

Abstract
Aims
Fabry disease (FD) is a rare X‐linked genetic disorder caused by α‐galactosidase A (AGALA) deficiency. Whereas ‘classic’ variant has multisystemic manifestation, the more recently described ‘later‐onset’ variant is characterized by predominant cardiac involvement that often mimics hypertrophic cardiomyopathy (HCM).
Methods and results
Consecutive unrelated patients with HCM were screened for FD in 16 (out of 17) cardiac centres in the Czech Republic covering specialized cardiology care from June 2017 to December 2018. AGALA activity and globotriaosylsphingosine (lyso‐Gb3) levels were measured in all subjects using the dry blood spot method. FD was suspected in male patients with AGALA activity <1.2 μmol/h/L and in females with either low AGALA activity or lyso‐Gb3 > 3.5 ng/mL. Positive screening results were confirmed by genetic testing. We evaluated 589 patients (390 males, 66%) with HCM (mean maximal myocardial thickness 19.1 ± 4.3 mm). The average age was 58.4 ± 14.7 years. In total, 17 patients (11 males, 6 females) had a positive screening result, and subsequently, six of them (four males and two females) had a genetically confirmed pathogenic GLA mutation (total prevalence of 1.02%). Five of these patients were carrying the p.N215S mutation known to cause a typical later‐onset cardiac FD.
Conclusions
We confirmed the prevalence of FD repeatedly reported in previous screening programmes (approximately 1% irrespective of gender) in a non‐selected HCM population in Central Europe. Our findings advocate a routine screening for FD in all adult patients with HCM phenotype including both genders. The dry blood spot method used led to identification of clearly pathogenic variants.
Keywords: Fabry disease, Hypertrophic cardiomyopathy, Screening, Alpha‐galactosidase, Lyso‐Gb3 , Genetic testing
Introduction
Fabry disease (FD) is a rare genetic disorder caused by mutations in the GLA gene encoding α‐galactosidase A (AGALA). Deficient AGALA activity leads to intralysosomal accumulation of neutral glycosphingolipids, predominantly globotriaosylceramide (Gb3), in various tissues, causing profound structural and functional cellular damage. 1 , 2 The inheritance pattern is X‐linked; hemizygous men are usually more severely affected as compared with heterozygous women. In the latter, the severity depends also on the X chromosome inactivation (females with skewed inactivation towards expression of the chromosome carrying the mutation may have a severe course of the disease). Homozygosity in women appears to be extremely rare.
The classical clinical phenotype is characterized by multiple organ involvement with age‐dependent manifestations. The disease is affecting peripheral and central nervous system (leading to neuropathic peripheral pain, white matter lesions, and premature stroke), skin (angiokeratomas, hypohidrosis), kidney (albuminuria and glomerular filtration impairment), gastrointestinal system (abdominal pain, diarrhoea), and heart. Cardiac involvement consists dominantly of ventricular hypertrophy, progressive left ventricular (LV) scarring, heart failure, tachyarrhythmias, bradycardia and AV conduction defects, and valvular involvement. Because LV hypertrophy is usually pronounced, the manifestation resembles sarcomeric hypertrophic cardiomyopathy (HCM). 3 Besides the classical phenotype, a large proportion of patients carry mutations causing a later‐onset phenotype with manifestations mostly limited to the heart. Moreover, a large number of non‐pathogenic gene variants or variants of unknown significance were described. 1 , 2 Early identification of affected patients with pathogenic mutations is of utmost importance as available specific treatment with enzyme replacement therapy and small molecular chaperone (migalastat) may slow down the disease progression and leads to some functional and structural improvements. 4 , 5
Several screening studies in patients with unexplained LV hypertrophy and/or HCM diagnosis have shown a relatively high prevalence of previously unrecognized FD in these cohorts. 6 , 7 , 8 In spite of this evidence, systematic testing for FD did not fully penetrate to the clinical routine mainly due to limited availability of enzyme activity assessment and costs related with gene sequencing. 9 Since about 10 years assays of AGALA activity measurements in dried blood spots (DBS) became available. Low AGALA activity measured from DBS is a reliable diagnostic tool in male patients. 10 However, in female heterozygotes, AGALA activity is often borderline or even within normal ranges, and the diagnosis is often feasible only by gene sequencing. 11 This method appears laborious, costly, and problematic due to identification of large numbers of non‐pathogenic variants and polymorphisms. 9 , 12
