

Abstract

Glucose, a common monosaccharide in nature, is dominated by the d-enantiomer. Meanwhile, the discovery of l-glucose-utilizing bacteria and the elucidation of their metabolic pathways 10 years ago suggests that l-glucose exists naturally. Most carbohydrates exist as glycosides rather than monosaccharides; therefore, we expected that nature also contains l-glucosides. Sequence analysis within glycoside hydrolase family 29 led us to identify two α-l-glucosidases, ClAgl29A and ClAgl29B, derived from Cecembia lonarensis LW9. ClAgl29A and ClAgl29B exhibited higher Km, kcat, and kcat/Km values for p-nitrophenyl α-l-glucoside than that for p-nitrophenyl α-l-fucoside. Structural analysis of ClAgl29B in complex with l-glucose showed that these enzymes have an active-site pocket that preferentially binds α-l-glucoside, but excludes α-l-fucoside. These results suggest that ClAgl29A and ClAgl29B evolved to hydrolyze α-l-glucoside, implying the existence of α-l-glucoside in nature. Furthermore, α-l-glucosidic linkages (α-l-Glc-(1 → 3)-l-Glc, α-l-Glc-(1 → 2)-l-Glc, and α-l-Glc-(1 → 6)-l-Glc) were synthesized by the transglucosylation activity of ClAgl29A and ClAgl29B. We believe that this study will lead to new research on α-l-glucosides, including determining the physiological effects on humans, and the discovery of novel α-l-glucoside-related enzymes.
Introduction
Carbohydrates are believed to be homochiral in nature. Primary carbohydrates including glucose, fructose, mannose, and galactose are present as d-sugars. Some l-sugars are also present in nature, including l-fucose, l-rhamnose, and l-arabinose. Both d- and l-sugars are present in nature in some cases. For example, l-arabinose is widespread as a component of polysaccharides in the gum and hemicellulose of plants, whereas d-arabinose is found in Mycobacterium polysaccharides and in Aloe sp. glycoside. The possibility that both enantiomers of glucose exist is suggested. The presence of an l-glucose-utilizing bacterium in soil (Paracoccus laeviglucosivorans)1 may indicate a break in glucose homochirality. However, it is possible that the bacterium only uses a syllo-inositol metabolic enzyme to catabolize l-glucose because l-glucose dehydrogenase, an initial enzyme for l-glucose catabolism, is more specific for syllo-inositol than l-glucose. The authors implicitly mention that l-glucose-utilizing bacteria are not directly related to the presence of l-glucose in nature.1
Carbohydrates often exist as oligosaccharides, polysaccharides, and glycosides through the formation of glycosidic linkages, and l-glucose is possibly present as l-glucoside. If l-glucoside is present, an enzyme could be present to break it down into monosaccharides. Organisms express a wide variety of glycoside hydrolases. Glycoside hydrolases are classified into glycoside hydrolase families (GH) based on their amino acid sequence similarities and are summarized in the carbohydrate-active enzyme database (CAZy, https://www.cazy.org/).2,3 Enzymes belonging to the same GH can be regarded as evolving from a common ancestor, and their structures and catalytic mechanisms are generally conserved. Meanwhile, there are frequent cases of divergent specificities within a GH. We may therefore be able to find enzymes with novel substrate specificities by examining molecular evolution within a GH.
We hypothesized that α-l-glucosidase may exist in GH29, which consists of α-l-fucosidase, α-1,3/1,4-l-fucosidase, α-1,2-l-fucosidase, and α-l-galactosidase. α-l-Glucoside differs from α-l-fucoside (6-deoxy l-galactoside) and α-l-galactoside in the orientation of the hydroxyl group at C4 (OH-4) (Figure 1).
Figure 1.

Structures of α-l-glucoside (1), α-l-fucoside (2), and α-l-galactoside (3).
We thought that if we could find the diversification of OH4 and OH6 recognition mechanisms within GH29, we would be able to find α-l-glucosidase in this family. We should also note the presence of an enzyme showing specificity for both α-l-fucoside and α-l-glucoside, such as GH1 β-glucosidase with high kcat/Km values for β-d-glucoside and β-d-fucoside.4 The catalytic domain of GH29 enzymes displays an atypical TIM (β/α)8-barrel fold and a missing part of the α-helix.5,6 Two His residues at the loop connecting β-strand 1 and α-helix 1 (β → α loop 1) and β → α loop 2 of the catalytic domain are involved in the stabilization of the axial OH-4 of α-l-fucoside through hydrogen bonds. Thermotoga maritima α-l-fucosidase (TmaFuc) is a representative GH29 enzyme with His34 and His128 responsible for its stabilization.5 TmaFuc has Phe32, His34, and Phe290 surrounding the C6 methyl group of l-fucose.
In this study, divergence of the residue corresponding to His34 of TmaFuc led to the discovery of α-l-glucosidases. Determination of the three-dimensional (3D) structure of α-l-glucosidases revealed an elaborate mechanism for their recognition, which is dissimilar to that of α-l-fucosidases. The synthesis of α-l-glucosidic linkages was further examined by exploiting transglycosylation of α-l-glucosidases. This is the first enzymatic synthesis of an α-l-glucosidic linkage. This study illustrates one aspect of the breakdown of homochirality in glucose and contributes to the development of a new field of glycoscience.
Results and Discussion
Candidates for α-l-Glucosidase
GH29 is a family of α-l-fucosidases, but we assumed that the family hides a diversity of recognition mechanisms for C4 and C6. This idea prompted us to search for α-l-glucosidase in GH29. We found two unique sequences encoded by the Cecembia lonarensis LW9 genome: GenBank accession numbers EKB48090.1 and EKB48091.1. GH29 α-l-fucosidases are equipped with His34 and His128 residues in TmaFuc to stabilize the axial OH-4 of α-l-fucoside. These His residues are invariant in α-l-fucosidases but are not completely conserved in GH29 proteins. The conservation of the His128 position was relatively high, whereas several variations (Ala, Arg, Asn, Asp, Cys, Gln, Glu, Ile, Lys, Phe, Ser, Thr, Tyr, and Val) are observed at the His34 position according to the HMM logo7 for Alpha_L_fuc (PF01120) in the Pfam database (https://pfam.xfam.org/family/PF01120). The position of His34 was replaced with Asp114 and Asp103 in EKB48090.1 and EKB48091.1, respectively. We anticipated that these Asp residues are involved in recognizing the equatorial OH-4 of α-l-glucoside. A conserved Asp residue is responsible for stabilizing equatorial OH-4 of α-d-glucoside through a hydrogen bond in GH13 and GH31 α-d-glucosidases.8 There is a proposed evolutionary relationship between GH27 and GH29,5 and between GH27 and GH13 or GH31,9−11 and we wondered if the expected α-l-glucosidase might have a substrate recognition machinery comparable to that of α-d-glucosidase.
EKB48090.1 and EKB48091.1 consist of 586 and 573 amino acid residues, respectively. They were predicted to have a signal peptide (Met1–Gly22) and a lipoprotein signal peptide (Met1–Ser18), respectively, by SignalP-5.0. Pairwise alignment of these proteins showed 56.1% identity and 70.3% similarity and contained a 19.8% gap. Sequences belonging to GH29-A,12 such as α-l-galactosidase from Bacteroides plebeius DSM 17153 (EDY95436.1) and TmaFuc (AE001712.1), were the top hits with similar sequences based on a FASTA search of these sequences using PDB as the target database. EKB48090.1 and EKB48091.1 are encoded in tandem in the genome: B879_03287 and B879_03288, respectively. The neighboring genes of B879_03288 encoded putative carbohydrate-metabolism enzymes, including a putative d-xylose transporter, a GntR family transcriptional regulator, a putative 1,5-d-anhydrofructose reductase, and a d-xylose isomerase-like TIM barrel protein. The putative 1,5-d-anhydrofructose reductase displays sequence similarity (31% identity) to l-glucose/scyllo-inositol dehydrogenase (EC 1.1.1.370), the first enzyme responsible for l-glucose oxidation.1 We chose to study EKB48091.1 (denoted as ClAgl29A) and EKB48090.1 (denoted as ClAgl29B) as potential candidates for α-l-glucosidase based on these sequence features.
Characterization of ClAgl29A and ClAgl29B
Recombinant ClAgl29A and ClAgl29B were produced in Escherichia coli without the signal peptides and their enzymatic properties were analyzed. The N-terminal Cys20 and Ser21 residues of mature ClAgl29B protein were deleted to prevent membrane anchoring. The hydrolytic rate constants were determined using 2 mM p-nitrophenyl α-l-fucoside (PNP α-l-Fuc), PNP α-l-glucoside (PNP α-l-Glc), and PNP α-l-galactoside (PNP α-l-Gal) for ClAgl29A and ClAgl29B (Table 1).
