

Summary
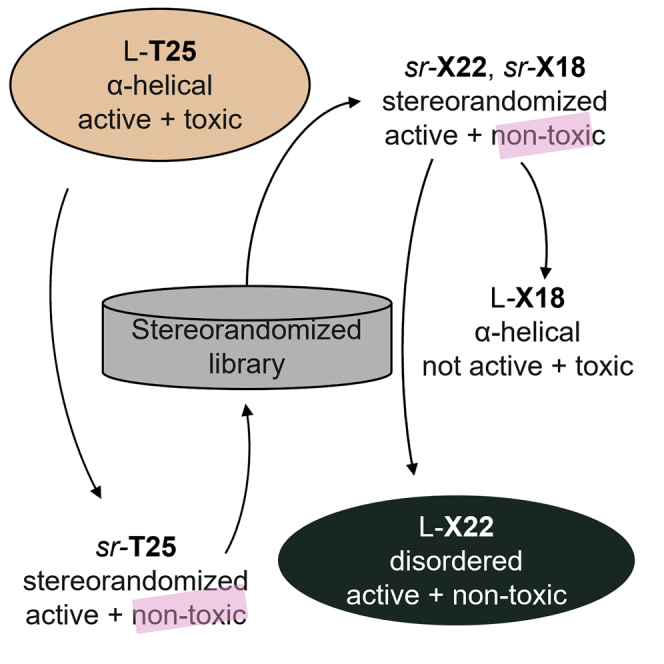
Membrane-disruptive amphiphilic antimicrobial peptides behave as intrinsically disordered proteins by being unordered in water and becoming α-helical in contact with biological membranes. We recently discovered that synthesizing the α-helical antimicrobial peptide dendrimer L-T25 ((KL)8(KKL)4(KLL)2KKLL) using racemic amino acids to form stereorandomized sr-T25, an analytically pure mixture of all possible diastereoisomers of L-T25, preserved antibacterial activity but abolished hemolysis and cytotoxicity, pointing to an intrinsically disordered antibacterial conformation and an α-helical cytotoxic conformation. In this study, to identify non-toxic intrinsically disordered homochiral antimicrobial peptide dendrimers (AMPDs), we surveyed sixty-three sr-analogs of sr-T25 selected by virtual screening. One of the analogs, sr-X18 ((KL)8(KLK)4(KLL)2KLLL), lost antibacterial activity as L-enantiomer and became hemolytic due to α-helical folding. By contrast, the L- and D-enantiomers of sr-X22 ((KL)8(KL)4(KKLL)2KLKK) were equally antibacterial, non-hemolytic, and non-toxic, implying an intrinsically disordered bioactive conformation. Screening stereorandomized libraries may be generally useful to identify or optimize intrinsically disordered bioactive peptides.
Keywords: antimicrobial peptides, dendrimers, stereorandomization, virtual screening, multidrug resistance, intrinsically disordered proteins, multi-drug resistant bacteria, solid-phase peptide synthesis, membrane disruption
Graphical abstract
Highlights
-
•
Stereorandomized antimicrobial peptide dendrimers are identified by virtual screening
-
•
A switch to L residues induces α-helical folding and strong toxicity in one sr-AMPD
-
•
Another AMPD remains intrinsically disordered, active, and non-toxic as L-AMPD
To discover intrinsically disordered bioactive peptides, Cai et al. screen libraries of stereorandomized peptides, each one obtained by synthesis using racemic amino acids to form an analytically pure mixture of all possible stereoisomers. This yields an intrinsically disordered non-toxic antimicrobial peptide dendrimer active against multidrug-resistant bacteria.
Introduction
Intrinsically disordered proteins (IDPs) are proteins that exist as random coils and whose bioactive conformation can either be unordered or become ordered in presence of their biological target.1,2,3 Many antimicrobial peptides (AMPs),4,5,6 which are being investigated as an attractive option to fight multidrug resistant (MDR) bacteria,7,8 formally belong to the second IDP class as they exist as random coils that fold to an amphiphilic α-helix in contact with the bacterial membrane, inducing membrane destabilization or pore formation and eventually killing the bacterium.9
In our own efforts to develop antibacterial agents,10,11,12,13 we recently discovered that AMP dendrimers (AMPDs; e.g. L-G3KL; Figure 1)14,15 are potent antibacterial agents acting by a membrane disruptive mechanism similar to AMPs,16 as do various cationic amphiphiles such as cyclic peptides,17 polymers,18 peptidomimetics,19 foldamers,20 and dendrimers.21,22,23,24 L-G3KL kills Gram-negative MDR bacteria, including polymyxin-resistant clinical isolates in a pH- and ionic-strength-dependent manner without hemolysis of human red blood cells,25,26,27 and shows antibiofilm28 and wound-healing properties29 and partial synergy with classical antibiotics,30 as well as retention of activity as chitosan conjugate.31
Figure 1.

Sequence of AMPDs, dendrimer virtual library, and structural formula of AMPD L-X22
Racemic residues are underlined. Branching lysines are L-chirality in L-sequences and racemic in sr sequences.
Circular dichroism (CD) and molecular dynamics (MD) studies with L-G3KL and analogs L-T7 and L-T25 showed that, similar to linear α-helical AMPs, these dendrimers fold into an α-helical conformation in contact with the membrane.32 However, their antibacterial activity was unaffected by stereorandomization, which consists of synthesizing the dendrimers using racemic amino acids to yield an analytically pure mixture of all possible diastereoisomers, suggesting that the bioactive antibacterial conformation of all L- or all D-AMPDs might not be α-helical but intrinsically disordered and therefore belong to the first class of IDPs.33
Here, we set out to identify an intrinsically disordered AMPD by screening a library of stereorandomized dendrimers to discover active stereorandomized (sr)-AMPDs and later testing their activity and conformational behavior in homochiral form. Starting with sr-T25, we composed a focused library of analogs (sr-X1 – sr-X63; Figure S1) by ligand-based virtual screening (LBVS)34,35 of a virtual library of 50,625 G3 peptide dendrimers featuring all possible permutations of up to three lysines or leucines in each generation.32 We computed similarities to sr-T25 using macromolecule-extended atom-pair fingerprint (MXFP), a molecular fingerprint counting atom pairs at increasing topological distances measured in bonds along the shortest path.36 Atom-pair fingerprints encode molecular shape and pharmacophores36,37,38,39,40 and were used previously to identify antimicrobial bicyclic peptides41,42 and AMPDs.32 As for most molecular fingerprints used in LBVS, MXFP is calculated from the two-dimensional (2D) structure without stereochemistry and is therefore suitable for stereorandomized sequences.
