

Abstract
Background
Alzheimer’s disease (AD) is the most common cause of dementia worldwide. In AD, abnormal tau accumulates within neurons of the brain, facilitated by extracellular β‐amyloid deposition, leading to neurodegeneration, and eventually, cognitive impairment. As this process is thought to be irreversible, early identification of abnormal tau in the brain is crucial for the development of new therapeutic interventions.
Aims
18F‐PI‐2620 is one of the second‐generation tau PET tracers with presumably less off‐target binding than its predecessors. Although a few clinical studies have recently reported the use of 18F‐PI‐2620 tau PET in patients with AD, its applicability to AD is yet to be thoroughly examined.
Methods
In the present pilot study, we performed 18F‐PI‐2620 tau PET in seven cases of probable AD (AD group) and seven healthy controls (HC group). Standardized uptake value ratios (SUVR) in regions of interest (ROIs) in the medial temporal region and neocortex were compared between the AD and HC groups. Furthermore, correlations between regional SUVR and plasma p‐tau181 as well as cognitive test scores were also analyzed.
Results
The uptake of 18F‐PI‐2620 was distinctly increased in the AD group across all the ROIs. SUVR in all the target ROIs were significantly correlated with plasma p‐tau181 levels, as well as with MMSE and ADAS‐cog scores.
Discussion & Conclusion
Our results add to accumulating evidence suggesting that 18F‐PI‐2620 is a promising tau PET tracer that allows patients with AD to be distinguished from healthy controls, although a study with a larger sample size is warranted.
Keywords: Alzheimer’s disease, MMSE, plasma p‐181, positron emission tomography, tau, tauADAS‐cog
The present pilot study examined the ability of 18F‐PI‐2620 tau PET tracer to distinguish between healthy control and Alzheimer's disease, as well as correlations with plasma p‐tau181 and cognitive test results. Though the sample size was small, seven for each group, its SUVR in the target brain regions showed distinct differences between them and high correlations with plasma p‐tau181 and cognitive test results.

1. INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of dementia worldwide. 1 It usually impairs multiple domains of cognition, including memory, attention, visuospatial ability, language, and executive function, with a negative impact on the activities of daily living and quality of life of both the individuals with the illness and the people providing care for these individuals.
The two characteristic pathological features of AD are the presence of β‐amyloid plaques and intraneuronal hyperphosphorylated tau‐rich neurofibrillary tangles in the brain. 2 The current amyloid hypothesis holds that β‐amyloid accumulation occurs first, followed by the appearance of tau pathology that eventually leads to neurodegeneration. 3 In this process, β‐amyloid is thought to be a trigger that facilitates the spread of tau beyond the medial temporal lobe. 2 However, abnormal tau protein may start to accumulate years before β‐amyloid deposition, 4 and its accumulation in the cerebral cortex begins from the transentorhinal region and then extends to the entorhinal cortex, hippocampus, and amygdala, followed by spread into the neocortex. 5 It has been reported that the progression of tau accumulation is not necessarily symmetrical. 6 To untangle the complex process of tau deposition, in vivo visualization of its spread in the human brain is crucial. In addition to the preexisting observations on postmortem autopsies, the advent of tau tracers for positron emission tomography (PET) has enabled visualization of tau deposition in the brain even prior to any clinical manifestation of AD. At present, tau PET imaging is widely used in academic settings for a variety of research on dementia, which has provided a greater understanding of the time‐course and spatial distribution of pathological tau protein. However, the usefulness of the so‐called first‐generation tau PET tracers was hampered by their off‐target binding to such structures as β‐amyloid plaques, monoamine oxidase A and B (MAO‐A and ‐B), and the choroid plexus (7,8). Second‐generation tau tracers have now been introduced, which are thought to show less off‐target binding. 7 One of the second‐generation tracers, 18F‐PI‐2620, has been demonstrated in vitro and in mouse studies, 9 to show selective binding to tau with no off‐target binding. In addition, studies 10 , 11 conducted in humans have also reported the usefulness of 18F‐PI‐2620 for detecting tau accumulation in AD. The first‐in‐human study 10 revealed higher SUVR in the medial temporal lobe regions, posterior cingulate cortex, and neocortex in participants with AD (N = 12) than in healthy controls (HC, N = 10). It also examined the relationship between those regions of interest (ROIs) and Alzheimer’s Disease Assessment Scale; Cognitive Behavior Section (ADAS‐cog) 12 scores and found moderate correlations among ROIs in the neocortex but not in the medial temporal lobe region. Another study 11 included participants with HC (N = 36) and AD with both typical (N = 7) and atypical features (N = 4). It found that participants with typical amnestic AD features showed consistent elevations in SUVR in the medial temporal lobe region but more varying degrees of elevations in the lateral parietal and posterior cingulate cortices. On the contrary, atypical AD participants showed a different uptake profile: relative sparing of the medial temporal lobe and robust elevations in the lateral parietal and posterior cingulate cortices. Correlation analyses were not performed in this study. Although these two studies favored the ability of 18F‐PI‐2620 to distinguish between HC and AD, more research is needed to fully understand its characteristics.
Plasma concentration of tau protein phosphorylated at residue 181 (p‐tau181), another potential AD biomarker, has also attracted attention as an accurate marker for exploring its disease course. For example, plasma p‐tau 181 levels have been reported to be strongly associated with the cerebrospinal fluid (CSF) concentrations of p‐tau181 and autopsy‐confirmed Braak stages. 13 Strong linear relationships have also been shown with standardized uptake value ratios (SUVR) in both Flortaucipir (FTP) tau PET and Pittsburgh Compound B (PiB) PET. 13 Another study 14 also reported an association between plasma levels of p‐tau181 and both FTP SUVR in the entorhinal cortex and PiB PET SUVRs. It is worthy of note that the correlation between the FTP SUVR in the entorhinal cortex and plasma p‐tau181 levels was only present in participants with β‐amyloid positivity. 14 No study so far has explored the association of its plasma level with 18F‐PI‐2620.
As hitherto mentioned, there has not yet been enough research on the characteristics of 18F‐PI‐2620. Thus, the present pilot study examined for the first time the relationship of its uptake in target brain regions to plasma p‐tau181 levels and cognitive test scores in participants with AD (AD group) and healthy controls (HC group) in a Japanese population. The difference in its uptake between the two groups was also assessed.
