

Abstract
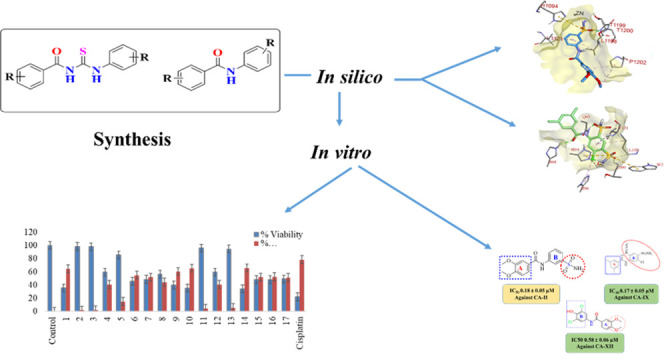
Series of sulfonamide-substituted amide (9–11), benzamide (12–15), and 1,3-disubstituted thiourea (17–26) derivatives were synthesized from a common precursor, i.e., substituted benzoyl chlorides. Structures of all of the synthesized compounds were characterized by spectroscopic techniques (1H nuclear magnetic resonance (NMR),13C NMR, and Fourier transform infrared spectroscopy (FTIR)). All of the amide (9–15) and thiourea (17–26) derivatives were screened against human carbonic anhydrases, hCA-II, hCA IX, and hCA-XII. Sulfonamide-substituted amides 9, 11, and 12 were found to be excellent selective inhibitors with IC50 values of 0.18 ± 0.05, 0.17 ± 0.05, and 0.58 ± 0.05 μM against hCA II, hCA IX, and hCA XII, respectively. Compound 9 was found to be highly selective for hCA II and about 6-fold more potent as compared to the standard antagonist, acetazolamide. Safe toxicity profiling of the most potent and selective compounds was determined against normal BHK-21 and HEK-293 T cells. Molecular docking studies were performed, which described the type of interactions between the synthesized compounds and enzyme proteins. In addition, in silico absorption, distribution, metabolism, and excretion (ADME) studies were performed, which showed that all of the synthesized molecules fulfilled the druggability criteria.
1. Introduction
Human carbonic anhydrases (EC 4.2.1.1, hCAs) are zinc-containing metalloenzymes found in numerous living organisms, and they catalyze the reversible hydration of carbon dioxide (CO2) to proton (H+) and bicarbonate (HCO3).1,2 Carbonic anhydrases (CAs) are involved in various physiological processes associated with the transport of CO2/bicarbonate in homeostasis, electrolyte secretion in a variety of tissues/organs, and biosynthetic reactions such as gluconeogenesis, ureagenesis, lipogenesis, respiration, calcification, bone resorption, and oncogenesis.1,2
There are eight genetically nonrelated families of CAs (α-, β-, γ-, δ-, ζ-, η-, θ-, and ι-CA) recognized to date.3,4 Among the human carbonic anhydrases, hCA-II plays an important role in angiogenesis through vascular endothelial growth factor receptor (VEGFR) signaling.5 Human carbonic anhydrases (hCA) IX and XII are tumor-associated CAs that belong to the α-family,6,7 and the overexpression of human carbonic anhydrase IX (hCA IX) and XII leads to various tumors. However, CA IX may act as a therapeutic and diagnostic biomarker for solid hypoxic tumors, whereas hCA XII is found in normal tissues and its overexpression is associated with certain types of noncombative tumors.8,9 Human carbonic anhydrase (hCA) IX and hCA XII isoforms have been reported as drug targets in various pathologies of cancers and metastasis, where both isoforms play a significant role in the growth, invasion, and migration of tumors.10−12 The vitality of these isoforms has attracted many medicinal chemists to develop selective inhibitors of these isoforms for anticancer drug development.
The overexpression of CAs could result in multiple diseases, including epilepsy, obesity, glaucoma, and various cancers.13,14 Therefore, inhibition of CAs could be a potential therapeutic target to reduce its hyperactivity in relevant disorders.6,15,16
Acetazolamide (1), dorzolamide (2), and brinzolamide (3) are commercially available CA standard inhibitors that have clinical applications. Nonselective inhibition by the reported CA inhibitors leads to serious undesirable side effects. Therefore, it is imperative to develop selective carbonic anhydrase inhibitors for the specific CA isoform.8,9 It was reported that phenols, aryl- or alkyl-substituted carboxylic acids, sulfonamides, sulfamoyl carbamates, and sulfamides may act as CA inhibitors.17 Among the known hCA inhibitors (CAIs), sulfonamide-based compounds have been reported as potent inhibitors because deprotonated nitrogen atoms of sulfonamide moiety bind with the catalytic zinc ion at the active site, thus blocking its enzymatic activity.17−22 Derivatives of sulfonamide also exhibit numerous biological activities, i.e., anticancer, antimicrobial, antiplatelet, and anti-inflammatory.23 Thiourea derivatives have been reported to have biological significance such as antibacterial, antifungal, insecticidal, anticancer, and anti-cholinesterase hCA IX inhibition activities.24 Primary sulfonamide and thiourea hybrids have been reported with fascinating pharmacological effects; among them, compound 4 was found standard inhibitor of hCA-II,25,26 while compound 5 was found potent a CA IX inhibitor27 (Figure 1).
Figure 1.
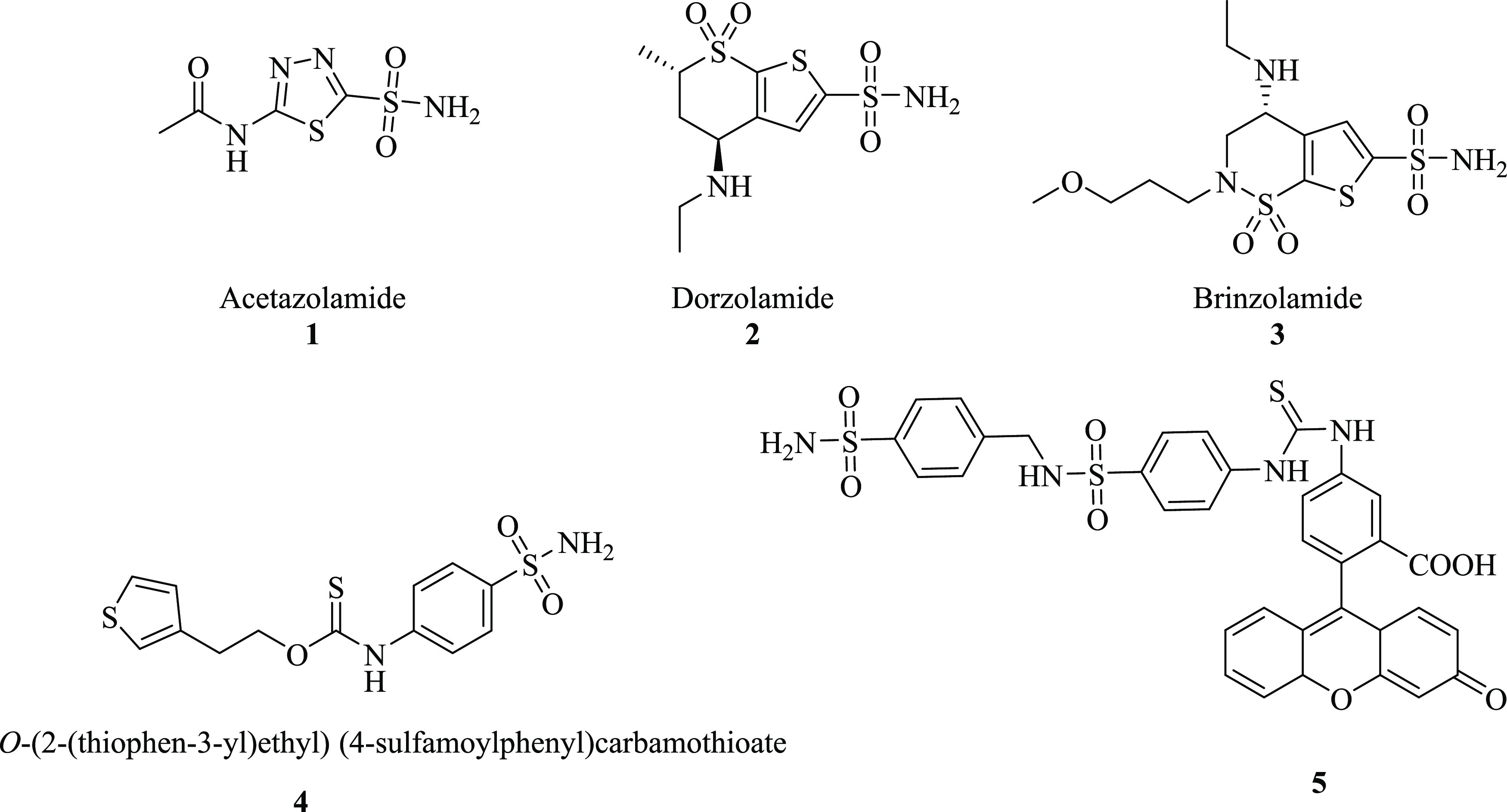
Chemical structures of standard inhibitors: (1–4) sulfonamide derivatives of CA-II and CA-IX and (5) potent inhibitor of thiourea derivative of CA-IX.
In continuation of our previous work,28 we synthesized secondary sulfonamides and screened for their inhibitory potential toward hCA II/IX/XII. In the present study, we have synthesized the sulfonamide-substituted benzamides (9–11), substituted benzamides (12–15), and 1,3-diarylthioureas (17–26). All of the synthesized compounds were analyzed for their CA inhibition activities toward hCA II, hCA IX, and hCA XII. Based on the screening assay, structure–activity relationships (SARs) were established, indicating the significance of functional moieties in the structure of synthesized derivatives. In addition, in silico studies and cytotoxicity or cell availability assay were performed for further confirmation to explore their safety profile against normal cells and their significance toward the development of effective treatment for various cancers.
2. Results and Discussion
2.1. Chemistry
Synthesis of our desired sulfonamide-substituted benzamides (9–11) was carried out by stirring substituted acid chlorides (6–7) with sulfonamide-substituted anilines (8a–b) at 0 °C in basic media for 3–4 h (Scheme 1).
Scheme 1. Synthesis of Sulfonamide-Substituted Amides (9–11).