In summary, 63 sr-AMPD analogs have been synthesized and tested and many of them are readily active and non-hemolytic, which leads to the identification of sr-X18 and sr-X22 as two particularly potent AMPDs. Structure-activity relationship, mechanistic, and modeling studies show that one of them, sr-X22, retains its high activity, low hemolysis, and low cell toxicity in the form of its pure L- or D-enantiomers, implying that its bioactive conformation is intrinsically disordered.
Results and discussion
Virtual screening, synthesis, and testing reveal potent non-hemolytic sr-AMPDs
To compose a focused library of sr-T25 analogs, we sorted our virtual dendrimer library using the MXFP pharmacophore fingerprint.36 We selected the 20 MXFP-nearest neighbors of sr-T25 and 43 additional sequences by clustering among the first 200 and 1,000 closest sequences (see experimental procedures for details). This selection sampled 2% of the virtual library covering a narrow range of the chemical space surrounding sr-T25, as illustrated by a principal-component analysis of the MXFP property space (Figures 2A and 2B). The selected dendrimers differed from sr-T25 in size (36–41 residues; Figure 2C) and overall positive charges (+3 to +24; Figure 2D), with increasingly different values as the similarity to sr-T25 decreased, as measured by an increase in the city-block distance (CBD)43 calculated from the MXFP values. These positive charges are contributed by the ε-amino groups of the free lysine side chains (pKa > 9) because the eight N termini of the peptide dendrimers have a lowered pKa of approximately 6.5 and occur as neutral amino groups at neutral pH (Figure S2).27
Figure 2.

Selecting a stereorandomized-focused library by virtual screening
(A) Principal-component analysis of the virtual library by MXFP values colored by similarity to sr-T25 as calculated using the city-block distance (CBD). See also https://tm.gdb.tools/map4/dendrimers_mxfp_pca/.
(B) Close-up view around sr-T25.
(C and D) Size distribution (C) and positive charges of dendrimers (D) in the focused stereorandomized library as function of CBD to sr-T25.
We synthesized the stereorandomized library of 63 sr-T25 analogs by high-temperature solid-phase peptide synthesis (SPPS) using racemic amino acid building blocks (sr-X1 – sr-X63; Table S1). All products were obtained as homogeneous products after preparative high-performance liquid chromatography (HPLC) purification. Most dendrimers were non-hemolytic yet showed very substantial activity against the Gram-negative bacterium Pseudomonas aeruginosa and, to a lesser extent, against Escherichia coli, Acinetobacter baumannii, and Klebsiella pneumoniae, in line with the activity profile of the parent AMPD sr-T25.33
The ratio of lysine to leucine side chains, which determines the ratio between positive charges and hydrophobic groups, was a key determinant of activity. From the 26 most active and least hemolytic dendrimers found at intermediate lysine (Lys)/leucine (Leu) ratio values (sr-X7 – sr-X32, Lys/Leu = 0.45–0.81; Figure S1), we selected two dendrimers for closer study due to their particularly good activity and interesting amino acid sequence. The first one was sr-X18, one of the most active close analogs of sr-T25 (CBD = 22) with a hydrophobic dendrimer core composed of only Leu residues in G1 and G0 and an inverted sequence in the G2 branch (KL → LK) compared with most AMPDs. The second one was sr-X22, one of the most distant yet very active analogs of sr-T25 (CBD = 144), in particular showing good activity against K. pneumoniae. Compared with sr-T25, sr-X22 featured a shorter and less cationic G2 branch (KL → L) but a more cationic G1-G0 core ((LL)2KKLL → (KLL)2KLKK).
sr-, L-, and D-X22 combine strong antibacterial activity with low toxicity
To gain a broader insight into the effect of stereorandomization on AMPD activity, we prepared all L and all D versions of sr-T25, sr-X18, and sr-X22, as well as analogs sr-aT25, sr-, L-, and D-aX18, and sr-, L-, and D-aX22, where N termini, which have an apparent pKa ∼ 6.5 (Figure S2), have been removed, an effect that may increase AMPD activity.27 We measured MICs (minimum inhibitory concentrations) of all compounds at a physiological pH of 7.4, as well as at pH 5, which occurs in skin infections, and at pH 8.0, relevant to the slightly alkaline conditions in chronic wounds.44,45 We considered four Gram-negative bacteria and additionally included methicillin-resistant Staphylococcus aureus (MRSA) since our AMPDs as well as polymyxin become active against this Gram-positive bacterium at pH 8.0 (Table 1).27 In these assays, the parent AMPD sr-T25, which is non-hemolytic compared with the pure enantiomers L-T25 or D-T25,33 was equally active against bacteria at all three pH values measured and was even more active than the pure enantiomers against MRSA at pH 8.0. Analog sr-aT25 with N termini removed was slightly more antibacterial than sr-T25 while retaining a low hemolysis.
Table 1.
