

Abstract

While cyclic polymers have intrigued researchers for their novel set of architecture-driven rheological interactions, the possibility of incorporating them in topological systems has been limited by the availability of large ring polymers. Thus, the need for scalable methods to produce ring polymers has become apparent. Here, a facile method to prepare polysiloxane ring polymers by means of Piers-Rubinsztajn chemistry is presented. The one-pot nature and commercial availability of reagents additionally confirm the applicability of the method for large-scale production. Furthermore, a highly efficient yet simple purification method was developed for the isolation of pure ring polymers without linear side products.
Introduction
Ring polymers have stoked the interest of researchers for decades because of the unique set of physical properties they exhibit due to their endless nature and constrained contours.1−7 Additionally, ring polymers feature a unique form of threading entanglements not present for linear polymers.8 The architecture-specific entanglements caused by the threading of ring polymers present a novel set of topological interactions that do not directly adhere to a well-defined state such as a knot or a link.5
Earlier studies have focused on investigating the properties of ring polymers mostly as an academic curiosity due to the limited quantities of ring polymers that could be prepared. However, incremental improvements in synthetic procedures have opened up the possibility to explore the beneficial attributes of cyclic topologies for practical applications such as increased biorepellency, lubrication,9 and drug release.6 The cyclic structure of ring polymers also presents the possibility to incorporate them in networks by means of spatially interlocked structures. More rigorous studies of ring polymers in topological networks have been constrained by the limited scalability and expensive synthetic methods for preparing ring polymers.10 Thus, further development of scalable and less arduous synthetic methods remains of great interest.
Recent studies have utilized the Piers-Rubinsztajn (PR) reaction to prepare high-molecular-weight linear poly(dimethylsiloxane) (PDMS) polymers from α,ω-hydride-terminated precursors.11,12 The method entails a two-step process, where a trace amount of water is first employed to hydrolyze a fraction of the hydride functional terminal groups. The formed silanols can then be coupled with the residual hydride groups to form a high-molecular-weight PDMS product. The appeal of this approach is that the same PR catalyst induces both the hydrolysis and coupling reactions, allowing for a one-pot preparation of the desired product. Furthermore, the reactions can be induced at room temperature with relatively low catalyst concentrations.13 The high efficiency and facile conditions of the PR chemistry make it an ideal candidate for the synthesis of PDMS ring polymers (Figure 1a). The former is especially important, considering the highly dilute conditions that are necessary for intramolecular coupling to dominate.
Figure 1.

(A) Synthesis of PDMS ring polymers by the hydrolysis (1) and condensation (2) of α,ω-hydride-terminated PDMS, both of which are driven by the PR reaction. (B) SEC curves of H-PDMS-H (Mn = 1000 g mol–1) starting material and ring mixtures produced at different polymer concentrations (cpolymer) in toluene. (C) Influence of water on the SiH terminal groups per PDMS chain as determined by 1H NMR. The reactions were conducted at a cpolymer = 3.2 g L–1 and a catalyst concentration of cPR = 34 ppm. (D) Influence of polymer concentration (cpolymer) at pseudo-dilute conditions on the size distribution of the reaction products.
Additionally, the initial hydrolysis has been found to be significantly slower than the subsequent condensation reaction.11 The stalled introduction of silanol groups in the system can thus be compared to gradual addition of coupling agents, which is a common method to avoid chain extension when end-to-end coupling of homo-functional chains.3 However, the increased speed of the condensation between the formed silanols and residual hydride groups should favor interactions between chain ends over completely capping the chains to create fully hydrolized “dead” chains. So while at first glance, the approach might resemble the method of linking homodifunctional chains with a coupling agent, there is the added benefit of kinetic disparity between the initial hydrolysis and subsequent coupling reactions. In this work, we investigate the viability of PR chemistry-based end-to-end coupling as a scalable yet straightforward procedure for the synthesis of PDMS ring polymers.
Results and Discussion
The PR catalyst-mediated end-to-end coupling method was first explored using relatively short linear PDMS precursors with a molecular weight of around 1000 g mol–1. The formation of ring polymers is commonly determined by the reduction in hydrodynamic volume when compared to analogous linear species of the same molecular weight.14,15
The decreased hydrodynamic volume is caused by the conformational constrictions of the cyclic architecture and can be clearly identified by size-exclusion chromatography as a shift toward higher retention volumes. This method of identification allows for a relatively fast validation method of both ring polymers and chain-extended linear byproducts (Figure 1B).
