

Abstract
Synthetic small molecules have been very effective in decimating cancer cells by targeting various aberrantly overexpressed oncogenic proteins. These small molecules target proteins involved in cell cycle regulation, cell division, migration, invasion, angiogenesis, and other regulatory proteins to induce apoptosis in cancer cells. In this study, we have synthesized a novel 1,2,5-trisubstituted benzimidazole chemical library of small molecules and unveiled their anticancer potential against a panel of cancer cell lines such as Jurkat, K-562, MOLT-4, HeLa, HCT116, and MIA PaCa-2 cancer cells. The MTT assay and Trypan blue dye exclusion assay clearly unveiled the cytotoxic effect of methyl 1-benzyl-2-(4-fluoro-3-nitrophenyl)-1H-benzo[d]imidazole-5-carboxylate (TJ08) and its potential to induce apoptosis with effective IC50 of 1.88 ± 0.51, 1.89 ± 0.55, 2.05 ± 0.72, 2.11 ± 0.62, 3.04 ± 0.8, and 3.82 ± 0.25 μM against Jurkat, K562, MOLT-4, HeLa, HCT116, and MIA PaCa-2 cancer cell lines, respectively. Altered mitochondrial membrane potential was observed in HeLa, HCT116, and Jurkat cells due to TJ08 treatment, which was unveiled by JC10 staining. Induction of early and late apoptosis by TJ08 treatment was also unveiled by apoptotic analysis and immunofluorescence imaging. Cell cycle analysis distribution confirms the accumulation of cells in the S-phase in a dose-dependent manner.
1. Introduction
Public health issues pose major challenges across the globe, especially cancer which increases every year and stands second in the causes of deaths across the globe. Hence, cancer is a major public health problem worldwide. Though many anticancer agents have been proven to be successful in the clinics, they have encountered various issues like low bioavailability and solubility. Cancer cells develop drug resistance to available drugs by various mechanisms, increasing drug efflux pumps (MDR1, P-gp) and drug transporters. Drug efflux by ATP binding cassette transporters is of major concern because it effectively eliminates the drug and inhibits the effective accumulation of drug inside the cells. Several cancer cells exhibit overexpression of these drug efflux transporters which results in developing drug resistance against various available drugs in the market. Hence multidrug resistance (MDR) and intolerable toxicities are a major concern for effective chemotherapeutic interventions against human malignancies. Hence, there is a constant demand to identify and develop structurally novel potent anticancer drugs to combat various cancers.
Benzimidazole is one of the most important heterocyclic compounds having structural similarity to nucleotides found in the human body, and it is an important pharmacophore in medicinal chemistry.1 The benzimidazole nucleus is a structural isostere of purine nucleosides. The structural resemblance of the benzimidazole with the purine nucleus in nucleotides makes benzimidazole derivatives, which are attractive ligands for interaction with biopolymers. The benzene ring, which is present in 1H-benzimidazole, functions as an electron-attracting group on imidazole and thereby decreases imidazole’s electronic density, which plays an important role in chemical interactions that have varied biological activities and thus are of great scientific interest nowadays. Substitution of the benzimidazole nucleus is an important synthetic strategy in the drug discovery process.2,3
The literature survey revealed that the benzimidazole derivatives have been extensively investigated as potential anticancer agents, and these benzimidazole derivatives exhibit anticancer effects through different mechanisms of action such as intercalating mode of action (DNA binding), nonintercalating mode of action (topoisomerases inhibition), DNA alkylation (DNA cleavage), acting on enzyme antimetabolites [DihydrofolateReductase (DHFR) inhibition], sirtuin inhibition (SIrt1 and SIrt2 inhibition), acting on structural proteins (tubulin protein inhibition), and protein kinase inhibition.4−30
Some of the benzimidazole scaffold-based US FDA approved anticancer drugs such as bendamustine (to treat chronic lymphocytic leukemia), binimetinib (protein kinase 1/2 (MEK 1/2) inhibitor to treat metastatic melanoma), selumetinib (MEK 1/2 inhibitor used in pediatric patients to treat neurofibromatosis type 1), glasdegib (hedgehog signaling inhibitor to treat acute myeloid leukemia), and abemaciclib (CDK inhibitor selective for CDK4 and CDK6 for the treatment of advanced or metastatic breast cancers) (Table 1) are currently available to treat cancer; some of the benzimidazole derivatives, such as pracinostat, carbendazim, galeterone, dovitinib, nazartinib etc. are currently in clinical trials; and some of the druggable benzimidazole derivatives were patented;2,4,31−35 hence, the benzimidazole scaffold is an important and promising pharmacophore for new anticancer drug discovery.
Table 1. US FDA Approved Drugs Having the Benzimidazole Scaffold.

In our previous findings, we have synthesized libraries of designed benzimidazole derivatives, evaluated their anticancer effects, and succeeded in identifying promising benzimidazole-based novel anticancer agents;36−38 further, we have developed a novel reagent for the synthesis of benzimidazole derivatives through N-formylation.39 Our previous benzimidazole derivative’s structure–activity relationship study results motivated us to synthesize designed novel 1,2,5-trisubstituted benzimdazole derivatives (Figure 1) to find structurally novel and potential benzimidazole-based anticancer agents.
Figure 1.

Chemical library designed based on our previous findings.
In light of the above facts, in the present study, we report the synthesis of a series of novel 1,2-disubstituted benzimidazole-5-carboxylates, 1,2-disubstituted benzimidazole-5-carboxylic acids, and 1,2-disubstituted benzimidazole-5-carboxamides (TJ01–TJ15) through multistep organic synthesis by reacting with different aromatic amines at the first position, reacting with different substituted aromatic acids at the second position, and introducing ester, acid, and amide at the fifth position of the benzimidazole core. The cytotoxic potency of newly synthesized 1,2,5-trisubstituted benzimidazole derivatives against various cancer cells reveals that the TJ08 compound is a promising hit which efficiently induces apoptosis in cancer cells. Here we enumerated the efficiency of the synthesized library of compounds against panels of cancer cells to unveil their anticancer potential.
2. Results and Discussion
2.1. Chemistry
The synthetic procedures that we followed for the synthesis of designed benzimidazole derivatives TJ01–TJ15 are outlined in Schemes 1–4. The structures of the substituents R1 and R2 of synthesized intermediate compounds 3a–c, 4a–c, and 5a–g are shown in Table 2. The structures of synthesized final compounds TJ01–TJ15 are displayed in Table 3.
Scheme 1. Synthesis of Benzimidazole Derivatives TJ01-TJ15.

Reagents and reaction conditions: (a) SOCl2 (1.5 equiv), MeOH (10 vol), 70 °C, 2 h, 98.0% yield; (b) K2CO3 (2 equiv), R1-NH2 (1.1 equiv), THF (10 vol), 0 °C to rt, 2 h, 95.0–98.6% yield; (c) iron powder (10% w/w), saturated aqueous NH4Cl solution (5 vol), IPA (10 vol), 90 °C, 2–3 h, 83.3–86.5% yield; (d) SOCl2 (1.5 equiv), toluene (20 vol), 50 °C, 2 h; (e) acyl chloride (1.1 equiv), THF (20 vol), DIPEA (2 equiv), 0 °C to rt, 3 h, 78.3–93.6% yield; (f) CH3COOH (10 vol), 100 °C, 3–5 h, 73.7–93.9% yield; (g) LiOH.H2O (1.5–3.1 equiv), CH3OH/THF/H2O (1:1:1), 0 °C to rt to 60 °C, 2–8 h, 57.2–98.3%; (h) CDI (1.5 equiv), aqueous NH3 (3 vol), THF (10 vol), 0 °C to rt, 6 h, 77.5%–90.2%.
Scheme 4. Synthesis of TJ10 from 5e.

Reagents and reaction conditions: (g) CH3COOH (16 V), 100 °C, 3 h; (h) LiOH.H2O (1.1 equiv), THF:CH3OH:H2O (1:1:l), 0 °C to rt, 2 h.
Table 2. Structures of R1 and R2 of Intermediate Compounds 3a–c, 4a–c, and 5a–g.

Table 3. Structures of the Final Compounds TJ01–TJ15.

The methyl 4-fluoro-3-nitrobenzoate (2) was obtained (98% yield) from 4-fluoro-3-nitrobenzoic acid (1) by an esterification reaction using thionyl chloride and DMF (catalytic amount) in methanol as a solvent under reflux conditions. Aromatic nucleophilic substitution reaction of compound 2 with different substituted aromatic amines in the presence of potassium carbonate as a base and tetrahydrofuran as solvent gives compounds 3a–c (95–98.6% yield).
Compounds 3a–c were converted to 4a–c (83.3–86.5% yield) by reduction of the nitro group to the corresponding amines. These conversions were performed by treatment with iron powder, saturated aqueous ammonium chloride solution, and isopropyl alcohol as a solvent under reflux conditions. The intermediate compounds 5a–g (78.3–93.6% yield) were obtained by reacting the compounds 4a–c with different aromatic acyl chlorides (acyl chlorides were prepared by reacting the corresponding carboxylic acids with thionyl chloride, a catalytic amount of DMF, and toluene as a solvent) in the presence of N,N-diisopropylethylamine as a base and tetrahydrofuran as a solvent.
The 1,2-disubstituted benzimidazole-5-carboxylates TJ03, TJ07, TJ08, and TJ13 were synthesized (73.7–93.9% yield) as described in Scheme 1 by heating intermediate compounds 5c, 5d, 5g, and 5f, respectively, in acetic acid under reflux conditions. The 1,2-disubstituted benzimidazole-5-carboxylic acids TJ04, TJ09, TJ11, and TJ14 were prepared (57.2–93.9% yield) from the compounds TJ03, TJ07, TJ08, and TJ13, respectively, by hydrolysis of the ester group of these compounds to the corresponding acids (during hydrolysis of ester of TJ07 and TJ08, in addition to hydrolysis of ester, the fluorine was also substituted by a hydroxyl group and produced compounds TJ09 and TJ11, respectively) by reacting with lithium hydroxide monohydrate, tetrahydrofuran, methanol, and water mixture as solvent. The compounds TJ01, TJ02, and TJ10 (84.1–98.3% yield) were obtained from intermediate compounds 5a, 5b, and 5e by heating in acetic acid under reflux conditions followed by hydrolysis of the ester to the corresponding acid.
The 1,2-disubstituted benzimidazole-5-carboxamides TJ05, TJ06, TJ12, and TJ15 were obtained (77.5–90.2% yield) from the compounds TJ04, TJ02, TJ10, and TJ14, respectively, by reacting with the carbonyldiimidazole (CDI) and aqueous ammonia (∼25% solution) in DMF as a solvent.
2.2. Biological Evaluation
2.2.1. Antiproliferative Effect of TJ08 on Various Cancer Cell Lines
All the synthesized compounds were tested against various cancer cells, including human leukemic cancer cells (Jurkat, K562, and Molt4), human cervical cancer cells (HeLa), human colorectal carcinoma cells (HCT116), and human pancreatic ductal adenocarcinoma (MIAPaCa-2) (Table S1). The results revealed that the compound TJ08 was very effective against various cells with IC50 ranging from 1.88 to 3.82 μM (Table 4), and doxorubicin displayed apoptotic activity against Jurkat cells at 4.89 μM. Jurkat cells were most sensitive, whereas MIA PaCa-2 cells were least sensitive to TJ08, which was unveiled by MTT assay (Figure 2a–f). Trypan Blue dye exclusion assay also unveiled a drastic reduction in the survival percentage of cancer cells. Interestingly, TJ08 displayed the least cytotoxic effect against normal kidney cell lines (Hek cells) (Figure 2g).
Table 4. IC50 Values of TJ08 against Various Cancer Cells and Normal Cellsa.
| IC50 (μM) |
|||||||
|---|---|---|---|---|---|---|---|
| compd | Jurkat | K562 | MOLT-4 | HeLa | HCT116 | MIA-PaCa-2 | HEK |
| TJ08 | 1.88 ± 0.51 | 1.89 ± 0.55 | 2.05 ± 0.72 | 2.11 ± 0.62 | 3.04 ± 0.8 | 3.82 ± 0.25 | >50 |
| DOXO | 4.89 ± 0.11 | 0.99 ± 0.21 | >1 | 0.21 ± 0.13 | 0.26 ± 0.19 | 0.059 ± 0.63 | >50 |
MTT assay, cell treated with TJ08 and incubated for 48 h. IC50 (μM) (mean ± SD, n = 3).
Figure 2.

