

Abstract
Hemophilia A and hemophilia B are rare congenital, recessive X-linked disorders caused by lack or deficiency of clotting factor VIII (FVIII) or IX (FIX), respectively. The severity of the disease depends on the reduction of coagulation FVIII or FIX activity levels, which is determined by the type of the pathogenic variants in the genes encoding the two factors ( F8 and F9 , respectively). Molecular genetic analysis is widely applied in inherited bleeding disorders. The outcome of genetic analysis allows genetic counseling of affected families and helps find a link between the genotype and the phenotype. Genetic analysis in hemophilia has tremendously improved in the last decades. Many new techniques and modifications as well as analysis softwares became available, which made the genetic analysis and interpretation of the data faster and more accurate. Advances in genetic variant detection strategies facilitate identification of the causal variants in up to 97% of patients. In this review, we discuss the milestones in genetic analysis of hemophilia and highlight the importance of identification of the causative genetic variants for genetic counseling and particularly for the interpretation of the clinical presentation of hemophilia patients.
Keywords: genetic analysis, hemophilia, carriers, next-generation sequencing
Zusammenfassung
Hämophilie A und B sind seltene angeborene, rezessive X-chromosomale Erkrankungen, die durch einen Mangel an Gerinnungsfaktor VIII (FVIII) bzw. IX (FIX) verursacht werden. Der Schweregrad der Erkrankung hängt von der Verringerung der Gerinnungsaktivitäten von FVIII bzw. FIX ab, die durch die Art der pathogenen Varianten in den Genen für die beiden Faktoren ( F8 bzw. F9 ) bestimmt wird. Die molekulargenetische Analyze wird häufig bei vererbten Blutungsstörungen eingesetzt. Die Ergebnisse ermöglichen eine genetische Beratung der betroffenen Familien und helfen dabei, eine Verbindung zwischen dem Genotyp und dem Phänotyp zu erstellen. Die genetische Analyze der Hämophilie hat sich in den letzten Jahrzehnten enorm verbessert. Viele neue Techniken und Modifikationen sowie Analysesoftware sind inzwischen verfügbar, so dass die genetische Analyze und die Interpretation der Daten schneller und genauer erfolgen kann. Fortschritte bei den Strategien zur Erkennung von genetische Varianten erleichtern die Identifizierung der ursächlichen Varianten bei bis zu 97% der Patienten. In dieser Übersicht werden die Meilensteine der genetischen Analyze der Hämophilie erörtert und die Bedeutung der Identifizierung ursächlicher genetischer Varianten für die genetische Beratung und insbesondere für die Interpretation des Krankheitsbildes von Hämophiliepatienten hervorgehoben.
Schlüsselwörter: genetische Untersuchung, Hämophilie, Konduktorinnen, “next-generation” Sequenzierung
Introduction
Hemophilia A (HA) and hemophilia B (HB) are rare bleeding disorders caused by genetic defects in the genes encoding coagulation factor VIII (FVIII) and factor IX (FIX), leading to deficiency or absence of either of the two coagulation proteins. Classification of the severity of hemophilia is based on the amount of residual FVIII or FIX activity and it is strongly dependent on the inherited genetic alterations. The severe form is defined as a factor level less than 1 IU/dL of normal, the moderate form as a factor level of 1 to 5 IU/dL, and the mild form with factor levels greater than 5 and less than 40 IU/dL. 1 Hemophilia is a leading disease model in the field of human molecular genetics. The identification of the causative genetic defects in affected individuals is important for family counseling and has a predictive value for bleeding tendency and inhibitor risk. Genetic testing has increased the number of families with an identified defect and improved carrier testing and prenatal diagnosis. This review will consider the role and the place of molecular genetic analysis in the diagnostic algorithm for hemophilia and highlights the benefits for clinical practice, especially with the introduction of next-generation sequencing (NGS) to the routine settings.
1. Inheritance
Since both F8 and F9 genes are located on the X chromosome, they are subject to the unique inheritance pattern of X-linked genes affecting almost exclusively males. 2 Such a male is termed hemizygous and has the full phenotype of the disease. Females carry two alleles for X chromosome genes; so, the defect in a single allele can be compensated by the normal allele and these heterozygous carriers remain with a healthy phenotype in most cases. In 1961, Mary Lyon proposed that in the cells of female mammals, one of the two X chromosomes is randomly inactivated in early embryonic stage, so that both males and females have a single active X chromosome. This hypothesis provided an improved understanding of the basic mechanisms responsible for X-linked diseases. 3 4 A male's X chromosome is transmitted to his daughters and the Y chromosome is transferred to his sons. If an affected male has a child with a healthy female, none of his male offspring will be affected, but all of his female offspring will be carriers, termed obligate carriers. Owing to the X-linked inheritance of the disease and in combination with a detailed family history, it is possible to identify obligate carriers. If a female carrier has a child with a healthy male, each male offspring has a 50% chance of being affected, and female offspring has a 50% chance of being a carrier. Thus, the disease can be transmitted from affected males to male grandchildren through the carrier daughter.
Approximately 30% of cases, in both HA and HB, are described as “sporadic” 5 as the newborn baby is the first hemophiliac in this family. The manifestation of the disease without a family history can be explained with the possibility of the occurrence of a de novo genetic defect in the F8/F9 genes, either in the affected child or in a female who gave birth to a hemophilia boy or her parents. Not rarely, family history remains unknown due to lack of information or the occurrence of few male births in the family. Maternal mosaicism can also contribute for the occurrence of such cases, where no mutation is detected in the mother by standard techniques. 6 Therefore, a negative family history is insufficient to rule out hemophilia when clinical presentation is suggestive of an excessive bleed tendency.
Rarely, females may also have symptomatic hemophilia designated as “hemophilic females.” These females are diagnosed as having hemophilia, with FVIII levels comparable to those of males. Approximately 250 women and girls with a severe or moderate phenotype have been reported in the literature. 7 8 The phenotype of such females may result from complex genetic causes, which can be summarized as follows: (1) homozygosity in a female (two identical hemophilia alleles due to consanguinity), (2) compound heterozygosity (two different hemophilia alleles inherited from each parent), (3) hemizygosity (one hemophilia allele and no normal allele due to chromosomal aberrations as in Turner syndrome or mosaic Turner syndrome) (4) heterozygosity with skewed X chromosome (a nonrandom inactivation in favor of the hemophilia allele, 9 10 11 and (5) genetic causes other than hemophilia —Women with decreased FVIII levels also may have combined factor V (FV) and FVIII deficiency, an autosomal recessive disorder characterized by mildly decreased levels of both FVIII and FV, caused by variations in the LMAN1 and MCFD2 genes. 12
2. Molecular Basis of HA and HB— F8/F9 Genes and Variants
HA and HB are monogenic disorders raised from pathogenic variants in either the F8 or F9 gene located at Xq28 and Xq27 of the X chromosome, respectively. 13 F8 gene is extremely large (∼ 180 kb) and structurally complex (26 exons), while F9 gene is considerably smaller (∼ 34 kb) and structurally simpler, containing only eight exons. Additionally, the F8 gene contains two nested genes, F8A and F8B , in the region of intron 22, which largely contribute significantly to the occurrence of the most common genetic defect in severe HA—intron 22 inversion ( Fig. 1 ).