Antibacterial activity and cytotoxicity of AMPD sr-X22, sr-X18, and analogs
| E. coli W3110 | MIC at pH 5.0/7.4/8.0a |
MHCb | IC50 HEK293c | ||||
|---|---|---|---|---|---|---|---|
| A. baumannii ATCC 19606 | P. aeruginosa PAO1 | K. pneumoniae NCTC 418 | S aureus COL | ||||
| sr-T25 | 8/4/2 | 8/8/2 | 8/8/2 | >64/64/8 | >64/>64/4 | 1,000 | 197 ± 27 |
| L-T25 | 32/8/8 | 16/4/4 | 32/8/8 | >64/32/16 | >64/>64/32 | 62.5 | 92 ± 3 |
| D-T25 | 32/8/4 | 32/8/4 | 32/8/8 | >64/16/16 | >64/>64/32 | 125 | 33 ± 3 |
| sr-aT25 | 2/2–4/2 | 2/2/2 | 8/8/2 | >64/32/4 | >64/>64/2 | 1,000 | 5.8 ± 0.3 |
| sr-X18 | 32/4/2 | 2/2/2 | 16/4/2 | >64/32/8 | >64/>64/2 | >2,000 | 117 ± 12 |
| L-X18 | 16/16/16 | 16/32/16 | 16/32/16 | >64/>64/32 | >64/>64/16 | 7.8 | 52 ± 4 |
| D-X18 | 32/64/32 | 32/64/32 | 32/64/32 | >64/>64/>64 | >64/>64/32 | 7.8 | 47 ± 12 |
| sr-aX18 | 4/4/2 | 2/2/2 | 16/8/4 | >64/16/8 | >64/16/2 | >2,000 | 18 ± 1 |
| L- aX18 | 32/>64/32 | 32/64/32 | 64/>64/64 | >64/>64/>64 | >64/>64/32 | <3.9 | 14 ± 0.6 |
| D- aX18 | 32/>64/32 | 32/>64/16 | 64/>64/64 | >64/>64/>64 | >64/>64/16 | <3.9 | 21 ± 2 |
| sr-X22 | 32/4/2 | 32/4/2 | 16/2/2 | >64/64/8 | >64/>64/16 | >2,000 | 575 ± 30 |
| L-X22 | 8/4/2 | 8/4/2 | 8/4/4 | >64/>64/8 | >64/>64/8 | >2,000 | 575 ± 23 |
| D-X22 | 8/4/4 | 16/8/4 | 16/4/4 | >64/>64/16 | >64/>64/4 | 2,000 | 514 ± 21 |
| sr-aX22 | 2/2/2 | 2/2/2 | 64/8/4 | >64/64/8 | 64/16/2 | >2,000 | 21 ± 3 |
| L- aX22 | 2/4/4 | 2/4/4 | 16/8/16 | 64/8/8 | 64/16/4 | >2,000 | 24 ± 3 |
| D- aX22 | 4/8/4 | 4/4/4 | 32/8/16 | 64/16/16 | >64/16/4 | >2,000 | 17 ± 2 |
| L-G3KL | 32/8/1–2 | 8/8/1 | 16/4/1 | >64/>64/4 | >64/>64/2 | >2,000 | 460 ± 17 |
| L-T7 | 16/4/2 | 16/8/2–4 | 16/8/2–4 | >64/32/8 | >64/>64/4 | >2,000 | 70 ± 6 |
| PMB | 0.02/0.25/0.13 | 1/0.25/0.25 | 0.03/0.5/0.5 | 8/0.25/0.25 | >64/>64/4 | >2,000 | 346 ± 20 |
MIC in μg/mL was measured in Müller-Hinton (MH) medium after incubation for 16–20 h at 37°C. Each result represents two independent experiments performed in duplicate.
MHC (minimum hemolytic concentration) in μg/mL was measured on human red blood cells in PBS (pH 7.4) at room temperature after incubation for 4 h at 37°C. Each result represents two independent experiments performed in duplicate.
IC50 (mean ± SD, μg/mL) was measured on HEK293 cell line after incubation for 48 h at 37°C. Data present results from three independent experiments in triplicates.
The non-hemolytic sr-X18 revealed a surprising effect because its pure enantiomers L-X18 and D-X18 were strongly hemolytic and twice as cytotoxic but showed much weaker antibacterial activities, implying that this sequence would not have been selected in a screen with pure enantiomers. This switch from an antibacterial non-hemolytic sr form to an inactive but strongly hemolytic L or D form occurred even more strongly with aX18 lacking N termini. In contrast to sr-X18, the antibacterial activity of sr-X22 remained constant or even slightly increased in the pure enantiomers L-X22 or D-X22, while hemolysis remained absent and cytotoxicity at a comparably low level. This activity pattern was also preserved upon removal of N termini to form sr-, L-, or D-aX22.
We also measured AMPD activity against HEK293 cells as an indication of toxicity, which, among previous AMPDs, was problematic for L-T7 and L- and D-T25 compared with L-G3KL. While toxicity was also quite high for dendrimers lacking N termini (IC50 = 5.8–24 μg/mL) and for L-, D-, and sr-X18 (IC50 = 47–117 μg/mL), L-, D-, and sr-X22 showed even lower toxicity (IC50 > 500 μg/mL) than L-G3KL and polymyxin B (PMB).
We performed additional profiling against MDR bacteria to compare the best AMPDs sr-X18 and L-, D-, and sr-X22 with our previous AMPDs sr-T25, L-G3KL, and L-T7 (Table 2). The dendrimers were all similarly active against Stenotrophomonas maltophilia (MIC = 4–8 μg/mL), P. aeruginosa PA14, and MDR clinical isolate ZEM-1A (MIC = 4–16 μg/mL), and all AMPDs except sr-X22 were active against Enterobacter cloacae (MIC = 8–32 μg/mL).
Table 2.
Activity of selected AMPDs against MDR bacteria
| S. maltophiliaa | P. aeruginosa PA14 | P. aeruginosa ZEM-1A | E. cloacae | K. pneumoniae OXA-48 | P. aeruginosa ZEM9A | B. cenocepacia | S. aureus Newman | |
|---|---|---|---|---|---|---|---|---|
| sr-X18 | 4 | 8 | 8 | 16 | 16 | 64 | >64 | >64 |
| sr-X22 | 4 | 4 | 4 | >64 | 32 | >64 | >64 | >64 |
| L-X22 | 4 | 4 | 4 | 4 | 8 | 32 | >64 | >64 |
| D-X22 | 4 | 4 | 4 | 8 | 4 | 8 | >64 | 64 |
| sr-T25 | 8 | 4 | 4 | 8 | >64 | 16 | >64 | >64 |
| L-G3KL | 8 | 2–4 | 4 | 8 | >64 | 16 | >64 | >64 |
| L-T7 | 4 | 8 | 4 | 8 | 16 | 8 | >64 | >64 |
| PMB | 4 | 0.5 | 0.5 | 0.5 | 1 | 64 | >64 | >64 |
MIC in μg/mL was measured in MH medium at pH 7.4 after incubation for 16–20 h at 37°C. Each result represents two independent experiments performed in duplicate.
Furthermore, all except sr-T25 and L-G3KL were active against the carbapenem-resistant K. pneumoniae strain OXA-48 (MIC = 4–32 μg/mL), with D-X22 standing out as the most active AMPD against this bacterium. Like the positive control PMB, none of these dendrimers showed significant activity against the clinical isolate P. aeruginosa ZEM9A, against Burkholderia cenocepacia, or against S. aureus Newman. Taken together, these data showed that sr-, L-, and D-X22 combined the best overall antibacterial effects with low toxicity compared with our previously best AMPDs L-G3KL, L-T7, and sr-T25. In terms of stability in human serum, degradation was acceptable for L-X22 (70% remaining at 24 h), very low for D-X22 (90% remaining at 24 h), and undetectable for sr-X22 (Figure S3).
