

Abstract
Cardiac sarcoidosis (CS) is a chameleon of cardiology, and it can mimic different cardiac diseases; among them is arrhythmogenic cardiomyopathy (ACM). We admitted a 70‐year‐old female patient with heart failure symptoms in 2015, who fulfilled all major ECG and non‐invasive imaging criteria of biventricular ACM. She was well with the recommended medications for 3 years, showing only isolated cardiac involvement, but in 2018, cervical and mediastinal lymphadenopathy appeared and cervical lymph node core biopsy histology, bronchoalveolar lavage flow cytometry strongly suggested extracardiac sarcoidosis. Therefore, our suspicion was that sarcoidosis is responsible for the cardiac involvement, which was not confirmed by PET‐CT and gallium scintigraphy examinations. At the end of 2018, she died in septicaemia with multiorgan failure, and only autopsy verified her CS. A new ECG algorithm published in 2021 for the differential diagnosis of CS and biventricular ACM, when applied on her ECGs recorded in 2015, suggested the diagnosis of CS.
Keywords: arrhythmogenic cardiomyopathy, electrocardiography, heart failure, sarcoidosis
1. Introduction
Approximately 5% of patients with sarcoidosis have clinically manifest cardiac involvement and two‐thirds with clinically manifest cardiac sarcoidosis (CS) have isolated CS. 1 CS can mimic different cardiac diseases, such as arrhythmogenic cardiomyopathy (ACM), dilated cardiomyopathy, amyloidosis, myocardial infarction, Chagas disease, and myocarditis; therefore, the diagnosis of isolated CS sometimes can be very difficult. 2 We present the case of a 70‐year‐old female patient, who was admitted to our department with signs and symptoms of heart failure in 2015, and during the initial work‐up, she fulfilled all major electrocardiographic and non‐invasive imaging criteria of biventricular ACM; therefore, we established the definitive diagnosis of ACM. In this patient, CS could be verified only by autopsy 3 years later, after clinical signs of extracardiac sarcoidosis appeared and the suspicion that cardiac involvement is also due to CS emerged, which could not be confirmed by in vivo examinations, and the patient ultimately died in septicaemia with multiorgan failure.
2. Case report
A 70‐year‐old female patient was admitted to our department in 2015 for dyspnoea on mild exertion, leg oedema and weakness. She had a history of hypertension since 2004 and several work‐ups for similar symptoms since 2012 at outpatient cardiology departments without revealing the underlying cause. In 2012, echocardiography, ECG, and dipyridamole stress perfusion imaging did not reveal any significant abnormality. In 2013, 2014, and 2015, repeated echocardiographies showed normal systolic left ventricular (LV) function, significant and progressive right ventricular (RV) dilation, grade II tricuspid insufficiency and an estimated pulmonary artery systolic pressure of 35 mmHg. Chest CT angiography in 2014 ruled out pulmonary embolism. Just before admission, the ECG recorded at a cardiology department initially showed a 105 b.p.m. ventricular tachycardia (VT) with 1:1 VA conduction (Figure 1 A ) and subsequently a sinus rhythm with wide QRS complex beats of identical morphology to the beats of the VT, arranged mostly in a bigeminal pattern (Figure 1 B ). The analysis of the ECG tracing shown in Figure 1 B revealed a potential underlying cause of her complaints. Looking at the narrow QRS sinus beats, first ԑ‐waves in leads V1 and V2, T wave inversions in leads V1–6 and aVF and a terminal activation delay of 280 ms from the S wave nadir to the end of the QRS complex in the absence of complete right bundle branch block in lead V2, low voltage and 1st degree AV block (PR interval: 210 ms) were observed. The first three ECG alterations corresponded to the major ECG criteria and one minor ECG criterion of arrhythmogenic cardiomyopathy (ACM) in effect at that time (Table 1 ). 3 The VT without left bundle branch block (LBBB) morphology, low voltage in the limb leads and T wave inversions not restricted to the right precordial leads, but present also in the inferolateral leads, suggest ACM with biventricular involvement, which is more prevalent (56%) than the classic form of ACM involving only the right ventricle (RV) (39%). 4 , 5
Figure 1.
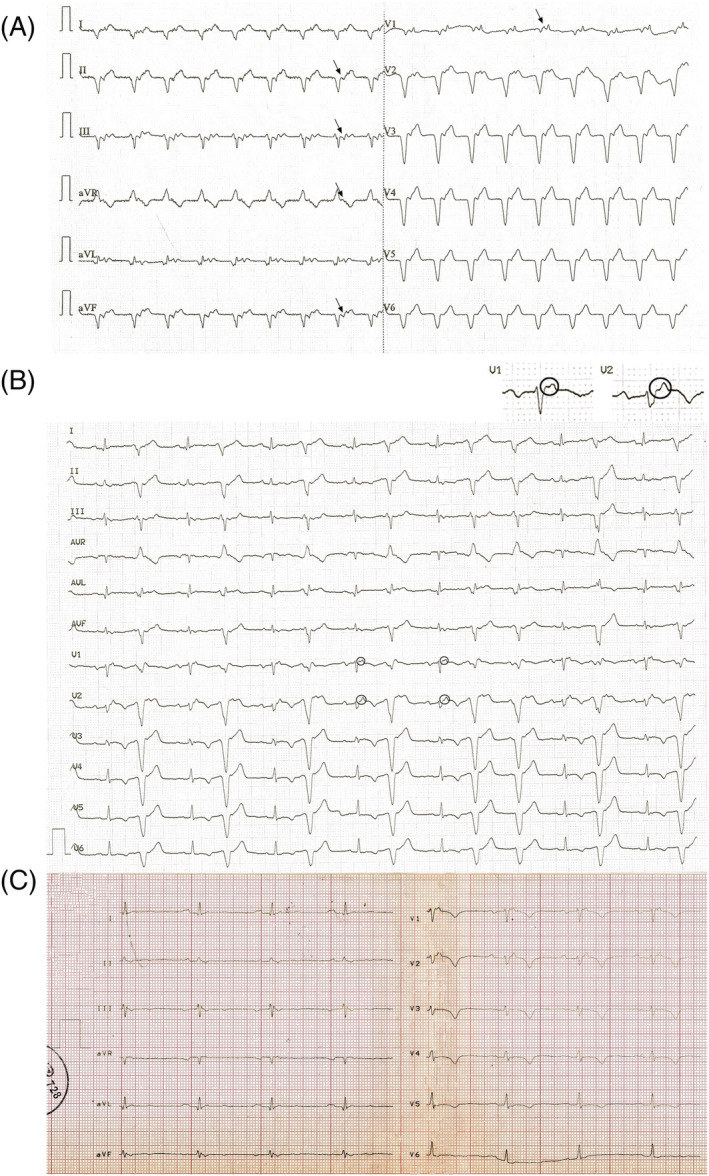
Preadmission ECGs and the ECG recorded at our department after admission. (A) Preadmission ECG tracing recorded right before admission to our department. A 105 b.p.m. ventricular rhythm with 1:1 VA conduction is shown. Arrows show the retrograde P waves. The VT diagnosis is based on the absence of RS complex in the chest leads (Brugada algorithm), the presence of initial R wave in lead aVR (Vereckei algorithm), the negative concordance pattern in the chest leads and the fact that the QRS morphology does not match that of a bundle branch block or a combination of bundle branch block and hemiblock. (B) ECG tracing recorded right before admission to our department. Sinus rhythm with ventricular premature beats (VPB) with an identical morphology to the beats of ventricular rhythm demonstrated in (A) arranged in a bigeminal pattern with the exception of two consecutive coupled VPBs is shown. Small circles denote the epsilon waves in leads V1 and V2, which are also highlighted in a magnified view. For further explanation, see text. (C) ECG tracing recorded at our department after admission, showing sinus rhythm without VPBs, otherwise essentially identical to the ECG tracing showed in (B), with the exception that the PR interval is longer: 230 ms.