Table 1. Kinetic Constants of ClAgl29A and ClAgl29B.
| k (s–1)a | Km (mM) | kcat (s–1) | kcat/Km (s–1 mM–1) | |
|---|---|---|---|---|
| ClAgl29A | ||||
| PNP α-l-Glc | 0.980 | 2.01 ± 0.26 | 2.00 ± 0.11 | 1.00 ± 0.08 |
| PNP α-l-Fuc | 0.0626 | 0.279 ± 0.005 | 0.0658 ± 0.0008 | 0.236 ± 0.005 |
| PNP α-l-Gal | 1.42 × 10–3 | |||
| ClAgl29B | ||||
| PNP α-l-Glc | 3.60 | 1.79 ± 0.18 | 7.76 ± 0.41 | 4.34 ± 0.22 |
| PNP α-l-Fuc | 0.108 | 0.240 ± 0.011 | 0.0844 ± 0.0019 | 0.352 ± 0.007 |
| PNP α-l-Gal | 7.83 × 10–3 |
The initial velocities, measured in McIlvaine buffer (pH 5.5) at 35 °C, were fitted with the Michaelis–Menten equation to obtain kinetic parameters. The rate constants were calculated from each hydrolytic rate at 2 mM substrate.
The values of TmaFuc were also determined as a control. ClAgl29A and ClAgl29B hydrolyzed PNP α-l-Glc 16 times faster and 33 times faster than PNP α-l-Fuc, respectively. These enzymes could hardly hydrolyze PNP α-l-Gal: the rate constant for the hydrolysis of PNP α-l-Gal was 1.5 × 10–3-fold (ClAgl29A) and 2.2 × 10–3-fold (ClAgl29B), compared to that for PNP α-l-Glc. In contrast, TmaFuc hydrolyzed PNP α-l-Fuc, but the rate of hydrolysis of PNP α-l-Glc was below the detection limit. This enzyme also hydrolyzes PNP α-l-Gal at 30% of the reaction rate for PNP α-l-Fuc. As discussed later, these two enzymes catalyze transglycosylation at 30 mM PNP α-l-Glc. If the reaction scheme includes transglycosylation, the rate equation will differ from the Michaelis–Menten kinetic equation. However, the reaction rate followed the Michaelis–Menten equation up to at least 6 mM PNP α-l-Glc (Figure S1), and thus we estimated Michaelis–Menten kinetic parameters. The kcat/Km values showed a specificity of these enzymes that favored PNP α-l-Glc over PNP α-l-Fuc (Table 1). ClAgl29A and ClAgl29B displayed higher kcat and Km values toward PNP α-l-Glc than PNP α-l-Fuc. Kinetic analysis indicated that ClAgl29A and ClAgl29B may have evolved as α-l-glucosidases which weakly bind α-l-glucoside to maximize the reaction rate, as described by Fersht.13
The pH effects on ClAgl29A and ClAgl29B were similar and the optimum pH for the hydrolysis of 2 mM PNP α-l-Glc of both enzymes was 5.5 (Figure 2). The pH stability was evaluated by maintaining the enzymes at various pH values for 24 h at 4 °C or 3 h at 35 °C and measuring their residual activity. The pH ranges with over 80% residual activity were wider at lower temperatures. The ranges showing >80% residual activity after 24 h were 4.8–11.2 for ClAgl29A and 3.3–11.2 for ClAgl29B; those after 3 h were 6.4–10.6 for ClAgl29A and 5.3–10.7 for ClAgl29B. The extensive stability in the alkaline range may be related to the nature of C. lonarensis LW9, which was originally isolated from a haloalkaline lake and can grow under alkaline conditions.14 ClAgl29A and ClAgl29B contain the predicted secretion signal sequence at their N-terminus and must be exposed to an alkaline environment. It is unclear why these enzymes seldom function in the alkaline range. ClAgl29A was more susceptible to heat than ClAgl29B. The optimal temperature for ClAgl29A and ClAgl29B was 45 and 55 °C, respectively (Figure 1). ClAgl29B maintained its activity at 55 °C for 15 min, whereas ClAgl29A lost 55% of its activity. ClAgl29B showed 80% residual activity after 15 min incubation at 60 °C, while ClAgl29A lost 90% of its activity. The determined structures of the two enzymes (stated below) are similar, and it is difficult to clearly explain the difference in temperature stability based on structural factors.
Figure 2.
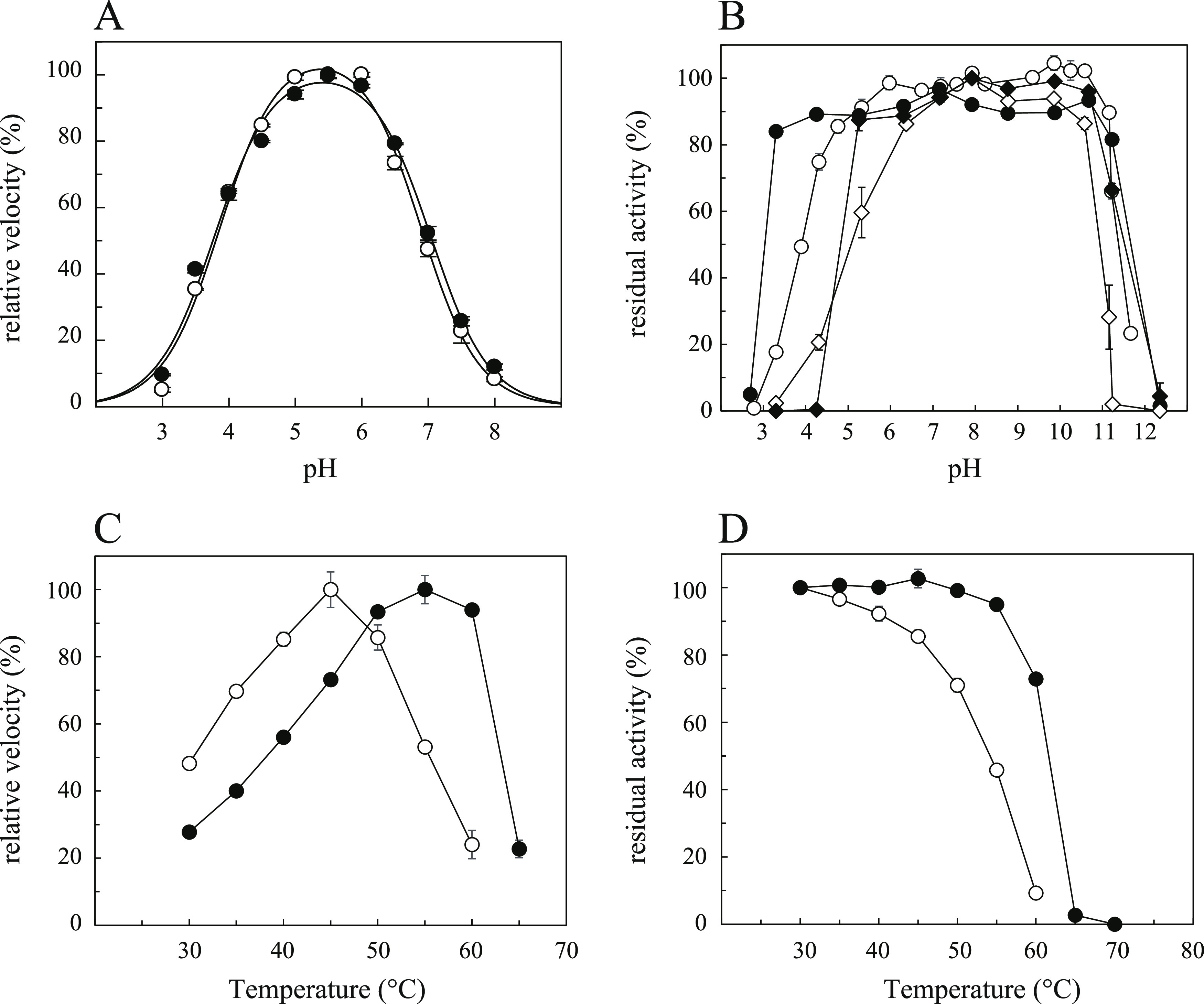
Effects of pH and temperature on the activity and stability of ClAgl29A and ClAgl29B. The relative rate and residual activity are expressed as percentages of the maximum values. The open and closed symbols represent the results for ClAgl29A and ClAgl29B, respectively. Two mM PNP α-l-Glc was used as a substrate in all of the experiments. Error bars represent the standard deviation of three independent experiments. (A) pH rate profile: data were fitted to the following equation: Relative rate = (Limit × log(pH–pKa1)/(log(2pH–pKa1–pKa2) + log(pH–pKa1) +1)). (B) pH stability: residual activity was examined after incubation for 24 h at 4 °C (circle) or 3 h at 35 °C (diamond) at various pH values. (C) Temperature–rate profile. (D) Temperature stability: residual activity was examined after incubation for 15 min at various temperatures.
Overall Structures of the α-l-Glucosidases
The crystal structures of ClAgl29A and ClAgl29B were determined at 2.0 Å (ClAgl29A) and 1.6 Å (ClAgl29B) resolution. Co-crystals of ClAgl29A with l-fucose were used for the structural analysis, but no electron density was observed in its active site. All residues were built based on electron density, except for residues 23 and 357–379 in ClAgl29A and residues 21–32 and 378–386 in ClAgl29B. Both asymmetric units contained two monomers, and this observation agreed with the results of gel filtration analysis, indicating that both ClAgl29A and ClAgl29B form dimers in solution, even though PISA15 suggested that a dimer composed of another crystal symmetry mate is a possible biological assembly in both cases.