The search for an effective biomarker of FD revealed that plasma globotriaosylsphingosine (lyso‐Gb3) is elevated in presence of pathogenic variants, particularly in classically affected patients. 13 , 14 Although lyso‐Gb3 levels are generally lower in women as compared with men and less elevated in later‐onset mutations, it has been proposed as a reliable diagnostic tool for identification of suspected FD patients in screening programmes. 15
Because new DBS technique combining AGALA and lyso‐Gb3 measurements allows a screening in both genders, we designed a large study focusing on a population of HCM patients diagnosed in tertiary centres and screened both men and women. The second objective of the study was to confirm that the selected diagnostic approach based on lyso‐Gb3 in females would be able to detect later‐onset variants of FD. 16
Methods
The study was organized by the Czech Society of Cardiology. In total, 16 out of the 17 specialized tertiary cardiovascular centres providing cardiology care in the Czech Republic (10.6 million people) participated in the screening. We included consecutive unrelated patients diagnosed or followed‐up for HCM. The screening took place from 1 June 2017 to 31 December 2018 (at least 12 months in each centre). HCM was defined by the presence of increased LV wall thickness (≥15 mm) that was not solely explained by abnormal loading conditions in one or more myocardial segments on echocardiography, MRI, or cardiac CT. 17 Patients with known FD and other HCM phenocopies including infiltrative diseases (e.g. amyloidosis) were excluded. All patients had to be older than 18 years and required to give a signed informed consent containing the information about Fabry disease and underlining the need of a subsequent investigation in a specialized centre to confirm the test positivity. The study was approved by multicentre Ethics Committee of the General University Hospital in Prague.
After completion of a brief questionnaire (age, family history of HCM, maximal wall thickness, LV outflow tract obstruction, implantable cardioverter‐defibrillator implantation, and possible extracardiac manifestation of FD), blood samples were taken and processed according to the DBS method described elsewhere. 16
All dried DBS kits were collected, anonymized and shipped to ARCHIMED Life laboratory in Vienna for processing. The samples were used for AGALA and lyso‐Gb3 measurement from the same filter paper card. Enzyme activity and lyso‐Gb3 were measured using a fully validated and accredited high‐sensitive electrospray ionization–liquid chromatography tandem mass spectrometry (ESI LC‐MS/MS) assay (ARCHIMED Life Science GmbH, Vienna, Austria; www.archimedlife.com). FD was suspected in male patients with AGALA activity <1.2 μmol/h/L and in females with either low AGALA activity or lyso‐Gb3 > 3.5 ng/mL. Positive screening results were confirmed by gene sequencing in the presence of a detectable mutation within the GLA gene.
Categorical variables are expressed as proportions (percentage) and continuous variables as mean and standard deviation (SD).
Results
Of 592 patients who underwent the screening, three were excluded (in two, DSB samples were insufficient, and one patient has withdrawn his consent with the study). The final screening population therefore included 589 patients, of which 390 (66.2%) were males. The average age was 58.4 ± 14.7 years, and the mean LV thickness was 19.1 ± 4.3 mm. According to clinical questionnaire, 124 (21.1%) of the patients had at least one potentially non‐cardiac FD manifestation such as proteinuria, renal insufficiency, stroke or transitory ischaemic attack history, neuropathic pain, angiokeratomas, or cornea verticillata. The baselines characteristics of our cohort are listed in Table 1 and study flowchart in Figure 1 .
Table 1.
Baseline characteristics of the entire cohort of screened patients with HCM diagnosis
| Age (years) | 58.4 ± 14.7 |
| Males (n; %) | 390; 66% |
| Maximal LV wall thickness (mm) | 19.1 ± 4.3 |
| Family history of hypertrophic cardiomyopathy (n; %) | 102; 17% |
| Presence of LVOT obstruction (n; %) | 259; 44% |
| ICD implantation (n; %) | 94; 16% |
| Fabry non‐cardiac manifestation (n; %) | 124; 21% |
| – Proteinuria or renal insufficiency | 61; 10% |
| – Acroparesthesia | 39; 7% |
| – Stroke/TIA | 38; 6% |
| – Angiokeratoma, cornea verticillata | 5; 1% |
ICD, implantable cardioverter‐defibrillator; LV, left ventricle; LVOT, left ventricular outflow tract; TIA, transitory ischaemic attack.
Figure 1.