2. METHODS
2.1. Participants
Participants of the present study comprised patients clinically diagnosed with AD recruited from the Memory Clinic at Keio University Hospital, and healthy controls enrolled from a patient recruitment agency (3H Medi Solution Inc., Tokyo, Japan). The diagnosis of probable AD was based on the 2011 NIA‐AA (the National Institute on Aging‐Alzheimer’s Association) guidelines. 15 The recruitment period was between July and December 2019. The main inclusion criteria at baseline were as follows: age 40–84 years and education years ≥12 for both the AD group and HC group; Mini‐Mental State Examination (MMSE) 16 score ≥ 24, Global Clinical Dementia Rating Score (CDR) 17 without an informant = 0, and Geriatric Depression Scale (GDS) 18 score <6 for the HC group; MMSE ≤23 and CDR with an informant = 0.5 or 1 for the AD group. The main exclusion criteria were as follows: concurrent diagnosis of neurodegenerative or neurological disease other than AD, history of major depressive disorder or bipolar disorder within the year prior to enrollment, history of any substance‐related, and/or addictive disorder within two years prior to enrollment, or history of schizophrenia diagnosis at any time.
All participants underwent comprehensive medical and neurological evaluation by a board‐certified neurologist, routine blood work, including complete blood count, blood chemistry, thyroid function tests, and vitamin B12/folate measurements, 3‐Tesla magnetic resonance imaging (MRI), and amyloid and tau PET scans. MRI, amyloid and tau PET, and cognitive tests were performed within 90 days of each other, except for one case with AD in which amyloid PET was delayed nine months.
The study design and protocol were approved by the Certified Review Board of Keio University (#N20170237), and the study was conducted in accordance with the Declaration of Helsinki. All participants and their proxies provided written informed consent for participation in the study.
The study was registered with the University Hospital Medical Information Network Clinical Trials Registry (UMIN‐CTR; https://www.umin.ac.jp/ctr/index.htm, ID# UMIN000032027) and Japan Registry of Clinical Trials (jRCT; https://jrct.niph.go.jp/, ID# jRCTs031180225).
2.2. MRI imaging
3D T1‐weighted imaging (3D BRAVO, repetition time 6.8 ms, echo time 3.0 ms, FOV 23.0 mm, voxel size 0.9 × 0.9 × 1.0 mm, Flip Angle 8°) was performed on a Discovery MR750 3.0 T scanner (GE Healthcare) at Keio University Hospital.
2.3. Amyloid PET imaging
Static 18F‐Florbetaben (FBB) PET 19 , 20 was performed in all the participants. FBB was manufactured according to good manufacturing practice at Keio University Hospital using an automated synthesizer (Synthera V2; IBA). PET imaging was performed in a PET/CT system (Siemens Biograph mCT or Siemens Biograph mCT flow, Munich, Germany; note that both equipments have equal competence, since we did not use the “flow‐motion” setting for the brain scans), with the images acquired at 90–110 min post‐injection of FBB (injected radioactivity, 308 ± 13 MBq, and molar activity, 221 ± 80 GBq/μmol), as previously described. 21 The acquired images were visually assessed as being β‐amyloid‐positive or β‐amyloid‐negative by a neuroradiologist who completed a required training. 22
2.4. Tau PET imaging
All the participants underwent dynamic 18F‐PI‐2620 PET in the Siemens Biograph mCT PET/CT system. After administration of a single dose of 18F‐PI‐2620 (injected radioactivity, 182 ± 9 MBq, and molar activity, 446 ± 156 GBq/μmol), PET images were acquired at 60–90 min post‐injection in list mode, and serial image data (5‐, 10‐, 15‐, and 20‐min images) were reviewed for the presence of head motion. Then, the PET data were reconstructed using an ordered subset expectation‐maximization algorithm.
2.5. Image processing of MRI/tau PET
The 3D T1‐weighted images were processed with FreeSurfer 23 (version 6.0.1) using the default automated reconstruction algorithm. Regions of interest (ROIs) in the present study were based on the FreeSurfer aseg 24 and aparc 25 atlases. Subsequent tau PET analyses used PMOD version 3.807 (PMOD Technologies Ltd). Each of the individual PET dataset was rigidly co‐registered to the participant’s 3D‐T1WI data and transformed into the PET native space. A manually drawn bilateral gray matter cerebellum ROI was used as reference to avoid potential spillover from the dural venous sinuses. 8 Tau deposition was represented as SUVR normalized to the cerebellum ROI. Target ROIs in the present study focused on the FreeSurfer‐defined medial temporal lobe regions (entorhinal cortex, hippocampus, amygdala, and parahippocampal cortex) and the neocortex (frontal, temporal, parietal, and occipital cortices) according to the mode of tau spread in Braak staging. 5
2.6. Plasma p‐tau181 measurement
Participants provided a 16‐mL fasting venous blood sample each (EDTA‐2 K, lavender‐top tube). Platelet‐rich plasma samples were obtained after the blood samples were centrifuged (1200 g for 10 min) within 2 h of the blood samples being drawn. Platelet‐free plasma was collected after further centrifugation in different tubes (2800 g for 10 min), aliquoted into polypropylene tubes, and stored at −80°C until the assay.
Plasma p‐tau181 was measured in‐house, using the commercial Quanterix® assay (Simoa® p‐Tau181 Advantage Kit or Simoa® NF‐light Kit) on a HD‐1 analyzer or SR–X, in accordance with the respective manufacturer’s instructions (Quanterix).
2.7. Apolipoprotein E (APOE) status
Genotyping for the APOE alleles (rs429358 and rs7412) that give rise to the three major isoforms (APOE ε2, APOE ε3, and APOE ε4) was performed in MCBI (Ibaraki, Japan).
2.8. Cognitive assessment
Cognitive function was assessed with CDR, MMSE, and ADAS‐cog.
2.9. Statistical analysis
To assess the differences in the baseline demographic characteristics between the AD and HC groups, Mann–Whitney’s U test was performed for continuous variables and the χ2 test for dichotomous data. Mann–Whitney’s U test was also used to compare regional PI‐2620 SUVR between the two groups. We examined each hemisphere separately since asymmetry in tau accumulation may exist. 6 The potential asymmetry in SUVR was assessed by calculating the hemispheric asymmetry index (AI) 26 , 27 for each ROI using the following formula:
where R = SUVR in a right ROI and L = SUVR in a left ROI.