The structure of each protein comprised two domains: an atypical catalytic TIM (β/α)8 barrel domain (residues 24–480, ClAgl29A; residues 32–492, ClAgl29B) and a β-sandwich domain (residues 481–573, ClAgl29A; residues 493–586, ClAgl29B) (Figure 3A,B) which is similar to the majority of GH29 proteins. ClAgl29A and ClAgl29B structures were similar, with the root-mean-square deviation (RMSD) calculated between the Cα-atoms of matched residues at the best 3D superposition, 0.376 Å. We searched PDBeFold (https://www.ebi.ac.uk/msd-srv/ssm/) for similar 3D structures of ClAgl29B and found that TmaFuc (PDB, 1HL9),5 α-l-fucosidase of Paenibacillus thiaminolyticus (PDB, 6GN6),16 and α-l-fucosidase of Bacteroides thetaiotaomicron (BT2970; PDB, 2XIB)17 were homologous enzymes with the highest Q-scores of 0.45, 0.40, and 0.39, respectively. The TIM (β/α)8-barrel fold of GH29 is unorthodox.5,6 There is a discontinuous barrel with an insufficient hydrogen bond between the fifth and sixth strands (β5 and β6) of the inner β-barrel and a lack of a typical helix structure between β5 and β6. The fifth α-helix (α5) is a feature of the GH29 TIM (β/α)8-barrel fold. ClAgl29A and ClAgl29B have no obvious hydrogen bonds between β5 and β6. ClAgl29B has a helix-loop-helix (HLH) structure (residues 362–396) instead of α5 (Figure 3C). α-Helices in a TIM (β/α)8 barrel generally surround the inner β-barrel to protect it from the solvent. However, the HLH structure exists independently of the barrel. We could not determine the corresponding part of ClAgl29A because of unclear electron density. ClAgl29A and ClAgl29B had no distinguishable α6, and a part of the inner barrel was exposed to the solvent; this was similar to that of TmaFuc and BT2970.5,17 ClAgl29A and ClAgl29B had singular structures at the β → α loop 3 and the N-terminus (Figure 3C). The β → α loop 3 is longer than that of other GH29 enzymes and consists of a small antiparallel β-sheet and a small helix (residues 232–289 for ClAgl29A and residues 244–301 for ClAgl29B). There was a structure containing a large α-helix (residues 46–74 for ClAgl29A and residues 57–85 for ClAgl29B) coiled around the TIM (β/α)8-barrel at the N-terminus. ClAgl29A and ClAgl29B showed Na+-binding at the same sites: ClAgl29A holds the ion via the O of Asp85, O of His 87, and Oδ1 of Asp386, whereas ClAgl29B holds the ion via the O of Lys96, O of His98, and Oδ1 of Asp398.
Figure 3.

Three-dimensional structures of ClAgl29A and ClAgl29B. Cartoon representations of ClAgl29A (A) and ClAgl29B (B). The sphere models show the bound sodium ions. (C) The characteristic structural elements found in ClAgl29B are indicated in green for the additional region of the N-terminus (33–108), cyan for the long insertion after β-strand 3 (244–301), and orange for the helix-loop-helix structure (362–396).
Active-Site Structure of α-l-Glucosidase
The ClAgl29B complex obtained with β-l-glucose by a soaking experiment was probably due to mutarotation in the solution, and was refined to 1.7 Å. The structure of ClAgl29B in complex with β-l-glucose allowed us to identify the active site, which was in a small pocket formed by loops connecting β-strands and α-helices of the (β/α)8-barrel domain (Figure 4). No structural rearrangements were observed between enzymes with and without ligand binding. Asp327 and Asp315 from ClAgl29B ClAgl29A, respectively, were the residues providing the catalytic nucleophile in comparison with other GH29 enzymes.5,18,19 The Oδ1 atom of Asp327 was at 2.8 Å to the O1 of β-l-glucose and 3.0 Å to the Nη1 atom of Arg361. Although these interactions should not be observed in the complex with a substrate, it might show that the Oδ1 atom is close enough to attack the anomeric carbon of the substrate and that the side chain of Arg361 affects the dissociation state of the catalytic nucleophile. The carboxy groups of Glu403 (ClAgl29B) and Glu391 (ClAgl29A) at pseudo β6 were located 5.5 Å from the catalytic nucleophile at the end of β4 and should act as a general acid/base catalyst. ClAgl29B tightly entrapped the sugar, where each hydroxy group was in direct contact with one or more amino acid side chains (Figure 4B), indicating that the enzyme explicitly recognizes l-glucose. C6 of l-glucose was surrounded by aromatic side chains derived from Phe112, Phe195, Tyr241, Trp325, and Phe425. O6 can form a hydrogen bond with Sγ of Cys418 at the β-strand 6 of the (β/α)8-barrel. O2 was hydrogen-bonded to Nε1 of Trp140 and Nε2 of His199. O3 was stabilized through hydrogen bonds with the Oδ1 of Asp139 and Nε2 of His198. Asp139 is also involved in the recognition of the equatorial O4 of l-glucose. The Oδ2 atom of Asp139 from O4 of l-glucose was 2.5 Å. We initially predicted that Asp114 (corresponding to His34 hydrogen bonding to the axial O4 of α-l-fucoside in TmaFuc) stabilized the equatorial O4 of α-l-glucoside. However, the side chain of Asp114 in ClAgl29B was positioned 3.8 Å from the equatorial O4, which is slightly too far away to form a hydrogen bond. Trp325 and Tyr137 were located within 4 Å of the ligand and were part of the active-site pocket formation. The amino acid residues of the active-site pocket of ClAgl29A are similar to those of ClAgl29B; therefore, the ligand-bound state of ClAgl29A and ClAgl29B is considered to be the same.
Figure 4.

Active-site pocket of ClAgl29B bound β-l-glucose. (A) Electron density observed in the active-site pocket fit to β-l-glucose. The contour level of the polder map is 5σ. Stick models show amino acid residues located within 4 Å of the ligand. (B) Diagram of the β-l-glucose–ClAgl29B interaction. Hydrogen bonds are shown as green dotted lines, while the spoked arcs represent protein residues making nonbonded contact with β-l-glucose. The diagram was drawn by LigPlot+.40
Comparison of the Active-Site Structures of α-l-Glucosidase and Other GH29 Enzymes
The active-site structure of ClAgl29B with l-glucose bound was compared to other GH29 enzymes to elucidate the specificity for α-l-glucoside (Figure 5A). We selected TmaFuc (PDB, 1ODU)5 as a representative of GH29-A and α-l-galactosidase BpGH29 from B. plebeius (PDB, 7LK7)20 as reference structures. The active-site pocket structures of these enzymes were similar, except for the recognition machinery for O6 and O4. No differences were apparent in the binding site of C6 among ClAgl29B, TmaFuc, and BpGH29, which have five aromatic side chains from β1, 2, 3, and 4 of the (β/α)8-barrel fold, as previously noted. BpGH29 has an Asp327 side chain that fixes O6, which is nearly identical to the Cys418 side chain of ClAgl29B. The corresponding residue in TmaFuc is Thr283 and could be hydrogen-bonded, which in line with weak α-l-galactosidase activity and structural features. The structures of ClAgl29B around the O4 atom of l-glucose were distinctly dissimilar to those of other GH29 enzymes (Figure 5B) and would be responsible for substrate specificity to α-l-glucoside. As mentioned above, Asp139 is responsible for the stabilization of equatorial O4, together with O3 of l-glucose in ClAgl29B. The corresponding residues were Glu66 in TmaFuc and Glu80 in BpGH29, and their Oε1 was hydrogen-bonded to O3 of l-fucose and l-galactose, respectively. His34 in TmaFuc and His68 in BpGH29 were responsible for stabilizing the axial O4 of l-fucose and l-galactose, whereas the corresponding Asp114 in ClAgl29B was separated from the substrate. The bulky side chain of Phe195 appeared to prevent its interaction with the axial O4 of α-l-fucoside and l-galactoside in ClAgl29B. The Phe residue is replaced by Thr125 and Val141 in TmaFuc and BpGH29, respectively, and they are sufficiently small side chains that do not interfere with α-l-fucoside and l-galactoside binding. Minor variations in the spatial positions of Phe290 in TmaFuc, Trp335 in BpGH29, and Phe425 in ClAgl29B are likely to affect substrate preferences. Phe290 undergoes a slight conformational change upon ligand binding and packing against C4, C5, and C6 of l-fucose, and interferes with binding of the equatorial hydroxy group at C4.5 BpGH29 and some α-l-fucosidases possess the corresponding Trp which also seems to play a role in excluding equatorial O4 from the active-site pocket. ClAgl29B has the counterpart Phe425, which has different spatial arrangements and does not hinder the binding of O4 to l-glucose. The spatial variation of these aromatic residues can be attributed to a difference in the main chain conformation: the φ and ψ angles are −92.0 and −48.5 of Phe290 in TmaFuc; −96.2 and −49.9 of Trp335 in BpGH29; and −92.9 and 107.1 of Phe425 in ClAgl29B. The disparate ψ angles of TmaFuc and BpGH29 are likely attributed to a hydrogen bond between the O atom of the aromatic residues and the Nδ2 atom of Asn residues at β7 and Asn324 in TmaFuc and Asn366 in BpGH29, while ClAgl29B has Val459, which cannot form a similar hydrogen bond. Alternatively, the O atom of Phe425 in ClAgl29B hydrogen bonds with the N atom of Asp139 in the small helix in the long insertion following β1. A subtle difference was also found at the pocket entrance. ClAgl29B and BpGH29 had Trp330 and Trp265 near the entrance, whereas TmaFuc had Met225 at the corresponding position. This difference resulted in a relatively narrower entrance in ClAgl29B and BpGH29, although it did not affect substrate specificity. We have discussed the comparison between ClAgl29B and other GH29 enzymes, where ClAgl29A and ClAgl29B have identical substrate recognition mechanisms.