FT-IR data of compounds 9–11 showed the signal for the CONH group in the range of 1635–1690 cm–1 that indicated the amide formation. For further structure confirmations, NMR spectroscopic techniques were used. In 1H NMR spectra, the appearance of singlet at δ 9.03–10.44 ppm, in addition to the characteristic signals of aromatic protons and sulfonamides groups, confirmed the synthesis of sulfonamide-substituted benzamides (9–11). The appearance of the CONH signal at 165.2–167.2 ppm further confirmed amide bond formation. The purity of the compound was determined by HPLC. The physical data of compounds (9–11) are presented in Table 1.
Table 1. Physical Data of Compounds 9–11.
| compound | yield (%) | M.P. (°C) | color | Rf |
|---|---|---|---|---|
| 9 | 84 | 217–219 | light yellow | 0.4a |
| 10 | 77 | 220–222 | light yellow | 0.4b |
| 11 | 73 | 212–214 | dark yellow | 0.5a |
n-hexane/EtOAc = 1:4.
n-hexane/EtOAc = 3:2.
For the synthesis of our desired product substituted benzamides (12–15), substituted acid chlorides (6–7) were stirred with substituted anilines (8c–f) at 0 °C in basic media for 3–4 h (Scheme 2).
Scheme 2. Synthesis of Substituted Amides (12–15).

In the FT-IR spectrum of the compounds (12–15), the appearance of the CONH signal in the range of 3290–3315 cm–1 indicated the NH stretching, while stretching bands at 1622–1690 cm–1 indicated NHC=O, confirming amide bond formation. For further structure confirmations, the NMR spectroscopic technique was used. In 1H NMR, the appearance of singlet at δ 9.09–11.54 ppm in addition to the characteristic signals of amide groups confirmed the synthesis of substituted benzamides (12–15). The appearance of the CONH signal at 164.9–168.6 ppm in 13C NMR further confirmed amide bond formation. The purity of the compounds was checked by HPLC, and physical data of compounds (12–15) are presented in Table 2.
Table 2. Physical Data of Compounds (12–15).
| compound | yield (%) | M.P. (°C) | color | Rf |
|---|---|---|---|---|
| 12 | 54 | 256–258 | light brown | 0.4a |
| 13 | 78 | 237–239 | yellow | 0.5b |
| 14 | 71 | 155–157 | brown | 0.4b |
| 15 | 60 | 180–182 | dark brown | 0.5b |
n-hexane/EtOAc = 1:4.
n-hexane/EtOAc = 3:2.
Compounds (17–26) were synthesized by following the synthetic Scheme 3 to obtain 1-aroyl-3-aryl thiourea derivatives. In situ condensation of freshly prepared benzoyl chlorides (6–7) with potassium thiocyanate (KSCN) was carried out to obtain pale yellow precipitates of benzyol isothiocyanate (16a–b). The addition of 1 equiv of substituted anilines (8a,c–f) in the reaction mixture led to the synthesis of 1-aroyl-3-aryl thioureas (17–26) in good yield (Table 3).
Scheme 3. Synthesis of 1,3-Diarylthiourea (17–26).

Table 3. Physical Data of Synthesized Compounds (17–26).
| compound | yield (%) | M.P. (°C) | color | Rf |
|---|---|---|---|---|
| 17 | 74 | 213–215 | brown | 0.3b |
| 18 | 88 | 238–240 | light yellow | 0.2a |
| 19 | 89 | 176–178 | yellow | 0.6c |
| 20 | 76 | 203–205 | off white | 0.4a |
| 21 | 87 | 227–229 | off white | 0.4d |
| 22 | 82 | 176–178 | yellow | 0.3e |
| 23 | 81 | 208–210 | off white | 0.5a |
| 24 | 79 | 256–258 | light brown | 0.4a |
| 25 | 87 | 228–230 | dark brown | 0.4a |
| 26 | 84 | 227–229 | off white | 0.4b |
n-hexane/EtOAc = 1:4.
n-hexane/EtOAc = 3:2.
n-hexane/EtOAc = 3:7.
n-hexane/EtOAc = 7:3.
n-hexane/EtOAc = 4:1.
According to the FT-IR spectra, stretching bands that appeared in the range of 3290–3450 cm–1 for the NH group, at 1480–1780 cm–1 for the (C=O) group, and at 1005–1090 cm–1 for the (C=S) group confirmed that 1-aroyl-3-arylthiourea derivatives have been synthesized. 1H NMR data showed the appearance of characteristic signals for NH protons of NHCSNH groups at δ 8.36–11.55 ppm (CONH) and δ 10.56–13.08 ppm (CSNH). In the 13C NMR spectrum, the characteristic peaks of the thiourea appear in the region of δ 175–180 ppm for thiocarbonyl carbon (C=S) and δ 164–170 ppm for carbonyl carbon. These two signals confirmed the formation of thiourea and amide linkage between the aromatic acid and the aniline ring. The purity of the compounds was checked by HPLC. Physical data of compounds (17–26) are presented in Table 3.
2.2. Carbonic Anhydrase Inhibition Activity
The newly synthesized sulfonamide-substituted amides (9–11), benzamides (12–15), and 1,3-disubstituted thioureas (17–26) were screened for their CA inhibitory potency against the human CA isoforms including hCA II, hCA IX, and CA XII, which have been defined as drug targets.29 Acetazolamide (AAZ) was used as the standard inhibitor of CA, and the results are summarized in Table 4. The inhibition exhibited by the screened compounds was compared with standard inhibitors, and SAR was established on the nature of the compounds and moieties attached to them. All of the compounds exhibited significant inhibition toward the prevalent cytosolic isoform of hCA-II in the range of 0.18–4.45 μM. Compounds 09, 22, and 25 (with IC50 value = 0.18 ± 0.05, 0.26 ± 0.03, and 0.38 ± 0.09 μM, respectively) were found to be more potent CA inhibitors when compared with the standard drug. In compounds 09 and 22, the methoxy (OCH3) group is present at the para and meta positions of ring A. In the case of compound 09, the sulfonamide group (SO2NH2) is present at the m-position as well, while compound 22 contains the chloro group at the para position and the nitro group at the m-position of ring B. Similarly, compound 25 has methyl groups at the o- and p-positions of A ring, while free carboxylic acid is present at the para position of B ring, as shown in Figure 5. Compound 10 is also considered a good inhibitor with an IC50 value of 0.93 ± 0.05 μM against hCA-II, having methyl (CH3) and sulfonamide groups (SO2NH2) at the m-position on rings A and B, respectively. It is considered that the activity of compound 10 might be less than that of the other sulfonamide analogues due to the replacement of the functionalities of methoxy (OCH3) with methyl (CH3). Compounds 11, 15, 19, and 24 showed inhibitions with IC50 values of 1.185 ± 0.143, 1.93 ± 0.15, 0.725 ± 0.068, and 1.24 ± 0.96 μM, respectively, and their inhibition is found to be equivalent to standard drug. Compounds 14, 17, 18, and 26 exhibited moderate inhibition, while compounds 12, 20, 21, and 23 did not show any inhibition toward CA.
Table 4. IC50 of hCA II and hCA IX and % Inhibitiona.
| IC50 ± SEM (μM) and % inhibition |
|||
|---|---|---|---|
| compounds | hCA II | hCA IX | hCA XII |
| 9 | 0.18 ± 0.05 | 22.31% | 37.49% |
| 10 | 0.93 ± 0.05 | 14.58 ± 1.07 | 9.17 ± 0.85 |
| 11 | 1.18 ± 0.14 | 0.17 ± 0.05 | 2.99 ± 0.32 |
| 12 | 34.31% | 32.14% | 0.58 ± 0.06 |
| 13 | 32.70% | 10.36 ± 1.45 | 37.78% |
| 14 | 6.49 ± 0.63 | 16.13% | 0.95 ± 0.08 |
| 15 | 1.93 ± 0.15 | 47.23% | 1.05 ± 0.11 |
| 17 | 11.51 ± 0.16 | 1.71 ± 0.65 | 44.48% |
| 18 | 4.17 ± 0.29 | 0.21 ± 0.09 | 1.68 ± 0.15 |
| 19 | 0.72 ± 0.06 | 1.01 ± 0.05 | 38.14% |
| 20 | 9.73% | 29.21% | 40.81% |
| 21 | 20.34% | 45.42% | 36.70% |
| 22 | 0.26 ± 0.03 | 4.93 ± 1.83 | 4.29 ± 0.24 |
| 23 | 13.28% | 28.21% | 35.76% |
| 24 | 1.24 ± 0.96 | 1.25 ± 0.02 | 9.90 ± 0.66 |
| 25 | 0.38 ± 0.01 | 9.76 ± 1.03 | 43.91% |
| 26 | 4.45 ± 0.11 | 1.28 ± 0.09 | 2.02 ± 0.28 |
| acetazolamide | 1.19 ± 0.04 | 1.08 ± 0.02 | 1.55 ± 0.03 |
All of the reported results are the mean of three independent experiments and compounds showing >50% inhibition were further analyzed for the IC50 value at different dilutions. The IC50 values were calculated through nonlinear regression analysis using GraphPad Prism version 8. SEM = standard error mean.
Figure 5.