Table 1.
2010 Task Force diagnostic criteria for ACM
| Major criteria | Minor criteria | |
|---|---|---|
| RV systolic function and structure | By 2D echo: | By 2D echo: |
|
► Regional RV akinesia, dyskinesia or aneurysm and one of the following (end diastole): PLAX RVOT ≥ 32 mm, PLAX‐RVOT/BSA ≥ 19 mm/m2 PSAX RVOT ≥ 36 mm, PSAX‐RVOT/BSA ≥ 21 mm/m2 Or fractional area change ≤33% |
Regional RV akinesia, dyskinesia or and 1 of the following (end diastole) PLAX RVOT ≥29 to <32 mm, PLAX‐RVOT/BSA ≥ 16 to <19 mm/m2 PSAX RVOT ≥32 to <36 mm, PSAX‐RVOT/BSA ≥ 18 to <21 mm/m2 Or fractional area change >33% to ≤40% |
|
| By MRI: | By MRI | |
| ► Regional RV akinesia, dyskinesia or dyssynchronous RV contraction and 1 of following: Ratio of RV end diastolic volume to BSA ≥ 110 ml/m2 (male) or ≥100 ml/m2 (female) (or RV EF ≤ 40%) | Regional RV akinesia, dyskinesia or dyssynchronous RV contraction and 1 of the following: Ratio of RV end‐diastolic volume to BSA ≥ 100 to <110 ml/m2 (male) or ≥90 to <100 ml/m2 (female) or RV EF > 40% to ≤ 45% | |
| By RV angiography: | ||
| Regional RV akinesia, dyskinesia or aneurysm | ||
| Tissue characterization | Residual myocytes <60% by morphometric analysis (or <50% if estimated) with fibrous replacement of the RV free wall myocardium in ≥1 sample, with or without fatty replacement of tissue on EMB | Residual myocytes 60% to 75% by morphometric analysis (or 50% to 65% if estimated), with fibrous replacement of the RV free wall myocardium in ≥1 sample, with or without fatty replacement of tissue on EMB |
| Repolarization’ abnormality | ► Inverted T waves in right precordial leads (V1–3) or beyond in individuals >14 years of age (in the absence of complete right bundle ‐ branch block QRS ≥ 120 ms | Inverted T waves in leads V1 and V2 in individuals >14 years of age (in the absence of complete right bundle branch block) or in V4–6 or inverted T waves in leads V1–V4 in individuals >14 years of age in the presence of complete right bundle branch block |
| Depolarization abnormality | ► Epsilon waves (reproducible low‐amplitude signals between the end of QRS complex to onset of the T wave) in the right precordial leads (V1–3) | Late potential by SAECG in ≥1 of 3 parameters in the absence of a QRS duration of ≥110 ms on the standard ECG; filtered QRS duration ≥114 ms; Duration of terminal QRS < 40 μV (low amplitude signal duration) or ≥38 ms; Root‐mean‐square voltage of terminal 40 ms ≤ 20 μV; Terminal activation duration of QRS ≥ 55 ms measured from the nadir of the S wave to the end of QRS, including R′, in V1–3, in the absence of complete right bundle‐branch block |
| Arrhythmias | Nonsustained or sustained ventricular tachycardia of left bundle‐branch morphology with superior axis (negative or indeterminate QRS in leads II, III and aVF and positive in lead aVL) |
Nonsustained or sustained ventricular tachycardia of RV outflow configuration, left bundle‐branch morphology with inferior axis (positive QRS in leads II, III, and aVF and negative in lead aVL) ► or >500 ventricular extrasystoles per 24 h (Holter) |
| Family history | ACM who meets Task Force criteria or confirmed pathologically in a first degree relative pathogenic mutation categorized as associated or probably associated with ACM in the patient under evaluation | History of ACM in a first degree relative or premature sudden death (<35 years of age) due to suspected ACM in a first degree relative or ACM confirmed pathologically or by current Task Force Criteria in second‐degree relative |
Note: Adapted from References 3, 4 with minor modifications. Definite diagnosis of ACM: 2 major, or 1 major and 2 minor, or 4 minor criteria, borderline diagnosis: 1 major and 1 minor, or 3 minor criteria, possible diagnosis: 1 major or 2 minor criteria from different categories. ► criteria that were present in our patient.