Figure 5.

Comparison of the active-site pocket of ClAgl29B with TmaFuc and BpGH29. The active sites of ClAgl29B (green), TmaFuc (magenta), and BpGH29 (cyan) were aligned using PyMOL molecular graphic system version 2.0 (Schrödinger, LLC). (A) Stereo images of the residues involved in ligand binding (β-l-glucose for ClAgl29B, β-l-fucose for TmaFuc, and α-l-galactose for BpGH29). The upper, middle, and lower residue numbers are for ClAgl29B, TmaFuc, and BpGH29, respectively. (B) Structural comparison of the hydroxy group at C4. Orange dashed lines represent hydrogen bonds.
Transglucosylation of ClAgl29A and ClAgl29B
Some glycoside hydrolases with a retaining mechanism have transglycosylation ability to form new glycosidic linkage in addition to hydrolysis. We investigated whether ClAgl29A and ClAgl29B can be used for the synthesis of novel α-l-glucosyl compounds via transglycosylation. The reaction of α-l-glucosidase with PNP α-l-Glc substrate results in cleavage of the glucosidic linkage, PNP release, and the glucosyl group forming a covalent intermediate with the enzyme. The reaction of the intermediate with a water molecule results in hydrolysis and l-glucose is released. Meanwhile, transglucosylation involves the reaction of the intermediate with the hydroxy group of another PNP α-l-Glc resulting in the formation of an α-l-glucosidic bond (Figure 6A). The transglucosylation ability was evaluated by measuring the ratio of the transglucosylation rate (vTG) to the total reaction rate (PNP release rate, vT) in the reaction of ClAgl29A and ClAgl29B with 30 mM PNP α-l-Glc as the substrate. The transglucosylation ratio (%) ((vTG/vT) × 100) was calculated from the equation ((vT – vH)/vT) × 100 since [vT = l-glucose release rate (hydrolysis rate, vH) + vTG] was established. The vT estimated from the released PNP was 1.05 μmol/min/mg (ClAgl29A) and 4.12 μmol/min/mg (ClAgl29B). The l-glucose release rate was 0.0361 μmol/min/mg (ClAgl29A) and 2.81 μmol/min/mg (ClAgl29B). The transglucosylation ratios calculated from these values were 60.3% (ClAgl29A) and 32.0% (ClAgl29B).
Figure 6.

Transglucosylation properties of ClAgl29A and ClAgl29B. (A) Schematic representation of hydrolysis and transglucosylation by α-l-glucosidase. G* indicates the covalent intermediate of enzyme and glucose. (B) Reversed-phase-HPLC analysis of the reaction mixtures containing 30 mM PNP α-l-Glc and ClAgl29A and ClAgl29B in McIlvaine buffer, pH 5.5, that were incubated at 35 °C for 33 h. (1) PNP trisaccharide; (2) α-l-Glcp-(1 → 2)-α-l-Glcp-O-PNP; (3) α-l-Glcp-(1 → 6)-α-l-Glcp-O-PNP; (4) PNP α-l-Glc; (5) α-l-Glcp-(1 → 3)-α-l-Glcp-O-PNP; (6) PNP. The area of the peaks marked with asterisks was unchanged before and after the reaction. (C) Time-dependent change of the concentrations of transglucosylation products and PNP α-l-Glc. The concentrations were calculated using a calibration curve prepared from chromatographic peak areas of PNP α-d-glucoside standards with known concentrations. Closed circle, α-l-Glcp-(1 → 3)-α-l-Glcp-O-PNP; open circle, α-l-Glcp-(1 → 2)-α-l-Glcp-O-PNP; closed diamond, α-l-Glcp-(1 → 6)-α-l-Glcp-O-PNP; open diamond, PNP trisaccharide; and open square, PNP α-l-Glc. The numbers in the diagram correspond to the indications in (B).
Next, we determined the structures of the transglucosylation products of PNP α-l-Glc. Both enzymes synthesized three types of PNP disaccharides: p-nitrophenyl (α-l-glucopyranosyl)-(1 → 3)-α-l-glucopyranoside (α-l-Glcp-(1 → 3)-α-l-Glcp-O-PNP), p-nitrophenyl (α-l-glucopyranosyl)-(1 → 2)-α-l-glucopyranoside (α-l-Glcp-(1 → 2)-α-l-Glcp-O-PNP), and p-nitrophenyl (α-l-glucopyranosyl)-(1 → 6)-α-l-glucopyranoside (α-l-Glcp-(1 → 6)-α-l-Glcp-O-PNP). ClAgl29B produced a small amount of PNP trisaccharide (Figure 6B). The formation of PNP trisaccharide can be attributed to one of the produced PNP disaccharides acting as an acceptor. The broad regioselectivity of transglucosylation of α-l-glucosidases is similar to that of TmaFuc, which can produce α-l-Fuc-(1 → 3)-α-l-Fuc, α-l-Fuc-(1 → 2)-β-d-Gal, and α-l-Fuc-(1 → 6)-β-d-Gal.21 ClAgl29A produced the highest amount of α-l-Glcp-(1 → 3)-α-l-Glcp-O-PNP, followed by α-l-Glcp-(1 → 2)-α-l-Glcp-O-PNP, and α-l-Glcp-(1 → 6)-α-l-Glcp-O-PNP based on the analysis of temporal changes in the concentration of the products (Figure 6C). ClAgl29B mainly produces α-l-Glcp-(1 → 3)-α-l-Glcp-O-PNP. It also yielded almost equal amounts of α-l-Glcp-(1 → 2)-α-l-Glcp-O-PNP and α-l-Glcp-(1 → 6)-α-l-Glcp-O-PNP at approximately one-third of the concentration of α-l-Glcp-(1 → 3)-α-l-Glcp-O-PNP. The amount of transglucosylation product (PNP disaccharide) was higher in ClAgl29A (6.2 mM) than in ClAgl29B (2.2 mM) after 33 h of reaction. At this point, 92 and 99% of PNP α-l-Glc were consumed in ClAgl29A and ClAgl29B, resulting in theoretical yields of 45 and 15%, respectively. The difference in the amounts produced by the two enzymes may reflect their different transglucosylation ratio.
The differences in the transglucosylation abilities of ClAgl29A and ClAgl29B were predicted by comparing the architecture of their subsite +1 with the structure of TmaFuc-bound α-l-fucopyranosyl-(1 → 2)-1-azide-1-deoxy-β-l-fucopyranose (PDB, 2WSP).22 Subsite +1 of ClAgl29A was presumed to consist of Thr121, Tyr125, Trp128, Leu263, Trp318, and Arg349. Subsite +1 of ClAgl29B was quite similar except that Gln275 replaced the Leu263 position. This difference may be responsible for the enhanced transglucosylation activity of ClAgl29A.
We succeeded in the enzymatic synthesis of an α-l-glucosidic linkage which was not previously reported. Oligosaccharides composed of d-series monosaccharides support human health by stimulating the growth and activity of beneficial bacteria in the intestines.23,24 Although it is not known whether α-l-glucosyl oligosaccharides have such a function, this study provides clues to reveal their hidden functionality.
Substrate Specificity in GH29
We demonstrated that ClAgl29A and ClAgl29B have elaborate α-l-glucoside recognition machinery that differs from that of α-l-fucosidases. The presence of ClAgl29A and ClAgl29B implies that α-l-glucoside-containing compounds exist in nature. Furthermore, α-l-glucosidases may be widespread. A BLAST search using ClAgl29B as the query sequence revealed that the top 45 amino acid sequences of the hits had substrate recognition mechanisms of α-l-glucosidases (Table S1). Thus, these sequences which were derived from the Bacteroidetes, Verrucomicrobia, Kiritimatiellaeota, Planctomycetes, Terrabacteria, and Armatimonadetes bacterial phyla, and Asgard archaea, can be regarded as α-l-glucosidases. A phylogenetic tree of representative proteins was constructed based on 15% co-membership thresholds (RP15) of PF01120 (Pfam accession number), 45 probable α-l-glucosidases, and the sequences annotated as characterized proteins in CAZy using the unweighted pair group method with arithmetic averages (UPGMA) (Figure S2). The tree indicates that a single cluster for α-l-glucosidase is embedded in GH29-A and diverged later than the cluster of GH29-B, a subfamily of α-l-fucosidases with different aglycone substrate specificities.
The discovery of α-l-glucosidase in the present study was based on the diversity of the conserved His residue that stabilizes OH-4 of α-l-fucoside in GH29 α-l-fucosidase. ClAgl29A and ClAgl29B, containing Asp instead of His, were identified as α-glucosidases in this study; however, we found substitutions for other amino acids in GH29. The genome of Coraliomargarita akajimensis encodes 11 GH29 enzymes. Five of these enzymes had the His residue substituted for Val (Caka_0522 and Caka_0523), Cys (Caka_0506 and Caka_0536), and Phe (Caka_0074). Using facilely purified recombinant enzymes, the activities toward PNP α-l-Fuc, PNP α-l-Glc, and PNP α-l-Gal were investigated. The activity was quite low for all of these substrates (Table 2).