Effect of TJ08 on cell proliferation and cell viability on a panel of cancer cell lines: Cells were seeded in a 96-well plate, incubated for 12 h, treated with an increased concentration of TJ08, incubated for 48 and 72 h, and subjected to MTT and Trypan blue dye exclusion assay to assess the cytotoxic potential of TJ08. DMSO-treated wells served as vehicle control. (a) Jurkat, (b) K562, (c) MOLT4, (d) HeLa, (e) HCT 116, (f) MIA PaCa-2, (g) HEK, and (h) Jurkat (doxorubicin). Each experiment has been repeated a minimum of 3 times. Error bars indicate the SEM and P-values were calculated by comparing the mean of control group with the mean of treated group.*, p < 0.05; **, p < 0.005.
2.2.2. TJ08 Treatment Diminishes Foci Formation and Mitigates Cell Migration and Invasion
Foci formation is the hallmark process of transformed cells; due to the loss of density-dependent growth they grow in multiple layers and form foci. The capability of cancer cells to form foci was examined with the TJ08 treatment. Interestingly, TJ08 treatment diminished the colony formation capability of HeLa, HCT116, and MIA PaCa-2 cells when compared to the DMSO-treated well in a concentration-dependent manner (Figure 3a–c). Metastasis plays a pivotal role in the development of secondary tumors, which was facilitated by the degradation of the extracellular matrix and promotes migration and invasion of cancer cells. TJ08 treatment diminishes migration and invasion of HeLa cells, which was unveiled by Roche’s cell migration and invasion kits (Figure 3d,e).
Figure 3.

Effect of TJ08 on Foci formation, migration, and invasion: Cells were seeded into a 6-well plate and treated with increased concentrations of TJ08, allowed for 2 weeks to form colonies, and stained with crystal violet. (a) HeLa cells, (b) HCT116, and (c) MIA PaCa-2 cells were used for the study. DMSO-treated wells served as the vehicle control; each experiment was repeated 3 times. (d, e) Effect of TJ08 on migration and invasion, respectively. HeLa cells were seeded and treated with TJ08, incubated, and subjected for assay as described in the procedure, and DMSO-treated wells served as the vehicle control; each experiment was repeated 3 times. Error bars indicate the SEM and P-values were calculated by comparing the mean of control group with the mean of TJ08-treated group. **, p < 0.05.
2.2.3. Induction of Apoptosis by TJ08 Treatment
Evasion of apoptosis is a characteristic feature of cancer cells to ensure rapid proliferation, but chemotherapeutic drugs forcibly induce apoptosis in cancer cells. TJ08-induced early and late apoptotic events were confirmed by Annexin V/PI and also by Hoechst and PI double staining. Accumulation of propidium iodide inside the cells confirms the induction of apoptosis upon TJ08 treatment, and the same was further confirmed by staining the cells with Annexin V/PI double stain. Early apoptotic cells were stained with green alone, and late apoptotic cells were stained with both green and red (Figure 4). Interestingly, we did not find any PI-stained cells, which confirms that TJ08 treatment induces cell death by apoptosis, not by necrosis, in a concentration-dependent manner.
Figure 4.

Effect of TJ08 on apoptosis. Jurkat cells were seeded in 6-well dishes, treated with increased concentrations of TJ08, and incubated for 24 h, and cells were harvested and incubated with Annexin V/PI. (a) Cytometric analysis of apoptotic cells. (b) Fluorescence microscopic imaging of apoptotic cells. (c, d) Histogram of percentage of apoptotic cells cytometric and fluorescence analysis. Cells marked with white arrow represents early apoptotic cells, and cells marked with red arrow represent late apoptotic cells.
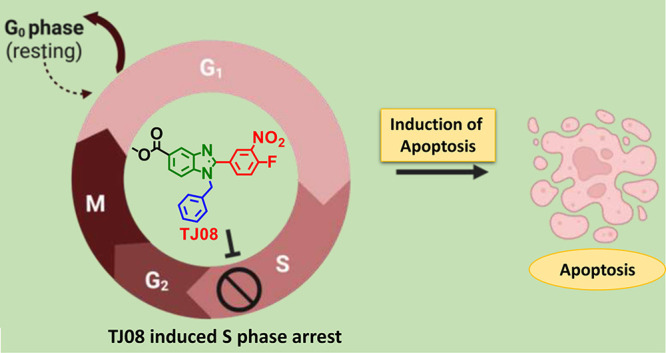
2.2.4. TJ08 Instigates S Phase Arrest and Abrogates Cancer Cell Progression
Interference in any protein pathway obstructs the progression of cell cycle and triggers cell cycle arrest. Competency to instigating cell cycle arrest by TJ08 is unveiled by cell cycle analysis which clearly unveils the accumulation of cells at the S phase in a concentration-dependent manner followed by increased accumulation of a sub-G1 population of cells (Figure 5). The results clearly demonstrate that the compound TJ08 induces cell cycle arrest upon treatment and promotes apoptosis of cancer cells.
Figure 5.

Impact of TJ08 on cell cycle distribution. Jurkat cells were seeded and treated with different concentrations of TJ08 (1, 5, and 10 μM) and incubated for 24 h. Cells were harvested and fixed with ethanol, stained with propidium iodide, and analyzed using flow cytometer. (a) Effect of TJ08 on cell cycle. (b) Percentage of cells distribution in different phases of cell the cycle. (c) Structure of TJ08. DMSO-treated cells served as the vehicle control.
2.2.5. TJ08 Treatment Permutes Mitochondrial Membrane Potential and Triggers Apoptosis
Membrane potential is a critical component to maintain the integrity of mitochondria. Alterations in the membrane potential result in release of cytochromes into the cytosol of cells, which actuates the apoptotic cascade events inside the cells by activating initiator caspases. Alteration of membrane potential by TJ08 treatment was analyzed using JC10 dye. The inherent negative potential of the intact mitochondrial membrane allows the positively charged JC10 dye to enter the mitochondria. TJ08 treatments alter the membrane potential and results in accumulation of JC10 monomers in the cytosol and exhibit intense green fluorescence in a dose-dependent manner (Figure 6a–c). The obtained result clearly confirms the alteration of mitochondrial membrane potential due to TJ 08 treatment.
Figure 6.

Effect of TJ08 on mitochondrial membrane potential. Cells were seeded and treated with an increased concentration of TJ08, incubated for 24 h, harvested, and subjected for MMP Assay according to manufacturer’s protocol. (a) HCT 116, (b) HeLa, and (c) Jurkat cells were used for the study. DMSO-treated cells served as the vehicle control.
2.2.6. TJ08 Alters the Expression of Pro-apoptotic and Anti-apoptotic Proteins
Pro-apoptotic and anti-apoptotic proteins play important roles in cell survival and cell death, specifically BCl2, a major antiapoptotic protein involved in cell survival. TJ08 treatment specifically alters the expression of these proteins and induces apoptosis in cancer cells. Western blot results clearly unveil the upregulation of cleaved caspase and downregulation of antiapoptotic BCl2 proteins. Cleavage of PARP proteins also indicates clear induction of cell death due to TJ08 treatment (Figure 7).
Figure 7.

