

Abstract
Transthyretin cardiomyopathy (ATTR‐CM) is an under‐recognized cause of heart failure, but it has received increasing attention due to the availability of treatment options. We present a case of hereditary transthyretin cardiomyopathy (A97S, an under‐represented variant in current clinical studies) who presented with heart failure. Timely diagnosis and intervention with tafamidis demonstrated reversed cardiac remodelling via multiple imaging techniques (echocardiography, cardiac magnetic resonance imaging and technetium‐99m pyrophosphate scintigraphy). The echocardiography and cardiac magnetic resonance imaging demonstrated improved global strain. Cardiac magnetic resonance imaging showed decreased extracellular volume. The technetium‐99m pyrophosphate scintigraphy demonstrated decreased heart‐to‐contralateral ratio. This case highlights the potential reversible effect of tafamidis on A97S amyloidosis cardiomyopathy.
Keywords: Cardiac amyloidosis, Tafamidis, Echocardiography, Cardiac magnetic resonance imaging, Technetium‐99m pyrophosphate scintigraphy
Introduction
Transthyretin (ATTR) amyloidosis is a systemic disease involving multiple organ systems. ATTR cardiomyopathy (ATTR‐CM) is an under‐recognized cause of heart failure (HF), but it has received increasing attention due to the availability of treatment options. The natural history of ATTR‐CM includes progressive HF complicated by arrhythmias and conduction system disease. Hereditary ATTR (hATTR) can often manifest simultaneously with multiple organ involvement. 1 The A97S genotype, although under‐represented in major ATTR‐CM studies, is the most common in Taiwan. 2
Since the ATTR‐ACT (Tafamidis in Transthyretin Cardiomyopathy Clinical Trial), treatment outcome and response of tafamidis on ATTR‐CM have received much attention. Most reports on treatment groups have demonstrated less deterioration rate or stable clinical course compared with the non‐treated group, whereas few demonstrated improvements in myocardial biomarkers. 3 , 4 , 5 , 6
In this case report, we present a case of hATTR‐CM of A97S variant, an under‐represented group in the current published data, with evidence of reverse cardiac remodelling demonstrated on multiple image modalities. This case report further demonstrated different treatment response of various variants of ATTR‐CM.
Case presentation
A 66‐year‐old man with history of hypertension, hyperlipidaemia, coronary artery disease, and paroxysmal atrial fibrillation was presented with progressive exertional dyspnoea and orthopnoea for 30 days, New York Heart Association (NYHA) functional class II. He also reported dysphagia, frequently changing bowel habits (alternating from diarrhoea to constipation every other day), and hypoesthesia of the right foot. At presentation, his blood pressure was 122/61 mmHg, and his heart rate was 89 beats/min. A cardiac examination revealed a systolic ejection murmur, which was loudest at the apex (grade II/VI), and no lower extremity oedema or jugular vein engorgement.
Chest radiography revealed a slightly enlarged cardiac silhouette with bilateral clear lung fields. Electrocardiography revealed low voltages in the limb leads with poor R‐wave progression, wide QRS, and premature atrial complexes. The troponin T level was 59.1 ng/L, and N‐terminal pro‐B‐type natriuretic peptide level was 2668 pg/mL. Transthoracic echocardiography (performed while the patient was in sinus rhythm) showed left ventricular hypertrophy with sparkling myocardium, with a left ventricular ejection fraction of 62.2%, moderate amount of pericardial effusion, and global longitudinal strain (GLS) of −9.9% with the cherry‐on‐top pattern (Figure 1 A and 1 B and Video S1 ). Cardiac amyloidosis was highly suspected.
Figure 1.
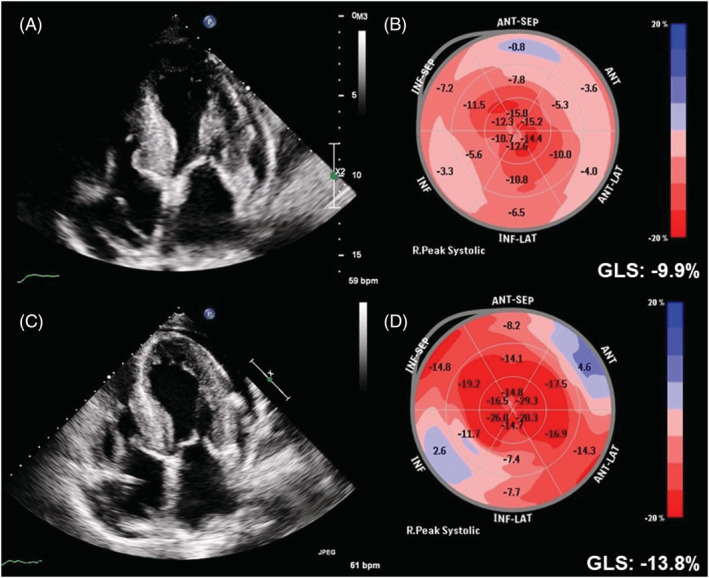
Echocardiography and global longitudinal strain (GLS) at baseline (A and B) and 1 year after tafamidis treatment (C and D).