Fig. 1.
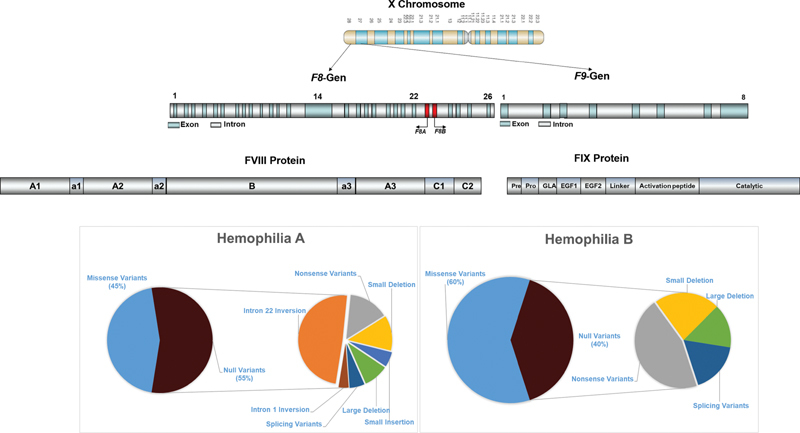
Prevalence and profile of genetic variants in hemophilia A and hemophilia B.
The molecular basis of HA and HB is extremely diverse which has been characterized in several thousands of patients. Although they occur with different frequency in HA and HB, nearly all types of genetic alterations are detected in both F8 and F9 genes—point variants, deletions, insertions, and rearrangements/inversions. Hemophilia is one of the few genetic diseases where a good correlation between genotype and phenotype is observed. The type of genetic defects strongly correlates with the plasma residual factor activity, bleeding tendency, and severity of symptoms in hemophilia patients.
Spectrum of Genetic Variants in HA
The type of genetic defect in the F8 gene predicts the severity of the disease. Defects that cause a significant disruption/absence of the FVIII protein or alteration of a major functional site are known as null variants (intron 22 and intron 1 inversions, large deletions, nonsense variants, non–A-run small deletions, and splice site variants). The second group summarizes the non-null variants, which are mainly missense variants and contribute to a moderate or mild phenotype in the majority of cases.
HA displays heterogeneity, with pathogenic variants identified throughout the whole gene. Depending on their localization, the pathogenic variants can affect FVIII protein by different mechanisms. They can interfere with the binding of FVIII to different ligands of the coagulation cascade and lead to decreased function or complete lack of FVIII synthesis or affect the expression, transport through the cell, activity, and stability of FVIII. The European Association for Hemophilia and Allied Disorders (EAHAD) variant database has recorded more than 3,000 causative genetic alterations in the F8 gene. In general, the most prevalent types of genetic defects across all disease severities are the missense variants. While for mild/moderate phenotypes, the highest prevalence of missense type is observed; in severe phenotypes they are much less presented. Severe HA phenotypes are associated with null variants in approximately 80% of index patients. The overall prevalence of genetic variants in HA can be summarized as follows: missense variants (45%), nonsense variants (8%), inversions (29%), small deletions (6%), duplications (3%), insertions (2%), and large deletions (7%). Although most genetic defects are irregularly distributed along the F8 , four hot spots can be identified accounting for recurrent occurrence of genetic changes: the two inversion hot spots, the CpG dinucleotide hotspots, caused by spontaneous methylation-induced deamination of the 5-methylcytosine, 14 15 and small deletions and insertions caused by polymerase slippage errors in series of adenine nucleotides (A stretches). 16 This subgroup of small deletions/duplications, located at stretches of adenines, is associated with a mitigated phenotype of severe HA and rare bleeding. Endogenous restoration of the reading frame by polymerase errors during DNA replication/RNA transcription, resulting in small amounts of endogenous FVIII protein, could explain the observed phenomena. 17
Intron 22 and Intron 1 Inversions
The two intronic regions of the F8 gene, intron 1 and intron 22, are of particular interest owing to their frequent involvement in pathological inversions resulting from recombination with homologous regions outside the F8 gene. The region that is homologous to intron 1 (int1h2) is located approximately 140 kb toward the telomere and is in the opposite orientation. Intrachromatid or intrachromosome homologous recombination of these two regions causes an inversion that displaces exon 1 of the F8 gene by approximately 140 kb toward the telomere. 18 This inversion causes 1 to 4% of all severe cases of HA.
However, the most frequent inversion involves intron 22. Approximately 45% of severe HA is accounted by an intrachromosomal inversion in intron 22 which occurs due to two regions homologous to the sequences in intron 22 that are positioned approximately 500 kb telomeric to the F8 gene. 19 20 This inversion event occurs spontaneously in male germ cells with 10-fold higher rate than in females. 21
Several gross complex rearrangements in F8 have been reported and due to their highly deleterious nature, these defects are associated with severe hemophilia phenotype. 22 23
HB Genetic Variant Spectrum
HB is also genetically heterogeneous, with a predominance of missense variants, without a common inversion event analogous to the inversions in the F8 gene. These variants account for approximately 65% of all unique genetic variants in HB. The pathogenic defects occur throughout the whole F9 gene including the promoter and 3′UTR but with the highest concentration in the largest exon 8. In contrast to the F8 gene, missense variants account for majority of cases with severe HB and for the larger proportion of pathogenic defects in mild/moderate phenotypes. The genetic variants are reported as nonsense (18%), small deletions (9%), small insertions (2%), splice variants (6%), and large deletions (5%). In severe HB, the proportion of null mutations is approximately 35%, while it is 80% in severe HA. The FIX EAHAD Variant Database currently reports more than 1,000 unique variants.
HB Leyden : Although rare, noteworthy is the existence of a particular unique variant of HB that initially is presented with severe or moderate phenotype (depending on the variant), followed by normalization of the FIX levels in the puberty—HB Leyden. 24 The molecular mechanism likely involves variants in the promoter region of the F9 disrupting the binding sites to one of three transcription factors—HNF4a (hepatic nuclear factor 4a), C/EBP (CCAAT enhancer-binding protein), or ONECUT1/2. This apparent “cure” of HB with age is thought to be related to the rising postpubertal growth hormone levels. Similar molecular mechanisms have not yet been identified in HA patients. 25
3. Genetic Analyses
In hemophilia, molecular testing remains the only way of providing an accurate diagnosis, overcoming the variability and inconsistent data obtained by the other methods based on the quantification of coagulation factors. Moreover, genetic analyses enhance the diagnostic precision, especially in milder bleeding phenotypes, contributing to correct treatment decisions.
Early genetic diagnosis of hemophilia was primarily focused on carrier detection 26 using a genetic marker that cosegregates with hemophilia in the family. Restriction fragment length polymorphism (RFLP) analysis was applied to trace the affected chromosome and predict the carrier status of females. Although this was the only existing approach, the procedure was not successful in many cases due to lack of informative evidence of the analyzed polymorphism, high proportion of sporadic cases, lack of index patients, and need of several family members for segregation analysis. 27 Additionally, RFLPs analyses were performed by Southern blotting, a laborious and time-consuming method.