The initial focus was on identifying the highest possible polymer concentration (cpolymer) where ring formation was dominant. It was found that dilute conditions well below the critical overlap concentration1 (c*) were necessary to facilitate intramolecular coupling. The appearance of chain-extended species could already be observed (Figure 1B, bottom) at a relatively low cpolymer = 9.6 g L–1 compared to c* (36 g L–1).
While limiting the amount of water was initially considered essential in avoiding the formation of fully hydrolyzed linear byproducts (Figure 1A), no significant change in the size distribution of the products could be observed by size-exclusion chromatography as the water content was altered between experiments (Figure S1). Furthermore, the concentration of silanols in the products was below the detection limit of infrared (IR), 1H NMR, and 29Si NMR, even when an excess of water was introduced to the system (Figure S3). These findings indicate that the kinetic disparity between the hydrolysis and condensation reactions was sufficient to counteract the formation of fully hydrolyzed linear byproducts regardless of the water concentration.
Thus, it was possible to boost the speed of the coupling reaction by increasing the water content without compromising the yield. When a stoichiometric excess of water (1:1 equiv SiH/H2O) was introduced, no unreacted hydride groups could be observed with 1H NMR after 30 min (Figure 1C). It should be noted that a slow decrease of hydride groups occurred even when no water was introduced to the reaction mixtures. Given the low concentration of the reactive terminal groups ([SiH] = 4.2 mM), it is probable that trace water in the solvents would be sufficient to account for the observed reaction.
The fast and efficient coupling reactions suggested the possibility of scaling up the ring polymer synthesis through stepwise addition of reagents. This method of achieving pseudo-dilute conditions has been proposed as a practical approach to avoid the use of large amounts of solvent and catalyst.18 The linear precursor was added at 1 h intervals to ensure that as the overall cpolymer in the reaction mixture increased, the concentration of chemically active precursors remained in the highly dilute regime necessary to avoid intermolecular reactions. As a result, no chain-extended byproducts could be observed even as the cpolymer reached values at which intermolecular coupling had previously been observed (Figure 1D). Furthermore, no significant increase in dispersity occurred between the steps, as it remained stable at ĐM ≈ 1.7.
While the disappearance of the hydride functional groups could be observed by both 1H NMR and IR analysis, neither method was sensitive enough to quantify unreacted silanol groups. Thus, end-group analysis could not be used to rule out the formation of linear byproducts, as the concentration of the silanol groups fell below the detection limit of the applied methods.
Although size-exclusion chromatography shows a significant shift toward larger retention volumes, a considerable overlap between linear starting materials and the reaction mixture remains. Therefore, this method cannot be used to unequivocally rule out the formation of linear byproducts. To gain further insight, an adsorption chromatography method was developed to separate oligomers of linear and ring polymers (Figure 2). The initial conditions were adapted from a study conducted by Durner et al.19 However, the use of an isocratic method allowed using a triple detector array consisting of a differential refractive index (DRI) detector, a light scattering detector (LS), and a viscosity detector. These detectors provide additional info on molecular mass and dimensions of the molecules in solutions, as discussed below.
Figure 2.

Chromatographic characterization of ring polymer mixtures and the linear precursors: (A) affinity chromatography with differential refractive index detection using a 7:93 V/V mixture of water and acetone at 35 °C, allowing oligomer separation. The numbers designate a counter (proportional to but not the actual degree of polymerization); (B) relative concentration of the separated peaks as determined by the area under the refractive index (RI area) detectors; (C) ratio between the signal from the right angle light scattering (RALS area) and the RI area detectors, which is proportional to the molecular weight16 as a function of peak number; and (D) ratio between the signal from the differential pressure and the refractive index detectors (DP area/RI area), which is proportional to the intrinsic viscosity16,17 as a function of the RALS area/RI area.
As Figure 2A shows, the chromatographic conditions used allowed reasonably good separation between individual oligomers both for linear and cyclic samples. The DRI detector provides information on the relative concentration of the eluted material, provided the refractive index increment (dn/dc) values of the individual oligomers are comparable (Figure 2B). This is expected for these chemically similar compounds, although some dependence on molecular weight may be expected.20 The chromatograms showed a difference in elution time for the individual oligomers of the linear and cyclic series, with the linear molecules generally eluting later than their cyclic counterparts. This indicated a higher end-group affinity between the hydride end group and the column material. Importantly, there appeared to be no signals originating from linear oligomers in the ring polymer mixture, which emphasizes the high cyclization efficiency.
