

Abstract
Introduction:
Antiretroviral therapy (ART) has transformed prognoses for HIV-1-infected individuals but requires lifelong adherence to prevent viral resurgence. Targeted elimination or permanent deactivation of the latently infected reservoir harboring integrated proviral DNA, which drives viral rebound, is a major focus of HIV-1 research.
Areas covered:
This review covers the current approaches to developing curative strategies for HIV-1 that target the latent reservoir. Discussed herein are shock and kill, broadly neutralizing antibodies (bNAbs), block and lock, Chimeric antigen receptor (CAR) T cells, immune checkpoint modulation, clustered regularly interspaced short palindromic repeats (CRISPR) / CRISPR-associated protein 9 (Cas9) coreceptor ablation, and CRISPR/Cas9 proviral excision. Emphasis in this review is placed on CRISPR/Cas9 proviral excision/inactivation. Recent advances and future directions towards discovery and translation of HIV-1 therapeutics are discussed.
Expert opinion:
CRISPR/Cas9 proviral targeting fills a niche amongst HIV-1 cure strategies by directly targeting the integrated provirus without the necessity of an innate or adaptive immune response. Each strategy discussed in this review has shown promising results with the potential to yield curative or adjuvant therapies. CRISPR/Cas9 is singular among these in that it addresses the root of the problem, integrated proviral DNA, with the capacity to permanently remove or deactivate the source of HIV-1 recrudescence.
Keywords: HIV-1, CRISPR/Cas9, proviral excision, shock and kill, bNAbs, CAR T, block and lock, immune blockade, coreceptor, CCR5, off-target, delivery
1. Introduction
Human immunodeficiency virus type 1 (HIV-1) infection remains a global pandemic now in its fourth decade. Combined anti-retroviral therapy (cART) has been transformative for prognoses, but ART regimens must be taken continuously to maintain viral suppression. Despite the success of cART at reducing viral load, cART does not cure HIV-1 infection and patients still experience disease manifestations including impaired immune response and HIV-1-associated neurocognitive disorders (HAND) [1,2]. In the absence of cART, patients experience a resurgence of viral load making ART regimens a lifelong necessity. Viral rebound is largely due to the latent reservoir of infected cells harboring integrated proviral HIV-1 DNA [3]. Accordingly, a major goal of HIV-1 research is to target the viral reservoir [4]. Given that the integrated provirus makes the latent reservoir a persistent problem, gene editing tools, specifically clustered regularly interspaced short palindromic repeats (CRISPR) / CRISPR-associated protein 9 (Cas9), are an ideal technology for removing the source of viral rebound and reducing the latent reservoir. In this review, we examine the progress that has been made to date on developing a therapeutic strategy using the CRISPR/Cas9 system. We also discuss the state of progress on the major complementary or alternative strategies for targeting the viral reservoir. A summary and comparison of each major strategy is presented in Table 1. Each strategy discussed has merit, and each has promising results making it worthy of pursuit. CRISPR/Cas9 in particular fills a niche that is separate from other strategies which denotes the possibility of a unique contribution to the existing range of therapeutic options.
Table 1:
Comparison of HIV-1 curative strategies currently in development
| Therapeutic strategy | HIV-1 Lifecycle Stage Targeted | Mechanism of Action | In vitro results | In vivo results | Advantages | Disadvantages |
|---|---|---|---|---|---|---|
| Shock and Kill | Transcription | Latency reversal to induce immune killing of reactivated cells | Ex vivo reservoir reduction when combined with CTLs and ABT-199[1] | Latency reversal; limited reservoir reduction [2] | HDACis are approved for human use; leverages innate immune response | Evidence that CD8+ T cells cannot efficiently clear infected cells [3] |
| bNAbs | Mature virions | Neutralization of virions via Env antigen recognition | Limit viral replication [4] | Transient viral suppression in RMs and humanized mice [5, 6] | Inhibit cell-free and cell-associated infection [7] | Transient effects[5, 6] |
| Block and Lock | Integration | dCA blocks Tat by binding to TAR | Prevents viral reactivation [8] | Delayed viral rebound in BLT mice [9] | Targets replication competent and incompetent virus [10] | Does not directly remove proviral DNA from reservoir |
| CAR T Cells | Infected cells | Recognizes and kills HIV-infected cells | Viral suppression [11] | Reduced infected cells by 97% in humanized mice [12] | Elimination of viral reservoir | Potential for sCRS and off-target effects; limited persistence [13–15] |
| Immune Checkpoint Modulation | Infected cells | Block immune-mediating receptors to reverse immune exhaustion | Reverses T-cell exhaustion; restore NK cells support [16] | Modulation of CTLA4 or OX40 enhances vaccine effectiveness in RM and mouse models [17] | Leverages innate immune response | Transient effects [18] |
| CRISPR/Cas9 Coreceptor Ablation | Binding | Ex vivo ablation of host coreceptor genes to create HIV-resistant cells for reinfusion | CCR5 ablation; CXCR4 ablation; CCR5 and CXCR4 ablation in tandem; all confer HIV-1 resistance [19–22] | R5-tropic resistance induced in humanized mice [23] | Recreates the only recognized cure of HIV-1-infected patients [24] | Does not prevent cell-associated infection; May not reach tissue resident cells [24] |
| CRISPR/Cas9 Proviral Targeting | Integrated provirus | In vivo targeting of integrated provirus for excision or inactivation | HIV-1 elimination in cell lines and primary cells [25–33] | Elimination of infection from a subset of humanized mice [34] | Targets the root cause of latency; directly removes proviral DNA [25–33] | Targeted delivery remains a challenge; adoptive transfer of T regulatory cells may be necessary to overcome adaptive immune response [35] |
2. Alternative strategies to CRISPR/Cas9 for targeting the latent viral reservoir
2.1. Shock and Kill
The principle of the shock and kill strategy is to use latency reversing agents (LRAs), to induce HIV-1 gene expression allowing host CD8+ T cells to find and eliminate HIV-1-infected cells in latent reservoirs (Figure 1 A) [5]. Classes of LRAs include histone methyltransferase (HMT) inhibitors, DNA methyltransferase inhibitors, bromodomain inhibitors, protein kinase C (PKC) agonists, and histone deacetylase inhibitors (HDACi), Several of these LRAs have advanced to clinical trials, including the HDACi vorinostat, panobinostat, and romidepsin [6]. Some successful preliminary results have been achieved with this strategy. HDACi have been shown to reverse latency and induce HIV-1 RNA expression in an SIV-infected rhesus macaque (RM) model [7], in ex vivo patient-derived CD4+ T cells [8], and in vivo in clinical trials (NCT01319383, (NCT01365065). [8,9]. HDACi reactivation has also been shown to induce immune clearance ex vivo [10]. The drawback of the shock and kill approach is that HDACi have been demonstrated, ex vivo, in vivo, and in clinical trials to have limited or no effect on the size of the latent reservoir [6,8,10,11]. One notable exception is a recently successful study utilizing B-cell lymphoma 2 (BCL-2) antagonist, ABT-199, based on the observation that BCL-2 overexpression is associated with CD4+ T cells that evade cytotoxic T lymphocytes (CTLs) [12]. Following LRA treatment ex vivo, the combination of CTLs and ABT-199 was shown to enable HIV-1 reservoir reduction. Whether this success can translate to in vivo reservoir reductions remains to be tested.
Figure 1: Each of the HIV-1 curative strategies currently being investigated fills a distinct mechanistic niche, acting at a separate point in the viral life cycle.
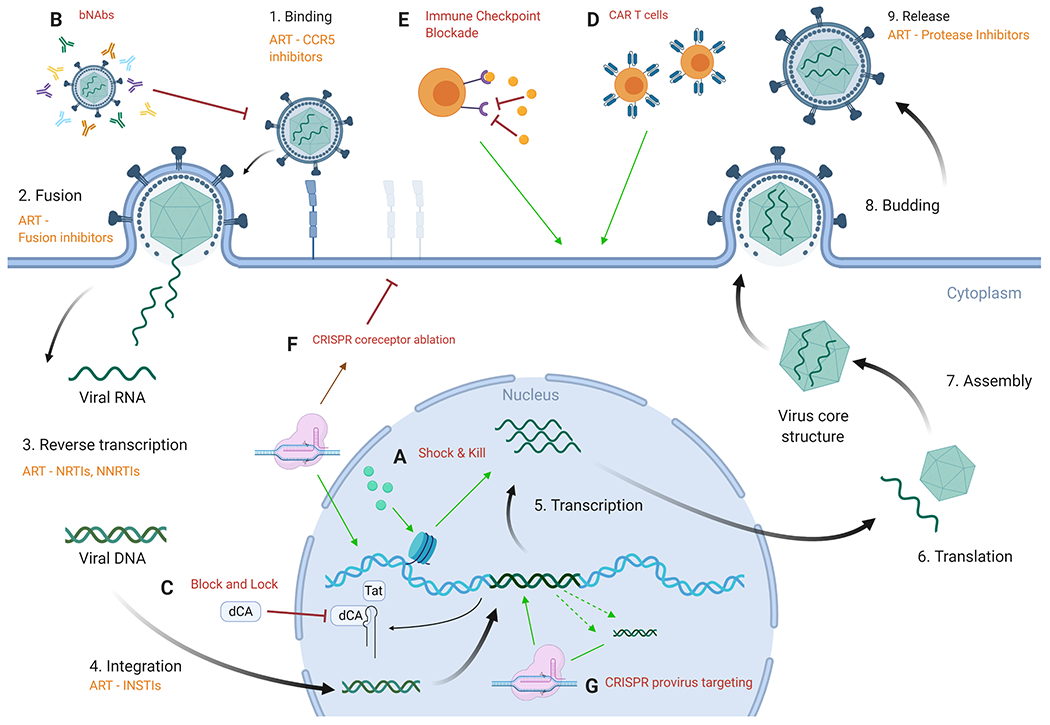
The nine main steps of HIV-1 viral replication are depicted. In orange are the ARTs that target that step in the replication cycle. This is overlaid with each of the major cure strategies labeled A to G. (A) Shock and kill uses LRAs to bring the virus out of latency, initiating transcription and subsequent translation in an effort to instigate immune clearance. (B) Use of bNAbs seeks to neutralize the virus by recognition of the extensive antigenic diversity found in the HIV-1 Env. (C) Block and lock attempts to block transcription of viral proteins. Using dCA in the block and lock strategy blocks Tat from binding to TAR. (D) CAR T cells are CD8+ T cells transfused with chimeric antigen receptors ex vivo and reinfused into the patient in order to recognize and kill HIV-1-infected cells. (E) Immune checkpoint blockade works by blocking receptors such as PD1 and CLTA4 to reverse immune exhaustion and kill HIV-1-infected cells. (F) CRISPR/Cas9 coreceptor ablation targets endogenous coreceptor genes to effect mutagenesis during endogenous repair thereby stopping coreceptor expression. (G) CRISPR/Cas9 targeting of the HIV-1 provirus can lead to excision of proviral DNA in the event of two simultaneous cleavage events or disruption of transcription via mutagenesis during the repair of singular cleavage events. Figure created with license for BioRender.
