

Abstract
Aims
Circulating inflammatory markers are associated with incident heart failure (HF), but prospective data on associations of immune cell subsets with incident HF are lacking. We determined the associations of immune cell subsets with incident HF as well as HF subtypes [with reduced ejection fraction (HFrEF) and preserved ejection fraction (HFpEF)].
Methods and results
Peripheral blood immune cell subsets were measured in adults from the Multi‐Ethnic Study of Atherosclerosis (MESA) and Cardiovascular Health Study (CHS). Cox proportional hazard models adjusted for demographics, HF risk factors, and cytomegalovirus serostatus were used to evaluate the association of the immune cell subsets with incident HF. The average age of the MESA cohort at the time of immune cell measurements was 63.0 ± 10.4 years with 51% women, and in the CHS cohort, it was 79.6 ± 4.4 years with 62% women. In the meta‐analysis of CHS and MESA, a higher proportion of CD4+ T helper (Th) 1 cells (per one standard deviation) was associated with a lower risk of incident HF [hazard ratio (HR) 0.91, (95% CI 0.83–0.99), P = 0.03]. Specifically, higher proportion of CD4+ Th1 cells was significantly associated with a lower risk of HFrEF [HR 0.73, (95% CI 0.62–0.85), <0.001] after correction for multiple testing. No association was observed with HFpEF. No other cell subsets were associated with incident HF.
Conclusions
We observed that higher proportions of CD4+ Th1 cells were associated with a lower risk of incident HFrEF in two distinct population‐based cohorts, with similar effect sizes in both cohorts demonstrating replicability. Although unexpected, the consistency of this finding across cohorts merits further investigation.
Keywords: Adaptive immunity, Innate immunity, Heart failure
Introduction
The role of inflammation in the development and progression of heart failure (HF) is well established. Observational studies have consistently shown an association between inflammatory cytokines [tumour necrosis factor (TNF) superfamily, interleukin (IL)‐1 family, and IL‐6] and incident HF as well as disease severity including mortality in adults with established HF. 1 , 2 , 3 , 4 , 5 , 6 However, prospective trials targeting these inflammatory cytokines in adults with existing HF have not shown benefit. 7 , 8 A more recent analysis from the Canakinumab Anti‐Inflammatory Thrombosis Outcomes Study (CANTOS) did show a decrease in incident HF with IL‐1β blockade in adults with prior myocardial infarction (MI) in secondary analyses, 9 and recent trials on colchicine following MI demonstrated significant reduction in major adverse cardiovascular events. 10 Although the mechanisms of these inflammation‐modulating interventions differ, the proof‐of‐concept findings highlight the importance of identifying specific immune mechanisms—including their cellular effectors—that may prevent or promote HF.
Immune pathways are broadly categorized into innate and adaptive responses. In persons with a high burden of vascular and metabolic risk factors, chronic innate and adaptive immune activation work in concert to drive inflammasome activation and elaboration of inflammatory cytokines such as TNF‐a, IL‐1β, and IL‐6, exacerbating myocardial damage and progression to clinical sequelae. 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 Experimental models and cross‐sectional studies in humans have implicated certain immune cell populations such as classical monocytes (CD14++CD16−) and γδ T cells in promoting myocardial inflammation and other populations such as intermediate monocytes (CD14++CD16+) and natural killer (NK) cells in regulating myocardial inflammation. 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 Although the pro‐inflammatory cytokines associated with HF are classically involved with innate immunity, there is growing appreciation that dysregulated adaptive immunity can cause myocardial dysfunction. 28 , 29 , 30 Several animal models have demonstrated a causative role of CD4+ T helper (Th) 1 and Th17 cells in development of HF. 31 , 32 In fact, CD4+ T regulatory cells, which generally have an inflammation‐resolving role, can phenotype‐switch and become dysfunctional in HF models, adopting a Th1 phenotype, 33 mirroring tissue‐level phenotype switches observed in atherosclerosis. 34 Chronic immune activation also results in a rise in subpopulations of antigen‐specific memory (CD45RO+) T cells, terminally differentiated effector memory T cells, and senescent (CD28−) T cells, which are associated with comorbidities such as age, hypertension, diabetes, and atherosclerosis. 35 , 36 , 37 , 38 , 39
No prospective population‐based studies to our knowledge have investigated the associations of innate and adaptive immune cell populations with the risk of incident HF. In this study, we measured 31 immune cell subsets using cryopreserved samples from participants enrolled in the population‐based Multi‐Ethnic Study of Atherosclerosis (MESA) and Cardiovascular Health Study (CHS). We evaluated the prospective relationships of these immune cell subsets with incident HF overall as well as pre‐specified subtypes of HF [with preserved ejection fraction (HFpEF) and with reduced ejection fraction (HFrEF)] in these cohorts. Based on prior experimental models and our cross‐sectional results in MESA, we hypothesized that higher proportions of classical monocytes (CD14++CD16−), γδ T cells, Th1, Th17, and CD4 + CD28− T cells, and lower proportions of intermediate monocytes (CD14 + CD16+), NK cells, and Treg (CD4 + CD25 + CD127−) cells would be associated with greater risk of incident HF.
Methods
Study cohorts
MESA is a prospective population‐based cohort study consisting of 6814 men and women from six U.S. communities (Chicago, IL; Los Angeles, CA; Baltimore, MD; St Paul, MN; New York, NY; and Forsyth County, NC). Adults aged 45–84 years were recruited for the baseline exam from 2000 to 2002 with follow‐up examinations in 2002–2004, 2004–2005, 2005–2007, 2010–2011, and 2016–2018. Patients with active cancer or history of clinical cardiovascular disease, including HF, were excluded from enrolment. Participants self‐identified as White, Black, Hispanic, or Chinese. Additional details on the design of the cohort have been previously described. 40
CHS is a prospective population‐based cohort study of adults aged 65 or older from four field centres (Washington County, MD; Pittsburgh, PA; Sacramento County, CA; and Forsyth County, NC). 41 In 1989–1990, 5201 adults were recruited, and an additional cohort of 687 primarily Black participants was recruited in 1992–1993. Participants had semi‐annual follow‐up via clinical examinations and telephone visits from 1990 to 1999 and biannual telephone follow‐ups after 1999.
The current study included 2106 MESA and 1860 CHS participants with cryopreserved peripheral blood mononuclear cell (PBMC) samples; these were obtained during the baseline exam in MESA (2000–2002) and the Year 11 exam (1998–1999) in CHS, which was defined as the analytic baseline for this study. Adults in CHS with HF at the 1998–1999 exam were excluded from the primary analysis, and no participants in MESA had HF at baseline. The samples were collected as part of two case‐cohort studies, one focused on MI (HL120854) and the other on HF (HL144484), both nested within MESA and CHS. 42 In each instance, all incident cases and a random cohort were sampled. Data from the two case cohorts were combined, with sampling weights incorporated as needed to permit valid estimates of associations with HF. All participants provided written informed consent for participation in the study and all procedures were conducted under institutionally approved protocols for human subjects research. The investigation conforms with the principles outlined in the Declaration of Helsinki.
Baseline clinical data
Clinical covariate data were obtained from the same examination as cryopreserved cells: 2000–2002 in MESA (baseline exam) and 1998–1999 in CHS (Year 11). In both cohorts, comprehensive standardized questionnaires were used to obtain self‐reported and measured baseline risk factors. Baseline covariates of interest included age, sex, race/ethnicity, education, alcohol use, smoking status, physical activity, body mass index (BMI), systolic blood pressure, use of anti‐hypertensive medications, diabetes, low‐density lipoprotein cholesterol (LDL‐C), high‐density lipoprotein cholesterol (HDL‐C), use of statin therapy, estimated glomerular filtration rate (eGFR), and cytomegalovirus (CMV) serostatus.
Race/ethnicity was categorized as White, Black, Hispanic, or Chinese in MESA and as Black or non‐Black in CHS. Education level was dichotomized into those with a bachelor's degree and above vs. those without in both cohorts. Alcohol use and smoking were defined as never, former (none within past 30 days), or current. In MESA, physical activity was determined using a 28‐item Typical Week Physical Activity Survey. In CHS, physical activity was assessed using a modified Minnesota Leisure‐Time Activities questionnaire, which evaluated the frequency and duration of 15 activities during a 2‐week period. For both cohorts, activities were assigned a metabolic equivalent task (MET) intensity score and then multiplied by minutes per week (MET‐min/week). 43 BMI was calculated as the ratio of weight to height squared (kg/m2). Diabetes was defined as a fasting blood glucose >125 mg/dL or the use of insulin or oral hypoglycaemic medications. Fasting lipid profile and serum creatinine were also obtained for each participant. The CKD‐EPI equation was used to calculate eGFR in both cohorts. 44 IgG antibodies to CMV were measured by enzyme immunoassay (Diamedix Corp., Miami Lakes, FL). The inter‐assay coefficients of variation were 5.1–6.8% for the CMV IgG immunoassay.