In principle, it should be possible to obtain absolute molecular weights of the individual oligomers by combining the data from the light scattering detector with the concentration obtained from the refractive index detector. However, a lack of available data on the refractive index increment (dn/dc) values for oligomeric PDMS and calibration standards in the solvent used makes such an assignment highly uncertain.16 Rather than determining the absolute molecular weights, the ratio between the integrated right angle light scattering signal (RALS) and the integrated refractive index signal is plotted in Figure 2C. This ratio can be assumed to be proportional to the absolute molecular weight (to a first approximation).16 Therefore, the correlation between the plots of the linear and cyclics (at least up to peak 16) indicates that despite the different elution behavior, the molecular weights are nearly-identical, as would be expected if only cyclization occurs. These results emphasized that the shift observed in the size-exclusion chromatograms (Figure 2A) is due to changes in molecular dimensions but not molecular masses (where the deviations for the higher peak indices are most likely due to their low concentration).
The intrinsic viscosity can be directly related to the dimensions of the polymer coils in solution.21 Therefore, plotting intrinsic viscosity as a function of molecular weight provides information on the evolution of the hydrodynamic volume with molecular weight. Since a cyclic polymer of a given molecular weight has a significantly smaller hydrodynamic volume than a linear polymer of the same molecular weight,22 a significant difference in the intrinsic viscosity is expected. This is confirmed by the plot in Figure 2D, where the ratio between the differential pressure of the viscosity detector and the refractive index, which is proportional to the intrinsic viscosity (to a first approximation),16,17 is plotted as a function of the RALS/RI ratio discussed above. Although there is a significant scattering of the data, distinctly different trends for the linear and cyclic materials are apparent, where the viscosity of the cyclic oligomers is generally lower (as seen by the Mark–Houwink–Sakurada plot: [η] = KMwa; K = 0.0091, a = 24.7, R2 = 0.48) than that of its linear counterparts (K = 0.0069, a = 20.0, R2 = 0.70).
The improved separation of the developed adsorption chromatography method allowed for an additional confirmation of the absence of linear byproducts. Consequently, eliminating the need for purification considerably streamlines the synthetic procedure for the preparation of PDMS rings.
Adapting Reaction Conditions for Larger PDMS Ring Polymers
One of the advantages of utilizing PR chemistry for the preparation of polysiloxane ring polymers is the abundance of commercially available hydride-terminated PDMS with various molecular weights. However, preparing ring polymers with longer chains presents practical hurdles due to the decrease in the effective concentration of terminal groups as the chain length increases.23 This entropic penalty is described by the Jacobson–Stockmayer theory, where the probability of cyclization was shown to diminish with the increase of the chain length to the power of N–5/2.24 Furthermore, the lower critical overlap concentration of longer polymers suggests the need for more dilute conditions to avoid chain extension.
Initial trials of ring polymerizing larger chains (Mn = 5000 g mol–1) at various polymer and catalyst concentrations resulted in no obvious signs of cyclization or chain extension, even as the terminal hydrides were consumed. The absence of condensation products or unreacted hydride groups in the samples indicated that the lower concentration of reactive termini ([SiH] = 0.5 mM) had shifted the balance in favor of the initial hydrolysis reaction.
A significant change in reaction products could be observed when the reaction medium was changed from toluene to heptane, which is a slightly better solvent for PDMS25 and poorer solvent for water26,27 (the saturated water concentration in toluene is 16 mM compared to 4 mM in heptane at 25 °C). General synthetic strategies for end-to-end coupling suggest that poorer solvents would be beneficial for intramolecular chain interactions as the contracted conformation of the polymer coils also results in lower average end-to-end distances.28−30 It was thus contradictory that ring coupling occurred only when the diluent was changed to a better solvent for PDMS. However, the catalyst deactivation and formation of nonreactive linear products are dependent on the absolute water content (Figure 1A).11,31 Changing to a less polar reaction medium offers an easy method to decrease the water content, thus lowering the amount of undesirable side reactions. Additionally, the zwitterionic nonactive catalyst–water adduct is expected to be more soluble in toluene than in the less polar heptane, shifting the equilibrium toward the active catalyst. The significantly lower catalyst concentrations (2.1 ppm, 0.5 mol %) at which it was possible to induce the coupling reactions in heptane agree well with this explanation.
When using the longer PDMS, purification of the ring polymer products was necessary as linear byproducts could be observed by size-exclusion measurements (Figure 3A) even after changing the reaction media. A simple procedure of treating the crude mixtures in heptane with silica gel was shown to be extremely efficient in binding the linear byproducts, as silanol chain ends adsorb to the silica surface (Figure 3B).32,33 In addition, unreacted linear chains can be bound to the silica surface by means of PR-mediated surface grafting, as illustrated. The purified products did not exhibit traces of linear species and had a yield of up to 43 wt %. The dispersity (ĐM) of the product decreased from 1.33 to 1.14 after the purification procedure (ĐM = 1.28 for the starting material).