AMPDs rapidly kill bacteria by membrane disruption
Time-kill experiments with P. aeruginosa confirmed that AMPDs sr-X18 and L-, D-, and sr-X22 rapidly killed bacteria, like the parent AMPD sr-T25 (Figures 3A and S4). Transmission electron microscopy (TEM) images showed damage to the bacterial membrane and partial emptying of cell contents (Figures 3B and S5–S12). To our surprise, both active (L-, D-, and sr-X22, sr-X18, L-, D-, and sr-T25) and inactive (L- and D-X18) dendrimers permeabilized the outer membrane of P. aeruginosa cells as measured by fluorescence assay with N-phenylnaphthylamine (NPN), a small molecule that cannot effectively cross the outer membrane but has strong fluorescence when binding to phospholipids (Figures 3C, 3F, and S13).46 The same AMPDs also depolarized the inner membrane as measured by fluorescence assay with the membrane potential sensitive dye DiSC3(5) (Figures 3D, 3G, and S14).47 However, only the active AMPDs fully permeabilized P. aeruginosa cells as measured by the uptake of propidium iodide (PI) using flow cytometry. PI is a membrane non-permeable compound that shows strong fluorescence when binding to DNA.30,48 By contrast, the inactive L-X18 and D-X18 had much lower permeabilizing effects (Figures 3E, 3H, and S15–S30).
Figure 3.

AMPDs rapidly kill bacteria by membrane disruption
(A) Bacteria-killing assay against P. aeruginosa PAO1 at 2× MIC. Data are presented in mean ± SD, n = 3. Experiments were performed ≥2 times in triplicates.
(B) TEM images of PAO1 (OD600 = 1) 2 h after treatment at 10× MIC in Müller-Hinton (MH) medium at pH 7.4. Scale bar is 200 nm. Blue arrows indicate disrupted membrane.
(C and F) NPN outer membrane permeability assay of PAO1 treated with X18 (C) and X22 (F) in the presence of 10 μM NPN. Fluorescent intensity (λexc = 340 nm, λem = 415 nm) was measured within 5 min after the treatment. Data are presented in mean ± SD, n = 3.
(D and G) DiSC3(5) inner membrane depolarization assay of PAO1 treated with X18 (D) and X22 (G) in the presence of 2 μM DiSC3(5). Fluorescence intensity (λexc = 610 nm, λem = 660 nm) was measured within 5 min after treatment. Data are presented in mean ± SD, n = 3.
(E and H) PI cell permeabilization assay with of PAO1 treated with X18 (E) and X22 (H). The percentage of PI-positive cells is indicated after incubating PAO1 (OD600 = 1) for 20 min.
By comparison, the control antibiotic PMB, which binds to lipid A of the outer bacterial membrane,49,50 permeabilized the outer membrane but did not depolarize the inner membrane. Nevertheless, PMB induced a strong uptake of PI. On the other hand, inactive PMB nonapeptide (PMBN) permeabilized the outer membrane,51,52 but had no effect on inner membranes, and showed no PI uptake (Figures 3F–3H).
Taken together, these experiments indicated that our AMPDs killed bacteria by disruption of the outer and inner membranes leading to permeabilization and partial emptying of the cell content. Most strikingly, the effects of sr-, L-, and D-X22 were indistinguishable, while in the case of X18, stereochemical purity controlled the overall permeabilization effect as measured by PI uptake, which only occurred with sr-X18.
Antibacterial dendrimer L-X22 and hemolytic dendrimer L-X18 show different folding and membrane interactions
To better understand the difference between the non-hemolytic, antibacterial L-X22 and the hemolytic, non-antibacterial L-X18, we investigated their conformational behavior. CD spectra of L-X18 (and its enantiomer D-X18) showed a transition from an unordered conformation in water to a more α-helical conformation in the presence of 20% v/v trifluoroethanol (TFE) as folding inducer or 5 mM n-dodecylphosphocholine (DPC) as a micelle-forming additive mimicking the membrane environment (Figures 4A, 4B, and S31).13,53,54 L-X22 (and its D-enantiomer) was also disordered in water and partially α-helical with TFE. However, its CD trace in the presence of DPC has a shape intermediate between the unordered trace in water and the α-helical trace with TFE, indicating a less extensive α-helical folding with DPC micelles (Figure 4B). These data showed that the antibacterial, non-hemolytic L-X22 was less prone to α-helical folding in a membrane environment than the hemolytic and non-antibacterial L-X18. We observed similar effects in the CD spectra of analogs L-/D-aX18 and L-/D-aX22 lacking the N termini (Figure S32).
Figure 4.

Antibacterial dendrimer L-X22 and hemolytic dendrimer L-X18 show different folding and membrane interactions
(A and B) CD spectra of L-/D-X18 (A) and L-/D-X22 (B) at 0.100 mg/mL in 8 mM phosphate (pH 7.4) with 20% TFE or 5 mM DPC.
(C) Time course of the distance from center of mass of dendrimers to center of mass of DPC micelle, and average radius of DPC micelle.
(D–G) MD simulation of L-X18 and L-X22 with a DPC micelle after 25 and 200 ns at 300 K. Initially, the dendrimers were placed at ∼3 nm of the preformed micelle surface and reached the micelle by passive diffusion.
We next performed MD using GROMACS55 to gain an insight into how the α-helical conformation of our AMPDs might look. We performed MD simulations starting with α-helically prefolded dendrimer models of L-X18 and L-X22 in either water, 20% TFE, or with a DPC micelle (Figures 4C–4G and S33). Both dendrimers were conformationally quite flexible under all three conditions; however, their branch comprising the α-peptide backbone of 12 residues in length extending from G0 to G3 mostly retained an α-helical conformation with 3 of the maximum possible 3.5 turns, thus showing a slightly stronger folding propensity than in our previous MD studies of AMPD L-G3KL, L-T7, and L-T25.32,33 The α-helix was less well formed in water compared with the simulation with TFE or with a DPC micelle, in line with the CD data (Figure S34).