Immune cell phenotyping
The flow cytometry gating strategies used for immune cell phenotyping and flow cytometry gating strategies have been previously described in detail. 42 , 45 , 46 , 47 PBMCs were isolated from blood collected in 8‐mL citrate CPT tubes during the baseline MESA exam in 2000–2002 and Year 11 CHS exam in 1998–1999. The isolated PBMCs were cryopreserved in media containing 90% foetal bovine serum and 10% dimethyl sulfoxide at −135°C. The cells were thawed for phenotyping in 2016–2018. A small pilot study demonstrated that cell viability was similar in cryopreserved and fresh samples.
Cell surface labelling was performed to identify monocyte subsets, NK cells, CD19+ B cells, γδ T cells, regulatory and helper CD4+ T cells, and naïve, memory, differentiated, activated, CD45RA+ re‐expressing effector memory (TEMRA) CD4+, and CD8+ T‐cell subsets in both cohorts. Cells were incubated with antibodies for 15 min at room temperature in the dark and refrigerated with 1% paraformaldehyde (Alfa Aesar, Tewksbury, MA) until flow cytometry was performed. All antibodies were from Miltenyi Biotec (San Diego, CA) and used manufacturer recommended dilutions (Table S1 ). The cell surface markers associated with each subset are listed in Table S2 .
In MESA, intracellular cytokine staining was used for CD4+ T helper cells. PBMCs were activated with phorbol myristic acid/ionomycin in the presence of brefeldin A. 48 After incubating for 3 hat 37°C, 5% CO2, cells were centrifuged, resuspended in phosphate buffered saline, pH 7.4 and incubated with a live dead stain (Thermo Fisher Cat# L34955) for 15 min at room temperature, and then centrifuged at 300 × g for 5 min. The PBMCs were resuspended in CD4 antibodies and incubated for 15 min at room temperature in the dark. Samples were centrifuged and the pellets were washed and fixed with 2% paraformaldehyde (Alfa Aesar Cat#43368) for 10 min. Paraformaldehyde was removed by centrifugation, and cells were incubated with antibodies to IL4 (Th2), IL17 (Th17), and interferon‐γ (Th1) in the presence of 1% saponin. After 15 min, cells are washed and resuspended in paraformaldehyde until flow cytometry was performed. T helper cells in CHS were determined by cell surface markers because samples from CHS lost viability during the intracellular staining protocol.
Flow cytometry was performed on a MQ10 flow cytometer and analysed with MACS Quantify software (Miltenyi Biotec). Calibration beads were used for daily calibration. Compensation was set using single‐colour compensation controls, and isotype controls were used to set negative gates for each assay. Cell phenotypes were expressed as percentages. Flow cytometry gating strategies are presented in Figures S1 – S6 . All data are presented as a percentage (CD4 subsets as % CD4+ T cells, CD8 subsets as % CD8+ T cells, monocyte subsets as % CD14+ monocytes, γδ T cells as % CD3+ T cells, B cell subsets as % CD19 B cells, and NK cells as % gated lymphocytes). Percentages, instead of absolute numbers, were used because PBMCs for this cohort were cryopreserved for many years. Due to sample manipulation from cryopreservation, our internal analyses have demonstrated percentages, rather than absolute counts, to be a better representation of circulating cells in the whole blood.
Heart failure adjudication
The co‐primary outcomes in the study were incident HF (overall) and HF subtypes HFrEF and HFpEF. In both cohorts, potential HF events were identified from hospitalizations reported at semi‐annual or annual participant interviews or from CMS claims data. In MESA, HF events were independently adjudicated by two physicians based on the presence of a clinical diagnosis of symptomatic HF with treatment, as well as objective signs of HF such as left ventricular systolic or diastolic dysfunction or pulmonary oedema. 40 In CHS, each event was confirmed through a review of available medical records by an expert adjudication panel and required a physician diagnosis along with symptoms and clinical findings or medical therapy for HF. 6 , 41 , 49 , 50 Adjudicated HF events were classified as HFrEF and HFpEF based on the measured left ventricular ejection fraction (LVEF) on cardiac imaging surrounding the time of HF diagnosis, using a cut‐off of 45%, or unclassified if LVEF was missing.
Statistical analysis
Our analyses included participants with at least one immune cell measure (n = 3966; MESA: 2106, CHS: 1860). The number of participants with data for each immune cell subset is available in Table S2 . Missing immune cell data were due to poor sample quality or technical errors, which occurred at random and were not biased by participant characteristics. Multiple imputation (100 imputations) with chained equations was used to impute missing immune cell and covariate data among the 3966 participants. All 3966 participants had data available on HF status.
Cox proportional hazard models were used to determine the associations between the immune cell subsets and HF (overall), HFrEF, and HFpEF. We analysed HF subtypes as co‐primary endpoints along with overall HF because HFpEF and HFrEF have distinct pathophysiological contributors and effective therapies for these different HF phenotypes likewise differ. All available participants in CHS had immune cell subsets measured, so no sampling weights were required. In MESA, the inverse sampling probabilities for inclusion in the immune cell studies were incorporated as weights in the model to obtain unbiased estimates; and robust standard error estimates were used. Each immune cell subset was modelled independently to avoid collinearity. Each cohort was analysed separately and then combined using fixed effects inverse‐variance‐weighted meta‐analysis. First, we adjusted for demographics including age, sex, race/ethnicity, education, clinical site, and immune cell batch. The primary model included HF risk factors including behaviours (alcohol use, smoking status, physical activity), clinical factors (BMI, diabetes, systolic blood pressure, use of anti‐hypertensive medications, LDL‐C, HDL‐C, use of statin therapy, and eGFR), and CMV serostatus. CMV serostatus was used as a covariate given prior findings in MESA that CMV titres were associated with T‐cell biasing and the high population prevalence as a chronic infection. 48 Multiple imputation was used for the primary analysis. Sensitivity analysis included complete‐case analysis that excluded any participants with missing immune cell or covariate data. Because the immune cell subsets in CHS were collected 9 years into the study, we also assessed the association between immune cell subsets and prevalent HF using logistic regression in a secondary analysis. Individuals with HF by 1998–1999 in CHS were otherwise excluded from the primary analysis of incidence. Bonferroni correction was used to account for multiple hypothesis testing. For the eight immune cell subsets included in the a priori hypotheses, P value threshold was set at ≤0.0063 (eight tests). For the remaining 20 immune cell subsets, P value threshold was set at ≤0.0018 (28 total tests). P values observed between the Bonferroni‐corrected P value and 0.05 were interpreted as ‘borderline significant’.
Results
The baseline demographics of participants from each cohort are described in Table 1 . The average age of the MESA cohort was 63.0 ± 10.4 years with 51% women and 28% Black. In contrast, the average age of the CHS cohort was 79.6 ± 4.4 years with 62% women and 18% Black. A higher proportion of MESA participants had an undergraduate or higher degree of education and performed more leisure‐time physical activity than the CHS participants. But there were more current smokers and drinkers in the MESA cohort. As expected, the older CHS cohort had a higher burden of hypertension than the MESA cohort. Other risk factors such as BMI, diabetes, and eGFR were relatively similar in both cohorts.
Table 1.
Baseline characteristics of participants from MESA and CHS
| MESA (n = 2106) | CHS (n = 1860) | |
|---|---|---|
| Age, years (mean ± SD) | 63.0 ± 10.4 | 79.6 ± 4.4 |
| Male, % | 49 | 38 |
| Race/ethnicity, % | ||
| White | 36 | 82 |
| Chinese | 14 | |
| Black | 28 | 18 |
| Hispanic | 22 | |
| Education, % (≥Bachelor's degree) | 34 | 21 |
| Leisure‐time physical activity, Met‐min/week (mean ± SD) | 1534 ± 2408 | 773 ± 1105 |
| Alcohol, % | ||
| Current | 53 | 7 |
| Former | 25 | 34 |
| Never | 22 | 59 |
| Smoking status, % | ||
| Current | 12 | 7 |
| Former | 38 | 44 |
| Never | 50 | 49 |
| BMI, kg/m2 (mean ± SD) | 28.3 ± 5.4 | 26.6 ± 4.5 |
| Diabetes, % | 15 | 13 |
| SBP, mmHg (mean ± SD) | 128.6 ± 21.8 | 135.2 ± 20.2 |
| Anti‐hypertensive use, % | 40 | 58 |
| Statin use, % | 16 | 16 |
| LDL cholesterol, mg/dL (mean ± SD) | 116.1 ± 30.8 | 129.4 ± 32.8 |
| HDL cholesterol, mg/dL (mean ± SD) | 50.1 ± 14.4 | 53.9 ± 14.3 |
| eGFR, mL/min/1.73 m2 (mean ± SD) | 77.2 ± 17.3 | 73.4 ± 18.3 |
| Log‐transformed CMV (mean ± SD) |
BMI, body mass index; CHS, Cardiovascular Health Study; CMV, cytomegalovirus; eGFR, estimated glomerular filtration rate; HDL, high‐density cholesterol; LDL, low‐density cholesterol; MESA, Multi‐Ethnic Study of Atherosclerosis; SBP, systolic blood pressure; SD, standard deviation.