Possible binding mode of inhibitor 22 inside hCA II and compound 18 inside the hCA IX active pocket.
All of the compounds exhibited adequate inhibition toward tumor-associated isoforms hCA-IX and hCA-XII, with the inhibition constant ranging from 0.17 to 14.58 μM. Among the synthesized derivatives, compound 11 with an IC50 value of 0.17 ± 0.05 μM was found to be a more potent hCA-IX inhibitor due to the electron-donating effect of the methyl substituent at the m-position on ring A and sulfonamide substituents at the o- and p-positions while the chloro group at the m-position of ring B.30 Compound 18 exhibited inhibition with an IC50 value of 0.21 ± 0.09 μM due to the presence of a methoxy substituent on ring A and a free carboxylic acidic group at the para position on ring B. Compounds 10, 13, 17, 19, 24, 22, 25, and 26 exhibited good inhibition with IC50 values of 14.58, 10.36, 1.71, 1.01, 1.25, 4.93, 9.76, and 1.28 μM, respectively, compared with that of the standard inhibitor. The rest of the compounds showed moderate or no inhibition activity. By looking at the CA inhibition activity and substitution pattern, it was found that compounds having electron-withdrawing groups substituted on their B ring exhibited excellent inhibition than compounds having electron-donating groups on B rings.31
Compounds 12 and 18 with the IC50 values of 0.58 ± 0.06 and 1.68 ± 0.15, respectively, were found to be potent inhibitors of hCA IX. In the case of thiourea, nonselectivity was observed between the two isoforms, while in the case of amide, selectivity was observed. Compounds 12 and 14 were found to be potent and selective inhibitors of hCA-XII containing the electron-donating OCH3 group at the m- and p-position of their ring A and two halogens (Cl) at the meta (m) and one hydroxyl (OH) group at the para position (p) on ring B, responsible for the better activity of the compound. The structure–activity relationship (SAR) from inhibition activity data showed that the amide derivatives were selective toward hCA-II, hCA-IX, and hCA-XII (shown in Figure 2).
Figure 2.
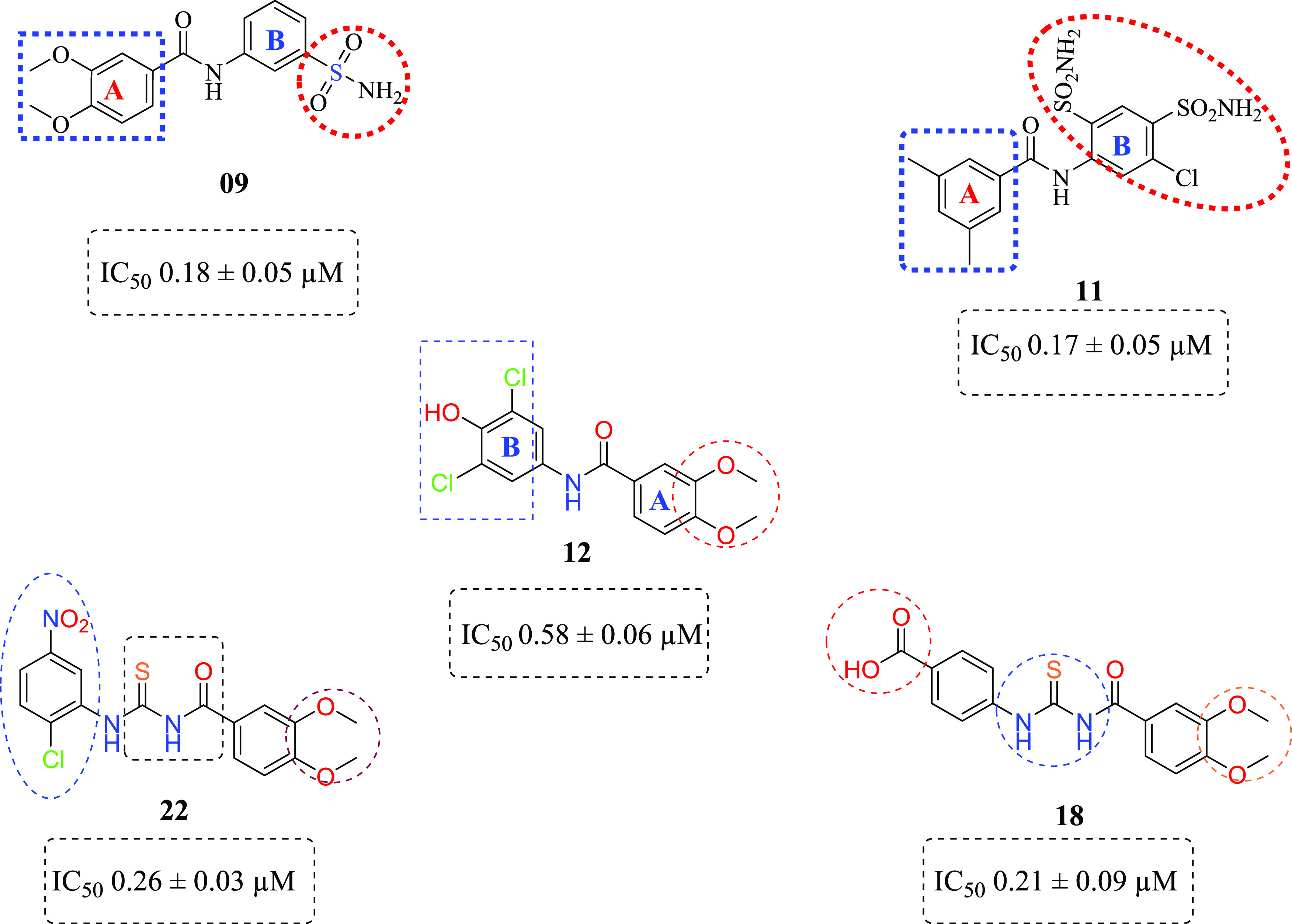
Structures of potent CA inhibitors contain amide and thiourea moieties.
2.3. Docking Studies of Human Carbonic Anhydrase II and IX Inhibitors
Molecular modeling studies were conducted on human carbonic anhydrase CA II (PDB: 3V7X), hCA-IX (PDB: 5FL4), and hCA-XII (PDB: 5MSA) enzymes. The relative selectivity and potency of our designed analogues were analyzed to clarify the trends observed and to ensure the accuracy and efficiency of modeling studies. For this, 3D structures of the enzymes were downloaded from the PDB server31https://www.rcsb.org/. A high-resolution crystal structure of hCA II, hCA IX, and hCA XII with 1.03, 1.8, and 1.2 Å were selected for the in silico studies of potent inhibitors. The crystal structure of hCA II contains the co-crystallized ligand N [2-(3,4-dimethoxyphenyl)ethyl]-4-sulfamoylbenzamide in its active site. The crystal structure of both hCA IX and hCA XII contains a homotetramer chain; therefore, chain A was selected for docking studies, which contain co-crystallized ligands 5-(1-naphthalen-1-yl-1,2,3-triazol-4-yl) thiophene-2-sulfonamide (9FK) and 2,3,5,6-tetrafluoro-4-(propylthio)benzenesulfonamide 3TV. The crystal structure of co-crystallized ligands was selected as a reference for docking, and the docking protocol was validated after successfully reproducing the co-crystallized pose. The RMSD values for the reference ligand of hCA II, hCA IX, and hCA XII were found to be 0.58, 0.83, and 1.19 Å, respectively, using MOE, LeadIT, and SeeSAR software.
Compounds 09 and 11 were docked inside the active pocket of hCA II and hCA IX. The HYDE assessment scores of the potent inhibitors were −44 and −38 kJ mol–1, which was comparable to the reference co-crystallized ligand. The cofactor (Zn++) binds with the deprotonated nitrogen (N) of primary sulfonamide to form a complex within the active side through a bond distance of 2.06 Å. Thr199 and Thr200 bind as a H-bond donor with the O atom of the SO2NH2 group with bond distances of 2.22 and 1.82 Å, respectively. Similarly, Val121, Pro, and Leu198 show π-alkyl and carbon–hydrogen bond interactions with the aromatic ring of compound 09.
The leading interaction is represented by coordination between the nitrogen (N) of the SO2NH2 with the Zn++ ion with a bond distance of 2.16 Å. Thr200 and Thr201 both bind with the oxygen atoms of the sulfonamide moiety with bond distances of 1.94 and 2.06 Å, respectively, while Gln92 also forms H-bonding with the carbonyl oxygen of amide via a bond distance of 1.74 Å; similarly, Val130, Leu91, and Val142,121 show π-alkyl and carbon–hydrogen bond interactions with compound 11 (Figure 3).
Figure 3.

Possible binding mode of inhibitor 9 inside CA-II and compound 11 inside the CA-IX active pocket.
The HYDE assessment scores for compounds 15 and 13 were −25 and −24 kJ mol–1. The conventional hydrogen bond was observed between Gln71 and the carbonyl of compound 13 with a bond distance of 2.22 Å. π–π stacking was observed between His94 and synthesized compound 13 via a bond distance of 3.75 Å. The residues His96 and Leu11 showed π-alkyl interactions with the compounds having bond distances of 5.48 and 5.03 Å, respectively (Figure 4).
Figure 4.

Possible binding mode of inhibitor 15 inside CA-II and compound 13 inside the CA-IX active pocket.
Among the thiourea derivatives, the most potent inhibitors were docked against hCA-II and hCA-IX. The HYDE assessment scores of the potent inhibitors were −28 and −24 kJ mol–1, respectively. The sulfur atom of compound 22 interacts with the proton of Asn66 via a bond distance of 2.2 Å. The Trp90 residue interacts through π–sulfur interactions with a bond distance of 4.91 Å. Leu199 and Val121 interact with the π-system of compound 22 through a bond distance of 4.79 and 4.44 Å, respectively. His94 shows π–π interactions with a bond distance of 4.70 Å. Thr200 shows conventional hydrogen bond interaction with compound 18 via a bond distance of 2.20 Å. The carbon–hydrogen interactions observed between His94, Pro202, Phe70, and Glu69 with compound 18 show bond distances of 2.60, 2.64, 2.54, and 2.89 Å, respectively. Leu57 and Leu91 interact with compound 18 through alkyl and π-alkyl interactions with bond distances of 5.03 and 5.35 Å, respectively. Glu69 interacts through π-anion interactions with a bond distance of 3.33 Å (Figure 5).
Within the hCA XII active site, the most active compounds were docked, and the binding affinity of both compounds was −27 and −35 kJ mol–1. Compound 12 showed potential H-bond with His 93, while Thr199 and Pro 200 form H-bonding through water molecule with the NH of the amide. Leu197 and 139, Val 197,141,206, and Trp206 show van der Waals interactions. Compound 18 forms H-bond with Thr200 and Gln71 with a bond distance of 2.93 and 4.24 Å, respectively. Zn metal complexed with the oxygen atom of carbonyl moiety (C=O) with a bond distance of 1.88 Å. Leu199 shows H-arene interactions with the phenyl ring with a distance of 3.7 Å (Figure 6).
Figure 6.