Another important shock and kill approach is the use of killer T cell vaccines. Inducing T cells to broadly target HIV proteins is an approach towards creating a functional cure [13,14]. This approach has shown favorable results in challenge studies with non-human primates and has yielded some promising outcomes in phase I clinical trials. For example, a recent phase I clinical trial using romidepsin to reverse latency and the MVA.HIVconsv vaccine, was able induce sustained viral control in 23% of treated patients. Although significant reservoir reduction was not achieved, sustained viral control in a portion of treated patients is a promising result and an important step towards a functional cure. Notably the patients selected for this study were early-treated individuals [15,16]. More work remains to bring this treatment strategy to a broad range of infected patients and demonstrate effectiveness against patients who are not early-treatment cases. Nonetheless, this approach may yield a functional cure strategy that allows cessation of ART in many cases.
2.2. Broadly Neutralizing Antibodies
Another strategy to reduce the HIV-1 proviral reservoir is to confer resistance to HIV-1 infection using broadly neutralizing antibodies (bNAbs) directed against HIV-1. Successful application of this approach would create a vaccine-like effect resulting in viral neutralization of an acute HIV-1 infection or following activation of latent infection in the reservoir (Figure 1 B). Progress has been made using this approach in that bNAbs have been identified that act against both cell-free and cell-associated infection [17] with bNAbs shown to limit viral replication in vitro [18]. In a rhesus macaque (RM) simian/human immunodeficiency virus (SHIV) model, bNAbs used to treat chronic infection have exhibited transient viral suppression [19,20]. It appears that the timing of bNAb treatment is important, with bNAbs administered early during acute infection leading to prolonged viral suppression [19]. This suggests a possible role for bNAbs in treatment regimens for early infection. Other successful results have been demonstrated in a humanized mouse model. For example, the use of adenovirus serotype 5 (Ad5) and adeno-associated virus serotype 1 (AAV1) vectors to deliver bNAbs, have shown efficacy in viral control in humanized mice [21]. It is important to highlight the fact that Abs in general have a finite half-life and thus the need for a suitable delivery system would be warranted.
Despite promising results, there are drawbacks to the use of bNAbs to target HIV-1. For instance, bNAbs that stop cell-free infection only partially stop cell-to-cell transmission for the same virus. The mechanism of this resistance remains unknown [22]. Furthermore, although transient viral suppression is a promising proof-of-concept that bNAbs can work to inhibit viral replication, the systemic half-life of bNAbs will limit their utility against chronic HIV-1 infection if this issue cannot be overcome [20]. A further confounding factor is that a host antibody response has been shown in RMs treated with anti-HIV-1 antibodies delivered by AAV. The anti-antibody response was observed in 17 out of 20 monkeys. This study did not report on the efficacy of the treatments with respect to viral control [23]. Additionally, the genetic diversity and corresponding antigenic diversity of HIV-1 complicates the use of bNAbs for HIV-1 and could inhibit broad applicability of this approach [24]. Lastly, even if these challenges can be addressed, successful implementation of bNAbs against the latent HIV-1 reservoir will not directly remove proviral DNA from the latent pool.
2.3. Block and Lock
The strategy of block and lock techniques for targeting HIV-1 is to block viral transcription and lock the virus in latency thereby preventing reactivation of the latent reservoir. Key examples of this technique utilize small molecule inhibitors to achieve this outcome (Figure 1 C). Examples include the Tat inhibitor didehydro-cortistatin A (dCA) and LEDGINs, small molecules that bind to the LEDGF/p75 binding pocket of HIV-1 integrase [25–27]. Tat inhibition with dCA works by adhering to the TAR-binding domain of Tat thereby inhibiting transactivation of the HIV-1 promoter [27]. LEDGINs work by inhibition of the interaction between LEDGF/p75 and integrase. This has been demonstrated in vitro to cause an increase in the fraction of integrated provirus having a transcriptionally silent phenotype.
The primary advantage of the block and lock approach with dCA is that it prevents viral reactivation [26]. Additionally, block and lock strategies can target both replication competent and incompetent virus [28]. This is significant because replication defective viruses have been shown to produce viral transcripts and toxic proteins [29,30]. The drawbacks to this strategy are similar to those of bNAbs. The effects are transient. Viral rebound was delayed by 19 days in BLT mice treated with dCA [31]. This is a promising demonstration of proof-of-concept for the block and lock approach but unless the effect can be sustained, this approach lacks clinical utility. In this way the block and lock strategy using dcA is more akin to ART than to other curative strategies. It would constitute another drug requiring lifelong adherence. In the case of LEDGINs, treatment would need to be administered early to be effective; there is a limited window of opportunity for LEDGINs to act [25]. Finally, as with bNAbs, a downside to the block and lock strategy is that it does not directly remove proviral DNA from the latent reservoir. Besides small molecule inhibitors, additional block and lock strategies include but are not limited to using RNA interference through siRNA or shRNA molecules, HIV-encoded antisense proteins, and Tat inhibitors [32]. A thorough treatment of the full range of block and lock strategies is beyond the scope of this review however we refer the reader to several high-quality in-depth reviews on the subject [25,28,32].
2.4. Chimeric antigen receptor (CAR) T-cell therapy
The principle of chimeric antigen receptor (CAR) T-cell therapy for HIV-1 treatment involves ex vivo transduction of patient CD8+ T cells with a CAR construct. Subsequent expansion of transduced cells and reinfusion into the patient then results in CAR T cells killing HIV-1-infected cells (Figure 1 D) thereby reducing the pool of infected cells [33]. Some experimental success has been achieved with this approach. HIV-1-targeting CAR T cells using bNAbs have shown HIV-1 suppression in vitro [34]. And a two-molecule CAR approach, referred to as duoCAR, introduced on a single lentiviral vector, has been demonstrated as highly effective in a humanized NOD scid gamma (NSG) mouse model. The two molecule duoCAR design has two Env binding sites to target differentially accessible gp120 binding sites for which accessibility depends on conformational changes that occur after CD4 and gp120 engagement. This approach reduced HIV-1-infected cells by over 97% in vivo. Furthermore, duoCAR was effective against strains of HIV-1 that were resistant to bNAbs [35]. CAR T-cell therapies are promising with clinical trials underway for HIV-1-targeting CAR T cells using bNAbs (NCT03240328) and zinc-finger nuclease (ZFN) disruption of CCR5 in CD4-CAR T cells (NCT03617198).
Nonetheless, there are aspects of the therapeutic development of CAR T cells for HIV-1 infection that may be difficult to overcome. CAR T-cell treatment has a potential to cause severe cytokine release syndrome (sCRS) [33]. In addition, CAR T cells in vitro do not eliminate follicular dendritic cells (FDCs) with surface-bound HIV-1, a component of the latent reservoir [36]. Also, difficulties with HIV-1-targeting CAR T-cell persistence and lack of expansion have been reported [37,38]. There is also a potential for problematic off-target effects as has been observed given the B-cell aplasia observed in studies using CAR T cells to treat leukemia by targeting the B-cell antigen CD19 [39]. Assuming these factors can be addressed, CAR T cells offer a promising approach to HIV-1 treatment.
2.5. Immune checkpoint blockade
Immune checkpoint modulation is another approach to HIV-1 treatment that shows promise as a curative strategy but has not yet been translated to a successful therapeutic strategy. Immune checkpoint modulation has been successfully implemented as a cancer therapeutic and that success makes its potential use against HIV-1 infection enticing [40]. HIV-1 infection leads to the exhaustion of HIV-1-specific T cells and the implementation of immune checkpoint blockades holds promise for reversing that effect (Figure 1 E). Immune checkpoint proteins such as programmed cell death protein (PD1) and cytotoxic T lymphocyte-associated protein 4 (CTLA4) mitigate pathogenic immune response but can also inhibit clearance of infection. Immune checkpoint blockade may enhance the adaptive immune response by increasing the functionality of HIV-1-specific T cells and it may also serve to increase the effectiveness of vaccines or any T- cell based approach.
For example, blockade of PD-1 or IL-10 has been shown to reinvigorate CD4+ T-cell function in vitro and restore support for NK cells [41]. This study provides proof-of-concept that immune exhaustion can be reversed and result in improvement of innate and adaptive immune function. In another study, examining the effect of immune checkpoint modulation on antibody induction in HIV-1 infection, antibodies targeting immune checkpoint proteins were administered to RM and bNAb immunoglobulin knock-in mice along with vaccines for HIV-1 envelope (Env) [42]. Results indicate that modulation of CTLA-4 or OX40 checkpoints enhances HIV-1 Env vaccine effectiveness. These studies and others suggest that immune checkpoint modulation strategies could be developed as important curative or adjuvant therapies for HIV-1 treatment. It is not clear whether using anti-PD-1 and anti-CTLA-4 antibodies would reduce latent reservoir since CD8+ T cells are excluded from B-cell follicles where the majority of cells harboring the virus reservoir include follicular helper T cells (Tfh).