To identify the aetiology of cardiac amyloidosis, a serological examination was performed, which did not suggest light chain disease. Technetium‐99m pyrophosphate (99mTc‐PYP) scintigraphy was further arranged, which showed grade 2 according to a semi‐quantitative visual grading system of the myocardium and anterior H/CL (heart to contralateral) ratio of 1.6055, with SPECT‐CT indicating myocardial uptake (Figure 2 A and 2 B ). Cardiac magnetic resonance imaging (CMR) showed subendocardial late gadolinium enhancement in both left and right ventricles, with an extracellular volume (ECV) of 51.9% (Figure 3 A ), global radial strain (GRS) of 17.5%, global circumferential strain (GCS) of −11.8%, and GLS of −6.9% (Figure 4 A ). An endomyocardial biopsy of the right ventricle revealed amyloid deposition with positive Congo red stain and positive transthyretin (TTR) immunohistochemical stain.
Figure 2.
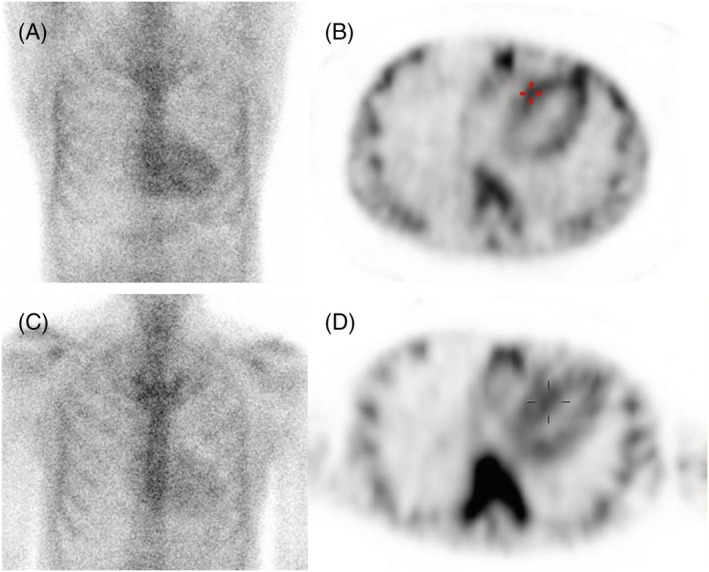
Technetium‐99m pyrophosphate (99mTc‐PYP) scintigraphy at baseline (A and B) and 1 year after tafamidis treatment (C and D).
Figure 3.
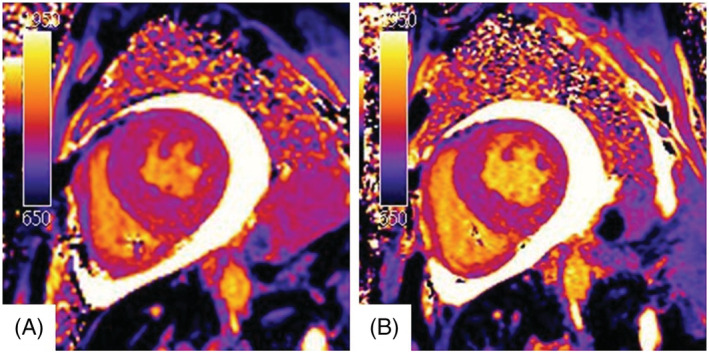
Native T1 image of cardiac magnetic resonance imaging at baseline (A) and 1 year after tafamidis treatment (B).
Figure 4.