Race/ethnicity was self‐identified.
The mean proportion of each immune cell subset in each cohort are included in Table S2 . The median duration of follow up was 16.3 years [interquartile range (IQR) 8.6–17.3 years] in MESA and 8.4 years (IQR 4.3–13.2 years) in CHS. There were a total of 933 HF events, 340 in MESA and 593 in CHS. Of those that had an LVEF at time of diagnosis (70%), 336 were categorized as HFpEF (129 in MESA and 207 in CHS), and 318 were categorized as HFrEF (168 in MESA and 150 in CHS). The median and IQR of LVEF in participants with HFrEF was 32.5% (IQR 27–39) in MESA and 35% (IQR 25–40) in CHS. The median and IQR of LVEF in participants with HFpEF was 60% (IQR 55–63) in MESA and 60% (IQR 55–63) in CHS.
The associations between the prespecified immune cell subsets and incident HF are shown in Figure 1 . In the primary model (Table 2 ), a higher proportion of CD4+ Th1 cells was associated with a borderline‐significantly lower risk of HF in the meta‐analysis [HR 0.91, (95% CI 0.83–0.99), P = 0.03], with similar HRs observed in MESA (0.92) and CHS (0.90). The direction of this association was opposite of our a priori hypothesis. The finding was driven by the association with HFrEF. When analysing HF sub‐phenotypes separately (HFrEF and HFpEF), a higher proportion of CD4+ Th1 cells was associated with a significantly lower risk of HFrEF in the meta‐analysis [HR 0.73, (95% CI 0.62–0.85), P < 0.001] even after Bonferroni correction (Table 3 ). The association was observed in both MESA [HR 0.80, (95% CI 0.65–0.99), P = 0.04] and CHS [HR 0.65, (95% CI 0.51–0.82), P < 0.001] cohorts. Of note, there was no significant difference observed in HF risk when categorized by overall CD4 quartiles Figure ( S7 ). A higher proportion of NK cells was marginally non‐significantly associated with a lower risk of HFrEF in MESA [HR 0.81, (95% CI 0.65–1.00), P = 0.06] but not in CHS [HR 1.00, (95% CI 0.81–1.22), P = 0.97]. No immune cell subsets, including CD4+ Th1 cells or NK cells, had a significant association with HFpEF in MESA or CHS (Table 4 ). In a sensitivity analysis restricting the cohort to only complete cases (Table S4 ), a higher proportion of CD4+ Th1 cells was borderline‐significantly associated with lower risk of HF in the meta‐analysis [HR 0.90, (95% CI 0.82–0.99), P = 0.03].
Figure 1.
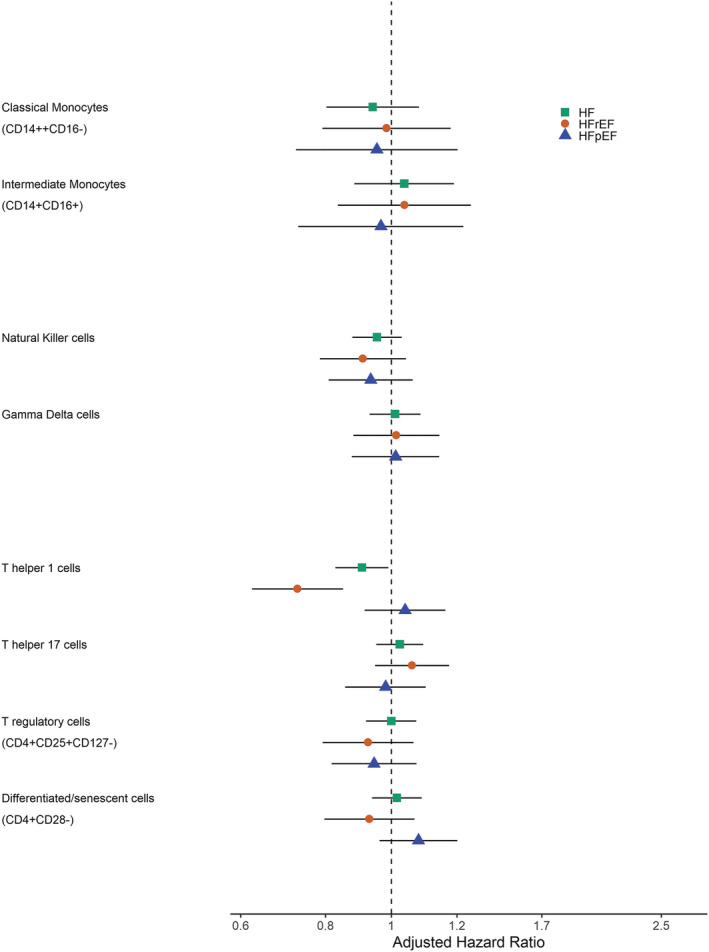
Association of pre‐specified immune cells with incident heart failure.
Table 2.
Association of immune cell subsets with incident heart failure
| Immune cell subsets | MESA | CHS | Meta‐analysis | |||
|---|---|---|---|---|---|---|
| HR (95% CI) | P value | HR (95% CI) | P value | HR (95% CI) | P value | |
| Monocytes | ||||||
| Classical monocytes | 0.94 (0.80–1.10) | 0.43 | ||||
| Intermediate monocytes | 1.05 (0.88–1.24) | 0.61 | ||||
| Non‐classical monocytes | 1.05 (0.90–1.22) | 0.56 | ||||
| Innate lymphocytes | ||||||
| Natural killer cells | 0.88 (0.76–1.03) | 0.11 | 0.99 (0.89–1.09) | 0.77 | 0.95 (0.88–1.04) | 0.26 |
| γδ T cells | 0.92 (0.78–1.10) | 0.36 | 1.05 (0.95–1.16) | 0.38 | 1.01 (0.93–1.10) | 0.77 |
| CD4+ T cells | ||||||
| Pan‐CD4+ T cells | 1.04 (0.89–1.21) | 0.66 | 0.97 (0.88–1.07) | 0.55 | 0.99 (0.91–1.07) | 0.79 |
| T helper 1 | 0.92 (0.79–1.08) | 0.31 | 0.90 (0.80–1.00) | 0.05 | 0.91 (0.83–0.99) | 0.03 |
| T helper 2 | 1.09 (0.93–1.27) | 0.29 | 1.02 (0.91–1.14) | 0.76 | 1.04 (0.95–1.14) | 0.39 |
| T helper 17 | 1.01 (0.88–1.16) | 0.90 | 1.04 (0.94–1.15) | 0.43 | 1.03 (0.95–1.11) | 0.47 |
| T regulatory cells | 1.04 (0.88–1.22) | 0.68 | 0.99 (0.89–1.09) | 0.81 | 1.00 (0.92–1.09) | 1.00 |
| Naive | 0.97 (0.84–1.10) | 0.61 | 1.04 (0.95–1.14) | 0.38 | 1.02 (0.94–1.10) | 0.66 |
| Memory | 1.00 (0.87–1.14) | 0.95 | 0.96 (0.87–1.07) | 0.49 | 0.98 (0.90–1.06) | 0.55 |
| Activated or T regulatory | 0.98 (0.83–1.16) | 0.79 | 1.06 (0.95–1.19) | 0.27 | 1.04 (0.94–1.14) | 0.44 |
| Activated or mature | 1.04 (0.95–1.14) | 0.38 | ||||
| Differentiated/senescent (CD4 + CD28−) | 0.98 (0.81–1.18) | 0.83 | 1.03 (0.94–1.13) | 0.54 | 1.02 (0.94–1.11) | 0.65 |
| Differentiated/senescent (CD4 + CD57+) | 0.95 (0.8–1.14) | 0.61 | 1.05 (0.96–1.16) | 0.27 | 1.03 (0.95–1.12) | 0.45 |
| Differentiated/senescent (CD4 + CD28 − CD57+) | 0.92 (0.77–1.11) | 0.39 | 1.02 (0.93–1.11) | 0.71 | 1.00 (0.92–1.08) | 0.96 |
| TEMRA | 0.93 (0.78–1.12) | 0.45 | 0.99 (0.90–1.08) | 0.81 | 0.98 (0.90–1.06) | 0.58 |
| CD8+ T cells | ||||||
| Pan‐CD8+ T cells | 1.02 (0.89–1.18) | 0.78 | 1.04 (0.94–1.14) | 0.47 | 1.03 (0.95–1.12) | 0.45 |