Effect of TJ08 on regulatory proteins. Jurkat cells were seeded in a 100 mm dish, treated with increased concentrations of TJ08, and incubated for 48 h. Cells were harvested and lysed by using RIPA buffer, electrophoresed, electroblotted, and incubated with respective antibodies, cyclin B1, PARP1, CASP 9, CASP3, BCL2, and tubulin.
3. Conclusion
In this study, we have synthesized 1,2,5-trisubstituted benzimidazole derivatives TJ01–TJ15 and evaluated their anticancer properties, and the compound TJ08 is identified as a structurally novel, potential anticancer agent among the synthesized compounds. Our results revealed that the compound TJ08 efficiently decreases the proliferation capability of various screened cancer cells which was unveiled by both cell proliferation and cell viability assays. The compound TJ08 also diminished the colony formation capabilities of the cancer cells and mitigates invasion and migratory capabilities. Efficient induction of apoptosis will be achieved by triggering cell cycle arrest in the cancer cells. The compound TJ08 efficiently induces G1/S phase arrest and promotes apoptosis in various cancer cells. The compound TJ08 instigates apoptosis by permuting mitochondrial membrane potential, which alters the mitochondrial charges and results in leakage of cytochromes into the cell cytosol which preliminarily activates the initiator caspases and executionary caspases to initiate the apoptotic cascade events. Compound TJ08 efficiently alters the pro- and anti-apoptotic proteins to induce cell death in cancer cells, cleavage of PARP proteins which was the critical evidence for the activation of caspase protein.
The structure–activity relationship results indicates that the key structural features, the 4-fluoro-3-nitrophenyl group at the R2 position and the benzyl group at the R1 position of the benzimidazole core, are crucial for the potential anticancer activity of TJ08. Replacement of these groups with other substituents abolished the anticancer potency, which clearly indicates the importance of these groups at the terminal ends. All consolidated results from in vitro assays unveil the potency of TJ08 as a promising structurally novel anticancer agent, and hence, we have selected this molecule to develop further to enhance its cytotoxic potential. The synthesis of designed TJ08 analogues and evaluation of their anticancer effect is in progress and will be published in the future.
4. Materials and Methods
4.1. Chemistry
Starting materials, reagents, and solvents were purchased from commercial sources and used as received unless stated otherwise. Moisture or air-sensitive reactions were conducted under nitrogen atmosphere in an oven-dried glass apparatus. Reaction progress was monitored by thin-layer chromatography (TLC) on precoated silica gel plates (silica gel 60 F254; Merck grade), and visualization was accomplished with UV light and potassium permanganate stains followed by heating. The solvents were removed under reduced pressure using rotary evaporator. Purification of intermediates and final compounds were carried out by silica gel 230–400 mesh (particle size 40–63 μm) flash column chromatography. The NMR spectra were recorded on Bruker 400 and 300 MHz and Agilent 100 MHz spectrometers, and chemical shifts are reported in ppm using the solvents (DMSO-d6). Chemical shifts are reported in ppm using TMS as the internal standard. Data are reported as s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet; coupling constant(s) in hertz. LC–MS analysis were performed on Agilent Technologies, and HRMS was performed on a Waters SYNAPTG2. FTIR spectra were recorded on a Cary 630 spectrometer and Bruker Alpha 2 spectrometer.
4.1.1. Synthesis of Methyl 4-Fluoro-3-nitrobenzoate (2)
To a stirred solution of 4-fluoro-3-nitrobenzoic acid (1) (5.0 g, 27.01 mmol, 1.0 equiv) in methanol (50 mL) was added thionyl chloride dropwise (3.0 mL, 41.51 mmol, 1.5 equiv) at 0 °C under nitrogen atmosphere, and then the reaction mixture was refluxed at 70 °C for 2 h. The reaction progress was monitored by TLC. Upon completion, the solvent was removed in vacuo to obtain the crude product. The crude product was further purified by recrystallization using a mixture of solvents diethyl ether and hexane (30 mL, 1:2 V/V) to obtain pure methyl 4-fluoro-3-nitrobenzoate (2) as an off-white solid with 98% yield (5.3 g). 1H NMR (400 MHz, DMSO-d6) δ: 8.56–8.59 (m, 1H, Ar-H), 8.31–8.36 (m, 1H, Ar-H), 7.72–7.78 (m, 1H, Ar-H), 3.91 (s, 3H, -OCH3). ESI/MS (m/z): Calculated for [C8H6FNO4] [M + H]+: 200.03. Found m/z: 199.9 [M + H]+; LCMS purity = 99.361%.
4.1.2. General Procedure A for the Synthesis of Compounds 3a–c
To a stirred solution of methyl 4-fluoro-3-nitro benzoate (2) (1.0 equiv) in tetrahydrofuran (10 V) was added potassium carbonate (2.0 equiv) followed by slow addition of different substituted amines (1.1 equiv) at 0 °C under nitrogen atmosphere. The reaction mixture was gradually allowed to warm to room temperature and stirred for additional 2 h. The reaction progress was monitored by TLC. Upon completion, the reaction mixture was poured into ice cold water, the organic phase was extracted with ethyl acetate (150 mL × 3), and the combined organic phase was washed with water and saturated brine. The organic phase was dried over anhydrous sodium sulfate, and the solvent was filtered and concentrated in vacuo to obtain crude product. It was further triturated with n-pentane (50 mL), filtered, and dried in vacuo to obtain pure compounds 3a–c.
4.1.2.1. Synthesis of Methyl 4-((2-Methoxyphenethyl) amino)-3-nitrobenzoate (3a)
Following the general procedure A, compound 3a was obtained from compound 2 (5.1 g, 25.61 mmol, 1.0 equiv) using 2-(2-methoxyphenyl)ethanamine (4.2 mL, 28.69 mmol, 1.1 equiv) as a yellow solid with 98.10% yield (8.3 g).
1H NMR (300 MHz, DMSO-d6) δ: 8.87–8.88 (d, J = 2.1 Hz, 1H, −N-H), 8.879 (s, 1H, Ar-H), 8.02–8.06 (m, 1H, Ar-H), 7.19–7.30 (m, 2H, Ar-H), 6.90–6.97 (m, 3H, Ar-H), 3.91–3.92 (d, 6H, −(OCH3)2), 3.56–3.63 (m, 2H, −CH2), 3.06–3.10 (t, J = 6.9 Hz, 2H, −CH2). ESI/MS (m/z): Calculated for [C17H18N2O5] [M + H]+: 331.12; Found m/z:331.20 [M + H]+; LCMS purity = 99.614%.
4.1.2.2. Synthesis of Methyl 4-((4-Methoxybenzyl) amino)-3-nitrobenzoate (3b)
Following the general procedure A, compound 3b was obtained from compound 2 (3.0 g, 15.06 mmol, 1.0 equiv) using (4-Methoxyphenyl) methanamine (2.2 mL, 16.83 mmol, 1.1equiv) as a yellow solid with 98.6% yield (4.7 g).
1H NMR (400 MHz, DMSO-d6) δ: 9.00–9.01 (d, J = 7.6 Hz, 1H, Ar-H), 8.62–8.63 (d, J = 1H, −N-H), 7.88–7.91 (m, 1H, Ar-H), 7.30–7.33 (d, J = 11.2 Hz, 2H, Ar-H), 7.02–7.05 (d, J = 12.4 Hz, 1H, Ar-H), 6.89–6.92 (d, J = 11.6 Hz, 2H, Ar-H), 4.60–4.62(d, J = 8.0 Hz, 2H, −CH2), 3.72–3.81(d, 6H, −(OCH3)2). ESI/MS (m/z): Calculated for [C16H16N2O5] [M + H]+: 317.11. Found m/z = 317.2 [M + H]+; LCMS purity = 99.303%.
4.1.2.3. Synthesis of Methyl 4-(Benzylamino)-3-nitrobenzoate (3c)
Following the general procedure A, compound 3c was obtained from compound 2 (3.0 g, 15.06 mmol, 1.0 equiv) using benzylamine (1.8 mL, 16.47 mmol, 1.1 equiv) as a yellow solid, with 95.0% yield (4.1 g).
1H NMR (400 MHz, DMSO-d6) δ: 9.10 (s, 1H, Ar-H), 8.64–8.64 (d, J = 2.0 Hz, 1H, Ar-H), 7.78–7.79 (m, 1H, Ar-H), 7.33–7.36 (m, 4H, Ar-H), 7.26–7.28 (t, J = 4.2 Hz, 1H, Ar-H), 6.99–7.02 (d, J = 1.2 Hz, 1H, Ar-H), 4.70–4.71 (d, J = 6.0 Hz, 2H, −CH2), 3.81 (s, 3H, −OCH3). ESI/MS (m/z): Calculated for [C15H14N2O4][M + H]+: 287.10. Found m/z = 287.0 [M + H]+; LCMS purity = 97.288%.
4.1.3. General Procedure B for the Synthesis of Compounds 4a–c
The suspensions of nitro compounds 3a–c (1.0 equiv) in isopropanol (10 V) taken in separate round-bottomed flasks were heated to 70 °C (reaction mixture turns to clear solution), and then iron powder (20% w/w) was added to the reaction mixtures followed by saturated aqueous ammonium chloride solution (5 V). Then the reaction mixtures were heated to 90 °C and stirred for 2–3 h. The reaction progress was monitored by TLC. Upon completion, the reaction mixtures were filtered through a Celite pad and thoroughly washed with isopropanol. The collected clear filtrate was concentrated in vacuo to obtain the crude product, which was further purified by flash column chromatography (using 25–40% ethyl acetate in petroleum ether mobile phase) to obtain pure amine compounds 4a–c, respectively.
4.1.3.1. Synthesis of Methyl 3-Amino-4-((2-methoxyphenylethyl)amino)benzoate (4a)
Following the general procedure B, compound 4a was obtained from compound 3a (8. 05 g, 24.37 mmol, 1.0 equiv) using iron powder (1.6 g, 20% w/w) and saturated aqueous ammonium chloride solution (40 mL, 5 V) as a pale brown solid with 83.3% yield (6.1 g).
1H NMR (400 MHz, DMSO-d6) δ: 7.18–7.25 (m, 4H, Ar-H), 6.98–7.00 (d, J = 8.8 Hz, 1H, Ar-H), 6.87–6.91 (t, J = 7.4 Hz, 1H, Ar-H), 6.53–6.56 (d, J = 8.4 Hz, 1H, Ar-H), 5.38–5.41 (t, J = 5.2 Hz,1H, −NH), 4.73 (s, 2H, -NH2), 3.83 (s, 3H, −OCH3), 3.72 (s, 3H, −OCH3), 3.26–3.33 (m, 2H, −CH2), 2.85–2.89 (t, J = 7.6 Hz,2H, −CH2). ESI/MS (m/z): Calculated for [C17H20N2O3][M + H]+: 301.15. Found m/z: 301.2 [M + H]+. LCMS purity = 98.764%.
4.1.3.2. Synthesis of Methyl 3-Amino-4-((4-methoxybenzyl)amino)benzoate (4b)
Following the general procedure B, compound 4b was obtained from compound 3b (4.60 g, 14.54 mmol, 1.0 equiv) using iron powder (0.92 g, 20% w/w) and saturated aqueous ammonium chloride solution (20 mL, 5 V) as an off-white solid with 84% yield (3.5 g).
1H NMR (300 MHz, DMSO-d6) δ: 7.25–7.32 (m, 2H, Ar-H), 7.18–7.18 (d, J = 1.8 Hz,1H, Ar-H), 7.10–7.13 (m, 1H, Ar-H), 6.87–6.90 (d, J = 8.7 Hz, 2H, Ar-H), 6.36–6.38(d, J = 12 Hz, 1H, Ar-H), 5.80–5.84 (t, 1H, −-NH), 4.81 (s, 2H, −NH2), 4.29–4.31(d, J = 5.4 Hz, 2H, −CH2), 3.77–3.79 (d, 6H, −(OCH3)2). ESI/MS (m/z): Calculated for [C16H18N2O3][M + H]+: 287.13. Found m/z: 287.0 [M + H]+. LCMS purity = 91.361%.
4.1.3.3. Synthesis of Methyl 3-Amino-4-(benzylamino)benzoate (4c)
Following the general procedure B, compound 4c was obtained from compound 3c (4.0 g, 13.97 mmol, 1.0 equiv) using iron powder (0.8 g, 20% w/w) and saturated aqueous ammonium chloride solution (20 mL, 5 V) as an off-white solid with 86.5% yield (3.1 g). 1H NMR (400 MHz, DMSO-d6) δ: 7.30–7.36 (m, 4H, Ar-H), 7.21–7.25 (m, 2H, Ar-H), 7.19–7.20 (d, J = 2.0 Hz, 1H, Ar-H), 6.34–6.36 (d, J = 5.2 Hz, 1H, Ar-H), 5.91–5.93 (t, J = 5.8 Hz, 1H, Ar-H), 4.83 (s, 2H, -NH2), 4.38–4.39 (d, J = 6.0 Hz, 2H, −CH2), 3.69 (s, 3H, −OCH3). ESI/MS (m/z): Calculated for [C15H16N2O2][M + H]+: 257.12. Found m/z: 257.2 [M + H]+. LCMS purity = 92.870%.
4.1.4. General Procedure C for the Synthesis of Compounds 5a–g
Substituted carboxylic acids 4a–c (1.1 equiv) were taken in separate round-bottomed flasks in toluene (10–20 V), thionyl chloride (1.5–1.8 equiv) was added at 0 °C under nitrogen atmosphere followed by a few drops of DMF, and then the reaction mixture was heated to 50 °C and stirred for 2 h. The reaction progress was monitored by TLC (esterification of aliquot reaction mixture with methanol). After completion of the reaction, the reaction mixture was concentrated in vacuo (toluene was added and redistilled to remove excess of thionyl chloride).
The obtained acid chlorides were further diluted with THF (10 V) and added to other round-bottom flasks containing the solutions of compounds 4a–c (1.0 equiv) in THF (10 V) at 0 °C under nitrogen atmosphere, N,N-diisopropylethylamine (DIPEA) base (2.0 equiv) was added to the reaction mixture at the same temperature, the reaction mixture (clear solution) was gradually warmed to room temperature and stirred for 3 h, and then the products were precipitated from the reaction mixtures.
Upon reaction completion, the reaction mixtures were concentrated partially (half of their volume) in vacuo and cooled at 0 °C, the products precipitated (5a and 5b) were filtered and washed with cold tetrahydrofuran (∼0.5 mL) and dried in vacuo, and other intermediates 5c, 5d, 5e, 5f, and 5g were obtained. Upon reaction completion, the reaction mixture was poured into ice-cold water, the organic product was extracted with ethyl acetate (15 mL × 3), and the combined organic phase was washed with water and brine and dried over anhydrous sodium sulfate. The solvent was filtered and concentrated invacuo, and the obtained crude intermediates 5a–g were directly used for the synthesis of final compounds TJ01–TJ15.
4.1.4.1. Synthesis of Methyl 3-(2-Bromo-6-methylbenzamido)-4-((2-methoxyphenethyl)amino)benzoate (5a)
Following the general procedure C, compound 5a was obtained from 4a (250 mg, 0.832 mmol, 1.0 equiv) using 2-bromo-6-methylbenzoic acid (200 mg, 0.930 mmol, 1.1 equiv) in toluene (4 mL), thionyl chloride (0.1 mL, 1.37 mmol, 1.6 equiv), and DIPEA (0.29 mL, 1.664 mmol, 2.0 equiv) as an off-white solid with 92.9% yield (430 mg).
4.1.4.2. Synthesis of Methyl 3-(4-Methoxy-3-nitrobenzamido)-4-((2-methoxyphenethyl)amino)benzoate (5b)
Following the general procedure C, compound 5b was obtained using 4a (138 mg, 0.459 mmol, 1.0 equiv) using 4-methoxy-3-nitrobenzoic acid (100 mg, 0.507 mmol, 1.1 equiv) in toluene (2 mL), thionyl chloride (0.06 mL, 0.826 mmol, 1.8 equiv), and DIPEA (0.16 mL, 0.918 mmol, 2.0 equiv) as an off-white solid with 91.7% yield (223 mg).
4.1.4.3. Synthesis of Methyl 3-(2-(4-Fluorophenyl)acetamido)-4-((2-methoxyphenethyl)amino)benzoate (5c)
Following the general procedure C, compound 5c was obtained from 4a (176 mg, 0.585 mmol. 1.0 equiv) using 2-(4-fluorophenyl)acetic acid (100 mg, 0.648 mmol, 1.1 equiv) in toluene (2 mL), thionyl chloride (0.08 mL, 1.10 mmol, 1.8 equiv), and DIPEA (0.2 mL, 1.148 mmol, 2.0 equiv) as a pale-brown solid with 93.6% yield (265 mg).
4.1.4.4. Synthesis of Methyl 3-(4-Fluoro-3-nitrobenzamido)-4-((4-methoxybenzyl) amino)benzoate (5d)
Following the general procedure C, compound 5d was obtained from 4b (209 mg, 0.729 mmol, 1.0 equiv) using 4-fluoro-3-nitrobenzoic acid (150 mg, 0.810 mmol, 1.1 equiv) in toluene (3 mL), thionyl chloride (0.1 mL, 1.37 mmol, 1.8 equiv), and DIPEA (0.26 mL, 1.492 mmol, 2.0 equiv) as a pale-brown solid with 88.4% yield (325 mg).
4.1.4.5. Synthesis of Methyl 3-(4-Methoxy-3-nitrobenzamido)-4-((4-methoxybenzyl)amino)benzoate (5e)
Following the general procedure C, compound 5e was obtained from 4b (130 mg, 0.454 mmol) using 4-methoxy-3-nitrobenzoic acid (100 mg, 0.507 mmol) in toluene (2 mL), thionyl chloride (0.06 mL, 0.826 mmol, 1.8 equiv), and DIPEA (0.16 mL, 0.918 mmol, 2.0 equiv) as a pale-brown solid with 78.3% yield (185 mg).
4.1.4.6. Synthesis of Methyl 4-((4-Methoxybenzyl)amino)-3-(4-(trifluoromethoxy)benzamido)benzoate (5f)
Following the general procedure C, compound 5f was obtained from 4b (188 mg, 0.656 mmol, 1.0 equiv) using 4-(Trifluoromethoxy) benzoic acid (150 mg, 0.727 mmol, 1.1 equiv) in toluene (3 mL), thionyl chloride (0.08 mL, 1.101 mmol, 1.7 equiv), and DIPEA (0.24 mL, 1.377 mmol, 2.0 equiv) as a pale-brown solid with 81.1% yield (280 mg).
4.1.4.7. Synthesis of Methyl 3-(4-Fluoro-3-nitrobenzamido)-4-((4-methoxybenzyl) amino) benzoate (5g)
Following the general procedure C, compound 5g was obtained from 4c (187 mg, 0.729 mmol, 1.0 equiv) using 4-Fluoro-3-nitrobenzoic acid (150 mg, 0.810 mmol, 1.1 equiv) in toluene (3 mL), thionyl chloride (0.1 mL, 1.37 mmol, 1.8 equiv), and DIPEA (0.26 mL, 1.492 mmol, 2.0 equiv) as a pale-brown solid with 90.3% yield (310 mg).
4.1.5. General Procedure D for the Synthesis of 1,2-Disubstituted Benzimidazole-5-carboxylates TJ03, TJ07, TJ08, and TJ13 from 5c, 5d, 5g, and 5f
The intermediate compounds 5c, 5d, 5g, and 5f were dissolved in glacial acetic acid (10–15 V) in separate round-bottom flasks and stirred for 3–5 h at 100 °C under nitrogen atmosphere. The reaction progress was monitored by TLC. Upon reaction completion it was cooled to 0 °C and quenched with ice-cold water during which time the products were precipitated and then sonicated for 30–60 s, filtered, washed with cold water, and dried in vacuo to obtain compounds TJ03, TJ07, TJ08, and TJ13, respectively.
4.1.6. General Procedure E for the Synthesis of 1,2-Disubstituted Benzimidazole-5-carboxylic Acids TJ04, TJ09, TJ11, and TJ14 from TJ03, TJ07, TJ08, and TJ13
The 1,2-disubstituted benzimidazole-5-carboxylates TJ03, TJ07, TJ08, and TJ13 were taken in separate round-bottomed flasks in a mixture of tetrahydrofuran:methanol (1:1 V/V, 10–20 V), a solution of lithium hydroxide monohydrate (1.1–3.2 equiv) in water was added at 0 °C, the reaction mixtures of TJ03 and TJ13 were gradually warmed to room temperature and stirred for 2 h, and the solutions of TJ07 and TJ08 (without methanol only with 1:1 tetrahedrofuran:water solvents) were stirred at 60 °C for 8 h. The reaction progress was monitored by TLC. Upon completion, the reaction mixture was concentrated partially (to remove tetrahydrofuran and methanol), and the residual aqueous layer was diluted with water and washed with 50% diethyl ether in n-pentane twice (10 mL × 2). The aqueous layer was acidified to pH ∼ 6 (by adding saturated citric acid solution and in some cases 1.0 N aqueous HCl solution) during which product was precipitated which was sonicated for a minute then filtered and washed with water and dried in vacuo to obtain final compounds TJ04, TJ09, TJ11, and TJ14, respectively (During hydrolysis of esters TJ07 and TJ08, along with hydrolysis of ester, the fluoro group was replaced by a hydroxyl group.)
4.1.7. General Procedure F for the Synthesis of 1,2-Disubstituted Benzimidazole-5-carboxamides TJ05, TJ06, TJ12, and TJ15 from Compounds TJ04, TJ02, TJ10, and TJ14
1,2-Disubstituted benzimidazole-5-carboxylic acids TJ04, TJ10, and TJ14 were taken in separate round-bottomed flasks, dissolved in DMF (10–15 V), and stirred at 0 °C under nitrogen atmosphere, carbonyldiimidazole (CDI, 1.5 equiv) was added, the reaction mixture was gradually warmed to room temperature and stirred for 1 h, and cooled at 0 °C, and aqueous ammonia (25% solution, 16–21 equiv) was added. After addition, the reaction mixture was warmed to room temperature and stirred for 6 h. The progress of the reaction was monitored by TLC. Upon completion of the reaction, ice-cold water was added to the reaction mixture, during which time product was precipitated out from the reaction mixture which was sonicated for 30–60 s, filtered, washed with water, and dried in vacuo to obtain final compounds TJ05, TJ06, TJ12, and TJ15, respectively.
4.1.8. Synthesis of Final Compounds TJ01–TJ15
Scheme 2. Synthesis of TJ01 from 5a.