Differences in the AI between the two groups were analyzed by the Mann–Whitney’s U test. Pearson’s correlation coefficients between SUVR and plasma p‐tau181 in the entire participants were determined. Correlations between SUVR and cognitive test scores were also explored. In addition, the same correlation analyses were performed in the AD group only to see whether similar relationships to those found in the entire participants exist. All p‐values were corrected using the Benjamini–Hochberg false discovery rate (FDR) method, with q‐values <0.05 reported as statistically significant. All the statistical analyses were conducted using R version 4.0.0 (https://www.r‐project.org/) and jamovi version 1.2.27.0 (https://www.jamovi.org).
3. RESULTS
3.1. Demographics
Of the 15 participants, eight participants had a clinical diagnosis of probable AD and seven participants were healthy controls. One participant with AD dropped out of the study, and all the remaining seven participants with a clinical diagnosis of AD showed β‐amyloid positivity in their amyloid PET scans. No amyloid pathology was observed in any of the seven healthy controls; thus, there were seven participants each in the AD and HC groups according to AT(N) classification system. 3 The participants ranged in age from 60 to 81 years at the time of enrollment. While no significant differences were seen in the distribution of sex, age, or education years between the AD and HC groups, cognitive test scores were significantly worse, and plasma p‐tau181 levels were significantly higher in the AD group than in the HC group (Table 1).
TABLE 1.
Demographics and cognitive test scores according to the diagnosis
| AD (N = 7) | HC (N = 7) | P value | |
|---|---|---|---|
| Mean (SD) or N (%) | |||
| Sex, No of male participants (%) | 4 (57.1%) | 4 (57.1%) | 1 |
| Age | 70.7 (9.11) | 71.1 (6.64) | 0.897 |
| APOE ɛ4, No of positive participants (%) | 5 (71.4%) | 1 (14.3%) | 0.031 |
| Education years | 13.7 (2.14) | 14.7 (3.30) | 0.674 |
| CDR_Global, No of participants (%) |
CDR = 0.5: 4 (57.1%) CDR = 1: 3 (42.9%) |
CDR = 0: 7 (100%) | N/A |
| MMSE score | 18.7 (5.12) | 28.4 (1.51) | 0.002 |
| ADAS‐Cog score | 24.1 (8.45) | 3.26 (1.24) | 0.002 |
| Plasma p‐tau181 (pg/mL) | 4.13 (0.993) | 1.12 (0.207) | < 0.001 |
Note: Differences in the baseline characteristics between the AD and HC groups were analyzed by the Mann–Whitney U test for continuous variables and the chi‐squared test for dichotomous variables.
Abbreviations: ADAS, Alzheimer’s Disease Assessment Scale Cognitive Behavior Section; AD, Alzheimer’s disease; CDR, Clinical Dementia Rating; HC, healthy control; MMSE, Mini‐Mental State Examination; N/A, not applicable; p‐tau181, phosphorylated tau at residue 181; SD, standard deviation.
3.2. Comparison of the mean 18F‐PI‐2620 SUVR
As shown in Table 2 and Figure 1, significant differences in 18F‐PI‐2620 binding in the target ROIs were observed between the AD and HC group. In general, participants with AD exhibited elevated values of SUVR in the medial temporal lobe region and neocortex bilaterally, as compared to the HC group. The highest SUVR among all the ROIs was found in the entorhinal cortices bilaterally. The average AI was within 7% in all the target ROIs in both the AD and HC groups, and no significant difference in the laterality was found in the respective ROIs between the two groups (Table 3).
TABLE 2.
Comparison of 18F‐PI‐2620 SUVR between the AD and HC groups
| AD (N = 7) | HC (N = 7) | P value | Effect size | |
|---|---|---|---|---|
| Mean (SD) | ||||
| Left hippocampus | 1.81 (0.219) | 1.42 (0.149) | 0.011 | 0.796 |
| Right hippocampus | 1.70 (0.198) | 1.38 (0.166) | 0.011 | 0.796 |
| Left amygdala | 2.07 (0.443) | 1.19 (0.149) | 0.001 | 0.959 |
| Right amygdala | 1.98 (0.316) | 1.14 (0.082) | <0.001 | 1 |
| Left entorhinal Cortex | 2.34 (0.601) | 1.42 (0.179) | 0.002 | 0.918 |
| Right entorhinal Cortex | 2.36 (0.555) | 1.52 (0.186) | 0.002 | 0.918 |
| Left parahippocampal cortex | 1.93 (0.553) | 1.30 (0.077) | 0.001 | 0.959 |
| Right parahippocampal cortex | 1.88 (0.404) | 1.28 (0.092) | 0.001 | 0.959 |
| Left frontal cortex | 1.69 (0.815) | 1.11 (0.078) | 0.001 | 0.959 |
| Right frontal cortex | 1.66 (0.606) | 1.15 (0.087) | 0.011 | 0.796 |
| Left parietal cortex | 1.94 (0.770) | 1.15 (0.030) | <0.001 | 1 |
| Right parietal cortex | 1.88 (0.738) | 1.16 (0.051) | <0.001 | 1 |
| Left temporal cortex | 2.17 (0.869) | 1.21 (0.087) | <0.001 | 1 |
| Right temporal cortex | 2.16 (0.730) | 1.24 (0.083) | <0.001 | 1 |
| Left occipital cortex | 1.85 (0.631) | 1.27 (0.042) | 0.011 | 0.796 |
| Right occipital cortex | 1.76 (0.622) | 1.27 (0.050) | <0.001 | 1 |
Note: Differences in the mean SUVR between the AD and HC groups were analyzed by the Mann–Whitney U test. The rank‐biserial correlation is reported as effect size.
Abbreviations: AD, Alzheimer’s disease; HC, healthy control; SD, standard deviation; SUVR, standardized uptake value ratio.
FIGURE 1.