3. Use of CRISPR/Cas9 to cure HIV-1 infection
CRISPR/Cas9 technology has given rise to two approaches to eliminating the latent HIV-1 reservoir. One is ex vivo ablation of genes expressing the CCR5 and CXCR4 coreceptors (Figure 1 F) [43]. In this approach, hematopoietic stem cells (HSCs) are extracted from patients and reinfused following CRISPR/Cas9 treatment to delete the receptors. The treated cells are then resistant to infection and if proliferation is successful, the resistant cells supplant HIV-1-susceptible cells. The other approach is to target the HIV-1 proviral DNA in the latent reservoir with the goal of either excising the provirus or portions of the provirus with two simultaneous CRISPR/Cas9 cleavage events or ablating viral promoters to silence transcription (Figure 1 G) [44].
3.1. CRISPR/Cas9 for ex vivo ablation of coreceptor genes
Conceptually, ex vivo ablation of coreceptor genes is an attempt to recreate the CCR5Δ32 mutation which has led to a functional cure for HIV-1 infection in two cases referred to as the Berlin and London patients respectively [33,43]. Notably, CXCR4 tropism and cell-to-cell transmission are not addressed by exclusive CCR5 ablation [33,43]. Yet the infusion of cells from patients carrying the CCR5Δ32 mutation has yielded success, albeit in a limited number of cases. Additionally, in vitro progress to recreate this effect has been achieved. CCR5 ablation using CRISPR/Cas9 has been demonstrated to produce HIV-1 resistance in vitro [45]. Other studies have focused on CXCR4 ablation using CRISPR/Cas9, which has also been demonstrated to produce HIV-1 resistance in vitro [46,47]. Additionally, simultaneous ablation of both CCR5 and CXCR4 co-receptors using CRISPR/Cas9 has been achieved in vitro [48]. Beyond that, CRISPR editing of CCR5 in HSCs engrafted into humanized mice has been shown to induce resistance to infection with R5-tropic virus [49]. Drawbacks to the approach of coreceptor ablation with CRISPR/Cas9 include the challenge of ex vivo editing efficiency and proliferation following transfusion of ex vivo edited cells. Furthermore, this strategy as it is designed, may not block infection in all latent reservoirs; tissue resident cells for example, may not be affected by this therapy. In addition, cell-associated viral infection is not addressed by coreceptor ablation [43]. Nonetheless this therapy may provide a valuable addition to the current range of available treatments and guarded optimism for a highly impactful treatment is fueled by the success of the Berlin and London patients.
3.2. Targeting the HIV-1 provirus in the latent reservoir using CRISPR/Cas9
Targeting the HIV-1 proviral DNA in the latent reservoir is a distinctly different use of CRISPR/Cas9 from coreceptor ablation. Although challenges remain in order to translate this technology to a viable therapy, the approach of targeting the provirus using CRISPR/Cas9 has seen significant progress. Proof-of-concept has been published for multiple aspects of this approach. For example, elimination of HIV-1 infection using this method has been achieved in cell lines and primary cells as well as animal models [50–58]. This makes a strong case for the translational capacity of this technology. Specifically, the most important progress to date is the elimination, without viral rebound, of HIV-1 infection from a subset of humanized mice using a combination of long-acting slow-effective release antiviral therapy (LASER ART) and CRISPR/Cas9 treatments delivered in vivo by AAV [59]. The study found that while neither LASER ART nor CRISPR/Cas9 were able to eliminate infection as individual treatments, the combination of the two was effective in a subset of treated humanized mice. CRISPR/Cas9 treatment involved a combination of guide RNAs (gRNAs) targeting the LTR and Gag regions. Adoptive transfer of immunocytes from animals cleared of virus into uninfected mice did not propagate HIV-1 infection. And in animals cleared of infection, virus was not detected by ddPCR in multiple tissue compartments, namely the blood, bone marrow, lymphoid tissue, and brain. The results of this study establish proof-of-concept that viral cure is possible using CRISPR/Cas9 in conjunction with LASER ART and that delivery of the CRISPR/Cas9 system in vivo is feasible using AAV vectors.
3.3. Optimal target sites for CRISPR/Cas9
An important aspect for the translational capacity of this technology is the choice of an optimal target for the CRISPR/Cas9 system. The length of the HIV-1 proviral genome is approximately 10,000 base pairs while the targeting mechanism of CRISPR/Cas9, the gRNA, is typically between 17 and 35 base pairs in length (usually 20). Potential targets, however, are limited to sequences containing a protospacer adjacent motif (PAM), necessary for Cas9 binding and subsequent cleavage. For Streptococcus pyogenes Cas9 (SpCas9) the PAM sequence is NGG. Evaluation of potential targets has demonstrated that the long-terminal repeat (LTR) region of the HIV-1 genome is an effective target for viral deactivation [60]. By targeting the LTR, translation of all HIV-1 proteins can be disrupted, because the LTR acts as the promoter for the virus. In addition, given the provirus has a copy of the LTR on each end, a portion of the events can excise the entire genome (the efficiency of this type of event is still under investigation). Accordingly, the LTR is the most common HIV-1 CRISPR/Cas9 target investigated thus far [50,52–59,61–71]
3.4. Mechanism of viral deactivation
The mechanism of action for viral deactivation has also been investigated. There are three possible outcomes following successful CRISPR/Cas9 cleavage: excision, mutation, and inversion [44]. Excision of the proviral genome is a desirable outcome involving two simultaneous CRISPR/Cas9 cleavage events leading to the removal of the segment of proviral DNA which is disconnected at both ends from its integration site. This requires a timing in which simultaneous CRISPR/Cas9 cleavage events both occur before the resultant double-strand breaks (DSBs) can be stabilized by a DNA damage response. This outcome is facilitated by targeting of the LTR because the identical LTR sequences which bookend the proviral genome allow two cleavage locations to be generated with a single gRNA sequence. Inversion occurs when an excised fragment is reinserted in the same site during endogenous DNA repair, but the fragment is rotated 180 degrees and the repaired ends are ligated to the wrong sides of the excision gap. Mutational outcomes leverage the nature of non-homologous end-joining (NHEJ). NHEJ has been shown to introduce insertions and deletions (indels) during repair which can disrupt the function of targeted genes and damage transcriptional start sites thereby inhibiting the expression of HIV-1 proteins when targeting the LTR.
A study measured relative rates for each outcome in SupT1 cells (a T-cell line line) infected with HIV-1 (LAI) or HIV-1 ( rtTA) and stably expressing Cas9 with one of two pairs of gRNAs [44]. Pairs of gRNAs targeted either Gag and TatRev or Gag and Env. This study reports that the relative frequencies for each outcome across the range of conditions tested were 1.2 – 10.2% for inversion, 1.5 – 39.3% for excision, and 34.7 – 76.5 % for mutation. Notably, outcome ratios were not correlated to target separation distance in base pairs. Inversion was always the least frequent outcome. Mutation was the most frequent outcome for most conditions except for the case where Gag and TatRev gRNAs were tested against HIV-rtTA. In that case, excision was observed at 38.3% and mutation at 34.7%. The same study tested a pair of gRNAs targeting two Gag locations. Results indicated that the mutation occurred most frequently, followed by excision, with negligible rates of inversion. Results of this study suggest that inversion is a rare event and does not likely contribute significantly to successful viral deactivation whereas excision and mutation are the more common mechanisms by which CRISPR/Cas9 gRNA pairs effect HIV-1 inactivation.
In their study demonstrating HIV-1 eradication from a subset of humanized mice, Dash et al. have also reported on excision rates using ddPCR on ex vivo tissue from treated mice [59] with variable excision rates reported between treated mice. Their highest reported excision rate in a single mouse was approximately 80% for a fragment between the 5’LTR and Gag and also approximately 80% for a fragment between Gag and the 3’LTR. They did not report comparative rates for other repair outcomes. Taken together the results of these studies indicated that repair outcome rates were dependent on multiple factors. More data from future studies will be necessary for reliable prediction of repair outcomes from dual targeting with CRISPR/Cas9.
4. Additional problems currently being addressed in the development of CRISPR/Cas9 HIV-1 therapy
4.1. Measuring the adaptive immune response to Cas9
Consideration of the adaptive immune response to Cas9 will be important to therapeutic translation of CRISPR/Cas9 technology as it may affect both safety and efficacy. Several reports examined the immune response to the Cas9 protein [72–74]. In a screening of 200 human serum samples using enzyme-linked immunosorbent assays (ELISA), the prevalence of Staphylococcus aureus Cas9 (saCas9) and SpCas9 antibodies was reported at 10% and 2.5% respectively [74]. A subsequent study similarly used ELISA to screen human serum samples finding a prevalence of 78% for SaCas9 antibodies and 58% for SpCas9 [72]. Additionally, this study found antigen-specific T cells for SaCas9 in 78% of human samples and SpCas9 in 67% of samples. The results of these studies, indicating that a portion of the population have preexisting antibodies to the Cas9 protein, are not surprising; Streptococcus pyogenes and Staphylococcus aureus are common infectious agents in the human population [75,76]. Consequently, there may be a need to evaluate patients for adaptive immunity to Cas9 in treatment regimens for in vivo gene therapy. Yet, despite the data showing the immunogenicity of both orthologs of the Cas9 protein, even therapies involving in vivo CRISPR/Cas9 delivery may elicit a negligible immune response provided that the protein is not expressed on the surface of the delivery vehicle or that Cas9 expression is transient. In any case, further study is warranted to examine this question in greater detail.
Regarding the cell-specific response, CD8+ T cells have been shown to recognize Cas9 epitopes presented on MHC-1 molecules resulting from intracellular Cas9 degradation. However, there is data to suggest that the response will be manageable. An examination of SpCas9-reactive T cells from healthy donors found CD4+ and CD8+ T cells specific to SpCas9, but the majority of the CD4+ T cells were T regulatory cells. Cells expressing SpCas9, indicated by GFP, were not eliminated by SpCas9-specific CD8+ T cells in vitro [73].