Reagents and reaction conditions (f): CH3COOH (13 V), 100 °C, 3 h; (g) LiOH·H2O (1.3 equiv), THF:CH3OH:H2O (2:2:l), 0 °C to rt, 2 h.
4.1.8.1. Synthesis of 2-(2-Bromo-6-methylphenyl)-1-(2-methoxyphenethyl)-1H-benzo[d]imidazole-5-carboxylic Acid (TJ01)
Following the general procedure D, the intermediate 5a1 was obtained from compound 5a (430 mg, 0.864 mmol, 1.0 equiv) using glacial acetic acid (4 mL) and refluxed at 100 °C for 3 h as a brown solid with 78% yield (325 mg). The obtained 5a1 converted to compound TJ01 by following the general procedure E using 5a1 (325 mg, 0.677 mmol, 1.0 equiv) in a mixture of tetrahydrofuran:methanol (4 mL, 12 V, 1:1 V/V) as the solvent and a solution of lithium hydroxide monohydrate (37 mg, 0.881 mmol, 1.3 equiv) in water (1 mL) at 0 °C to rt for 2 h. The pure compound TJ01 was obtained as a white solid with 98.3% yield (310 mg). (TLC Rf = 0.43 in dichloromethane:methanol 9:1 V/V.)
1H NMR (400 MHz, DMSO-d6) δ: 12.8 (s, 1H, −COOH), 8.24 (s, 1H, Ar-H), 7.95–7.97 (d, J = 8.4 Hz, 1H, Ar-H), 7.78–7.80 (d, J = 8.4 Hz, 1H, Ar-H), 7.63–7.65 (d, J = 8.0 Hz, 1H, Ar-H), 7.31–7.33 (d, J = 8.0 Hz, 1H, Ar-H), 7.16–7.20 (t, J = 7.4 Hz, 1H, Ar-H), 6.94 (s, 1H, Ar-H), 6.77–6.79 (d, J = 8.0 Hz, 1H, Ar-H), 6.66–6.67 (t, J = 6.4 Hz, 1H, Ar- H), 6.55–6.56 (d, J = 7.2 Hz, 1H, Ar-H), 4.30 (s, 2H, −CH2), 3.37 (s, 3H, −OCH3), 2.9 (s, 2H, −CH2), 1.95 (s, 3H, −CH3). 13C NMR (100 MHz, DMSO-d6) δ: 168.39, 157.63, 153.70, 142.68, 137.94, 137.19, 133.00, 132.58, 132.54, 132.08, 130.56, 128.83, 125.44, 125.31, 124.24, 121.48, 120.60, 118.80, 111.41, 110.98, 60.22, 55.11, 30.40, 19.08. FTIR νmax (cm–1): 3298.7, 3019.1, 2940.9, 2840.2, 1681.0, 1248.7. ESI/HRMS (m/z): Calculated for [C24H21BrN2O3][M + H]+ (79Br): 465.07, [M + H]+ (81Br): 467.07; Found m/z: 465.0783 [M + H]+ (79Br), 467.0755 [M + H]+ (81Br). ESI/MS (m/z): Calculated for [C24H21BrN2O3][M + H]+ (79Br): 465.07, [M + H]+ (81Br): 467.07. Found m/z: 465.0 [M + H]+ (79Br), 467.07 [M + H]+ (81Br). LCMS purity = 98.773%.
4.1.8.2. Synthesis of 2-(4-Methoxy-3-nitrophenyl)-1-(2-methoxyphenethyl)-1H-benzo[d]imidazole-5-carboxylic Acid (TJ02)
Scheme 3. Synthesis of TJ02 from 5b.