Abbreviations: ACM, arrhythmogenic cardiomyopathy; BSA, body surface area; ECG, electrocardiogram; EMB, endomyocardial biopsy; LV, left ventricle; MRI; magnetic resonance imaging; PLAX, parasternal long axis; PSAX, parasternal short axis; RV, right ventricular; RVOT, right ventricular outflow tract; SAECG, signal averaged ECG.
On physical examination, her blood pressure was 120/80 mmHg, pulse 120 b.p.m., respiratory rate 14 breaths per minute, and oxygen saturation 95%. Obesity was present, and oedema was not palpable. The heart was enlarged 1.5 cm both to the left and right, a right‐sided third heart sound and a 2/6 grade protomesosystolic murmur over the left parasternal area and inspiratory crackles over a territory of 1 cm above the diaphragms were heard. The ECG recorded at our department (Figure 1 C ) showed a 57 b.p.m. sinus bradycardia and was practically identical to the ECG shown in Figure 1 B , but ventricular premature beats were not present and the PR interval was 230 ms. Echocardiography revealed minimally thickened mitral valve, minimally thickened and calcified, tricuspid aortic valve, otherwise normal heart valves, significantly dilated RV and right atrium. The longitudinal systolic RV function was significantly decreased; the global systolic left ventricular (LV) function was slightly decreased (LVEF: 49%); the anterior and mid ventricular septum was hypo‐, akinetic and more echodense; and the basal half of the LV anterior free wall was also hypo‐, akinetic and other LV regions were slightly hypokinetic. The anterior and lateral RV free wall, with the exception of the lower one third of the lateral wall, was akinetic, and on the lateral wall, an aneurysm‐like bulge was also seen. Minimal concentric pericardial effusion, not causing impression or tamponade, and grade I mitral and aortic, grade II tricuspid and slight pulmonary insufficiencies were present. The pulmonary artery systolic pressure was 27–32 mmHg. The cardiac MRI (Figure 2 ) showed moderately decreased systolic LV function (LVEF: 43%), with normal LV volumes, significantly decreased systolic RV function (RVEF: 14.25%) with significantly increased RV volumes. In addition to the diffuse RV hypokinesis, regional RV akinesis and dyskinesis and thinned RV wall were also present. Concentric small amount (6–7 mm) pericardial effusion and in the apical one third of the RV a 25 × 13 × 28 mm size adherent thrombus were seen. Late gadolinium enhancement (LGE) was present in the subepicardial and partly in the subendo‐midmyocardial LV in the anteroseptal segment, in the subepicardial LV inferior wall and in the RV myocardium located in the vicinity of the thrombus. The 24 h ambulatory ECG monitoring showed sinus rhythm with a significant number (9978, 11.99%) of ventricular premature beats, non‐sustained or sustained VTs were not detected.
Figure 2.
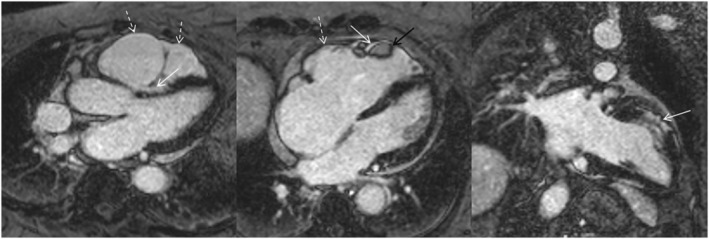
Long axis delayed contrast enhancement cardiac MRI images. Right ventricular dilation and bulging are present, dashed white arrows show the right ventricular wall thinning and akinetic/dyskinetic regions. White arrows show the left and right ventricular delayed contrast enhancement, the black arrow shows the right ventricular thrombus. For further explanation, see text.
The ECG, echocardiography and cardiac MRI findings corresponded to all major non‐invasive imaging and electrocardiographic criteria of ACM (Table 1 ) 1 ; therefore, we set up the definitive diagnosis of biventricular ACM with predominant RV involvement. A VVI ICD implantation was performed for primary prevention, as she belonged to the high arrhythmic risk category. 6 From 2015 to 2018, we saw the patient regularly as an outpatient, and she was quite well, with her recommended medication (40 mg/day furosemide, 12.5 mg/day hydrochlorothiazide, 300 mg/day irbesartan, 5‐0‐2.5 mg/day bisoprolol, and 1 mg/day acenocoumarol).
In March 2017, a peau d'orange alteration on the lower part of the right breast skin appeared, repeated mammography, breast ultrasound and fine needle aspiration biopsy (FNAB) examinations in 2017 and 2018 at an outpatient facility revealed neither any significant alterations nor any sign suggesting malignancy, but in April 2018 a cervical soft tissue ultrasound examination at an outpatient facility showed a 14 × 16 × 29 mm size pathological soft tissue mass in the right supraclavicular region lying against the jugular vein. Therefore, in June 2018, when we became aware of this finding, we admitted her to our department for further work‐up. On physical examination, pathological lymph nodes in the supraclavicular area or elsewhere were not palpable. The peau d'orange skin alteration was present not only on the lower part of the right breast but also on the right side of the mid and lower part of the abdominal skin. A moderate bilateral lower limb oedema (the right leg oedema was somewhat more conspicuous) and a mild redness and tenderness of her right shin skin were present (Figure 3 ). Her temperature was 37.2°C. These alterations suggested the presence of a potential right‐sided lymph flow obstruction, and the redness and tenderness of the lower third of the right shin skin a secondary cellulitis. Physical examination showed signs of moderate heart failure. Laboratory examinations showed normal leukocyte count with neutrophilia and lymphopenia, mild normocytic anaemia, slightly increased CRP and procalcitonin. The IgA, IgG and IgM levels were normal, and the detailed autoimmune disease blood test panel did not indicate the possibility of any specific immunological disease. The secondary cellulitis on the right leg recovered after oral cefuroxime treatment.
Figure 3.