It is important to note that these results were obtained in vitro and that more in-depth research is surely warranted. However, such results are indeed promising for the potential translational capacity of CRISPR/Cas9. These results indicate that the adaptive immune system does not selectively kill SpCas9-expressing cells. This has important implications for potential CRISPR/Cas9 therapy. For CD8+ T cells targeting SpCas9-expressing cells, the effector function of these cells can be dampened by T regulatory cells. These results also suggest a potential solution for an adaptive immune response to CRISPR/Cas9 therapy. Adoptive transfer of T regulatory cells reactive to Cas9 may be able to mitigate an adaptive immune response to CRISPR/Cas9 therapy (Figure 2 A) [73]. This possibility is supported by clinical data. For example, safe, well-tolerated adoptive transfer of T regulatory cells has been shown to mitigate inflammation in patients receiving kidney transplants [77]. This approach could preclude the need for an approach based on global immune suppression. However, tissue-resident immune responses differ from peripheral blood [78]. Further study is necessary to address this aspect of therapeutic development.
Figure 2: The potential pitfalls of the CRISPR/Cas9 system have solutions.
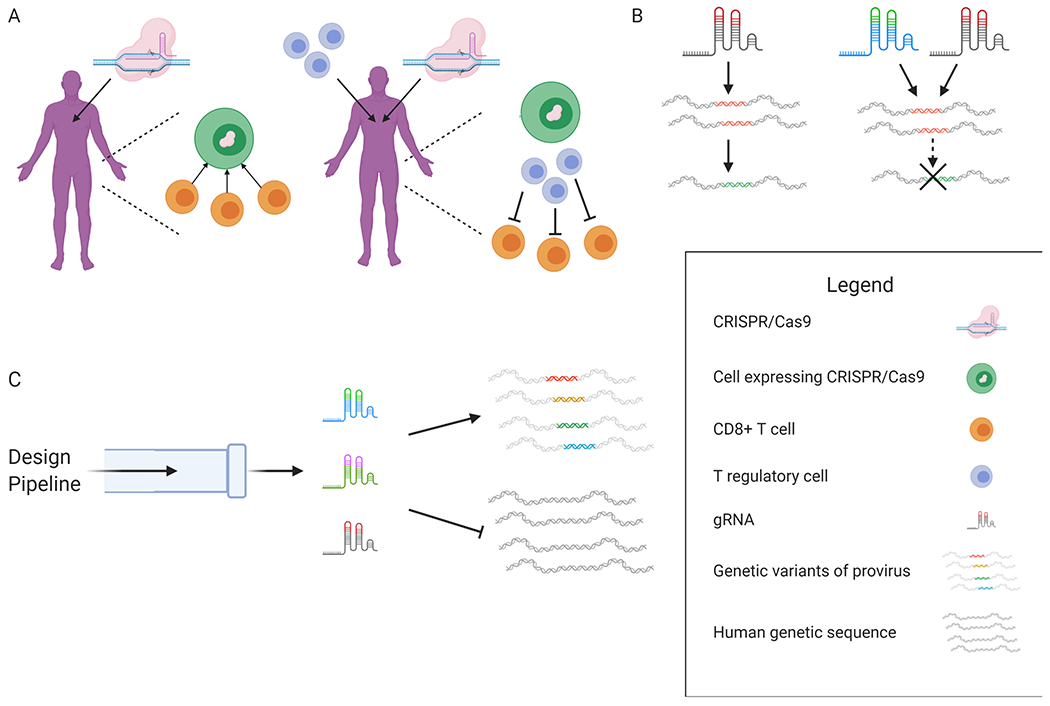
(A) Delivery of CRISPR/Cas9 to patients may elicit an adaptive immune response with CD8+ T cells targeting cells expressing CRISPR/Cas9. A potential solution is adoptive transfer of Cas9-specific T regulatory cells in conjunction with CRISPR/Cas9 treatment to dampen the effector function of Cas9-specific CD8+ T cells. (B) Use of a single gRNA to target the latent infection creates escape mutants. Using multiple gRNAs to target latent infection prevents viral escape. (C) The design pipeline [91] generates a package of multiple gRNAs capable of recognizing and cleaving genetic variants of proviral DNA found within and between patients. The gRNA package does not exhibit off-target cleavage in the human genome. Figure created with license for BioRender.
The immunogenicity of the delivery vehicle for a potential CRISPR/Cas9 therapeutic has also been investigated. The AAV vector is a promising means for CRISPR/Cas9 delivery with successful delivery demonstrated in humanized mice [59]. A study investigating AAV for delivery of CRISRP/Cas9 found that AAV does trigger a host immune response in humanized mice [79]. However, the study also reported that tissue damage was not detected in muscles treated with AAV-delivered Cas9 in vivo. Overall, this area of therapeutic development needs more study, but the current state of understanding indicates that the problems related to the immunogenic properties of the CRISPR/Cas9 system may be surmountable.
4.2. What animal models can be used to test the CRISPR/Cas9 therapeutic strategy
There are two predominant animal models used to test HIV-1 cure strategies: the rhesus macaque (RM) model using simian immunodeficiency virus (SIV) and humanized mouse models using HIV-1. The RM model is supported by numerous reports and has been shown to recapitulate human disease [20,80,81]. Additionally, the RM latent reservoir is seeded rapidly following infection [82]. However, to date, there are no published reports of SIV-targeting CRISPR/Cas9 being tested in an RM model. However, the CRISPR/Cas9 system using the RM model would require testing CRISPR against SIV beginning with ex vivo experiments using RM CD4+ T cells. This is a viable strategy; SIV Gag can be detected by ELISA allowing efficient quantitation of CRISPR/Cas9 effectiveness in vitro. For these experiments, the SIVmac239 strain could be used. Furthermore, this experimental paradigm could also be applied to infected RM PBMCs suppressed into latency by ART. Following ex vivo experiments, the next step would be to establish proof of principle in vivo in the RM model. Notably, a limitation of the RM model in validating CRISPR/Cas9 strategies is imposed by the differences between SIV and HIV-1 sequences. The sequence specificity of CRISPR/Cas9 gRNAs would necessitate design of SIV-specific gRNAs for use in the RM model and in vivo assessment would be for SIV-specific targets not necessarily present in the HIV-1 genome.
Humanized mouse models are also widely used in testing HIV-1 cure strategies. One distinct advantage of the humanized mouse model is that HIV-1 is used rather than SIV, as in the RM model. Several recent advances have been made in murine models. [21,54,59] Currently, the widely accepted model is the bone marrow, live, thymus model (BLT mouse) [83]. The BLT model can reconstitute multiple lineages of human immune cells, making it a versatile mouse model for HIV-1 infection and the ensuing immune response. Nonetheless, there are drawbacks to the use of humanized mice for HIV-1 study. For example, microglia and macrophages in the brain are not reconstituted in this model which may impact how entrenched the viral reservoir is in these animals. Also, the approximate time to graft-versus-host disease (GVHD) in a BLT mouse is approximately six months. Conversely, the RM model does not have this issue; RMs can be suppressed with ART for the animals’ lifespan. It is noteworthy that despite the usefulness of animal models, a gRNA/CRISPR/Cas9 package that is capable of translation as a therapeutic in human disease will necessarily differ from those tested in animal models. On balance, neither model can offer a complete picture of human HIV-1 infection, but both are valuable tools which will be necessary for the clinical translation of CRISPR/Cas9 therapy for HIV-1 infection.
4.3. HIV-1 resistance to CRISPR therapy
In vitro studies have shown that targeting the HIV-1 provirus with a single gRNA leads to escape mutants [55,62,84–86]. However, a viable solution to this problem is simply to target the provirus simultaneously at multiple sites (Figure 2 B). Multiplexed gRNAs have been shown to prevent escape mutants [58,84–89]. To this end, our group developed a package of HIV-1-targeting gRNAs capable of targeting the full breadth of genetic variation in HIV-1 proviral DNA within and between patients infected with subtype B [70,71,90,91] and across subtypes [91]. Resistance studies for these highly effective gRNA packages have yet to be conducted, however the success of other multiplex gRNAs suggests a promising outcome. Furthermore, our gRNAs target highly conserved regions (i.e. those with low Shannon entropy) which decreases the likelihood of high viral fitness that would result in the development of escape mutants [70,88].
4.4. Will off-target events preclude the CRISPR/Cas9 system from being used in patients?
The potential for off-target cleavage by CRISPR/Cas9 in the human genome is a concern for the development of safe therapeutic strategies. The specificity of the CRISPR/Cas9 system is a function of complementarity between gRNA and target sequences as well as cellular factors, specifically chromatin architecture and target-site accessibility [70,71,90–94]. The mismatch tolerance between gRNA and target, allowing cleavage between sites with multiple mismatches necessitates careful design and rigorous screening to ensure the safety profile of CRISPR/Cas9 therapeutics. Some HIV-1-specific gRNAs have been designed to avoid off-target specificity, but the design processes have lacked rigor in light of the current predictive computational tools for gRNA design [54,55,59,61,62,65,67,85]. In this regard, we have designed an in silico design pipeline for HIV-1-targeting gRNAs which accounts for gRNA specificity by utilizing the cutting frequency determination (CFD) matrix and also accounts for the genetic diversity within and between patients (Figure 2 C) [70,71,90–92,95].
Avoiding off-target proclivity in the CRISPR/Cas9 system requires a two-fold approach. Beyond gRNA design, off-target screening is also necessary. To date, some off-target screening has been performed with HIV-1-specific gRNAs but these analyses have lacked rigor. For published HIV-1-targeting gRNAs, the off-target detection methods that have been used are targeted amplicon sequencing, whole genome sequencing (WGS), SURVEYOR assay, T7E1 assay, and tracking of indels by decomposition (TIDE) [49–53,56,58,59,64,67–69,84,85,96]. Each of these methods has utility and has provided valuable data regarding the targeting fidelity of HIV-1-targeting gRNAs. None of these methods are currently considered state-of-the-art off-target detection methodologies. However, establishing proof of principle for HIV-1-targeting gRNAs does not necessarily require the degree of targeting fidelity necessary for clinical application. The limited application of off-target detection methods thus far has been appropriate to the experimental paradigms of CRISPR/Cas9 HIV-1-targeting studies.