Reagents and reaction conditions (f): CH3COOH (13 V), 100 °C, 5 h; (g) LiOH·H2O (1.3 equiv), THF:CH3OH (1:1), 0 °C to rt, 2 h.
Following the general procedure D, the compound 5b1 was obtained from 5b (223 mg, 0.465 mmol, 1.0 equiv) using glacial acetic acid (3 mL) at 100 °C for 5 h as a brown solid with yield 80.0% (172 mg). The obtained compound 5b1 converted to TJ02 by following the general procedure E using 5b1 (172 mg, 0.372 mmol), a mixture of tetrahydrofuran:methanol (3 mL, 17 V, 1:1 V/V) as solvent, and lithium hydroxide monohydrate (21 mg 0.50 mmol) at 0 °C to rt for 2 h. The pure compound TJ02 obtained as a pale-yellow solid with 84.1% yield (140 mg). (TLC Rf = 0.39 in dichloromethane:methanol 9:1 V/V).
1H NMR (400 MHz, DMSO-d6) δ: 12.67 (s, 1H, −COOH), 8.24 (s, 1H, Ar-H), 7.94–7.96 (d, J = 9.6 Hz, 1H, Ar-H), 7.77–7.83 (m, 3H, Ar-H), 7.45–7.48 (d, J = 9.2 Hz, 1H, Ar-H), 7.05–7.08 (t, J = 7.6 Hz, 1H, Ar-H), 6.65–6.68 (m, 2H, Ar-H), 6.55–6.63 (d, J = 7.2 Hz, 1H, Ar-H), 4.56–4.59 (t, J = 6.2 Hz, 2H, −CH2), 4.03 (s, 3H, -OCH3), 3.46 (s, 3H, -OCH3), 2.91–2.94 (t, J = 6.2 Hz, 2H, −CH2). 13C NMR (100 MHz, DMSO-d6) δ: 168.32, 157.43, 153.36, 153.16, 142.49, 139.35, 138.95, 135.08, 130.53, 128.65, 125.97, 125.58, 125.29, 124.34, 122.43, 121.32, 120.48, 114.78, 111.26, 110.53, 57.52, 55.25, 44.44, 30.62. FTIR νmax (cm–1): 3067.6, 2937.1, 2836.5, 1684.8, 1244.9. ESI/HRMS (m/z): Calculated for [C24H21N3O6][M + H]+: 448.14; Found m/z: 448.1496 [M + H]+. ESI/MS (m/z): Calculated for [C24H21N3O6][M + H]+: 448.14. Found m/z: 448.20 [M + H]+. LCMS purity = 92.339%.
4.1.8.3. Synthesis of Methyl 2-(4-Fluorobenzyl)-1-(2-methoxyphenethyl)-1H-benzo[d]imidazole-5-carboxylate (TJ03)
Following the general procedure D, the compound TJ03 was obtained from the intermediate compound 5c (265 mg, 0.605 mmol, 1.0 equiv) using glacial acetic acid (3 mL), refluxed at 100 °C for 3 h. The obtained crude pale-brown solid was triturated with diethyl ether (3 mL) cooled to 0 °C, filtered, washed with cold diethyl ether, dried under vacuo. Pure TJ03 obtained as an off-white solid with 93.9% yield (230 mg). (TLC Rf = 0.2 in petroleum ether:ethyl acetate 6:4 V/V).
1H NMR (400 MHz, DMSO-d6) δ: 8.15–8.16 (d, J = 1.2 Hz, 1H, Ar-H), 7.83–7.85 (t, J = 7.2 Hz, 1H, Ar-H), 7.51–7.53 (d, J = 8.4 Hz, 1H, Ar-H), 7.14–7.28 (m, 5H, Ar-H), 6.92–6.94 (d, J = 8.0 Hz, 2H, Ar-H), 6.80–6.84 (t, J = 7.4 Hz, 1H, Ar- H), 4.33–4.37 (t, J = 7.0 Hz, 2H, CH2), 4.08 (s, 2H, −CH2), 3.86 (s, 3H, -OCH3), 3.69 (s, 3H, -OCH3), 2.83–2.87 (t, J = 7.0 Hz, 2H, −CH2). 13C NMR (100 MHz, DMSO-d6) δ: 167.24, 162.74 (d, JC–F = 242 Hz), 157.76, 156.01, 142.25, 139.05, 133.05, 131.18, 131.10, 130.90, 128.82, 125.90, 123.43, 123.39, 120.81, 120.62, 115.84, 115.63, 111.06, 110.48, 55.63, 52.38, 43.85, 32.36, 30.60. FTIRνmax (cm–1): 3063.9, 2989.3, 2944.6, 2836.5, 1710.8, 1297.1. ESI/HRMS (m/z): Calculated for [C25H23FN2O3][M + H]+: 419.17; Found m/z: 419.1801 [M + H]+. ESI/MS (m/z): Calculated for [C25H23FN2O3][M + H]+: 419.17. Found m/z: 419.0 [M + H]+. LCMS purity = 99.539%.
4.1.8.4. Synthesis of 2-(4-Fluorobenzyl)-1-(2-methoxyphenethyl)-1H-benzo[d]imidazole-5-carboxylic acid (TJ04)
Following the general procedure E, the compound TJ04 was obtained from compound TJ03 (190 mg, 0.45 mmol, 1.0 equiv) using a mixture of tetrahydrofuran:methanol (2 mL, 10 v, 1:1 V/V) as solvent and lithium hydroxide monohydrate (25 mg, 0.595 mmol, 1.3 equiv) in water (1 mL) at 0 °C to rt for 2 h. The pure compound TJ04 was obtained as a white solid with 93.9% yield (171 mg). (TLC Rf = 0.41, dichloromethane:methanol 9:1 V/V).
1H NMR (400 MHz, DMSO-d6) δ: 12.59 (s, 1H, −COOH), 8.12 (s, 1H, Ar-H), 7.81–7.83 (d, J = 8.4 Hz, 1H, Ar-H), 7.48–7.50 (d, J = 8.4 Hz, 1H, Ar-H), 7.20–7.27 (m, 3H, Ar-H), 7.13–7.17 (t, J = 8.8 Hz, 2H, Ar-H), 6.91–6.95 (t, J = 6.8 Hz, 2H, Ar-H), 6.79–6.83 (t, J = 7.2 Hz, 1H, Ar-H), 4.31–4.35 (t, J = 7.0 Hz, 2H, −CH2), 4.06 (s, 2H, −CH2), 3.70 (s, 3H, −-OCH3), 2.82–2.85 (t, J = 7.0 Hz, 2H, −CH2). 13C NMR (100 MHz, DMSO-d6) δ: 168.40, 162.76 (d, JC–F = 241 Hz), 157.79, 155.75, 142.25, 138.81, 133.14, 131.20, 131.12, 130.94, 128.86, 125.93, 124.57, 123.73, 120.85, 120.79, 115.88, 115.67, 111.09, 110.27, 55.68, 43.82, 32.39, 30.66. FTIR νmax (cm–1): 3447.8, 3011.7, 2937.1, 1677.3, 1308.3. ESI/HRMS (m/z): Calculated for [C24H21FN2O3][M + H]+: 405.15; Found m/z: 405.1613 [M + H]+. ESI/MS (m/z): Calculated for [C24H21FN2O3][M + H]+: 405.15; Found m/z:405.0 [M + H]+. LCMS purity = 98.938%.
4.1.8.5. Synthesis of 2-(4-Fluorobenzyl)-1-(2-methoxyphenethyl)-1H-benzo[d]imidazole-5-carboxamide (TJ05)
Following the general procedure F, the compound TJ05 was obtained from compound TJ04 (110 mg, 0.271 mmol, 1.0 equiv) in DMF (1.5 mL) as solvent, CDI (67 mg, 0.413 mmol, 1.5 equiv), and aqueous ammonia (∼25% solution, 0.4 mL, 16 equiv) at 0 °C to rt for 7 h. The compound TJ05 was obtained as a white solid with 86.7% yield (95 mg) after purification (TLC Rf = 0.47, dichloromethane:methanol 9:1 V/V).
1H NMR (400 MHz, DMSO-d6) δ: 8.14 (s, 1H, Ar-H), 7.92 (s, 1H, −NH2), 7.76–7.78 (t, J = 2.2 Hz, 1H, Ar-H), 7.46–7.48 (d, J = 8.0 Hz, 1H, Ar-H), 7.13–7.27 (m, 5H, Ar-H), 6.91–6.97 (m, 2H, Ar-H), 6.80–6.84 (t, J = 7.2 Hz, 1H, Ar-H), 4.31–4.34 (t, J = 7.2 Hz, 2H, −CH2), 4.05 (s, 2H, −CH2), 3.73 (s, 3H, −OCH3), 2.83–2.86 (t, J = 7.2 Hz, 2H, −CH2). 13C NMR (100 MHz, DMSO-d6) δ: 168.85, 162.69 (d, JC–F = 241 Hz), 157.74, 155.16, 142.17, 137.59, 131.12, 131.05, 130.90, 128.80, 128.20, 125.96, 122.23, 120.81, 118.68, 115.80, 115.59, 111.08, 109.98, 55.68, 43.68, 32.35, 30.68. FTIR νmax (cm–1): 3309.9, 3134.7, 2978.1, 2836.5, 1677.3, 1244.9. ESI/HRMS (m/z): Calculated for [C24H22FN3O2][M + H]+: 404.17. Found m/z: 404.1691 [M + H]+. ESI/MS (m/z): Calculated for [C24H22FN3O2][M + H]+: 404.17. Found m/z: 404.0 [M + H]+. LCMS purity = 99.876%.
4.1.8.6. Synthesis of 2-(4-Methoxy-3-nitrophenyl)-1-(2-methoxyphenethyl)-1H-benzo[d]imidazole-5-carboxamide (TJ06)
Following the general procedure F, the compound TJ06 was obtained from compound TJ02 (90 mg, 0.201 mmol, 1.0 equiv) in DMF (1.2 mL) as a solvent, CDI (49 mg, 0.302 mmol, 1.5 equiv), and aqueous ammonia (∼25% solution, 0.3 mL, 21 equiv) at 0 °C to rt for 7 h. TJ06 was obtained as a white solid with 90.2% yield (81 mg). (TLC Rf = 0.47, dichloromethane: methanol 9:1 V/V).
1H NMR (400 MHz, DMSO-d6) δ: 8.23 (s, 1H, Ar-H), 8.01 (s, 1H, −NH2); 7.88–7.90 (d, J = 8.4 Hz, 1H, −Ar-H), 7.72–7.80 (m, 2H, Ar-H), 7.43–7.46 (d, J = 8.4 Hz, 1H, Ar-H), 7.29 (s, 1H, Ar-H), 7.04–7.07 (t, J = 7.4 Hz,1H, Ar-H), 6.61–6.67 (m, 2H, Ar-H), 6.52–6.54 (d, J = 7.2 Hz,1H, −Ar-H), 4.53–4.56 (t, J = 6.8 Hz, 2H, −CH2), 4.01 (s, 3H, −OCH3), 3.46 (s, 3H, −OCH3), 2.89–2.92 (t, J = 6.2 Hz, 2H, −CH2). 13C NMR (100 MHz, DMSO-d6) δ: 168.89, 157.28, 153.40, 153.21, 142.54, 139.40, 138.98, 135.12, 130.58, 128.69, 126.01, 125.70, 125.34, 124.39, 122.48, 121.37, 120.53, 114.81, 111.28, 110.57, 57.55, 55.28, 44.47, 30.65. FTIR νmax (cm–1): 3451.5, 3332.2, 3283.8, 3179.4, 2944.6, 1669.8, 1244.9. ESI/HRMS (m/z): Calculated for [C24H22N4O5] [M + H]+: 447.16. Found m/z: 447.1580 [M + H]+. ESI/MS (m/z): Calculated for [C24H22N4O5] [M + H]+: 447.16. Found m/z: 447.0 [M + H]+. LCMS Purity = 98.866%.
4.1.8.7. Synthesis of Methyl 2-(4-Fluoro-3-nitrophenyl)-1-(4-methoxybenzyl)-1H-benzo[d]imidazole-5-carboxylate (TJ07)
Following the general procedure D, the compound TJ07 was obtained from intermediate compound 5d (325 mg, 0.716 mmol, 1.0 equiv) using glacial acetic acid (4 mL) and stirred at 100 °C for 3 h. The compound TJ07 was obtained as a pale yellow solid with 73.7% yield (230 mg). (TLC Rf = 0.25, petroleum ether:ethyl acetate 6:4 V/V).
1H NMR (300 MHz, DMSO-d6) δ: 8.47–8.49 (t, J = 3.6 Hz, 1H, Ar-H), 8.47 (s, 1H, Ar-H), 8.18–8.19 (d, J = 4.5 Hz, 1H, Ar-H), 7.91–7.94 (d, J = 8.7 Hz, 1H, Ar-H), 7.69–7.75 (t, J = 9.45 Hz, 2H, Ar-H), 6.94–6.97(d, J = 8.7 Hz, 2H, Ar-H), 6.82–6.85 (t, J = 8.4 Hz, 2H, Ar-H), 5.60 (s, 2H, −CH2), 3.88 (s, 3H, −OCH3), 3.68 (s, 3H, -OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 167.01, 159.18, 157.23 (d, JC–F = 264 Hz), 152.71, 142.46, 139.77, 137.52, 137.44, 137.37, 137.27, 128.53 (d, J = 35 Hz), 127.47(d, J = 32 Hz), 127.46, 127.19, 124.65, 121.62, 119.96, 119.75. 114.72, 112.08, 55.52, 52.59, 47.80. FTIR νmax (cm–1): 3082.5, 3004.2, 2832.8, 1714.6, 1297.1, 1215.1. ESI/HRMS (m/z): Calculated for [C23H18FN3O5] [M + H]+: 436.12. Found m/z: 436.1216 [M + H]+. ESI/MS (m/z): Calculated for [C23H18FN3O5] [M + H]+: 436.12. Found m/z: 436.0 [M + H]+. LCMS purity = 93.457%.
4.1.8.8. Synthesis of Methyl 1-Benzyl-2-(4-fluoro-3-nitrophenyl)-1H-benzo[d]imidazole-5-carboxylate (TJ08)
Following the general procedure D, the final compound TJ08 was obtained from intermediate compound 5g (310 mg, 0.732 mmol, 1.0 equiv) using glacial acetic acid (4 mL), stirred at 100 °C for 3 h. TJ08 was obtained as a pale-yellow solid with 75.8% yield (225 mg) after purification of crude product by flash column chromatography using 25–33% ethyl acetate in petroleum ether as a mobile phase. (TLC Rf = 0.31, petroleum ether: ethyl acetate 6:4 V/V).