| Naïve | 1.07 (0.93–1.23) | 0.35 | 1.04 (0.94–1.14) | 0.45 | 1.05 (0.97–1.13) | 0.25 |
| Memory | 0.90 (0.77–1.04) | 0.16 | 0.95 (0.87–1.05) | 0.33 | 0.94 (0.87–1.02) | 0.11 |
| Activated or mature | 1.06 (0.96–1.17) | 0.25 | ||||
| Differentiated/senescent (CD8 + CD28−) | 1.05 (0.89–1.23) | 0.58 | 1.05 (0.96–1.16) | 0.29 | 1.05 (0.97–1.14) | 0.24 |
| Differentiated/senescent (CD8 + CD57+) | 1.02 (0.88–1.19) | 0.76 | 0.98 (0.90–1.07) | 0.70 | 0.99 (0.92–1.07) | 0.86 |
| Differentiated/senescent (CD8 + CD28 − CD57+) | 1.06 (0.90–1.24) | 0.47 | 0.98 (0.90–1.08) | 0.73 | 1.00 (0.93–1.09) | 0.96 |
| TEMRA | 1.05 (0.91–1.21) | 0.54 | 1.01 (0.92–1.10) | 0.91 | 1.02 (0.94–1.10) | 0.67 |
| B cells | ||||||
| Pan‐CD19+ B cells | 1.05 (0.90–1.21) | 0.56 | 0.98 (0.89–1.09) | 0.71 | 1.00 (0.92–1.09) | 0.98 |
| Memory | 1.03 (0.87–1.23) | 0.72 | 0.97 (0.86–1.10) | 0.66 | 0.99 (0.90–1.10) | 0.88 |
Analysis adjusted for age, sex, race/ethnicity, education, clinical site, immune cell analytical batch, alcohol use, smoking status, physical activity, BMI, diabetes, systolic blood pressure, use of anti‐hypertensive medications, LDL‐C, HDL‐C, use of statin therapy, eGFR, and CMV serostatus. Multiple imputation method used.
Table 3.
Association of immune cell subsets with incident heart failure with reduced ejection fraction
| Immune cell subsets | MESA | CHS | Meta‐analysis | |||
|---|---|---|---|---|---|---|
| HR (95% CI) | P value | HR (95% CI) | P value | HR (95% CI) | P value | |
| Monocytes | ||||||
| Classical monocytes | 0.98 (0.79–1.22) | 0.89 | ||||
| Intermediate monocytes | 1.05 (0.83–1.31) | 0.69 | ||||
| Non‐classical monocytes | 0.98 (0.79–1.21) | 0.83 | ||||
| Innate lymphocytes | ||||||
| Natural killer cells | 0.81 (0.65–1.00) | 0.06 | 1.00 (0.82–1.23) | 0.97 | 0.91 (0.78–1.05) | 0.20 |
| γδ T cells | 0.86 (0.67–1.10) | 0.23 | 1.12 (0.93–1.35) | 0.23 | 1.02 (0.88–1.18) | 0.82 |
| CD4 + T cells | ||||||
| Pan‐CD4+ T cells | 1.13 (0.91–1.39) | 0.28 | 0.93 (0.77–1.12) | 0.45 | 1.01 (0.88–1.16) | 0.87 |
| T helper 1 | 0.80 (0.65–0.99) | 0.04 | 0.65 (0.51–0.82) | <0.001 | 0.73 (0.62–0.85) | <0.001 |
| T helper 2 | 0.99 (0.79–1.24) | 0.94 | 1.16 (0.96–1.41) | 0.13 | 1.08 (0.94–1.25) | 0.27 |
| T helper 17 | 0.99 (0.81–1.20) | 0.89 | 1.14 (0.96–1.35) | 0.12 | 1.07 (0.95–1.22) | 0.27 |
| T regulatory cells | 0.90 (0.72–1.13) | 0.36 | 0.95 (0.76–1.18) | 0.63 | 0.92 (0.79–1.08) | 0.32 |
| Naive | 1.04 (0.86–1.25) | 0.68 | 1.05 (0.87–1.25) | 0.61 | 1.04 (0.92–1.19) | 0.52 |
| Memory | 0.94 (0.77–1.15) | 0.55 | 0.86 (0.70–1.05) | 0.13 | 0.90 (0.78–1.03) | 0.13 |
| Activated or T regulatory | 0.95 (0.75–1.19) | 0.64 | 1.08 (0.86–1.37) | 0.51 | 1.01 (0.86–1.19) | 0.90 |
| Activated or mature | 1.19 (1.00–1.41) | 0.05 | ||||
| Differentiated/senescent (CD4 + CD28−) | 0.87 (0.67–1.13) | 0.31 | 0.96 (0.79–1.16) | 0.68 | 0.93 (0.80–1.08) | 0.35 |
| Differentiated/senescent (CD4 + CD57+) | 0.84 (0.65–1.07) | 0.16 | 0.96 (0.79–1.16) | 0.65 | 0.91 (0.79–1.06) | 0.22 |
| Differentiated/senescent (CD4 + CD28 − CD57+) | 0.83 (0.65–1.06) | 0.13 | 0.92 (0.76–1.11) | 0.37 | 0.88 (0.76–1.02) | 0.10 |
| TEMRA | 0.90 (0.71–1.15) | 0.41 | 0.91 (0.75–1.10) | 0.33 | 0.91 (0.78–1.05) | 0.20 |
| CD8 + T cells | ||||||
| Pan‐CD8+ T cells | 0.95 (0.80–1.13) | 0.54 | 0.89 (0.73–1.08) | 0.24 | 0.92 (0.81–1.05) | 0.21 |
| Naïve | 1.08 (0.87–1.33) | 0.48 | 1.00 (0.83–1.20) | 0.99 | 1.03 (0.90–1.19) | 0.65 |
| Memory | 0.94 (0.76–1.18) | 0.61 | 0.94 (0.77–1.13) | 0.50 | 0.94 (0.81–1.08) | 0.39 |
| Activated or mature | 0.99 (0.81–1.20) | 0.90 | ||||
| Differentiated/senescent (CD8 + CD28−) | 1.08 (0.86–1.36) | 0.52 | 0.98 (0.81–1.19) | 0.84 | 1.02 (0.88–1.18) | 0.80 |
| Differentiated/senescent (CD8 + CD57+) | 1.02 (0.84–1.25) | 0.83 | 0.93 (0.78–1.11) | 0.41 | 0.97 (0.85–1.10) | 0.63 |
| Differentiated/senescent (CD8 + CD28 − CD57+) | 1.06 (0.85–1.32) | 0.61 | 0.91 (0.76–1.10) | 0.34 | 0.97 (0.84–1.12) | 0.68 |
| TEMRA | 1.06 (0.86–1.30) | 0.58 | 1.00 (0.83–1.20) | 1.00 | 1.03 (0.90–1.17) | 0.72 |
| B cells | ||||||
| Pan‐CD19+ B cells | 1.07 (0.89–1.29) | 0.47 | 1.07 (0.88–1.30) | 0.52 | 1.07 (0.93–1.22) | 0.33 |
| Memory | 1.02 (0.81–1.27) | 0.88 | 0.93 (0.73–1.20) | 0.60 | 0.98 (0.83–1.16) | 0.81 |
Analysis adjusted for age, sex, race/ethnicity, education, clinical site, immune cell analytical batch, alcohol use, smoking status, physical activity, BMI, diabetes, systolic blood pressure, use of anti‐hypertensive medications, LDL‐C, HDL‐C, use of statin therapy, eGFR, and CMV serostatus. Multiple imputation method used.
Table 4.
