

Abstract
RNA detection is important in diverse diagnostic and analytical applications. RNAs can be rapidly detected using molecular beacons, which fluoresce upon hybridizing to a target RNA but require oligonucleotides with complex fluorescent dye and quencher conjugations. Here we describe a simplified method for rapid fluorescence detection of a target RNA using simple unmodified DNA oligonucleotides. To detect RNA, we developed Lettuce, a fluorogenic DNA aptamer that binds and activates the fluorescence of DFHBI-1T, an otherwise nonfluorescent molecule that resembles the chromophore found in green fluorescent protein (GFP). Lettuce was selected from a randomized DNA library based on binding to DFHBI-agarose. We further show that Lettuce can be split into two separate oligonucleotide components, which are nonfluorescent on their own but become fluorescent when their proximity is induced by a target RNA. We designed several pairs of split Lettuce fragments that contain an additional 15–20 nucleotides that are complementary to adjacent regions of the SARS-CoV-2 RNA, resulting in Lettuce fluorescence only in the presence of the viral RNA. Overall, these studies describe a simplified RNA detection approach using fully unmodified DNA oligonucleotides that reconstitute the Lettuce aptamer templated by RNA.
Graphical Abstract
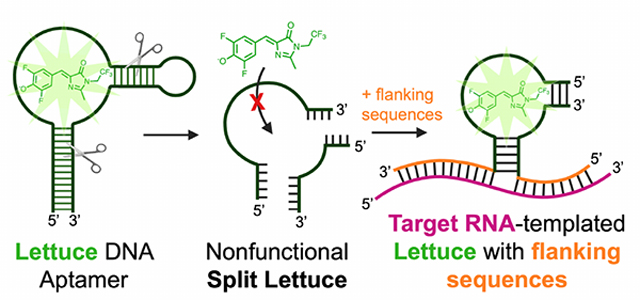
INTRODUCTION
RNA detection is critical for diverse types of analytical and diagnostic applications. A major method for RNA detection is reverse transcription followed by quantitative PCR (RT-qPCR). This method is highly effective and sensitive but does not provide immediate detection of the target RNA. This is because the sample needs to undergo an enzymatic step to convert the RNA to DNA, followed by PCR steps. Other assays have been developed to detect RNA in a much more rapid manner. The most well-known are the molecular beacons, which are hairpin DNA oligonucleotides containing a fluorescent dye and a quencher at the 5’ and 3’ ends1. The hairpin structure places the fluorescent dye and quencher in proximity, thus quenching the fluorescence of the dye. However, when the loop region of the hairpin hybridizes with a target RNA, the hairpin stem unwinds, disrupting the proximity between the fluorescent dye and the quencher. This results in a “turn on” of fluorescence when the molecular beacon hybridizes with the target RNA. This is a powerful approach that enables simple and rapid detection of target RNA sequences.
The problem with the molecular beacon method is that it requires the synthesis of a hairpin oligonucleotide containing two different small molecule conjugates – the fluorescent dye and the quencher. These are expensive, and therefore it is impractical to make large numbers of different molecular beacons to identify optimal regions in a target RNA that efficiently hybridize to the molecular beacon and do not hybridize to other closely related RNA sequences in a sample. A strategy to simplify the use of unmodified DNA oligonucleotides to detect target RNA would make RNA detection much simpler and less expensive.
Several groups have taken advantage of fluorogenic RNA aptamers to create RNA-detection assays. Fluorogenic RNA aptamers bind and activate the fluorescence of otherwise nonfluorescent small molecules2. To detect a target RNA, an RNA fluorogenic aptamer such as Spinach or Broccoli3,4 is modified so that it contains flanking sequences on the 5’ and 3’ ends that hybridize to a target RNA sequence. The fluorogenic aptamer is modified so that a critical helical stem is shortened and made thermodynamically unstable, making it unable to fold into a helix. However, when the flanking sequences bind to the target RNA, the helical stem can hybridize allowing the aptamer to fold, resulting in fluorescence. This system can be made even more specific by splitting the aptamer into two separate RNAs, each with a single flanking sequence5. When each of these two RNAs bind the target RNA, they are placed in proximity to each other and can interact to reconstitute the fluorogenic aptamer. These approaches allow RNA detection without molecular beacons. The problem with these approaches is that the fluorogenic aptamers are composed of RNA, and are therefore susceptible to degradation by RNases. The instability of the RNA makes RNA-based RNA sensors difficult to use in diverse settings.
Here we describe RNA detection using unmodified DNA oligonucleotides. To do this, we developed Lettuce, a fluorogenic DNA aptamer. Lettuce was developed using the SELEX approach6 in which a large combinatorial DNA library was screened for its ability to bind DFHBI-1T, a small molecule analogue of the chromophore found in green fluorescent protein. Lettuce binds and induces the fluorescence of DFHBI, DFHBI-1T and related fluorophores. Lettuce lacks sequence similarity to Spinach, but appears to have a G-quadruplex based on the presence of G-quadruplex sequence motifs and the ability to bind to dyes that have affinity for G-quadruplexes. We developed and optimized an approach to split Lettuce into two separate oligonucleotide strands which each have a flanking sequence that hybridizes to contiguous sequences in the SARS-CoV-2 genomic RNA. Using a series of split Lettuce pairs that bind to different regions of the SARS-CoV-2 RNA, we show that we can reconstitute the Lettuce aptamer on a SARS-CoV-2 RNA fragment. As a result, the split Lettuce aptamer pairs can readily detect and quantify the levels of a synthetic SARS-CoV-2 target RNA. Overall, these results show a highly simplified approach for light-up detection of a target RNA using fully unmodified DNA oligonucleotides based on RNA-induced reconstitution of the Lettuce DNA aptamer.
RESULTS
Selecting DNA mimics of green fluorescent protein
We first sought to identify DNA aptamers that bind and activate the fluorescence of DFHBI-1T (4-(3,5-difluoro-4-hydroxybenzylidene)-2-methyl-1-(2,2,2-trifluoroethyl)-1H-imidazol-5(4 H)-one)3. DFHBI-1T and its derivatives are highly useful fluorophores since they exhibit low background fluorescence in solution or biological extracts3. However, their intrinsic fluorescence can be markedly increased upon binding RNA aptamers such as Spinach or Broccoli3,4. Spinach and Broccoli have been used as RNA imaging tags and have been adapted into RNA-based metabolite and protein sensors using DFHBI-1T4,7–9. Since these RNA aptamers were developed using the SELEX (systematic evolution of ligands by exponential enrichment) method10,11, we reasoned a similar approach could be used to develop DNA aptamers that bind DFHBI-1T and activate its fluorescence.
For the SELEX experiments, we designed three different ssDNA libraries (Figure 1a). Each library contained unique, fixed sequences on their 5’ and 3’ ends to facilitate PCR amplification. The first library, termed the “random library” contained a thirty nucleotide-long region comprising random nucleotides, flanked by fixed sequences on either end (Figure 1a). The second library was the “stem-loop library,” which contained an internal, fixed four-base pair stem and a three-nucleotide loop, surrounded by two twenty-nucleotide random regions (Figure 1a). The third library was the “G-rich” library. Since Broccoli, Spinach, and other fluorogenic RNA aptamers contain G-quadruplexes12–15, a library with fixed guanine residues may be more prone to form G-quadruplexes and may therefore be particularly useful for developing fluorogenic DNA aptamers. Therefore, the G-rich library contained six fixed guanine nucleotides interspersed within a thirty-nucleotide random region (Figure 1a).
Figure 1. Characterization of DNA aptamers that bind DFHBI-1T.
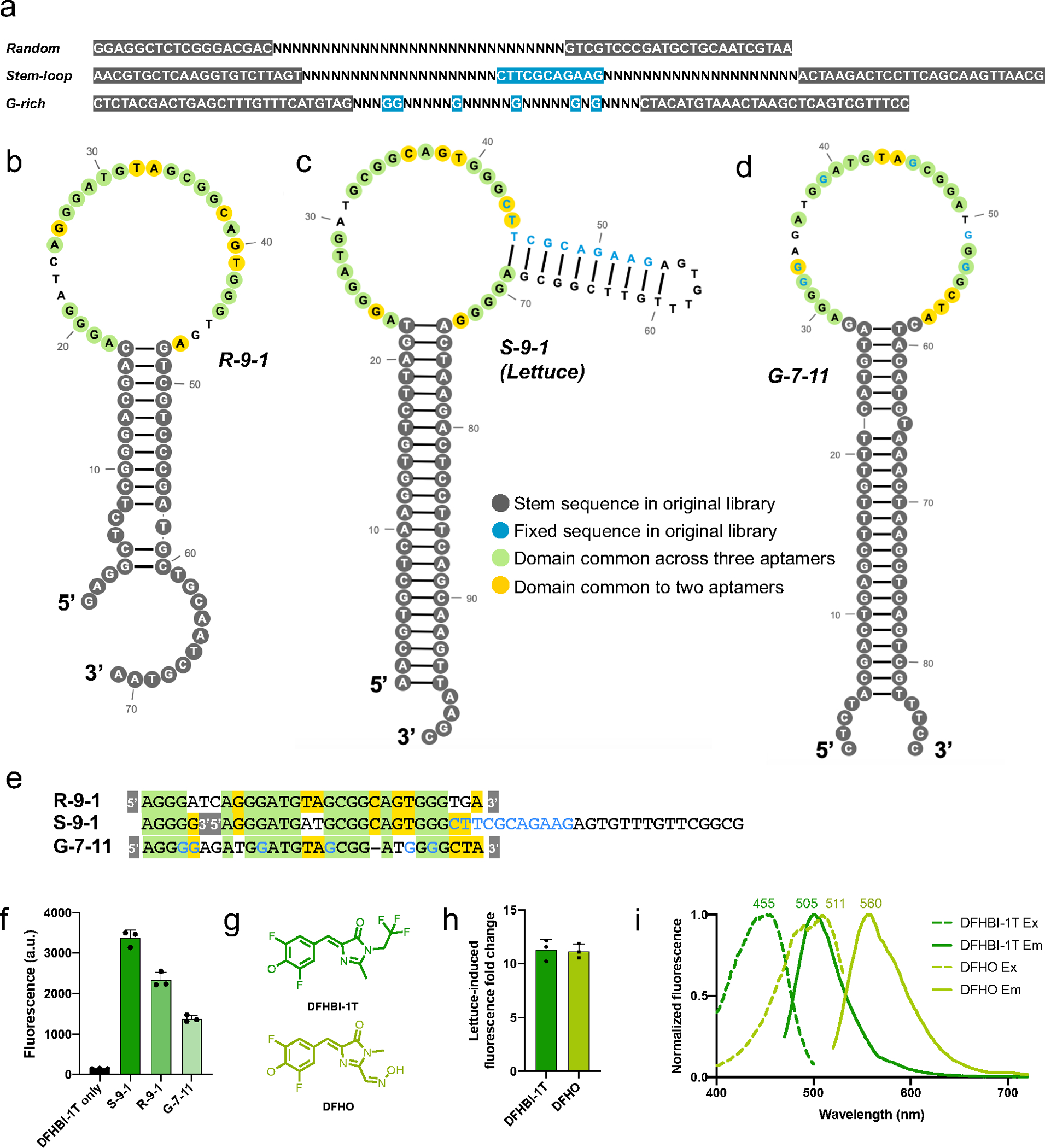
a) SELEX libraries used to discover DFHBMT-1T-binding DNA aptamers. The “random” library contains 5’ and 3’ flanking sequences (grey) that are complementary to each other and provide a fixed sequence to facilitate amplification between SELEX rounds. In between these fixed regions is a 40-nt random region. “N” represents a nucleotide added from a phosphoramidite mixture containing an equal proportion of A’s, G’s, C’s, and U’s. The “stem-loop” library contains eleven nucleotides (shown in blue) in the middle of the randomized region which form a 4-bp stem with a 3-nt loop. The “G-rich” library contains six fixed guanine nucleotides (blue) interspersed throughout the variable region. b–d) Secondary structure prediction of b) R-9-1, c) S-9-1 and d) G-7-11. Green and yellow shaded nucleotides represent conserved positions across all three and two of the three aptamers, respectively, when their sequences are aligned as shown in Figure 1e. Dark grey shaded nucleotides represent nucleotides that were part of the stem-forming flanking regions in the original library (Figure 1a). Fixed nucleotides are shown in blue. e) Sequence alignment for aptamers R-9-1, S-9-1, and G-7-11. The grey-shaded 5’ and 3’ represent the constant 5’ and 3’ sequences of each aptamer. Fixed nucleotides are shown in blue. f) DFHBI-1T fluorescence enhancement by aptamers discovered from each library. S-9-1, R-9-1, and G-7-11 originated from the stem-loop, random, and G-rich libraries, respectively. Fluorescence was measured using 20 μM DNA and 2 μM DFHBI-1T on a Horiba fluorometer (excitation 460 nm, emission 505 nm). The S-9-1 aptamer exhibits the highest fluorescence enhancement of DFHBI-1T and was thus chosen for further study using the name “Lettuce”. The mean and SEM values are shown (n=3). g) Chemical structures of DFHBI-1T and DFHO fluorophores. h) Lettuce activates the fluorescence of DFHBI-1T and DFHO to a similar extent. Each fluorophore (2 μM) was incubated with and without Lettuce (20 μM). Shown is the fold increase between the fluorescence of the fluorophore alone and the signal of the fluorophore when activated by Lettuce. The mean and SEM values are shown (n=3). i) Lettuce-DFHO displays red-shifted fluorescence excitation and emission spectra compared to Lettuce-DFHBI-1T. Excitation and emission maximal wavelengths are indicated. Spectra were measured using 20 μM DNA and 2 μM of the indicated compound.