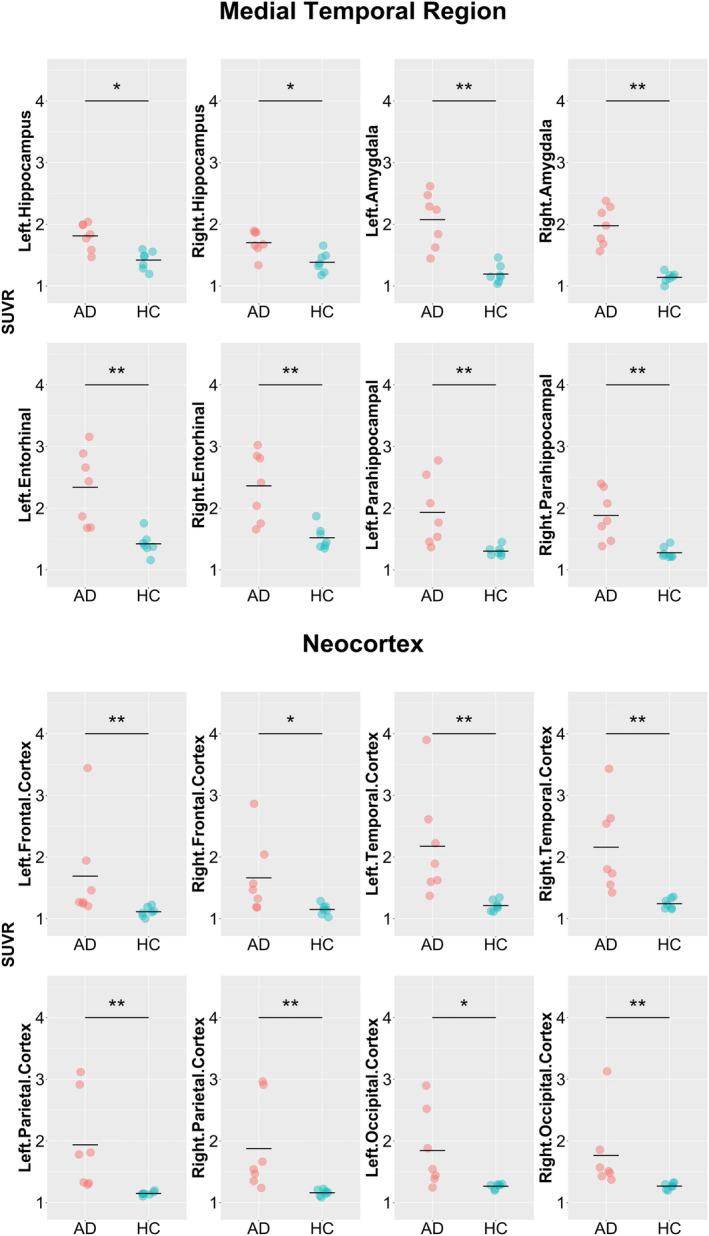
Comparison of 18F‐PI‐2620 SUVR between the AD and HC groups. AD, Alzheimer’s disease; HC, healthy control; SUVR, standardized uptake value ratio. Comparison of SUVR in the target regions of interest. The dots in the graph indicate the SUVR value in each participant and the horizontal bar indicates the mean SUVR value. AD and HC participants are expressed in red and green dots, respectively. Asterisks (*) and (**) represent significant P‐values after false discovery rate (FDR) correction at q = 0.05 and 0.01, respectively
TABLE 3.
Laterality (AI) difference in 18F‐PI‐2620 SUVR between the AD and HC groups
| AD (N = 7) | HC (N = 7) | P value | |
|---|---|---|---|
| AI (%) (SD) | |||
| Hippocampus | −6.24 (12.99) | −2.69 (5.2) | 0.383 |
| Amygdala | −3.87 (12.42) | −4.25 (7.4) | 1 |
| Entorhinal cortex | 1.41 (10.93) | 6.74 (5.44) | 0.165 |
| Parahippocampal cortex | −1.17 (13.01) | −2.13 (3.49) | 0.902 |
| Frontal cortex | 0.9 (12.23) | 3.24 (5.99) | 0.805 |
| Parietal cortex | −2.85 (13.97) | 1.02 (2.46) | 0.383 |
| Temporal cortex | 0.53 (18.26) | 2.46 (3.04) | 0.62 |
| Occipital cortex | −3.94 (17.08) | 0.09 (1.38) | 0.902 |
Note: Differences in the AI between the AD and HC groups were analyzed by the Mann–Whitney U test.
Abbreviations: AD, Alzheimer’s disease; AI, asymmetry index; HC, healthy control; SD, standard deviation; SUVR, standardized uptake value ratio.
3.3. Correlation between 18F‐PI‐2620 SUVR and plasma p‐tau181 levels
Correlation coefficients between 18F‐PI‐2620 SUVR and plasma p‐tau181 levels were moderate‐to‐high across all the target ROIs, ranging from 0.62 to 0.95 in the medial temporal region and from 0.42 to 0.68 in the neocortex. All of the ROIs in the medial temporal region had statistical significance, whereas the bilateral temporal and right parietal cortices survived the FDR correction in the neocortex (Figure 2). However, when limiting the analyses to the seven AD participants, most of the ROIs showed only low correlations except for the bilateral hippocampus and amygdala. None of the results were statistically significant (Figure S1).
FIGURE 2.