The MD simulation with the DPC micelle was most interesting. AMPD L-X18 and L-X22 both rapidly approached the micelle surface (Figure 4C); however, the two dendrimers interacted very differently with the micelle in a manner consistent with their different folding behavior. The more α-helical L-X18 remained rather compact, sitting at the water interface, with its α-peptide backbone as a fully folded, extensively solvent exposed, α-helix spanning residues G3-G0. The α-helix formed a binding motive that quickly extracted a DPC molecule from the micelle (<20 ns) before releasing it to the solvent (>165 ns) (Figures 4D and 4E). The binding mode involved lateral hydrophobic contacts between the DPC lipid tail and the Leu side chains in G0, G1, and G2, combined with hydrogen bonding and electrostatic interactions involving the phosphocholine head group and the N-terminal Lys residue in G3.
On the other hand, AMPD L-X22, which was less α-helical than L-X18 as seen by CD, became flattened at the micelle surface and sank below the micelle surface (Figure 4C), with its α-helix directly in contact with the DPC micelle, engaging in multiple hydrophobic and H-bonding interactions over an extended patch of the micelle surface (Figures 4F and 4G). Despite these extensive contacts, the interaction between L-X22 and the micelle was quite stable and did not result in the extraction of a DPC molecule as seen with L-X18.
Why does α-helical folding favor hemolysis and why does conformational disorder favor antibacterial effects?
The switch from the hemolytic, non-antibacterial L- and D-X18 to a non-hemolytic, antibacterial sr-X18 must be triggered by a conformational effect. Our MD studies indicate α-helical folding along the α-peptide backbone of L-X18 combining a strongly hydrophobic dendrimer core (KLL)2KLLL with cationic outer branches (KL)8(KLK)4. This α-helix seems capable of extracting a lipid from the zwitterionic DPC micelles, which could be interpreted as a model for the destabilization of eukaryotic membranes (hemolysis). The DPC extraction in the MD with L-X18 might represent a general model for hemolytic AMPs presenting hydrophobic patches on their surface.56
The more generally accepted hypothesis to explain hemolysis involves aggregation of α-helically folded AMPs at the membrane surface leading to pore formation.9 This effect has been observed by atomic force microscopy on supported lipid bilayers and did not occur with peptide mixtures containing both L and D residues.57 Similarly, the exposed hydrophobic patch formed by the α-helix in L-X18 could trigger aggregation, which would be blocked in sr-X18. We previously observed that peptide dendrimer aggregation can be controlled by stereochemistry with siRNA transfection dendrimers.58 Although we did not detect any aggregation of L-, D-, or sr-X18 using Nile red,59 the close analogs aX18 lacking N termini, which similarly switched from non-hemolytic in sr form to hemolytic in L or D form, indeed showed aggregation as pure enantiomers but not in sr form (Figures 5A, 5B, and S35). In addition, TEM images of L- and D-aX18 showed filamentous aggregates, which were not observed with sr-aX18 solutions (Figures 5C and S36).
Figure 5.

Aggregation of peptide dendrimers
(A and B) AMPD sr-, L-, and D-aX18 (A) and sr-, L-, and D-aX18 (B) in PBS (pH 7.4) in presence of 0.2 μM Nile red. Fluorescence measured at λex = 540 nm, λem = 615 nm. Data are presented in mean ± SD, n = 3. Experiments were performed ≥2 times in triplicates.
(C) TEM images of AMPD (10 mg/mL) in PBS deposited on glow-discharged 400 mesh copper grids, dried, and stained by uranyl acetate. Scale bars are 100 nm.
The above models linking hydrophobic patches to hemolysis, either by enabling the extraction of lipid molecules from the membrane or via aggregation and pore formation, do not account for the loss of antibacterial effects when converting sr-X18 to pure L- or D-enantiomers or for the antibacterial activity of the non-hemolytic L-, D- and sr-X22. The above mentioned study with L-/D-peptide mixtures57 proposes that bacterial membrane destabilization occurs by the carpet model, i.e., homogeneous spreading on the membrane surface, a generally accepted mechanism for AMPs.9 This mechanism would be accessible to sr-AMPD in similar manner to L-/D-peptide mixtures but also to the homochiral L- or D-X22, which would act independently of folding in an intrinsically disordered bioactive conformation. The more extensive membrane coverage and deeper insertion predicted by MD for L-X22 could contribute to a better membrane perturbation via the carpet model and thus its higher antimicrobial activity compared with L-X18 while remaining unaffected by stereorandomization. The MD study showing that L-X18 folds but interacts less extensively with the DPC micelle than L-X22 might indicate that L-X18 cannot homogeneously coat the membrane, explaining its lack of antibacterial activity by the carpet model. On the other hand, the absence of a hydrophobic dendrimer core in sr-X22 probably prevents it from becoming hemolytic in the homochiral form L-X22.
To conclude, we explored the possibility of discovering an intrinsically disordered AMPD by investigating a library of stereorandomized analogs of AMPD sr-T25 selected by virtual screening using a fingerprint approach that does not take stereochemistry into account. HPLC and high-resolution mass spectrometry (HRMS) spectra are available in Figures S37–S190. Many of the sr-dendrimers were strongly antibacterial and non-hemolytic, confirming the favorable effect of stereorandomization on the therapeutic index previously discovered with sr-T25.
Investigating two of the most active AMPDs, sr-X18 and sr-X22, revealed unexpected effects. In the first case, sr-X18 lost its antibacterial activity while becoming strongly hemolytic and cytotoxic as pure enantiomers L- or D-X18, probably because α-helical folding of the α-peptide backbone of the dendrimer enabled a lipid extraction as observed by MD or aggregation at the membrane surface. This hypothesis was supported by observing aggregates with the close and similarly active analogs L- and D-aX18. In the second case, L- and D-X22 were similarly antibacterial and non-hemolytic as sr-X22. Although CD spectra of L- and D-X22 show partial α-helical content in a membrane-like environment, the similar activity of sr-, L-, and D-X22 suggests that their bioactive antibacterial conformation is intrinsically disordered and probably favors the carpet model of membrane disruption as suggested by MD simulations with DPC micelles.