Interaction of compound 12 with hCA XII and compound 18 in the active pocket of hCA XII.
2.4. Pharmacokinetic Studies
Many potential enzyme inhibitors have poor pharmacokinetic properties, making it difficult for them to reach clinical trials. To understand the basic properties, it is necessary to study the pharmacokinetics of compounds. Using in silico techniques, a web-based module called SwissADME was used to obtain the absorption, distribution, metabolism, and excretion profiles (ADME profile) of synthesized molecules 09–15 and 17–26. The resulting data are summarized in Table 5. SwissADME was used to predict the absorption of a compound passively through the gastrointestinal tract (GI).32,33 SwissADME includes a highly intuitive method known as the boiled-egg plot. The egg-shaped plot provides an overview of the gastrointestinal absorption of the compound. The white part of the egg represents the gastrointestinal absorption of the new compound, while the yolk portion represents its BBB penetration. Based on the plot, none of the synthesized compounds can penetrate the BBB, but all can cross the gastrointestinal tract, representing good oral absorption (Figure 6).
Table 5. Evaluation of Synthesized Compounds Using In Silico ADME.
| compounds | MW | no. of H-bond acceptors | no. of H-bond donors | WLOGP | GI absorption | BBB permeant | Lipinski no. of violations | PAINS no. of alerts |
|---|---|---|---|---|---|---|---|---|
| 9 | 336.36 | 6 | 2 | 2.49 | high | no | 0 | 0 |
| 10 | 304.36 | 4 | 2 | 3.09 | high | no | 0 | 0 |
| 11 | 417.89 | 7 | 3 | 3.47 | low | no | 0 | 0 |
| 12 | 342.17 | 4 | 2 | 3.78 | high | yes | 0 | 0 |
| 13 | 269.3 | 3 | 2 | 3.06 | high | yes | 0 | 0 |
| 14 | 304.73 | 3 | 1 | 3.93 | high | yes | 0 | 0 |
| 15 | 372.18 | 4 | 1 | 6.3 | high | no | 1 | 0 |
| 17 | 508.98 | 9 | 4 | 2.75 | low | no | 2 | 0 |
| 18 | 360.38 | 5 | 3 | 2.34 | high | no | 0 | 0 |
| 19 | 474.71 | 5 | 2 | 4.49 | low | no | 0 | 0 |
| 20 | 332.37 | 4 | 3 | 2.35 | high | no | 0 | 0 |
| 21 | 358.41 | 4 | 2 | 2.84 | high | no | 0 | 0 |
| 22 | 395.82 | 5 | 2 | 3.2 | low | no | 0 | 0 |
| 23 | 463.27 | 6 | 2 | 5.57 | high | no | 0 | 0 |
| 24 | 300.38 | 2 | 3 | 2.95 | high | no | 0 | 0 |
| 25 | 328.39 | 3 | 3 | 2.94 | high | no | 0 | 0 |
| 26 | 369.27 | 2 | 3 | 4.25 | high | no | 0 | 0 |
All scaffolds were consistent with Lipinski’s rule of drug-likeness.34 Furthermore, the PAINS alerts (Pan-assay interference compounds) filter was used to assess the promiscuity of the prepared hits. All of the tested compounds (Table 5) showed druggable properties with no PAINS alerts except for compound 17. In addition, all of the compounds showed high gastrointestinal absorption and topological polar surfaces ranging from 113.17 to 92.69 Å. As a result of all of these parameters, it can be predicted that synthesized hits could serve as potentially promising agents for the oral treatment of diseases caused by the overexpression of carbonic anhydrase.
2.5. Cell Viability Assay
For evaluating the cell viability/cytotoxicity, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay was performed against BHK-21 and HEK-293 T normal cells.34 Comparing the most selective and potent compounds, the cell viability or safety profiling of compounds 09–15 and 17–26 was determined. All compounds were screened at a final concentration of 100 μM (Figure 7 and Table 6). The results were compared with the standard drug cisplatin. Most of the compounds were found to be noncytotoxic and safe at 100 μM concentration.
Figure 7.

Boiled-egg plot of thiourea derivates and amide derivatives.
Table 6. Analysis of Selective and Potent Compounds at a Final Concentration (100 μM) Cell Viability in BHK-21 and HEK-293 Normal Cells Compared to Untreated Control and Cisplatin (a Cytotoxic Agent).
| samples | % cell viability ± SD(BHK-21 cells) | % cytotoxicity ± SD(BHK-21 cells) | % cell viability ± SD(HEK-293 T cells) | % cytotoxicity ± SD(HEK-293 T cells) |
|---|---|---|---|---|
| control | 100 | 0 | 100 | 0 |
| 9 | 98 ± 0.06 | 1.8 ± 0.06 | 72 ± 0.2 | 27.0 ± 0.2 |
| 10 | 34 ± 0.1 | 65.70 ± 0.07 | 86 ± 0.3 | 14 ± 0.010 |
| 11 | 48 ± 0.06 | 51 ± 0.005 | 72 ± 0.6 | 27 ± 0.06 |
| 12 | 35 ± 0.08 | 64 ± 0.03 | 73 ± 0.1 | 26.6 ± 0.06 |
| 13 | 94 ± 0.2 | 05.50 ± 0.23 | 86 ± 0.6 | 14 ± 0.09 |
| 14 | 47 ± 0.09 | 52 ± 0.14 | 85 ± 0.10 | 15 ± 0.4 |
| 15 | 48 ± 0.04 | 51.2 ± 0.23 | 82 ± 0.6 | 18 ± 0.6 |
| 17 | 97 ± 0.05 | 2.16 ± 0.05 | 97 ± 0.3 | 03 ± 0.04 |
| 18 | 59 ± 0.03 | 40.7 ± 0.04 | 75. ± 0.5 | 24 ± 0.07 |
| 19 | 85 ± 0.2 | 14.7 ± 0.01 | 49 ± 0.6 | 50 ± 0.08 |
| 20 | 45 ± 0.09 | 54.6 ± 0.02 | 86 ± 0.07 | 13 ± 0.03 |
| 21 | 48 ± 0.1 | 51.5 ± 0.06 | 86 ± 0.6 | 13 ± 0.05 |
| 22 | 39 ± 0.08 | 60.0 ± 0.05 | 91 ± 0.6 | 09 ± 0.050 |
| 23 | 34 ± 0.1 | 65.0 ± 0.06 | 74 ± 0.4 | 26 ± 0.06 |
| 24 | 56 ± 0.2 | 43.8 ± 0.01 | 81 ± 0.1 | 18 ± 0.03 |
| 25 | 95 ± 0.05 | 04.3 ± 0.09 | 98 ± 0.6 | 02 ± 0.02 |
| 26 | 59 ± 0.01 | 40.70 ± 0.1 | 98 ± 0.4 | 02 ± 0.06 |
| cisplatin | 21.0 ± 0.04 | 78.0 ± 0.06 | 23 ± 0.8 | 77 ± 0.09 |
Compounds 09, 23, 25, and 13 showed the highest cell viability profile of more than 90% in BHK-21 cells, while 09, 22, 25, and 26 showed the highest safety profile/viability in HEK-293 T cells (>90%) compared to the control after 24–48 h incubation, followed by cisplatin cell viability in BHK-21 (21%) and HEK-293 (23%) T cells compared to the control after 24–48 h incubation. However, compounds 12, 22, 23, and 10 showed the lowest cell viability in BHK-293 normal cells but expressed the highest cell viability in HEK-293 T compared to the untreated control. So, it is concluded that these selective and potent compounds could be evaluated for any bioassays (Figure 8).
Figure 8.

Analysis of cell viability/cytotoxicity using BHK-21 normal cells treated with compounds 1–17 at a final concentration of 100 μM. Statistics indicating significant values *p < 0.05, **p < 0.01, ***p < 0.001 vs the untreated group. Cisplatin (100 μM) is used as a positive control.
3. Conclusions
Synthesis of sulfonamide-substituted amides (9–11), benzamides (12–15), and 1,3-disubstituted thioureas (17–26) was carried out to check their potential role as selective CA inhibitors. It was found that compounds 9–26 exhibited their activity as CA inhibition activity in comparison with the standard drug acetazolamide. Docking studies of the targeted compounds further confirmed the mode of binding with our targeted protein. Among the three series compounds, 9 and 11 showed maximum selective CA inhibition activity with IC50 values of 0.18 and 0.17 μM against hCA II and hCA IX, respectively. Docking studies further confirmed the binding affinities of the lead compounds against the targeted proteins. As the overexpression of hCA XII and hCA IX was known to be involved in cancer disease, all of the synthesized compounds, especially the most potent compounds 9 and 11, were subjected to cell viability studies in comparison to the cisplatin drug. It was found that compounds 9, 23, 25, and 13 showed potential activity against BHK-21 and HEK-293 T cells. Based on our investigations, it is proposed that derivatives 9 and 11 may serve as a lead structure to design potentially more active carbonic anhydrase inhibitors.
4. Experimental Section
All of the reactants purchased from Sigma-Aldrich were used without further purification. Commercially available solvents were purified by standard methods, and reaction progress and product purity were checked by a TLC silica gel plate (0.2 mm, 60 HF254, Merck). The melting points of all of the synthesized compounds were noted in capillary tubes with an electrothermal melting point instrument, Stuart digital melting point (SMP 10) apparatus. 1H NMR spectra were obtained from a spectrophotometer of 400 MHz using a DMSO-d6 (deuterated) solvent, while internal reference was TMS (tetramethylsilane), and 13C was carried out at 100 MHz using the DMSO-d6 (deuterated) solvent.
4.1. General Procedure of Synthesis of Sulfonamide-Substituted Benzamides (9–11)
For the synthesis of our desired product sulfonamide-substituted benzamide (9–11), substituted acid chlorides (6–7) were stirred with sulfonamide-substituted anilines (8a–b) at room temperature in basic media for 16–18 h (Scheme 1).23
4.1.1. 3,4-Dimethoxy-N-(3-sulfamoylphenyl) Benzamide (09)