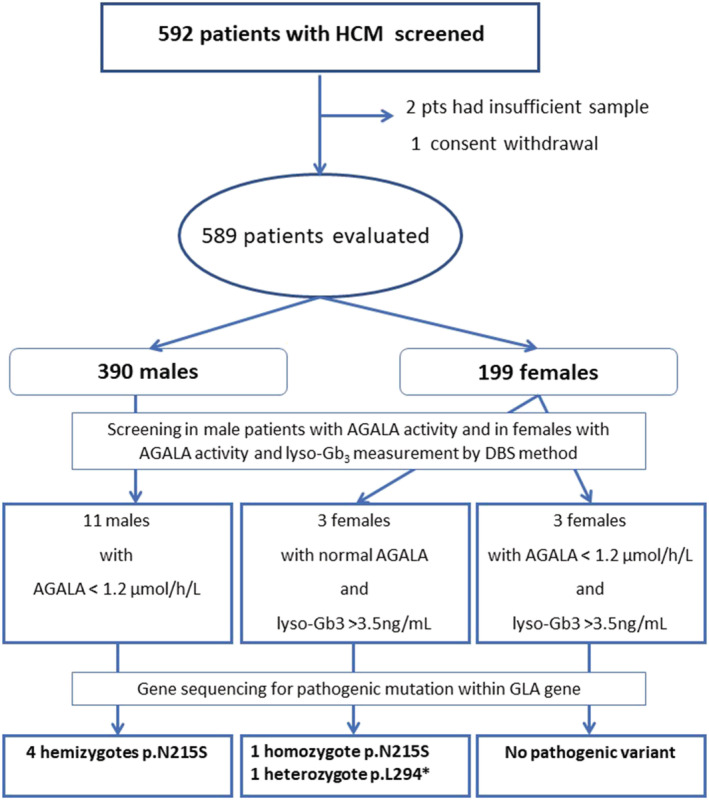
The flowchart of the study showing stepwise diagnostic approach results based on AGALA activity, lyso‐Gb3 measurements, and GLA gene sequencing.
Out of the 589 patients, we identified 14 patients in whom FD diagnosis was suspected based on AGALA activity measurement (11 males, 3 females). Furthermore, three other females had normal AGALA activity but elevated lyso‐Gb3. Of these 17 patients, the diagnosis of FD was confirmed by genetic testing in six cases (four males and two females). In the remaining 11, two had negative second sample analysis, and in nine individuals, the gene sequencing did not identify any variant within the GLA gene.
Based on the screening results, FD was suspected in six females. Three of them presented with low AGAL activity, in two cases associated with increased level of lyso‐Gb3. These two females were subsequently confirmed by genetic testing as carriers of a pathogenic mutation. The last female patient with low AGALA activity had normal lyso‐Gb3 and negative genetic testing. Three other female patients had normal AGAL activity with increased lyso‐Gb3 and had also a negative genetic testing result.
Among patients with confirmed FD, five patients (four males and one female) had the p.N215S pathogenic variant (Table 2 ). This mutation is known for causing typically the later‐onset isolated cardiac disease. The last positive patient was female with a nonsense p.Leu294* mutation in exon 6. The characteristics of these patients are summarized in Table 2 . The female patient carrying the p.N215S variant was found to have an extremely rare homozygous configuration, which explains the severity of her manifestation. Of note, this patient has been previously treated by a successful alcohol septal ablation due to significant obstruction of LV outflow tract and implanted with an implantable cardioverter‐defibrillator; four other members of her family were identified as mutation carriers, one of them having a severe cardiac disease already.
Table 2.
Characteristics of patients with genetically confirmed Fabry disease
| Sex | Age | Gene variant | Variant type | AGALA activity (μmo/L/h) | Lyso‐Gb3 (ng/mL) | Septal thickness (mm) | Posterior wall thickness (mm) | LVMi (g/m2) | LVOTO | ECG | ICD | Positive family history | Non‐cardiac Fabry disease manifestation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M | 56 | p.N215S | Cardiac | 0.4 | ‐ | 20 | 17 | 107 | Yes | Atrial fibrillation, LBBB | No | No | No |
| M | 57 | p.N215S | Cardiac | 0.7 | ‐ | 23 | 21 | 131 | No |
RBBB + LAH LV hypertrophy |
No | No | No |
| M | 55 | p.N215S | Cardiac | 0.3 | ‐ | 27 | 12 | 112 | No | RBBB + LAH | No | No | No |
| M | 66 | p.N215S | Cardiac | 0.3 | ‐ | 22 | 14 | 96 | No |
RBBB + LAH LV hypertrophy |
No | Yes | Proteinuria |
| F | 56 | p.N215S | Cardiac | 0.3 | 10.0 | 17 | 23 | 111 | Yes | LV hypertrophy | Yes | Yes |
Proteinuria Neuropathic pain |
| F | 53 | p.L294* | Novel | 0.6 | 16.0 | 18 | 14 | 116 | No | LV hypertrophy | No | No |
Proteinuria Angiokeratomas Neuropathic pain |
AGAL, α‐galactosidase; ECG, electrocardiography; ICD, implantable cardioverter‐defibrillator; LAH, left anterior hemiblock; LBBB, left bundle branch block; LV, left ventricle; LVMi, left ventricular mass index; LVOTO, left ventricular outflow tract obstruction; Lyso‐Gb3, plasma globotriaosylsphingosine; RBBB, right bundle branch block.