Table 2. Enzyme Activity of GH29 Enzymes from C. akajimensisa.
| caka_0074 | caka_0506 | caka_0522 | caka_0523 | caka_0536 | |
|---|---|---|---|---|---|
| PNP α-l-Fuc | 1.4 ± 0.3 | 12 ± 0.4 | 2.1 ± 0.6 | 1.3 ± 0.8 | 3.1 ± 1.5 |
| PNP α-l-Glc | 1.3 ± 0.6 | 7.0 ± 0.2 | 8.2 ± 0.1 | <0.12 | 6.0 ± 0.3 |
| PNP α-l-Gal | 1.4 ± 0.3 | 5.5 ± 1.9 | 7.5 ± 0.3 | <0.12 | 4.5 ± 0.8 |
Hydrolysis rates were determined using 2 mM substrate (PNP α-l-Fuc, PNP α-l-Glc, and PNP α-l-Gal). The unit of the values is nmol/min/mg.
GH29-B enzymes have strict substrate specificity on the aglycone side and hardly hydrolyze PNP α-l-Fuc.12 However, phylogenetic analysis showed that the five enzymes from C. akajimensis were classified as GH29-A with relaxed specificity (Figure S3). Caka_0506 prefers PNP α-l-Fuc, Caka_0522 prefers PNP α-l-Glc and PNP α-l-Gal, while Caka_0536 accepts all substrates (PNP α-L-Glc, PNP α-L-Fuc, and PNP α-L-Gal). However, detailed evaluation of their substrate specificity was not performed due to the relatively slow reaction rates. The specificity of GH29 enzymes may be more divergent than known to date and the substrate specificity of these enzymes requires further investigation.
This study identified the α-l-glucosidases ClAgl29A and ClAgl29B in GH29 by analyzing an expanded sequence space enabled by genome analysis. The active-site architectures exclude α-l-fucoside but allow α-l-glucoside, suggesting that ClAgl29A and ClAgl29B may have evolved as α-l-glucosidases. On the other hand, further analysis of substrate specificity for α-l-glycosides, including α-l-xylosides and α-l-quinovosides (6-deoxy l-glucosides), will be necessary to demonstrate that these enzymes are true α-l-glucosidases. It is unclear whether these enzymes evolved under selection pressure to utilize l-glucoside, or if they were neutrally created and retained by chance. The discovery of α-l-glucosidase raises the possibility that α-l-glucoside exists in nature.
Experimental Procedures
Chemicals
l-Galactose was prepared from d-galacturonic acid (Fujifilm Wako Pure Chemical Corporation, Osaka, Japan) according to a previous method except that Amberlite IR120 (H+) was used instead of sulfuric acid to adjust the pH during the reduction of l-galactono-1,4-lactone.25 The PNP group addition reaction was performed as previously described.261H NMR (500 MHz) spectra were recorded using a Bruker AVANCE I spectrometer (Bruker Corporation, Billerica, MA). PNP α-l-Glc: 1H NMR (500 MHz, D2O) δ (ppm): 8.26 (d, J = 9.3 Hz, 2H, PNP), 7.30 (d, J = 9.3 Hz, 2H, PNP), 5.82 (d, J = 3.7 Hz, 1H, H1), 3.94 (t, J = 9.5 Hz, 1H, H3), 3.78 (dd, J = 3.7 and 9.8 Hz, 1H, H2), 3.74 (m, 2H, H6), 3.68 (dd, J = 3.5 and 6.3 Hz, 1H, H5), 3.53 (t, J = 9.5 Hz, 1H, H4). PNP α-l-Gal: 1H NMR (500 MHz, D2O) δ (ppm): 8.26 (d, J = 9.3 Hz, 2H, PNP), 7.30 (d, J = 9.3 Hz, 2H, PNP), 5.86 (d, J = 3.8 Hz, 1H, H1), 4.11 (dd, J = 3.3 and 10.3 Hz, 1H, H3), 4.06 (d, J = 3.3 Hz, 1H, H4), 4.04 (dd, J = 3.8 and 10.3 Hz 1H, H2), 3.98 (dd, J = 4.8 and 7.5 Hz, 1H, H5), 3.72 (dd, J = 7.5 and 11.9 Hz, 1H, H6a), 3.68 (dd, J = 4.8 and 11.9 Hz, 1H, H6b). l-glucose was purchased from TCI (Tokyo, Japan). PNP α-l-Fuc was obtained from Sigma-Aldrich (St. Louis, MA).
Sequence Analysis
Full-length amino acid sequences and RP15 belonging to the alpha_L_fucos family (PF01120) were obtained from the Pfam database (https://pfam.xfam.org/family/PF01120). The 8,266 obtained sequences were aligned using MAFFT version 7 (https://mafft.cbrc.jp/alignment/software/).27 Refinement of the multiple sequence alignment involved excluding sequences with 220 amino acids or less. FASTA and BLAST searches were performed using GenomeNet (https://www.genome.jp/tools/fasta/) and NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi) databases, respectively. Signal peptide prediction was performed using SignalP-5.028 (https://services.healthtech.dtu.dk/service.php?SignalP-5.0). Phylogenetic trees were constructed using the UPGMA and neighbor-joining method using the Jones–Taylor–Thornton model on the MAFFT version 7 website. iTOL v6 (https://itol.embl.de/)29 was used to visualize the tree.
Cloning
Genes encoding ClAgl29A (EKB48091.1) and ClAgl29B (EKB48090.1), whose locus_tags were B879_03288 and B879_03287, respectively, were synthesized at Eurofins Genomics K.K. (Tokyo, Japan). The genes were amplified by PCR and cloned into the Novagen pET28a vector (Merck; Darmstadt, Germany) using an iVEC3 in vivo E. coli cloning system.30 The gene encoding TmaFuc (TM0306; nucleotide 326116 to 327465 of AE000512.1) in T. maritima NBRC 100826T genomic DNA was amplified by PCR and cloned into pET28a using NdeI and HindIII restriction sites. Genes encoding Caka_0074, Caka_0506, Caka_0522, Caka_0523, and Caka_0536 were cloned based on the C. akajimensis DSM 45221 complete genome (CP001998.1): 79209..80672, 649949..651400, 670716..672635, 672662..674473, and 688656..690170, respectively. The regions were amplified by PCR using genomic DNA of C. akajimensis JCM 23193T as a template which was provided by RIKEN BRC through the National BioResource Project of MEXT, Japan. The PCR products were cloned into pET28a using a T5 exonuclease DNA assembly method.31 PrimeSTAR MAX DNA Polymerase (Takara Bio Inc., Shiga, Japan) was used for all PCR amplifications, and reaction conditions were followed according to the attached protocol. The primers used for the PCR are listed in Table S2.
Production and Purification of Recombinant Enzymes
ClAgl29A and ClAgl29B: E. coli Rosetta (DE3) (Novagen-Merck) transformants with pET28a-derived plasmids were inoculated in ZYP-5052 medium32 supplemented with 100 μg·mL–1 kanamycin and 30 μg·mL–1 chloramphenicol and cultured at 30 °C for 24 h. Cells collected by centrifugation were resuspended in 10 mM sodium phosphate buffer (pH 7.5) containing 300 mM sodium chloride (buffer A), and sonicated. Cell debris was removed by centrifugation, and the supernatant was loaded onto a Ni-chelating Sepharose Fast Flow column (Cytiva; Tokyo, Japan) preequilibrated with buffer A containing 10 mM imidazole. After washing the column with buffer A containing 30 mM imidazole, the absorbed proteins were eluted using 300 mM imidazole in buffer A. The active fractions were dialyzed against 10 mM sodium phosphate buffer (pH 7.5) and concentrated using Amicon Ultra-15 centrifugal filter units (30,000 NMWL, Millipore, Merck).
TmaFuc: The procedure followed a previously published method;5 however, 50 mM sodium phosphate buffer (pH 8.0) containing 300 mM sodium chloride was used as the initial buffer for Ni2+ affinity column chromatography, and the purified enzyme obtained by Ni2+ affinity chromatography was dialyzed against 50 mM sodium phosphate buffer (pH 8.0) and concentrated using a Vivaspin 20 10,000 MWCO ultrafiltration unit (Sartorius AG; Göttingen, Germany).
Caka_0074, Caka_0506, Caka_0522, Caka_0523, and Caka_0536: E. coli BL21 (DE3) for Caka_0506 and Caka_0523 and Rosetta-gami 2 (DE3) (Novagen-Merck) for Caka_0074 Caka_0522, and Caka_0536 were transformed with the appropriate pET28a-derived plasmids. Each transformant was inoculated in LB medium supplemented with 100 μg·mL–1 kanamycin. Rosetta-gami 2 (DE3) transformants also contained 30 μg·mL–1 chloramphenicol in the medium. Expression of recombinant proteins was induced by incubation at 18 °C (Caka_0074, Caka_0522, and Caka_0536) or 30 °C (Caka_0506 and Caka_0523) without the addition of IPTG. The recombinant proteins were purified using the Amicon Pro purification system (30,000 NMWL, Millipore–Merck). Sodium phosphate buffer (20 mM, pH 7.5) containing 500 mM sodium chloride was used as the basic buffer during purification and was subsequently replaced with 20 mM sodium phosphate buffer, pH 7.5.