Yield: 84%, Rf = 0.3, (n-hexane/ethyl acetate = 1:4), M.P.: 217–219 °C, color: light yellow. FT-IR (ύ, cm–1): 3535 (NH, stretching), 1635 (C=O, stretching), 1396 (C=C, stretching), 1108 (C–O, stretching) cm–1. 1H NMR (400 MHz, DMSO-d6) δ (NH, s, 1H), 10.35 (s, 1H, Ar–H), 8.32 (t, J = 1.4 Hz, 1H, Ar–H), 7.97 (dtd, J = 6.2, 3.9, 2.0 Hz, 1H, Ar–H), 7.76–7.65 (m, 1H, Ar–H), 7.67–7.60 (m, 1H, Ar–H), 7.62–7.50 (m, 2H, Ar–H), 7.38 (s, 2H, Ar–H), 7.11 (dd, J = 8.6, 5.6 Hz, 1H, Ar–H), 3.86 (s, 6H, OCH3) ppm. 13C NMR (100 MHz, DMSO-d6) δ 165.2 (C=O), 152.0, 148.4, 144.6, 139.8, 129.4, 126.5, 123.4, 121.3, 120.6, 117.5, 111.1, 111.1 (ArCs), 55.8, 55.7 (OCH3) ppm.
4.1.2. 3,5-Dimethyl-N-(3-sulfamoylphenyl) Benzamide (10)

Yield: 77%, Rf = 0.4, (n-hexane/ethyl acetate = 1:4), M.P.: 220–222 °C, color: light yellow. FT-IR (ύ, cm–1): 3304 (NH, stretching), 1654 (C=O, stretching), 1337 (C=C, stretching), 1236 (C–O, stretching) cm–1. 1H NMR (400 MHz, DMSO-d6) δ 10.44 (NH, s, 1H), 8.36 (s, 1H, Ar–H), 7.96 (dd, J = 6.7, 3.0 Hz, 1H, Ar–H), 7.64–7.57 (m, 3H, Ar–H), 7.57–7.53 (m, 3H, Ar–H), 7.24 (s, 1H, Ar–H), 3.35 (CH3, s, 6H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 166.1 (C=O), 144.6, 139.7, 137.7, 134.6, 133.2, 129.4, 125.5, 123.2, 120.7, 117.3 (ArCs), 21.0, 20.8 (CH3) ppm.
4.1.3. N-(5-Chloro-2,4-disulfamoylphenyl)-3,5-dimethylbenzamide (11)

Yield: 73%, Rf = 0.5, (n-hexane/ethyl acetate = 3:2), M.P.: 212–214 °C, color: dark yellow. FT-IR (ύ, cm–1): 3670 (NH, stretching), 1690 (C=O, stretching), 1445 (C=C, stretching), 1370 (C–N, stretching), 970 (C–Cl, stretching). 1H NMR (400 MHz, DMSO-d6) δ 9.03 (NH, s, 1H), 8.00 (d, J = 13.4 Hz, 2H, Ar–H), 7.60–7.39 (m, 5H, Ar–H), 7.04 (d, J = 8.5 Hz, 1H, Ar–H), 6.89 (s, 1H, Ar–H), 2.50 (CH3, s, 6H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 167.2 (C=O), 152.7, 148.6, 148.4, 145.7, 134.4, 127.9, 125.7, 123.3, 112.0, 111.1 (ArCs), 28.5, 28.5 (CH3) ppm.
4.2. General Procedure of Synthesis of Substituted Benzamides (12–15)
For the synthesis of our desired product substituted benzamides (12–15), substituted acid chlorides (6–7) were stirred with substituted anilines (8c–f) at room temperature in basic media for 16–18 h (Scheme 1).23
4.2.1. N-(3,5-Dichloro-4-hydroxyphenyl)-3,4-dimethoxybenzamide (12)

Yield: 54%, Rf = 0.4, (n-hexane/ethyl acetate = 3:2), M.P.: 256–258 °C, color: light brown. FT-IR (ύ, cm–1): 3450 (NH, stretching), 2991 (OH), 1629 (C=O, stretching), 1446 (C=C, stretching), 1257 (C–O, stretching), 990 (C–Cl, stretching) cm–1. 1H NMR (400 MHz, DMSO-d6) δ 9.90 (NH, s, 1H), 7.82 (d, J = 1.9 Hz, 2H, Ar–H), 7.65–7.55 (m, 1H, Ar–H), 7.51 (d, J = 2.1 Hz, 1H, Ar–H), 7.12–7.05 (m, 1H, Ar–H), 3.83 (s, 6H, OCH3) ppm. 13C NMR (100 MHz, DMSO-d6) δ 164.9 (C=O), 151.9, 148.4, 145.1, 132.5, 126.5, 122.1, 121.1, 120.4, 111.0, 111.0 (ArCs), 55.8, 55.7 (OCH3) ppm.
4.2.2. 3-(2,4-Dimethylbenzamido) Benzoic Acid (13)

Yield: 78%, Rf = 0.5, (n-hexane/ethyl acetate = 1:4), M.P.: 237–239 °C, color: yellow. FT-IR (ύ, cm–1): 3460 (NH, stretching), 3060 (OH, stretching), 1640 (C=O, stretching), 1448 (C=C, stretching), 1272 (C–N, stretching) cm–1. 1H NMR (400 MHz, DMSO-d6) δ 12.8 (OHacidic, s, 1H) 11.54 (NH, s, 1H), 8.36 (s, 1H, Ar–H), 7.96 (ddd, J = 6.7, 5.5, 3.0 Hz, 1H, Ar–H), 7.64–7.57 (m, 3H, Ar–H), 7.57–7.53 (m, 3H, Ar–H), 7.24 (s, 1H, Ar–H), 3.35 (CH3, s, 6H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 168.6 (C=O), 168.1, 141.9, 139.5, 139.4, 135.6, 134.1, 132.3, 131.3, 130.6, 129.0, 128.6, 126.5, 124.3, 120.5, 40.0, 21.4, 21.0 ppm.
4.2.3. N-(2-Chloro-5-nitrophenyl)-3,5-dimethylbenzamide (14)
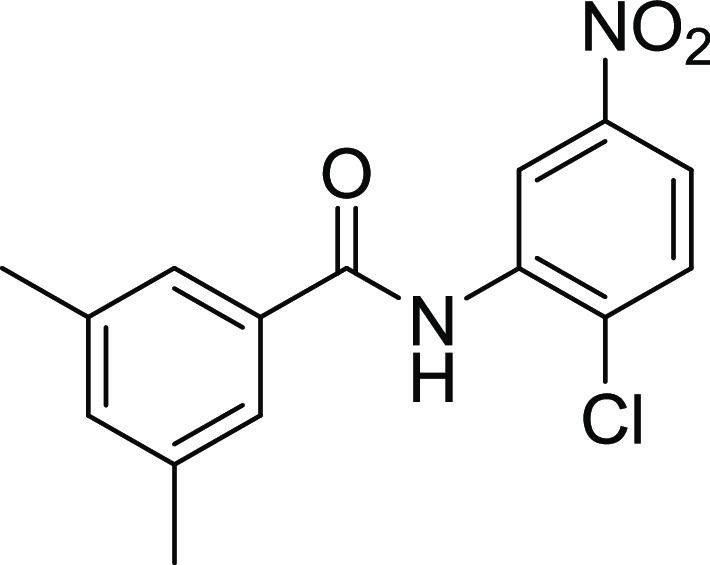
Yield: 71%, Rf = 0.4, (n-hexane/ethyl acetate = 3:2), M.P.: 155–157 °C, color: brown. FT-IR (ύ, cm–1): 3670 (NH, stretching), 1680 (C=O, stretching), 1454 (C=C, stretching), 962 (C–Cl, stretching) cm–1. 1H NMR (400 MHz, DMSO-d6) δ 10.25 (NH, s, 1H), 8.53 (d, J = 2.8 Hz, 1H, Ar–H), 8.11 (dd, J = 8.9, 2.8 Hz, 1H, Ar–H), 7.86 (d, J = 8.9 Hz, 2H, Ar–H), 7.62 (d, J = 1.6 Hz, 2H, Ar–H), 7.27 (s, 1H, Ar–H), 3.35 (CH3, s, 3H), 2.37 (CH3, s, 3H) ppm. 13C NMR (101 MHz, DMSO-d6) δ 165.9 (C=O), 146.4, 137.9, 136.4, 135.8, 133.6, 133.5, 130.9, 125.7, 122.0, 121.6 (ArCs), 20.9 (CH3) ppm.
4.2.4. N-(3-Bromo-5-(trifluoromethyl) phenyl)-3,5-dimethylbenzamide (15)

Yield: 60%, Rf = 0.5, (n-hexane/ethyl acetate = 3:2), M.P.: 180–182 °C, color: dark brown. FT-IR (ύ, cm–1): 3488 (NH, stretching), 3055 (C–H, stretching), 1622 (C=O, stretching), 1446 (C=C, stretching), 1328 (C–N, stretching), 740 (C–Br, stretching), 1159 (C–F, stretching) cm–1. 1H NMR (400 MHz, DMSO-d6) δ 9.09 (NH, s, 1H), 8.37 (t, J = 1.9 Hz, 1H, Ar–H), 8.22 (d, J = 1.8 Hz, 1H, Ar–H), 7.66 (d, J = 1.9 Hz, 1H, Ar–H), 7.58 (d, J = 1.7 Hz, 2H, Ar–H), 7.25 (d, J = 1.8 Hz, 1H, Ar–H), 2.36 (CH3, s, 6H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 166.4 (C=O), 141.6, 137.9, 134.1, 133.5, 131.4, 131.0, 125.9, 125.5, 124.6, 122.3, 121.9, 115.4, 115.3 (ArCs), 20.9 (CH3) ppm.
4.3. General Procedure of Synthesis of 1,3-Diaryl Thioureas (17–26)
In the synthesis of thiourea derivatives, the first step was acid chloride formation. Acid chloride formation was carried out in a two-neck round bottom flask (100 mL). 3,4-Dimethoxybenzoic acid (0.0011 mmol) and thionyl chloride (0.119 mL) were fused at 178–183 °C for 6 h, and then the reaction mixture was cooled at room temperature. The suspension of potassium thiocyanate (KSCN) in acetone was added to the reaction mixture and was continued to stir. A solution of aniline in acetone was added to the reaction mixture, and the reaction was refluxed for about 3–4 h at 65 °C. Progress of the reaction was monitored by TLC until the reaction was completed, and isothiocyanate was completely converted into thiourea. After completion of the reaction, the solvent was evaporated under reduced pressure using a rotary evaporator at 50 °C. About 30 mL of chilled distilled water was added to the reaction mixture to remove the unreacted reactants from the reaction mixture. To purify the products, recrystallization in ethanol or methanol was done according to their solubilities.24
4.3.1. N-((5-Chloro-2,4-disulfamoylphenyl) carbamothioyl)-3,4-dimethoxybenzamide (17)