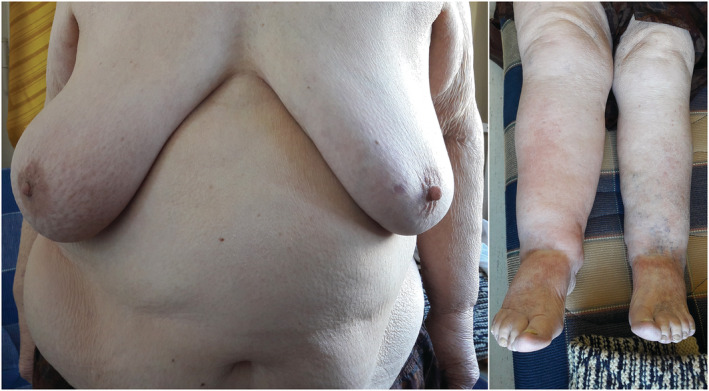
The right‐sided peau d'orange alterations on the breast and abdominal skin (left panel) and the more conspicuous oedema on the right leg with redness and tenderness of the lower third of the right shin skin, corresponding a secondary cellulitis (right panel), suggested the presence of a potential right‐sided lymph flow obstruction. For further explanation, see text.
Detailed examinations (chest‐abdominal‐pelvic CT, PET‐CT, abdominal and transvaginal ultrasound, colonoscopy, gastroscopy and gynaecological examination) could not identify any primary neoplasm. PET‐CT revealed cervical and mediastinal lymphadenopathy that showed FDG accumulation, and lymphadenopathy elsewhere (in the left axilla, retroperitoneal, mesenterial, right‐sided parailiacal and inguinal regions) without FDG accumulation. The CT examination revealed bilateral pathological lymph node conglomerates lying against and mildly compressing the internal jugular veins and propagating into the upper mediastinal space. Echocardiography findings were similar to those in 2015. We released her from the hospital in July 2018. In August 2018, an ultrasound‐guided core biopsy was performed from the right parajugular lymph nodes, and its histological examination revealed a non‐necrotizing, non‐caseating granulomatous inflammation without signs suggesting malignancy, corresponding mostly to sarcoidosis. The serum angiotensin convertase enzyme (SACE) levels were mildly to moderately elevated. Bronchoscopy did not reveal any significant alteration. The bronchoalveolar lavage (BAL) flow cytometry analysis revealed lymphocytosis (63% lymphocyte, 3% monocyte and 30% granulocyte), 92% of the lymphocytes were T‐lymphocyte and the CD4+/CD8+ ratio was 9.04. The Quantiferon test and the BAL fluid Ziehl–Neelsen staining and Mycobacterium tuberculosis culture were negative. Gallium scintigraphy showed isotope accumulation only in the parajugular and upper mediastinal lymph nodes, but not in the heart. Dipyridamole myocardium perfusion SPECT imaging revealed severe, medium to great extent, fixed perfusion defect in the basal two third of LV anterior wall and septum, with significantly decreased wall thickening at these territories, with only slight degree and small extent of reversibility at its side area at the inferoseptal transition with questionable significance. Molecular genetic analysis was also performed, 103 cardiomyopathy genes, including core ACM genes (PKP2, DSP, DSG2, DSC2, JUP, TMEM3, PLN and DES) were analysed by next‐generation sequencing. No pathogenic or likely pathogenic variants were identified in the patient's sample.
The histology of the core biopsy sample, the increased SACE levels, the lymphocytosis and increased CD4+/CD8+ ratio of 9.04 in the BAL fluid sample (a CD4+/CD8+ ratio >3.5 strongly suggests sarcoidosis) strongly suggested the presence of extracardiac sarcoidosis. Therefore, we thought that cardiac involvement is also due to sarcoidosis, which we tried to verify. However, neither the PET‐CT examination, recommended as the first choice test for CS, nor the Gallium scintigraphy revealed FDG or isotope accumulation in the heart. The molecular genetic analysis did not find pathogenic gene mutations characteristic to ACM; however, these are present in approximately 50% of patients with ACM, therefore a negative genetic test does not rule out ACM. 3 Thus, although it was very likely, we could not definitely verify the presence of CS. After these examinations, we planned to start glucocorticoid therapy (30–40 mg/day prednisone) for extracardiac sarcoidosis and the likely CS, but right after the administration of gallium isotope, diarrhoea and fever (38°C) occurred on 14 November 2018. Her condition deteriorated, and she had dyspnoea on minimal exertion, mild to moderate leg swelling and diarrhoea four to five times daily. Therefore, on 16 November 2018 we admitted her to our department. After admission, her condition rapidly deteriorated and died within a few days in septicaemia of unidentified origin and multiorgan (heart, respiratory, liver and kidney) failure, despite the complementation of initial fluid resuscitation, empirical antibiotic (IV imipenem and PO vancomycin), loop diuretic, methylprednisolone therapy with mechanical ventilation, high dose combined vasopressor and inotropic agent administration.
3. Discussion
3.1. Brief summary of arrhythmogenic cardiomyopathy
Arrhythmogenic cardiomyopathy (ACM) is a rare genetic cardiomyopathy, mostly caused by mutations in genes encoding proteins of desmosomal complex characterized by myocyte loss with fibro‐fatty replacement, which affects the RV, the LV or both, and underlies global and/or regional ventricular dysfunction and predisposes to potentially lethal ventricular arrhythmias regardless of the severity of systolic ventricular dysfunction. It is usually inherited in an autosomal dominant pattern 4 , 5 , 7 The current classification of ACM includes three phenotypic variants: (1) the classic ‘dominant‐right’ variant (39% of cases) characterized by predominant RV involvement with no or minor LV abnormalities; (2) the ‘biventricular disease’ variant (56%) characterized by parallel involvement of the RV and LV; (3) the ‘dominant‐left’ variant (5%) characterized by predominant LV involvement with no or minor RV abnormalities. 4 , 7 In young adults and athletes, ACM has been reported as the second most common cause of sudden cardiac death and the first most common cause in the Veneto region. It typically manifests in the second‐fourth decade of life and rarely manifests before the age of 12 or after the age of 60 years but can occur in any age (occurs in 20% after the age of 50 years). 4 , 5 Myocyte loss starts from the epicardium and eventually extends to become transmural. The gross pathognomonic features of ACM consist of RV dilation and RV aneurysm(s) located in the so called ‘triangle of dysplasia’ (The RV inflow, apex and outflow tract) and with disease progression further involvement of the RV free wall. LV involvement also occurs, typically limited to the subepicardial, midmyocardial layers of the posterolateral free wall and usually sparing the septum, but septal involvement can also occur. 4 , 5 , 8 In adolescents or young adults ACM usually presents with palpitations, syncope, cardiac arrest and ventricular arrhythmias. Less common presentations, rather in more elderly patients, are RV or biventricular dilation with or without heart failure symptoms. However, not infrequently non‐specific clinical presentation mimicking myocarditis or myocardial infarction can occur.