The human F8 gene was cloned and characterized in 1982. In 1991, Higuchi et al performed the first systematic analysis of complete F8 coding region, using denaturing gradient gel electrophoresis as a variant screening method but no causative variant was detected in about half of the severely affected patients. 28 Shortly after, the F8 intron 22 inversion was discovered, which accounts for approximately 45% of the severe HA cases. Initially, the analyses were performed using Southern blotting and long-distance PCR until a simplified technique of inverse PCR (inverse shifting PCR [IS-PCR]) was established. 29 30
The advent of sequencing technologies that allow direct characterizing of variations has helped overcome many of the limitations of previous techniques. Direct sequencing, either Sanger or NGS, has increased the sensitivity of screening techniques and is currently the gold standard for molecular genetic analysis.
Sanger sequencing was the first applied method that involved amplification of F8/F9 genes by polymerase chain reaction (PCR) as short DNA segments (200–1,000 nucleotides) followed by sequencing and alignment of resulting sequences with the human genome consensus sequence using dedicated software. Currently, Sanger sequencing is considered a time-consuming and rather expensive method with low throughput.
In recent years, NGS became available in diagnostic laboratories and began to be used for genetic analysis of bleeding disorders. The implementation of NGS in routine diagnostics allowed the parallel analysis of a large number of genes, which greatly reduced the workload and costs compared with Sanger sequencing and provided the result within a short and clearly defined time period. 31 32 33 Moreover, the NGS technique enables the analysis of large deletions and duplications without a need for the conventional gene dosage analysis using multiplex ligation-dependent probe amplification (MLPA). As the correct interpretation of genetic variants and their pathogenicity evaluation is crucial for diagnosis and appropriate genetic counseling, standards and guidelines for the interpretation and classification of gene variants have been suggested. The identified genetic variations are categorized as “pathogenic,” “likely pathogenic,” “uncertain significance,” “likely benign,” and “benign” based on evidence for a set of criteria and the strength of those criteria. 34
Currently, sequencing of coagulation F8 or F9 genes in the hemophilia patient and the determination of carrier status is a fairly trivial procedure, which allows for adequate genetic counseling. The inclusion of NGS in the routine diagnostic algorithm improved the molecular testing in terms of speed, costs, and efficiency. 35
In severe HA patients, molecular diagnosis starts with screening for intron 22 and intron 1 inversions, followed by direct sequencing of coding regions and splice sites of the F8 in patients negative for the inversions ( Fig. 2 ). In mild/moderate HA and HB patients, the genetic analyze is initiated directly with detection of molecular defects by direct sequencing ( Figs. 2 , 3 ).
Fig. 2.
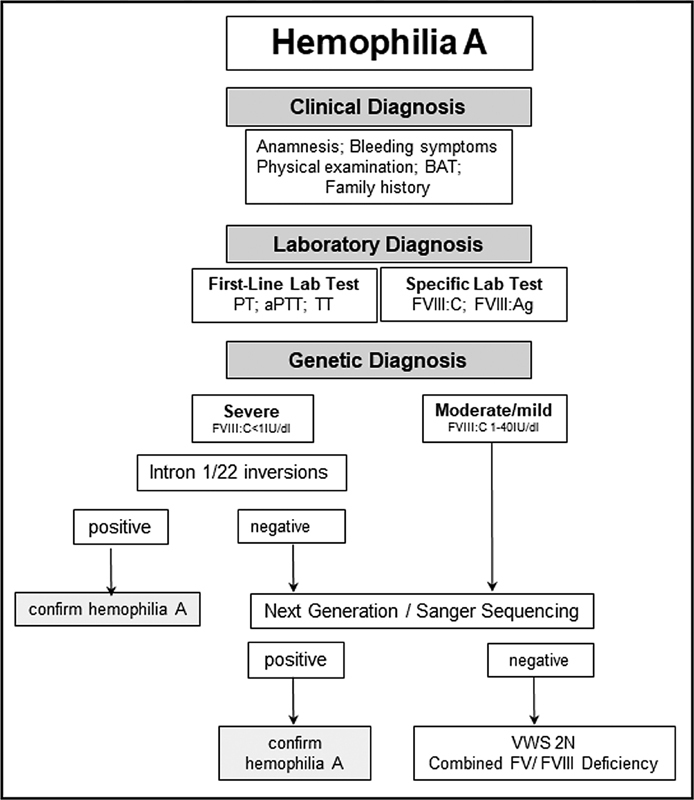
Diagnostic flow chart for hemophilia A. PT, prothrombin time; aPTT, activated partial thromboplastin time; TT, thrombin time; BAT, bleeding assessment tool; VWS 2N, von Willebrand syndrome type 2 Normandy.
Fig. 3.
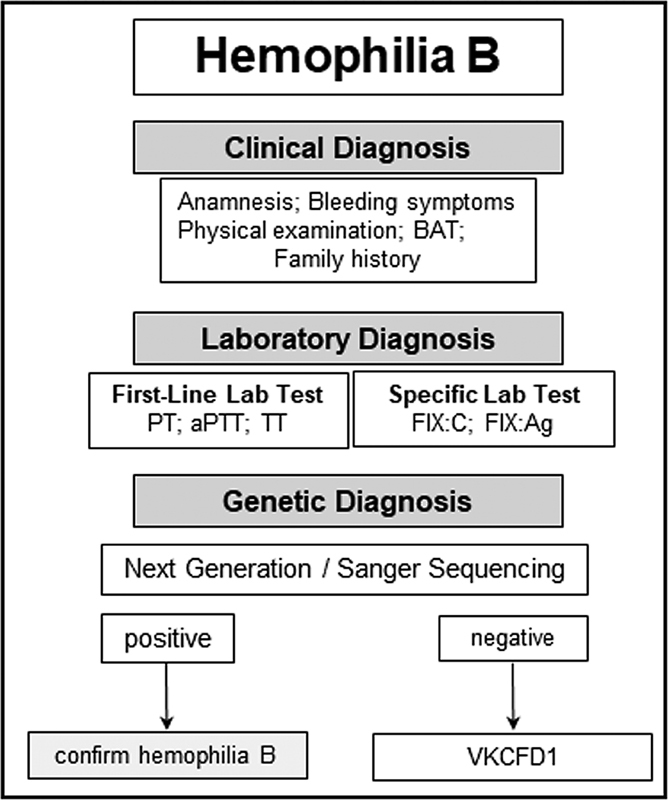
Diagnostic flow chart for hemophilia B. PT, prothrombin time; APTT, activated partial thromboplastin time; TT, thrombin time; BAT, bleeding assessment tool; VKCFD1, vitamin K–dependent clotting factors-1.