Figure 3.

(A) SEC chromatograms of PDMS ring polymer (Mn = 5000 g mol–1) mixtures before and after purification with silica gel. (B) Binding of linear byproducts on the surface of silica gel due to chain-end hydrogen-bond-type interactions and covalent grafting of unreacted linear chains by utilizing the PR reaction.
While the yield of the purified long-chain PDMS ring polymers was considerably lower when compared to the shorter rings (94 wt %), it was still sufficient for the production of the isolated ring polymers on a preparative scale. The high purity achieved using relatively simple synthetic procedures has already affirmed the feasibility of the method, although further development would be necessary to attain additional insights into the nature of the competing hydrolysis and condensation reactions.
The characteristic shift in the hydrodynamic size of the cyclic species is also reflected in the apparent molecular weight determined by size-exclusion methods (Tables S1–S3). The reduced hydrodynamic size caused by the conformational constrictions of the closed contours of the polymer chains results in a lower apparent molecular weight when compared with linear standards.22,34 Thus, alternative methods are required for determining the molecular weight distribution of ring polymers.
The use of size-exclusion chromatography coupled with triple detection is a common method to distinguish branched polymers based on hydrodynamic size and to determine the absolute molecular weight based on light scattering.35,36 An absolute molecular weight can similarly be determined for ring polymers without the need for well-defined cyclic standards. The discrepancy between the apparent and absolute molecular weights of the cyclic and linear PDMS samples was significantly more pronounced for larger chain lengths (Figure 4). This would suggest the potential of developing a screening method for larger cyclic structures of undetermined molecular weight.
Figure 4.
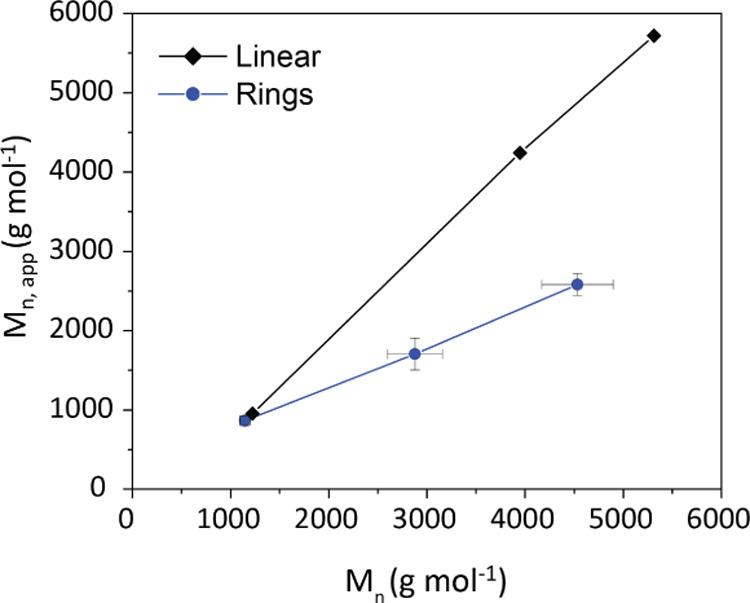
Apparent molecular weight as determined by standard calibration as a function of the absolute molecular weight determined by light scattering of linear and cyclic PDMS.
Triple detection could also be useful for detecting high-molecular-weight impurities that are present only in small quantities, such as concatenated structures.37
In summary, the efficiency and robustness of the PR catalyst-mediated hydrolysis and condensation reactions were presented as an alternative to the more rigorous end-to-end synthetic methods previously reported. The one-pot nature and commercial availability of reagents additionally increase the applicability of the method for production at a preparative scale. Furthermore, the production of ring polymers with a well-defined molecular weight by simple purification methods attested the efficiency and procedural simplicity of this approach.
Experimental Section
Materials
α,ω-hydride-terminated poly(dimethylsiloxane) (M ≈ 1000 g mol–1, 7–10 cSt; M ≈ 3000 g mol–1, 50 cSt; M ≈ 5000 g mol–1, 100 cSt) was purchased from Gelest and used as received. Tris(pentafluorophenyl)borane, B(C6F5)3, was acquired from TCI Chemicals and used as received. Toluene (HPLC grade, ≥99.8%) and n-heptane (HPLC grade, ≥99%) were purchased from VWR and dried over activated 4 Å molecular sieves (10 wt/vol %). Acetone (99+%, extra pure) was acquired from Acros Organics and used as received. PDMS standards were purchased from PSS Polymer Standards Service GmbH. All other chemicals were acquired from Aldrich and used as received unless otherwise specified. All glassware was dried in a 130 °C oven overnight before use.