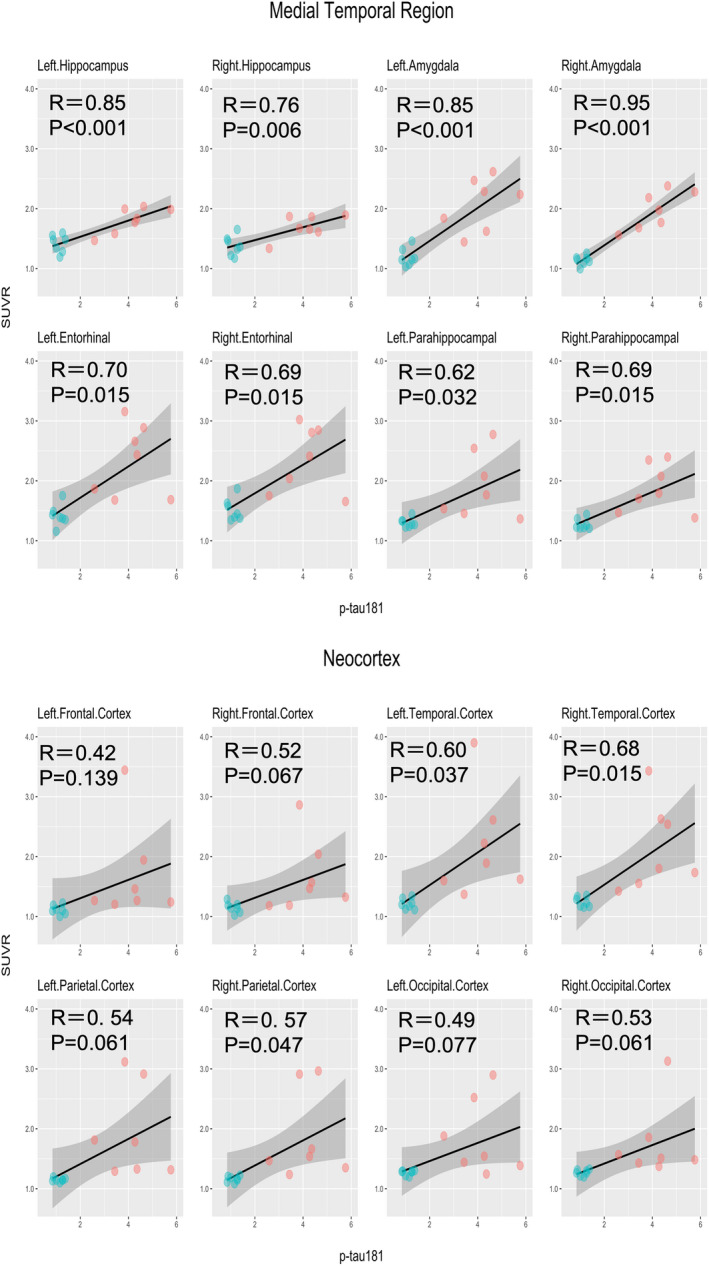
Correlations between 18F‐PI‐2620 SUVRs and plasma p‐tau181. p‐tau181, phosphorylated tau at residue 181; P, P‐values after false discovery rate (FDR) correction; R, Pearson’s correlation coefficient; SUVR, standardized uptake value ratio. Correlations between SUVR in the target regions of interest and plasma p‐tau181 levels. AD and HC participants are expressed in red and green dots, respectively
3.4. Correlation between 18F‐PI‐2620 SUVR and cognitive test scores
Both the MMSE and ADAS‐cog results had moderate‐to‐high correlations with 18F‐PI‐2620 SUVR. All the correlations but one (i.e., the correlation between the right hippocampus and MMSE) were statistically significant (Figures 3 and 4). Correlation coefficients for the MMSE were between −0.48 and − 0.83 in the medial temporal lobe region and between −0.65 and − 0.84 in the neocortex. Those for the ADAS‐cog were between 0.60 and 0.86 in the medial temporal lobe region and between 0.63 and 0.89 in the neocortex. When limiting the analyses to the AD participants only, these cognitive tests had comparable correlations with SUVR in the target ROIs to those in the entire participants, except for the lower correlations with the bilateral hippocampus and amygdala. However, none of the results were significant after FDR correction (Figures S2 and S3).
FIGURE 3.
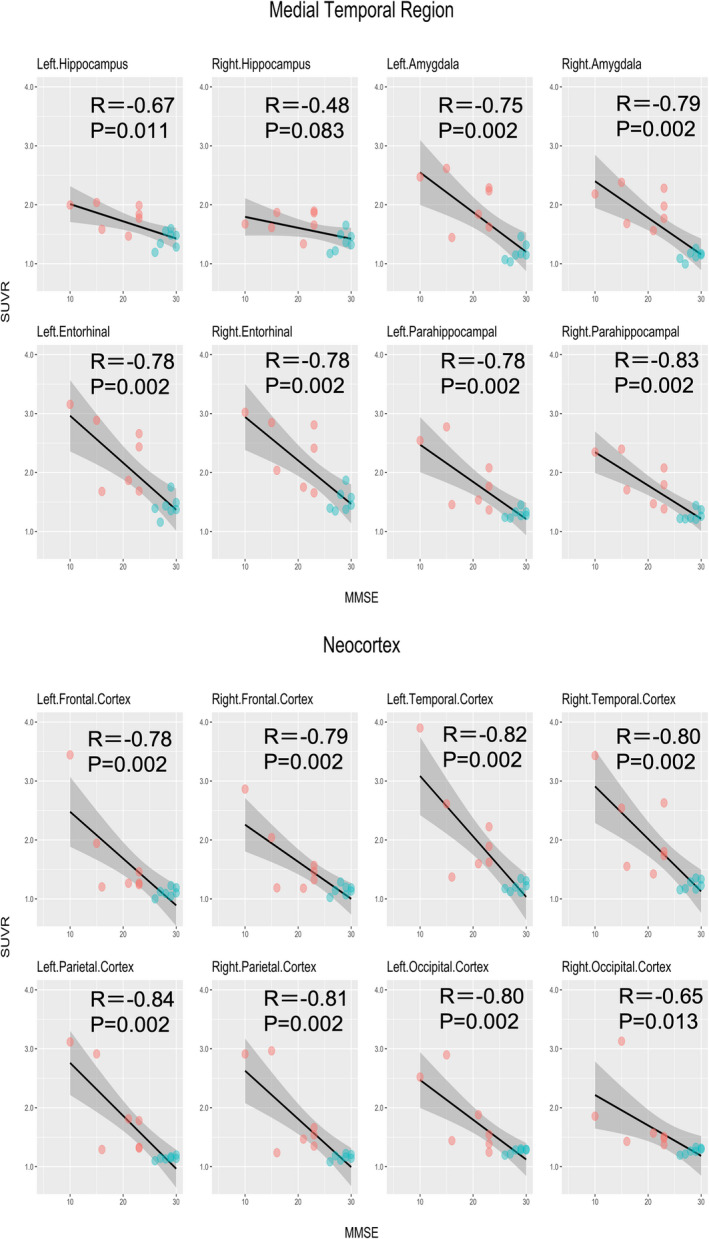
Correlations between 18F‐PI‐2620 SUVR and MMSE. MMSE, Mini‐Mental State Examination; P, P‐values after false discovery rate (FDR) correction; R, Pearson’s correlation coefficient; SUVR, standardized uptake value ratio. Correlations between SUVR in the target regions of interest and the MMSE scores. AD and HC participants are expressed in red and green dots, respectively
FIGURE 4.