To select for DFHBI-1T-binding ssDNA aptamers, we performed ssDNA SELEX using a previously described protocol6. In brief, the library was PCR amplified using primers that incorporated a 5’ phosphate on one strand of the PCR product. After PCR, which generates double-stranded DNA, the strand of DNA containing the 5’ phosphate was selectively degraded with lambda phosphatase. This results in ssDNA that was gel purified before incubation with DFHBI-1T-agarose beads. After washing the beads to remove nonspecifically bound ssDNA, specifically bound ssDNA aptamers were eluted by adding 10 μM DFHBI-1T. The eluted DNA was PCR amplified for the subsequent round of SELEX. We assessed the ability of the DNA pool to activate DFHBI-1T fluorescence after each SELEX round. By the ninth round of selection, we observed that the pool of DNA from each of the three libraries weakly activated the fluorescence of DFHBI-1T (Figure S1a).
To identify individual DNA aptamers that accounted for DFHBI-1T fluorescence, we cloned the ssDNA from round 9 of SELEX into bacteria. We found one ssDNA aptamer from each of the three DNA libraries that activated the fluorescence of DFHBI-1T, which we designated R-9-1, S-9-1, and G-7-11 in reference to the random, stem-loop, and guanine-rich libraries, respectively (Figure 1b–d). Although these aptamers originated from separate DNA libraries, aligning their sequences reveals that these aptamers are highly similar, and likely have structural similarities as well (Figure 1e).
We next sought to further characterize the fluorescence and binding properties of the aptamers in complex with DFHBI-1T. The spectra of the three DNA aptamers were similar, with emission peaks around 505 nm and excitation peaks from 474–480 nm (Table 1, Figure S1b). The quantum yields of the aptamer-DFHBI-1T complexes ranged from 0.045 to 0.105 (Table 1). The dissociation constants (KD) ranged from 350–960 nM (Table 1, Figure S1c). The S-9-1 aptamer was superior to the other DNA aptamers in terms of quantum yield, KD, and brightness (Table 1). Thus, we focused the rest of our studies on S-9-1, which we termed Lettuce, in keeping with the vegetable nomenclature system used for other nucleic acid aptamers.
Table 1.
Characterization of DFHBI-1T-binding DNA aptamers
| Max ex (nm) | Max em (nm) | Extinction coefficient (M−1 cm−1) | Quantum yield | Brightness | KD (nM) | |
|---|---|---|---|---|---|---|
| DFHBI-1T | 426 | 495 | 35400 | .00098 | 1 | - |
| R-9-1 | 469 | 501 | 33800 | .056 | 59 | 830 |
| S-9-1 | 455 | 505 | 32400 | .109 | 100 | 350 |
| G-7-11 | 456 | 502 | 30400 | .045 | 39 | 960 |
We next tested whether Lettuce can activate the fluorescence of other, structurally related fluorophores. We used DFHO (Figure 1f), a molecule that mimics the natural fluorophore in red fluorescent proteins and has known fluorogenic activity when bound to RNA aptamers, such as Squash16 and Corn17. Lettuce activated the fluorescence of DFHO to a similar extent as DFHBI-1T (Figure 1g), indicating that the fluorophore-binding pocket of Lettuce can tolerate different substituents on the 1 and 2 positions of the imidazolinone ring (Figure 1f). Notably, the Lettuce-DFHO complex was characterized by red-shifted excitation and emission spectra, with maximal emission at 510 nm and 560 nm for DFHBI-1T and DFHO, respectively (Figure 1h). These emission maxima are similar to that of the RNA aptamer Squash which also binds both DFHBI-1T and DFHO16.
Next, we examined the photostability of the Lettuce-DFHBI-1T complex. RNA aptamers (Spinach and Broccoli) that activate DFHBI-1T fluorescence rapidly lose fluorescence after illumination with a high-powered excitation light source17. This process is due to light-induced isomerization of the DFHBI-1T molecule from the bright cis form to the dim trans form18. In order to overcome the inherent lack of photostability of DFHBI-1T with Spinach and Broccoli, newer fluorophore derivatives have been developed19,20. Therefore, we asked whether DFHBI-1T similarly exhibits poor photostability with the DNA-based Lettuce aptamer. Surprisingly, Lettuce in combination with DFHBI-1T maintained high levels of fluorescence upon excitation compared to Broccoli-DFBHI-1T and Corn-DFHO complexes (Figure S1d). Because Lettuce exhibits high photostability but has relatively low fluorescence, we sought to improve its overall brightness.
Conserved nucleotides in DNA aptamers indicate a DFHBI-1T binding region
We wanted to optimize Lettuce to improve its brightness upon binding DFHBI-1T. To accomplish this, we used directed evolution. In this approach, we prepared a randomized ssDNA library based on Lettuce. This library is designed so that each library member contains one or more mutations relative to the parental aptamer, i.e., Lettuce. This method is previously described4. In brief, these libraries are made by chemical synthesis using phosphoramidite mixtures that predominantly contain either A, C, T, or G, but also contain low levels of the other three phosphoramidites. As a result, each position in the library has a fixed probability of having a mutation at each position. The library is designed so that each possible single, double, triple, up to seven-nucleotide mutant combination is represented in the library4. After synthesis of the Lettuce-templated library, we performed two rounds of SELEX to enrich for mutants that bind DFHBI-1T. We then tested individual sequences as described earlier. We found several aptamers that bind and activate the fluorescence of DFHBI-1T, but none of these sequences were brighter than Lettuce.
Nonetheless, the directed evolution experiment revealed important insights into the functional domains of Lettuce that are required for DFHBI-1T fluorescence activation. By aligning the sequences of the winning Lettuce-based fluorogenic aptamers, we identified several conserved and non-conserved nucleotide positions in these aptamers (Figure 2a, b). The single-stranded region of Lettuce was almost exclusively conserved, while the majority of the non-conserved residues were found in the double-stranded P2 stem (Figure 2b). Because the non-conserved nucleotide positions are unlikely to make sequence-specific contacts with DFHBI-1T, we reasoned that DFHBI-1T may bind in the single-stranded, conserved region of the aptamer. Thus, the directed evolution experiments identified nucleotides that are likely to be important for the binding and fluorescence activation of DFHBI-1T.
Figure 2. Lettuce likely contains a G-quadruplex in its DFHBI-1T-binding region.
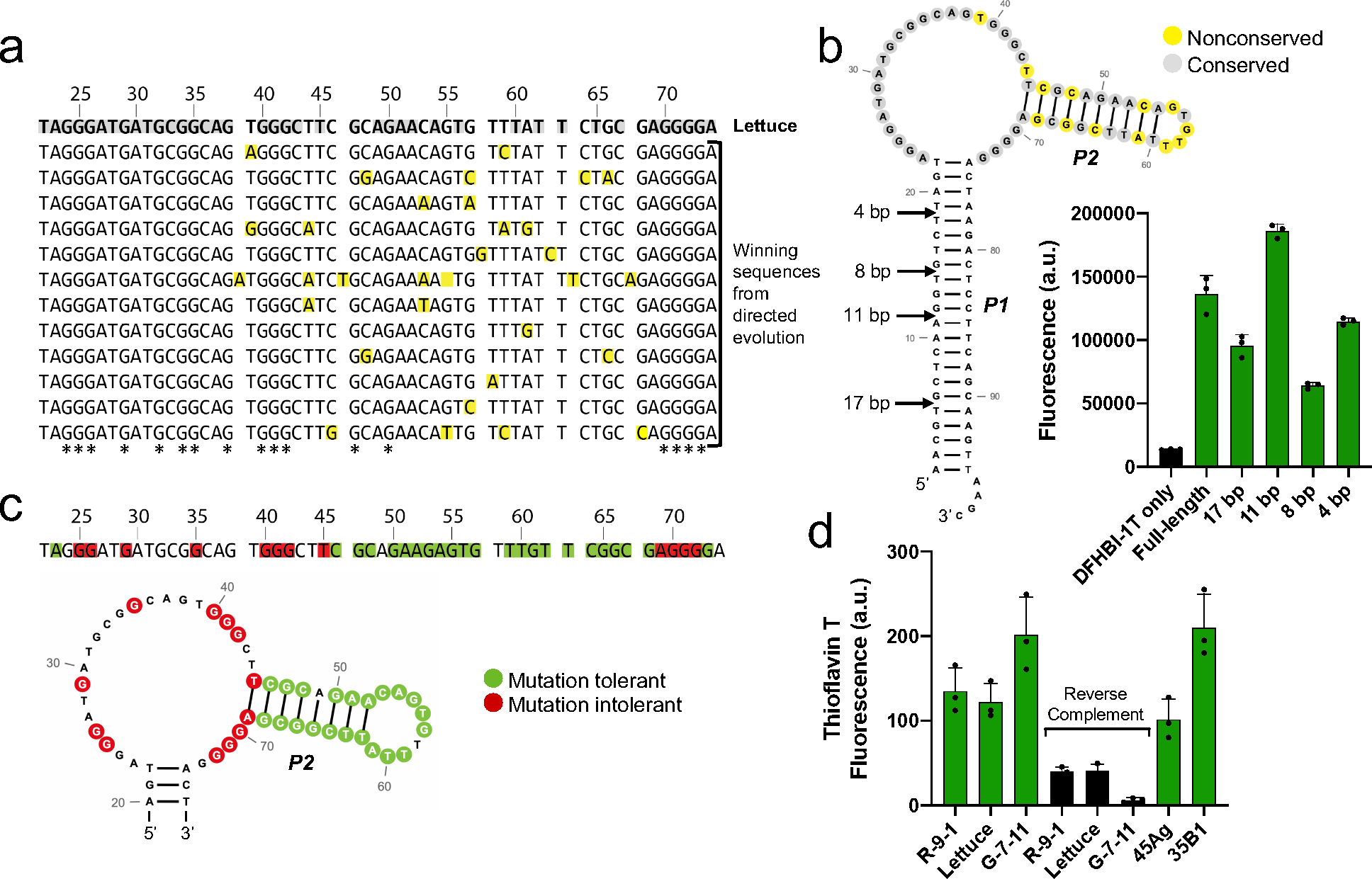
a) Sequence alignment of aptamers that retained the ability to induce DFHBI-1T fluorescence after directed evolution of Lettuce. All aptamers exhibited similar fluorescence signal to that of Lettuce. The top sequence is Lettuce, and all sequences below are sequences of functionally active aptamers discovered from directed evolution of Lettuce. Yellow-shaded nucleotides differ from the sequence of Lettuce. Grey-shaded nucleotides in the Lettuce sequence were conserved across all functionally active aptamers from the direct evolution experiment. G residues that are conserved across all functionally active aptamers are indicated in the bottom row with an asterisk. Fluorescence emission was measured at (excitation 460 nm, emission 480–560 nm) for all mutants using 10 μM DNA and 20 μM DFHBI-1T. b) Lettuce’s long stem is nonessential for fluorescence activation of DFHBI-1T. Arrows indicate the lengths to which the stem was truncated. Fluorescence was measured (excitation 460 nm, emission 505 nm) of each truncated construct using 1 μM DNA and 5 μM DFHBI-1T. The secondary structure prediction of Lettuce is color coded as in Figure 2a, where yellow represents nonconserved positions and grey represents conserved positions, c) Summary of mutational analysis. To identify essential and nonessential residues for fluorescence, Lettuce was mutated at single positions and the fluorescence of each mutant was assessed, where mutants that retained fluorescence within 10% of Lettuce were considered tolerant, and mutants with fluorescence values below 30% of Lettuce were considered to be intolerant. Green and red shaded nucleotides indicate positions at which a mutation was tolerated or not tolerated, respectively. Unshaded positions were not tested. Fluorescence emission was measured (excitation 460 nm, emission 480–560 nm) for 35 mutants of Lettuce using 10 μM DNA and 20 μM DFHBI-1T. d) Fluorescence activation of thioflavin T by DNA aptamers. R-9-1, Lettuce, and G-7-11 were incubated with thioflavin T, a compound known to bind G-quadruplexes. Reverse complement sequences which are not expected to form G-quadruplexes were tested, along with two sequences known to form G-quadruplexes (45Ag and 35B1). Fluorescence (excitation 430 nm, emission 485 nm) was measured using 1 μM DNA and 1 μM thioflavin T.