Characterization
Size-exclusion chromatography was carried out on a chromatographic system consisting of a Waters Acquity solvent delivery module and column oven connected to a Waters Photodiode Array TaperSlit (PDA TS) detector and Malvern Omnisec Reveal triple detector array (refractive index, light scattering, viscosity detectors). The columns used were a 150 mm × 4.6 mm Acquity APC XT 450 2.5 μm, a 150 mm × 4.6 mm Acquity APC XT 125 μm × 2.5 μm, a 150 mm × 4.6 mm Acquity APC XT 45 μm × 1.7 μm, and a 30 mm × 4.6 mm Acquity APC XT 900 μm × 2.5 μm organic size-exclusion chromatography columns connected in series. This column combination provides a separation of polymers with molecular weights from 200 to 2,000,000 Daltons. The detectors were calibrated with a narrow disperse polystyrene standard (Malvern PolyCal). All samples and calibration standards were analyzed using a flow rate of 0.7 mL/min of HPLC grade toluene containing 2% V/V absolute ethanol at a column temperature of 40 °C. Sample solutions were made up to approximately 5 mg/mL and filtered through a 0.45 μm PTFE filter. Injection volume was 20 μL. Molecular mass averages and hydrodynamic radii were calculated using a dn/dc value of 0.0913. Additionally, a standard calibration curve was calculated by fitting a third-order polynomial with PDMS standards of a narrow molecular weight distribution (Mp = 305,000 g mol–1, ĐM = 1.19; Mp = 107,000 g mol–1, ĐM = 1.13; Mp = 53,600 g mol–1, ĐM = 1.08; Mp = 35,700 g mol–1, ĐM = 1.04; Mp = 19,000 g mol–1, ĐM = 1.15; Mp = 5470 g mol–1, ĐM = 1.20; Mp = 1100 g mol–1, ĐM = 1.37; Mp = 311 g mol–1, ĐM = 1.00).
The critical overlap concentrations of the precursors were estimated by calculating the inverse of the intrinsic viscosity of the polymer chains (c* = 1/kMa, where M, k, and a represent the molecular weight of the polymer and the Mark–Houwink constants, respectively).
Polymer adsorption chromatography was carried out on a chromatographic system consisting of a Waters Acquity solvent delivery module and column oven connected to an Acquity ultra-performance liquid chromatography (UPLC) evaporative light scattering detector (ELSD). The column used was an XBridge BEH C18 2.5 μm 4.6 mm × 100 mm. Sample solutions were prepared to a concentration of approximately 50 mg/mL in acetone (99+% purity) and filtered through a 0.45 μm PTFE filter. An injection volume of 100 μL was used, and the samples were eluted at a flow rate of 0.8 mL, at a column temperature of 25 °C. A linear solvent gradient was applied for 40 min, starting with a mixture of methanol/water (75:25, v/v) as the adsorption-promoting solvent and toward pure acetone (99+% purity) as the desorption-promoting solvent. Pure acetone was then eluted for 5 min, followed by a sharp 1-minute linear gradient to the methanol/water mixture and 2 min of elution with the methanol/water mixture.
1H NMR spectra were acquired on a Bruker 300 MHz spectrometer with the residual solvent peak used as the internal standard. 29Si NMR spectra were acquired on a Bruker AVANCE 400 MHz spectrometer system at room temperature. Samples were prepared in CDCl3 at a sample concentration of 2% (w/v) for 1H and 80% w/v for 29Si NMR measurements.
Fourier transform infrared spectroscopy (FTIR) was performed on a PerkinElmer Spectrum One Fourier transform infrared system equipped with a universal attenuated total reflection (ATR) accessory on a ZnSe/diamond composite. Spectra were recorded in the range of 4000–650 cm–1 with 4 cm–1 resolution with 16 scans.
Differential scanning calorimetry (DSC Q1000; TA Instruments) measurements were performed on the samples from −180 to 200 °C at a heating rate of 10 °C min–1 under a helium atmosphere.