Implementation of NGS in the routine diagnosing settings for HA/HB has both strengths and limitations. 36 A limitation may be considered the inability of NGS to detect gross genetic rearrangements, such as large inversions, which conventionally require specific techniques for the analyses. Large deletions/duplications of one or more exons can be as efficiently identified with NGS as with MLPA. 37
Although NGS is of great benefit for hemophilia patients and their family, the inevitable discovery of genetic variants not related to the hemophilia, the so-called unsolicited, incidental or secondary findings, presents a potential challenge. This is a complex issue with many confounding factors, involving ethics and legitimacy. 38 Nowadays, there is a consensus that if these variants are not known to be connected with any phenotype they are of no further concern. If these variants are associated with a clinical phenotype, particularly a severe one, then these incidental findings become relevant and should be reported. Particularly important and difficult is the decision whether to report or not pathogenic variants in genes ( RUNX1 , ETV6 , and ANKRD26 ) associated with increased risk of malignancy development. Thus, it is very essential to note that using multigene panel analyses, HA/HB patients should be explicitly informed about the possibility of unexpected findings, giving them the choice to decide whether to be informed about such variants or not. 39
The success rate in identifying pathogenic variants in inherited bleeding disorders depends on both the type of molecular analysis performed and the genetic heterogeneity of the disease. Despite remarkable improvements in screening technologies, the detection rate of causative defects in both F8 and F9 genes is approximately 95%. Still, in 5% of cases with reduced FVIII/FIX levels, the genetic defect remains unclear. 40 The missing 5% may be in part due to phenotypic misdiagnosis of HA and von Willebrand disease type 2N (VWD 2N) as the clinical presentation is quite similar. In rare cases, combined FV/FVIII deficiency is associated with pathogenic variants in the MCFD2 and LMAN1 genes that contribute to the low FVIII levels. Sporadic cases of multiple vitamin K–dependent coagulation factor deficiency (VKCFDs) involving prothrombin, FVII, FIX, and FX might be misdiagnosed as HB. 41 Pathogenic HA/HB variants outside the region of the F8/F9 genes might remain undetectable with the current testing procedures. Although additional molecular and informatics research is needed, NGS is expected to elucidate cases without “detected” genetic alterations in the F8/F9 genes, leading to better understanding of genotype/phenotype correlation and more effective clinical care.
4. Genetic Counseling and Reproductive Options
Genetic Counseling
HA and HB, as congenital disorders, have a significant medical, psychological, and emotional impact on both affected males and female carriers. The possibility of transmitting the genetic disorders to offspring may lead to feelings of guilt and self-blame in parents and may have a strong impact on reproductive decision-making. 42 43 Genetic counseling, as a process that provides individuals and families with information about how genetic disorders are inherited and helps them make informed medical and personal decisions, has become an integral part of comprehensive care for hemophilia. This is of extreme importance especially for girls and women who carry the disease and are emotionally burdened during reproductive decision-making. In that context, genetic counseling of hemophilia involves three main aspects: identifying females who are at increased risk of either having hemophilia or passing it to the offspring; providing knowledge about the genetic aspects of the disease and its consequences for offspring; and offering information and education of prenatal diagnosis and reimplantation genetic diagnosis. As a consequence, the final decision of partners can be based on genetic evidence and counseling from one side and from their own experience with the disease on the other, as the severity of hemophilia remains stable within an individual family. 44
Nowadays, the causative genetic variant is routinely identified in hemophilia families, followed by genetic counseling, determination of carrier status in women at risk, so that informed reproductive decisions can be made in affected families. Large studies exploring the attitude of women from hemophilia families to become pregnant and childbearing showed quite controversial opinions that depended firmly on women's experiences with hemophilia in their families. The quality of life in hemophilia family strongly influenced this decision. In families that experienced a satisfactory quality of life, the women were more readily to have children of their own compared with women coming from families that faced difficulties with acceptance and managements of the disease. 45 46 47 In general, women wished to be well informed before making a reproductive decision.
Genetic counseling is of great importance, as it offers women the opportunity to be professionally informed about the existing probability of having a hemophilic son and the current prevention procedures. Accurate knowledge of the genetic defect in the family allows prenatal diagnosis to be performed at different stages: on the germ line of the female carrier for preimplantation genetic diagnosis (PGD), on chorionic villi during the first trimester of pregnancy, or on amniocytes at later stages of pregnancy. 48 49 50 The optimal time to determine genetic risk, clarify carrier status, and discuss the availability of prenatal/preimplantation genetic testing is before pregnancy. Males with hemophilia also need genetic counseling, including education about the genetics of their disease, their risk of affecting grandchildren, and the possibility that their daughters may have bleeding symptoms. Therefore, it is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to both hemophilia males and female carriers.
Special attention is required when counseling families with newly diagnosed hemophilia without family history of disease, especially when the mother is not a carrier. In these cases, the disease can be attributed either to a de novo variant in the F8/F9 genes or to the presence of a mosaicism in the mother. 51 52 Mosaicism is the presence of cell lineages with different genotypes in different tissues of an individual. It may be somatic or germline specific or both; both may have an impact on reproductive outcomes. In this case, the pathogenic variant is present only in some of the germline cells and therefore cannot be detected in peripheral white blood cells, which are typically used to perform genetic analysis. 53 54 The possibility of mosaicism in a family with a hemophilia patient without a family history can never be excluded. Genetic counseling has to be made with extreme caution and the parents must be informed that there is a possibility of having a second child with hemophilia, even if their own genetic test is negative.
Prenatal Diagnosis
Prenatal diagnostics are primarily used to guide obstetric management and to enable parents to be psychologically prepared for raising an affected child. 55 Prenatal testing is indicated in families with severe or moderate forms of hemophilia, while in families with the mild phenotype such indication is rare. Knowledge of fetal status can guide management decisions during childbirth, including avoidance of procedures and instrumentation that can cause neonatal trauma, such as forceps or vacuum-assisted extraction in vaginal and caesarean deliveries. Molecular genetic testing of at-risk female relatives to determine their genetic status is most informative if the pathogenic variant has been already identified in the proband.
In families with severe hemophilia, prenatal diagnosis is sometimes performed with the aim of terminating a pregnancy in the case of an affected child. 45 Quite several studies revealed that the choices regarding prenatal diagnosis is again based on experiences with the clinical severity, complications, and management of hemophilia within the family. Religious beliefs and prenatal diagnosis experiences of family members could also affect the decision-making. In some women, there are concerns about prenatal diagnosis when the potential risk of the procedure is taken into consideration and the fear when results became known. 45 56 From emotional relief and happiness in confirmation of expecting a healthy child to sadness, disappointment, and burden of subsequent decisions in diagnosis of an affected child could be disclosed.
Prenatal diagnosis from chorionic villus biopsy or amniocentesis is widely available. It can be performed between the 11th and 14th weeks of gestation and enables early genetic diagnosis with subsequent possibility of termination of pregnancy. There are cases in which pregnant women undergo prenatal diagnosis only to know whether or not can expect a male fetus affected with hemophilia. In this situation, mothers should be well informed about the procedure-related risk (miscarriages). Recently, noninvasive testing of fetus cells present in maternal plasma has been performed as early as 10 weeks of pregnancy, although it has been more successful later in pregnancy. 57 These tests may reduce the number of women undergoing invasive procedures when pregnancy termination is not their option, but it does not yet provide a definitive diagnosis. Invasive testing for prenatal diagnosis is becoming less common and is often performed to help with psychological preparation rather than to help the choice for terminating the pregnancy if the fetus is affected by severe hemophilia.
Improvements in care and clinical outcomes of hemophilia have been paralleled not only with advances in methods of prenatal diagnosis but also in attitudes to decisions about carrying out prenatal diagnostic tests for hemophilia. PGD and noninvasive prenatal testing are among the latest methods of prenatal diagnosis available to couples. PGD offers the possibility to select unaffected embryos but implies that the pregnancy occurs through in vitro fertilization procedures. One extensive study showed that hemophilia carriers have positive attitude toward PGD, except those who ejected it based on religious beliefs.