3.2. Brief summary of sarcoidosis
Sarcoidosis is an inflammatory multisystemic disease of unknown aetiology, which may be caused by a dysregulated immunological response to an unidentified antigenic trigger in genetically predisposed individuals, characterized by the formation of non‐caseating granulomas. Sarcoidosis can affect any organ in the body, predominantly the lungs, lymphatic system, skin and eyes. 2 , 9 , 11 While >90% of patients with sarcoidosis have lung involvement, only approximately 5% have clinically manifest cardiac involvement. Another 20%–25% of patients have asymptomatic, clinically silent cardiac involvement. Most patients with clinically manifest CS have minimal extracardiac disease and up to two thirds have isolated CS. 1 Most disease occurs in patients between 25 and 60 years of age, and sarcoidosis is unusual in people under the age of 15 or older than 70 years, the disease affects both sexes, with a slight predominance in women. 1 , 9 , 11 CS can present with conduction abnormalities, ventricular arrhythmias, heart failure or sudden cardiac death. 10 CS is the second leading cause of death among affected patients after pulmonary sarcoidosis. 11 For this reason, the early diagnosis of CS is important, because early treatment may prevent death from heart failure or sudden cardiac death. Approximately 30% of patients <60 years presenting with unexplained high degree AV block (Mobitz type II second degree or third degree AV block) or VT were diagnosed with CS, therefore CS should be strongly considered in such patients. 10 , 12 CS is a chameleon of cardiology, and it can mimic different cardiac diseases; among them is ACM. 2
3.3. Distinguishing ACM from CS in our patient
Our patient fulfilled all major ECG and non‐invasive imaging modality criteria of ACM that were in effect at the time of initial presentation and of the newest Padua criteria as well (Tables 1 and 2 ) 3 , 7 ; therefore, we established the definitive diagnosis of biventricular ACM. The only atypical findings not characteristic to ACM, which could have suggested sarcoidosis, were the older age (67 years) at the onset of the symptoms, involvement of the ventricular septum, 1st degree AV block and negative family history, but these findings may also occur in ACM, therefore do not rule out the diagnosis of ACM. 1 , 7 , 10 , 12 ACM is typically a disease of younger male patients; the male/female ratio is 3:1 and typically manifests in 20‐ to 60‐year‐old patients. It is rare in <12‐year‐old and >60‐year‐old patients. Therefore, the older age of our patient at the onset of clinical symptoms and her gender were more suggestive of CS. The involvement (hypo‐akinesis, increased echogenicity and LGE) of the ventricular septum is most characteristic to CS, but it can also occur in ACM with LV involvement. 4 , 5 , 8 , 10 Otherwise, the findings of the imaging studies were not helpful in distinguishing ACM from CS in our patient. Although regional RV akinesis, dyskinesis, aneurysm(s), thinning with RV dilation and dysfunction are more characteristic to ACM, they can be present in end stage CS as well. The typical pattern of LGEs is subepicardial and midmyocardial in both ACM and CS. The involvement of the inferolateral LV free wall is characteristic to biventricular or left‐dominant ACM, but any part of the LV free wall can be involved in CS as well. 8 , 13 After the initial cardiac symptoms in 2012, our patient presented with isolated cardiac involvement for 5 years until the first signs of extracardiac sarcoidosis appeared. When the examinations strongly suggested extracardiac sarcoidosis, we thought that the cardiac involvement might also be due to sarcoidosis instead of ACM. However, we could not verify in vivo the diagnosis of CS with certainty either with the recommended FDG PET‐CT or with gallium scintigraphy. A potential reason why FDG PET‐CT (which has a reported 89% sensitivity and 78% specificity in the diagnosis of CS 13 ) and gallium scintigraphy, which has a lower sensitivity than FDG PET‐CT in the diagnosis of CS, did not reveal FDG or isotope accumulation in the heart might be that there were predominantly inactive, burnt‐out and fibrotic granulomas in the heart instead of active, inflammatory ones. This argumentation is supported by the great extent of scar tissue both in the RV and in the LV at autopsy and the finding of myocardial perfusion SPECT imaging showing a predominant fixed perfusion defect. Originally, we did not plan to perform endomyocardial biopsy, because, due to the disease's focal nature, endomyocardial biopsy has a low diagnostic yield of approximately 25%. 10 However, after the negative FDG PET‐CT and gallium scintigraphy, due to the uncertain clinical picture, because our patient fulfilled both the diagnostic criteria of biventricular ACM (Tables 1 and 2 ) and probable CS (Table 3 ), an imaging (cardiac MRI)‐guided endomyocardial biopsy would have been the last possible examination, which could have established the in vivo diagnosis of CS. But, because the patient died right after the gallium scintigraphy, only histological examination (Figure 4 ) of the heart during autopsy verified the presence of CS in our patient, illustrating the great difficulty to diagnose CS in some patients. However, very recently, an ECG algorithm including a PR interval of ≥220 ms, the presence of R′ wave and the surface area of the maximum R′ wave in leads V1–3 ≥ 1.65 mm 2 was developed to distinguish CS with RV and LV involvement from ACM (Figure 5 A ). 14 It was either negative or positive for CS in our patient in its first step, as the PR interval of the ECG shown in Figure 1 B was 210 ms; however, the PR interval of the ECG shown in Figure 1 C was 230 ms. R′ wave was present in V1–3 and the surface area of maximum R′ wave in V1–3 was ≥1.65 mm2 (Figure 5 B ) in lead V2 in our patient, which established the diagnosis of CS. Thus, if we had known this ECG algorithm in 2015, we could have suggested the diagnosis of CS at the initial presentation, which we could not verify in vivo by the application of the most sophisticated imaging modalities. The suspicion of CS based on the ECG algorithm at the initial presentation could have changed the course of her disease, because we could have diagnosed CS 3 years before the verification of extracardiac sarcoidosis, if imaging (cardiac MRI)‐guided endomyocardial biopsy performed because of the uncertain clinical picture, as described above (assuming that FDG PET‐CT and gallium scintigraphy had been negative at that time as well), would have yielded a positive result for CS.