But gRNAs that will be moved towards clinical trials will need to be screened using genome-wide, unbiased methods. There are currently a variety of published methods fulfilling that criteria (Table 2). Some are better suited to characterization of HIV-1-targeting gRNAs than others. In vitro techniques such as in vitro nuclease-digested genome sequencing (Digenome-seq), selective enrichment and identification of tagged genomic DNA ends by sequencing (SITE-Seq) and circularization for in vitro reporting of cleavage effects by sequencing (CIRCLE-seq) are sensitive but also prone to abundant false positives and the CRISPR/Cas9 cleavage sites they detect lack cellular context [97–99]. Linear-amplification-mediated polymerase chain reaction high-throughput genome-wide translocation sequencing (LAM-PCR HTGTS) requires significant quantities of input DNA, roughly 100 μg of genomic DNA per sample, a substantial input quantity [100,101]. In addition, in situ end-capture methods such as breaks labeling in situ and sequencing (BLISS) utilize complex protocols and capture only the DSBs present at a single time-point when cells are fixed for processing [102]. Genome-wide, unbiased identification of DSBs enabled by sequencing (GUIDE-seq), discovery of in situ cas off-targets and verification by sequencing (DISCOVER-Seq), and verification of in vivo off-targets (VIVO) are more suited for characterization of HIV-1 targeting gRNAs though each has advantages and drawbacks.
Table 2:
Comparison of off-target detection methods for CRISPR/Cas9 cleavage
| Off-target Methods | Detection State | Target Enrichment Method | Edits Detected |
|---|---|---|---|
| Digenome-seq [36] | In vitro | None | DSBs |
| SITE-seq [37] | In vitro | Streptavidin selection | DSBs |
| CIRCLE-seq [38] | In vitro | Linearization of cicularized library | DSBs |
| HTGTS [39] | Ex vivo | Anchored primer amplification | translocations |
| BLISS [40] | In situ | Transcription | unrepaired DSBs |
| GUIDE-seq [41] | Ex vivo | Anchored primer amplification | NHEJ repair sites |
| DISCOVER-seq [42] | In vivo | Chromatin Immunoprecipitation | unrepaired DSBs |
| VIVO [43] | In vivo | Targeted sequencing | Repair site mutations |
GUIDE-seq is more sensitive than DISCOVER-Seq and it is capable of off-target detection of ex vivo cleavage events. It is however, cytotoxic to some primary cells due to the transfection of short oligonucleotides necessary for cleavage-site detection and enrichment [103,104]. VIVO is more sensitive than GUIDE-seq or DISCOVER-Seq, but it has a high false discovery rate due to the in vitro CIRCLE-seq step and therefore requires a selection of identified off-target sequences for targeted amplicon validation [105]. In a head-to-head comparison with DISCOVER-Seq, evaluating the Pcsk9-gP gRNA, 17 bona fide off-target sites that were identified with DISCOVER-Seq were also detected in the CIRCLE-seq phase of VIVO but overlooked for validation amongst the many false positives. DISCOVER-Seq and VIVO can be applied to in vivo CRISPR/Cas9 editing, which makes DISCOVER-Seq more ubiquitously applicable than GUIDE-seq. DISCOVER-Seq is also less prone to false positives than VIVO. However, DISCOVER-Seq is the least sensitive of the three. Due to the chromatin immunoprecipitation (ChIP)-based nature of cleavage-site capture and enrichment, DISCOVER-Seq captures only DSBs bound by the MRE11-RAD50-NBS1 (MRN) complex during the early DNA damage response, when cells are crosslinked with formaldehyde [104]. Whereas the output of GUIDE-seq and VIVO represent the accumulation of cleavage events through the duration of the assay. In summary, there is no single best assay for off-target detection in every context. Full characterization of gRNAs for clinical translation of CRISPR/Cas9 may require the application of multiple techniques from the repertoire of current state-of-the-art, genome-wide, unbiased methodologies.
All of this is not to say that off-target effects are rampant amongst published HIV-1-targeting gRNAs. Recommendations for in-depth off-target analysis are motivated by a push for rigor and an abundance of caution. CRISPR/Cas9 treatments are tested in cell viability assays in tandem with assays for efficacy. Cytotoxic gRNAs can be identified without the application of genome-wide, unbiased off-target detection methods [58]. Downstream off-target effects have not been observed in in vivo HIV-1-targeting CRISPR studies [52,59]. Recently, we have used a modified GUIDE-seq assay to evaluate the off-target profile of certain gRNAs. No off-target cleavage events were detected from the gRNAs designed with our pipeline [106]. These results support the efficacy of our design pipeline and indicate that off-target proclivity can be avoided through rigorous design and screening methodology.
4.5. How can HIV-1 CRISPR/Cas9 therapy be delivered to target tissues
The question of delivery for CRISPR/Cas9 targeting of the HIV-1 provirus is important for clinical translation of the therapy. The CRISPR/Cas9 system must reach tissue and cellular compartments harboring latent infection including the brain, gut, lymph nodes, and spleen in order to be effective. This has been a major criticism of the development of CRISPR/Cas9 therapy approaches that will require administration in vivo: how will this therapy be delivered?
Recent advances have demonstrated the feasibility of delivery in vivo in murine models. Several studies have demonstrated that the administration of AAV by tail-vein injection results in delivery of the CRISPR/Cas9 system to target tissues and successful CRISPR/Cas9 cleavage has been validated using ddPCR to confirm excision of proviral fragments [52,59]. These studies serve as proof-of-concept that in vivo delivery of CRISPR/Cas9 is possible, although further development will be necessary. For example, targeted delivery specifically to tissues harboring latent infection and more specifically to latently infected cells would be optimal. Progress has been made on this front as well. For example, astrocyte transduction using the novel vector AAV9P1 was demonstrated in vitro [67]. The AAV9P1 vector contains a synthetic peptide on the surface for astrocyte transduction. The AAV9P1 vector was shown to preferential transduce astrocytes when administered in cultures of mixed brain cells with astrocytes and neurons.
Another example of progress in CRISPR/Cas9 delivery is the development of a system for delivery of CRISPR/Cas9 across the blood-brain-barrier (BBB) [68]. The system has been demonstrated to work in vitro for the delivery of a nano-formulation (NF) consisting of Cas9 and gRNA bound to magneto-electric nanoparticles (MENPs). The system allows release of the Cas9 and gRNA payload by ac-magnetic field stimulation and it accomplishes cell uptake using nano-electroporation. The same group previously demonstrated MENP delivery to the brain in a murine model [107]. These proof-of-concept studies may lead to a system whereby targeted delivery of Cas9 to latently infected cells in the brain may be possible.
The significance of the challenge of CRISPR/Cas9 delivery for effective therapeutic translation should not be understated. It is a critically important aspect of the technology. Identification of a latency-specific biomarker would facilitate a solution to identify the cellular reservoir of latently infected cells, however to-date no such biomarker has been reported. Methods of delivery used in vitro to establish proof-of-concept for CRISPR/Cas9 action against the HIV-1 provirus are not considered viable options for delivery in patients. This aspect of CRISPR/Cas9 therapy must remain a high-priority field of investigation if a clinical translation is to be realized.
4.6. Capturing HIV diversity with CRISPR/Cas9
The genetic diversity of HIV-1 demands consideration in the development of therapeutics. Genetic diversity within and between patients can be addressed by gRNA design [55,65,70,71,90,91,95]. However, for a therapy to be broadly effective it must also be applicable to the range of subtypes presented by HIV-1. We have developed a pipeline for the design of CRISPR/Cas9 gRNAs [70,90,91]. The pipeline is specifically tailored to the design of gRNAs with broad-spectrum targeting capacity in order to cope with genetic variation within and between patients by targeting conserved regions of the LTR. The currently designed package is specific to subtype B [91]. A multiplexed package of four of the top-performing gRNAs is predicted to cleave target sequences for all viral quasispecies observed by publications to date within subtype B. In silico predictions indicate that our top performing gRNA for subtype B will also target 96% of other subtypes [91]. Given that all training data for the current gRNA package was derived from subtype B sequence, it is reasonable to expect that given adequate data our pipeline could design an equally effective package of gRNAs for each of the other subtypes.
5. Conclusion
In conclusion, the major curative strategies being pursued for HIV-1 infection all have encouraging early results and may lead to cures or adjuvant therapies. CRISPR/Cas9 stands out among them as particularly exciting. Recent in vivo results in humanized mice are a major step forward serving as proof-of-concept that viral eradication is possible and CRISPR/Cas9 delivery by AAV is feasible [59]. There are aspects of therapeutic development that still need to be addressed including the potential for an adaptive immune response, potential escape mutations, potential off-target cleavage, the question of efficient delivery, and applicability across subtypes. But results published on each of these aspects suggest that all of these potential problems have solutions. Future studies will further elucidate the solutions to those problems and drive CRISPR/Cas9 towards clinical translation for HIV-1 treatment.
6. Expert opinion
HIV-1 remains a global pandemic. Although a cure remains elusive, great strides have been made in the past 35 plus years towards treatment of HIV-1 and AIDS. ART has been transformative for the lives, livelihoods, and health of infected patients with access to appropriate drug regimens. Yet HIV-1 infection still severely impacts the health and well-being of even the most well-suppressed patients. Viral resurgence in the absence of ART forces a life-long dependence on drug regimens and HAND is still prevalent among HIV-1-infected patients. Hence development of a therapeutic strategy to eliminate or neutralize the latent viral reservoir, allowing safe cessation of ART concurrent with sustained viral suppression, is a highly sought-after achievement. Each of the approaches discussed in this review, shock and kill, bNAbs, block and lock, CAR T cells, immune checkpoint modulation, coreceptor ablation, has merit and encouraging progress has been made with respect to each approach. Yet no strategy to date has achieved elimination or neutralization of the latent viral reservoir.
In this review, we have addressed the question: Why CRISPR? Why in the midst of so many options, each encouraging in its progress, should CRISPR be investigated as a potential solution to this problem? The answer is multi-faceted. The CRISPR/Cas9 gene editing system offers some key advantages over other cure strategies currently being investigated for HIV-1. The basis of these advantages is that CRISPR is the approach that addresses the root of the problem. Viral recrudescence is ultimately due to the harboring of proviral DNA in small but critical populations of cells. CRISPR/Cas9 targets this root directly. CRISPR/Cas9 is the technology capable of directly targeting the HIV-1 provirus with two potential outcomes: elimination by excision and inactivation by ablation.