Screening stereorandomized libraries might be generally useful to identify homochiral peptides possessing an intrinsically disordered bioactive conformation or to optimize disordered peptides. Note that stereorandomization is distinct from using achiral amino acids, which may stabilize helical conformations (α-amino-isobutyric acid) or not (glycine).20 Our proof of concept focused on AMPD sr-T25 because folding was known to be unnecessary for its antibacterial effects and the cause of its toxicity. The approach might be further applicable to optimize the sequence of intrinsically disordered linear peptides, for example the AMP indolicidin,60,61,62,63 or Leu-Lys oligomers known to tolerate mixed chirality arrangements for membrane disruption.13,64,65,66 The discovery of homochiral, intrinsically disordered AMPDs such as L- and D-X22 matching the high activity and low toxicity of the parent sr-AMPD overcomes the intrinsic difficulty of characterizing the billions of stereoisomers composing sr-sequences (68,719,476,736 in the case of sr-X22) and therefore opens the way for further development of these compounds.
Experimental procedures
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Prof. Dr. Jean-Louis Reymond (jean-louis.reymond@unibe.ch).
Materials availability
All materials generated in this study are available from the lead contact.
Acknowledgments
This work was supported by the Swiss National Science Foundation (200020_178998) and the European Research Council (885076). Electron microscopies were performed by the University of Bern Microscopy Imaging Center.
Author contributions
X.C. performed the peptide synthesis and biotests and wrote the paper. M.O. performed the visualization of the virtual library and wrote the paper. A.C. built the virtual library, performed the nearest neighbour searching, and wrote the paper. S.J. performed MD simulations and wrote the paper. T.K. and C.v.D. supervised antibacterial assays and wrote the paper. J.-L.R. supervised the whole study and wrote the paper.
Declaration of interests
The authors declare no conflict of interest.
Inclusion and diversity
One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community.
Published: December 1, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrp.2022.101161.
Supplemental information
Data and code availability
All data mentioned in this study will be made available upon request from the research community. The source code and dataset used for this study are available at GitHub: https://github.com/reymond-group/T25_analogs.
References
- 1.Babu M.M., van der Lee R., de Groot N.S., Gsponer J. Intrinsically disordered proteins: regulation and disease. Curr. Opin. Struct. Biol. 2011;21:432–440. doi: 10.1016/j.sbi.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 2.Baul U., Chakraborty D., Mugnai M.L., Straub J.E., Thirumalai D. Sequence effects on size, shape, and structural heterogeneity in intrinsically disordered proteins. J. Phys. Chem. B. 2019;123:3462–3474. doi: 10.1021/acs.jpcb.9b02575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kulkarni P., Bhattacharya S., Achuthan S., Behal A., Jolly M.K., Kotnala S., Mohanty A., Rangarajan G., Salgia R., Uversky V. Intrinsically disordered proteins: critical components of the wetware. Chem. Rev. 2022;122:6614–6633. doi: 10.1021/acs.chemrev.1c00848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mojsoska B., Jenssen H. Peptides and peptidomimetics for antimicrobial drug design. Pharmaceuticals. 2015;8:366–415. doi: 10.3390/ph8030366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magana M., Pushpanathan M., Santos A.L., Leanse L., Fernandez M., Ioannidis A., Giulianotti M.A., Apidianakis Y., Bradfute S., Ferguson A.L., et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020;20:e216–e230. doi: 10.1016/S1473-3099(20)30327-3. [DOI] [PubMed] [Google Scholar]
- 6.Mookherjee N., Anderson M.A., Haagsman H.P., Davidson D.J. Antimicrobial host defence peptides: functions and clinical potential. Nat. Rev. Drug Discov. 2020;19:311–332. doi: 10.1038/s41573-019-0058-8. [DOI] [PubMed] [Google Scholar]
- 7.De Oliveira D.M.P., Forde B.M., Kidd T.J., Harris P.N.A., Schembri M.A., Beatson S.A., Paterson D.L., Walker M.J. Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 2020;33:001811-19. doi: 10.1128/CMR.00181-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murray C.J., Ikuta K.S., Sharara F., Swetschinski L., Robles Aguilar G., Gray A., Han C., Bisignano C., Rao P., Wool E., et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet. 2022;399:629–655. doi: 10.1016/S0140-6736(21)02724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen L.T., Haney E.F., Vogel H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011;29:464–472. doi: 10.1016/j.tibtech.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Fluxà V.S., Maillard N., Page M.G.P., Reymond J.L. Bead diffusion assay for discovering antimicrobial cyclic peptides. Chem. Commun. 2011;47:1434–1436. doi: 10.1039/c0cc04670a. [DOI] [PubMed] [Google Scholar]
- 11.Michaud G., Visini R., Bergmann M., Salerno G., Bosco R., Gillon E., Richichi B., Nativi C., Imberty A., Stocker A., et al. Overcoming antibiotic resistance in Pseudomonas aeruginosa biofilms using glycopeptide dendrimers. Chem. Sci. 2016;7:166–182. doi: 10.1039/C5SC03635F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He R., Di Bonaventura I., Visini R., Gan B.-H., Fu Y., Probst D., Lüscher A., Köhler T., van Delden C., Stocker A., et al. Design, crystal structure and atomic force microscopy study of thioether ligated d, l-cyclic antimicrobial peptides against multidrug resistant Pseudomonas aeruginosa. Chem. Sci. 2017;8:7464–7475. doi: 10.1039/c7sc01599b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baeriswyl S., Personne H., Di Bonaventura I., Köhler T., van Delden C., Stocker A., Javor S., Reymond J.-L. A mixed chirality α-helix in a stapled bicyclic and a linear antimicrobial peptide revealed by X-ray crystallography. RSC Chem. Biol. 2021;2:1608–1617. doi: 10.1039/D1CB00124H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stach M., Siriwardena T.N., Köhler T., van Delden C., Darbre T., Reymond J.L. Combining topology and sequence design for the discovery of potent antimicrobial peptide dendrimers against multidrug-resistant Pseudomonas aeruginosa. Angew. Chem., Int. Ed. Engl. 2014;53:12827–12831. doi: 10.1002/anie.201409270. [DOI] [PubMed] [Google Scholar]