Yield: 74%, Rf = 0.3 (n-hexane/ethyl acetate = 3:2), M.P.: 213–215 °C, color: brown. FT-IR (ύ, cm–1): 3444 (NH, stretching), 1691 (C=O, stretching), 1377 (C=C, stretching), 1216 (C=S, stretching) cm–1. 1H NMR (400 MHz, DMSO-d6) δ 11.77 (NH, s, 1H), 10.55 (NH, s, 1H), 8.16 (s, 2H, Ar–H), 7.44 (d, J = 2.0 Hz, 1H, Ar–H), 6.97 (s, 2H, Ar–H), 6.61 (s, 4H, Ar–H), 3.83 (s, 6H, OCH3) ppm. 13C NMR (100 MHz, DMSO-d6) δ 178.4 (C=S), 167.2 (C=O), 152.7, 148.6, 134.5, 130.4, 126.6, 123.3, 123.0, 121.6, 117.6, 112.0, 111.1 (ArCs), 55.7, 55.5 (OCH3) ppm.
4.3.2. 4-(3-(3,4-Dimethoxybenzoyl) thioureido) Benzoic Acid (18)

Yield: 88%, Rf = 0.2, (n-hexane/ethyl acetate = 1:4), M.P.: 238–240 °C, color: light yellow. FT-IR (ύ, cm–1): 3571 (NH, stretching), 1685 (C=O, stretching), 1472 (C=C, stretching), 1290 (C=S, stretching), 1075 (C–O, stretching) cm–1. 1H NMR (400 MHz, DMSO-d6) δ 12.81 (NH, s, 1H), 11.53 (NH, s, 1H), 10.34 (s, 1H, Ar–H), 8.01–7.92 (m, 1H, Ar–H), 7.92 (d, J = 6.8 Hz, 3H), 7.92–7.86 (m, 1H, Ar–H), 7.54 (d, J = 2.1 Hz, 1H, Ar–H), 7.10 (dd, J = 8.6, 4.4 Hz, 1H, Ar–H), 3.85 (OCH3, s, 6H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 179.3 (C=S), 167.1 (C=O), 165.4 (C=Oacidic), 152.0, 148.4, 143.5, 130.3, 130.0, 126.7, 121.4, 119.6, 111.2, 111.0 (ArCs), 55.8, 55.7 (OCH3) ppm.
4.3.3. N-((2-Bromo-6-chloro-4-nitrophenyl) carbamothioyl)-3,4-dimethoxybenzamide (19)

Yield: 89%, Rf = 0.6, (n-hexane/ethyl acetate = 3:7), M.P.: 176–178 °C, color: yellow. FT-IR (ύ, cm–1): 3290 (N–H, stretching), 3031 (Ar=CH), 2359 (C–N of −(CO)NH), 1703 (C=O), 1591 (N–C=S), 1445 (C–N), 1328 (C–O), 1267 (C=S), 1076 (C–O–C), 743–604 (Ar–H), 700 (C–Br), 680 (C–Cl) cm–1. 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 11.90 (s, 1H), 8.49 (dd, J = 26.8, 2.5 Hz, 1H), 8.00–6.96 (m, 3H), 3.95–3.73 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 148.56, 136.04, 127.61, 124.88, 116.33, 105.79.
4.3.4. N-((2-Hydroxyphenyl) carbamothioyl)-3,4-dimethoxybenzamide (20)

Yield: 76%, Rf = 0.4, (n-hexane/ethyl acetate = 1:4), M.P.: 203–205 °C, color: off-white. FT-IR (ύ, cm–1): 3450 (NH, stretching), 1612 (C=O, stretching), 1450 (C=CAr, stretching), 1320 (C=S, stretching), 1080 (C–O, stretching) cm–1. 1H NMR (400 MHz, DMSO-d6) δ 13.08 (NH, s, 1H), 11.33 (NH, s, 1H), 10.20 (s, 1H), 8.55 (s, 1H, Ar–H), 7.72 (s, 1H, Ar–H), 7.62 (s, 1H, Ar–H), 7.10 (d, J = 8.5 Hz, 2H, Ar–H), 6.96 (d, J = 9.4 Hz, 1H, Ar–H), 6.84 (t, J = 8.4 Hz, 1H, Ar–H), 3.87 (s, 6H, OCH3) ppm. 13C NMR (100 MHz, DMSO-d6) δ 177.8 (C=S), 167.5 (C=O), 153.1, 149.0, 148.3, 126.5, 126.1, 123.8, 123.4, 123.0, 118.4, 115.2, 111.6, 111.1 (ArCs), 55.9, 55.8 (OCH3) ppm.
4.3.5. 3-(3-(3,4-Dimethoxybenzoyl) thioureido) Benzoic Acid (21)

Yield: 87%, Rf = 0.4, (n-hexane/ethyl acetate = 7:3), M.P.: 227–229 °C, color: off-white. FT-IR (ύ, cm–1): 3433 (NH, stretching), 3040 (OH, stretching), 1612 (C=O, stretching), 1446 (C=C, stretching), 1278 (C=S, stretching) cm–1. 1H NMR (400 MHz, DMSO-d6) δ 13.08 (NH, s, 1H), 11.33 (NH, s, 1H), 10.20 (OH acidic, s, 1H), 8.55 (s, 1H, Ar–H), 7.72 (s, 1H, Ar–H), 7.62 (s, 1H, Ar–H), 7.10 (d, J = 8.5 Hz, 2H, Ar–H), 6.96 (d, J = 9.4 Hz, 1H, Ar–H), 6.84 (t, J = 8.4 Hz, 1H, Ar–H), 3.87 (OCH3, s, 6H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 179.7 (C=S), 167.3 (C=O), 165.1 (C=Oacidic), 153.2, 148.4, 139.6, 138.5, 131.3, 127.1, 125.3, 124.3, 123.2, 121.3, 111.6, 111.1 (ArCs), 55.9, 55.7 (OCH3) ppm.
4.3.6. N-((2-Chloro-5-nitrophenyl) carbamothioyl)-3,4-dimethoxybenzamide (22)

Yield: 82%, Rf =0.3, (n-hexane/ethyl acetate = 4:1), M.P.: 176–178 °C, color: yellow. FT-IR (ύ, cm–1): 3427 (NH), 1643 (C=O), 1423 (C=C), 1272 (C=S), cm–1. 1H NMR (400 MHz, DMSO-d6) δ 11.88 (Ns, 1H), 9.17 (NH, s, 1H), 8.37–8.06 (m, 2H, Ar–H), 8.02–7.83 (m, 1H, Ar–H), 7.77–7.59 (m, 2H, Ar–H), 7.12 (d, J = 8.6 Hz, 1H, Ar–H), 3.8H, 7 (OCH3, s, 6H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 180.6 (C=S), 168.0 (C=O), 153.4, 148.3, 145.9, 136.7, 130.8, 129.9, 128.1, 123.4, 122.3, 122.0, 111.7, 111.2 (ArCs), 55.9, 55.8 (OCH3) ppm.
4.3.7. N-((3-Bromo-5-(trifluoromethyl) phenyl) carbamothioyl)-3,4-dimethoxybenzamide (23)

Yield: 81%, Rf =0.5, (n-hexane/ethyl acetate = 1:4), M.P.: 208–210 °C, color: off-white (cream). FT-IR (ύ, cm–1): 3431 (N–H, stretching), 1639 (C=O, stretching), 1515 (C=C, stretching), 1359 (C=S, stretching) cm–1. 1H NMR (400 MHz, DMSO-d6) δ 12.81 (NH, s, 1H), 11.65 (NH, s, 1H), 8.28 (s, 1H, Ar–H), 8.14 (s, 1H, Ar–H), 7.86 (s, 1H, Ar–H), 7.78–7.61 (m, 2H, Ar–H), 7.11 (d, J = 8.6 Hz, 1H, Ar–H), 3.86 (CH3, s, 6H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 180.3 (C=S), 167.3 (C=O), 153.3, 148.3, 140.5, 131.3, 131.1, 130.8, 124.4, 123.5, 123.3, 121.8, 121.7, 118.3, 111.6, 111.2 (ArCs), 55.9, 55.8 (OCH3) ppm.
4.3.8. N-((2-Hydroxyphenyl) carbamothioyl)-3,5-dimethylbenzamide (24)

Yield: 79%, Rf =0.4, (n-hexane/ethyl acetate = 1:4), M.P.: 256–259 °C, color: light brown. FT-IR (ύ, cm–1): 3583 (NH, stretching), 3008 (OH, stretching), 1780 (C=O, stretching), 1385 (C=C, stretching), 1309 (C=S, stretching) cm–1. 1H NMR (400 MHz, DMSO-d6) δ 12.67 (NH, s, 1H), 11.51 (NH, s, 1H), 8.30 (t, J = 1.9 Hz, 1H, Ar–H), 7.91–7.80 (m, 2H, Ar–H), 7.63 (s, 2H, Ar–H), 7.55 (t, J = 7.9 Hz, 1H, Ar–H), 7.29 (s, 1H, Ar–H), 2.35 (OCH3, s, 6H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 179.6 (C=S), 168.4 (C=O), 166.9, 138.4, 137.8, 137.8, 134.6, 132.0, 131.5, 129.1, 128.8, 127.1, 126.4 (ArCs), 30.8, 20.8 (OCH3) ppm.
4.3.9. 4-(3-(2,4-Dimethylbenzoyl) thioureido) Benzoic Acid (25)