According to the questionnaire, non‐cardiac manifestations of FD were present in two out of the six identified patients. Both patients—one male with p.N215S and one female homozygous for p.N215S—had proteinuria with preserved renal function. In addition, the homozygous female reported a typical neuropathic pain. Moreover, a systematic careful evaluation during hospitalization after the screening found non‐cardiac manifestation (angiokeratomas, neuropathic pain, and proteinuria) in the female patient with nonsense GLA mutation p.L294*.
Discussion
Our study represents another approach for screening in patients with unexplained LV hypertrophy or HCM by analysing cases diagnosed and followed up in highly specialized tertiary centres. Our cohort represents a nationwide sample because all but one centres agreed to recruit their HCM patients. As compared with previous screening programme performed in the Czech Republic, this study included both male and female patients and confirms that FD may mimic HCM findings in both genders. 7
The main novelty of our diagnostic approach is the use of screening strategy proposed by Kasper and colleagues and used by ARCHIMED Life laboratory based on measurements of AGALA activity in men and both AGALA activity and lyso‐Gb3 in women. In male patients, the presence of pathogenic GLA variant is always associated with depressed AGALA activity to an extent that a false negative diagnosis seems almost excluded including cases with later‐onset variants in whom a certain residual activity is always present. 18 In contrast in female heterozygotes, AGALA activities may be close to normal or even within normal ranges. It has been documented that up to 40% of females may be missed by an enzymatic screening alone. 19 Therefore, we used a second‐line screening method based on lyso‐Gb3. 20 However, this approach may also fail to detect FD particularly in women carrying a later‐onset disease causing mutations. As shown by Nowak et al. using the same method for DBS‐measured lyso‐Gb3, the values of lyso‐Gb3 from DBS and sera are in perfect correlation, but DBS‐derived values are about 50% lower. In later‐onset variant heterozygous females (one of them on ERT), Nowak et al. reported a median DBS lyso‐Gb3 level 1.3 (range: 1.1–2.6) ng/mL, therefore below the detection threshold used in our study. Using the same method, these authors reported slightly higher levels in more symptomatic later‐onset mutation carrying women reaching 4.9 (1.6–4.9) ng/mL, thus overlapping with the threshold used in our study. 21 Smid et al. have also demonstrated that later‐onset variants in female patients are associated with a statistically significant elevation of lyso‐Gb3. However, values in their cohort were also overlapping with normal control subjects. 22 Nevertheless, the recently published analysis in female patients yielded a high overall positive predictive value (97%) for the same diagnostic algorithm used in our study. Unfortunately, the studied cohort comprised a low number of later‐onset variant carriers, and the few missed by this diagnostic approach were arising from this group. 16
On the one hand, our study demonstrated that the strategy based on lyso‐Gb3 assessments allows identification of women carrying pathogenic mutations for FD; on the other, it did not give an answer whether heterozygous women with mutations causing later‐onset variants would be successfully identified in screening programmes investigating populations with cardiac manifestations.
One of the females identified by our study is carrying the p.N215S variant. However, she was shown to represent an extremely rare case of homozygosity, therefore manifesting a disease severity and biomarker levels similar to male patients. The second female is carrying a p.Leu294* nonsense mutation with clinical picture suggesting a classical disease. Although the latter mutation was not described in the literature yet, its character and the presence of extracardiac manifestations along with relatively high lyso‐Gb3 suggest the classical phenotype. Therefore, our study clearly demonstrates that screening strategies in HCM patients should be focusing not only on male but also on female patients. However, our data are unable to confirm that in female patients, the method based on lyso‐Gb3 is sufficient as we may have missed females with later‐onset variants in whom enzymatic assay may show AGALA levels within normal ranges and minimal lyso‐Gb3 elevation.