A distinct advantage to the CRISPR/Cas9 system is that antigen expression is unnecessary, therefore patients would not need to stop ART. Shock and kill, bNAb and CAR T cell therapies all rely on an intact immune system to clear infected cells. This is disadvantageous given the immunocompromising nature of HIV-1 infection. There is evidence that CD8+ T cells are unable to clear infected cells [78,108,109]. These studies have shown that HIV-1 can easily escape from immunodominant epitopes, while leading to increased CD8+ T-cell exhaustion. The CRISPR/Cas9 system does not rely on an intact immune system nor antigen expression. Additionally, the CRISPR/Cas9 system can directly attack the provirus, therefore expression does not need to be sustained.
Until a viable therapy for HIV-1 cure is found, which eliminates the need for sustained cART and is broadly applicable to all of the infected cell populations, each of the approaches discussed in this review should be pursued. This is especially true for CRISPR/Cas9 which is capable of addressing the root of the problem: integrated proviral DNA. The possibility of a cure for HIV-1 is no longer fanciful speculation. CRISPR/Cas9 has been shown to be a viable strategy for eliminating HIV-1 infection based on success in humanized mice. No other strategy for elimination of the latent HIV-1 reservoir has ever been so close to realization. At this point, even the harshest critics of the CRISPR/Cas9 strategy should be swayed towards cautious optimism.
7. Article highlight box.
HIV-1 curative strategies focus on elimination or deactivation of the latent viral reservoir.
HIV-1 curative strategies include CRISPR/Cas9 proviral excision, CRISPR/Cas9 coreceptor ablation, shock and kill, bNAbs, CAR T cells, block and lock, and immune blockade.
CRISPR/Cas9 proviral excision fills a unique niche among HIV-1 curative strategies by targeting proviral DNA directly.
Proof-of-concept that HIV-1 viral elimination is possible using the CRISPR/Cas9 system has been demonstrated in a murine model.
Problems surrounding the adaptive immune response, HIV-1 escape mutations, and delivery can all be solved for the CRISPR/Cas9 system as an HIV-1 therapy.
Off-target proclivity of the CRISPR/Cas9 system targeting HIV-1 can be avoided with an appropriate design and screening paradigm.
9. Acknowledgment
This work was supported by National Institute of Mental Health (NIMH) R01 MH110360 (Contact PI, BW), NIMH Comprehensive NeuroAIDS Center (CNAC) P30 MH092177 (Kamel Khalili, PI; Brian Wigdahl, PI of the Drexel subcontract involving the Clinical and Translational Research Support Core, Drexel Component PI, BW), NIMH T32 MH079785 (Drexel Component PI, BW) and the Ruth L. Kirschstein National Research Service Award T32 MH079785 (Dr. Brian Wigdahl, Principal Investigator of the Drexel University College of Medicine component and Drs. Olimpia Meucci and Michael Nonnemacher as Co-Directors). The contents of the paper were solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
This work was performed in Philadelphia, Pennsylvania USA
Declaration of interest
The authors claim they have no financial interest, direct or indirect, in the subject matter or materials discussed in the manuscript (such as consultancies, employment, expert testimony, honoraria, speakers’ bureaus, retainers, stock options or ownership) that may affect the conduct or reporting of the work submitted.
Works Cited
- 1.Marino J, Wigdahl B, Nonnemacher MR. Extracellular HIV-1 Tat Mediates Increased Glutamate in the CNS Leading to Onset of Senescence and Progression of HAND. Front Aging Neurosci. 2020;12:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marino J, Maubert ME, Mele AR, et al. Functional impact of HIV-1 Tat on cells of the CNS and its role in HAND. Cell Mol Life Sci. 2020. Jun 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arainga M, Edagwa B, Mosley RL, et al. A mature macrophage is a principal HIV-1 cellular reservoir in humanized mice after treatment with long acting antiretroviral therapy. Retrovirology. 2017. Mar 9;14(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barouch DH, Deeks SG. Immunologic strategies for HIV-1 remission and eradication. Science. 2014. Jul 11;345(6193):169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deeks SG. HIV: Shock and kill. Nature. 2012. Jul 25;487(7408):439–40. [DOI] [PubMed] [Google Scholar]
- 6.Kim Y, Anderson JL, Lewin SR. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe. 2018. Jan 10;23(1):14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Policicchio BB, Xu C, Brocca-Cofano E, et al. Multi-dose Romidepsin Reactivates Replication Competent SIV in Post-antiretroviral Rhesus Macaque Controllers. PLoS Pathog. 2016. Sep;12(9):e1005879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Archin NM, Kirchherr JL, Sung JA, et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J Clin Invest. 2017. Aug 1;127(8):3126–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barton K, Hiener B, Winckelmann A, et al. Broad activation of latent HIV-1 in vivo. Nat Commun. 2016. Sep 8;7:12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu G, Swanson M, Talla A, et al. HDAC inhibition induces HIV-1 protein and enables immune-based clearance following latency reversal. JCI Insight. 2017. Aug 17;2(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai P, Wu G, Baker CE, et al. In vivo analysis of the effect of panobinostat on cell-associated HIV RNA and DNA levels and latent HIV infection. Retrovirology. 2016. May 21;13(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren Y, Huang SH, Patel S, et al. BCL-2 antagonism sensitizes cytotoxic T cell-resistant HIV reservoirs to elimination ex vivo. J Clin Invest. 2020. May 1;130(5):2542–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanke T Aiming for protective T-cell responses: a focus on the first generation conserved-region HIVconsv vaccines in preventive and therapeutic clinical trials. Expert Rev Vaccines. 2019. Oct;18(10):1029–1041. [DOI] [PubMed] [Google Scholar]
- 14.Korber B, Fischer W. T cell-based strategies for HIV-1 vaccines. Hum Vaccin Immunother. 2020. Mar 3;16(3):713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mothe B, Manzardo C, Sanchez-Bernabeu A, et al. Therapeutic Vaccination Refocuses T-cell Responses Towards Conserved Regions of HIV-1 in Early Treated Individuals (BCN 01 study). EClinicalMedicine. 2019;11:65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mothe B, Rosas-Umbert M, Coll P, et al. HIVconsv Vaccines and Romidepsin in Early-Treated HIV-1-Infected Individuals: Safety, Immunogenicity and Effect on the Viral Reservoir (Study BCN02). Front Immunol. 2020;11:823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malbec M, Porrot F, Rua R, et al. Broadly neutralizing antibodies that inhibit HIV-1 cell to cell transmission. J Exp Med. 2013. Dec 16;210(13):2813–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang J, Kang BH, Ishida E, et al. Identification of a CD4-Binding-Site Antibody to HIV that Evolved Near-Pan Neutralization Breadth. Immunity. 2016. Nov 15;45(5):1108–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishimura Y, Gautam R, Chun TW, et al. Early antibody therapy can induce long-lasting immunity to SHIV. Nature. 2017. Mar 23;543(7646):559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gautam R, Nishimura Y, Gaughan N, et al. A single injection of crystallizable fragment domain-modified antibodies elicits durable protection from SHIV infection. Nat Med. 2018. May;24(5):610–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Badamchi-Zadeh A, Tartaglia LJ, Abbink P, et al. Therapeutic Efficacy of Vectored PGT121 Gene Delivery in HIV-1-Infected Humanized Mice. J Virol. 2018. Apr 1;92(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Zony C, Chen P, et al. Reduced Potency and Incomplete Neutralization of Broadly Neutralizing Antibodies against Cell-to-Cell Transmission of HIV-1 with Transmitted Founder Envs. J Virol. 2017. May 1;91(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Navio JM, Fuchs SP, Pedreno-Lopez S, et al. Host Anti-antibody Responses Following Adeno-associated Virus-mediated Delivery of Antibodies Against HIV and SIV in Rhesus Monkeys. Mol Ther. 2016. Feb;24(1):76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stefic K, Bouvin-Pley M, Braibant M, et al. Impact of HIV-1 Diversity on Its Sensitivity to Neutralization. Vaccines (Basel). 2019. Jul 25;7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Debyser Z, Vansant G, Bruggemans A, et al. Insight in HIV Integration Site Selection Provides a Block-and-Lock Strategy for a Functional Cure of HIV Infection. Viruses. 2018. Dec 26;11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mediouni S, Chinthalapudi K, Ekka MK, et al. Didehydro-Cortistatin A Inhibits HIV-1 by Specifically Binding to the Unstructured Basic Region of Tat. mBio. 2019. Feb 5;10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spector C, Mele AR, Wigdahl B, et al. Genetic variation and function of the HIV-1 Tat protein. Med Microbiol Immunol. 2019. Apr;208(2):131–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vansant G, Bruggemans A, Janssens J, et al. Block-And-Lock Strategies to Cure HIV Infection. Viruses. 2020. Jan 10;12(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Imamichi H, Dewar RL, Adelsberger JW, et al. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proceedings of the National Academy of Sciences. 2016;113(31):8783–8788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paiardini M, Muller-Trutwin M. HIV-associated chronic immune activation. Immunol Rev. 2013. Jul;254(1):78–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kessing CF, Nixon CC, Li C, et al. In Vivo Suppression of HIV Rebound by Didehydro-Cortistatin A, a “Block-and-Lock” Strategy for HIV-1 Treatment. Cell Rep. 2017. Oct 17;21(3):600–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahlenstiel CL, Symonds G, Kent SJ, et al. Block and Lock HIV Cure Strategies to Control the Latent Reservoir. Front Cell Infect Microbiol. 2020;10:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qi J, Ding C, Jiang X, et al. Advances in Developing CAR T-Cell Therapy for HIV Cure. Front Immunol. 2020;11:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hale M, Mesojednik T, Romano Ibarra GS, et al. Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol Ther. 2017. Mar 1;25(3):570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anthony-Gonda K, Bardhi A, Ray A, et al. Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Sci Transl Med. 2019. Aug 7;11(504). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ollerton MT, Berger EA, Connick E, et al. HIV-1-Specific Chimeric Antigen Receptor T Cells Fail To Recognize and Eliminate the Follicular Dendritic Cell HIV Reservoir In Vitro. J Virol. 2020. May 4;94(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brodie SJ, Lewinsohn DA, Patterson BK, et al. In vivo migration and function of transferred HIV-1-specific cytotoxic T cells. Nat Med. 1999. Jan;5(1):34–41. [DOI] [PubMed] [Google Scholar]