1H NMR (400 MHz, DMSO-d6) δ: 8.45–8.48 (m, 1H, Ar-H), 8.363–8.365 (d, J = 0.8 Hz, 1H, Ar- H), 8.16–8.20 (m, 1H, Ar-H), 7.92–7.95 (m, 1H, Ar-H), 7.75–7.78 (m, 1H, Ar-H), 7.69–7.71 (d, J = 8.4 Hz,1H, Ar-H), 7.25–7.32 (m, 3H, Ar-H), 7.01–7.03 (d, J = 6.8 Hz, 2H, Ar-H), 5.70 (s, 2H, −CH2), 3.89 (s, 3H, −OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 167.00, 157.23 (d, JC–F = 264 Hz), 152.75, 142.45, 139.86, 137.48 (d, JC–F = 25 Hz), 137.40, 137.33, 136.72, 129.37, 128.20, 127.39, 127.37, 127.11, 127.07, 126.69, 124.74, 124.71, 121.66, 119.95 (d, JC–F = 21 Hz), 111.99, 52.57, 48.28. FTIR νmax (cm–1): 3029.5, 2952.8, 2844.0, 1716.68, 1545.95, 1285.01. ESI/HRMS (m/z): Calculated for [C22H16FN3O4] [M + H]+: 406.11. Found m/z: 406.1218 [M + H]+. ESI/MS (m/z): Calculated for [C22H16FN3O4] [M + H]+: 406.11. Found m/z: 406.0 [M + H]+. LCMS purity = 98.344%.
4.1.8.9. Synthesis of 2-(4-Hydroxy-3-nitrophenyl)-1-(4-methoxybenzyl)-1H-benzo[d]imidazole-5-carboxylic Acid (TJ09)
Following the general procedure E, the compound TJ09 was obtained from compound TJ07 (100 mg, 0.229 mmol, 1.0 equiv) using THF (1 mL) as a solvent and a solution of lithium hydroxide monohydrate (31 mg, 0.738 mmol, 3.2 equiv) in water (1 mL) at 60 °C and stirred for 8 h. The pure TJ09 was obtained as a pale-yellow solid with 57.2% yield (55 mg) after purification of crude TJ09 by flash column chromatography using 1–2% methanol in dichloromethane as a mobile phase. (TLC Rf = 0.21, dichloromethane:methanol 9:1 V/V).
1H NMR (400 MHz, DMSO-d6) δ: 12.81 (s, 1H, −COOH), 11.65 (s, 1H, −OH); 8.25–8.28 (d, J = 15.2 Hz, 2H, Ar- H), 7.87–7.95 (m, 2H, Ar-H), 7.61–7.64 (d, J = 8.8 Hz, 1H, Ar-H), 7.26–7.28 (d, J = 8.4 Hz, 1H, Ar-H), 6.95–6.97 (d, J = 8.4 Hz, 2H, Ar-H), 6.84–6.86 (d, J = 8.0 Hz, 2H, Ar-H), 5.58 (s, 2H, −CH2), 3.68 (s, 3H, −OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 168.14, 159.07, 153.77, 153.53, 142.49, 139.58, 137.35, 135.77, 128.70, 128.00, 126.47, 125.54, 124.48, 121.37, 120.87, 120.10, 114.67, 111.50, 55.48, 47.70. FTIR νmax (cm–1): 3092.3, 3068.4, 2964.51, 1629.42, 1257.23. ESI/HRMS (m/z): Calculated for [C22H17N3O6] [M + H]+: 420.11; Found m/z: 420.1067 [M + H]+. ESI/MS (m/z): Calculated for [C22H17N3O6][M + H]+: 420.11; Found m/z: 420.2 [M + H]+. LCMS purity = 87.992%.
4.1.8.10. Synthesis of 2-(4-Methoxy-3-nitrophenyl)-1-(4-methoxybenzyl)-1H-benzo[d]imidazole-5-carboxylic Acid (TJ10)
Following the general procedure D, the intermediate compound 5e1 was obtained from compound 5e (185 mg, 0.397 mmol, 1.0 equiv) using glacial acetic acid (3 mL) at 100 °C for 3 h as a pale-brown solid with 87.2% yield (155 mg). The obtained 5e1 was converted to TJ10 by following the general procedure E using the precursor ester compound 5e1 (155 mg, 0.346 mmol, 1.0 equiv) in a mixture of tetrahydrofuran:methanol (3 mL, 20 V, 1:1 V/V) as a solvent and a solution of lithium hydroxide monohydrate (16 mg, 0.381 mmol, 1.1 equiv) in water (1 mL) at 0 °C to rt for 2 h. The crude pale-brown solid was further purified by flash column chromatography using 2–3% methanol in dichloromethane mobile phase, and pure compound TJ10 was obtained as a yellow solid with 88.0% yield (132 mg). (TLC Rf = 0.30, dichloromethane:methanol 9:1 V/V).
1H NMR (400 MHz, DMSO-d6) δ: 12.80 (s, 1H, −COOH), 8.26–8.28 (d, J = 9.6 Hz, 2H, Ar-H), 8.02–8.04 (d, J = 9.2 Hz, 1H, Ar-H), 7.81–7.84 (d, J = 8.4 Hz, 1H, Ar-H), 7.52–7.59 (m, 2H, Ar-H), 6.93–6.95 (d, J = 8.0 Hz, 2H, Ar-H), 6.83–6.85 (d, J = 8.4 Hz, 2H, Ar-H), 5.59 (s, 2H, −CH2), 4.01 (s, 3H, -OCH3), 3.68 (s, 3H, -OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 168.12, 159.08, 153.54, 153.22, 142.48, 139.56, 137.35, 135.72, 135.24, 128.66, 127.98, 126.17, 125.62, 124.58, 122.23, 121.44, 115.35, 114.68, 111.48, 57.49, 55.48, 47.70. 27.25. FTIR νmax (cm–1): 3075.2, 2938.46, 2838.5, 1686.27, 1247.17. ESI/HRMS (m/z): Calculated for [C23H19N3O6] [M + H]+: 434.13. Found m/z: 434.1151 [M + H]+. ESI/MS (m/z): Calculated for [C23H19N3O6][M + H]+: 434.13. Found m/z: 434.2 [M + H]+. LCMS purity = 99.309%.
4.1.8.11. Synthesis of 1-Benzyl-2-(4-hydroxy-3-nitrophenyl)-1H-benzo[d]imidazole-5-carboxylic Acid (TJ11)
Following the general procedure E, the compound TJ11 was obtained from compound TJ08 (100 mg, 0.246 mmol, 1.0 equiv) using THF (1 mL) as a solvent and a solution of lithium hydroxide monohydrate (30 mg, 0.714 mmol, 2.9 equiv) in water (1 mL) at 60 °C and stirred for 8 h. The obtained crude TJ11 was further purified by flash column chromatography using 50–70% ethyl acetate in dichloromethane, and pure TJ11 was obtained as a pale-yellow solid with 65.7% (63 mg). (TLC Rf = 0.32, dichloromethane:methanol 9:1 V/V).
1H NMR (300 MHz, DMSO-d6) δ: 12.80 (s, 1H, −COOH), 11.62 (s, 1H, OH), 8.30 (s, 1H, Ar-H), 8.22 (s, 1H, Ar- H), 7.86–7.94 (m, 2H, Ar-H), 7.60–7.63 (d, J = 8.4 Hz, 1H, Ar-H), 7.25–7.30 (t, J = 8.4 Hz, 4H, Ar-H), 7.00–7.03 (d, J = 6.6 Hz, 2H, −CH2), 5.66 (s, 2H, −CH2). 13C NMR (100 MHz, DMSO-d6) δ: 168.17, 153.80, 153.60, 142.52, 139.70, 137.36, 136.94, 135.80, 129.35, 128.89, 128.11, 127.12, 126.60, 126.45, 125.66, 124.59, 121.44, 120.86, 120.14, 111.48, 48.22. FTIR νmax (cm–1): 3091.7, 3064.9, 3029.8, 2934.32, 1661.64, 1214.55. ESI/HRMS (m/z): Calculated for [C21H15N3O5] [M + H]+: 390.10. Found m/z: 390.1065 [M + H]+. ESI/MS (m/z): Calculated for [C21H15N3O5] [M + H]+: 390.10. Found m/z: 390.1 [M + H]+. LCMS purity = 88.982%.
4.1.8.12. Synthesis of 2-(4-Methoxy-3-nitrophenyl)-1-(4-methoxybenzyl)-1H-benzo[d]imidazole-5-carboxamide (TJ12)
Following the general procedure F, the final compound TJ12 was obtained from TJ10 (70 mg, 0.161 mmol, 1.0 equiv) using DMF (1.0 mL) as a solvent, CDI (40 mg, 0.246 mmol, 1.5 equiv), and aqueous ammonia (∼25% solution, 0.2 mL, 18 equiv) at 0 °C to rt and stirred for 7 h. The compound TJ12 was obtained as a pale-yellow solid with 77.5% yield (54 mg). (TLC Rf = 0.52, dichloromethane:methanol 9:1 V/V).
1H NMR (400 MHz, DMSO-d6) δ: 8.27–8.28 (d, J = 3.2 Hz, 2H, -NH2), 7.99–8.03 (t, J = 8.2 Hz, 2H, −Ar-H), 7.81–7.84 (d, J = 15.6 Hz, 1H, Ar-H), 7.57–7.59 (d, J = 8.4 Hz, 1H, Ar-H), 7.51–7.53 (d, J = 8.8 Hz, 1H, Ar-H), 7.29 (s, 1H, Ar- H), 6.93–6.95 (d, J = 8.0 Hz, 2H, Ar-H), 6.82–6.84 (d, J = 8.0 Hz, 2H, Ar-H), 5.57 (s, 2H, −CH2), 4.00 (s, 3H, −OCH3), 3.67 (s, 3H, −OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 168.66, 159.11, 153.49, 152.79, 142.50, 139.60, 138.50, 135.24, 129.33, 128.85, 128.02, 126.13, 123.22, 122.49, 119.37, 115.38, 114.70, 111.19, 57.52, 55.51, 47.66. FTIR νmax (cm–1): 3452.96, 3185.60, 2949.51, 1669.33, 1247.30. ESI/HRMS (m/z): Calculated for [C23H20N4O5] [M + H]+: 433.14. Found m/z: 433.1477 [M + H]+. ESI/MS (m/z): Calculated for [C23H20N4O5] [M + H]+: 433.14. Found m/z: 433.2 [M + H]+. LCMS purity = 92.826%.
4.1.8.13. Synthesis of Methyl 1-(4-methoxybenzyl)-2-(4-(trifluoromethoxy) phenyl)-1H-benzo[d]imidazole-5-carboxylate (TJ13)
Following the general procedure D, the compound TJ13 was obtained from intermediate compound 5f (280 mg, 0.590 mmol, 1.0 equiv) using glacial acetic acid (3 mL) and stirred at 100 °C for 3 h. TJ13 was obtained as a pale-brown solid with 81.6% yield (220 mg). (TLC Rf = 0.25, petroleum ether: ethyl acetate 5:5 V/V).
1H NMR (400 MHz, DMSO-d6) δ: 8.33 (s, 1H, Ar-H), 7.90–7.92 (d, J = 6.4 Hz, 1H, Ar-H), 7.72–7.77 (m, 2H, Ar-H), 7.66–7.68 (d, J = 8.4 Hz, 1H, Ar-H), 7.57–7.63 (m, 2H, Ar- H), 6.86–6.88 (d, J = 8.3 Hz, 2H, Ar- H), 6.77–6.79 (d, J = 7.6 Hz, 2H, Ar- H), 5.33 (s, 2H, −CH2), 3.88 (s, 3H, −OCH3), 3.67 (s, 3H, −OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.46, 164.56, 158.72, 147.12, 145.41, 132.30, 131.35, 131.11, 130.66, 129.25, 128.71, 128.17, 128.01, 122.34, 122.14, 116.46, 114.45, 114.23, 110.75, 55.47, 51.87, 45.84. FTIR νmax (cm–1): 2960.23, 1649.21, 1245.78, 1176.00. ESI/HRMS (m/z): Calculated for [C24H19F3N2O4] [M + H]+: 457.13. Found m/z: 457.1238 [M + H]+. ESI/MS (m/z): Calculated for [C24H19F3N2O4] [M + H]+: 457.13. Found m/z: 457.1 [M + H]+. LCMS purity = 97.681%.
4.1.8.14. Synthesis of 1-(4-Methoxybenzyl)-2-(4-(trifluoromethoxy) phenyl)-1H-benzo[d]imidazole-5-carboxylic Acid (TJ14)
Following the general procedure E, the compound TJ14 was obtained from TJ13 (170 mg, 0.372 mmol, 1.0 equiv) using a mixture of tetrahydrofuran:methanol (2 mL, 12 V, 1:1 V/V) as solvent and a solution of lithium hydroxide monohydrate (21 mg, 0.500 mmol, 1.3 equiv) in water (1 mL) at 0 °C to rt for 2 h. The pure TJ14 obtained as a white solid with 89.7% yield (148 mg). (TLC Rf = 0.39, dichloromethane:methanol 9:1 V/V).
1H NMR (300 MHz, DMSO-d6) δ: 12.73 (s, 1H, −COOH), 8.30 (s, 1H, Ar-H), 7.88–7.90 (d, J = 8.4 Hz, 1H, Ar-H), 7.70–7.79 (m, 2H, Ar-H), 7.56–7.64 (m, 3H, Ar-H), 6.85–6.88 (d, J = 8.7 Hz, 2H, Ar- H), 6.76–6.79 (d, J = 8.4 Hz, 2H, Ar- H), 5.32 (s, 2H, −CH2), 3.66 (s, 3H, −OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 168.14, 159.16, 151.07, 146.84, 146.83, 142.81, 138.41, 133.09, 133.06, 128.47, 128.21, 125.53, 124.57, 124.16 (q, JC–F = 257 Hz, 257 Hz, 256 Hz), 123.84, 121.74, 121.59, 121.46, 119.02, 116.46, 114.45, 111.79, 55.49, 47.44. FTIR νmax(cm–1): 3093.7, 2959.5, 2937.1, 2840.2, 1692.2, 1244.9. ESI/HRMS (m/z): Calculated for [C23H17F3N2O4] [M + H]+: 443.11. Found m/z: 443.1077 [M + H]+. ESI/MS (m/z): Calculated for [C23H17F3N2O4] [M + H]+: 443.11. Found m/z: 443.2 [M + H]+. LCMS purity = 97.377%.
4.1.8.15. Synthesis of 1-(4-Methoxybenzyl)-2-(4-(trifluoromethoxy) phenyl)-1H-benzo[d]imidazole-5-carboxamide (TJ15)
Following the general procedure F, the compound TJ15 was obtained from TJ14 (80 mg, 0.180 mmol, 1.0 equiv) using DMF (1.0 mL) as a solvent, CDI (44 mg, 0.271 mmol, 1.5 equiv), and aqueous ammonia (∼25% solution, 0.2 mL, 16 equiv) at 0 °C to rt and stirred for 7 h. Pure TJ15 compound was obtained as an off-white solid with 78.7% yield (63 mg) after purification. (TLC Rf = 0.47, dichloromethane:methanol 9:1 V/V).