Association of immune cell subsets with incident heart failure with preserved ejection fraction
| Immune cell subsets | MESA | CHS | Meta‐analysis | |||
|---|---|---|---|---|---|---|
| HR (95% CI) | P value | HR (95% CI) | P value | HR (95% CI) | P value | |
| Monocytes | ||||||
| Classical monocytes | 0.95 (0.72–1.25) | 0.72 | ||||
| Intermediate monocytes | 0.97 (0.73–1.28) | 0.80 | ||||
| Non‐classical monocytes | 1.10 (0.86–1.40) | 0.43 | ||||
| Innate lymphocytes | ||||||
| Natural killer cells | 1.00 (0.79–1.26) | 0.98 | 0.90 (0.75–1.07) | 0.24 | 0.93 (0.81–1.08) | 0.34 |
| γδ T cells | 0.94 (0.70–1.26) | 0.68 | 1.04 (0.88–1.24) | 0.63 | 1.01 (0.88–1.18) | 0.85 |
| CD4 + T cells | ||||||
| Pan‐CD4+ T cells | 0.99 (0.78–1.27) | 0.95 | 0.93 (0.79–1.10) | 0.40 | 0.95 (0.83–1.09) | 0.46 |
| T helper 1 | 1.06 (0.83–1.34) | 0.65 | 1.04 (0.88–1.24) | 0.63 | 1.05 (0.91–1.20) | 0.50 |
| T helper 2 | 1.10 (0.84–1.45) | 0.47 | 0.87 (0.70–1.08) | 0.21 | 0.96 (0.81–1.13) | 0.61 |
| T helper 17 | 0.98 (0.79–1.22) | 0.86 | 0.98 (0.82–1.18) | 0.83 | 0.98 (0.86–1.12) | 0.78 |
| T regulatory cells | 1.01 (0.80–1.29) | 0.92 | 0.91 (0.75–1.09) | 0.28 | 0.94 (0.82–1.09) | 0.43 |
| Naive | 0.88 (0.71–1.10) | 0.27 | 1.08 (0.93–1.26) | 0.33 | 1.01 (0.89–1.14) | 0.87 |
| Memory | 1.06 (0.85–1.32) | 0.59 | 0.96 (0.81–1.15) | 0.66 | 1.00 (0.87–1.14) | 1.00 |
| Activated or T regulatory | 0.92 (0.71–1.21) | 0.56 | 1.01 (0.84–1.22) | 0.90 | 0.98 (0.84–1.14) | 0.81 |
| Activated or mature | 1.04 (0.89–1.22) | 0.61 | ||||
| Differentiated/senescent (CD4 + CD28−) | 1.16 (0.90–1.50) | 0.24 | 1.07 (0.92–1.25) | 0.38 | 1.10 (0.96–1.25) | 0.17 |
| Differentiated/senescent (CD4 + CD57+) | 1.10 (0.85–1.42) | 0.45 | 1.04 (0.88–1.22) | 0.64 | 1.06 (0.92–1.21) | 0.42 |
| Differentiated/senescent (CD4 + CD28 − CD57+) | 1.08 (0.84–1.39) | 0.54 | 1.04 (0.90–1.21) | 0.58 | 1.05 (0.93–1.20) | 0.43 |
| TEMRA | 1.00 (0.77–1.29) | 0.97 | 1.02 (0.88–1.19) | 0.79 | 1.01 (0.89–1.15) | 0.84 |
| CD8+ T cells | ||||||
| Pan‐CD8+ T cells | 1.09 (0.84–1.41) | 0.50 | 1.00 (0.86–1.18) | 0.95 | 1.03 (0.90–1.18) | 0.68 |
| Naïve | 1.13 (0.93–1.38) | 0.23 | 1.04 (0.89–1.22) | 0.61 | 1.08 (0.95–1.22) | 0.25 |
| Memory | 0.84 (0.68–1.05) | 0.12 | 0.95 (0.81–1.12) | 0.54 | 0.91 (0.80–1.04) | 0.15 |
| Activated or mature | 1.12 (0.95–1.31) | 0.18 | ||||
| Differentiated/senescent (CD8 + CD28−) | 1.05 (0.80–1.37) | 0.71 | 1.14 (0.97–1.33) | 0.11 | 1.11 (0.97–1.27) | 0.12 |
| Differentiated/senescent (CD8 + CD57+) | 1.04 (0.82–1.33) | 0.72 | 1.03 (0.89–1.20) | 0.65 | 1.04 (0.91–1.18) | 0.57 |
| Differentiated/senescent (CD8 + CD28 − CD57+) | 1.10 (0.85–1.43) | 0.47 | 1.04 (0.89–1.21) | 0.64 | 1.05 (0.92–1.20) | 0.44 |
| TEMRA | 1.07 (0.87–1.32) | 0.53 | 1.04 (0.89–1.21) | 0.62 | 1.05 (0.93–1.18) | 0.44 |
| B cells | ||||||
| Pan‐CD19+ B cells | 1.01 (0.77–1.31) | 0.96 | 1.06 (0.90–1.26) | 0.47 | 1.05 (0.91–1.20) | 0.53 |
| Memory | 1.06 (0.80–1.39) | 0.69 | 0.95 (0.78–1.17) | 0.65 | 0.99 (0.84–1.16) | 0.90 |
Analysis adjusted for age, sex, race/ethnicity, education, clinical site, immune cell analytical batch, alcohol use, smoking status, physical activity, BMI, diabetes, systolic blood pressure, use of anti‐hypertensive medications, LDL‐C, HDL‐C, use of statin therapy, eGFR, and CMV serostatus. Multiple imputation method used.
Because PBMCs were collected in CHS 9 years into the study, there were 68 cases of prevalent HF in the cohort. In secondary analyses, we determined the association of immune cell subsets with prevalent HF (Table S5 ). In the final model, adjusted for the same covariates as the above primary model, we observed an association between CD4+ Th17 cells and increased risk of prevalent HF [OR 1.36, (95% CI 1.08–1.71), P = 0.008]. There was no association between CD4+ Th1 or NK cells with prevalent HF.
Discussion
In this study, we evaluated the prospective associations of circulating immune cell subsets with the risk of incident HF in two longitudinal population‐based cohorts. We observed consistent associations of CD4+ Th1 cells with lower risk of overall HF, driven by a significantly lower risk for incident HFrEF. Specifically, one standard deviation increase in circulating Th1 cells as a proportion of total circulating CD4+ T cells was associated with an approximately 25% lower risk for incident HFrEF. Associations of Th1 cells with HF and HFrEF were similar in CHS and MESA, demonstrating replication across two distinct populations with important differences in demographics, baseline risk factor profiles, and follow‐up duration. Congruent findings across both cohorts add to the robustness of the results as Th immune cell subsets were identified using cell surface markers in CHS and cytokine profiles in MESA. No immune cell subsets were associated with risk for incident HFpEF.
Our central finding that a higher proportion of CD4+ Th1 cells was associated with a lower risk of incident HFrEF was not expected and thus is hypothesis‐generating and requires validation by future experimental studies. Prior human studies have shown that CD4+ Th1 cells and its inflammatory maker, interferon‐γ, are elevated in patients with prevalent HFrEF and associated with worse disease severity. 51 Animal models of HFrEF have also demonstrated expansion and infiltration of CD4+ Th1 cells, along with CD4+ Th2 and Th17 cells, into the myocardium. 31 , 32 , 33 Importantly, our analysis differs from prior studies in that we examined prospective associations of cell subsets with incident (not prevalent) HF, and we focused on circulating immune cells rather than cells entering into tissue‐specific spaces such as the myocardium. This association was not driven by interval MI, as prior analyses from these cohorts showed no association between CD4+ Th1 cells and MI. 42
First, the distinction between prevalent and incident disease—as well as the potentially divergent roles and functions of Th1 cells in individuals who are healthy at baseline compared with those who have existing disease—merits discussion. Unlike adults who have already developed HF, which creates a pro‐inflammatory environment and contributes to immune activation, participants in our cohorts were free of HF at the time of PBMC collection. 52 Thus, the make‐up and role of immune cell subsets may substantially differ before and after development of HF. Given the surprising but consistent protective association we observed of Th1 cells with HFrEF, these cells may represent entirely different processes in largely healthy individuals compared with individuals with existing HFrEF: In healthy individuals, higher Th1 proportions may reflect adaptive immune competence in response to invading pathogens (consistent with canonical Th1 functions 53 ) whereas if the overwhelming inflammatory stimulus is chronic, non‐resolving HF, Th1 cells may simply reflect HF severity. Furthermore, although animal models can help understand the role of immune cell subsets prior to development of HF, the model of cardiac injury is more acute and rapid than the natural history of HF in adults with competing co‐morbidities and chronic risk factors, which unfolds over years. When categorized by quartiles of Th1 proportions, baseline characteristics of participants in MESA and CHS did show some differences in demographics, such as greater Th1 proportions in women in CHS, but no significant differences in risk factor profiles (Tables S6 and S7 ).