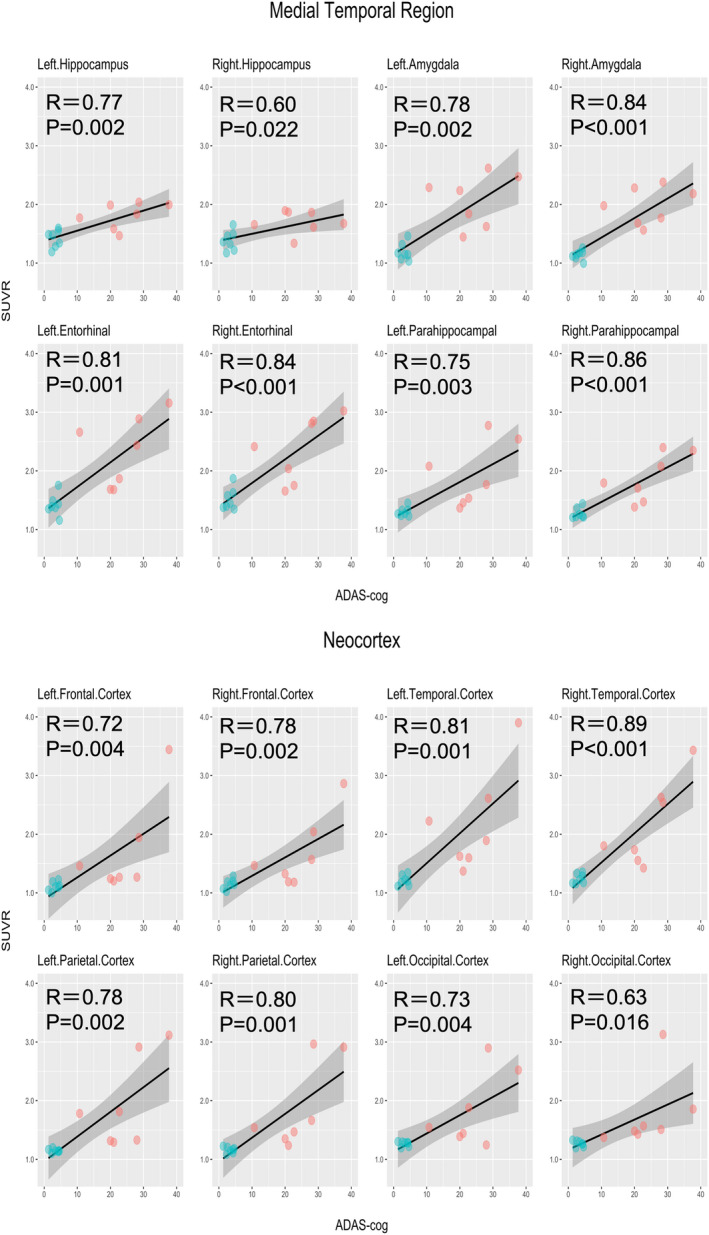
Correlations between 18F‐PI‐2620 SUVR and ADAS‐cog. ADAS‐cog, Alzheimer’s Disease Assessment Scale Cognitive Behavior Section; P, P‐values after false discovery rate (FDR) correction; R, Pearson’s correlation coefficient; SUVR, standardized uptake value ratio. Correlations between SUVR in the target regions of interest and the ADAS‐cog scores. AD and HC participants are expressed in red and green dots, respectively
4. DISCUSSION
In recent years, tau imaging has become rapidly accessible in academic settings. Multiple tau PET tracers have been developed for use in various tauopathies, especially AD. However, the so‐called first‐generation tau PET tracers had the drawback of off‐target binding, 7 which made interpretation of the results difficult. On the contrary, the second‐generation tau PET tracers, of which 18F‐PI‐2620 is one, are thought to show decreased off‐target binding, although their usefulness has yet to be thoroughly examined. Thus, we conducted a pilot study to examine the ability of 18F‐PI‐2620 to distinguish between AD and HC, with particular focus on its relationship with plasma p‐tau181, which previous reports did not explore.
We noted increased 18F‐PI‐2620 SUVR in the AD group as compared to the HC group bilaterally in both the medial temporal lobe region and neocortex. Despite the small sample size of 14 participants in this study, the differences in SUVR between the two groups were distinct, with large effect sizes. Our results corroborate a first‐in‐humans study 10 published in 2020, which showed higher 18F‐PI‐2620 SUVR in the medial temporal lobe region and neocortex in patients with AD than in healthy controls. While the age range of the participants in that study was relatively broad, it was narrower in our study. Similar results were reported from another 18F‐PI‐2620 tau PET study 11 that examined the differences in SUVR between participants with AD continuum and HC participants.
We also observed robust associations between plasma p‐tau181 levels and 18F‐PI‐2620 SUVR across all the target ROIs, with statistical significance in all the ROIs in the medial temporal lobe region and nearly half of the ROIs in the neocortex. In our power analyses, to reach statistical significance for a one‐sided correlation test with a power of 0.8 and a coefficient of 0.4 or 0.5, sample size must be greater than 36 or 22, respectively. Thus, our sample size of 14 is clearly insufficient. Nevertheless, to the best of our knowledge, this is the first report of the association of 18F‐PI‐2620 SUVR with plasma p‐tau181 levels in patients with AD and healthy controls. Recently, measurement of this blood‐based biomarker has been in the spotlight as an accurate marker of tau accumulation in the brains of patients with AD, 13 , 14 , 28 and elevation of the plasma p‐tau 181 levels is considered to be specific to AD. 29 Previous studies have demonstrated good correlations between plasma p‐tau181 levels and the degree of tau deposition in the brain as measured by FTP tau PET. 13 , 14 The consistent and robust correlations between 18F‐PI‐2620 SUVR and plasma p‐tau181 levels found in the present study align with these reports and reinforce the usefulness of the new tracer in detecting AD. These correlations were not statistically significant when limited to AD participants only, unlike the previous study that showed significant correlations between plasma p‐tau181 and FTP tau PET SUVR in most ROIs in the medial temporal lobe region and the neocortex. The different results might be due to our study’s much smaller sample size of only seven.