1H NMR (300 MHz, DMSO-d6) δ: 8.29 (s, 1H, Ar-H), 7.99 (s, 1H, −NH), 7.69–7.83 (m, 3H, Ar-H), 7.59–7.62 (d, J = 9 Hz, 3H, Ar-H), 7.29 (s, 1H, −NH), 6.85–6.88 (d, J = 8.7 Hz, 2H, Ar-H), 6.76–6.79 (d, J = 8.7 Hz, 2H, Ar-H), 5.30 (s, 2H, −CH2), 3.66 (s, 3H, −OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 168.67, 159.13, 150.56, 146.85, 142.75, 137.29, 133.09, 132.95, 129.16, 128.45, 128.42, 128.22, 124.17 (q, JC–F = 257 Hz, 256 Hz, 257 Hz), 124.07, 123.19, 121.60, 121.53, 119.57, 119.04, 116.47, 114.43, 111.39, 55.49, 47.36. FTIR νmax (cm–1): 3567.1, 3306.1, 3134.7, 2952.1, 2832.8, 1613.9, 1162.9. ESI/HRMS (m/z): Calculated for [C23H18F3N3O3] [M + H]+: 442.13. Found m/z: 442.1346 [M + H]+. ESI/MS (m/z): Calculated for [C23H18F3N3O3] [M + H]+: 442.13. Found m/z: 442.0 [M + H]+. LCMS purity = 96.380%.
4.2. Biological Activities
4.2.1. Cell Lines and Culture Conditions
Human chronic myeloid leukemia cells, K-562; human leukemic T cell lymphoblast cell lines, Jurkat; human acute lymphoblastic leukemia cells, MOLT-4; human colorectal carcinoma cells, HCT116; and human pancreatic adenocarcinoma cells, MIA PaCa-2 were purchased from the National Centre for Cell Sciences (NCCS), Pune, India. Human cervical cancer cell lines, HeLa, and human normal kidney cell lines, HEK-293 were generously gifted by Prof. Sathees C. Raghavan, Dept of Biochemistry, Indian Institute of Sciences (IISc), Bengaluru. Cells were cultured in RPMI 1640, McCoy’s 5A, DMEM low glucose media, and DMEM high glucose media, respectively, with 2 mM l-glutamine (Thermo Fisher Scientific, Inc.; Waltham, MA USA) containing 10% FBS (Gibco; Grand Island, NY, USA) and some are supplemented with 2 mM Sodium pyruvate (Thermo Fisher Scientific, Inc.; Waltham, MA USA). Cells were cultured in humidified Incubator with 5% CO2 at 37 °C.
4.2.2. MTT Cell Proliferation Assay
MTT assay was performed as described previously.40 Cells treated with an increased concentration of TJ08 were subjected to MTT assay after 48 and 72 h of incubation with addition of thiazolyl blue tetrazolium bromide (MTT; Sigma; St Louis, MO, USA) and incubated additionally for 4 h. Insoluble formazan crystals were solubilized using 100 μL of DMSO, and absorbance was recorded at 570 nm using a Tecan Microplate Reader (Tecan Instruments, Switzerland). Error bars were calculated based on a minimum of three independent experiments and data represented as histogram.
4.2.3. Trypan Blue Dye Exclusion Assay
TBDE assay was performed as described. Cells treated with an increased concentration of TJ08 were incubated for 48 and 72 h, and cells were harvested and subjected to trypan blue dye exclusion assay. Error bars were calculated based on a minimum of three independent experiments and data represented as histogram.
4.2.4. Colony Formation Assay
CFA was performed as described. HeLa, HCT116, and MiaPaca2 cells were used to assess the colony formation capability after TJ08 treatment. Cells were incubated for 2 weeks and allowed to form colonies and cells were washed with PBS, fixed in methanol:acetic acid (7:1) and stained with 0.5% crystal violet stain and allowed to dry and imaged.
4.2.5. Cell Migration and Cell Invasion Assay
4.2.5.1. Cell Migration
HeLa cells were seeded at 5 × 104 per 96 well plate, allowed to attach overnight, treated with increased concentration of TJ08, incubated for 12 h, and subjected for migration assay according to the manufacturer’s protocol (ECM510, Merck, Milipore).
4.2.5.2. Cell Invasion
HeLa cells were serum starved for 18 h, cells were seeded into a 96-well plate and incubated with increased concentration of TJ08 for 12 h, and the lower chamber was supplemented with and without FBS (Chemo attractant) and incubated for 8 h and then subjected for cell invasion assay according to the manufacturer’s protocol (ECM555, Merck, Milipore).
4.2.6. Mitochondrial Membrane Potential Assay
HeLa, Jurkat, and HCT116 cells were used for the assay, and the assay was performed as described previously.40 The treated cells were allowed to incubate for 24 h and subjected for MMP assay as per th emanufacturer’s protocol (MAK159, Sigma-Aldrich). Spectrophotometric absorbance of the plate was recorded at (λex = 490/λem = 525 nm) and (λex = 540/λem = 590 nm) for ratio analysis using Tecan Microplate Reader (Tecan Instruments, Switzerland). Cells were centrifuged at 800 rpm and imaged using an Olympus CKX53 inverted fluorescence microscope.
4.2.7. Annexin V-FITC/PI Staining
Jurkat cells were seeded at 1 × 105 cells/mL in 6-well dishes and allowed to grow overnight. Cells were treated with different concentrations of TJ08 10 and 20 μM and allowed to incubate for 48 h. Cells were harvested, washed with cold PBS, and subjected for Annexin V FITC/PI double staining assay. Samples were resuspended in 1× binding buffer and incubated with Annexin V FITC and PI according to the manufacturer’s protocol (Roche) and imaged using Olympus CKX53 inverted fluorescence microscope.
4.2.8. Cell Cycle Analysis
Cell cycle analysis by flow cytometer was performed as described previously. Synchronized cells by serum starvation were treated with different concentrations of TJ08, incubated for 24 h, harvested, fixed with 70% ethanol, subjected to propidium iodide stating, and analyzed using flow cytometer Beckman cell counter at an excitation with 488 nm laser and emission at 560/570 nm.
4.2.9. Western Blotting
Western blot was performed as described previously. Proteins were separated in different percentage gels and subjected to Western blotting to identify expressed proteins. Blots were blocked and treated with primary and secondary antibodies to detect pro-apoptotic, anti-apoptotic and loading control proteins. HRP-conjugated secondary antibodies were developed by using chemiluminescent solution and scanned using chemi doc (UViTech, UK).
4.2.10. Statistical Analysis
Data was analyzed by GraphPad prism software (GraphPad Software Inc., San Diego, CA, USA) for elucidating IC50 of synthesized small molecules. Each experiment was repeated a minimum of 3 times. Error bars indicate the SEM and P values and were calculated by comparing the mean of control group with the mean of TJ08-treated group p < 0.05.
Acknowledgments
We thank Vision Group on Science and Technology (VGST), Government of Karnataka, for financial support under the research project-SMYSR (No. VGST/SMYSR/GRD-426/2014-15). We thank Prof. Sathees C. Raghavan, IISc, Bangalore, for gifting HeLa and HEK cell lines. This work was supported by the Adichunchanagiri University internal funding scheme. We thank Prof. Dr. Chandrashekar Shetty and Dr. M. G. Shivaramu for their kind support.
Glossary
Abbreviations
- CDI
carbonyldiimidazole
- THF
tetrahydrofuran
- DIPEA
N,N-diisopropylethylamine
- DMSO
dimethyl sulfoxide-d6
- Rf
retention factor
- rt
room temperature
- V
volume
- V/V
volume/volume
- W/W
weight/weight
- equiv
equivalent
- h
hours
- ESI
electron spray ionization
- HRMS
high resolution mass spectrometry
- LCMS
Liquid chromatography-mass spectrometry
- MHz
megahertz
- NMR
nuclear magnetic resonance
- FTIR
Fourier transform infrared
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c06057.
1H NMR spectra of intermediates and final compounds 2, 3a, 3b, 3c, 4a, 4b, 4c, and TJ01–TJ15; 13C NMR spectra of the final compounds TJ01–TJ15; HRMS spectra of the final compounds TJ01–TJ15; LC–MS spectra of intermediates and final compounds 2, 3a, 3b, 3c, 4a, 4b, 4c, and TJ01–TJ15; FT-IR spectra of the final compounds TJ01–TJ15 (PDF)
Author Contributions
# J.G.S. and S.M.S. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Yadav S.; Narasimhan B. Perspectives of benzimidazole derivatives as anticancer agents in the new era. Anti-Cancer Agents Med. Chem. 2016, 16, 1403–1425. 10.2174/1871520616666151103113412. [DOI] [PubMed] [Google Scholar]
- Shrivastava N.; Naim M.-J.; Alam M.-J.; Nawaz F.; Ahmed S.; Alam O. Benzimidazole scaffold as anticancer agent: synthetic approaches and structure–activity relationship. Arch. Pharm. 2017, 350, e201700040 10.1002/ardp.201700040. [DOI] [PubMed] [Google Scholar]
- Barot K.-P.; Nikolova S.; Ivanov I. D.; Ghate M. Novel research strategies of benzimidazole derivatives. Mini-Rev. Med. Chem. 2013, 13, 1421–1447. 10.2174/13895575113139990072. [DOI] [PubMed] [Google Scholar]
- Akhtar M.-J.; Yar M.-S.; Sharma V.-K.; Khan A.-A.; Ali Z.; Haider M.-D.; Pathak A. Recent progress of Benzimidazole hybrids for anticancer potential. Curr. Med. Chem. 2020, 27, 5970–6014. 10.2174/0929867326666190808122929. [DOI] [PubMed] [Google Scholar]
- Murray J.-M.; Sweeney Z.-K.; Chan B.-K.; Balazs M.; Bradley E.; Castanedo G.; Sutherlin D.-P. Potent and highly selective benzimidazole inhibitors of PI3-kinase delta. J. Med. Chem. 2012, 55, 7686–7695. 10.1021/jm300717c. [DOI] [PubMed] [Google Scholar]
- El Rashedy A.-A.; Aboul-Enein H.-Y. Benzimidazole derivatives as potential anticancer agents. Mini-Rev. Med. Chem. 2013, 13, 399–407. 10.2174/138955713804999847. [DOI] [PubMed] [Google Scholar]
- Shimomura I.; Yokoi A.; Kohama I.; Kumazaki M.; Tada Y.; Tatsumi K.; Yamamoto Y. Drug library screen reveals benzimidazole derivatives as selective cytotoxic agents for KRAS-mutant lung cancer. Cancer Lett. 2019, 451, 11–22. 10.1016/j.canlet.2019.03.002. [DOI] [PubMed] [Google Scholar]