A separate potential reason for our findings relates to the distinction between circulating and tissue‐infiltrating immune cells. Indeed, analyses of human myocardial tissue demonstrate that peripheral blood may not reflect the immune composition within different tissues, including myocardium and nearby lymph nodes. 54 The importance of these findings is underscored by the plasticity of CD4+ T‐cell subsets. It is becoming increasingly appreciated that CD4 T‐cell subsets are not terminally differentiated cells and in fact can acquire new properties and change functions over time depending on the local microenvironment. Specifically, CD4+ Th1 cells can switch from pro‐inflammatory effector cells to regulatory‐like T cells, with co‐morbidity‐ and local microenvironment‐specific determinants of these phenotype switches. 34 , 55 Follow‐up investigation of the role of T‐cell plasticity and related determinants, including T‐cell antigen specificity and clonal expansion, 56 , 57 warrants further study but was beyond the scope of this analysis.
A related possible explanation for our findings relates to differential migration into tissues of circulating cells. Recently, we observed a similarly surprising association whereby higher proportions of a canonically pro‐inflammatory cell subset (in this case, classical monocytes) were associated with carotid intima‐media thickness regression in MESA. 58 One hypothesized explanation for this finding was that specific cell subsets (such as classical monocytes, which commonly migrate across the endothelium into arteriosclerotic plaque) may actually have higher proportions in the blood if they are less effective at migrating out of the vascular space and into tissues of interest. Of course, as applies to Th1 cells and our HF findings, this is a hypothesis that is beyond the scope of the present study but could be investigated further in models evaluating circulating and myocardial‐specific T‐cell migration and differentiation.
Other associations we observed were inconsistent and not replicated across cohorts or analytic models. We observed an association between higher proportion of NK cells and lower risk of incident HFrEF. However, this was only observed in MESA and was not significant after correction for multiple testing. Although NK cells have regulatory functions and may be cardioprotective, this association needs to be corroborated in additional cohorts. No other CD4+ T cell, CD8+ T cells, monocyte, or B‐cell subsets were associated with incident HF in our study. This again contrasts from animal models of HF and human studies in participants who have already developed HF. Overall, our null findings suggest that although dysregulation of different immune cell subsets may play an important role in development of HF after acute myocardial injury, peripheral blood immune cells measured prior to development of cardiovascular disease are of limited use as HF biomarkers.
In an exploratory analysis of CHS patients with existing HF, we observed an association with CD4+ Th17 cells but not CD4+ Th1 cells. Prior studies have also shown an increase in the relative proportion of CD4+ Th17 cells in patients with HF, regardless of HF subtype. 59 Although performed in a limited number of participants, the lack of association with CD4+ Th1 argues against reverse causation in the primary analysis with incident HF.
Study limitations
Our study has limitations. The monocyte subsets were measured in MESA but not available in CHS, and data for each immune cell subset were not available for every participant. Although a relatively comprehensive list of immune cell phenotypes was measured, several subcategories of immune cell subsets such as central and effector memory T cells and severe B‐cell phenotypes were not measured. Additionally, cell surface markers may be affected by cryopreservation, although there were no significant differences when compared with fresh samples in our cohort. 42 Although this is the first longitudinal study to evaluate the associations of different immune cell subsets with incident HF, future studies will need to investigate changes in immune cell subsets over time. Furthermore, in addition to cell surface markers, assessing cytokine profiles and antigen specificities will be key to understanding the function of each immune cell's roles in HF. Relative proportions of peripheral immune cells are likely not representative of myocardial resident immune cells, and thus, better characterization of immune cell phenotypes in the myocardium is also needed. Given this is an observational study with a case‐cohort design, there may be residual confounding and bias despite adjustment for clinically relevant covariates and use of sampling weights in our models. Furthermore, additional echocardiographic data or levels of natriuretic peptides were not available at the time of the HF diagnosis.
Conclusions
In this meta‐analysis, we measured the association of immune cell subsets with incident HF in two large longitudinal observational cohorts. Our primary finding was a significant association between a higher proportion of CD4+ Th1 cells and lower risk of incident HF, specifically HFrEF. Although CD4+ Th1 cells are inflammatory in acute settings, this finding suggests that higher Th1 proportions may reflect adaptive immune competence in adults without HF. This finding is hypothesis‐generating and needs further investigation including understanding of temporal changes in the abundance and functionality of CD4+ Th1 cells and their association with incident HFrEF. Identifying pathways underlying this finding may provide targets for immunomodulatory therapy aimed at HF prevention in high‐risk populations.
Conflict of interest
Dr Shah has received research grants from Actelion, AstraZeneca, Corvia, Novartis, and Pfizer and has received consulting fees from Abbott, Actelion, AstraZeneca, Amgen, Axon Therapeutics, Bayer, Boehringer Ingelheim, Bristol‐Myers Squibb, Cardiora, CVRx, Cytokinetics, Eisai, GSK, Ionis, Ironwood, Lilly, Merck, MyoKardia, Novartis, Novo Nordisk, Pfizer, Regeneron, Sanofi, Shifamed, Tenax, and United Therapeutics. Dr Psaty serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. JSF has consulted for Shionogi Inc. The remaining authors have no disclosures to report.
Funding
MESA: This research was supported by contracts 75N92020D00001, HHSN268201500003I, N01‐HC‐95159, 75N92020D00005, N01‐HC‐95160, 75N92020D00002, N01‐HC‐95161, 75N92020D00003, N01‐HC‐95162, 75N92020D00006, N01‐HC‐95163, 75N92020D00004, N01‐HC‐95164, 75N92020D00007, N01‐HC‐95165, N01‐HC‐95166, N01‐HC‐95167, N01‐HC‐95168, and N01‐HC‐95169 and grants R01 HL98077 and R01 HL156792 from the National Heart, Lung, and Blood Institute and by grants KL2TR001424, UL1‐TR‐000040, UL1‐TR‐001079, and UL1‐TR‐001420 from the National Center for Advancing Translational Sciences.
CHS: This research was supported by NHLBI contracts HHSN268201200036C, HHSN268200800007C, HHSN268201800001C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, and 75N92021D00006 and NHLBI grants U01HL080295, R01HL087652, R01HL103612, R01HL120393, R01HL120854, R01144483, and U01HL130114 with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided through R01AG023629 from the National Institute on Aging (NIA). A full list of principal CHS investigators and institutions can be found at CHS‐NHLBI.org.
Supporting information
Table S1. Flow Cytometry Antibody Isotypes and Fluorophores (Miltenyi Biotec).
Table S2. Summary of Immune Cell Subsets in MESA and CHS.
Table S3. Association of Immune Cell Subsets with Incident Heart Failure.
Table S4. Association of Immune Cell Subsets with Incident Heart Failure Complete Case Analysis.
Table S5. Association of Immune Cell Subsets with Prevalent Heart Failure.
Table S6. Baseline Characteristics of Participants from MESA by Th1 Quartile.
Table S7. Baseline Characteristics of Participants from CHS by Th1 Quartile.
Figure S1. Gating Strategy for CD4 Memory, Naïve, CD28, CD57, CD38 (Same Strategy used for CD8 Subsets).
Figure S2. Gating Strategy for CHS CD4+ T Helper Cells.
Figure S3. Gating Strategy for MESA CD4+ T Helper Cells.
Figure S4. Gating Strategy for CD4+ T Regulatory Cells.
Figure S5. Gating Strategy for T Cells, B cells, NK cells, γδ T cells.
Figure S6. Gating Strategy for Monocytes.
Acknowledgement
The authors thank the other investigators, the staff, and the participants of the MESA and CHS studies for their valuable contributions.
Sinha, A. , Sitlani, C. M. , Doyle, M. F. , Fohner, A. E. , Buzkova, P. , Floyd, J. S. , Huber, S. A. , Olson, N. C. , Njoroge, J. N. , Kizer, J. R. , Delaney, J. A. , Shah, S. S. , Tracy, R. P. , Psaty, B. , and Feinstein, M. (2022) Association of immune cell subsets with incident heart failure in two population‐based cohorts. ESC Heart Failure, 9: 4177–4188. 10.1002/ehf2.14140.