The question remains whether the risk of missing a female carrying a later‐onset variant by this approach is clinically relevant. First, female patients with advanced LV hypertrophy mimicking HCM have elevated lyso‐Gb3 levels as compared with healthy subjects. 14 Second, it seems that the proportion of females in the cardiac screening programmes carrying a later‐onset variant is lower as compared with the classic phenotype carriers. The analysis of published screening programmes performed by Doheny et al. has shown that the ratio between the classic and later‐onset phenotype in male patients is usually 1:3 or 1:4, whereas in females, the ratio is reversed ranging from 1:1 up to 4:1. 18
The main advantage of the selected approach is elimination of frequently encountered benign variants such as p.E66Q, p.R118C, p.S126G, p.D313Y, or p.A143T. These patients are usually not presenting with severe clinical phenotype and have very low lyso‐Gb3 values, and their inclusion into positively identified cases falsely increased previously reported prevalence of FD in high‐risk populations. The bias induced by misinterpretation of pathogenic mutations was analysed by Doheny et al. in 16 studies of cardiac screening published till 2017. The authors demonstrated that after elimination of potentially benign variants, the prevalence of FD in cardiomyopathy patients decreased from 1.21% to 0.94% in males and from 0.90% to 0.56% in females. This number is in agreement with the results of our study, although we are finding a lower ratio between classical and late‐onset variants 1:5 although others were reporting a ratio 1:2 in Caucasian patients in both genders. The need for a careful evaluation of detected variants based on clinical assessment, family history, AGALA and Lyso‐Gb3 levels, literature and database search, in vitro predicting tools, assessments of the prevalence of the allele in the population, and segregation analysis was recently ascertained by Germain et al. In agreement with previous evidence, their review is also suggesting the benign character of the above listed variants. 23
Five out of six cases identified in our study were carrying the p.N215S variant, which appears to be the most common mutation in Western countries. 24 Affected patients may have significant levels of residual AGALA activity (2–20% of normal values). Most of p.N215S carriers do not report early symptoms of Fabry disease (hypohidrosis, neuropathic pain, febrile crises, gastrointestinal complaints), and manifestations in older patients are usually limited to the heart (LV hypertrophy, cardiac fibrosis, conduction disturbances, arrhythmias). However, a subset of patients may present with proteinuria and mildly decreased renal function similarly to two patients from our cohort. 25 Rarely, a progressive worsening to end‐stage renal disease has been reported. 26 The late‐onset nature of disease development and absence of typical signs of FD may lead to misdiagnosis of FD for HCM delaying a possibility of early specific treatment initiation. Late stages with irreversible ventricular fibrosis seem to respond poorly to ERT and were shown to be associated with arrhythmic events. 27 Therefore, a systematic screening for FD should become part of diagnostic workup in adult HCM patients regardless of the presence or absence of associated extracardiac signs and symptoms. As shown by our study, the systematic screening cannot be replaced by clinically oriented screenings. Of note, 21% of our HCM cohort had at least one of the signs or symptoms reported in classical FD phenotype. On the other hand, as shown by our case carrying the p.Leu294* mutation, her clinical signs of FD were overlooked by cardiology routine follow‐up.
The suggested systematic screening for Fabry disease among HCM patients should allow identification of index cases and open the possibility for cascade family genetic testing to make a timely diagnosis in affected family members. By applying this approach, we were able to detect additional patients among the relatives of our probands and initiate a prompt targeted therapy if needed. However, as suggested by the recent literature review, several cultural, legislative, and logistic barriers exist in different countries, and in many of them, this approach remains difficult or impossible. 28
It should be emphasized that DBS may be associated with a false positivity (e.g. due to sampling errors) and always requires confirmatory testing. This is clearly demonstrated by our result, where 17 cases had positive screening results, but the disease was confirmed only in six of them.
The major limitation of our work is a potentially limited sensitivity of the diagnostic algorithm based on AGALA activity and lyso‐Gb3 measurements in later‐onset mutation carriers in females. In absence of a more sensitive biomarker for FD, this may be solved only by a systematic gene sequencing. Because many HCM patients undergo a complex search of sarcomeric genes responsible for the disease anyway, GLA gene should be included in the diagnostic panel. However, this approach mandates a careful evaluation of pathogenicity as many common variants within the GLA gene were shown to be non‐pathogenic.
The second limitation may be a pre‐selected character of our population eliminating patients that were previously diagnosed in major cardiovascular centres by DBS kits distributed widely for clinical use. It should be also noted that in a substantial proportion of patients with late‐onset FD, the myocardial thickening does not exceed the 15 mm threshold required for HCM diagnosis. 29 Because our study was on purpose focusing on those with suspected HCM, our data do not elucidate the prevalence of FD in patients with milder LVH.