5. Benefits of Genetic Testing in Clinical Practice
Reaching a definitive molecular diagnosis is important for a variety of reasons. Currently, molecular diagnosis of HA and HB allows prediction of likelihood of inhibitor development associated with specific pathogenic variants and response to immune tolerance induction. It enables the clinician to tailor clinical management and discuss the prognosis with the patient. Identification of family-specific genetic variant enables carrier testing of female relatives. Without genetic testing, it can be challenging to establish the exact cause of a bleeding in mild phenotype with similar clinical and laboratory features (HA vs. VWD 2N).
Inhibitor Prediction
Alloantibodies (inhibitors) against FVIII or FIX represent the major complication in hemophilia care, as they render classical substitution therapy ineffective. Inhibitors occur at a frequency of 20 to 30% in severe HA and approximately 3 to 5% of those with severe HB. 58 59 The frequency of inhibitor formation varies depending on various environmental (treatment regimen, type of concentrate) and genetic factors (race/ethnicity, family history, type of genetic defect). 60 The F8 gene variant is a major contributor to genetic risk factors for inhibitor development. 61 62 The correlation between the genetic variant and inhibitor development is well established. Variants predicted to cause complete absence of FVIII are associated with a higher risk, whereas variants that lead to some protein synthesis are associated with a lower risk. Moreover, patients carrying large multidomain deletions are at the highest risk (up to 80%), while patients bearing missense variants have the lowest risk (<5%). Interestingly, all other types of genetic variants are associated with a median risk of inhibitor formation ranging between 17 and 41%.
Molecular analysis may predict the risk of inhibitor development, not only in the case of severe HA but also in mild HA. It is well known that some missense variants are associated with occurrence of inhibitors in mild HA, while this has never been reported in patients with mild HB. 63 An alternative pathologic mechanism may underlie inhibitor development in these patients. Certain missense variants may alter the immunogenicity of the FVIII protein and induce an inhibitor response against the altered epitope. 64 The higher prevalence of less severe genetic variants (missense) in HB and the less common presence of truncated protein compared with HA may partly explain the lower prevalence of inhibitors in severe HB. Inhibitors in nonsevere HB are almost absent. 59 65 Identification of underlying genetic variant, especially in HB, is important not only for the prediction of the risk of inhibitor formation but also the possibility of anaphylactic reaction occurring after infused replacement therapy. 62
Carriers
Due to the recessive X-linked inheritance pattern, women in hemophilia families may be heterozygous for the genetic defect referred to as “carriers of hemophilia.” Knowing mother's carrier status and genotype related to disease severity can help in evaluating the mother's personal bleeding risk and in planning treatment. Approximately one-third of hemophilia carriers bleed and require treatment. They experience more spontaneous and provoked hemorrhages than noncarriers experience, and are at higher risk of prolonged bleeding after operations, tooth extractions, and tonsillectomy. The risk is highest in those with the lowest clotting factor levels.
Owing to the X-inactivation phenomena, pedigree analysis and clotting FVIII or FIX levels alone are not sufficient to diagnose carriership for hemophilia. While genetic testing is not mandatory for women who are obligate carriers, it is the only way to identify carriers in other scenarios. Genetic testing for carriers is a complex issue, raising several ethical and cultural concerns. Main concerns include disclosure of genetic confidentiality, psychosocial stigma, and emotional burden of future disability.
Severe Hemophilia with Mild Bleeding Phenotype
Although the severity of hemophilia is defined by overall bleeding manifestations, the individual clinical phenotype varies within each group. However, the severity and frequency of bleeding may be different in hemophiliacs with the same factor activity and genetic defect. 66 A mild bleeding tendency is reported in 10 to 15% of severe hemophilia patients. The basis for this heterogeneity in the clinical expression of severe hemophilia is still poorly understood. One possible explanation is the nature and location of the genetic defect. Small deletion/insertions within polyA runs of exon 14 of the F8 are theoretically associated with null variants and FVIII:C of less than 1%, but the hemophilia phenotype is associated with reduced bleeding tendency due to restoration of the reading frame in these specific nucleotide sequences. 16 The evidence that some F8/F9 genetic variants are independent predictors of mild bleeding pattern in severe hemophiliacs may have implications for patient management, as it would influence the choice and timing of onset of prophylaxis and/or the adoption of individualized dose-escalating regimens. These findings are in addition to a lower risk of inhibitor development associated with less severe gene defects, and strengthen the recommendation for early molecular diagnosis to plan different therapeutic strategies in children.
Another possible explanation for the mitigation of the bleeding phenotype in severe hemophilia includes the effect of genetic variants in other loci. By simultaneously detecting pathogenic variants in many genes in a single test, it has been possible to detect genetic variants in one or more other genes along with those in the F8/F9 genes which may lead to phenotypes with milder symptoms. It has been suggested that the mild hypercoagulable state associated with gain-of-function thrombophilic variants such as FV Leiden or prothrombin G20210A may protect hemophiliacs from excessive bleeding; however, the clinical significance of these variants is still controversial. 67 Other coagulation abnormalities, related to natural anticoagulants (deficiencies of antithrombin, protein C, protein S), platelet disorders, and fibrinolytic factors, have been assumed to modulate the bleeding phenotype. 68 69 70 71 With the application of NGS in routine diagnostic, identification of such coincidental inheritance is rather straightforward due to the use of multiple gene panels. Such findings need to be interpreted carefully and might provide more evidence to elucidate this phenomenon.
Differential Diagnosis between Mild/Moderate Hemophilia A and VWD
Bleeding phenotypes in VWD are heterogeneous, and most of the forms can be distinguished from HA by measuring VWF antigen and activity levels. However, these functional assays frequently display considerable interlaboratory variability, and a definitive diagnosis cannot be achieved. Differentiating VWD-2N from mild/moderate HA is crucial for effective genetic counseling and therapy. 72 These important findings prompted a change in the therapeutic approach and genetic counseling. 73 Based on these and previously published results, it is necessary to consider VWD 2N in patients with low FVIII:C in the context of the failure to identify a hemophilic variant, FVIII:C/VWF activity ratio less than 0.9, or inadequate response to recombinant FVIII treatment. 35 74 Simultaneous genetic analyses of the F8/VWF with NGS yield a definitive diagnosis in such cases.
Discrepancy between Factor VIII Activity Assays
Regardless of the scenario that leads to the suspicion of hemophilia, the definite diagnosis is made by measurement of residual FVIII and FIX clotting activity (FVIII:C and FIX:C). These measurements can be performed using either one-stage or chromogenic coagulation assays. Results from the various FVIII:C assays are usually equivalent in patients with HA, although several reports have revealed systematic assay discrepancies in FVIII:C in one-third of patients with nonsevere HA as part of HA phenotype. This phenomenon becomes clinically significant when it affects diagnosis of HA or assessment of disease severity. 75 76 The reported data confirm that the discrepancy in FVIII:C assay is a fairly common event in nonsevere HA and has genetic background. The profile of genetic defects associated with FVIII:C discrepancy is linked to the class of missense defects. All genetic variants associated with discrepancy phenomena, where one-stage assay shows higher values then the chromogenic assay, are localized in the A1, A2, and A3 domains at the interface between the subunits (A1–A2, A1–A3, A2–A3). These defects lead to a decreased stability of the A2 domain in the activated FVIII protein, resulting in destabilization of the FVIII protein and premature loss of cofactor activity. Discrepancies characterized by higher FVIII:C measured by chromogenic assay involved genetic defects affecting either thrombin, VWF, or FIX binding sites.