Protein concentrations of ClAgl29A and ClAgl29B were estimated based on the protein hydrolysate (6 M hydrochloric acid, 110 °C, 24 h) using an L-8900 amino acid analyzer (Hitachi High-Tech, Tokyo, Japan) equipped with a ninhydrin-detection system. The molar extinction coefficient values calculated from the amino acid analysis were 1.38 × 105 M–1cm–1 for ClAgl29A and 1.34 × 105 M–1cm–1 for ClAgl29B. The concentration of C. akajimensis-derived proteins was computed from the theoretical extinction coefficient at 280 nm (ProtoParam tool, https://web.expasy.org/protparam/) derived from the amino acid sequence.
Enzyme Assay
α-l-Glucosidase activity was determined by measuring the increase in PNP during its hydrolysis. The standard reaction was performed in McIlvaine buffer (pH 5.5) containing 2 mM PNP α-l-Glc and an appropriately diluted enzyme at 35 °C for 10 min. The enzyme was diluted with McIlvaine buffer (pH 5.5) containing 0.1 mg/mL bovine serum albumin (BSA). The reaction was terminated by the addition of two volumes of 1 M sodium carbonate. The amount of PNP released was measured based on the absorption at 400 nm using a 1 cm cuvette with a molar extinction coefficient of 5,560 M–1 cm–1. One unit of enzyme was defined as the amount of enzyme that produced 1 μmol of PNP per minute under the above conditions. The hydrolysis rates for 2 mM PNP α-l-Gal and 2 mM PNP α-l-Fuc were measured under the same conditions as described above.
The optimal pH for hydrolysis of 2 mM PNP α-l-Glc was measured at various pH values using McIlvaine buffer (pH 3.0–8.0) with 67 nM ClAgl29A and 55 nM ClAgl29B. The pH stability was assessed by incubating ClAgl29A (1.34 μM) and ClAgl29B (1.37 μM) in twofold-diluted Britton–Robinson buffer (pH 2.5–11.5) and 50 mM glycine–NaOH (pH 11.5–12.0) containing 0.1 mg/mL BSA at 4 °C for 24 h or at 35 °C for 3 h, followed by measurement of the residual activity. The optimal reaction temperature was determined by measuring the hydrolysis rate at various temperatures using 67 nM ClAgl29A and 16 nM (ClAgl29B). The thermal stability of ClAgl29A (680 nM) and ClAgl29B (540 nM) was measured by diluting each enzyme with an equal volume of McIlvaine buffer (pH 5.5) containing 0.5 mg/mL BSA and incubation at each temperature (30–70 °C) for 15 min, followed by measurement of their residual activities.
The initial velocities of PNP α-l-Glc (0.5–6 mM for ClAgl29A, 0.25–4 mM for ClAgl29B) and those of PNP α-l-Fuc (0.04–0.6 mM for ClAgl29A, 0.05–0.5 mM for ClAgl29B) were measured under standard conditions. The enzyme concentrations were 13 nM ClAgl29A for PNP α-l-Glc, 890 nM ClAgl29A for PNP α-l-Fuc, 8.2 nM ClAgl29B for PNP α-l-Glc, and 550 nM ClAgl29B for PNP α-l-Fuc. Kinetic parameters were obtained by fitting data from the reactions to the Michaelis–Menten equation using the computer program KaleidaGraph v. 3.6 (Synergy Software; Reading, PA).
Crystallization, Data Collection, Structure Determination, and Refinement
The enzymes used for crystallization were purified via anion exchange column chromatography using DEAE Sepharose Fast Flow (Cytiva) and Ni-affinity column chromatography. ClAgl29B was further purified by gel filtration column chromatography using a Sephacryl S-200 HR column (Cytiva). Chromatography was performed using 10 mM sodium phosphate buffer (pH 7.5) as the mobile phase. Anion exchange column chromatography involved the elution of proteins by linearly increasing the ionic strength with sodium chloride. The active fractions were dialyzed against 10 mM sodium phosphate buffer (pH 7.5) and concentrated to 10 mg/mL using an Amicon Ultra-15 centrifugal filter unit (30,000 NMWL).
All crystallization procedures were performed at 20 °C. Initial crystallization trials were performed using the sitting-drop vapor diffusion method in 96-well plates using PEGRx1, PEGRx2, SaltRx1, and SaltRx2 screen kits (Hampton Research, Aliso Viejo, CA). Microcrystals appeared under several conditions of PEGRx1 and PEGRx2. The crystallization conditions were optimized by varying the concentration of poly(ethylene glycol) (PEG) using the hanging-drop vapor diffusion method. ClAgl29A crystals obtained in a drop composed of 2 μL of ClAgl29A (10 mg·mL–1), 2 μL of reservoir solution [0.1 M sodium citrate tribasic dihydrate (pH 5.5), 16% (w/v) PEG 3,350], and 1 μL of 1 mM l-fucose. Crystals were soaked in a cryoprotectant [0.1 M sodium citrate tribasic dihydrate (pH 5.5), 18% (w/v) PEG 3,350, 10% (v/v) glycerol, 1 mM l-fucose]. ClAgl29B crystals were obtained in a drop composed of 2 μL of ClAgl29B (5 mg·mL–1), 2 μL of reservoir solution [0.1 M citric acid (pH 3.5), 5% (v/v) 2-propanol, 5% (w/v) PEG 20,000], and 1 μL of 1 mM l-glucose. The crystals were cryoprotected with 0.1 M citric acid (pH 3.5), 5% (v/v) 2-propanol, 6% (w/v) PEG 20,000, 20% (v/v) glycerol, and 1 mM l-Glc for the ligand-free from, and 0.1 M citric acid (pH 3.5), 5% (v/v) 2-propanol, 6% (w/v) PEG 20,000, 20% (v/v) glycerol, and 30 mM l-Glc for the l-glucose-bound form. All crystals were flash-frozen in liquid nitrogen. The diffraction datasets were collected at the X06SA beamline of the Swiss Light Source, Switzerland, for ClAgl29A, and at the AR-NE3A beamline of the Photon Factory, KEK, Japan, for ClAgl29B using an EIGER 16M detector (Dectris; Dättwil AG, Switzerland) and Pilatus 2m-F pixel detector (Dectris), respectively, at a wavelength of 1.00000 Å. The dataset was collected from a single crystal under a stream of nitrogen at a temperature of 100 K. The diffraction dataset was indexed, integrated, and scaled using the XDS program.33
The structure of ClAgl29A was determined by the molecular replacement method with Phenix. automr software34,35 using the protein coordinates of TmaFuc (PDB, 2XSD) as a search model. The refinement was converged by several cycles of manual model corrections with Coot software36,37 and further refined using Refmac538 and phenix.refine39 programs. The data processing and refinement statistics are presented in Table 3.
Table 3. Summary of Data Collection and Refinement Statisticsa.
| ClAgl29B/l-glucose | ClAgl29B | ClAgl29A | |
|---|---|---|---|
| PDB ID | 7XSH | 7XSG | 7XSF |
| data collection | |||
| beamline | PF AR-NE3A | PF AR-NE3A | SLS X06SA |
| wavelength | 1.00000 | 1.00000 | 1.00000 |
| space group | P212121 | P212121 | P1211 |
| unit cell | |||
| a, b, c (Å) | 72.39 121.58 166.37 | 73.13 123.41 166.60 | 74.58 95.52 83.55 |
| β = 97.33° | |||
| resolution range (Å) | 46.60–1.71 (1.81–1.71) | 45.42–1.61 (1.71–1.61) | 47.81–2.01 (2.13–2.01) |
| total no. of reflections | 1,058,673 (157,759) | 1,292,170 (201,560) | 539,809 (86368) |
| no. of unique reflections | 159,024 (25,291) | 194,275 (30357) | 76,908 (12,196) |
| multiplicity | 6.7 (6.2) | 6.7 (6.6) | 7.0 (7.1) |
| completeness (%) | 99.8 (99.1) | 99.5 (97.0) | 98.9 (97.3) |
| mean I/σ(I) | 17.44 (2.37) | 22.17 (4.51) | 14.80 (2.45) |
| Rmean | 10.1 (94.2) | 6.9 (45.9) | 9.0 (96.2) |
| Rsym | 9.3 (86.3) | 6.3 (42.3) | 8.3 (89.1) |
| CC1/2 | 0.999 (0.810) | 0.999 (0.942) | 0.999 (0.90) |
| refinement | |||
| Rwork | 17.9 | 17.2 | 20.5 |
| Rfree | 20.2 | 18.8 | 24.3 |
| number of atoms | |||
| macromolecules | 8938 | 8985 | 8634 |
| ligands | 76 | 40 | 15 |
| solvent | 1008 | 203 | |
| RMSD values from ideal | |||
| bonds | 0.009 | 0.009 | 0.007 |
| angles | 1.39 | 1.43 | 1.25 |
| Ramachandran (%) | |||
| favored | 96.88 | 97.23 | 96.56 |
| allowed | 3.12 | 2.77 | 3.44 |
| outliers | 0.00 | 0.00 | 0.00 |
| rotamer outliers (%) | 1.13 | 0.82 | 0.76 |
| clashscore | 1.42 | 1.80 | 2.19 |
Values in parentheses are for the highest-resolution shell. RMSD, root-mean-square deviation.