- 15.Reymond J.-L. Peptide dendrimers: from enzyme models to antimicrobials and transfection reagents. Chimia. 2021;75:535–538. doi: 10.2533/chimia.2021.535. [DOI] [PubMed] [Google Scholar]
- 16.Gan B.-H., Siriwardena T.N., Javor S., Darbre T., Reymond J.-L. Fluorescence imaging of bacterial killing by antimicrobial peptide dendrimer G3KL. ACS Infect. Dis. 2019;5:2164–2173. doi: 10.1021/acsinfecdis.9b00299. [DOI] [PubMed] [Google Scholar]
- 17.Gan B.H., Gaynord J., Rowe S.M., Deingruber T., Spring D.R. The multifaceted nature of antimicrobial peptides: current synthetic chemistry approaches and future directions. Chem. Soc. Rev. 2021;50:7820–7880. doi: 10.1039/D0CS00729C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ergene C., Yasuhara K., Palermo E.F. Biomimetic antimicrobial polymers: recent advances in molecular design. Polym. Chem. 2018;9:2407–2427. doi: 10.1039/C8PY00012C. [DOI] [Google Scholar]
- 19.Molchanova N., Hansen P.R., Franzyk H. Advances in development of antimicrobial peptidomimetics as potential drugs. Molecules. 2017;22:1430. doi: 10.3390/molecules22091430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yokoo H., Hirano M., Misawa T., Demizu Y. Helical antimicrobial peptide foldamers containing non-proteinogenic amino acids. ChemMedChem. 2021;16:1226–1233. doi: 10.1002/cmdc.202000940. [DOI] [PubMed] [Google Scholar]
- 21.Lee C.C., MacKay J.A., Fréchet J.M.J., Szoka F.C. Designing dendrimers for biological applications. Nat. Biotechnol. 2005;23:1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]
- 22.Crespo L., Sanclimens G., Pons M., Giralt E., Royo M., Albericio F. Peptide and amide bond-containing dendrimers. Chem. Rev. 2005;105:1663–1681. doi: 10.1021/cr030449l. [DOI] [PubMed] [Google Scholar]
- 23.Sapra R., Verma R.P., Maurya G.P., Dhawan S., Babu J., Haridas V. Designer peptide and protein dendrimers: a cross-sectional analysis. Chem. Rev. 2019;119:11391–11441. doi: 10.1021/acs.chemrev.9b00153. [DOI] [PubMed] [Google Scholar]
- 24.Dhumal D., Maron B., Malach E., Lyu Z., Ding L., Marson D., Laurini E., Tintaru A., Ralahy B., Giorgio S., et al. Dynamic self-assembling supramolecular dendrimer nanosystems as potent antibacterial candidates against drug-resistant bacteria and biofilms. Nanoscale. 2022;14:9286–9296. doi: 10.1039/D2NR02305A. [DOI] [PubMed] [Google Scholar]
- 25.Pires J., Siriwardena T.N., Stach M., Tinguely R., Kasraian S., Luzzaro F., Leib S.L., Darbre T., Reymond J.L., Endimiani A. In vitro activity of the novel antimicrobial peptide dendrimer G3KL against multidrug-resistant acinetobacter baumannii and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2015;59:7915–7918. doi: 10.1128/aac.01853-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ben Jeddou F., Falconnet L., Luscher A., Siriwardena T., Reymond J.-L., van Delden C., Köhler T. Adaptive and mutational responses to peptide dendrimer antimicrobials in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2020;64 doi: 10.1128/AAC.02040-19. e02040020400-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai X., Javor S., Gan B.H., Köhler T., Reymond J.-L. The antibacterial activity of peptide dendrimers and polymyxin B increases sharply above pH 7.4. Chem. Commun. 2021;57:5654–5657. doi: 10.1039/D1CC01838H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han X., Liu Y., Ma Y., Zhang M., He Z., Siriwardena T.N., Xu H., Bai Y., Zhang X., Reymond J.-L., Qiao M. Peptide dendrimers G3KL and TNS18 inhibit Pseudomonas aeruginosa biofilms. Appl. Microbiol. Biotechnol. 2019;103:5821–5830. doi: 10.1007/s00253-019-09801-3. [DOI] [PubMed] [Google Scholar]
- 29.Abdel-Sayed P., Kaeppeli A., Kaeppli A., Siriwardena T., Darbre T., Perron K., Jafari P., Reymond J.L., Pioletti D.P., Applegate L.A. Anti-Microbial dendrimers against multidrug-resistant P. aeruginosa enhance the angiogenic effect of biological burn-wound bandages. Sci. Rep. 2016;6:22020–22110. doi: 10.1038/srep22020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gan B.-H., Cai X., Javor S., Köhler T., Reymond J.-L. Synergistic effect of propidium iodide and small molecule antibiotics with the antimicrobial peptide dendrimer G3KL against gram-negative bacteria. Molecules. 2020;25:5643. doi: 10.3390/molecules25235643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patrulea V., Gan B.-H., Perron K., Cai X., Abdel-Sayed P., Sublet E., Ducret V., Nerhot N.P., Applegate L.A., Borchard G., et al. Synergistic effects of antimicrobial peptide dendrimer-chitosan polymer conjugates against Pseudomonas aeruginosa. Carbohydr. Polym. 2022;280:119025. doi: 10.1016/j.carbpol.2021.119025. [DOI] [PubMed] [Google Scholar]
- 32.Siriwardena T.N., Capecchi A., Gan B.H., Jin X., He R., Wei D., Ma L., Köhler T., van Delden C., Javor S., Reymond J.L. Optimizing antimicrobial peptide dendrimers in chemical space. Angew. Chem., Int. Ed. Engl. 2018;57:8483–8487. doi: 10.1002/anie.201802837. [DOI] [PubMed] [Google Scholar]
- 33.Siriwardena T.N., Gan B.-H., Köhler T., van Delden C., Javor S., Reymond J.-L. Stereorandomization as a method to probe peptide bioactivity. ACS Cent. Sci. 2021;7:126–134. doi: 10.1021/acscentsci.0c01135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Venkatraman V., Pérez-Nueno V.I., Mavridis L., Ritchie D.W. Comprehensive comparison of ligand-based virtual screening tools against the DUD data set reveals limitations of current 3D methods. J. Chem. Inf. Model. 2010;50:2079–2093. doi: 10.1021/ci100263p. [DOI] [PubMed] [Google Scholar]