- 38.Tan R, Xu X, Ogg GS, et al. Rapid death of adoptively transferred T cells in acquired immunodeficiency syndrome. Blood. 1999. Mar 1;93(5):1506–10. [PubMed] [Google Scholar]
- 39.Porter DL, Levine BL, Kalos M, et al. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011. Aug 25;365(8):725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wykes MN, Lewin SR. Immune checkpoint blockade in infectious diseases. Nat Rev Immunol. 2018. Feb;18(2):91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porichis F, Hart MG, Massa A, et al. Immune Checkpoint Blockade Restores HIV-Specific CD4 T Cell Help for NK Cells. J Immunol. 2018. Aug 1;201(3):971–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bradley T, Kuraoka M, Yeh CH, et al. Immune checkpoint modulation enhances HIV-1 antibody induction. Nat Commun. 2020. Feb 19;11(1):948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allen AG, Chung CH, Atkins A, et al. Gene Editing of HIV-1 Co-receptors to Prevent and/or Cure Virus Infection. Front Microbiol. 2018;9:2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Binda CS, Klaver B, Berkhout B, et al. CRISPR-Cas9 Dual-gRNA Attack Causes Mutation, Excision and Inversion of the HIV-1 Proviral DNA. Viruses. 2020. Mar 18;12(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang W, Ye C, Liu J, et al. CCR5 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS One. 2014;9(12):e115987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Z, Chen S, Jin X, et al. Genome editing of the HIV co-receptors CCR5 and CXCR4 by CRISPR-Cas9 protects CD4(+) T cells from HIV-1 infection. Cell Biosci. 2017;7:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Q, Chen S, Xiao Q, et al. Genome modification of CXCR4 by Staphylococcus aureus Cas9 renders cells resistance to HIV-1 infection. Retrovirology. 2017. Nov 15;14(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu S, Yao Y, Xiao H, et al. Simultaneous Knockout of CXCR4 and CCR5 Genes in CD4+ T Cells via CRISPR/Cas9 Confers Resistance to Both X4- and R5-Tropic Human Immunodeficiency Virus Type 1 Infection. Hum Gene Ther. 2018. Jan;29(1):51–67. [DOI] [PubMed] [Google Scholar]
- 49.Xu L, Yang H, Gao Y, et al. CRISPR/Cas9-Mediated CCR5 Ablation in Human Hematopoietic Stem/Progenitor Cells Confers HIV-1 Resistance In Vivo. Mol Ther. 2017. Aug 2;25(8):1782–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu W, Kaminski R, Yang F, et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci U S A. 2014. Aug 5;111(31):11461–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ophinni Y, Inoue M, Kotaki T, et al. CRISPR/Cas9 system targeting regulatory genes of HIV-1 inhibits viral replication in infected T-cell cultures. Sci Rep. 2018. May 17;8(1):7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaminski R, Bella R, Yin C, et al. Excision of HIV-1 DNA by gene editing: a proof-of-concept in vivo study. Gene Ther. 2016. Aug;23(8-9):690–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaminski R, Chen Y, Fischer T, et al. Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci Rep. 2016. Mar 4;6:22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bella R, Kaminski R, Mancuso P, et al. Removal of HIV DNA by CRISPR from Patient Blood Engrafts in Humanized Mice. Mol Ther Nucleic Acids. 2018. Sep 7;12:275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Darcis G, Binda CS, Klaver B, et al. The Impact of HIV-1 Genetic Diversity on CRISPR-Cas9 Antiviral Activity and Viral Escape. Viruses. 2019. Mar 13;11(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Campbell LA, Coke LM, Richie CT, et al. Gesicle-Mediated Delivery of CRISPR/Cas9 Ribonucleoprotein Complex for Inactivating the HIV Provirus. Mol Ther. 2019. Jan 2;27(1):151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ebina H, Misawa N, Kanemura Y, et al. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep. 2013;3:2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lebbink RJ, de Jong DC, Wolters F, et al. A combinational CRISPR/Cas9 gene-editing approach can halt HIV replication and prevent viral escape. Sci Rep. 2017. Feb 8;7:41968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dash PK, Kaminski R, Bella R, et al. Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat Commun. 2019. Jul 2;10(1):2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liao HK, Gu Y, Diaz A, et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat Commun. 2015. Mar 10;6:6413. [DOI] [PubMed] [Google Scholar]
- 61.Yin C, Zhang T, Li F, et al. Functional screening of guide RNAs targeting the regulatory and structural HIV-1 viral genome for a cure of AIDS. AIDS. 2016. May 15;30(8):1163–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Z, Pan Q, Gendron P, et al. CRISPR/Cas9-Derived Mutations Both Inhibit HIV-1 Replication and Accelerate Viral Escape. Cell Rep. 2016. Apr 19;15(3):481–489. [DOI] [PubMed] [Google Scholar]
- 63.Su H, Sravanam S, Gorantla S, et al. Amplification of Replication Competent HIV-1 by Adoptive Transfer of Human Cells From Infected Humanized Mice. Front Cell Infect Microbiol. 2020;10:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saayman SM, Lazar DC, Scott TA, et al. Potent and Targeted Activation of Latent HIV-1 Using the CRISPR/dCas9 Activator Complex. Mol Ther. 2016. Mar;24(3):488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roychoudhury P, De Silva Feelixge H, Reeves D, et al. Viral diversity is an obligate consideration in CRISPR/Cas9 designs for targeting the HIV reservoir. BMC Biol. 2018. Jul 11;16(1):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Limsirichai P, Gaj T, Schaffer DV. CRISPR-mediated Activation of Latent HIV-1 Expression. Mol Ther. 2016. Mar;24(3):499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kunze C, Borner K, Kienle E, et al. Synthetic AAV/CRISPR vectors for blocking HIV-1 expression in persistently infected astrocytes. Glia. 2018. Feb;66(2):413–427. [DOI] [PubMed] [Google Scholar]
- 68.Kaushik A, Yndart A, Atluri V, et al. Magnetically guided non-invasive CRISPR-Cas9/gRNA delivery across blood-brain barrier to eradicate latent HIV-1 infection. Sci Rep. 2019. Mar 8;9(1):3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaminski R, Chen Y, Salkind J, et al. Negative Feedback Regulation of HIV-1 by Gene Editing Strategy. Sci Rep. 2016. Aug 16;6:31527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dampier W, Sullivan NT, Chung CH, et al. Designing broad-spectrum anti-HIV-1 gRNAs to target patient-derived variants. Sci Rep. 2017. Oct 31;7(1):14413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dampier W, Nonnemacher MR, Sullivan NT, et al. HIV Excision Utilizing CRISPR/Cas9 Technology: Attacking the Proviral Quasispecies in Reservoirs to Achieve a Cure. MOJ Immunol. 2014. Oct 17;1(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Charlesworth CT, Deshpande PS, Dever DP, et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med. 2019. Feb;25(2):249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wagner DL, Amini L, Wendering DJ, et al. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat Med. 2019. Feb;25(2):242–248. [DOI] [PubMed] [Google Scholar]
- 74.Simhadri VL, McGill J, McMahon S, et al. Prevalence of Pre-existing Antibodies to CRISPR-Associated Nuclease Cas9 in the USA Population. Mol Ther Methods Clin Dev. 2018. Sep 21;10:105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brouwer S, Barnett TC, Rivera-Hernandez T, et al. Streptococcus pyogenes adhesion and colonization. FEBS Lett. 2016. Nov;590(21):3739–3757. [DOI] [PubMed] [Google Scholar]
- 76.Jenul C, Horswill AR. Regulation of Staphylococcus aureus Virulence. Microbiol Spectr. 2018. Feb;6(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chandran S, Tang Q, Sarwal M, et al. Polyclonal Regulatory T Cell Therapy for Control of Inflammation in Kidney Transplants. Am J Transplant. 2017. Nov;17(11):2945–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reuter MA, Del Rio Estrada PM, Buggert M, et al. HIV-Specific CD8(+) T Cells Exhibit Reduced and Differentially Regulated Cytolytic Activity in Lymphoid Tissue. Cell Rep. 2017. Dec 19;21(12):3458–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chew WL, Tabebordbar M, Cheng JK, et al. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat Methods. 2016. Oct;13(10):868–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sugimoto C, Merino KM, Hasegawa A, et al. Critical Role for Monocytes/Macrophages in Rapid Progression to AIDS in Pediatric Simian Immunodeficiency Virus-Infected Rhesus Macaques. J Virol. 2017. Sep 1;91(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu K, Acharya P, Kong R, et al. Epitope-based vaccine design yields fusion peptide-directed antibodies that neutralize diverse strains of HIV-1. Nat Med. 2018. Jun;24(6):857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Whitney JB, Hill AL, Sanisetty S, et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature. 2014. Aug 7;512(7512):74–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marsden MD, Zack JA. Humanized Mouse Models for Human Immunodeficiency Virus Infection. Annu Rev Virol. 2017. Sep 29;4(1):393–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang Z, Wang W, Cui YC, et al. HIV-1 Employs Multiple Mechanisms To Resist Cas9/Single Guide RNA Targeting the Viral Primer Binding Site. J Virol. 2018. Oct 15;92(20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yoder KE, Bundschuh R. Host Double Strand Break Repair Generates HIV-1 Strains Resistant to CRISPR/Cas9. Sci Rep. 2016. Jul 12;6:29530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang G, Zhao N, Berkhout B, et al. A Combinatorial CRISPR-Cas9 Attack on HIV-1 DNA Extinguishes All Infectious Provirus in Infected T Cell Cultures. Cell Rep. 2016. Dec 13;17(11):2819–2826. [DOI] [PubMed] [Google Scholar]