With regard to the relationships between cognitive test scores and 18F‐PI‐2620 SUVR, we found strong correlations with statistical significance in almost all the target ROIs. The correlations with the MMSE scores and ADAS‐cog scores were similar. These results are concordant with those of the aforementioned first‐in‐humans study 10 of 18F‐PI‐2620 tau PET but with much stronger correlations across all the ROIs. This may be partly because the MMSE scores of some participants with AD in the first‐in‐human study were almost within the normal range, which might have weakened the correlation with the tracer uptake. When limiting the analyses to the seven AD participants, these correlations lost statistical significance. Again, a larger sample size is needed to draw a practical inference.
As stated above, findings in the present study favor the usefulness of 18F‐PI‐2620 tau PET in distinguishing AD from HC. However, it should be noted that participants in the AD group in our study were relatively homogenous, with CDR of 0.5 or 1. The more severe or milder (i.e., mild cognitive impairment) cases were not included. Although we speculate that the uptake of 18F‐PI‐2620 in the brain would tend to follow the progression of the disease, it is still unclear how the actual tracer uptake would be across the AD continuum. The inclusion of participants with all stages of AD is desirable in future studies.
In terms of the spatial distribution of tau, asymmetric lateralization has been reported in AD patients with atypical presentations. In addition, even patients with typical AD might show some lateralization, particularly in the later phase of the disease. 30 However, in the present study, no significant laterality in any of the target ROIs was observed in the AD group. The reasons, in addition to the small sample size, could be that only AD participants with typical clinical presentations were included in the present study, and that all the participants were in the relatively early phase of the disease.
We did not perform analyses on the involvement of APOE ɛ4 status because its carriers were skewed toward the AD group (i.e., five out of seven in the AD group and one out of seven in the HC group). However, APOE ɛ4 has been suggested to have a role toward accumulating tau in the brain in people with 31 or without 32 , 33 cognitive impairment. It may even facilitate tau accumulation in the medial temporal lobe in people with neither dementia nor β‐amyloid accumulation. 34 On the contrary, another report did not observe the effect. 35 Therefore, its role in tau accumulation in the brain is to be further explored.
Our work has several limitations. First and foremost, the results of the study cannot be generalized because of the small number of study participants, and it is, therefore, premature to draw any definitive conclusions. That being said, we still observed distinct differences in 18F‐PI‐2620 SUVR between the AD and HC groups, along with the strong correlations of 18F‐PI‐2620 SUVR with the plasma p‐tau181 levels and cognitive test scores. Other limitations include the lack of diversity in the AD stages, and the study’s cross‐sectional nature. Future research should be longitudinal and include more participants with various disease stages to assess the ability of 18F‐PI‐2620 to detect the earliest tau deposition among healthy older adults and characterize longitudinal changes in its accumulation as the disease progresses.
5. CONCLUSION
The increased uptake of 18F‐PI‐2620 in the AD group as compared to the HC group was evident in the brain areas affected by AD pathophysiology. The tracer uptake was also correlated with the plasma p‐tau181 levels and cognitive test scores. The results of the present study add to accumulating evidence of the usefulness and applicability of 18F‐PI‐2620 in the field of AD research. Further validation of 18F‐PI‐2620 tau PET is warranted in a larger sample of individuals in various phases of the disease.
AUTHOR CONTRIBUTIONS
MM, DI, and HT conceived and designed the study. SB, SM, TT, SN, DI, and MM drafted the manuscript and figures. All authors participated in data collection and analysis.
CONFLICT OF INTEREST
Author YS is employed by Eisai Co., Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ETHICAL APPROVAL
The study design and protocol were approved by the Certified Review Board of Keio University (#N20170237), and the study was conducted in accordance with the Declaration of Helsinki.
PATIENT CONSENT STATEMENT
All participants and their proxies provided written informed consent for participation in the study.
REGISTRY AND THE REGISTRATION NO. OF THE STUDY
The study was registered with the University Hospital Medical Information Network Clinical Trials Registry (UMIN‐CTR; https://www.umin.ac.jp/ctr/index.htm, ID# UMIN000032027) and Japan Registry of Clinical Trials (jRCT; https://jrct.niph.go.jp/, ID# jRCTs031180225).
ANIMAL STUDIES
Not applicable.
Supporting information
Figure S1
Figure S2
Figure S3
Table S1
ACKNOWLEDGEMENTS
The authors would like to thank Mr. Kiyotaka Nakajima, Mr. Kouki Oumi, Mr. Yosinori Taniguchi, Mr. Kazuya Minamishima, Mr. Yoshiki Oowaki, and the staff of the Division of Nuclear Medicine and the Department of Radiology for their help with the PET examinations and image processing.
Bun S, Moriguchi S, Tezuka T, Sato Y, Takahata K, Seki M, Findings of 18F‐PI‐2620 tau PET imaging in patients with Alzheimer’s disease and healthy controls in relation to the plasma P‐tau181 levels in a Japanese sample. Neuropsychopharmacol Rep. 2022;42:437–448. 10.1002/npr2.12281
Funding information
This research was supported by AMED Grant Number 17pc0101006
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in the Table S1.