Transglucosylation
The reaction mixture consisted of 30 mM PNP α-l-Glc, diluted enzymes (1.34 μM for ClAgl29A and 0.274 μM for ClAgl29B), and 0.1 mg/mL BSA in McIlvaine buffer (pH 5.5) and was incubated at 35 °C. An aliquot of the reaction solution was collected at 3, 6, 9, and 12 min, and the reaction was stopped by treatment at 100 °C for 2 min. Two volumes of 1.0 M sodium carbonate were added to the collected sample, and the absorbance value of the solution was measured at 400 nm to determine the PNP concentration. The l-glucose concentration was determined by high-performance anion exchange chromatography–pulsed amperometric detection (HPAEC-PAD) using a Dionex ICS-3000 system (Dionex-Thermo Fisher Scientific; Waltham, MA) equipped with a CarboPac PA1 analytical column (4 mm × 250 mm; Dionex/Thermo Fisher Scientific). The column was preequilibrated with the eluent before chromatographic analysis. Separation conditions included a flow rate of 0.8 mL.min–1, sample injection of 10 μL (containing sorbitol internal standard) under isocratic elution with 160 mM sodium hydroxide, which was prepared from super special grade 50% sodium hydroxide solution (Fujifilm Wako Pure Chemical Corporation). The concentration was calculated using a calibration curve prepared from the chromatographic peak areas of sorbitol and known l-glucose standards. Chromatograms were evaluated using Chromeleon software (Dionex/Thermo Fisher Scientific).
PNP glycosides were quantified using 1.34 μM ClAgl29A and 0.274 μM ClAgl29B. The sample was desalted using Amberlite MB-4 resin (Organo Co., Tokyo, Japan) and separated by reversed-phase HPLC (RP-HPLC) using a Jasco system (Tokyo, Japan) equipped with a Cosmosil 5C18-PAQ analytical column (4 mm × 150 mm; Nacalai Tesque, Kyoto, Japan). The separation was performed at 40 °C, the mobile phase consisted of methanol/water = 20/80 (v/v), and the flow rate was 1.0 mL.min–1. Each PNP glycoside was detected by measuring the absorbance at 313 nm, and the concentration was calculated using a calibration curve prepared from the chromatographic peak areas of PNP α-d-glucoside standard.
The molecular masses of the products were confirmed by electrospray ionization (ESI)-MS using an Exactive spectrometer (Thermo Fisher Scientific, Inc.). 1H NMR (500 MHz) and 13C (126 MHz), COSY, HSQC, HSQC-TOCSY, H2BC, and HMBC NMR spectra were recorded with a Bruker AVANCE I system. p-Nitrophenyl trisaccharide was only detected by MS because of the low yield; MS: m/z calcd: 648.17 [M + Na]+; found: 648.18.
p-Nitrophenyl (α-l-glucopyranosyl)-(1 → 3)-α-l-glucopyranoside [α-l-Glcp-(1→3)-α-l-Glcp-O-PNP] (Figure S4)
1H NMR (500 MHz, D2O) δ (ppm): 8.25 (d, J = 9.0 Hz, 2H, PNP), 7.31 (d, J = 9.0 Hz, 2H, PNP), 5.85 (s, 1H, H-1), 5.45 (d, J = 3.4 Hz, 1H, H-1′), 4.12 (t, J = 9.2, 1H, H-3), 4.08 (m, 1H, H-5′), 3.91 (d, J = 11.3 Hz, 1H, H-2), 3.90 (m, 1H, H-6a′), 3.83 (m, H, H-6b′), 3.80 (m, 1H, H-3′), 3.79 (m, 1H, H-4), 3.76 (brd, 2H, H-6), 3.71 (m, 1H, H-5), 3.63 (dd, J = 3.4 and 10.1, 1H, H-2′), 3.50 (t, J = 9.5, 1H, H-4′); 13C NMR (126 MHz, D2O) δ (ppm): 162.1 (PNP), 143.2 (PNP), 126.9 (PNP), 117.6 (PNP), 100.1 (C-1′), 97.6 (C-1), 80.4 (C-3), 73.7 (C-3′), 73.5 (C-5), 72.7 (C-5′), 72.6 (C-2′), 70.5 (C-4), 70.4 (C-2) 70.3 (C-4′), 61.3 (C-6′), 60.9 (C-6); MS: m/z calcd: 486.12 [M + Na]+; found: 486.12.
p-Nitrophenyl (α-l-glucopyranosyl)-(1→2)-α-l-glucopyranoside [α-l-Glcp-(1→2)-α-l-Glcp-O-PNP] (Figure S5)
1H NMR (500 MHz, D2O) δ (ppm): 8.26 (d, J = 8.5 Hz, 2H, PNP), 7.35 (d, J = 8.5 Hz, 2H, PNP), 6.05 (s, 1H, H-1), 5.12 (d, J = 3.2 Hz, 1H, H-1′), 4.09 (t, J = 9.5, 1H, H-3), 3.99 (d, J = 9.8, 1H, H-5′), 3.92 (d, J = 12.4 Hz, 1H, H-2), 3.91 (m, 1H, H-6a′), 3.91 (m, H, H-6a), 3.83 (m, 1H, H-3′), 3.83 (m, 1H, H-6b′), 3.80 (m, 1H, H-6b), 3.75 (d, J = 13.0, 1H, H-5), 3.62 (t, J = 9.4, 1H, H4), 3.55 (dd, J = 3.2 and 9.6, 1H, H-2′), 3.48 (t, J = 9.6, 1H, H-4′); 13C NMR (126 MHz, D2O) δ (ppm): 162.3 (PNP), 143.3 (PNP), 126.9 (PNP), 117.8 (PNP), 96.9 (C-1′), 94.9 (C-1), 75.6 (C-2), 73.6 (C-5′), 73.5 (C-3′), 72.1 (C-3), 71.9 (C-2′), 70.2 (C-4′), 69.9 (C-4), 61.2 (C-6′), 61.1 (C-6); MS: m/z calcd: 486.12 [M + Na]+; found: 486.12.
p-Nitrophenyl (α-l-glucopyranosyl)-(1→6)-α-l-glucopyranoside [α-l-Glcp-(1→6)-α-l-Glcp-O-PNP] (Figure S6)
1H NMR (500 MHz, D2O) δ (ppm): 8.31 (d, J = 8.4 Hz, 2H, PNP), 7.34 (d, J = 8.4 Hz, 2H, PNP), 5.87 (d, J = 2.3 Hz, 1H, H-1), 4.90 (s, 1H, H-1′), 3.98 (t, J = 9.4, 1H, H-3), 3.94 (m, 1H, H-6a), 3.91 (d, J = 11.3 Hz, 1H, H-2), 3.91 (m, 1H, H-5), 3.91 (m, H, H-6a′), 3.77 (dd, J = 4.6 and 12.0, 1H, H-6b′), 3.73 (m, 1H, H-6b), 3.69 (m, 1H, H-5′), 3.63 (dd, J = 9.1 and 9.4, 1H, H-4), 3.63 (m, 1H, H-3′), 3.55 (d, J = 11.0, 1H, H-2′), 3.43 (t, J = 9.3, 1H, H-4′); 13C NMR (126 MHz, D2O) δ (ppm): 162.1 (PNP), 143.3 (PNP), 126.9 (PNP), 117.7 (PNP), 98.5 (C-1′), 97.4 (C-1), 74.0 (C-3), 73.9 (C-3′), 72.6 (C-5), 72.2 (C-2′), 71.7 (C-2), 70.3 (C-4′), 70.2 (C-4), 66.3 (C-6), 61.3 (C-6′); MS: m/z calcd: 502.10 [M + Na]+; found: 502.09.
Acknowledgments
The authors thank the staff of the Instrumental Analysis Division of the Creative Research Institution at Hokkaido University for conducting amino acid analysis and mass spectrometry analysis and the staff of the GC-MS & NMR Laboratory at the Research Faculty of Agriculture at Hokkaido University for NMR analysis. They acknowledge the Paul Scherrer Institute (Villigen, Switzerland) and Photon Factory (Tsukuba, Japan) for providing synchrotron radiation beamtime at beamline X06SA of the SLS (proposal numbers: 20191094 and 20191134) and AR-NE3A, respectively, and thank all beamline staff for assistance. They also thank Editage (www. editage. com) for English language editing.
Glossary
Abbreviations
- ESI
electrospray ionization
- HPAEC-PAD
high-performance anion exchange chromatography–pulsed amperometric detection
- UPGMA
unweighted pair group method with arithmetic averages
Data Availability Statement
The atomic coordinates were deposited in the Protein Data Bank with accession codes 7XSF, 7XSG, and 7XSH. The authors declare that all data are contained within the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c06991.
Michaelis–Menten analysis (Figure S1); UPGMA phylogenetic tree of RP15 of Alpha_L_fuc (PF01120) (Figure S2); neighbor-joining phylogenetic tree of structure-known α-l-fucosidases, five GH29 sequences from C. akajimensis, ClAgl29A, and ClAgl29B (Figure S3); NMR spectra of α-l-Glcp-(1 → 3)-α-l-Glcp-O-PNP (Figure S4), α-l-Glcp-(1→2)-α-l-Glcp-O-PNP (Figure S5); and α-l-Glcp-(1 → 6)-α-l-Glcp-O-PNP (Figure S6); list of putative α-l-glucosidases (Table S1); and oligonucleotide sequences used in this study (Table S2) (PDF)
Author Contributions
‡ R.S. and H.K. contributed equally to this work.
This work was partly supported by the Japanese Society for the Promotion of Science KAKENHI (grant no. 18K19159).
The authors declare no competing financial interest.