We next wondered if the P1 stem that was fixed in the original and directed evolution libraries contributes to the fluorescence of Lettuce. For example, it could provide structural support for the DFHBI-1T binding site, or it could make direct contacts with DFHBI-1T. To test whether this stem is essential for Lettuce’s fluorescence, we generated truncated Lettuce sequences and tested each of their abilities to activate fluorescence of DFHBI-1T (Figure 2b). We truncated Lettuce to four different lengths: 17 bp, 11 bp, 8 bp, and 4 bp. While the first truncation to 17 bp decreased the fluorescence of Lettuce, the second truncation to 11 bp surprisingly resulted in a brighter version of Lettuce (Figure 2b). Even when the long stem was truncated to just 4 base pairs, Lettuce retained most of its fluorescence (Figure 2b). This supports the idea that the DFHBI-1T binding site is in the single-stranded region, since a significant portion of the Lettuce’s long stem can be truncated while still retaining fluorescent activity.
DFHBI-1T-binding DNA aptamers likely contain G-quadruplexes
Fluorogenic RNA aptamers frequently contain G-quadruplexes as critical structural elements15, which usually comprise four stretches containing two or more consecutive G’s21. We noticed that 17 guanine nucleotides were maintained in all fluorescent aptamers in the directed evolution experiments, ten of which appear in clusters of at least three consecutive guanines (Figure 2a). Additionally, these patterns of guanines are similar across the Lettuce, R-9-1, and G-7-11 aptamers (Figure 1e). It is less surprising that the G-7-11 aptamer contains these stretches of G’s, since it was derived from a G-rich library. However, the presence of similar G stretches in aptamers derived from libraries that were not biased to contain guanines indicates that these conserved G’s may be essential for the fluorescence of these aptamers. Taken together, these observations support the idea that each of the three DFHBI-1T-binding DNA aptamers may contain a G-quadruplex.
We asked if we could identify essential guanine nucleotides that may make up a G-quadruplex in Lettuce. We systematically mutated guanine residues in Lettuce and tested whether the guanine-to-adenine mutant could activate the fluorescence of DFHBI-1T. We found ten guanines that did not tolerate the G-A mutation (Figure 2c). Several other residues tolerated mutation, including all of the stem residues that were tested (Figure 2c). Notably, three separate stretches had two or more consecutive essential guanines. The presence of guanines that are essential for fluorescence provides additional support for the existence of a G-quadruplex in Lettuce.
To further test whether our fluorogenic DNA aptamers contain G-quadruplexes, we assessed their ability to activate fluorescence of thioflavin T – a dye that is known to exhibit enhanced fluorescence upon binding to G-quadruplexes22. Upon addition of either of the three fluorogenic DNA aptamers R-9-1, Lettuce, or G-7-11, thioflavin T showed a large increase in fluorescence (Figure 2d). As a control, we used known G-quadruplex sequences (45Ag and 35B1). 45Ag is a 45-nt DNA sequence from the human telomere, and 35B1 is a 35-nt DNA sequence from the promoter of K-ras22. Upon addition of thioflavin T to these control G-quadruplexes, thioflavin T similarly showed a substantial increase in fluorescence (Figure 2d)22. To test the specificity of these interactions, we measured thioflavin T fluorescence activation upon addition of DNA sequences that comprised the reverse complement sequences of the three DFHBI-1T-binding DNA aptamers. These DNAs would not be expected to contain G-quadruplexes. When these DNAs were added to thioflavin T, no substantial change in thioflavin T fluorescence was observed (Figure 2d). Overall, these binding and sequence data indicate that the three DNA aptamers may each contain G-quadruplexes.
In-gel imaging of Lettuce-fluorophore complex
Typically, DNAs are detected using Southern blotting, which is a relatively time-consuming method. However, we reasoned that DNAs tagged with a Lettuce aptamer could be directly visualized within the gel after removing any denaturing agent to allow the DNA to fold, and then staining with DFHBI-1T. Simplified detection of single-stranded DNAs would be useful for visualizing phage DNA, viral DNA, primers, or components of DNA nanotechnology applications. A similar approach using Broccoli has proven to be useful for visualizing Broccoli-tagged RNAs of interest on a gel by staining with DFHBI-1T7. Thus, we sought to develop a method for imaging Lettuce-tagged ssDNAs in gels.
We first asked if Lettuce exhibits fluorescence in gels that are stained with DFHBI-1T. We resolved Lettuce by denaturing polyacrylamide gel electrophoresis, which allows nucleic acids to be resolved based on their size. For these experiments, we used urea-PAGE gels containing 7 M urea. After electrophoresis, the gel was washed with water to remove urea, and the gel was incubated in buffer containing DFHBI-1T. In this buffer, Lettuce can refold and bind DFHBI-1T. To test the detection limit of DFHBI-1T staining for imaging Lettuce DNA, we resolved linearly increasing concentrations of Lettuce in different lanes of the polyacrylamide gel. We found that the detection limit of DFHBI-1T staining for imaging Lettuce DNA is ~0.15 pmol (Figure 3a). Thus, Lettuce can fold and fluoresce in a gel upon addition of DFHBI-1T.
Figure 3. Gel staining using Lettuce.
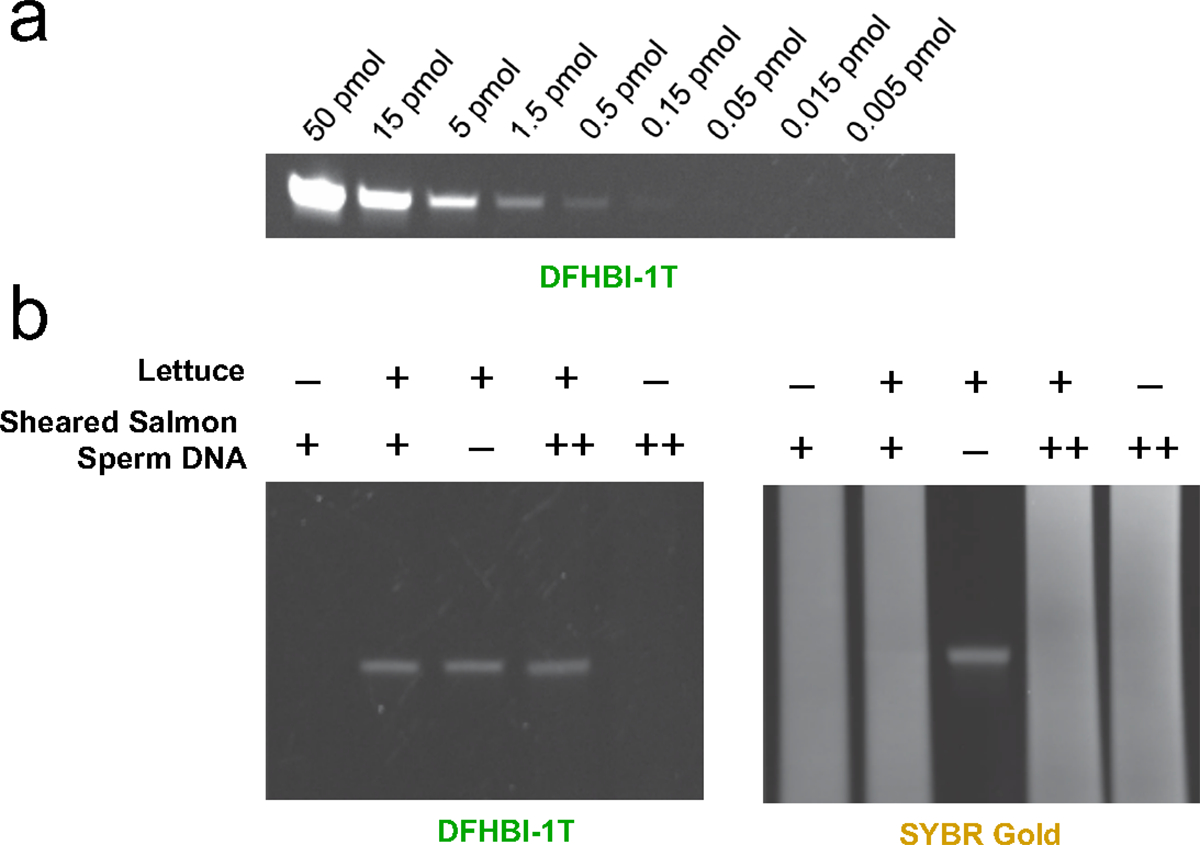
a) Titration of Lettuce on a gel. To test the amount required to visualize Lettuce on a gel, we prepared samples containing known amounts of Lettuce. The amount of Lettuce specified was loaded into each lane of a 10% TBE-urea polyacrylamide gel. After polyacrylamide gel electrophoresis, the gel was washed to remove urea, and then stained with DFHBI-1T (20μM) in a solution containing 140 mM KCl and 1 mM MgCl2. Fluorescence was measured using the FITC protocol in a Bio-Rad gel doc imager. b) Lettuce can be observed on a gel in a mix of other DNA with DFHBI-1T staining. The gel was also stained with SYBR-Gold (right) to observe total DNA. Samples were prepared with Lettuce (2 pmol) and sheared salmon sperm DNA (“+” corresponds to 8 μg, “++” corresponds to 40 μg). Samples were loaded into a 10% TBE-urea polyacrylamide gel. After polyacrylamide gel electrophoresis, the gel was stained with DFHBI-1T (20 μM) or SYBR Gold in a solution containing 140 mM KCl and 1 mM MgCl2. Fluorescence was measured using the FITC imaging channel in a Bio-Rad gel doc imager.