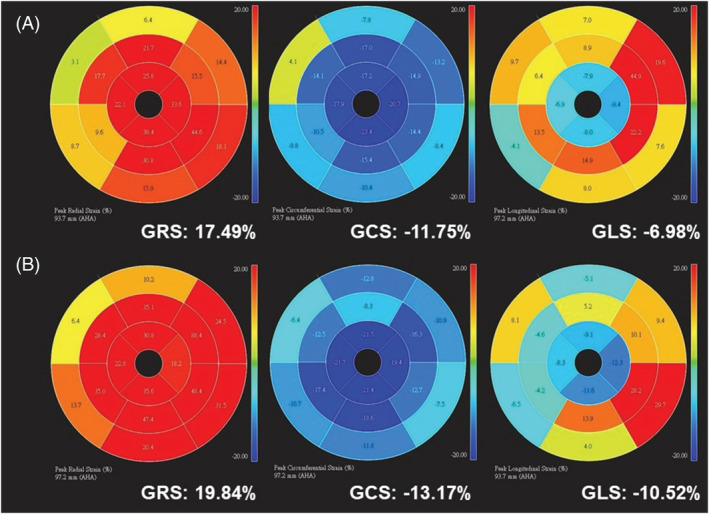
Global radial strain (GRS), global circumferential strain (GCS), and global longitudinal strain (GLS) at baseline (A) and 1 year after tafamidis treatment (B).
Further history taking revealed that his elder sister had been diagnosed with cardiac amyloidosis and had recently passed away due to sudden cardiac death. Genetic testing revealed the A97S transthyretin mutation, and hereditary transthyretin amyloidosis with cardiomyopathy (hATTR‐CM) was diagnosed. Further investigations also revealed neurological and gastrointestinal involvement.
The patient received tafamidis 61 mg per day in addition to furosemide 40 mg per day. After 1 year of treatment, his Kansas City cardiomyopathy questionnaire (KCCQ‐23) score improved from 67 (after 7 months of treatment) to 84. He reported improved exercise capacity and improved exertional dyspnoea, and his N‐terminal pro‐B‐type natriuretic peptide level decreased from 2668 to 1505 pg/mL. Follow‐up echocardiography (performed while the patient was in sinus rhythm) at 1 year after treatment (Figure 1 C and Video S2 ) revealed a significant improvement in GLS compared with baseline (Figure 1 D , from −9.9% to −13.8%). CMR further indicated a decrease in ECV from 51.9 to 38.6% (Figure 3 B ), with improvements in GRS (from 17.5 to 19.8%), GCS (from −11.8 to −13.2%), and GLS (from −6.9 to −10.5%) (Figure 4 B ). In addition, 99mTc‐PYP scintigraphy showed a decrease in anterior H/CL ratio from 1.6055 to 1.3957 (Figure 2 B ). These results suggested clinical, functional, and structural improvements with tafamidis treatment. No further adjustment of HF medication was required during the follow‐up 1 year period.
Discussion
The ATTR‐ACT demonstrated that tafamidis treatment improved all‐cause mortality and reduced the decline in functional capacity in patients with ATTR‐CM. 7 The long‐term findings of the ATTR‐ACT trial further highlight the importance of an early diagnosis and initiation of tafamidis treatment to improve patient outcomes. 8 Tafamidis improves survival in ATTR‐CM patient mainly by slowing the rate of clinical progression. However, the degree of treatment response in patients with various genotypes and the effect of tafamidis on diseased myocardial substrate is unclear, and patients with genotype A97S were not included in the ATTR‐ACT trial or any cohort studies. A phase 2, open‐label study showed that treatment with tafamidis did not result in significant changes in echocardiographic measures of wall thickness, left ventricular ejection fraction, and tissue Doppler diastolic assessment between baseline and after 12 months of treatment in patients with V122I or wild‐type ATTR‐CM. 9 Speckle tracking evaluation of the wild‐type ATTR‐CM cohort treated with tafamidis showed less deterioration in GLS, myocardial work index, and efficiency compared with the control group. 3 In addition, MRI ECV analysis of the tafamidis‐treated ATTR‐CM cohort (61 mg once daily) showed stable measurements and even improvement compared with the treatment‐naïve group after 1 year of follow‐up. In the same study, although the 20 mg once daily group also showed stable measurements, no improvement was observed compared with the treatment‐naïve group. 4 These results further highlight the heterogeneous treatment response to tafamidis among ATTR‐CM patients.