Conclusions
High-throughput NGS has revolutionized DNA sequencing by allowing the simultaneous rapid exploration of multiple genes at manageable cost. 32 In hemophilia, molecular testing remains the only way of providing an accurate diagnosis, overcoming the variability and inconsistent data obtained by the conventional methods based on the quantification of coagulation factors. Understanding the basic genetics of hemophilia has contributed to clinical care by providing information leading to advanced treatment, better testing, and more accurate information for patients and families. Genetic testing is indicated for family planning, carrier testing, and prenatal diagnosis.
Footnotes
Conflict of Interest The authors declare that they have no conflict of interest.
References
- 1.Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders of the Scientific and Standardization Committee of the International Society on Thrombosis and Hemostasis . Blanchette V S, Key N S, Ljung L R, Manco-Johnson M J, van den Berg H M, Srivastava A. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935–1939. doi: 10.1111/jth.12672. [DOI] [PubMed] [Google Scholar]
- 2.Berntorp E, Shapiro A D.Modern haemophilia care Lancet 2012379(9824):1447–1456. [DOI] [PubMed] [Google Scholar]
- 3.Lyon M F. Gene action in the X-chromosome of the mouse (Mus musculus L.) Nature. 1961;190:372–373. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- 4.Harper P S. Mary Lyon and the hypothesis of random X chromosome inactivation. Hum Genet. 2011;130(02):169–174. doi: 10.1007/s00439-011-1013-x. [DOI] [PubMed] [Google Scholar]
- 5.Fischer K, Ljung R, Platokouki H. Prospective observational cohort studies for studying rare diseases: the European PedNet Haemophilia Registry. Haemophilia. 2014;20(04):e280–e286. doi: 10.1111/hae.12448. [DOI] [PubMed] [Google Scholar]
- 6.Gomez K, Laffan M, Keeney S, Sutherland M, Curry N, Lunt P. Recommendations for the clinical interpretation of genetic variants and presentation of results to patients with inherited bleeding disorders. A UK Haemophilia Centre Doctors' Organisation Good Practice Paper. Haemophilia. 2019;25(01):116–126. doi: 10.1111/hae.13637. [DOI] [PubMed] [Google Scholar]
- 7.Miller C H, Bean C J. Genetic causes of haemophilia in women and girls. Haemophilia. 2021;27(02):e164–e179. doi: 10.1111/hae.14186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pavlova A, Brondke H, Müsebeck J, Pollmann H, Srivastava A, Oldenburg J. Molecular mechanisms underlying hemophilia A phenotype in seven females. J Thromb Haemost. 2009;7(06):976–982. doi: 10.1111/j.1538-7836.2009.03346.x. [DOI] [PubMed] [Google Scholar]
- 9.Acquila M, Caprino D, Bicocchi P, Mori P G, Tagliaferri A R. A skewed lyonization phenomenon as cause of hemophilia A in a female patient. Blood. 1995;85(02):599–600. [PubMed] [Google Scholar]
- 10.Bennett C M, Boye E, Neufeld E J. Female monozygotic twins discordant for hemophilia A due to nonrandom X-chromosome inactivation. Am J Hematol. 2008;83(10):778–780. doi: 10.1002/ajh.21219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radic C P, Rossetti L C, Abelleyro M M. Phenotype-genotype correlations in hemophilia A carriers are consistent with the binary role of the phase between F8 and X-chromosome inactivation. J Thromb Haemost. 2015;13(04):530–539. doi: 10.1111/jth.12854. [DOI] [PubMed] [Google Scholar]
- 12.Palla R, Peyvandi F, Shapiro A D. Rare bleeding disorders: diagnosis and treatment. Blood. 2015;125(13):2052–2061. doi: 10.1182/blood-2014-08-532820. [DOI] [PubMed] [Google Scholar]
- 13.Gitschier J, Wood W I, Goralka T M.Characterization of the human factor VIII gene Nature 1984312(5992):326–330. [DOI] [PubMed] [Google Scholar]
- 14.Youssoufian H, Wong C, Aronis S, Platokoukis H, Kazazian H H, Jr, Antonarakis S E. Moderately severe hemophilia A resulting from Glu–Gly substitution in exon 7 of the factor VIII gene. Am J Hum Genet. 1988;42(06):867–871. [PMC free article] [PubMed] [Google Scholar]
- 15.El-Maarri O, Olek A, Balaban B. Methylation levels at selected CpG sites in the factor VIII and FGFR3 genes, in mature female and male germ cells: implications for male-driven evolution. Am J Hum Genet. 1998;63(04):1001–1008. doi: 10.1086/302065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oldenburg J, Schröder J, Schmitt C, Brackmann H H, Schwaab R. Small deletion/insertion mutations within poly-A runs of the factor VIII gene mitigate the severe haemophilia A phenotype. Thromb Haemost. 1998;79(02):452–453. [PubMed] [Google Scholar]
- 17.Chatterjee N, Walker G C. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58(05):235–263. doi: 10.1002/em.22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bagnall R D, Waseem N, Green P M, Giannelli F. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood. 2002;99(01):168–174. doi: 10.1182/blood.v99.1.168. [DOI] [PubMed] [Google Scholar]
- 19.Naylor J A, Buck D, Green P, Williamson H, Bentley D, Giannelli F. Investigation of the factor VIII intron 22 repeated region (int22h) and the associated inversion junctions. Hum Mol Genet. 1995;4(07):1217–1224. doi: 10.1093/hmg/4.7.1217. [DOI] [PubMed] [Google Scholar]
- 20.Lakich D, Kazazian H H, Jr, Antonarakis S E, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat Genet. 1993;5(03):236–241. doi: 10.1038/ng1193-236. [DOI] [PubMed] [Google Scholar]
- 21.Rossiter J P, Young M, Kimberland M L. Factor VIII gene inversions causing severe hemophilia A originate almost exclusively in male germ cells. Hum Mol Genet. 1994;3(07):1035–1039. doi: 10.1093/hmg/3.7.1035. [DOI] [PubMed] [Google Scholar]
- 22.Tavassoli K, Eigel A, Horst J. A deletion/insertion leading to the generation of a direct repeat as a result of slipped mispairing and intragenic recombination in the factor VIII gene. Hum Genet. 1999;104(05):435–437. doi: 10.1007/s004390050981. [DOI] [PubMed] [Google Scholar]
- 23.Vidal F, Farssac E, Tusell J, Puig L, Gallardo D. First molecular characterization of an unequal homologous Alu-mediated recombination event responsible for hemophilia. Thromb Haemost. 2002;88(01):12–16. [PubMed] [Google Scholar]
- 24.Briet E, Bertina R M, van Tilburg N H, Veltkamp J, Veltkamp J.Hemophilia B Leyden: A sex-linked hereditary disorder that improves after pubertyN Engl J Med, 306 (1982), p. 788 [DOI] [PubMed]