REFERENCES
- 1. WHO . Global Action Plan on the Public Health Response to Dementia 2017–2025 [Internet]. Geneva: World Health Organization; 2021. [Google Scholar]
- 2. van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid‐β‐independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci. 2020;21(1):21–35. [DOI] [PubMed] [Google Scholar]
- 3. Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70(11):960–9. [DOI] [PubMed] [Google Scholar]
- 5. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82(4):239–59. [DOI] [PubMed] [Google Scholar]
- 6. King A, Bodi I, Nolan M, Troakes C, Al‐Sarraj S. Assessment of the degree of asymmetry of pathological features in neurodegenerative diseases. What is the significance for brain banks? J Neural Transm. 2015;122(10):1499–508. [DOI] [PubMed] [Google Scholar]
- 7. Leuzy A, Chiotis K, Lemoine L, Gillberg P‐G, Almkvist O, Rodriguez‐Vieitez E, et al. Tau PET imaging in neurodegenerative tauopathies—still a challenge. Mol Psychiatry. 2019;24(8):1112–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lemoine L, Leuzy A, Chiotis K, Rodriguez‐Vieitez E, Nordberg A. Tau positron emission tomography imaging in tauopathies: the added hurdle of off‐target binding. Alzheimers Dement. 2018;10:232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kroth H, Oden F, Molette J, Schieferstein H, Capotosti F, Mueller A, et al. Discovery and preclinical characterization of [18F]PI‐2620, a next‐generation tau PET tracer for the assessment of tau pathology in Alzheimer’s disease and other tauopathies. Eur J Nucl Med Mol Imaging. 2019;46(10):2178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mueller A, Bullich S, Barret O, Madonia J, Berndt M, Papin C, et al. Tau PET imaging with 18F‐PI‐2620 in patients with Alzheimer disease and healthy controls: a first‐in‐humans study. J Nucl Med. 2020;61(6):911–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mormino EC, Toueg TN, Azevedo C, Castillo JB, Guo W, Nadiadwala A, et al. Tau PET imaging with 18F‐PI‐2620 in aging and neurodegenerative diseases. Eur J Nucl Med Mol Imaging. 2021;48(7):2233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry. 1984;141(11):1356–64. [DOI] [PubMed] [Google Scholar]
- 13. Thijssen EH, La Joie R, Wolf A, Strom A, Wang P, Iaccarino L, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat Med. 2020;26(3):387–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mielke MM, Hagen CE, Xu J, Chai X, Vemuri P, Lowe VJ, et al. Plasma phospho‐tau181 increases with Alzheimer’s disease clinical severity and is associated with tau‐ and amyloid‐positron emission tomography. Alzheimers Dement. 2018;14(8):989–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging‐Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”: a practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research. 1975;12(3):189–98. [DOI] [PubMed] [Google Scholar]
- 17. Berg L. Clinical dementia rating (CDR). Psychopharmacol Bull. 1988;24(4):637–9. [PubMed] [Google Scholar]
- 18. Yesavage JA, Sheikh JI. 9/Geriatric Depression Scale (GDS). Clin Gerontologist. 1986;5(1–2):165–79. [Google Scholar]
- 19. Sabri O, Seibyl J, Rowe C, Barthel H. Beta‐amyloid imaging with florbetaben. Clin Transl Imaging. 2015;3(1):13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sabri O, Sabbagh MN, Seibyl J, Barthel H, Akatsu H, Ouchi Y, et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer’s disease: phase 3 study. Alzheimer’s & Dementia. 2015;11(8):964–74. [DOI] [PubMed] [Google Scholar]
- 21. Mashima K, Ito D, Kameyama M, Osada T, Tabuchi H, Nihei Y, et al. Extremely low prevalence of amyloid positron emission tomography positivity in Parkinson’s disease without dementia. Eur Neurol. 2017;77(5–6):231–7. [DOI] [PubMed] [Google Scholar]
- 22. Bullich S, Catafau A, Senda M, Khodaverdi‐Afaghi V, Stephens A. Performance of 18F‐florbetaben PET image reading training in japanese language. J Nuclear Med. 2016;57(2):1822. [Google Scholar]
- 23. Fischl B. FreeSurfer. NeuroImage. 2012;62(2):774–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33(3):341–55. [DOI] [PubMed] [Google Scholar]
- 25. Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31(3):968–80. [DOI] [PubMed] [Google Scholar]
- 26. Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain. 2016;139(5):1551–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rabinovici GD, Jagust WJ, Furst AJ, Ogar JM, Racine CA, Mormino EC, et al. Aβ amyloid & glucose metabolism in three variants of primary progressive aphasia. Ann Neurol. 2008;64(4):388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frings L, Hellwig S, Spehl TS, Bormann T, Buchert R, Vach W, et al. Asymmetries of amyloid‐β burden and neuronal dysfunction are positively correlated in Alzheimer’s disease. Brain. 2015;138(10):3089–99. [DOI] [PubMed] [Google Scholar]
- 29. Simrén J, Leuzy A, Karikari TK, et al. The diagnostic and prognostic capabilities of plasma biomarkers in Alzheimer’s disease. Alzheimers Dement. 2021;17:1145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moscoso A, Grothe MJ, Ashton NJ, Karikari TK, Lantero Rodríguez J, Snellman A, et al. Longitudinal associations of blood phosphorylated Tau181 and Neurofilament light chain with neurodegeneration in Alzheimer disease. JAMA Neurol. 2021;78:396–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vogel JW, Iturria‐Medina Y, Strandberg OT, Smith R, Levitis E, Evans AC, et al. Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat Commun. 2020;11(1):2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jack CR, Wiste HJ, Weigand SD, Therneau TM, Lowe VJ, Knopman DS, et al. Predicting future rates of tau accumulation on PET. Brain. 2020;143(10):3136–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Salvadó G, Grothe MJ, Groot C, Moscoso A, Schöll M, Gispert JD, et al. Differential associations of APOE‐ε2 and APOE‐ε4 alleles with PET‐measured amyloid‐β and tau deposition in older individuals without dementia. Eur J Nucl Med Mol Imaging. 2021;48(7):2212–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tan M‐S, Yang Y‐X, Wang H‐F, Xu W, Tan C‐C, Zuo C‐T, et al. PET amyloid and tau status are differently affected by patient features. J Alzheimers Dis. 2020;78(3):1129–36. [DOI] [PubMed] [Google Scholar]
- 35. Weigand AJ, Thomas KR, Bangen KJ, Eglit GML, Delano‐Wood L, Gilbert PE, et al. APOE interacts with tau PET to influence memory independently of amyloid PET in older adults without dementia. Alzheimer’s & Dementia. 2021;17(1):61–9. [DOI] [PubMed] [Google Scholar]
- 36. Ossenkoppele R, Smith R, Mattsson‐Carlgren N, Groot C, Leuzy A, Strandberg O, et al. Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal Alzheimer disease: a head‐to‐head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 2021;78:961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Table S1
Data Availability Statement
The data that support the findings of this study are available in the Table S1.