References
- 1. Vasan RS, Sullivan LM, Roubenoff R, Dinarello CA, Harris T, Benjamin EJ, Sawyer DB, Levy D, Wilson PW, D'Agostino RB, Framingham Heart Study . Inflammatory markers and risk of heart failure in elderly subjects without prior myocardial infarction: The Framingham heart study. Circulation. 2003; 107: 1486–1491. [DOI] [PubMed] [Google Scholar]
- 2. Ohkuma T, Jun M, Woodward M, Zoungas S, Cooper ME, Grobbee DE, Hamet P, Mancia G, Williams B, Welsh P, Sattar N, Shaw JE, Rahimi K, Chalmers J, ADVANCE Collaborative Group . Cardiac stress and inflammatory markers as predictors of heart failure in patients with type 2 diabetes: The ADVANCE trial. Diabetes Care. 2017; 40: 1203–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kalogeropoulos A, Georgiopoulou V, Psaty BM, Rodondi N, Smith AL, Harrison DG, Liu Y, Hoffmann U, Bauer DC, Newman AB, Kritchevsky SB, Harris TB, Butler J, Health ABC Study Investigators . Inflammatory markers and incident heart failure risk in older adults: The health ABC (health, aging, and body composition) study. J Am Coll Cardiol. 2010; 55: 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990; 323: 236–241. [DOI] [PubMed] [Google Scholar]
- 5. Hage C, Michaëlsson E, Linde C, Donal E, Daubert JC, Gan LM, Lund LH. Inflammatory biomarkers predict heart failure severity and prognosis in patients with heart failure with preserved ejection fraction: A holistic proteomic approach. Circ Cardiovasc Genet. 2017; 10: e001633. [DOI] [PubMed] [Google Scholar]
- 6. al‐Kindi SG, Buzkova P, Shitole SG, Reiner AP, Garg PK, Gottdiener JS, Psaty BM, Kizer JR. Soluble CD14 and risk of heart failure and its subtypes in older adults. J Card Fail. 2020; 26: 410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, Djian J, Drexler H, Feldman A, Kober L, Krum H, Liu P, Nieminen M, Tavazzi L, van Veldhuisen DJ, Waldenstrom A, Warren M, Westheim A, Zannad F, Fleming T. Targeted anticytokine therapy in patients with chronic heart failure: Results of the randomized etanercept worldwide evaluation (RENEWAL). Circulation. 2004; 109: 1594–1602. [DOI] [PubMed] [Google Scholar]
- 8. Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Randomized, double‐blind, placebo‐controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor‐alpha, in patients with moderate‐to‐severe heart failure: Results of the anti‐TNF therapy against congestive heart failure (ATTACH) trial. Circulation. 2003; 107: 3133–3140. [DOI] [PubMed] [Google Scholar]
- 9. Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, Libby P, Glynn RJ, Ridker PM. Anti‐inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. 2019; 139: 1289–1299. [DOI] [PubMed] [Google Scholar]
- 10. Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, Berry C, López‐Sendón J, Ostadal P, Koenig W, Angoulvant D, Grégoire JC, Lavoie MA, Dubé MP, Rhainds D, Provencher M, Blondeau L, Orfanos A, L'Allier PL, Guertin MC, Roubille F. Efficacy and safety of low‐dose colchicine after myocardial infarction. N Engl J Med. 2019; 381: 2497–2505. [DOI] [PubMed] [Google Scholar]
- 11. Mian MO, Paradis P, Schiffrin EL. Innate immunity in hypertension. Curr Hypertens Rep. 2014; 16: 413. [DOI] [PubMed] [Google Scholar]
- 12. Van Beusecum JP, Moreno H, Harrison DG. Innate immunity and clinical hypertension. J Hum Hypertens. 2022; 36: 503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schütz G, Lumeng CN, Mortensen RM. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest. 2010; 120: 3350–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lumeng CN. Innate immune activation in obesity. Mol Aspects Med. 2013; 34: 12–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fernández‐Real JM, Pickup JC. Innate immunity, insulin resistance and type 2 diabetes. Trends Endocrinol Metab. 2008; 19: 10–16. [DOI] [PubMed] [Google Scholar]
- 16. Graves DT, Kayal RA. Diabetic complications and dysregulated innate immunity. Front Biosci. 2008; 13: 1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frantz S, Falcao‐Pires I, Balligand JL, Bauersachs J, Brutsaert D, Ciccarelli M, Dawson D, de Windt LJ, Giacca M, Hamdani N, Hilfiker‐Kleiner D, Hirsch E, Leite‐Moreira A, Mayr M, Thum T, Tocchetti CG, van der Velden J, Varricchi G, Heymans S. The innate immune system in chronic cardiomyopathy: A European Society of Cardiology (ESC) scientific statement from the working group on myocardial function of the ESC. Eur J Heart Fail. 2018; 20: 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adamo L, Rocha‐Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol. 2020; 17: 269–285. [DOI] [PubMed] [Google Scholar]
- 19. Sager HB, Hulsmans M, Lavine KJ, Moreira MB, Heidt T, Courties G, Sun Y, Iwamoto Y, Tricot B, Khan OF, Dahlman JE, Borodovsky A, Fitzgerald K, Anderson DG, Weissleder R, Libby P, Swirski FK, Nahrendorf M. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ Res. 2016; 119: 853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: Critical importance of the cardiosplenic axis. Circ Res. 2014; 114: 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, Ornitz DM, Randolph GJ, Mann DL. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A. 2014; 111: 16029–16034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wrigley BJ, Shantsila E, Tapp LD, Lip GY. CD14++CD16+ monocytes in patients with acute ischaemic heart failure. Eur J Clin Invest. 2013; 43: 121–130. [DOI] [PubMed] [Google Scholar]
- 23. Wrigley BJ, Shantsila E, Tapp LD, Lip GY. Increased expression of cell adhesion molecule receptors on monocyte subsets in ischaemic heart failure. Thromb Haemost. 2013; 110: 92–100. [DOI] [PubMed] [Google Scholar]
- 24. Ong S, Rose NR, Čiháková D. Natural killer cells in inflammatory heart disease. Clin Immunol. 2017; 175: 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huber S, Shi C, Budd RC. Gammadelta T cells promote a Th1 response during coxsackievirus B3 infection in vivo: Role of Fas and Fas ligand. J Virol. 2002; 76: 6487–6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huber SA. T cells expressing the gamma delta T cell receptor induce apoptosis in cardiac myocytes. Cardiovasc Res. 2000; 45: 579–587. [DOI] [PubMed] [Google Scholar]
- 27. Sinha A, Rivera AS, Doyle MF, Sitlani C, Fohner A, Huber SA, Olson NC, Lima JAC, Delaney JA, Feinstein MJ, Shah SJ, Tracy RP, Psaty BM. Association of immune cell subsets with cardiac mechanics in the multi‐ethnic study of atherosclerosis. JCI Insight. 2021; 6: e149193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blanton RM, Carrillo‐Salinas FJ, Alcaide P. T‐cell recruitment to the heart: Friendly guests or unwelcome visitors? Am J Physiol Heart Circ Physiol. 2019; 317: H124–h40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramos GC, van den Berg A, Nunes‐Silva V, Weirather J, Peters L, Burkard M, Friedrich M, Pinnecker J, Abeßer M, Heinze KG, Schuh K, Beyersdorf N, Kerkau T, Demengeot J, Frantz S, Hofmann U. Myocardial aging as a T‐cell‐mediated phenomenon. Proc Natl Acad Sci U S A. 2017; 114: E2420–e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nevers T, Salvador AM, Grodecki‐Pena A, Knapp A, Velázquez F, Aronovitz M, Kapur NK, Karas RH, Blanton RM, Alcaide P. Left ventricular T‐cell recruitment contributes to the pathogenesis of heart failure. Circ Heart Fail. 2015; 8: 776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, Prabhu SD. Activated T lymphocytes are essential drivers of pathological remodeling in ischemic heart failure. Circ Heart Fail. 2017; 10: e003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Laroumanie F, Douin‐Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C, Delage C, Calise D, Dutaur M, Parini A, Pizzinat N. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation. 2014; 129: 2111–2124. [DOI] [PubMed] [Google Scholar]