- 25.Funnell A PW, Crossley M. Hemophilia B Leyden and once mysterious cis-regulatory mutations. Trends Genet. 2014;30(01):18–23. doi: 10.1016/j.tig.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 26.Schwaab R, Oldenburg J, Higuchi M. Haemophilia A: carrier detection by DNA analysis. Blut. 1988;57(02):85–90. doi: 10.1007/BF00319731. [DOI] [PubMed] [Google Scholar]
- 27.Lin S-Y, Su Y-N, Hung C-C. Mutation spectrum of 122 hemophilia A families from Taiwanese population by LD-PCR, DHPLC, multiplex PCR and evaluating the clinical application of HRM. BMC Med Genet. 2008;9:53. doi: 10.1186/1471-2350-9-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Higuchi M, Kazazian H H, Jr, Kasch L. Molecular characterization of severe hemophilia A suggests that about half the mutations are not within the coding regions and splice junctions of the factor VIII gene. Proc Natl Acad Sci U S A. 1991;88(16):7405–7409. doi: 10.1073/pnas.88.16.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossetti L C, Radic C P, Larripa I B, De Brasi C D. Genotyping the hemophilia inversion hotspot by use of inverse PCR. Clin Chem. 2005;51(07):1154–1158. doi: 10.1373/clinchem.2004.046490. [DOI] [PubMed] [Google Scholar]
- 30.Rossetti L C, Radic C P, Larripa I B, De Brasi C D. Developing a new generation of tests for genotyping hemophilia-causative rearrangements involving int22h and int1h hotspots in the factor VIII gene. J Thromb Haemost. 2008;6(05):830–836. doi: 10.1111/j.1538-7836.2008.02926.x. [DOI] [PubMed] [Google Scholar]
- 31.Lu J T, Campeau P M, Lee B H. Genotype-phenotype correlation–promiscuity in the era of next-generation sequencing. N Engl J Med. 2014;371(07):593–596. doi: 10.1056/NEJMp1400788. [DOI] [PubMed] [Google Scholar]
- 32.Sikkema-Raddatz B, Johansson L F, de Boer E N. Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics. Hum Mutat. 2013;34(07):1035–1042. doi: 10.1002/humu.22332. [DOI] [PubMed] [Google Scholar]
- 33.Bastida J M, Del Rey M, Lozano M L. Design and application of a 23-gene panel by next-generation sequencing for inherited coagulation bleeding disorders. Haemophilia. 2016;22(04):590–597. doi: 10.1111/hae.12908. [DOI] [PubMed] [Google Scholar]
- 34.ACMG Laboratory Quality Assurance Committee . Richards S, Aziz N, Bale S. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(05):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bastida J M, González-Porras J R, Jiménez C. Application of a molecular diagnostic algorithm for haemophilia A and B using next-generation sequencing of entire F8, F9 and VWF genes. Thromb Haemost. 2017;117(01):66–74. doi: 10.1160/TH16-05-0375. [DOI] [PubMed] [Google Scholar]
- 36.Ver Donck F, Downes K, Freson K. Strengths and limitations of high-throughput sequencing for the diagnosis of inherited bleeding and platelet disorders. J Thromb Haemost. 2020;18(08):1839–1845. doi: 10.1111/jth.14945. [DOI] [PubMed] [Google Scholar]
- 37.Zhao M, Wang Q, Wang Q, Jia P, Zhao Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics. 2013;14 11:S1. doi: 10.1186/1471-2105-14-S11-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolf S M. The continuing evolution of ethical standards for genomic sequencing in clinical care: restoring patient choice. J Law Med Ethics. 2017;45(03):333–340. doi: 10.1177/1073110517737531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ver Donck F, Labarque V, Freson K. Hemostatic phenotypes and genetic disorders. Res Pract Thromb Haemost. 2021;5(08):e12637. doi: 10.1002/rth2.12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pezeshkpoor B, Pavlova A, Oldenburg J, El-Maarri O. F8 genetic analysis strategies when standard approaches fail. Hamostaseologie. 2014;34(02):167–173. doi: 10.5482/HAMO-13-08-0043. [DOI] [PubMed] [Google Scholar]
- 41.Czogalla K J, Watzka M, Oldenburg J. VKCFD2 - from clinical phenotype to molecular mechanism. Hamostaseologie. 2016;36 02:S13–S20. [PubMed] [Google Scholar]
- 42.McLintock C. Women with bleeding disorders: clinical and psychological issues. Haemophilia. 2018;24 06:22–28. doi: 10.1111/hae.13501. [DOI] [PubMed] [Google Scholar]
- 43.Lavery S. Preimplantation genetic diagnosis of haemophilia. Br J Haematol. 2009;144(03):303–307. doi: 10.1111/j.1365-2141.2008.07391.x. [DOI] [PubMed] [Google Scholar]
- 44.Punt M C, Aalders T H, Bloemenkamp K WM. The experiences and attitudes of hemophilia carriers around pregnancy: a qualitative systematic review. J Thromb Haemost. 2020;18(07):1626–1636. doi: 10.1111/jth.14825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gillham A, Greyling B, Wessels T-M. Uptake of genetic counseling, knowledge of bleeding risks and psychosocial impact in a South African cohort of female relatives of people with hemophilia. J Genet Couns. 2015;24(06):978–986. doi: 10.1007/s10897-015-9834-8. [DOI] [PubMed] [Google Scholar]
- 46.von der Lippe C, Frich J C, Harris A, Solbrække K N. “It was a lot tougher than I thought it would be”. A qualitative study on the changing nature of being a hemophilia carrier. J Genet Couns. 2017;26(06):1324–1332. doi: 10.1007/s10897-017-0112-9. [DOI] [PubMed] [Google Scholar]
- 47.Kadir R A, Sabin C A, Goldman E, Pollard D, Economides D L, Lee C A. Reproductive choices of women in families with haemophilia. Haemophilia. 2000;6(01):33–40. doi: 10.1046/j.1365-2516.2000.00353.x. [DOI] [PubMed] [Google Scholar]
- 48.Laurie A D, Hill A M, Harraway J R. Preimplantation genetic diagnosis for hemophilia A using indirect linkage analysis and direct genotyping approaches. J Thromb Haemost. 2010;8(04):783–789. doi: 10.1111/j.1538-7836.2010.03768.x. [DOI] [PubMed] [Google Scholar]
- 49.Ljung R C. Prenatal diagnosis of haemophilia. Haemophilia. 1999;5(02):84–87. doi: 10.1046/j.1365-2516.1999.00295.x. [DOI] [PubMed] [Google Scholar]
- 50.Cutler J, Chappell L C, Kyle P, Madan B. Third trimester amniocentesis for diagnosis of inherited bleeding disorders prior to delivery. Haemophilia. 2013;19(06):904–907. doi: 10.1111/hae.12247. [DOI] [PubMed] [Google Scholar]
- 51.Ljung R C, Sjörin E. Origin of mutation in sporadic cases of haemophilia A. Br J Haematol. 1999;106(04):870–874. doi: 10.1046/j.1365-2141.1999.01631.x. [DOI] [PubMed] [Google Scholar]