In conclusion, our study demonstrated that the most frequently encountered pathogenic variant in Caucasian population with previous diagnosis of HCM in the Czech Republic is the p.N215S known to cause the later‐onset cardiac FD variant. Our data support recommendation for routine screening in all adult HCM patients for FD. This screening could be effectively done by evaluation AGALA activity in males and AGALA activity and lyso‐Gb3 levels in females from DBS. Despite the successful detection of pathogenic mutations in women, we cannot exclude that a small proportion of females with late‐onset variants may be missed due to low lyso‐Gb3 values and AGALA activity above threshold limits. Therefore, in HCM patients in whom gene testing is indicated, GLA gene should be included in the tested panel.
Conflict of interest
DZ received the travel grant from the Czech Society of Cardiology and speaker's honoraria from Sanofi‐Genzyme. GD received honoraria and travel funding from Sanofi‐Genzyme, Shire/Takeda, Protalix, and Greenovation Biotech GmbH. AL received speaker's and consultancy honoraria from Sanofi‐Genzyme and Takeda and consultancy honoraria from Amicus Therapeutics and Avrobio.
Funding
This work was supported by PROGRES Q38/LF1 grant from the Ministry of Education, Youth and Sports of Czech Republic and grant from Czech Society of Cardiology and Sanofi‐Genzyme.
Acknowledgements
The authors thank investigators from other centres such as Eva Krčová, Markéta Peterková, Věra Šťastná, Marian Felšoci, Jiří Štastný, Jakub Walder, Patrik Keprt, and Alexandra Vodzinská. We also thank study coordinators Denisa Ebelova and Petra Černohousová whose work was essential for success of the study. Special thanks to Peter Križalkovič and Václav Kuda from Sanofi‐Genzyme and Charles University in Prague for their support.
Zemánek, D. , Januška, J. , Honěk, T. , Čurila, K. , Kubánek, M. , Šindelářová, Š. , Zahálková, L. , Klofáč, P. , Laštůvková, E. , Lichnerová, E. , Aiglová, R. , Lhotský, J. , Vondrák, J. , Dostálová, G. , Táborský, M. , Kasper, D. , and Linhart, A. (2022) Nationwide screening of Fabry disease in patients with hypertrophic cardiomyopathy in Czech Republic. ESC Heart Failure, 9: 4160–4166. 10.1002/ehf2.14135.
References
- 1. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010; 5: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Machann W, Breunig F, Weidemann F, Sandstede J, Hahn D, Köstler H, Neubauer S, Wanner C, Beer M. Cardiac energy metabolism is disturbed in Fabry disease and improves with enzyme replacement therapy using recombinant human galactosidase A. Eur J Heart Fail. 2011; 13: 278–283. [DOI] [PubMed] [Google Scholar]
- 3. Linhart A, Kampmann C, Zamorano JL, Sunder‐Plassmann G, Beck M, Mehta A, Elliott PM, European FOS Investigators . Cardiac manifestations of Anderson‐Fabry disease: Results from the international Fabry outcome survey. Eur Heart J. 2007; 28: 1228–1235. [DOI] [PubMed] [Google Scholar]
- 4. Germain DP, Elliott PM, Falissard B, Fomin VV, Hilz MJ, Jovanovic A, Kantola I, Linhart A, Mignani R, Namdar M, Nowak A, Oliveira JP, Pieroni M, Viana‐Baptista M, Wanner C, Spada M. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: A systematic literature review by a European panel of experts. Mol Genet Metab Rep. 2019; 19: 100454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hughes DA, Nicholls K, Shankar SP, Sunder‐Plassmann G, Koeller D, Nedd K, Vockley G, Hamazaki T, Lachmann R, Ohashi T, Olivotto I, Sakai N, Deegan P, Dimmock D, Eyskens F, Germain DP, Goker‐Alpan O, Hachulla E, Jovanovic A, Lourenco CM, Narita I, Thomas M, Wilcox WR, Bichet DG, Schiffmann R, Ludington E, Viereck C, Kirk J, Yu J, Johnson F, Boudes P, Benjamin ER, Lockhart DJ, Barlow C, Skuban N, Castelli JP, Barth J, Feldt‐Rasmussen U. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18‐month results from the randomised phase III ATTRACT study. J Med Genet. 2017; 54: 288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Monserrat L, Gimeno‐Blanes JR, Marín F, Hermida‐Prieto M, García‐Honrubia A, Pérez I, Fernández X, de Nicolas R, de la Morena G, Payá E, Yagüe J, Egido J. Prevalence of Fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2007; 50: 2399–2403. [DOI] [PubMed] [Google Scholar]
