellular carcinoma. J Immunother. 2017;40:175–186. doi: 10.1097/CJI.0000000000000166. [DOI] [PubMed] [Google Scholar]
- 49.Chen K., Wu Z., Zhao H., Wang Y., Ge Y., Wang D., Li Z., An C., Liu Y., Wang F., Bi X., Wang H., Cai J., Ma C., Qu C. XCL1/glypican-3 fusion gene immunization generates potent antitumor cellular immunity and enhances anti-PD-1 efficacy. Cancer Immunol Res. 2020;8:81–93. doi: 10.1158/2326-6066.CIR-19-0210. [DOI] [PubMed] [Google Scholar]
- 50.Baumgart J., Nilsson K., Stavreus Evers A., Kunovac Kallak T., Kushnir M.M., Bergquist J., Sundström Poromaa I. Androgen levels during adjuvant endocrine therapy in postmenopausal breast cancer patients. Climacteric. 2014;17:48–54. doi: 10.3109/13697137.2013.800039. [DOI] [PubMed] [Google Scholar]