- 33. Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, Prabhu SD. Dysfunctional and proinflammatory regulatory T‐lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation. 2019; 139: 206–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ali AJ, Makings J, Ley K. Regulatory T cell stability and plasticity in atherosclerosis. Cell. 2020; 9: 2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Olson NC, Doyle MF, Jenny NS, Huber SA, Psaty BM, Kronmal RA, Tracy RP. Decreased naive and increased memory CD4(+) T cells are associated with subclinical atherosclerosis: The multi‐ethnic study of atherosclerosis. PLoS ONE. 2013; 8: e71498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Giubilato S, Liuzzo G, Brugaletta S, Pitocco D, Graziani F, Smaldone C, Montone RA, Pazzano V, Pedicino D, Biasucci LM, Ghirlanda G, Crea F. Expansion of CD4+CD28null T‐lymphocytes in diabetic patients: Exploring new pathogenetic mechanisms of increased cardiovascular risk in diabetes mellitus. Eur Heart J. 2011; 32: 1214–1226. [DOI] [PubMed] [Google Scholar]
- 37. Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY, Choi YS, Lee SH, Kang SM, Jang Y, Yoo OJ, Shin EC, Park S. Immunosenescent CD8+ T cells and C‐X‐C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension. 2013; 62: 126–133. [DOI] [PubMed] [Google Scholar]
- 38. Lee YH, Kim SR, Han DH, Yu HT, Han YD, Kim JH, Kim SH, Lee CJ, Min BH, Kim DH, Kim KH, Cho JW, Lee WW, Shin EC, Park S. Senescent T cells predict the development of hyperglycemia in humans. Diabetes. 2019; 68: 156–162. [DOI] [PubMed] [Google Scholar]
- 39. Bergström I, Backteman K, Lundberg A, Ernerudh J, Jonasson L. Persistent accumulation of interferon‐γ‐producing CD8+CD56+ T cells in blood from patients with coronary artery disease. Atherosclerosis. 2012; 224: 515–520. [DOI] [PubMed] [Google Scholar]
- 40. Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, Greenland P, Jacobs DR Jr, Kronmal R, Liu K, Nelson JC. Multi‐ethnic study of atherosclerosis: Objectives and design. Am J Epidemiol. 2002; 156: 871–881. [DOI] [PubMed] [Google Scholar]
- 41. Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, Kuller LH, Manolio TA, Mittelmark MB, Newman A, O'Leary DH, Psaty B, Rautaharju P, Tracy RP, Weiler PG. The cardiovascular health study: Design and rationale. Ann Epidemiol. 1991; 1: 263–276. [DOI] [PubMed] [Google Scholar]
- 42. Olson NC, Sitlani CM, Doyle MF, Huber SA, Landay AL, Tracy RP, Psaty BM, Delaney JA. Innate and adaptive immune cell subsets as risk factors for coronary heart disease in two population‐based cohorts. Atherosclerosis. 2020; 300: 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pandey A, LaMonte M, Klein L, Ayers C, Psaty BM, Eaton CB, Allen NB, de Lemos JA, Carnethon M, Greenland P, Berry JD. Relationship between physical activity, body mass index, and risk of heart failure. J Am Coll Cardiol. 2017; 69: 1129–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Inker LA, Schmid CH, Tighiouart H, Eckfeldt JH, Feldman HI, Greene T, Kusek JW, Manzi J, van Lente F, Zhang YL, Coresh J, Levey AS, CKD‐EPI Investigators . Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med. 2012; 367: 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Olson NC, Doyle MF, Sitlani CM, de Boer IH, Rich SS, Huber SA, Landay AL, Tracy RP, Psaty BM, Delaney JA. Associations of innate and adaptive immune cell subsets with incident type 2 diabetes risk: The MESA study. J Clin Endocrinol Metab. 2020; 105: e848–e857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Delaney JAC, Olson NC, Sitlani CM, Fohner AE, Huber SA, Landay AL, Heckbert SR, Tracy RP, Psaty BM, Feinstein M, Doyle MF. Natural killer cells, gamma delta T cells and classical monocytes are associated with systolic blood pressure in the multi‐ethnic study of atherosclerosis (MESA). BMC Cardiovasc Disord. 2021; 21: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Feinstein MJ, Doyle MF, Stein JH, Sitlani CM, Fohner AE, Huber SA, Landay AL, Heckbert SR, Rice K, Kronmal RA, Hedrick C, Manichaikul A, McNamara C, Rich S, Tracy RP, Olson NC, Psaty BM, Delaney JAC. Nonclassical monocytes (CD14dimCD16+) are associated with carotid intima‐media thickness progression for men but not women: The multi‐ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. 2021; 41: 1810–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tracy RP, Doyle MF, Olson NC, Huber SA, Jenny NS, Sallam R, Psaty BM, Kronmal RA. T‐helper type 1 bias in healthy people is associated with cytomegalovirus serology and atherosclerosis: The multi‐ethnic study of atherosclerosis. J Am Heart Assoc. 2013; 2: e000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ives DG, Fitzpatrick AL, Bild DE, Psaty BM, Kuller LH, Crowley PM, Cruise RG, Theroux S. Surveillance and ascertainment of cardiovascular events: The cardiovascular health study. Ann Epidemiol. 1995; 5: 278–285. [DOI] [PubMed] [Google Scholar]
- 50. Psaty BM, Delaney JA, Arnold AM, Curtis LH, Fitzpatrick AL, Heckbert SR, McKnight B, Ives D, Gottdiener JS, Kuller LH, Longstreth WT Jr. Study of cardiovascular health outcomes in the era of claims data: The cardiovascular health study. Circulation. 2016; 133: 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fukunaga T, Soejima H, Irie A, Sugamura K, Oe Y, Tanaka T, Nagayoshi Y, Kaikita K, Sugiyama S, Yoshimura M, Nishimura Y, Ogawa H. Relation between CD4+ T‐cell activation and severity of chronic heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 2007; 100: 483–488. [DOI] [PubMed] [Google Scholar]
- 52. Zhang Y, Bauersachs J, Langer HF. Immune mechanisms in heart failure. Eur J Heart Fail. 2017; 19: 1379–1389. [DOI] [PubMed] [Google Scholar]
- 53. Glimcher LH, Murphy KM. Lineage commitment in the immune system: The T helper lymphocyte grows up. Genes Dev. 2000; 14: 1693–1711. [PubMed] [Google Scholar]
- 54. Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, Epelman S, Kreisel D, Liu Y, Itoh A, Shankar TS, Selzman CH, Drakos SG, Lavine KJ. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med. 2018; 24: 1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Geginat J, Paroni M, Maglie S, Alfen JS, Kastirr I, Gruarin P, de Simone M, Pagani M, Abrignani S. Plasticity of human CD4 T cell subsets. Front Immunol. 2014; 5: 630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Petrelli A, Mijnheer G, van Konijnenburg DPH, van der Wal MM, Giovannone B, Mocholi E, Vazirpanah N, Broen JC, Hijnen D, Oldenburg B, Coffer PJ, Vastert SJ, Prakken BJ, Spierings E, Pandit A, Mokry M, van Wijk F. PD‐1+CD8+ T cells are clonally expanding effectors in human chronic inflammation. J Clin Invest. 2018; 128: 4669–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tian Y, Babor M, Lane J, Schulten V, Patil VS, Seumois G, Rosales SL, Fu Z, Picarda G, Burel J, Zapardiel‐Gonzalo J, Tennekoon RN, de Silva AD, Premawansa S, Premawansa G, Wijewickrama A, Greenbaum JA, Vijayanand P, Weiskopf D, Sette A, Peters B. Unique phenotypes and clonal expansions of human CD4 effector memory T cells re‐expressing CD45RA. Nat Commun. 2017; 8: 1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Feinstein MJ, Doyle MF, Stein JH, Sitlani CM, Fohner AE, Huber SA, Landay AL, Heckbert SR, Rice K, Kronmal RA, Hedrick C, Manichaikul A, McNamara C, Rich S, Tracy RP, Olson NC, Psaty BM, Delaney JAC. Nonclassical monocytes (CD14dimCD16+) are associated with carotid intima‐media thickness progression for men but not women: The multi‐ethnic study of atherosclerosis‐brief report. Arterioscler Thromb Vasc Biol. 2021; 41: 1810–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li N, Bian H, Zhang J, Li X, Ji X, Zhang Y. The Th17/Treg imbalance exists in patients with heart failure with normal ejection fraction and heart failure with reduced ejection fraction. Clin Chim Acta. 2010; 411: 1963–1968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Flow Cytometry Antibody Isotypes and Fluorophores (Miltenyi Biotec).
Table S2. Summary of Immune Cell Subsets in MESA and CHS.
Table S3. Association of Immune Cell Subsets with Incident Heart Failure.
Table S4. Association of Immune Cell Subsets with Incident Heart Failure Complete Case Analysis.
Table S5. Association of Immune Cell Subsets with Prevalent Heart Failure.
Table S6. Baseline Characteristics of Participants from MESA by Th1 Quartile.
Table S7. Baseline Characteristics of Participants from CHS by Th1 Quartile.
Figure S1. Gating Strategy for CD4 Memory, Naïve, CD28, CD57, CD38 (Same Strategy used for CD8 Subsets).
Figure S2. Gating Strategy for CHS CD4+ T Helper Cells.
Figure S3. Gating Strategy for MESA CD4+ T Helper Cells.
Figure S4. Gating Strategy for CD4+ T Regulatory Cells.
Figure S5. Gating Strategy for T Cells, B cells, NK cells, γδ T cells.
Figure S6. Gating Strategy for Monocytes.