Table 2.
The ‘Padua criteria’ for diagnosis of ACM
| Category | Right ventricle (upgraded 2010 ITF diagnostic criteria) | Left ventricle (new diagnostic criteria) |
|---|---|---|
| I. Morpho‐functional ventricular abnormalities | By echocardiography, CMR or angiography: | By echocardiography, CMR or angiography: Minor |
| ► Major | ||
| specific | • Regional RV akinesia, dyskinesia, or bulging plus one of the following: | ► • Global LV systolic dysfunction (depression of LV EF or reduction of echocardiographic global longitudinal strain), with or without LV dilatation (increase of LV EDV according to the imaging test nomograms for age, sex, and BSA) |
| ‐ Global RV dilatation (increase of RV EDV according to the imaging test specific nomograms) | ||
| ‐ Global RV systolic dysfunction (reduction of RV EF according to the imaging test specific nomograms) | Minor | |
| • Regional LV hypokinesia or akinesia of LV free wall, septum, or both | ||
| II. Structural myocardial abnormalities | Minor | |
| • Regional RV akinesia, dyskinesia or aneurysm of RV free wall | ||
| By CE‐CMR:Major | By CE‐CMR:Major | |
|
• Transmural LGE (stria pattern) of ≥1 RV region(s) (inlet, outlet, and apex in 2 orthogonal views) By EMB (limited indications): Major |
► • LV LGE (stria pattern) of ≥1 Bull's Eye segment(s) (in 2 orthogonal views) of the free wall (subepicardial or midmyocardial), septum or both (excluding septal junctional LGE) | |
| • Fibrous replacement of the myocardium in ≥1 sample, with or without fatty tissue | ||
| III. Repolarization abnormalities | Major | Minor |
| ► • Inverted T waves in right precordial leads (V1, V2 and V3) or beyond in individuals with complete pubertal development (in the absence of complete RBBB) | ||
| Minor | ||
|
• Inverted T waves in leads V1 and V2 in individuals with completed pubertal development (in the absence of complete RBBB) • Inverted T waves in V1, V2, V3 and V4 in individuals with completed pubertal development in the presence of complete RBBB. |
► • Inverted T waves in left precordial leads (V4–V6) (in the absence of complete LBBB) | |
| IV. Depolarization abnormalities | Minor | Minor |
|
► • Epsilon wave (reproducible low‐amplitude signals between end of QRS complex to onset of the T wave) in the right precordial leads (V1 to V3) ► •Terminal activation duration of QRS ≥ 55 ms measured from the nadir of the S wave to the end of the QRS, including R′, in V1, V2 or V3 (in the absence of complete RBBB) |
• Low QRS voltages (<0.5 mV peak to peak) in limb leads (in the absence of obesity, emphysema, or pericardial effusion) | |
| V. Ventricular arrhythmias | Major | Minor |
| • Frequent ventricular extrasystoles (>500 per 24 h), non‐sustained or sustained ventricular tachycardia of LBBB morphology | •Frequent ventricular extrasystoles (>500 per 24 h), non‐sustained or sustained ventricular tachycardia with a RBBB morphology (excluding the ‘fascicular pattern’) | |
| Minor | ||
| • Frequent ventricular extrasystoles (500 per 24 h), non‐sustained or sustained ventricular tachycardia of LBBB morphology with inferior axis (‘RVOT pattern’) | ||
| VI. Family history/genetics | Major | |
|
• ACM confirmed in a first‐degree relative who meets diagnostic criteria • ACM confirmed pathologically at autopsy or surgery in a first degree relative • Identification of a pathogenic or likely pathogenetic ACM mutation in the patient under evaluation |
||
| Minor | ||
|
•History of ACM in a first‐degree relative in whom it is not possible or practical to determine whether the family member meets diagnostic criteria •Premature sudden death (<35 years of age) due to suspected ACM in a first‐degree relative •ACM confirmed pathologically or by diagnostic criteria in a second‐degree relative |
||
Note: Adapted from References 7, 10 with minor modifications. Any diagnosis of ‘right‐dominant’ or biventricular ACM requires that at least 1 criterion (RV criterion for ‘right‐dominant’ both RV and LV criterion for biventricular ACM), either major or minor from category I (Morpho‐functional ventricular abnormalities) or II (Structural myocardial abnormalities) be fulfilled. ‘Right‐dominant ‘ACM definite diagnosis: 2 major, or 1 major and 2 minor, or 4 minor RV criteria, borderline diagnosis: 1 major and 2 minor, or 3 minor RV criteria, possible diagnosis: 2 minor RV criteria,. Biventricular ACM definite diagnosis: 2 major, or 1 major and 2 minor, or 4 minor RV or LV criteria, borderline diagnosis: 1 major and 2 minor, or 3 minor RV or LV criteria, possible diagnosis: 2 minor RV or LV criteria. ‘Left‐dominant’ ACM diagnosis: a major structural LV criterion plus pathogenic or likely pathogenic ACM‐causing gene mutation must be present. ► criteria, that were present in our patient.
Abbreviations: ACM, arrhythmogenic cardiomyopathy; BSA, body surface area; EDV, end diastolic volume; EF, ejection fraction; ITF=International Task Force; LBBB, left bundle‐branch block; LGE, late gadolinium enhancement; LV, left ventricle; RBBB, right bundle‐branch block; RV, right ventricle; RVOT, right ventricular outflow tract.
Table 3.
Heart Rhythm Society Expert Consensus Recommendations on criteria for the diagnosis of cardiac sarcoidosis (2014)
| There are two pathways to a diagnosis of cardiac sarcoidosis (CS): |
| 1. Histological diagnosis from myocardial tissue |
| CS is diagnosed if an endomyocardial biopsy shows non‐caseating granuloma with no alternative cause for the histological findings identified |
| 2. Clinical diagnosis from invasive and non‐invasive studies: |
| CS is probable a if |
| (a) There is a histological diagnosis of extra‐cardiac sarcoidosis ◄ |
| and |
| (b) One or more of following is present: |
|
|
|
|
|
|
|
| and |
| (c) Other causes for the cardiac manifestation(s) have been reasonably excluded |
Note: Adapted from Reference 1 with minor modifications. ◄ criteria that were present in our patient.