- 87.Gao Z, Fan M, Das AT, et al. Extinction of all infectious HIV in cell culture by the CRISPR-Cas12a system with only a single crRNA. Nucleic Acids Res. 2020. Apr 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang G, Zhao N, Berkhout B, et al. CRISPR-Cas9 Can Inhibit HIV-1 Replication but NHEJ Repair Facilitates Virus Escape. Mol Ther. 2016. Mar;24(3):522–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhao N, Wang G, Das AT, et al. Combinatorial CRISPR-Cas9 and RNA Interference Attack on HIV-1 DNA and RNA Can Lead to Cross-Resistance. Antimicrob Agents Chemother. 2017. Dec;61(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dampier W, Sullivan NT, Mell JC, et al. Broad-Spectrum and Personalized Guide RNAs for CRISPR/Cas9 HIV-1 Therapeutics. AIDS Res Hum Retroviruses. 2018. Nov;34(11):950–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sullivan NT, Dampier W, Chung CH, et al. Novel gRNA design pipeline to develop broad-spectrum CRISPR/Cas9 gRNAs for safe targeting of the HIV-1 quasispecies in patients. Sci Rep. 2019. Nov 19;9(1):17088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Doench JG, Fusi N, Sullender M, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016. Feb;34(2):184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hsu PD, Scott DA, Weinstein JA, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013. Sep;31(9):827–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chung CH, Allen AG, Sullivan NT, et al. Computational Analysis Concerning the Impact of DNA Accessibility on CRISPR-Cas9 Cleavage Efficiency. Mol Ther. 2020. Jan 8;28(1):19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Link RW, Nonnemacher MR, Wigdahl B, et al. Prediction of Human Immunodeficiency Virus Type 1 Subtype-Specific Off-Target Effects Arising from CRISPR-Cas9 Gene Editing Therapy. CRISPR J. 2018. Aug;1:294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hou P, Chen S, Wang S, et al. Genome editing of CXCR4 by CRISPR/cas9 confers cells resistant to HIV-1 infection. Sci Rep. 2015. Oct 20;5:15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cameron P, Fuller CK, Donohoue PD, et al. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat Methods. 2017. Jun;14(6):600–606. [DOI] [PubMed] [Google Scholar]
- 98.Kim D, Bae S, Park J, et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods. 2015. Mar;12(3):237–43, 1 p following 243. [DOI] [PubMed] [Google Scholar]
- 99.Tsai SQ, Nguyen NT, Malagon-Lopez J, et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods. 2017. Jun;14(6):607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Frock RL, Hu J, Meyers RM, et al. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol. 2015. Feb;33(2):179–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hu J, Meyers RM, Dong J, et al. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat Protoc. 2016. May;11(5):853–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yan WX, Mirzazadeh R, Garnerone S, et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat Commun. 2017. May 12;8:15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tsai SQ, Zheng Z, Nguyen NT, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015. Feb;33(2):187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wienert B, Wyman SK, Yeh CD, et al. CRISPR off-target detection with DISCOVER-seq. Nat Protoc. 2020. May;15(5):1775–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Akcakaya P, Bobbin ML, Guo JA, et al. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature. 2018. Sep;561(7723):416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chung CH, Allen AG, Atkins AJ, Sullivan NT, Homan G, Costello R, Madrid R, Nonnemacher MR, Dampier W, Wigdahl B Safe CRISPR-Cas9 Inhibition of HIV-1with High Specificity and Broad-Spectrum Activity by Targeting LTR NF-kB Binding Sites. Molecular Therapy: Nucleic Acids. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kaushik A, Jayant RD, Nikkhah-Moshaie R, et al. Magnetically guided central nervous system delivery and toxicity evaluation of magneto-electric nanocarriers. Sci Rep. 2016. May 4;6:25309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Clayton KL, Collins DR, Lengieza J, et al. Resistance of HIV-infected macrophages to CD8(+) T lymphocyte-mediated killing drives activation of the immune system. Nat Immunol. 2018. May;19(5):475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huang SH, Ren Y, Thomas AS, et al. Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J Clin Invest. 2018. Feb 1;128(2):876–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tables 1 and 2 References
- 1.Ren Y, et al. , BCL-2 antagonism sensitizes cytotoxic T cell-resistant HIV reservoirs to elimination ex vivo. J Clin Invest, 2020. 130(5): p. 2542–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim Y, Anderson JL, and Lewin SR, Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe, 2018. 23(1): p. 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang SH, et al. , Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J Clin Invest, 2018. 128(2): p. 876–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang J, et al. , Identification of a CD4-Binding-Site Antibody to HIV that Evolved Near-Pan Neutralization Breadth. Immunity, 2016. 45(5): p. 1108–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gautam R, et al. , A single injection of crystallizable fragment domain-modified antibodies elicits durable protection from SHIV infection. Nat Med, 2018. 24(5): p. 610–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishimura Y, et al. , Early antibody therapy can induce long-lasting immunity to SHIV. Nature, 2017. 543(7646): p. 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malbec M, et al. , Broadly neutralizing antibodies that inhibit HIV-1 cell to cell transmission. J Exp Med, 2013. 210(13): p. 2813–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mediouni S, et al. , Didehydro-Cortistatin A Inhibits HIV-1 by Specifically Binding to the Unstructured Basic Region of Tat. mBio, 2019. 10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kessing CF, et al. , In Vivo Suppression of HIV Rebound by Didehydro-Cortistatin A, a “Block-and-Lock” Strategy for HIV-1 Treatment. Cell Rep, 2017. 21(3): p. 600–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vansant G, et al. , Block-And-Lock Strategies to Cure HIV Infection. Viruses, 2020. 12(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hale M, et al. , Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol Ther, 2017. 25(3): p. 570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anthony-Gonda K, et al. , Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Sci Transl Med, 2019. 11(504). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brodie SJ, et al. , In vivo migration and function of transferred HIV-1-specific cytotoxic T cells. Nat Med, 1999. 5(1): p. 34–41. [DOI] [PubMed] [Google Scholar]
- 14.Porter DL, et al. , Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med, 2011. 365(8): p. 725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi J, et al. , Advances in Developing CAR T-Cell Therapy for HIV Cure. Front Immunol, 2020. 11: p. 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porichis F, et al. , Immune Checkpoint Blockade Restores HIV-Specific CD4 T Cell Help for NK Cells. J Immunol, 2018. 201(3): p. 971–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradley T, et al. , Immune checkpoint modulation enhances HIV-1 antibody induction. Nat Commun, 2020. 11(1): p. 948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wykes MN and Lewin SR, Immune checkpoint blockade in infectious diseases. Nat Rev Immunol, 2018. 18(2): p. 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang W, et al. , CCR5 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS One, 2014. 9(12): p. e115987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z, et al. , Genome editing of the HIV co-receptors CCR5 and CXCR4 by CRISPR-Cas9 protects CD4(+) T cells from HIV-1 infection. Cell Biosci, 2017. 7: p. 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Q, et al. , Genome modification of CXCR4 by Staphylococcus aureus Cas9 renders cells resistance to HIV-1 infection. Retrovirology, 2017. 14(1): p. 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu S, et al. , Simultaneous Knockout of CXCR4 and CCR5 Genes in CD4+ T Cells via CRISPR/Cas9 Confers Resistance to Both X4- and R5-Tropic Human Immunodeficiency Virus Type 1 Infection. Hum Gene Ther, 2018. 29(1): p. 51–67. [DOI] [PubMed] [Google Scholar]
- 23.Xu L, et al. , CRISPR/Cas9-Mediated CCR5 Ablation in Human Hematopoietic Stem/Progenitor Cells Confers HIV-1 Resistance In Vivo. Mol Ther, 2017. 25(8): p. 1782–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen AG, et al. , Gene Editing of HIV-1 Co-receptors to Prevent and/or Cure Virus Infection. Front Microbiol, 2018. 9: p. 2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bella R, et al. , Removal of HIV DNA by CRISPR from Patient Blood Engrafts in Humanized Mice. Mol Ther Nucleic Acids, 2018. 12: p. 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campbell LA, et al. , Gesicle-Mediated Delivery of CRISPR/Cas9 Ribonucleoprotein Complex for Inactivating the HIV Provirus. Mol Ther, 2019. 27(1): p. 151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Darcis G, et al. , The Impact of HIV-1 Genetic Diversity on CRISPR-Cas9 Antiviral Activity and Viral Escape. Viruses, 2019. 11(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ebina H, et al. , Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep, 2013. 3: p. 2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu W, et al. , RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci U S A, 2014. 111(31): p. 11461–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaminski R, et al. , Excision of HIV-1 DNA by gene editing: a proof-of-concept in vivo study. Gene Ther, 2016. 23(8-9): p. 690–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaminski R, et al. , Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci Rep, 2016. 6: p. 22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lebbink RJ, et al. , A combinational CRISPR/Cas9 gene-editing approach can halt HIV replication and prevent viral escape. Sci Rep, 2017. 7: p. 41968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ophinni Y, et al. , CRISPR/Cas9 system targeting regulatory genes of HIV-1 inhibits viral replication in infected T-cell cultures. Sci Rep, 2018. 8(1): p. 7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dash PK, et al. , Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat Commun, 2019. 10(1): p. 2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagner DL, et al. , High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat Med, 2019. 25(2): p. 242–248. [DOI] [PubMed] [Google Scholar]
- 36.Kim D, et al. , Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods, 2015. 12(3): p. 237–43, 1 p following 243. [DOI] [PubMed] [Google Scholar]
- 37.Cameron P, et al. , Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat Methods, 2017. 14(6): p. 600–606. [DOI] [PubMed] [Google Scholar]
- 38.Tsai SQ, et al. , CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods, 2017. 14(6): p. 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frock RL, et al. , Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol, 2015. 33(2): p. 179–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan WX, et al. , BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat Commun, 2017. 8: p. 15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsai SQ, et al. , GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol, 2015. 33(2): p. 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wienert B, et al. , CRISPR off-target detection with DISCOVER-seq. Nat Protoc, 2020. 15(5): p. 1775–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akcakaya P, et al. , In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature, 2018. 561(7723): p. 416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]