In order to function as an in-gel imaging tag, Lettuce should be able to be detected when loaded in a gel as part of a complex mixture of nucleic acids. To test if Lettuce can be detected within a complex mixture, we incubated Lettuce with salmon sperm DNA and loaded the sample on a gel. We again detected the Lettuce aptamer by staining the gel with DFHBI-1T and using fluorescent gel imaging (Figure 3b). Notably, the Lettuce aptamer could not be detected using the non-specific DNA stain SYBR Gold (Figure 3b), confirming that Lettuce was present at very low concentrations relative to the other DNA in this sample. Thus DFHBI-1T staining of Lettuce can be used to detect Lettuce DNA specifically within a highly heterogeneous sample of nucleic acids.
Design of a split Lettuce aptamer
Previous studies of Spinach have described a “split” Spinach system for detecting target RNAs in vitro5. In this system, Spinach was split into two RNA oligonucleotides, with each RNA fragment being nonfunctional with respect to Spinach fluorescence. However, the two RNA oligonucleotides contained additional flanking sequences that are complementary to two adjacent sequences in a target RNA. This results in the juxtaposition of each split Spinach strand. The two adjacent oligonucleotides interact with each other to re-form the Spinach aptamer. The problem with this system is that it utilizes RNA, which is generally susceptible to ribonuclease degradation and therefore difficult to use. We therefore wanted to develop a “split Lettuce” system to detect target RNAs.
In order to split Lettuce into two strands, we needed to first identify a site in Lettuce that would serve as the break point that divides one strand from the other. Importantly, many sites might be critical for the folding and function of Lettuce, and splitting Lettuce at that position could prevent refolding of Lettuce from each of the resulting strands. Alternatively, other split sites might split Lettuce, but one of the strands could still retain Lettuce’s fluorogenic activity. We chose a split site in the mutation-tolerant P2 stem (Figure 4a). Importantly, this split site would separate guanines essential for Lettuce’s fluorescence activation (Figure 4a). We predicted this would disrupt the putative G-quadruplex where DFHBI-1T may bind, since essential guanines would be present on either strand. When Lettuce was split at this position (designated S1), Lettuce retained its fluorescence at levels comparable to the non-split original aptamer (Figure 4a). The S1 split site did not cause a loss of fluorescence presumably because the various predicted helical stems in Lettuce are thermodynamically stable and therefore remained intact despite the split site at S1. Thus, the S1 site is a location where Lettuce can be split into two separate strands, both of which spontaneously interact to form a functional Lettuce.
Figure 4. Development of conditionally fluorescent split Lettuce.
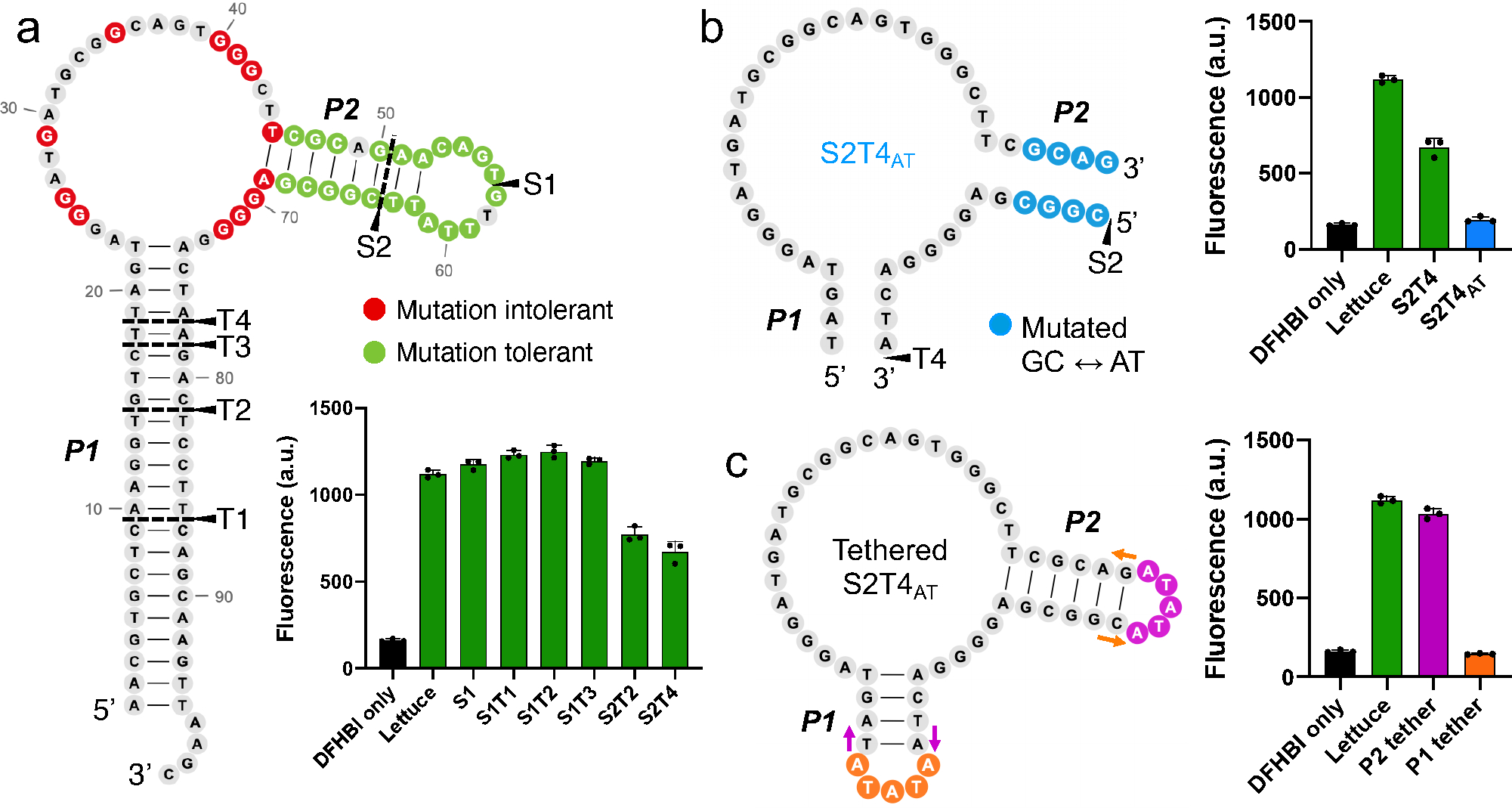
a) Map of Lettuce split and truncation sites and their resulting fluorescence activation. The two helices (P1 and P2) that were modified are shown. Split sites (S) indicate positions where Lettuce was broken into two strands. Truncation sites (T) mark the length to which the P1 stem was truncated. Constructs that were both split at site X and truncated at site Y are designated SXTY. Residues that are tolerant (green) or intolerant (red) to mutation are shown. Fluorescence activation of each pair of Lettuce strands was measured (excitation 460 nm, emission 505 nm) using 10 μM DNA and 5 μM DFHBI on a Gen5 fluorescence plate reader. Error bars indicate s.d. (n=3). b) Decreasing the G/C content of the P2 stem inactivates Lettuce fluorescence. The four terminal base pairs in the P2 stem of S2T4 (Figure 4A) were mutated from G/C to A/T or vice versa, forming a new, inactive version with an AT-rich stem, S2T4AT. Fluorescence activation was measured as described in Figure 4a. Error bars indicate s.d. (n=3). c) Forced proximity of the two S2T4AT strands reactivates fluorescence. A five-nucleotide tether loop was placed on either the P1 (orange) or P2 (pink) stem to test if the two strands could activate fluorescence when brought together. The P2 tether was successful at restoring fluorescence activity. The 5’ and 3’ ends of the strand are indicated by colored arrows that correspond to each tethered construct. Error bars indicate s.d. (n=3).
In order for the two strands of Lettuce to “turn on” upon binding a target RNA, the two strands need to show no interaction in the absence of RNA. To decrease the background fluorescence of the Lettuce strands, we kept S1 as the split site while truncating P1, a long stem that confers favorable thermodynamics for hybridization (Figure 4a). By progressively shortening P1, we hoped to find an optimal length of P1 that prevents the two strands from spontaneously hybridizing and forming a functional aptamer. We truncated P1 down to 13 base pairs (T1), 8 base pairs (T2) and five base pairs (T3) while maintaining the split site at S1 and did not observe any decrease in fluorescence from the original Lettuce (Figure 4a). Thus, the long stem of P1 is not responsible for the spontaneous association of the two split Lettuce strands.
We next truncated the other stem, P2, which was originally split by S1. To shorten this stem, we moved the split site to S2, which yielded a P2 stem that was 6 base pairs long (Figure 4a). This resulted in a considerable decrease in fluorescence when combined with a P1 stem of 8 base pairs (S2T2) and when combined with a P1 stem of 4 base pairs (S2T4) (Figure 4a). However, the background fluorescence of both constructs was still more than half that of the original Lettuce. Thus, while P2 seemed to be more sensitive to truncation than P1, the binding energy of both stems was still enough to induce some reassembly of the two strands.
We then asked whether we could fine-tune Lettuce self-assembly by altering the G/C content of the stems. Since the P2 stem is tolerant to mutation, we replaced the P2 stem of S2T4 with an equally long A/T rich stem (S2T4AT), reducing the G/C content from 66% to 33% (Figure 4b). This had a dramatic effect, reducing the fluorescence to near the DFHBI-only background level (Figure 4b). Thus, besides changing stem length, altering stem G/C content allowed us to reduce the overall thermodynamic stability of the interaction of the two split strands.
We next tested whether the minimally fluorescent S2T4AT split construct could become fluorescent when its strands are forcibly brought into proximity. In an RNA detection system, the strands would be brought into proximity by 5’- and 3’-appended sequences which hybridize to a target RNA sequence, bringing each inactive strand close enough to reform the DFHBI-binding site and restore fluorescent activity. To mimic this, we tethered the S2T4AT strands together with five-nucleotide loops on either stem (Figure 4c). The P1 tether was unsuccessful in restoring fluorescence, but when the P2 stem was tethered and the P1 stem was left open, fluorescence was nearly fully recovered. This indicates that the Lettuce-based S2T4AT core has the potential to function as a “turn-on” system, where the “on” state is only induced when strands are brought in proximity through interactions of the P2 stem.