Yield: 87%, Rf = 0.4, (n-hexane/ethyl acetate = 3:2), M.P.: 228–230 °C, color: dark yellow. FT-IR (ύ, cm–1): 3467 (NH, stretching), 3074 (OH, stretching), 1608 (C=O, stretching), 1443 (C=C, stretching), 1170 (C=S, stretching) cm–1. 1H NMR (400 MHz, DMSO-d6) δ 12.94 (NH, s, 1H), 12.77 (NH, s, 1H), 11.70 (OHacidic, s, 1H), 8.03–7.87 (m, 4H, Ar–H), 7.44 (d, J = 7.7 Hz, 1H, Ar–H), 7.16–7.08 (m, 2H, Ar–H), 2.37 (CH3, s, 6H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 179.0 (C=S), 170.6 (C=O), 166.8 (C=Oacidic), 142.0, 141.2, 136.7, 131.5, 130.9, 130.0, 128.7, 128.1, 126.2, 123.5 (ArCs), 19.7–20.23 (CH3) ppm.
4.3.10. N-((3,5-Dichloro-4-hydroxyphenyl) carbamothioyl)-3,5-dimethylbenzamide (26)

Yield: 84%, Rf = 0.3, (n-hexane/ethyl acetate = 1:4), M.P.: 227–229 °C, color: off-white (cream). FT-IR (ύ, cm–1): 3394 (NH, stretching), 1502 (C=O, stretching), 1321 (C=CAr, stretching), 1207 (C=S, stretching) cm–1. 1H NMR (400 MHz, DMSO-d6) δ 12.39 (NH, s, 1H), 11.50 (NH, s, 1H), 10.30 (OHAr, s, 1H), 8.93 (s, 1H, Ar–H), 7.57–7.64 (m, 3H, Ar–H), 7.60 (s, 1H, Ar–H), 7.28 (s, 1H, Ar–H), 2.45 (CH3, s, 6H) ppm. 13C NMR (100 MHz, DMSO-d6) δ180.0 (C=S), 168.3 (C=O), 137.8, 134.6, 133.6, 131.9, 131.3, 130.8, 126.4, 125.5, 121.7, 109.7 (ArCs), 20.8, 20.2 (CH3) ppm.
4.4. Protein Expression
The plasmids of CA II, CA IX, and CA XII were transformed into competent Escherichia coli bacteria,35 and the plasmid was extracted using the SanPrep column plasmid miniprep kit as directed in the kit instructions obtained from Sangon Biotech. Steps for transfection included seeding of HEK-293 cells in a 96-well plate by the addition of a mixture of plasmid (0.1 μg) and Lipofectamine 3000 (0.2 μL). The next step was to grow the cells in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% pen/strep mixture, and to perform transfection, the mixture was incubated at 37 °C and 5% CO2.36 After 6 h, the cells were aspirated, and the freshly prepared medium (+10% FBS and 1% pen/strep) was added. On the next day, new media were added for the selection procedure, which contained DMEM, FBS, and 200 g/mL Hygromycin B. Under a fluorescent microscope, the cells transfected with the off-spark plasmids were checked to ensure protein production after they reached 70–80% confluency in the selective media. Using the method described by ref (37), cell lysate was prepared by centrifuging the transfected cells at 2000 rpm for 5 min, washing them with PBS, and centrifuging them again. Using cell lysis buffer, the pellet was suspended, incubated on ice for 15 min, and then centrifuged again at 10000 g for 15 min at 4 °C, and then the supernatant was collected for purification with Ni-NTA.38 The eluted solution was assessed using the protein quantification technique Biuret assay and Bradford reagent.
4.5. Carbonic Anhydrase Inhibition Assay
Carbonic anhydrase inhibition activity was carried out using the previously developed method with slight modification. The principle of the current method involved hCA hydrolyzing the p-nitrophenyl acetate to p-nitrophenol, which is determined spectrophotometrically. The reaction mixture contained 60 μL of 50 mM Tris-sulfate buffer (pH 7.6 containing 0.1 mM ZnCl2), 10 μL (0.5 mM) of test compound in 1% DMSO, and 10 μL of carbonic anhydrase. All of the ingredients were blended and preincubated at 25 °C for 10 min. A 96-well plate reader was used to read the plate at 348 nm. Preparation of p-nitrophenyl acetate was done by taking 6 mM stock using <5% acetonitrile in buffer and was used fresh. To assay the well, about 20 μL of substrate solution was added to attain 0.6 mM concentration. The total assay volume was made to 100 μL. After 30 min of incubation at 37 °C, all ingredients were blended, and a reading was taken at 348 nm. Acetazolamide was used as a positive control, while 1% DMSO was used as a negative control. The results reported are the mean of three independent experiments (±SEM) and expressed as percent inhibitions calculated by the formula
IC50 values of selected compounds exhibiting >50% inhibition activity at 1.0 mM were further evaluated to measure the IC50 value through serial dilution.
4.6. In Silico Studies
4.6.1. Compound Preparation
All of the structures of the synthesized compounds are drawn using Chemdraw professional 15.0. The 3D structures of the compounds were generated using Moe software with the standard parameters, and the gradient value for the minimization of energy was selected as 0.00001 (RMS, kcal/A2). Then, the structures of the synthesized compounds were saved as Mol2 forms for further molecular docking studies.
4.6.2. Docking Studies
Docking studies have been performed on the most potent inhibitors 09, 11, 15, 13, 22, and 18 of the series using PDB IDs 3V7X(1) (CA II) (http://doi.org/10.2210/pdb3V7X/pdb), 5FL4 (CA IX)39 (http://doi.org/10.2210/pdb5FL4/pdb), and 5MSA CAXII http://doi.org/10.2210/pdb5MSA/pdb. Molecular docking was performed using Moe, LeadIT program’s Flexx utility.40 We prepared the receptor for docking using the default parameters of the protein preparation. We selected the co-crystallized inhibitors in the isozymes of carbonic anhydrase from the protein database as our reference ligand. Using Flexx docking, enthalpy and entropy approaches were used to score and rank the docking poses of the potent inhibitors after validation of the docking protocol. Liquid crystals of the co-crystallized inhibitors were used for redocking. HYDE assessment of the top-ranking poses was carried out next, followed by the selection of a possible binding mode.41
4.7. Cell Viability Assay
The cell viability assay 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was performed with slight modification in previously reported protocol34 to test selective and potent compounds for cell viability. For cell viability assay, HEK-293 T and BHK-21 normal cells were grown in DMEM containing 10% fetal bovine serum at 37 °C and 5% CO2. HEK-293 T cells (10 000/well) were seeded in a 96-well plate for 24 h. After 24 h, incubation media were removed, and compounds were added at a final concentration of 100 μM in serum-free media in triplicate, and further incubation was done for 24–48 h at 37 °C. MTT reagent was added at 4 mg/mL concentration, 100 μL per well, and left for 4–6 h until formazan crystals were formed. By adding pure DMSO, the formed crystals were dissolved. Using a microplate reader (Omega star), absorbance was taken at 590 nm, and 630 nm background absorbance was subtracted. Cell viability was determined using the following formula: absorbance of sample/absorbance of control × 100.
Acknowledgments
The authors gratefully acknowledge the financial support for this research provided by the Higher Education Commission of Pakistan (HEC) via NRPU Project No. 20-15846/NRPU/R&D/HEC/2021, German–Pakistani Research Collaboration Programme and Equipment Grant funded by DAAD, Germany.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c06513.
FTIR, 1H NMR, and 13C NMR, bioassay graphs including carbonic anhydrase graph, cell viability, and in silico docking studies (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Zaib S.; Saeed A.; Stolte K.; Flörke U.; Shahid M.; Iqbal J. New aminobenzenesulfonamide–thiourea conjugates: synthesis and carbonic anhydrase inhibition and docking studies. Eur. J. Med. Chem. 2014, 78, 140–150. 10.1016/j.ejmech.2014.03.023. [DOI] [PubMed] [Google Scholar]
- Aggarwal M.; McKenna R. Update on carbonic anhydrase inhibitors: a patent review (2008–2011). Expert Opin. Ther. Pat. 2012, 22, 903–915. 10.1517/13543776.2012.707646. [DOI] [PubMed] [Google Scholar]
- Carta F.; Supuran C. T. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin. Ther. Pat. 2013, 23, 681–691. 10.1517/13543776.2013.780598. [DOI] [PubMed] [Google Scholar]
- a Maren T. H. Carbonic anydrase: chemistry, physiology, and inhibition. Am. J. Physiol. 1992, 232, 292–297. [DOI] [PubMed] [Google Scholar]; b Kumar A.; Siwach K.; Supuran C. T.; Sharma P. K. A decade of tail-approach based design of selective as well as potent tumor associated carbonic anhydrase inhibitors. Bioorg. Chem. 2022, 126, 105920 10.1016/j.bioorg.2022.105920. [DOI] [PubMed] [Google Scholar]