- 52.Leuer M, Oldenburg J, Lavergne J M. Somatic mosaicism in hemophilia A: a fairly common event. Am J Hum Genet. 2001;69(01):75–87. doi: 10.1086/321285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kasper C K, Buzin C H. Mosaics and haemophilia. Haemophilia. 2009;15(06):1181–1186. doi: 10.1111/j.1365-2516.2009.02003.x. [DOI] [PubMed] [Google Scholar]
- 54.Lannoy N, Hermans C. Genetic mosaicism in haemophilia: a practical review to help evaluate the risk of transmitting the disease. Haemophilia. 2020;26(03):375–383. doi: 10.1111/hae.13975. [DOI] [PubMed] [Google Scholar]
- 55.Mårtensson A, Tedgård U, Ljung R. Prenatal diagnosis of haemophilia in Sweden now more commonly used for psychological preparation than termination of pregnancy. Haemophilia. 2014;20(06):854–858. doi: 10.1111/hae.12516. [DOI] [PubMed] [Google Scholar]
- 56.Boardman F K, Young P J, Warren O, Griffiths F E. The role of experiential knowledge within attitudes towards genetic carrier screening: a comparison of people with and without experience of spinal muscular atrophy. Health Expect. 2018;21(01):201–211. doi: 10.1111/hex.12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hudecova I, Jiang P, Davies J, Lo Y MD, Kadir R A, Chiu R WK. Noninvasive detection of F8 int22h -related inversions and sequence variants in maternal plasma of hemophilia carriers . Blood. 2017;130(03):340–347. doi: 10.1182/blood-2016-12-755017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.PedNet Study Group . van den Berg H M, Fischer K, Carcao M. Timing of inhibitor development in more than 1000 previously untreated patients with severe hemophilia A. Blood. 2019;134(03):317–320. doi: 10.1182/blood.2019000658. [DOI] [PubMed] [Google Scholar]
- 59.Male C, Andersson N G, Rafowicz A. Inhibitor incidence in an unselected cohort of previously untreated patients with severe haemophilia B: a PedNet study. Haematologica. 2021;106(01):123–129. doi: 10.3324/haematol.2019.239160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Astermark J. FVIII inhibitors: pathogenesis and avoidance. Blood. 2015;125(13):2045–2051. doi: 10.1182/blood-2014-08-535328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gouw S C, van den Berg H M, Oldenburg J. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta-analysis. Blood. 2012;119(12):2922–2934. doi: 10.1182/blood-2011-09-379453. [DOI] [PubMed] [Google Scholar]
- 62.Oldenburg J, Pavlova A. Genetic risk factors for inhibitors to factors VIII and IX. Haemophilia. 2006;12 06:15–22. doi: 10.1111/j.1365-2516.2006.01361.x. [DOI] [PubMed] [Google Scholar]
- 63.INSIGHT Study Group . Eckhardt C L, Loomans J I, van Velzen A S. Inhibitor development and mortality in non-severe hemophilia A. J Thromb Haemost. 2015;13(07):1217–1225. doi: 10.1111/jth.12990. [DOI] [PubMed] [Google Scholar]
- 64.Castaman G, Fijnvandraat K. Molecular and clinical predictors of inhibitor risk and its prevention and treatment in mild hemophilia A. Blood. 2014;124(15):2333–2336. doi: 10.1182/blood-2014-02-546127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bardi E, Astermark J. Genetic risk factors for inhibitors in haemophilia A. Eur J Haematol. 2015;94 77:7–10. doi: 10.1111/ejh.12495. [DOI] [PubMed] [Google Scholar]
- 66.Pavlova A, Oldenburg J. Defining severity of hemophilia: more than factor levels. Semin Thromb Hemost. 2013;39(07):702–710. doi: 10.1055/s-0033-1354426. [DOI] [PubMed] [Google Scholar]
- 67.Lee D H, Walker I R, Teitel J. Effect of the factor V Leiden mutation on the clinical expression of severe hemophilia A. Thromb Haemost. 2000;83(03):387–391. [PubMed] [Google Scholar]
- 68.Preisler B, Pezeshkpoor B, Banchev A. Familial multiple coagulation factor deficiencies (FMCFDs) in a large cohort of patients - a single-center experience in genetic diagnosis. J Clin Med. 2021;10(02):347. doi: 10.3390/jcm10020347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Escuriola Ettingshausen C, Halimeh S, Kurnik K. Symptomatic onset of severe hemophilia A in childhood is dependent on the presence of prothrombotic risk factors. Thromb Haemost. 2001;85(02):218–220. [PubMed] [Google Scholar]
- 70.van Dijk K, van der Bom J G, Fischer K, de Groot P G, van den Berg H M. Phenotype of severe hemophilia A and plasma levels of risk factors for thrombosis. J Thromb Haemost. 2007;5(05):1062–1064. doi: 10.1111/j.1538-7836.2007.02447.x. [DOI] [PubMed] [Google Scholar]
- 71.Shetty S, Vora S, Kulkarni B. Contribution of natural anticoagulant and fibrinolytic factors in modulating the clinical severity of haemophilia patients. Br J Haematol. 2007;138(04):541–544. doi: 10.1111/j.1365-2141.2007.06693.x. [DOI] [PubMed] [Google Scholar]
- 72.Hemophilia Inhibitor Research Study Investigators . Boylan B, Rice A S, De Staercke C. Evaluation of von Willebrand factor phenotypes and genotypes in hemophilia A patients with and without identified F8 mutations. J Thromb Haemost. 2015;13(06):1036–1042. doi: 10.1111/jth.12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gupta M, Lillicrap D, Stain A M, Friedman K D, Carcao M D. Therapeutic consequences for misdiagnosis of type 2N von Willebrand disease. Pediatr Blood Cancer. 2011;57(06):1081–1083. doi: 10.1002/pbc.23120. [DOI] [PubMed] [Google Scholar]
- 74.Mazurier C, Goudemand J, Hilbert L, Caron C, Fressinaud E, Meyer D. Type 2N von Willebrand disease: clinical manifestations, pathophysiology, laboratory diagnosis and molecular biology. Best Pract Res Clin Haematol. 2001;14(02):337–347. doi: 10.1053/beha.2001.0138. [DOI] [PubMed] [Google Scholar]
- 75.Pavlova A, Delev D, Pezeshkpoor B, Müller J, Oldenburg J. Haemophilia A mutations in patients with non-severe phenotype associated with a discrepancy between one-stage and chromogenic factor VIII activity assays. Thromb Haemost. 2014;111(05):851–861. doi: 10.1160/TH13-08-0690. [DOI] [PubMed] [Google Scholar]
- 76.Keeling D M, Sukhu K, Kemball-Cook G, Waseem N, Bagnall R, Lloyd J V. Diagnostic importance of the two-stage factor VIII:C assay demonstrated by a case of mild haemophilia associated with His1954–>Leu substitution in the factor VIII A3 domain. Br J Haematol. 1999;105(04):1123–1126. doi: 10.1046/j.1365-2141.1999.01460.x. [DOI] [PubMed] [Google Scholar]