Reconstitution of functional Lettuce with target RNA
We sought to design an RNA detection strategy based on the split S2T4AT Lettuce core. We designed each S2T4AT strand so that it has short sequences of nucleotides on the 5’ and 3’ ends of the P2 stem, which we refer to as flanking sequences. These flanking sequences are complementary to a target RNA region and recruit the two DNA strands into juxtaposition and thus facilitate folding and reconstitution of the S2T4AT Lettuce core (Figure 5a). Previous reports of a split Spinach aptamer use an asymmetrical flanking sequence approach, in which the longer side anchors its strand to the target site through a stronger binding interaction, while the short side is able to move with rapid on/off kinetics until it finds its designated binding site next to the long half5. Therefore, we designed the left and right flanking sequences to be longer (15 or 20 nt) and shorter (7, 10, or 15 nt), respectively (Figure 5a). By testing different combinations of left and right flanking sequence length, we hoped to find optimal parameters for the RNA detection system.
Figure 5. Optimization of split Lettuce for detection of target RNA.
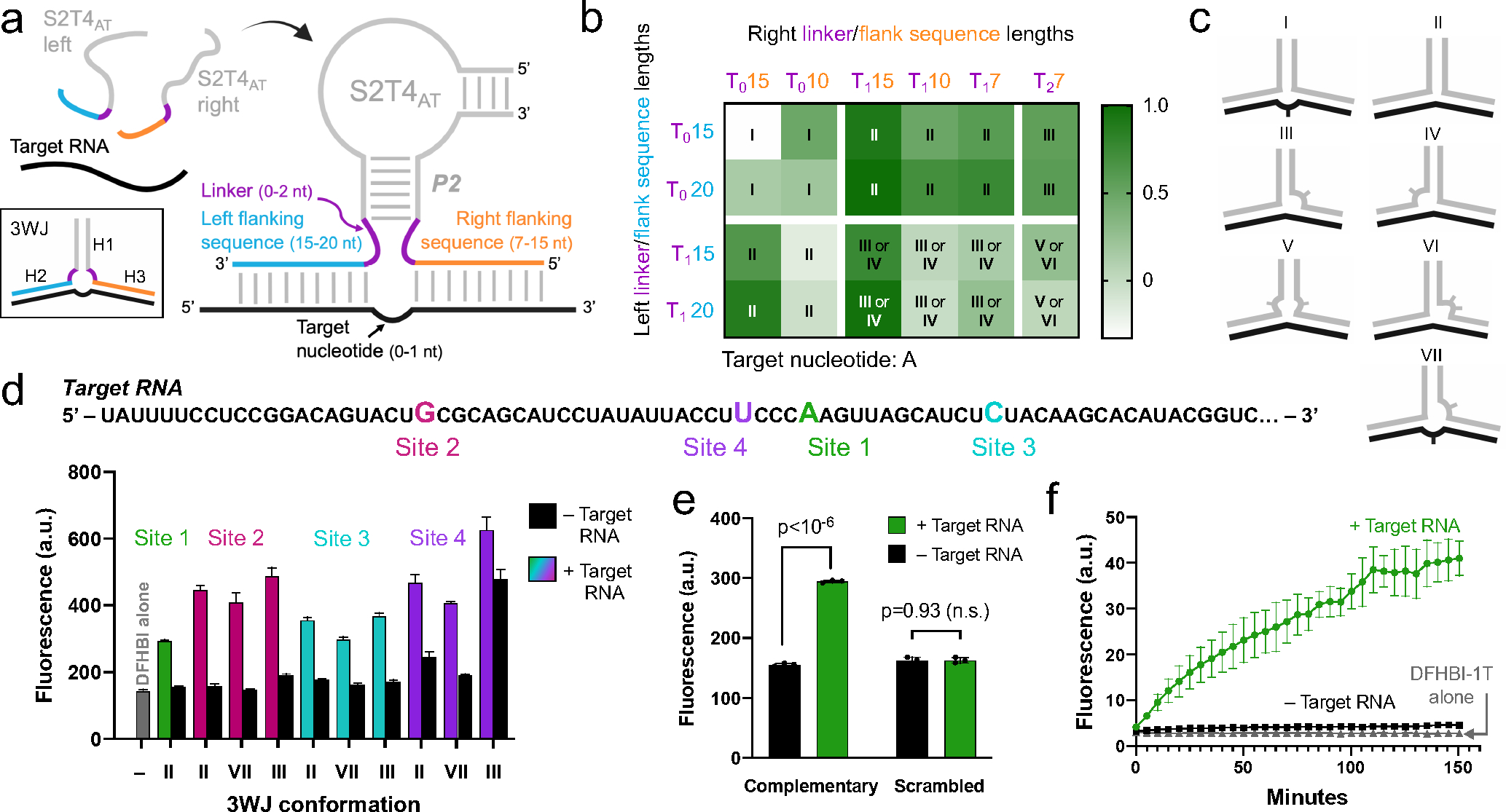
a) Diagram of the split Lettuce RNA sensor and regions to be optimized. Each half of S2T4at can assemble on a target RNA sequence via flanking sequences (orange and blue) that are complementary to the target RNA (black), forming a three-way junction (3WJ) between the two Lettuce oligonucleotides and the target RNA. The three helices (H1-H3) of the 3WJ are indicated. The left and right strands we tested had 7, 10, 15, or 20-nt flanking sequences and 0, 1, or 2 linker residues (purple). We tested RNA target sites with either one or zero unpaired target nucleotides, b) Sensor pairs have various 3WJ conformations and activate a range of fluorescence signal. Left and right strands with the indicated lengths of thymine linkers (purple) and flanking sequences (blue and orange) were designed to target a single site on a target RNA. Fluorescence of each pair was measured with and without target RNA. Heat map values were obtained by subtracting the fluorescence of samples without target RNA from samples with target RNA, and values over 0.5 were considered successful. Roman numerals refer to each pair’s 3WJ structure, which are shown in Figure 5c. The 3WJ assignments take into account that the target nucleotide, adenine, can base pair with thymines in the linkers. Type II and III 3WJs were the most successful on this particular target site (Figure 5d, Site 1). c) Diagrams of a selection of the 3WJs that can form between target RNA (black) and the two Lettuce sensor strands (grey). The location of unpaired nucleotides is indicated by curves, where one and two tick marks correspond to one and two unpaired nucleotides, d) Sensor designs that were successful on one target RNA site also work on three other target RNA sites. Sensor pairs of type II, type III and type VII 3WJs were designed to target three other RNA sites (Site 2, Site 3, and Site 4) which each have a different target nucleotide (G, C, and U, respectively). For comparison, the highest-performing sensor from Figure 1b, which targets Site 1, is included. The majority of the sensors exhibited signal-to-background ratios as high or higher than the original Site 1 sensor, showing that these sensor designs can be generalized to detect other RNA targets of interest. Fluorescence was measured (excitation 460 nm, emission 505 nm) using 10 μM DNA, 5 μM DFHBI, and 5 μM RNA on a Gen5 fluorescence plate reader. Error bars indicate s.d. (n=3). e) Split Lettuce RNA detection is sequence-dependent. To confirm the specific base pairing between flanking sequences and the target RNA sequence, we randomly scrambled the flanking sequences of the Site 1 sensor shown in Figure 5d. No increase in fluorescence is observed when target RNA is present. With fully complementary flanking sequences, the sensor exhibits an increase in fluorescence when target RNA is present. Fluorescence was measured as described in Figure 5d. Error bars indicate s.d. (n=3). f) Time dependence of split Lettuce sensor fluorescence. Split Lettuce halves and DFHBI-1T were incubated with or without target RNA and fluorescence was measured as described in Figure 5d at different time points for a total of 150 minutes. Error bars indicate s.d. (n=3).
We also wanted to optimize the spacing of the junction formed by the target RNA and the two Lettuce oligonucleotides. We hypothesized that the success of the sensors would depend largely on the stability of this DNA/RNA three-way junction (3WJ) (Figure 5a). Naturally occurring DNA 3WJ structures often include unpaired nucleotides between the three helical arms, which can have a major effect on the folding of the junction23,24. Accordingly, we designed left and right Lettuce strands with inserted unpaired thymine residues linking the flanking sequences to the S2T4AT core (Figure 5a). We termed these inserted residues linkers, and designated them T0, T1, and T2, indicating 0, 1, or 2 thymine residues, respectively. We reasoned that when these various left and right strands are combined with target RNA, the 3WJ that forms will fold differently, and in certain cases may form a 3WJ that is more conducive to Lettuce folding. While it is impossible to exhaustively cover all the possible 3WJ conformations, sampling several of them may reveal general principles that could inform the design of the split Lettuce RNA sensor.
While the linkers address the spacing on either side of the S2T4AT helix (H1), we were also interested in the spacing between the two target RNA-based helices (H2 and H3) (Figure 5a). We developed sensors that targeted an adenine residue (Site 1) on the target RNA (Figure 5d), and tested designs in which this target nucleotide was either unpaired or base paired to one of the thymine linker residues.
We combined various combinations of left and right sensor strands in the presence and absence of target RNA in order to optimize split Lettuce for RNA detection (Figure 5b). For each tested combination, the background signal in the absence of RNA was subtracted, and fluorescence of each combination was plotted as a heat map, where values above 0.5 were considered successful (Figure 5b). Successful pairs had an increase in fluorescence when target RNA was added, and little to no fluorescence without target RNA (Figure S2). Some unsuccessful pairs showed fluorescence in the absence of target RNA, suggesting that the flanking nucleotides caused spontaneous assembly (Figure S2). Other unsuccessful pairs remained nonfluorescent after addition of target RNA, suggesting that the sensor could not assemble into a functional form on the target RNA.
Considering the possible base pairing between the linker thymines and the target adenine, we predicted the 3WJ conformations resulting from each combination of left and right strands (Figure 5b, c). 3WJs with no unpaired nucleotides (type II) performed well, succeeding in 8 out of 10 cases (Figure 5b). When the target adenine was the only unpaired nucleotide in the junction (type I), all four constructs failed in the sense that fluorescence was induced in the absence of target RNA (Figure 5b, Figure S2). All other failed pairs were unable to induce fluorescence in the presence of target RNA. Constructs which had either two unpaired thymines on the right (type IV) or one unpaired thymine on the left and right (type V) were generally unsuccessful, indicating that too much space in those regions of the junction may be detrimental to 3WJ stability. We conclude from these results that type II or III 3WJs are good candidates for the design of split Lettuce sensors.
We were able to extract general trends from these data, but we also note that there are complex factors involved in 3WJ folding that make it difficult to predict success solely based on locations of unpaired nucleotides around a junction. For example, in certain cases flanking sequence length made a substantial difference, while in other cases it did not seem to be a factor in sensor success. Specifically, when the left strand had a T1 linker, the right flanking sequences with 15 nt greatly outperformed the 10-nt and 7-nt versions, both in type II 3WJs and in type III/IV 3WJs (Figure 5b). However, when the left strand had a T0 linker, there wasn’t a significant difference between right flanking sequence lengths of 7, 10, or 15 nt in the type II 3WJ. Given the tendency for 15-nt right flanking sequences to function better than shorter sequences, we settled on a final design with 15-nt right flanking sequences and 20-nt left flanking sequences. While in general we identified arrangements that are likely to work, it is not assured that a given design will always result in a functional sensor, and even sensors with the most optimal design should always be tested before use.
We next asked if the same design principles that were effective for one target RNA site would still work on other target RNA sites that have different sequences and a different individual target nucleotide. All experiments up to this point were performed with flanking sequences that hybridize to either side of Site 1 (Figure 5d) and had a target adenine connecting H2 and H3 (Figure 5a). To address whether type II and III 3WJs, which were the highest-performing 3WJs on Site 1, work uniquely with the particular sequence of Site 1 or if they can be generalized to other RNA target sequences, we chose three alternate sites on which to test new sensors (Figure 5d). We chose to test sensors that have type II and III 3WJs as well as an additional 3WJ design, type VII (Figure 5c). Interestingly, many of these pairs worked better than the best sensor from the original Site 1 (Figure 5d). Thus, while there are some exceptions, all of these 3WJ types seem to be generalizable to other target RNA sites.