- Suk F.-M.; Liu C.-L.; Hsu M.-H.; Chuang Y.-T.; Wang J.-P.; Liao Y.-J. Treatment with a new benzimidazole derivative bearing a pyrrolidine side chain overcomes sorafenib resistance in hepatocellular carcinoma. Sci. Rep. 2019, 9, 1–10. 10.1038/s41598-019-53863-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu B.; Liu F.; Li L.; Ding C.; Chen K.; Sun Q.; Jiang Y. A benzimidazole derivative exhibiting antitumor activity blocks EGFR and HER2 activity and upregulates DR5 in breast cancer cells. Cell Death Dis 2015, 6, e1686–e1686. 10.1038/cddis.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y.; Wang Y.; Li G.; Zhang Z.; Ma L.; Cheng B.; Chen J. Discovery of novel benzimidazole and indazole analogues as tubulin polymerization inhibitors with potent anticancer activities. J. Med. Chem. 2021, 64, 4498–4515. 10.1021/acs.jmedchem.0c01837. [DOI] [PubMed] [Google Scholar]
- Kim M.-K.; Shin H.; Park K.-S.; Kim H.; Park J.; Kim K.; Chong Y. Benzimidazole derivatives as potent JAK1-selective inhibitors. J. Med. Chem. 2015, 58, 7596–7602. 10.1021/acs.jmedchem.5b01263. [DOI] [PubMed] [Google Scholar]
- Eldehna W.-M.; El Hassab M.-A.; Abo-Ashour M.-F.; Al-Warhi T.; Elaasser M.-M.; Safwat N.-A.; El-Haggar R. Development of isatin-thiazolo [3, 2-a] benzimidazole hybrids as novel CDK2 inhibitors with potent in vitro apoptotic anti-proliferative activity: Synthesis, biological and molecular dynamics investigations. Bioorg. Chem. 2021, 110, 104748. 10.1016/j.bioorg.2021.104748. [DOI] [PubMed] [Google Scholar]
- El-Hameed R.-H.; Fatahala S.-S.; Sayed A.-I. Synthesis of Some Novel Benzimidazole Derivatives as Anticancer Agent and Evaluation for CDK2 Inhibition Activity. Med. Chem. 2022, 18, 238–248. 10.2174/1573406417666210304100830. [DOI] [PubMed] [Google Scholar]
- Cheong J.-E.; Zaffagni M.; Chung I.; Xu Y.; Wang Y.; Jernigan F.-E.; Sun L. Synthesis and anticancer activity of novel water soluble benzimidazole carbamates. Eur. J. Med. Chem. 2018, 144, 372–385. 10.1016/j.ejmech.2017.11.037. [DOI] [PubMed] [Google Scholar]
- Hegde M.; Kumar K.S.-S.; Thomas E.; Ananda H.; Raghavan S.-C.; Rangappa K.-S. A novel benzimidazole derivative binds to the DNA minor groove and induces apoptosis in leukemic cells. RSC Adv. 2015, 5, 93194–93208. 10.1039/C5RA16605E. [DOI] [Google Scholar]
- Elgawish M.-S.; Nafie M.-S.; Yassen A.-S.; Yamada K.; Ghareb N. The design and synthesis of potent benzimidazole derivatives via scaffold hybridization and evaluating their antiproliferative and proapoptotic activity against breast and lung cancer cell lines. New J. Chem. 2022, 46, 4239–4256. 10.1039/D1NJ05655G. [DOI] [Google Scholar]
- Ali Y.; Abd Hamid S. Insights into the structure and drug design of benzimidazole derivatives targeting the epidermal growth factor receptor (EGFR). Chem. Biol. Drug Des. 2022, 100, 921–934. 10.1111/cbdd.13974. [DOI] [PubMed] [Google Scholar]
- El-Gohary N.-S.; Shaaban M.-I. Synthesis and biological evaluation of a new series of benzimidazole derivatives as antimicrobial, antiquorum-sensing and antitumor agents. Eur. J. Med. Chem. 2017, 131, 255–262. 10.1016/j.ejmech.2017.03.018. [DOI] [PubMed] [Google Scholar]
- Yoon Y.-K.; Ali M.-A.; Wei A.-C.; Choon T.-S.; Osman H.; Parang K.; Shirazi A.-N. Synthesis and evaluation of novel benzimidazole derivatives as sirtuininhibitors with antitumor activities. Bioorg. Med. Chem. 2014, 22, 703–710. 10.1016/j.bmc.2013.12.029. [DOI] [PubMed] [Google Scholar]
- Satija G.; Sharma B.; Madan A.; Iqubal A.; Shaquiquzzaman M.; Akhter M.; Alam M.-M. Benzimidazole based derivatives as anticancer agents: Structure activity relationship analysis for various targets. J. Heterocycl. Chem. 2022, 59, 22–66. 10.1002/jhet.4355. [DOI] [Google Scholar]
- Wang Z.; Deng X.; Xiong S.; Xiong R.; Liu J.; Zou L.; Tang G. Design, synthesis and biological evaluation of chrysin benzimidazole derivatives as potential anticancer agents. Nat. Prod Res. 2018, 32, 2900–2909. 10.1080/14786419.2017.1389940. [DOI] [PubMed] [Google Scholar]
- Bansal Y.; Minhas R.; Singhal A.; Arora R. K.; Bansal G. Benzimidazole: A multifactednucelus for anticancer agents. Curr. Org. Chem. 2021, 25, 669–694. 10.2174/1385272825666210208141107. [DOI] [Google Scholar]
- Hsieh C.-Y.; Ko P. W.; Chang Y.-J.; Kapoor M.; Liang Y.-C.; Chu H.-L.; Hsu M.-H. Design and synthesis of benzimidazole-chalcone derivatives as potential anticancer agents. Molecules. 2019, 24, 3259. 10.3390/molecules24183259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues F.-S.; de Almeida H.-B.; Bortoluzzi A.-J.; Cuin A.; de Almeida E.-T.; de Gois E.-P.; Camargo M.-A. Unexpected synthesis of benzimidazole from Schiff base mediated by lanthanide chloride and citotoxic activities. J. Mol. Struct. 2022, 1252, 132140. 10.1016/j.molstruc.2021.132140. [DOI] [Google Scholar]
- Hassan A.; Heakal B.-H.; Khamis H.; Hassan G. A.-N.; Marzouk E.; Abdelmoaz M.-A.; Younis A. Design, Synthesis, DFT Studies and Anticancer Activity of Novel Metal Complexes Containing 1, 3, 5-triazino [1, 2-a] benzimidazole Moiety Using Microwave as an Approach for Green Chemistry. Egypt. J. Chem. 2020, 64, 323–340. 10.21608/ejchem.2020.29618.2639. [DOI] [Google Scholar]
- Singla P.; Luxami V.; Paul K. Benzimidazole-biologically attractive scaffold for protein kinase inhibitors. RSC Adv. 2014, 4, 12422–12440. 10.1039/c3ra46304d. [DOI] [Google Scholar]
- Feng L.-S.; Su W. Q.; Cheng J.-B.; Xiao T.; Li H.-Z.; Chen D.-A.; Zhang Z.-L. Benzimidazole hybrids as anticancer drugs: An updated review on anticancer properties, structure–activity relationship, and mechanisms of action (2019–2021). Arch Pharm. 2022, 355, e2200051 10.1002/ardp.202200051. [DOI] [PubMed] [Google Scholar]
- Kwegyir-Afful A.-K.; Ramalingam S.; Ramamurthy V.-P.; Purushottamachar P.; Murigi F.-N.; Vasaitis T.-S.; Njar V.-C. Galeterone and the next generation galeterone analogs, VNPP414 and VNPP433–3β exert potent therapeutic effects in castration-/drug-resistant prostate cancer preclinical models in vitro and in vivo. Cancers. 2019, 11, 1637. 10.3390/cancers11111637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija G.; Sharma B.; Madan A.; Iqubal A.; Shaquiquzzaman M.; Akhter M.; Alam M.-M. Benzimidazole based derivatives as anticancer agents: Structure activity relationship analysis for various targets. J. Heterocycl. Chem. 2022, 59, 22–66. 10.1002/jhet.4355. [DOI] [Google Scholar]
- Sontakke V.-A.; Kate A.-N.; Ghosh S.; More P.; Gonnade R.; Kumbhar N. M.; Shinde V.-S. Synthesis, DNA interaction and anticancer activity of 2-anthryl substituted benzimidazole derivatives. New J. Chem. 2015, 39, 4882–4890. 10.1039/C4NJ02415J. [DOI] [Google Scholar]
- Tahlan S.; Kumar S.; Kakkar S.; Narasimhan B. Benzimidazole scaffolds as promising antiproliferative agents. BMC Chem. 2019, 13, 1–16. 10.1186/s13065-019-0579-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goud N. S.; Kumar P.; Bharath R. D.. Recent developments of target-based benzimidazole derivatives as potential anticancer agents. Heterocycles-Synthesis and Biological Activities; Intech Open: London, 2020; 10.5772/intechopen.90758. [DOI] [PubMed] [Google Scholar]
- Sivaramakarthikeyan R.; Iniyaval S.; Saravanan V.; Lim W.-M.; Mai C.-W.; Ramalingan C. Molecular hybrids integrated with benzimidazole and pyrazole structural motifs: design, synthesis, biological evaluation, and molecular docking studies. ACS omega. 2020, 5, 10089–10098. 10.1021/acsomega.0c00630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheson B.-D.; Brugger W.; Damaj G.; Dreyling M.; Kahl B.; Kimby E.; Ogura M.; Weidmann E.; Wendtner C.-M.; Zinzani P.-L. Optimal use of bendamustine in hematologic disorders: Treatment recommendations from an international consensus panel–an update. Leuk. Lymphoma. 2016, 57, 766–782. 10.3109/10428194.2015.1099647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njar V.-C.; Brodie A.-M. Discovery and development of Galeterone (TOK-001 or VN/124–1) for the treatment of all stages of prostate cancer. J. Med. Chem. 2015, 58, 2077–2087. 10.1021/jm501239f. [DOI] [PubMed] [Google Scholar]
- Gowda N.-T.; Kavitha C.-V.; Chiruvella K.-K.; Joy O.; Rangappa K.-S.; Raghavan S.-C. Synthesis and biological evaluation of novel 1-(4-methoxyphenethyl)-1H-benzimidazole-5-carboxylic acid derivatives and their precursors as antileukemic agents. Bioorg. Med. Chem. Lett. 2009, 19, 4594–4600. 10.1016/j.bmcl.2009.06.103. [DOI] [PubMed] [Google Scholar]
- Thimmegowda N.-R.; Swamy S.-N.; Kumar C.-A.; Kumar Y.-S.; Chandrappa S.; Yip G.-W.; Rangappa K.-S. Synthesis, characterization and evaluation of benzimidazole derivative and its precursors as inhibitors of MDA-MB-231 human breast cancer cell proliferation. Bioorg. Med. Chem. Lett. 2008, 18, 432–435. 10.1016/j.bmcl.2007.08.078. [DOI] [PubMed] [Google Scholar]
- Kim S.-O.; Sakchaisri K.; NR T.; Soung N.-K.; Jang J.-H.; Kim Y. S.; Kim B.-Y. STK295900, a dual inhibitor of topoisomerase 1 and 2, induces G2 arrest in the absence of DNA damage. PloS one. 2013, 8, e53908 10.1371/journal.pone.0053908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagadeesha G.-S.; Mantelingu K.; Thimmegowda N.-R.; Rangappa K.-S. Microwave-Assisted, Metal-Free, Chemoselective N-Formylation of Amines using 2-Formyl-1, 3-dimethyl-1H-imidazol-3-ium Iodide and In Situ Synthesis of Benzimidazole and Isocyanides. SynOpen. 2022, 6, 132–140. 10.1055/s-0041-1737605. [DOI] [Google Scholar]
- Shwetha B.; Sudhanva M.-S.; Jagadeesha G.-S.; Thimmegowda N.-R.; Hamse V.-K.; Sridhar B.-T.; Rangappa K.-S. Furan-2-carboxamide derivative, a novel microtubule stabilizing agent induces mitotic arrest and potentiates apoptosis in cancer cells. Bioorg. Chem. 2021, 108, 104586. 10.1016/j.bioorg.2020.104586. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.