- 7. Paleček T, Honzíková J, Poupětová H, Vlasková H, Kuchynka P, Goláň L, Magage S, Linhart A. Prevalence of Fabry disease in male patients with unexplained left ventricular hypertrophy in primary cardiology practice: Prospective Fabry cardiomyopathy screening study (FACSS). J Inherit Metab Dis. 2014; 37: 455–460. [DOI] [PubMed] [Google Scholar]
- 8. Elliott P, Baker R, Pasquale F, Quarta G, Ebrahim H, Mehta AB, Hughes DA, ACES study group . Prevalence of Anderson‐Fabry disease in patients with hypertrophic cardiomyopathy: The European Anderson‐Fabry disease survey. Heart. 2011; 97: 1957–1960. [DOI] [PubMed] [Google Scholar]
- 9. Newman DB, Miranda WR, Matern D, Peck DS, Geske JB, Maleszewski JJ, Ommen SR, Ackerman MJ. Cost efficacy of α‐galactosidase a enzyme screening for Fabry disease. Mayo Clin Proc. 2019; 94: 84–88. [DOI] [PubMed] [Google Scholar]
- 10. Sachdev B, Takenaka T, Teraguchi H, Tei C, Lee P, McKenna WJ, Elliott PM. Prevalence of Anderson‐Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation. 2002; 105: 1407–1411. [DOI] [PubMed] [Google Scholar]
- 11. Havndrup O, Christiansen M, Stoevring B, Jensen M, Hoffman‐Bang J, Andersen PS, Hasholt L, Nørremølle A, Feldt‐Rasmussen U, Køber L, Bundgaard H. Fabry disease mimicking hypertrophic cardiomyopathy: Genetic screening needed for establishing the diagnosis in women. Eur J Heart Fail. 2010; 12: 535–540. [DOI] [PubMed] [Google Scholar]
- 12. Ortiz A, Germain DP, Desnick RJ, Politei J, Mauer M, Burlina A, Eng C, Hopkin RJ, Laney D, Linhart A, Waldek S, Wallace E, Weidemann F, Wilcox WR. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018; 123: 416–427. [DOI] [PubMed] [Google Scholar]
- 13. Aerts JM, Groener JE, Kuiper S, Donker‐Koopman WE, Strijland A, Ottenhoff R, van Roomen C, Mirzaian M, Wijburg FA, Linthorst GE, Vedder AC, Rombach SM, Cox‐Brinkman J, Somerharju P, Boot RG, Hollak CE, Brady RO, Poorthuis BJ. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A. 2008; 105: 2812–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nowak A, Mechtler TP, Hornemann T, Gawinecka J, Theswet E, Hilz MJ, Kasper DC. Genotype, phenotype and disease severity reflected by serum LysoGb3 levels in patients with Fabry disease. Mol Genet Metab. 2018; 123: 148–153. [DOI] [PubMed] [Google Scholar]
- 15. Maruyama H, Miyata K, Mikame M, Taguchi A, Guili C, Shimura M, Murayama K, Inoue T, Yamamoto S, Sugimura K, Tamita K, Kawasaki T, Kajihara J, Onishi A, Sugiyama H, Sakai T, Murata I, Oda T, Toyoda S, Hanawa K, Fujimura T, Ura S, Matsumura M, Takano H, Yamashita S, Matsukura G, Tazawa R, Shiga T, Ebato M, Satoh H, Ishii S. Effectiveness of plasma lyso‐Gb3 as a biomarker for selecting high‐risk patients with Fabry disease from multispecialty clinics for genetic analysis. Genet Med. 2019; 21: 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Balendran S, Oliva P, Sansen S, Mechtler TP, Streubel B, Cobos PN, Lukacs Z, Kasper DC. Diagnostic strategy for females suspected of Fabry disease. Clin Genet. 2020; 97: 655–660. [DOI] [PubMed] [Google Scholar]