In general, ‘probable involvement’ is considered adequate to establish a clinical diagnosis of CS.
Figure 4.
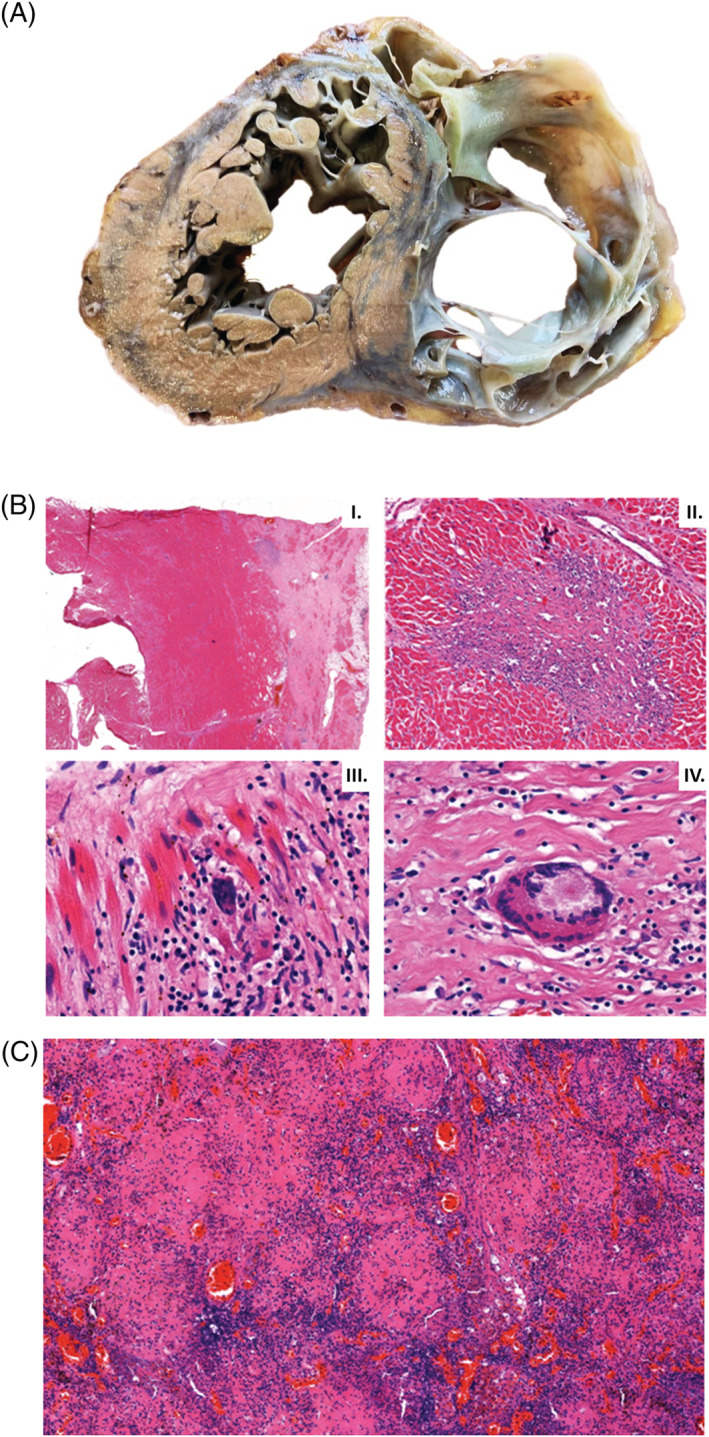
Macroscopic appearance of the heart and histological findings in the heart and a mediastinal lymph node during autopsy. (A) Photograph of a transverse slice of the heart at midventricular level with extremely thin right ventricular wall. The right ventricular myocardium is mostly replaced by fibrotic tissue. The appearance is similar to arrhytmogenic right ventricular cardiomyopathy. (B) Left ventricular tissues showing fibrosis advancing from the epicardium towards the endocardium (I.). There is non‐necrotizing granulomatous inflammation (II.) with epithelioid histiocytes (III.) and multinucleated giant cells (IV.) supporting the diagnosis of sarcoidosis. (C) The histology of a firm mediastinal lymph node shows non‐necrotizing granuloma‐like structures with a few giant cells. Mycobacteria were not confirmed by Ziehl–Neelsen staining, excluding tuberculosis.
Figure 5.
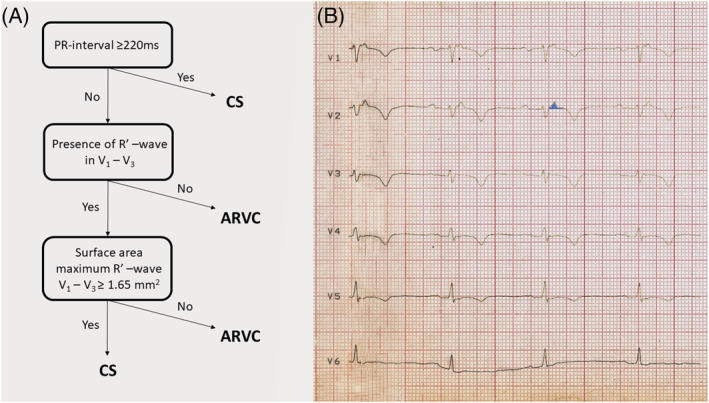
The algorithm devised to distinguish sarcoidosis with left and right ventricular involvement from ACM and its application on our patient's ECG. (A) The ECG algorithm. (B) The application of the algorithm on our patient's ECG recorded after the first admission. The surface area of the maximum R′ wave in lead V2 marked by light blue colour was ≥1.65 mm2. R′ wave was defined as any positive deflection after an S wave. ARVC, arrhythmogenic right ventricular cardiomyopathy; CS, cardiac sarcoidosis. For further explanation, see text.