- Mohamed M. M.; Sloane B. F. Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discovery 2008, 7, 168–181. 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Nada H.; Elkamhawy A.; Abdellattif M. H.; Angeli A.; Lee C. H.; Supuran C. T.; Lee K. 4-Anilinoquinazoline-based benzenesulfonamides as nanomolar inhibitors of carbonic anhydrase isoforms I, II, IX, and XII: design, synthesis, in-vitro, and in-silico biological studies. J. Enzyme Inhib. Med. Chem. 2022, 37, 994–1004. 10.1080/14756366.2022.2055553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorekova S.; Gillies R. J. The role of carbonic anhydrase IX in cancer development: links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019, 38, 65–77. 10.1007/s10555-019-09799-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau J.; Lin K.-S.; Bénard F. Past, present, and future: development of theranostic agents targeting carbonic anhydrase IX. Theranostics 2017, 7, 4322. 10.7150/thno.21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran C. T.; Alterio V.; Di Fiore A.; D’Ambrosio K.; Carta F.; Monti S. M.; De Simone G. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med. Res. Rev. 2018, 38, 1799–1836. 10.1002/med.21497. [DOI] [PubMed] [Google Scholar]
- Lou Y.; McDonald P. C.; Oloumi A.; Chia S.; Ostlund C.; Ahmadi A.; Kyle A.; Leung S.; Huntsman D.; Clarke B. Targeting Tumor Hypoxia: Suppression of Breast Tumor Growth and Metastasis by Novel Carbonic Anhydrase IX InhibitorsTargeted Inhibition of CAIX Suppresses Breast Cancer ProgressionTargeted Inhibition of CAIX Suppresses Breast Cancer Progression. Cancer Res. 2011, 71, 3364–3376. 10.1158/0008-5472.CAN-10-4261. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrases and metabolism. Metabolites 2018, 8, 25 10.3390/metabo8020025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran C. T. Diuretics: from classical carbonic anhydrase inhibitors to novel applications of the sulfonamides. Curr. Pharm. Des. 2008, 14, 641–648. 10.2174/138161208783877947. [DOI] [PubMed] [Google Scholar]
- Bao B.; Groves K.; Zhang J.; Handy E.; Kennedy P.; Cuneo G.; Supuran C. T.; Yared W.; Rajopadhye M.; Peterson J. D. In vivo imaging and quantification of carbonic anhydrase IX expression as an endogenous biomarker of tumor hypoxia. PLoS One 2012, 7, e50860 10.1371/journal.pone.0050860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- T Supuran C. Inhibition of bacterial carbonic anhydrases and zinc proteases: from orphan targets to innovative new antibiotic drugs. Curr. Med. Chem. 2012, 19, 831–844. 10.2174/092986712799034824. [DOI] [PubMed] [Google Scholar]
- Supuran C. T.; Fiore A. D.; Simone G. D. Carbonic anhydrase inhibitors as emerging drugs for the treatment of obesity. Expert Opin. Emerg. Drugs 2008, 13, 383–392. 10.1517/14728214.13.2.383. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3467–3474. 10.1016/j.bmcl.2010.05.009. [DOI] [PubMed] [Google Scholar]
- Carta F.; Scozzafava A.; Supuran C. T. Sulfonamides: a patent review (2008–2012). Expert Opin. Ther. Pat. 2012, 22, 747–758. 10.1517/13543776.2012.698264. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio K.; Smaine F.-Z.; Carta F.; De Simone G.; Winum J.-Y.; Supuran C. T. Development of potent carbonic anhydrase inhibitors incorporating both sulfonamide and sulfamide groups. J. Med. Chem. 2012, 55, 6776–6783. 10.1021/jm300818k. [DOI] [PubMed] [Google Scholar]
- Marini A. M.; Maresca A.; Aggarwal M.; Orlandini E.; Nencetti S.; Da Settimo F.; Salerno S.; Simorini F.; La Motta C.; Taliani S.; et al. Tricyclic sulfonamides incorporating benzothiopyrano [4, 3-c] pyrazole and pyridothiopyrano [4, 3-c] pyrazole effectively inhibit α-and β-carbonic anhydrase: X-ray crystallography and solution investigations on 15 isoforms. J. Med. Chem. 2012, 55, 9619–9629. 10.1021/jm300878g. [DOI] [PubMed] [Google Scholar]
- T Supuran C. Editorial [Hot Topic: Carbonic Anhydrases: Again, and Again, and Again (ExecutiveEditor: Claudiu T. Supuran)]. Curr. Pharm. Des. 2010, 16, 3231–3232. 10.2174/138161210793429779. [DOI] [PubMed] [Google Scholar]
- Schulze Wischeler J.; Innocenti A.; Vullo D.; Agrawal A.; Cohen S. M.; Heine A.; Supuran C. T.; Klebe G. Bidentate Zinc Chelators for α-Carbonic Anhydrases that Produce a Trigonal Bipyramidal Coordination Geometry. ChemMedChem 2010, 5, 1609–1615. 10.1002/cmdc.201000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan K. M.; Rahim F.; Wadood A.; Taha M.; Khan M.; Naureen S.; Ambreen N.; Hussain S.; Perveen S. MI Choudhary. Bioorg. Med. Chem. Lett. 2014, 24, 1825. 10.1016/j.bmcl.2014.02.015. [DOI] [PubMed] [Google Scholar]
- Congiu C.; Onnis V.; Balboni G.; Supuran C. T. Synthesis and carbonic anhydrase I, II, IX and XII inhibition studies of 4-N, N-disubstituted sulfanilamides incorporating 4, 4, 4-trifluoro-3-oxo-but-1-enyl, phenacylthiourea and imidazol-2 (3H)-one/thione moieties. Bioorg. Med. Chem. Lett. 2014, 24, 1776–1779. 10.1016/j.bmcl.2014.02.030. [DOI] [PubMed] [Google Scholar]
- Mahdavi M.; Shirazi M. S.; Taherkhani R.; Saeedi M.; Alipour E.; Moghadam F. H.; Moradi A.; Nadri H.; Emami S.; Firoozpour L.; et al. Synthesis, biological evaluation and docking study of 3-aroyl-1-(4-sulfamoylphenyl) thiourea derivatives as 15-lipoxygenase inhibitors. Eur. J. Med. Chem. 2014, 82, 308–313. 10.1016/j.ejmech.2014.05.054. [DOI] [PubMed] [Google Scholar]
- Connor D. T.; Roark W. H.; Sorenson R. J.. Indole and Benzimidazole 15-Lipoxygenase Inhibitors. U.S. Patent US20040038943A1, 2005.
- Cecchi A.; Hulikova A.; Pastorek J.; Pastoreková S.; Scozzafava A.; Winum J.-Y.; Montero J.-L.; Supuran C. T. Carbonic anhydrase inhibitors. Design of fluorescent sulfonamides as probes of tumor-associated carbonic anhydrase IX that inhibit isozyme IX-mediated acidification of hypoxic tumors. J. Med. Chem. 2005, 48, 4834–4841. 10.1021/jm0501073. [DOI] [PubMed] [Google Scholar]
- Khushal A.; Mumtaz A.; Shadoul W. A.; Zaidi S. H. M.; Rafique H.; Munir A.; Maalik A.; Shah S. J. A.; Baig A.; Khawaja W.; et al. Synthesis, Carbonic Anhydrase II/IX/XII Inhibition, DFT, and Molecular Docking Studies of Hydrazide-Sulfonamide Hybrids of 4-Methylsalicyl-and Acyl-Substituted Hydrazide. BioMed Res. Int. 2022, 2022, 5293349 10.1155/2022/5293349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimori I.; Vullo D.; Innocenti A.; Scozzafava A.; Mastrolorenzo A.; Supuran C. T. Carbonic anhydrase inhibitors. The mitochondrial isozyme VB as a new target for sulfonamide and sulfamate inhibitors. J. Med. Chem. 2005, 48, 7860–7866. 10.1021/jm050483n. [DOI] [PubMed] [Google Scholar]
- Swain B.; Sahoo S. K.; Singh P.; Angeli A.; Yaddanapudi V. M.; Supuran C. T.; Arifuddin M. Exploration of 2-phenylquinoline-4-carboxamide linked benzene sulfonamide derivatives as isoform selective inhibitors of transmembrane human carbonic anhydrases. Eur. J. Med. Chem. 2022, 234, 114247 10.1016/j.ejmech.2022.114247. [DOI] [PubMed] [Google Scholar]
- Goodsell D. S. Computational docking of biomolecular complexes with AutoDock. Cold Spring Harbor Protoc. 2009, 2009, pdb.prot5200 10.1101/pdb.prot5200. [DOI] [PubMed] [Google Scholar]
- Daina A.; Zoete V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016, 11, 1117–1121. 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Thomas P.; Smart T. G. HEK293 cell line: a vehicle for the expression of recombinant proteins. J. Pharmacol. Toxicol. Methods 2005, 51, 187–200. 10.1016/j.vascn.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Wang L.; Yan J.; Yan J.; Xu H.; Zhang D.; Wang X.; Sheng J. Expression and purification of the human epidermal growth factor receptor extracellular domain. Protein Expr. Purif. 2018, 144, 33–38. 10.1016/j.pep.2017.11.009. [DOI] [PubMed] [Google Scholar]
- Buran K.; Bua S.; Poli G.; Önen Bayram F. E.; Tuccinardi T.; T Supuran C. Novel 8-substituted coumarins that selectively inhibit human carbonic anhydrase IX and XII. Int. J. Mol. Sci. 2019, 20, 1208. 10.3390/ijms20051208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader P.; Pecina A.; Cígler P.; Lepšík M.; Šícha V.; Hobza P.; Grüner B.; Fanfrlík J.; Brynda J.; Řezáčová P. Carborane-based carbonic anhydrase inhibitors: insight into CAII/CAIX specificity from a high-resolution crystal structure, modeling, and quantum chemical calculations. BioMed Res. Int. 2014, 2014, 389869 10.1155/2014/389869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buran K.; Bua S.; Poli G.; Önen Bayram F. E.; Tuccinardi T.; Supuran C. T. Novel 8-substituted coumarins that selectively inhibit human carbonic anhydrase IX and XII. Int. J. Mol. Sci. 2019, 20, 1208. 10.3390/ijms20051208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudutienė V.; Zubrienė A.; Smirnov A.; Gylytė J.; Timm D.; Manakova E.; Gražulis S.; Matulis D. 4-Substituted-2, 3, 5, 6-tetrafluorobenzenesulfonamides as inhibitors of carbonic anhydrases I, II, VII, XII, and XIII. Bioorg. Med. Chem. 2013, 21, 2093–2106. 10.1016/j.bmc.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Schneider N.; Lange G.; Hindle S.; Klein R.; Rarey M. A consistent description of HYdrogen bond and DEhydration energies in protein–ligand complexes: methods behind the HYDE scoring function. J. Comput.-Aided Mol. Des. 2013, 27, 15–29. 10.1007/s10822-012-9626-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.