We next asked if the increase in fluorescence that we observe in the presence of target RNA is sequence specific. To rule out the possibility that nonspecific interactions with RNA are causing the two strands of S2T4AT to fold into the functional form, we scrambled the flanking sequences of one of our sensors. When tested in the presence of target RNA, only the sensor with complementary flanking sequences produced an increase in fluorescence (Figure 5e). The scrambled sensor only produced background-level fluorescence both in the presence and absence of target RNA. Thus, the RNA-induced fluorescence is a result of specific base pairing interactions between the flanking sequences and the target RNA.
We next sought to characterize the time dependence of the fluorescence signal. We combined the type VII sensor targeting Site 4 with target RNA and measured the fluorescence of DFHBI-1T at various time points up to 150 minutes. The majority of the fluorescence signal occurs within 2 hours, after which the signal remains fairly constant (Figure 5f). Thus, for maximal signal-to-noise ratios the sensors may be incubated for 2 hours, but significant fluorescence enhancement is seen in less than 1 hour.
Lastly, we asked how the sensors respond to higher temperatures. A temperature gradient from 25°C to 65°C was applied, with 5-minute incubations at each temperature to allow for equilibration. The fluorescence signal decreased moderately with each 10°C increase, until all signal was lost at 65°C (Figure S3). Thus, RNA detection can be performed at room temperature, but the fluorescence signaling will remain to some degree up to 55°C.
Detection of SARS-CoV-2 RNA using split Lettuce sensors
We next asked if SARS-CoV-2 viral RNA sequences could be detected using the split Lettuce system. We chose target RNA regions within the SARS-CoV-2 N gene and the viral 3’ UTR (Figure 6a, Table S1). The sites were chosen so that the resulting flanking sequences would not contain complementary regions to one another. We designed 12 sensors using the type VII 3WJ (Figure 5c), in which the left strand had no linker (T0) and a 20-nt flanking sequence and the right side had a single thymine linker (T1) and a 15-nt flanking sequence. The unpaired RNA target nucleotide varied depending on the target site chosen (Table S1). Of these original twelve pairs, nine sensors were able to activate fluorescence in the presence of target RNA, five of which induced a >4-fold increase in fluorescence over the RNA-absent background (Figure 6b). Thus, while each sensor should be checked for functionality before use, in general Lettuce split sensors can easily be designed to successfully detect target RNAs, including SARS CoV-2 viral RNA.
Figure 6. Detection of SARS-CoV-2 RNA using split Lettuce sensors.
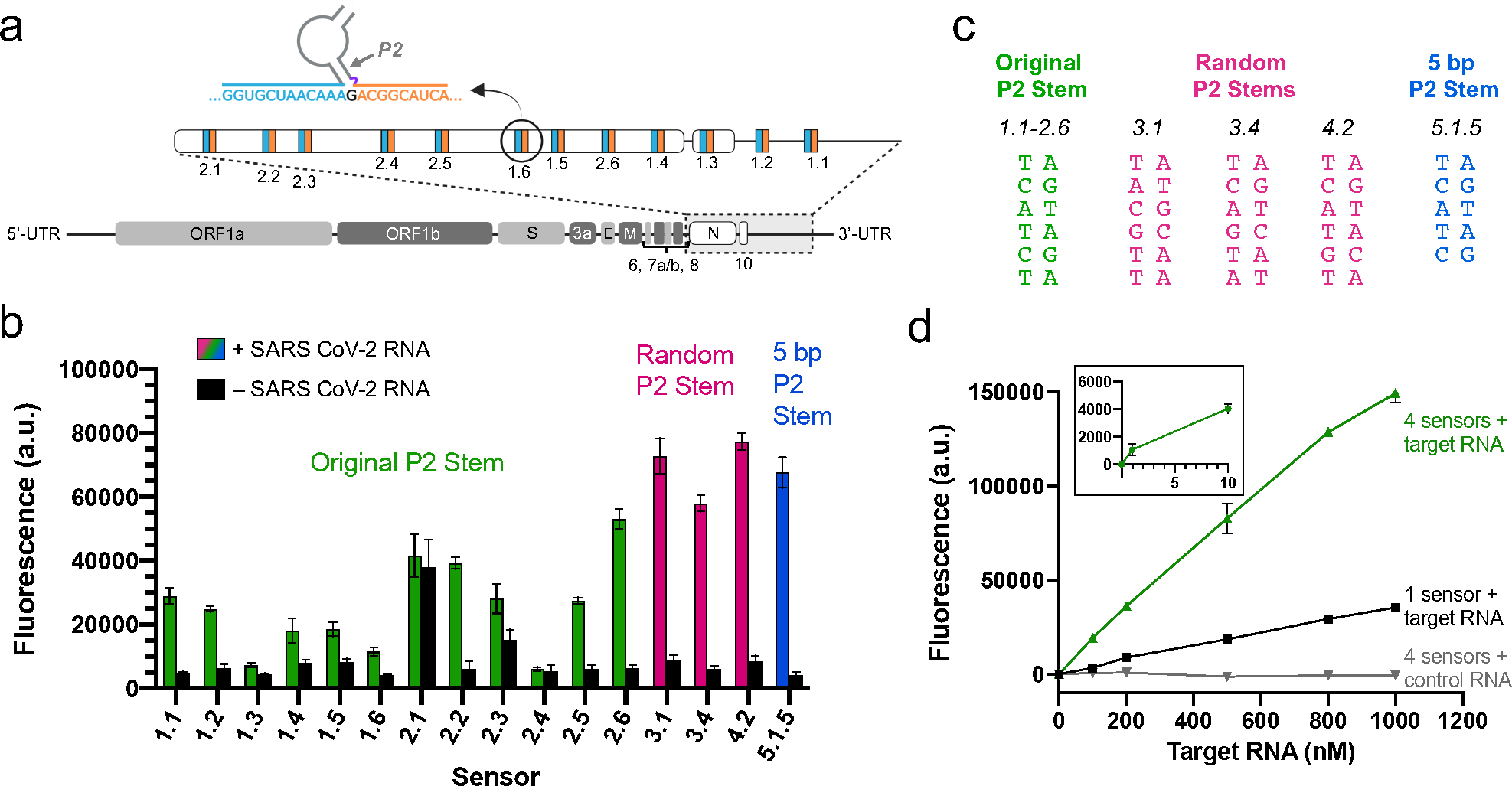
a) SARS-CoV-2 RNA target sites of split Lettuce sensors. An outline of the SARS-CoV-2 viral genome is shown. The N gene, ORF10, and 3’-UTR were amplified, and sites were chosen as targets for left (blue) and right (orange) sensor flanking sequences. Sites for sensors 1.1–2.6 are shown. Diagram is not to scale. b) Fluorescence of SARS-CoV-2 sensors. Sensors were designed with the original Lettuce P2 stem sequence (green), a randomized P2 stem sequence (pink) and a 5 bp stem sequence (blue). Buffer-only background fluorescence was subtracted from all samples. Sensor 5.1.5 targets the same site as sensor 1.5, but has one deleted base pair in its P2 stem. All other sensors target unique RNA sites. Specific P2 stem sequences for these sensors are shown in Figure 6c. Fluorescence was measured (excitation 460 nm, emission 505 nm) with a SpectraMax M-series fluorescence plate reader using 1.5 μM DNA, 0.5 μM RNA, and 2 μM DFHBI-1T. Error bars indicate s.d. (n=3). c) Sequences of the P2 stem in each sensor. P2 stems are spatially oriented as shown in Figure 6a such that the bottom of the stem connects to the flanking sequences of the sensor and the top connects to the rest of the Lettuce sequence. The 5-bp P2 stem is the same as the original P2 stem, with the bottom A/T base pair deleted. d) Increasing the number of split Lettuce sensors results in higher sensitivity of SARS-CoV-2 RNA detection. The fluorescence activation of four split Lettuce sensors compared to one split Lettuce sensor was measured at a range of SARS-CoV-2 RNA concentrations. The background signal (DNA and DFHBI-1T only) was subtracted from each data point. Beta-actin RNA was used as a control to test for off-target fluorescence activation. Fluorescence was measured as described in Figure 6b. Error bars indicate s.d. (n=3).
While the original design of the split Lettuce SARS CoV-2 sensors works well, we found that there is room for optimization in the S2T4AT core, specifically in its P2 stem. For example, to develop sensor 5.1.5 we altered the P2 stem of sensor 1.5 by deleting the AT base pair that was closest to the flanking sequences (Figure 6c), causing the adjacent G/C stem base pair to be exposed to the 3WJ (Figure S4). Surprisingly, this minor adjustment resulted in a 3.6-fold increase in fluorescence of sensor 5.1.5 over sensor 1.5 (Figure 6b, Figure S4). We hypothesize this is due to the specific composition of nucleotides around the 3WJ and their unique non-canonical interactions with one another that may stabilize the complex. Thus, the specific architecture of the 3WJ may be an important area of optimization in future work.
We also wondered whether the entire P2 stem sequence in the S2T4AT core needed to be the same as the sequence from Lettuce, or if that too could be optimized. We developed several sensors with scrambled P2 stem sequences, keeping the G/C content the same as the original stem (Figure 6c and Figure S5). We found that many of these constructs were as bright or brighter than the winning sensor from the original set of 12, but some also had higher background fluorescence (Figure S5). The best four sensors out of all those tested in the current study (sensors 3.1, 3.4, 4.2, and 5.1.5 in Figure 6b) had been adjusted from the original design by either changing the sequence of P2 or decreasing the length of P2 to five base pairs (Figure 6c). This suggests that the base pairing in the P2 stem can be modified from the original Lettuce sequence, and that there may be certain ways of adjusting the P2 stem to get optimal fluorescence signal.
We next asked if higher sensitivity could be achieved by simultaneously using multiple sensors targeting different sites on the same SARS CoV-2 transcript. We chose four target sites within a roughly 1.6-kb long RNA encoding the SARS-CoV-2 N gene followed by the 3’ UTR and measured the fluorescence induced by one sensor compared to four sensors. To determine if these signals were linear and correlated with viral RNA concentration, we tested in vitro transcribed SARS-CoV-2 RNAs ranging from 1 nM to 1000 nM. At every concentration, the group with four sensors had roughly four times higher fluorescence than the group with one sensor (Figure 6d). With four sensors, RNA concentrations as low as 10 nM can be reliably detected, whereas when only one sensor was used, the detection limit is closer to 100 nM (Figure 6d). Importantly, the background signal in the group with four sensors was comparable to the group with one sensor (Figure S6). Therefore, low nanomolar concentrations of RNA can be detected with just four sensors, with the potential for higher sensitivity as more sensors are added.
We finally sought to determine whether the sensors produce off-target fluorescence when incubated with an unrelated RNA of similar size to SARS CoV-2. We used the same four sensors designed to target SARS CoV-2 viral RNA, but instead incubated them with a roughly 1.8-kb long mCh-ACTB in vitro transcribed RNA at concentrations ranging from 100 nM to 1000 nM. There was no increase in fluorescence over background levels at every concentration tested (Figure 6d). This indicates that there is no off-target fluorescence when these sensors are exposed to a long sequence of unrelated RNA.