Conflicts of interest
None declared.
Vereckei, A. , Katona, G. , Révész, K. , Vágó, H. , Müller, V. , Nagy, B. , Nagy, P. , Sepp, R. , and Suvarna, K. (2022) Cardiac sarcoidosis completely mimicking biventricular arrhythmogenic cardiomyopathy. ESC Heart Failure, 9: 4304–4314. 10.1002/ehf2.14123.
References
- 1. Birnie DH, Kandolin R, Nery PB, Kupari M. Cardiac manifestations of sarcoidosis: Diagnosis and management. Eur Heart J. 2017; 38: 2663–2670. [DOI] [PubMed] [Google Scholar]
- 2. Sedaghat‐Hamedani F, Kayvanpour E, Hamed S, Frankenstein L, Riffel J, Gi W‐T, Amr A, Samani OS, Haas J, Miersch T, Herpel E, Kreusser MM, Ehlermann P, Katus HA, Meder B. The chameleon of cardiology: Cardiac sarcoidosis before and after heart transplantation. ESC Heart Fail. 2020; 7: 692–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marcus FI, McKernna WJ, Sherill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MGPJ, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Pronotarios N, Saffitz JE, Sanborn DMY, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Proposed modification of the task force criteria. Circulation. 2010; 121: 1533–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pinamonti B, Brun F, Mestroni L, Sinagra G. Arrhythmogenic right ventricular cardiomyopathy: From genetics to diagnostic and therapeutic challenges. World J Cardiol. 2014; 6: 1234–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pilichou K, Thiene G, Bauce B, Rigato I, Lazzarini E, Migliore F, Marra MP, Rizzo S, Zorzi A, Daliento L, Corrado D, Basso C. Arrhythmogenic cardiomyopathy. Orphanet J Rare Dis. 2016; 11: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corrado D, Wichter T, Link MS, Hauer R, Marchlinski F, Anastasakis A, Bauce B, Basso C, Brunckhorst C, Tsatsopoulou A, Tandri H, Paul M, Schmied C, Peliccia A, Duru F, Pronotarios N, Mark Estes III, NA, McKenna WJ, Thiene G, Marcus FI, Calkins H. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: An international task force consensus statement. Eur Heart J. 2015; 36: 3227–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Corrado D, Perazzolo Marra M, Zorzi A, Beffagna G, Cipriani A, De Lazzari M, Migliore F, Pilichou K, Rampazzo A, Rigato I, Rizzo S, Thiene G, Anastasakis A, Asimaki A, Bucciarelli‐Ducci C, Haugaa KH, Marchlinski FE, Mazzanti A, McKenna WJ, Pantazis A, Peliccia A, Schmied C, Sharma S, Wichter T, Bauce B, Basso C. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int J Cardiol. 2020; 319: 106–114. [DOI] [PubMed] [Google Scholar]
- 8. Corrado D, Zorzi A, Cipriani A, Bauce B, Bariani R, Beffagna G, De Lazzari M, Migliore F, Pilichou K, Rampazzo A, Rigato I, Rizzo S, Thiene G, Marra P, Basso C. Evolving diagnostic criteria for arrhythmogenic cardiomyopathy. J Am Heart Assoc. 2021; 10: e021987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Terasaki F, Azuma A, Anzai T, Ishizaka N, Ishida Y, Isobe M, Inomata T, Ishibashi‐Ueda H, Eishi Y, Kitakaze M, Kusano K, Sakata Y, Shijubo N, Tsuchida A, Tsutsui H, Nakajima T, Nakatani S, Horii T, Yazaki Y, Yamaguchi E, Yamaguchi T, Ide T, Okamura H, Kato Y, Goya M, Sakakibara M, Soejima K, Nagai T, Nakamura H, Noda T, Hasegawa T, Morita H, Ohe T, Kihara Y, Saito Y, Sugiyama Y, Morimoto SI, Yamashina A. JCS 2016 guideline on diagnosis and treatment of cardiac sarcoidosis. Digest version Circ J. 2019; 83: 2329–2388. [DOI] [PubMed] [Google Scholar]
- 10. Krohn J, Ellenbogen KA. Cardiac sarcoidosis: contemporary review. J Cardiovasc Electrophysiol. 2015; 26: 104–109. [DOI] [PubMed] [Google Scholar]
- 11. Drent M, Crouser ED, Grunewald J. Challenges of sarcoidosis and its management. N Engl J Med. 2021; 385: 1018–1032. [DOI] [PubMed] [Google Scholar]
- 12. Philips B, Madhavan S, James CA, te Riele ASJM, Murray B, Tichnell C, Bhonsale A, Nazarian S, Judge DP, Calkins H, Tandri H, Cheng A. Arrhythmogenic right ventricular dysplasia/cardiomyopathy and cardiac sarcoidosis. Circ:Arrhythm Electrophysiol. 2014; 7: 230–236. [DOI] [PubMed] [Google Scholar]
- 13. Slart RHJA, Glaudermans AWJM, Lancelotti P, Hyafil F, Blankstein R, Schwartz RG, Jaber WA, Russell R, Gimelli A, Rouzet F, Hacker M, Gheysens O, Plein S, Miller EJ, Dorbala S, Donal E. A joint procedural position statement on imaging in cardiac sarcoidosis: From the cardiovascular and Inflammation & Infection Committees of the European Association of Nuclear Medicine, the European Association of Cardiovascular Imaging and the American society of nuclear cardiology. Eur Heart J‐Cardiovasc Imaging. 2017; 18: 1073–1089. [DOI] [PubMed] [Google Scholar]
- 14. Hoogendoorn JC, Venlet J, Out YNJ, Man S, Kumar S, Sramko M, Dechering DG, Nakajima I, Siontis KC, Watanabe M, Nakamura Y, Tedrow UB, Bogun F, Eckardt L, Peichl P, Stevenson WG, Zeppenfeld K. The precordial R′ wave: A novel discriminator between cardiac sarcoidosis and arrhythmogenic right ventricular cardiomyopathy in patients presenting with ventricular tachycardia. Heart Rhythm. 2021; 18: 1539–1547. [DOI] [PubMed] [Google Scholar]