Supplementary Material
References
- Shimizu T.; Takaya N.; Nakamura A. An l-glucose catabolic pathway in Paracoccus species 43P. J. Biol. Chem. 2012, 287, 40448–40456. 10.1074/jbc.M112.403055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrissat B.; Davies G. Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 1997, 7, 637–644. 10.1016/S0959-440X(97)80072-3. [DOI] [PubMed] [Google Scholar]
- Drula E.; Garron M.-L.; Dogan S.; Lombard V.; Henrissat B.; Terrapon N. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. 10.1093/nar/gkab1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempton J. B.; Withers S. G. Mechanism of Agrobacterium β-glucosidase: Kinetic studies. Biochemistry 1992, 31, 9961–9969. 10.1021/bi00156a015. [DOI] [PubMed] [Google Scholar]
- Sulzenbacher G.; Bignon C.; Nishimura T.; Tarling C. A.; Withers S. G.; Henrissat B.; Bourne Y. Crystal structure of Thermotoga maritima α-L-fucosidase: Insights into the catalytic mechanism and the molecular basis for fucosidosis. J. Biol. Chem. 2004, 279, 13119–13128. 10.1074/jbc.M313783200. [DOI] [PubMed] [Google Scholar]
- Summers E. L.; Moon C. D.; Atua R.; Arcus V. L. The structure of a glycoside hydrolase 29 family member from a rumen bacterium reveals unique, dual carbohydrate-binding domains. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2016, 72, 750–761. 10.1107/S2053230X16014072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster-Böckler B.; Schultz J.; Rahmann S. HMM Logos for visualization of protein families. BMC Bioinf. 2004, 5, 7 10.1186/1471-2105-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuyama M.; Saburi W.; Mori H.; Kimura A. α-Glucosidases and α-1,4-glucan lyases: structures, functions, and physiological actions. Cell. Mol. Life Sci. 2016, 73, 2727–2751. 10.1007/s00018-016-2247-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst H. A.; Lo Leggio L.; Willemoës M.; Leonard G.; Blum P.; Larsen S. S. Structure of the Sulfolobus solfataricus α-glucosidase: implications for domain conservation and substrate recognition in GH31. J. Mol. Biol. 2006, 358, 1106–1124. 10.1016/j.jmb.2006.02.056. [DOI] [PubMed] [Google Scholar]
- Okuyama M.; Kaneko A.; Mori H.; Chiba S.; Kimura A. Structural elements to convert Escherichia coli α-xylosidase (YicI) into α-glucosidase. FEBS Lett. 2006, 580, 2707–2711. 10.1016/j.febslet.2006.04.025. [DOI] [PubMed] [Google Scholar]
- Janeček Š.; Svensson B.; MacGregor E. A. A remote but significant sequence homology between glycoside hydrolase clan GH-H and family GH31. FEBS Lett. 2007, 581, 1261–1268. 10.1016/j.febslet.2007.02.036. [DOI] [PubMed] [Google Scholar]
- Sakurama H.; Tsutsumi E.; Ashida H.; Katayama T.; Yamamoto K.; Kumagai H. Differences in the substrate specificities and active-site structures of two α-l-fucosidases (glycoside hydrolase family 29) from Bacteroides thetaiotaomicron. Biosci., Biotechnol., Biochem. 2012, 76, 1022–1024. 10.1271/bbb.111004. [DOI] [PubMed] [Google Scholar]
- Fersht A.Structure and Mechanism in Protein Science: a Guide to Enzyme Catalysis and Protein Folding; W. H. Freeman and Company: New York, 1998; pp 349–376. [Google Scholar]
- Kumar P. A.; Srinivas T. N. R.; Madhu S.; Sravan R.; Singh S.; Naqvi S. W. A.; Mayilraj S.; Shivaji S. Cecembia lonarensis gen. nov., sp. nov., a haloalkalitolerant bacterium of the family Cyclobacteriaceae, isolated from a haloalkaline lake and emended descriptions of the genera Indibacter, Nitritalea and Belliella. Int. J. Syst. Evol. Microbiol. 2012, 62, 2252–2258. 10.1099/ijs.0.038604-0. [DOI] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Kovaľová T.; Kovaľ T.; Benešová E.; Vodičková P.; Spiwok V.; Lipovová P.; Dohnálek J. Active site complementation and hexameric arrangement in the GH family 29; a structure–function study of α-l-fucosidase isoenzyme 1 from Paenibacillus thiaminolyticus. Glycobiology 2019, 29, 59–73. 10.1093/glycob/cwy078. [DOI] [PubMed] [Google Scholar]
- Coyle T.; Wu L.; Debowski A. W.; Davies G. J.; Stubbs K. A. Synthetic and crystallographic insight into exploiting sp2 hybridization in the development of α-l-fucosidase inhibitors. ChemBioChem 2019, 20, 1365–1368. 10.1002/cbic.201800710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobucci-Ponzano B.; Trincone A.; Giordano A.; Rossi M.; Moracci M. Identification of the catalytic nucleophile of the family 29 α-l-fucosidase from Sulfolobus solfataricus via chemical rescue of an inactive mutant. Biochemistry 2003, 42, 9525–9531. 10.1021/bi035036t. [DOI] [PubMed] [Google Scholar]
- van Bueren A. L.; Ardèvol A.; Fayers-Kerr J.; Luo B.; Zhang Y.; Sollogoub M.; Blériot Y.; Rovira C.; Davies G. J. Analysis of the reaction coordinate of α-l-fucosidases: a combined structural and quantum mechanical approach. J. Am. Chem. Soc. 2010, 132, 1804–1806. 10.1021/ja908908q. [DOI] [PubMed] [Google Scholar]
- Robb C. S.; Hobbs J. K.; Pluvinage B.; Reintjes G.; Klassen L.; Monteith S.; Giljan G.; Amundsen C.; Vickers C.; Hettle A. G.; Hills R.; Nitin; Xing X.; Montina T.; Zandberg W. F.; Abbott D. W.; Boraston A. B. Metabolism of a hybrid algal galactan by members of the human gut microbiome. Nat. Chem. Biol. 2022, 18, 501–510. 10.1038/s41589-022-00983-y. [DOI] [PubMed] [Google Scholar]
- Osanjo G.; Dion M.; Drone J.; Solleux C.; Tran V.; Rabiller C.; Tellier C. Directed evolution of the α-l-fucosidase from Thermotoga maritima into an α-l-transfucosidase. Biochemistry 2007, 46, 1022–1033. 10.1021/bi061444w. [DOI] [PubMed] [Google Scholar]
- Cobucci-Ponzano B.; Conte F.; Bedini E.; Corsaro M. M.; Parrilli M.; Sulzenbacher G.; Lipski A.; Piaz F. D.; Lepore L.; Rossi M.; Moracci M. β-Glycosyl azides as substrates for α-glycosynthases: preparation of efficient α-l-fucosynthases. Chem. Biol. 2009, 16, 1097–1108. 10.1016/j.chembiol.2009.09.013. [DOI] [PubMed] [Google Scholar]
- Slavin J. Fiber and prebiotics: mechanisms and health benefits. Nutrients 2013, 5, 1417–1435. 10.3390/nu5041417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lordan C.; Thapa D.; Ross R. P.; Cotter P. D. Potential for enriching next-generation health-promoting gut bacteria through prebiotics and other dietary components. Gut Microbes 2020, 11, 1–20. 10.1080/19490976.2019.1613124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuber M.; Bevill R. D.; Osborn M. J. Aldoheptoses in the O-antigenic lipopolysaccharide of Salmonella typhimurium and other gram-negative bacteria. I. Chemical synthesis of l-glycero-d-mannoheptose and β-l-glycero-d-mannoheptopyranosyl 1-phosphate. Biochemistry 1968, 7, 3303–3308. 10.1021/bi00849a038. [DOI] [PubMed] [Google Scholar]
- Okuyama M.; Yoshida T.; Hondoh H.; Mori H.; Yao M.; Kimura A. Catalytic role of the calcium ion in GH97 inverting glycoside hydrolase. FEBS Lett. 2014, 588, 3213–3217. 10.1016/j.febslet.2014.07.002. [DOI] [PubMed] [Google Scholar]
- Kuraku S.; Zmasek C. M.; Nishimura O.; Katoh K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, W22–W28. 10.1093/nar/gkt389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenteros J. J. A.; Tsirigos K. D.; Sønderby C. K.; Petersen T. N.; Winther O.; Brunak S.; Von Heijne G.; Nielsen H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. 10.1038/s41587-019-0036-z. [DOI] [PubMed] [Google Scholar]
- Letunic I.; Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki S.; Niki H. Exonuclease III (XthA) enforces in vivo DNA cloning of Escherichia coli to create cohesive ends. J Bacteriol. 2019, 201, e00660-18 10.1128/JB.00660-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y.; Li K.; Li J.; Wang T.; Gu L.; Xun L. T5 exonuclease-dependent assembly offers a low-cost method for efficient cloning and site-directed mutagenesis. Nucleic Acids Res. 2019, 47, e15 10.1093/nar/gky1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F. W. Protein production by auto-induction in high density shaking cultures. Protein Expression Purif. 2005, 41, 207–234. 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/s0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P. V.; Grosse-Kunstleve R. W.; Echols N.; Headd J. J.; Moriarty N. W.; Mustyakimov M.; Terwilliger T. C.; Urzhumtsev A.; Zwart P. H.; Adams P. D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 352–367. 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; Swindells M. B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates were deposited in the Protein Data Bank with accession codes 7XSF, 7XSG, and 7XSH. The authors declare that all data are contained within the manuscript.