DISCUSSION
Here we describe a simple and efficient approach for RNA detection using unlabeled DNA oligonucleotides. To achieve this, we developed Lettuce, a fluorogenic DNA aptamer that binds DFHBI-1T and activates its fluorescence. Unlike other DNA-activated dyes such as ethidium bromide, which show relatively nonspecific fluorescence activation by DNA, DFHBI-1T and its derivatives are highly specific for the Lettuce aptamer, enabling only Lettuce-tagged DNAs to exhibit fluorescence. We show that Lettuce can be split into two separate strands that each fail to activate DFHBI-1T fluorescence on their own and cannot reconstitute the Lettuce aptamer when mixed together. However, when each of the split Lettuce strands contain additional sequence elements that allow them to hybridize to adjacent regions in an RNA, the split Lettuce strands are brought into proximity, allowing the strands to re-form the Lettuce aptamer. We show that this approach can be used to detect a SARS-CoV-2 RNA standard using a set of four pairs of split Lettuce strands, enabling detection of as little as 10 nM viral RNA.
Although we do not have a crystal structure of Lettuce, the structural mechanism for fluorescence activation of DFHBI-1T may involve DFHBI-1T binding to a G-quadruplex in Lettuce. G-quadruplexes have been seen in Spinach and Corn, which are RNA aptamers that bind DFHBI-1T or DFHO, a structurally related fluorophore12,13. Our studies show that Lettuce has sequence features of a G-quadruplex, including three separate stretches of three consecutive guanines. Additionally, thioflavin T, a small fluorescent ‘light up’ probe for G-quadruplexes22 is fluorescently activated upon incubation with Lettuce. These data suggest that Lettuce contains G-quadruplex. Mutation of putative guanines in the G-quadruplex abolishes Lettuce-induced fluorescence of DFHBI-1T, suggesting that the G-quadruplex is required for fluorescence activation of DFHBI-1T. A crystal structure will be needed to fully confirm this hypothesis and to understand how the G-quadruplex, if it exists, contributes to fluorescence activation of DFHBI-1T and related fluorophores.
Notably, our split Lettuce system is designed so each strand contains part of the G-quadruplex. The G-quadruplex would only fold if the two split strands are brought in proximity. Thus, the target RNA likely induces G-quadruplex formation, which subsequently enables binding and fluorescence activation of DFHBI-1T.
Exceptions to sensor success may occur for a variety of reasons, so it is necessary to test each sensor pair before use. For example, the type II and III sensors designed for Site 4 had fairly high background signal (see Figure 5d). This high background may have been due to the flanking sequences. When adding flanking sequences to each end of the S2T4AT stem, there is the potential for the flanking sequences to be partially complementary to one another, adding enough binding energy to the two strands that they are able to self-assemble without target RNA. To avoid this, target RNA sites should be carefully chosen so that corresponding flanking sequences do not contain significant stretches of complementary sequences, especially near the S2T4AT stem.
It is also possible that certain target RNA sites will impede the functional assembly of the two strands causing the sensor to remain “off”. Thus, while most sensors function as expected, each sensor must be tested to ensure functionality on the particular target RNA site chosen.
Our results suggest a general route for designing split Lettuce DNA oligonucleotides that can bind and detect a target RNA. First, target RNA sites should be chosen such that the resulting complementary flanking sequences attached to split Lettuce have minimal complementarity to each other. The left and right flanking sequences of split Lettuce sensors are designed to hybridize to the 5’ and 3’ sides of the RNA target site, respectively (see Figure 5a). We show that successful sensors include a left flanking sequence of 20 nt that is directly attached to the left split Lettuce core strand. The right flanking sequence has a length of 15 nt and is attached to the right split Lettuce core strand via a 1-nt linker which remains unpaired. Our design also includes one unpaired nucleotide in the target RNA that lies between the DNA/RNA hybrid helices created by the flanking sequences and target RNA. Using this strategy, numerous DNA oligonucleotides can be simply designed to bind multiple 36-nt long RNA sequences along an RNA of interest with few, if any, optimization steps.
Overall, our approach allows RNA detection with DNA oligonucleotides and commercially available fluorophores. Although we used a synthetic SARS-CoV-2 RNA, future studies can focus on detecting viral RNA in clinical samples and comparison of this detection approach with current PCR-based methods. Further optimization of this approach will likely involve further improvements of Lettuce or the design of new fluorogenic DNA aptamers with improved quantum yield and a concomitant increase in overall brightness. This is expected to increase the sensitivity of RNA detection for applications in which low levels target RNA are present.
METHODS
Reagents and equipment.
Unless otherwise stated, all reagents were purchased from Sigma Aldrich. Commercially available reagents were used without further purification. Fluorophores used in this study were obtained from Lucerna Technologies (New York, NY) or were synthesized as described previously17,25. All DNA sequences were purchased from Integrated DNA Technologies unless otherwise indicated. Absorbance spectra were recorded using a cuvette on a Thermo Scientific Nanodrop 2000 spectrophotometer. Fluorescence excitation and emission spectra were measured with a Fluoromax-4C fluorometer (Horiba Scientific). Photobleaching was measured using a CoolSnap HQ2 CCD camera through a 60x oil objective mounted on an Eclipse TE2000-E microscope (Nikon) and analyzed with the NIS-Elements software. ChemiDoc MP imager (BioRad) was used to record fluorescence in gels. Fluorescence measurements for Figure 2b and Figure 6 were collected using a SpectraMax M-series plate reader (Molecular Devices). Fluorescence measurements for Figures 4 and 5 were collected using a Gen5 plate reader (BioTek Instruments). All other fluorescence measurements were collected using a Fluoromax-4C fluorometer (Horiba Scientific).
SELEX library preparation.
DNA oligonucleotide libraries were designed to contain fixed 5’ and 3’ ends to facilitate PCR amplification. Random, stem-loop, and G-rich libraries contained random regions produced using an equal phosphoramidite mixture of A, G, C, and T.
The library used for directed evolution of Lettuce was produced so that each sequence resembles Lettuce, except for containing on average 2.5 mutations. To obtain this library, every position was chemically synthesized with phosphoramidite nucleoside mixtures that contain primarily the nucleotide found in the parent aptamer, but also contains very low levels of the other three nucleotides. The mixtures for each nucleotide are as follows: A-mix (95.9%A, 1.3%T, 1.1%G, 1.7%C); T-mix (2.1%A, 94.5%T, 1.4%G, 2.1%C); G-mix (2.5%A, 2.0%T, 93.1%G 2.5%C), C-mix (1.7%A, 1.3%T, 1.1%G, 95.9%C). The 5’ and 3’ ends (nts 1–22 and 74–99, respectively) were chemically synthesized using only the parent nucleotide to produce fixed ends to facilitate PCR amplification (numbering as in Figure 1c). DNA libraries were prepared by the W.M. Keck Oligonucleotide Synthesis Facility using standard cleavage and deprotection, and cartridge purification.
SELEX of DNA aptamers.
The affinity matrix for SELEX (DFHBI-agarose) was prepared as described previously3. Random, stem-loop, and G-rich ssDNA libraries or Lettuce directed evolution library (3 nmoles) were diluted in selection buffer containing 40 mM HEPES pH 7.4, 100 mM KCl, 1.25 mM MgCl2, and 0.1% DMSO. First, ssDNA sequences capable of binding to the agarose matrix were removed by incubating the library with “mock” resin consisting of aminohexyl linker bound to agarose. The ssDNA solution that did not bind to the “mock” resin was then incubated with DFHBI-1T-coupled resin. The DNA bound to DFHBI-1T resin was washed with 3×3 ml selection buffer during rounds 1–2 and during the directed evolution SELEX rounds, 4X3ml for round 3–5, 6×3ml for rounds 6–9. Bound RNA was eluted with free DFHBI-1T (1mM) in selection buffer.
The eluted DNA was then amplified by PCR using a 5’-phosphorylated reverse primer and a 5’-OH reverse primer. Following amplification with Taq DNA polymerase (New England Biosciences), the antisense DNA strand was degraded using lambda exonuclease (New England Biosciences). The resulting ssDNA was purified via ethanol precipitation to yield the pool for the next round. The presence of fluorescent DNA sequences in each pool was assayed by measuring fluorescence of samples containing 0.5 μM DNA and 1 μM DFHBI-1T on a fluorometer in comparison with samples containing fluorophore mixed with the original library.
To identify ssDNA aptamers during SELEX, we screened for individual aptamers after nine rounds of SELEX and after two rounds of directed evolution SELEX. The ssDNA isolated after round 9 of the original SELEX and after round 2 of the directed evolution SELEX was cloned into bacteria and plated on LB-AMP plates so that each colony contained a plasmid containing a single aptamer sequence. Colonies were then pooled in groups of 30 to screen for aptamers that bind and activate DFHBI-1T fluorescence. The 30 colonies were pooled and plasmid DNA was isolated, and the mixture of plasmids was subjected to PCR to amplify the encoded aptamer sequences. The ssDNA aptamers were generated by treating the PCR products with lambda exonuclease. We analyzed 24 pools of ssDNA from each of the three original DNA libraries and identified three pools (one from each library) that activated DFHBI-1T fluorescence. We then tested each of the individual 30 colonies that made up each of these three fluorescent pools. We incubated these individual ssDNA sequences with DFHBI-1T to identify aptamers responsible for activating DFHBI-1T fluorescence. These experiments resulted in one aptamer per library which activated DFHBI-1T fluorescence.
Preparation of DNA sensors and RNA targets.
SARS-CoV-2-targeted Lettuce sensors were purchased from Thermo Fisher. Mutants of S-9-1 from directed evolution and all split Lettuce oligonucleotides were used in fluorescence measurements directly after dilution.
The 87-nt target RNA used in sensor optimization experiments was in vitro transcribed using a dsDNA template that was designed using a random sequence generator with a specified 50% G/C content, on to which a 5’ T7 promoter sequence was added. mCh-ACTB RNA used as a negative control was generated using in vitro transcription from a linearized plasmid. Previously created19 pcDNA-mCh-ACTB-24x F30-Broccoli plasmid was linearized by BamHI and overhangs were removed using T4 DNA polymerase. Following purification using QIAquick PCR purification kit (Qiagen), the linearized template was in vitro transcribed using mMESSAGE mMACHINE™ T7 Transcription kit (Invitrogen) for 3 h at 37°C. After treating with DNase I for 30 min at 37°C, the RNA was purified using RNA Clean and Concentrator 25 columns (Zymo Research).
The SARS CoV-2 RNA fragment that included the N gene, ORF10, and 3’ UTR was generated using a dsDNA template created using PCR. The plasmid26 (pCC1-CoV2-F7) for generating the template was obtained from World Reference Center for Emerging Viruses and Arboviruses and amplified by PCR using Phusion High Fidelity DNA Polymerase (New England Biolabs). The PCR product was verified for correct size on 1% TAE-agarose gel. PCR product was purified with the MinElute PCR Purification kit (Qiagen). The target RNAs was prepared by in vitro transcription using AmpliScribe T7-Flash Transcription Kit (Lucigen) by incubating at 40°C for at least 4 hours. After treating with DNase I for 30 min at 37°C, the RNA was purified using RNA Clean and Concentrator 25 columns (Zymo Research).
In vitro characterization of aptamers.