Preparation of Polysiloxane Ring Polymers
Synthesis of Small (M ≈ 1000 g mol–1) PDMS Ring Polymers
α,ω-hydride-terminated PDMS (Mn = 1100 g mol–1, 0.1 g, 0.1 mmol) was weighed into a round-bottom flask that was dried in a 130 °C oven overnight and subjected to three cycles of evacuation and backfilling with dry N2. Dry toluene (21.7 mL) and a stock solution of wet toluene (9.2 mL, cH2O = 14 mM) were transferred to the reaction flask by syringe, resulting in a total water concentration of 4.2 mM (1 equiv relative to hydride groups) and a polymer concentration of 3.2 g L–1. A stock solution of B(C6F5)3 in toluene (296 μL, 6.7 mM) was then added, resulting in a catalyst concentration of 32 ppm. The reaction mixture was left stirring under nitrogen for 1 h at room temperature before being quenched by the addition of activated neutral alumina (approximately half a gram). The product was then retrieved by filtering off the alumina and evaporation of the solvent in vacuo as a transparent oil (81 mg, yield 81 wt %).
Scaled-Up Synthesis of Small (M ≈ 1000 g mol–1) PDMS Ring Polymers
α,ω-hydride-terminated PDMS (Mn = 1100 g mol–1, 3.0 g, 2.0 mmol) was weighed into a predried round-bottom flask and subjected to three cycles of evacuation and backfilling with dry N2. Dry toluene (940 mL) was transferred to the reaction flask with a measuring cylinder, and 71 μL of water was added by pipette, resulting in a water concentration of 4.2 mM (based on added amount). The reaction flask was flushed with dry N2 while stirring for approximately 10 min. A stock solution of B(C6F5)3 in toluene (8.1 mL, 6.5 mM) was then added, resulting in a catalyst concentration of 32 ppm. The reaction mixture was left stirring at room temperature. After 2 h, 3.0 g of α,ω-hydride-terminated PDMS was dissolved in 3 mL of toluene and then added to the reaction mixture with an additional 35 μL of water and left with stirring for 2 h. The addition of PDMS and water was repeated once more, and after 2 h, the reaction was quenched with activated neutral alumina. The final product was isolated by filtration and evaporation in vacuo and retrieved as a transparent oil (8.5 g, yield 94 wt %).
Synthesis of Large (M ≈ 5000 g mol–1) PDMS Ring Polymers
α,ω-hydride-terminated PDMS (Mn = 5000 g mol–1, 50 mg, 0.01 mmol) was weighed into a predried round-bottom flask and subjected to three cycles of evacuation and backfilling with dry N2. Dry heptane (15.6 mL) was transferred to the reaction flask, and after approximately 15 min of stirring, a stock solution of B(C6F5)3 in toluene (23.6 μL, 7.2 mM) was added, resulting in a catalyst concentration of 8.3 ppm. The reaction mixture was left stirring overnight at room temperature before being quenched with activated neutral alumina (approximately 1 g). The product was then retrieved by filtration and evaporation in vacuo as a transparent oil (20 mg, yield 40 wt %).
Acknowledgments
This work was supported by DTU’s alliance stipend program.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c05996.
SEC chromatographs of PDMS ring polymers prepared at various water concentrations; standard calibration curve; 1H and 29Si NMR and IR spectra of PDMS ring polymers, and experimental parameters of PDMS ring polymers of various molecular weights (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- de Gennes P. G.; Witten T. A. Scaling Concepts in Polymer Physics. Phys. Today 1980, 33, 51–54. 10.1063/1.2914118. [DOI] [Google Scholar]
- Semlyen J. A.Cyclic Polymers; Kluwer Academic Publishers: Dordrecht, 2002. [Google Scholar]
- White B. M.; Watson W. P.; Barthelme E. E.; Beckham H. W. Synthesis and Efficient Purification of Cyclic Poly(Dimethylsiloxane). Macromolecules 2002, 35, 5345–5348. 10.1021/ma0255027. [DOI] [Google Scholar]
- Williams R. J.; Dove A. P.; O’Reilly R. K. Self-Assembly of Cyclic Polymers. Polym. Chem. 2015, 6, 2998–3008. 10.1039/C5PY00081E. [DOI] [Google Scholar]
- Michieletto D.; Marenduzzo D.; Orlandini E.; Turner M. S. Ring Polymers: Threadings, Knot Electrophoresis and Topological Glasses. Polymers 2017, 9, 349 10.3390/polym9080349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romio M.; Trachsel L.; Morgese G.; Ramakrishna S. N.; Spencer N. D.; Benetti E. M. Topological Polymer Chemistry Enters Materials Science: Expanding the Applicability of Cyclic Polymers. ACS Macro Lett. 2020, 9, 1024–1033. 10.1021/acsmacrolett.0c00358. [DOI] [PubMed] [Google Scholar]