- 17. Cardim N, Galderisi M, Edvardsen T, Plein S, Popescu BA, D'Andrea A, Bruder O, Cosyns B, Davin L, Donal E, Freitas A, Habib G, Kitsiou A, Petersen SE, Schroeder S, Lancellotti P, Camici P, Dulgheru R, Hagendorff A, Lombardi M, Muraru D, Sicari R. Role of multimodality cardiac imaging in the management of patients with hypertrophic cardiomyopathy: An expert consensus of the European Association of Cardiovascular Imaging Endorsed by the Saudi Heart Association. Eur Heart J Cardiovasc Imaging. 2015; 16: 280. [DOI] [PubMed] [Google Scholar]
- 18. Doheny D, Srinivasan R, Pagant S, Chen B, Yasuda M, Desnick RJ. Fabry Disease: Prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995‐2017. J Med Genet. 2018; 55: 261–268. [DOI] [PubMed] [Google Scholar]
- 19. Linthorst GE, Poorthuis BJ, Hollak CE. Enzyme activity for determination of presence of Fabry disease in women results in 40% false‐negative results. J Am Coll Cardiol. 2008; 51: 2082 author reply 2082‐3. [DOI] [PubMed] [Google Scholar]
- 20. Yamashita S, Saotome M, Satoh H, Kajihara J, Mochizuki Y, Mizuno K, Nobuhara M, Miyajima K, Kumazawa A, Tominaga H, Takase H, Tawarahara K, Wakahara N, Matsunaga M, Wakabayashi Y, Matsumoto Y, Terada H, Sano M, Ohtani H, Urushida T, Hayashi H, Ishii S, Maruyama H, Maekawa Y. Plasma globotriaosylsphingosine level as a primary screening target for Fabry disease in patients with left ventricular hypertrophy. Circ J. 2019; 83: 1901–1907. [DOI] [PubMed] [Google Scholar]
- 21. Nowak A, Mechtler T, Kasper DC, Desnick RJ. Correlation of lyso‐Gb3 levels in dried blood spots and sera from patients with classic and later‐onset Fabry disease. Mol Genet Metab. 2017; 121: 320–324. [DOI] [PubMed] [Google Scholar]
- 22. Smid BE, van der Tol L, Biegstraaten M, Linthorst GE, Hollak CE, Poorthuis BJ. Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. J Med Genet. 2015; 52: 262–268. [DOI] [PubMed] [Google Scholar]
- 23. Germain DP, Levade T, Hachulla E, Knebelmann B, Lacombe D, Seguin VL, Nguyen K, Noël E, Rabès JP. Challenging the traditional approach for interpreting genetic variants: Lessons from Fabry disease. Clin Genet. 2022; 101: 390–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Germain DP, Brand E, Burlina A, Cecchi F, Garman SC, Kempf J, Laney DA, Linhart A, Maródi L, Nicholls K, Ortiz A, Pieruzzi F, Shankar SP, Waldek S, Wanner C, Jovanovic A. Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: A multicenter Fabry registry study. Mol Genet Genomic Med. 2018; 6: 492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lavalle L, Thomas AS, Beaton B, Ebrahim H, Reed M, Ramaswami U, Elliott P, Mehta AB, Hughes DA. Phenotype and biochemical heterogeneity in late onset Fabry disease defined by N215S mutation. PLoS ONE. 2018; 13: e0193550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sugarman M, Choudhury J, Jovanovic A. An atypical p.N215S variant of Fabry disease with end‐stage renal failure. Mol Genet Metab Rep. 2018; 15: 43–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weidemann F, Niemann M, Breunig F, Herrmann S, Beer M, Störk S, Voelker W, Ertl G, Wanner C, Strotmann J. Long‐term effects of enzyme replacement therapy on fabry cardiomyopathy: Evidence for a better outcome with early treatment. Circulation. 2009; 119: 524–529. [DOI] [PubMed] [Google Scholar]
- 28. Germain DP, Moiseev S, Suárez‐Obando F, Al Ismaili F, Al Khawaja H, Altarescu G, Barreto FC, Haddoum F, Hadipour F, Maksimova I, Kramis M, Nampoothiri S, Nguyen KN, Niu DM, Politei J, Ro LS, Vu Chi D, Chen N, Kutsev S. The benefits and challenges of family genetic testing in rare genetic diseases‐Lessons from Fabry disease. Mol Genet Genomic Med. 2021; 9: e1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oder D, Liu D, Hu K, Üçeyler N, Salinger T, Müntze J, Lorenz K, Kandolf R, Gröne HJ, Sommer C, Ertl G, Wanner C, Nordbeck P. α‐Galactosidase A genotype N215S induces a specific cardiac variant of Fabry disease. Circ Cardiovasc Genet. 2017; 10: e001691. [DOI] [PubMed] [Google Scholar]