Characterization of S-9-1, R-9-1, and G-7-11 as seen in Table 1 and Figure 1e was done using purified ssDNAs diluted to 20 μM in a buffer containing 40 mM HEPES (pH 7.5), 100 mM KCl, and 1 mM MgCl2. DNAs were purified by running samples on 10% TBE-urea gel, excising the band of the full-length sequence, and dissolving the ssDNA into H2O prior to use. DNAs were then heated up to 75°C for 5 min and then cooled to room temperature. DNA solutions were then mixed with an equal volume of the same buffer containing 2 μM DFHBI-1T. After 1-hour incubation, fluorescence of each sample was measured with excitations and emissions specified, 5 nm slit widths, and 0.1 s integration time. Background signal was detected with the DNA-free sample containing 1 μM DFHBI-1T in 40 mM HEPES (pH 7.5), 100 mM KCl, and 1 mM MgCl2.
Extinction coefficients were calculated based on the absorbance spectrum and the Beer-Lambert-Bouguer law. For quantum yield calculations, the fluorescence signal of Lettuce-DFHBI-1T complex was compared to that of equally absorbing Broccoli-DFHBI-1T complex, for which the quantum yield was previously determined4. Excitation and emission spectra were measured for solutions using “excess DNA” conditions and limiting amount of fluorophore to ensure that no free fluorophore contributes to the absorbance or fluorescence signal. This approach gives a fixed concentration of DNA-fluorophore complex which is equal to the concentration of the fluorophore that was initially added.
Gel staining of fluorescent DNAs.
10 μl samples were prepared using the specified amount of ssDNA in H2O mixed and loading buffer containing bromophenol blue. DNA samples were loaded into wells of precast 10% TBE-Urea Gel (Life Technologies) and ran at 250 V in 1xTBE buffer.
The gel was run until bromophenol blue passed the gel end. The gel was then washed for 3×5 min with water and stained for 30 min with 10 μM DFHBI-1T in buffer containing 40 mM HEPES (pH 7.4), 100 mM KCl, and 1 mM MgCl2. The gel was imaged using a ChemiDoc MP with 470±30 nm excitation and 532±28 nm emission. In order to see all DNA in the sample, the gel was washed for 3×5 min with water. Then the gel was stained for 20 min with SYBR Gold fluorophore (Life Technologies) diluted 1/10000 in TBE buffer. The SYBR Gold-stained gel was imaged with the same instrument using preset SYBR Gold settings (302 nm excitation and 590±110 nm emission).
Split Lettuce fluorescence and RNA detection assays.
Development of the conditionally fluorescent S2T4AT construct was performed by prediction of Lettuce’s secondary structure using RNAfold online software, based on which split sites and truncation sites were chosen. Modified RNAfold secondary structures are shown for S-9-1, R-9-1, and G-7-11, in which short (< 3 bp) stems present in the original structure prediction were removed. To measure fluorescence, nonsplit (10 μM) or left and right (10 μM each) Lettuce strands were incubated with DFHBI (5 μM) in buffer (40 mM HEPES pH 7.5, 140 mM KCl, 5 mM MgCl2) at a final volume of 30 μl. Samples were heated in a thermocycler at 70°C for 5 min and cooled at room temperature for 1 hr before being transferred to a clear-bottom 364-well plate. Fluorescence was measured using excitation at 460 nm and emission at 505 nm. Background signal was measured using the DNA-free sample (5 μM DFHBI in 1x buffer).
RNA detection was carried out with the addition of random target RNA (5 μM) or SARS-CoV-2 target RNA (0.5 μM unless otherwise indicated) to samples containing split sensor strands (10 μM each for random target RNA detection, 1.5 μM each for SARS-CoV-2 and mCh-ACTB RNA detection) and DFHBI-1T (5 μM for random target RNA detection and 2 μM for SARS-CoV-2 RNA detection) in buffer (40 mM HEPES pH 7.5, 140 mM KCl, 5 mM MgCl2) at a final volume of 30 μl. mCh-ACTB RNA was used as a negative control at the concentrations specified. All samples were heated in a thermocycler at 70°C for 5 min and cooled at room temperature for 1 hr. Samples were then transferred to a clear-bottom 364-well plate and fluorescence was measured using excitation at 460 nm and emission at 505 nm. To measure the time dependence of fluorescence, reagents were combined directly in a 364-well plate except for DFHBI-1T. Samples were kept at room temperature and brought to the fluorescence plate reader, after which DFHBI-1T was quickly added and fluorescence was measured.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Jaffrey lab for helpful comments and suggestions. We thank X. Li for synthesizing DFHBI-1T and DFHO. This work was supported by NIH grants R01NS064516 and R35NS111631 to S.R.J. J.D.M. was supported by a Medical Scientist Training Program grant from the National Institute of General Medical Sciences of the National Institutes of Health under award number T32GM007739 to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD- PhD Program.
Footnotes
Declaration of Competing Interests
The authors declare the following competing financial interest(s): S.R.J. is the cofounder of Lucerna Technologies and Chimerna Therapeutics and has equity in these companies. Lucerna has licensed technology related to Spinach and other RNA-fluorophore complexes.
SUPPORTING INFORMATION
Additional figures and notes, SARS-CoV-2 sensor and RNA target sequence information, and photographs of representative fluorescence data.
REFERENCES
- (1).Tyagi S; Kramer FR Molecular Beacons: Probes That Fluoresce upon Hybridization. Nature Biotechnology 1996, 14 (3). 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- (2).You M; Jaffrey SR Structure and Mechanism of RNA Mimics of Green Fluorescent Protein. Annual Review of Biophysics 2015, 44 (1), 187–206. 10.1146/annurev-biophys-060414-033954. [DOI] [PubMed] [Google Scholar]
- (3).Paige JS; Wu KY; Jaffrey SR RNA Mimics of Green Fluorescent Protein. Science 2011, 333 (6042). 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Filonov GS; Moon JD; Svensen N; Jaffrey SR Broccoli: Rapid Selection of an RNA Mimic of Green Fluorescent Protein by Fluorescence-Based Selection and Directed Evolution. Journal of the American Chemical Society 2014, 136 (46). 10.1021/ja508478x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kikuchi N; Kolpashchikov DM Split Spinach Aptamer for Highly Selective Recognition of DNA and RNA at Ambient Temperatures. ChemBioChem 2016, 1589–1592. 10.1002/cbic.201600323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Huizenga DE; Szostak JW A DNA Aptamer That Binds Adenosine and ATP. Biochemistry 1995, 34 (2), 656–665. 10.1021/bi00002a033. [DOI] [PubMed] [Google Scholar]
- (7).Filonov GS; Kam CW; Song W; Jaffrey SR In-Gel Imaging of RNA Processing Using Broccoli Reveals Optimal Aptamer Expression Strategies. Chemistry & Biology 2015, 22 (5), 649–660. 10.1016/j.chembiol.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Paige JS; Nguyen-Duc T; Song W; Jaffrey SR Fluorescence Imaging of Cellular Metabolites with RNA. Science (New York, N.Y.) 2012, 335 (6073), 1194. 10.1126/science.1218298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Su Y; Hickey SF; Keyser SGL; Hammond MC In Vitro and In Vivo Enzyme Activity Screening via RNA-Based Fluorescent Biosensors for S -Adenosyl- l -Homocysteine (SAH). Journal of the American Chemical Society 2016, 138 (22), 7040–7047. 10.1021/jacs.6b01621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Tuerk C; Gold L Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science 1990, 249 (4968), 505–510. 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- (11).Ellington AD; Szostak JW In Vitro Selection of RNA Molecules That Bind Specific Ligands. Nature 1990, 346 (6287), 818–822. 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- (12).Warner KD; Sjekloća L; Song W; Filonov GS; Jaffrey SR; Ferré-D’Amaré AR A Homodimer Interface without Base Pairs in an RNA Mimic of Red Fluorescent Protein. Nature chemical biology 2017, 13 (11), 1195–1201. 10.1038/nchembio.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Warner KD; Chen MC; Song W; Strack RL; Thorn A; Jaffrey SR; Ferré-D’Amaré AR Structural Basis for Activity of Highly Efficient RNA Mimics of Green Fluorescent Protein. Nature Structural & Molecular Biology 2014, 21 (8), 658–663. 10.1038/nsmb.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Trachman RJ; Autour A; Jeng SCY; Abdolahzadeh A; Andreoni A; Cojocaru R; Garipov R; Dolgosheina EV; Knutson JR; Ryckelynck M; et al. Structure and Functional Reselection of the Mango-III Fluorogenic RNA Aptamer. Nature Chemical Biology 2019, 15 (5), 472–479. 10.1038/s41589-019-0267-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Trachman RJ; Truong L; Ferré-D’Amaré AR Structural Principles of Fluorescent RNA Aptamers. Trends in Pharmacological Sciences 2017, 38 (10), 928–939. 10.1016/J.TIPS.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dey SK; Filonov GS; Olarerin-George AO; Jackson BT; Finley LWS; Jaffrey SR Repurposing an Adenine Riboswitch into a Fluorogenic Imaging and Sensing Tag. Nature Chemical Biology 2021 2021, 1–11. 10.1038/s41589-021-00925-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Song W; Filonov GS; Kim H; Hirsch M; Li X; Moon JD; Jaffrey SR Imaging RNA Polymerase III Transcription Using a Photostable RNA–Fluorophore Complex. Nature Chemical Biology 2017, 13 (11). 10.1038/nchembio.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wang P; Querard J; Maurin S; Nath SS; Le Saux T; Gautier A; Jullien L Photochemical Properties of Spinach and Its Use in Selective Imaging. Chemical Science 2013, 4 (7), 2865. 10.1039/c3sc50729g. [DOI] [Google Scholar]
- (19).Li X; Kim H; Litke JL; Wu J; Jaffrey SR Fluorophore-Promoted RNA Folding and Photostability Enables Imaging of Single Broccoli-Tagged MRNAs in Live Mammalian Cells. Angewandte Chemie International Edition 2020, 59 (11). 10.1002/anie.201914576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Li X; Mo L; Litke JL; Dey SK; Suter SR; Jaffrey SR Imaging Intracellular S -Adenosyl Methionine Dynamics in Live Mammalian Cells with a Genetically Encoded Red Fluorescent RNA-Based Sensor. Journal of the American Chemical Society 2020, 142 (33). 10.1021/jacs.0c02931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Dvorkin SA; Karsisiotis AI; Webba da Silva M Encoding Canonical DNA Quadruplex Structure. Science Advances 2018, 4 (8). 10.1126/sciadv.aat3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Renaud de la Faverie A; Guédin A; Bedrat A; Yatsunyk LA; Mergny J-L Thioflavin T as a Fluorescence Light-up Probe for G4 Formation. Nucleic acids research 2014, 42 (8), e65. 10.1093/nar/gku111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Welch JB; Walter F; Lilley DMJ Two Inequivalent Folding Isomers of the Three-Way DNA Junction with Unpaired Bases: Sequence-Dependence of the Folded Conformation. Journal of Molecular Biology 1995, 251 (4). 10.1006/jmbi.1995.0452. [DOI] [PubMed] [Google Scholar]
- (24).Leontis NB; Kwok W; Newman JS Stability and Structure of Three-Way DNA Junctions Containing Unpaired Nucleotides. Nucleic Acids Research 1991, 19 (4). 10.1093/nar/19.4.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Song W; Strack RL; Svensen N; Jaffrey SR Plug-and-Play Fluorophores Extend the Spectral Properties of Spinach. Journal of the American Chemical Society 2014, 136 (4). 10.1021/ja410819x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Xie X; Muruato A; Lokugamage KG; Narayanan K; Zhang X; Zou J; Liu J; Schindewolf C; Bopp NE; Aguilar P v.; et al. An Infectious CDNA Clone of SARS-CoV-2. Cell Host & Microbe 2022, 27 (5). 10.1016/j.chom.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.