- Haque F. M.; Grayson S. M. The Synthesis, Properties and Potential Applications of Cyclic Polymers. Nat. Chem. 2020, 12, 433–444. 10.1038/s41557-020-0440-5. [DOI] [PubMed] [Google Scholar]
- Cosgrove T.; Turner M. J.; Griffiths P. C.; Hollingshurst J.; Shenton M. J.; Semlyen J. A. Self-Diffusion and Spin-Spin Relaxation in Blends of Linear and Cyclic Polydimethylsiloxane Melts. Polymer 1996, 37, 1535–1540. 10.1016/0032-3861(96)83701-3. [DOI] [Google Scholar]
- Morgese G.; Trachsel L.; Romio M.; Divandari M.; Ramakrishna S. N.; Benetti E. M. Topological Polymer Chemistry Enters Surface Science: Linear versus Cyclic Polymer Brushes. Angew. Chem., Int. Ed. 2016, 55, 15583–15588. 10.1002/anie.201607309. [DOI] [PubMed] [Google Scholar]
- Kricheldorf H. R. Cyclic Polymers: Synthetic Strategies and Physical Properties. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 251–284. 10.1002/pola.23755. [DOI] [Google Scholar]
- Liao M.; Schneider A. F.; Laengert S. E.; Gale C. B.; Chen Y.; Brook M. A. Living Synthesis of Silicone Polymers Controlled by Humidity. Eur. Polym. J. 2018, 107, 287–293. 10.1016/j.eurpolymj.2018.07.023. [DOI] [Google Scholar]
- Liao M.; Chen Y.; Brook M. A. When Attempting Chain Extension, Even Without Solvent, It Is Not Possible to Avoid Chojnowski Metathesis Giving D-3. Molecules 2021, 26, 231 10.3390/molecules26010231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook M. A. New Control Over Silicone Synthesis Using SiH Chemistry: The Piers–Rubinsztajn Reaction. Chem. - Eur. J. 2018, 24, 8458–8469. 10.1002/chem.201800123. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos G.; Bilalis P.; Hadjichristidis N. Well-Defined Cyclic Triblock Terpolymers: A Missing Piece of the Morphology Puzzle. ACS Macro Lett. 2016, 5, 1242–1246. 10.1021/acsmacrolett.6b00807. [DOI] [PubMed] [Google Scholar]
- Foston M.; Beckham H. W. Cyclic Poly(Dimethylsiloxane) via Ring-Closing Dehydrocoupling of α,ω-Dihydroxy-PDMS with α,ω-Dihydrido-PDMS in Dilute Solution. Polymer 2010, 51, 2515–2519. 10.1016/j.polymer.2010.04.006. [DOI] [Google Scholar]
- Striegel A. M. There’s Plenty of Gloom at the Bottom: The Many Challenges of Accurate Quantitation in Size-Based Oligomeric Separations. Anal. Bioanal. Chem. 2013, 405, 8959–8967. 10.1007/s00216-013-7198-1. [DOI] [PubMed] [Google Scholar]
- Trathnigg B.Size-Exclusion Chromatography of Polymers. In Encyclopedia of Separation Science; John Wiley & Sons, 2000. [Google Scholar]
- Beckham H. W. Ring Polymers: Effective Isolation and Unique Properties. Complex Macromol. Archit. 2011, 791–821. 10.1002/9780470825150.ch26. [DOI] [Google Scholar]
- Durner B.; Ehmann T.; Matysik F. M. Separation of Linear and Cyclic Poly(Dimethylsiloxanes) with Polymer High-Performance Liquid Chromatography. Monatsh. Chem. - Chem. Mon. 2019, 150, 1603–1610. 10.1007/s00706-019-02389-4. [DOI] [Google Scholar]
- Itakura M.; Sato K.; Lusenkova M. A.; Matsuyama S.; Shimada K.; Saito T.; Kinugasa S. Molecular Weight Dependency of Refractive Index Increment of Polystyrene Determined by Uniform Oligomers. J. Appl. Polym. Sci. 2004, 94, 1101–1106. 10.1002/app.21006. [DOI] [Google Scholar]
- Dawkins J. V.; Maddock J. W.; Coupe D. Gel-Permeation Chromatography: Examination of Universal Calibration Procedures for Polydimethylsiloxane in a Poor Solvent. J. Polym. Sci., Part A-2 1970, 8, 1803–1821. 10.1002/pol.1970.160081015. [DOI] [Google Scholar]
- Higgins J. S.; Ma K.; Nicholson L. K.; Hayter J. B.; Dodgson K.; Semlyen J. A. Studies of Cyclic and Linear Poly(Dimethyl Siloxanes): 12. Observation of Diffusion Behaviour by Quasielastic Neutron Scattering. Polymer 1983, 24, 793–799. 10.1016/0032-3861(83)90191-X. [DOI] [Google Scholar]
- Coqueret X.; Wegner G. Synthesis of Silicone Macrocycles Having a Single Functional Side Group by Hydrosilylation under High Dilution. Die Makromol. Chem. 1992, 193, 2929–2943. 10.1002/macp.1992.021931118. [DOI] [Google Scholar]
- Jacobson H.; Stockmayer W. H. Intramolecular Reaction in Polycondensations. I. The Theory of Linear Systems. J. Chem. Phys. 1950, 18, 1600–1606. 10.1063/1.1747547. [DOI] [Google Scholar]
- Lee J. N.; Park C.; Whitesides G. M. Solvent Compatibility of Poly(Dimethylsiloxane)-Based Microfluidic Devices. Anal. Chem. 2003, 75, 6544–6554. 10.1021/ac0346712. [DOI] [PubMed] [Google Scholar]
- Louisiana State University; The Macromolecular Studies Group. Toluene Solvent Properties. https://macro.lsu.edu/howto/solvents/toluene.htm (accessed November 15, 2021).