New therapies for ATTR‐CM could potentially demonstrate beneficial modifications of substrate. After 1 year of tafamidis treatment, an ATTR‐CM patient with an unspecified genotype was shown to have decreased uptake of hydroxymethylene diphosphonate technetium‐99m (HMDP‐99mTc) in the diseased myocardium, which was confirmed on SPECT‐CT. 5 In addition, treatment with patisiran, a double‐stranded small interfering RNA that targets a sequence within the TTR messenger RNA, has been shown to reduce cardiac MRI ECV in ATTR‐CM patients. 6
To the best of our knowledge, this is the first reported case of clinical, functional, and structural improvements in a patient with A97S ATTR‐CM after tafamidis treatment. This was demonstrated through multimodality imaging modalities (echocardiography, 99mTc‐PYP scintigraphy, and MRI) and suggest that diseased amyloid cardiomyopathy is potentially reversible after tafamidis treatment. The treatment effect of new therapies on various ATTR‐CM genotypes remains to be elucidated, and further studies are warranted to clarify this issue.
Conflict of interest
None declared.
Funding
None declared.
Supporting information
Video S1. Supporting Information.
Video S2. Supporting Information.
Acknowledgements
We thank the staff of the Second Core Lab in the Department of Medical Research in National Taiwan University Hospital for technical assistance.
Wu, Y.‐K. (A). , Tsai, C.‐H. , Su, M.‐Y. , Chao, C.‐C. , Cheng, M.‐F. , Shun, C.‐T. , Hsieh, S.‐T. , and Lin, Y.‐H. (2022) Reverse cardiac remodelling and dysfunction in A97S transthyretin cardiac amyloidosis after tafamidis treatment. ESC Heart Failure, 9: 4335–4339. 10.1002/ehf2.14165.
References
- 1. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2019; 73: 2872–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hsueh HW, Chao CC, Chang K, Jeng YM, Katsuno M, Koike H, Hsieh ST. Unique phenotypes with corresponding pathology in late‐onset hereditary transthyretin amyloidosis of A97S vs. V30M. Front Aging Neurosci. 2021; 13: 786322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Giblin GT, Cuddy SAM, Gonzalez‐Lopez E, Sewell A, Murphy A, Dorbala S, Falk RH. Effect of tafamidis on global longitudinal strain and myocardial work in transthyretin cardiac amyloidosis. Eur Heart J Cardiovasc Imaging. 2022; 23: 1029–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rettl R, Mann C, Duca F, Dachs TM, Binder C, Ligios LC, Schrutka L, Dalos D, Koschutnik M, Donà C, Kammerlander A, Beitzke D, Loewe C, Charwat‐Resl S, Hengstenberg C, Kastner J, Eslam RB, Bonderman D. Tafamidis treatment delays structural and functional changes of the left ventricle in patients with transthyretin amyloid cardiomyopathy. Eur Heart J Cardiovasc Imaging. 2021; 23: 767–780. [DOI] [PubMed] [Google Scholar]
- 5. Bellevre D, Bailliez A, Marechaux S, Manrique A, Mouquet F. First follow‐up of cardiac amyloidosis treated by tafamidis, evaluated by absolute quantification in bone scintigraphy. JACC Case Rep. 2021; 3: 133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fontana M, Martinez‐Naharro A, Chacko L, Rowczenio D, Gilbertson JA, Whelan CJ, Strehina S, Lane T, Moon J, Hutt DF, Kellman P, Petrie A, Hawkins PN, Gillmore JD. Reduction in CMR derived extracellular volume with patisiran indicates cardiac amyloid regression. JACC Cardiovasc Imaging. 2021; 14: 189–199. [DOI] [PubMed] [Google Scholar]
- 7. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington‐Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, Drachman BM, Shah SJ, Hanna M, Judge DP, Barsdorf AI, Huber P, Patterson TA, Riley S, Schumacher J, Stewart M, Sultan MB, Rapezzi C. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018; 379: 1007–1016. [DOI] [PubMed] [Google Scholar]
- 8. Elliott P, Drachman BM, Gottlieb SS, Hoffman JE, Hummel SL, Lenihan DJ, Ebede B, Gundapaneni B, Li B, Sultan MB, Shah SJ. Long‐term survival with tafamidis in patients with transthyretin amyloid cardiomyopathy. Circ Heart Fail. 2022; 15: e008193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maurer MS, Grogan DR, Judge DP, Mundayat R, Packman J, Lombardo I, Quyyumi AA, Aarts J, Falk RH. Tafamidis in transthyretin amyloid cardiomyopathy: Effects on transthyretin stabilization and clinical outcomes. Circ Heart Fail. 2015; 8: 519–526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Supporting Information.
Video S2. Supporting Information.