- Louisiana State University; The Macromolecular Studies Group. Heptane Solvent Properties. https://macro.lsu.edu/HowTo/solvents/heptane.htm (accessed November 15, 2021).
- Yu G. E.; Sinnathamby P.; Price C.; Booth C. Preparation of Large Cyclic Poly(Oxyethylene)S. Chem. Commun. 1996, 1, 31–32. 10.1039/cc9960000031. [DOI] [Google Scholar]
- Winnik M. A.; Redpath T.; Richards D. H. The Dynamics of End-to-End Cyclization in Polystyrene Probed by Pyrene Excimer Formation. Macromolecules 1980, 13, 328–335. 10.1021/ma60074a023. [DOI] [Google Scholar]
- Winnik M. A. End-to-End Cyclization of Polymer Chains. Acc. Chem. Res. 1985, 18, 73–79. 10.1021/ar00111a002. [DOI] [Google Scholar]
- Schneider A. F.; Chen Y.; Brook M. A. Trace Water Affects Tris(Pentafluorophenyl)Borane Catalytic Activity in the Piers–Rubinsztajn Reaction. Dalton Trans. 2019, 48, 13599–13606. 10.1039/C9DT02756D. [DOI] [PubMed] [Google Scholar]
- Boonstra B. B.; Cochrane H.; Dannenberg E. M. Reinforcement of Silicone Rubber By Particulate Silica. Rubber Chem. Technol. 1975, 48, 558–576. 10.5254/1.3539660. [DOI] [Google Scholar]
- Peng Z.; Qin Y.; Song J.; Zhu D.; Chen S.; Ren J.; Li Z.; Zou Z. Structuring of Hydroxy-Terminated Polydimethylsiloxane Filled by Fumed Silica. e-Polymers 2021, 21, 131–139. 10.1515/epoly-2021-0012. [DOI] [Google Scholar]
- Dodgson K.; Semlyen J. A. Studies of Cyclic and Linear Poly(Dimethyl Siloxanes): 1. Limiting Viscosity Number-Molecular Weight Relationships. Polymer 1977, 18, 1265–1268. 10.1016/0032-3861(77)90291-9. [DOI] [Google Scholar]
- Hadjichristidis N.; Xenidou M.; Iatrou H.; Pitsikalis M.; Poulos Y.; Avgeropoulos A.; Sioula S.; Paraskeva S.; Velis G.; Lohse D. J.; Schulz D. N.; Fetters L. J.; Wright P. J.; Mendelson R. A.; García-Franco C. A.; Sun T.; Ruff C. J. Well-Defined, Model Long Chain Branched Polyethylene. 1. Synthesis and Characterization. Macromolecules 2000, 33, 2424–2436. 10.1021/ma991670w. [DOI] [Google Scholar]
- Lederer A.; Burchard W.; Khalyavina A.; Lindner P.; Schweins R. Is the Universal Law Valid for Branched Polymers?. Angew. Chem., Int. Ed. 2013, 52, 4659–4663. 10.1002/anie.201209228. [DOI] [PubMed] [Google Scholar]
- Liu X. M.; Maziarz E. P.; Heiler D. J. Characterization of Implant Device Materials Using Size-Exclusion Chromatography with Mass Spectrometry and with Triple Detection. J. Chromatogr. A 2004, 1034, 125–131. 10.1016/j.chroma.2004.02.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.