- 35.Geppert H., Vogt M., Bajorath J. Current trends in ligand-based virtual screening: molecular representations, data mining methods, new application areas, and performance evaluation. J. Chem. Inf. Model. 2010;50:205–216. doi: 10.1021/ci900419k. [DOI] [PubMed] [Google Scholar]
- 36.Capecchi A., Awale M., Probst D., Reymond J.L. PubChem and ChEMBL beyond lipinski. Mol. Inform. 2019;38:1900016. doi: 10.1002/minf.201900016. [DOI] [PubMed] [Google Scholar]
- 37.Carhart R.E., Smith D.H., Venkataraghavan R. Atom pairs as molecular features in structure-activity studies: definition and applications. J. Chem. Inf. Comput. Sci. 1985;25:64–73. doi: 10.1021/ci00046a002. [DOI] [Google Scholar]
- 38.Schneider G., Neidhart W., Giller T., Schmid G. Scaffold-hopping” by topological pharmacophore search: a contribution to virtual screening. Angew. Chem., Int. Ed. Engl. 1999;38:2894–2896. [PubMed] [Google Scholar]
- 39.Awale M., Reymond J.L. Atom pair 2D-fingerprints perceive 3D-molecular shape and pharmacophores for very fast virtual screening of ZINC and GDB-17. J. Chem. Inf. Model. 2014;54:1892–1907. doi: 10.1021/ci500232g. [DOI] [PubMed] [Google Scholar]
- 40.Capecchi A., Probst D., Reymond J.-L. One molecular fingerprint to rule them all: drugs, biomolecules, and the metabolome. J. Cheminf. 2020;12:43. doi: 10.1186/s13321-020-00445-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Di Bonaventura I., Jin X., Visini R., Probst D., Javor S., Gan B.H., Michaud G., Natalello A., Doglia S.M., Köhler T., et al. Chemical space guided discovery of antimicrobial bridged bicyclic peptides against Pseudomonas aeruginosa and its biofilms. Chem. Sci. 2017;8:6784–6798. doi: 10.1039/c7sc01314k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Bonaventura I., Baeriswyl S., Capecchi A., Gan B.-H., Jin X., Siriwardena T.N., He R., Köhler T., Pompilio A., Di Bonaventura G., et al. An antimicrobial bicyclic peptide from chemical space against multidrug resistant gram-negative bacteria. Chem. Commun. 2018;54:5130–5133. doi: 10.1039/c8cc02412j. [DOI] [PubMed] [Google Scholar]
- 43.Al Khalifa A., Haranczyk M., Holliday J. Comparison of nonbinary similarity coefficients for similarity searching, clustering and compound selection. J. Chem. Inf. Model. 2009;49:1193–1201. doi: 10.1021/ci8004644. [DOI] [PubMed] [Google Scholar]
- 44.Jones E.M., Cochrane C.A., Percival S.L. The effect of pH on the extracellular matrix and biofilms. Adv. Wound Care. 2015;4:431–439. doi: 10.1089/wound.2014.0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koo H., Allan R.N., Howlin R.P., Stoodley P., Hall-Stoodley L. Targeting microbial biofilms: current and prospective therapeutic strategies. Nat. Rev. Microbiol. 2017;15:740–755. doi: 10.1038/nrmicro.2017.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Helander I.M., Mattila-Sandholm T. Fluorometric assessment of Gram-negative bacterial permeabilization. J. Appl. Microbiol. 2000;88:213–219. doi: 10.1046/j.1365-2672.2000.00971.x. [DOI] [PubMed] [Google Scholar]
- 47.Te Winkel J.D., Gray D.A., Seistrup K.H., Hamoen L.W., Strahl H. Analysis of antimicrobial-triggered membrane depolarization using voltage sensitive dyes. Front. Cell Dev. Biol. 2016;4:29. doi: 10.3389/fcell.2016.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Netuschil L., Auschill T.M., Sculean A., Arweiler N.B. Confusion over live/dead stainings for the detection of vital microorganisms in oral biofilms - which stain is suitable? BMC Oral Health. 2014;14:2–14. doi: 10.1186/1472-6831-14-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deris Z.Z., Swarbrick J.D., Roberts K.D., Azad M.A.K., Akter J., Horne A.S., Nation R.L., Rogers K.L., Thompson P.E., Velkov T., Li J. Probing the penetration of antimicrobial polymyxin lipopeptides into gram-negative bacteria. Bioconjugate Chem. 2014;25:750–760. doi: 10.1021/bc500094d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Velkov T., Thompson P.E., Nation R.L., Li J. Structure-activity relationships of polymyxin antibiotics. J. Med. Chem. 2010;53:1898–1916. doi: 10.1021/jm900999h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vaara M. Agents that increase the permeability of the outer membrane. Microbiol. Rev. 1992;56:395–411. doi: 10.1128/mr.56.3.395-411.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pi H., Nguyen H.T., Venter H., Boileau A.R., Woolford L., Garg S., Page S.W., Russell C.C., Baker J.R., McCluskey A., et al. In vitro activity of robenidine analog NCL195 in combination with outer membrane permeabilizers against gram-negative bacterial pathogens and impact on systemic gram-positive bacterial infection in mice. Front. Microbiol. 2020;11:1556. doi: 10.3389/fmicb.2020.01556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jasanoff A., Fersht A.R. Quantitative determination of helical propensities from trifluoroethanol titration curves. Biochemistry. 1994;33:2129–2135. doi: 10.1021/bi00174a020. [DOI] [PubMed] [Google Scholar]
- 54.Arunkumar A.I., Kumar T.K., Yu C. Specificity of helix-induction by 2, 2, 2-trifluoroethanol in polypeptides. Int. J. Biol. Macromol. 1997;21:223–230. doi: 10.1016/s0141-8130(97)00064-0. [DOI] [PubMed] [Google Scholar]
- 55.Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. Software. 2015;1-2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 56.Mourtada R., Herce H.D., Yin D.J., Moroco J.A., Wales T.E., Engen J.R., Walensky L.D. Design of stapled antimicrobial peptides that are stable, nontoxic and kill antibiotic-resistant bacteria in mice. Nat. Biotechnol. 2019;37:1186–1197. doi: 10.1038/s41587-019-0222-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hayouka Z., Bella A., Stern T., Ray S., Jiang H., Grovenor C.R.M., Ryadnov M.G. Binary encoding of random peptide sequences for selective and differential antimicrobial mechanisms. Angew. Chem., Int. Ed. Engl. 2017;56:8099–8103. doi: 10.1002/anie.201702313. [DOI] [PubMed] [Google Scholar]
- 58.Heitz M., Javor S., Darbre T., Reymond J.-L. Stereoselective pH responsive peptide dendrimers for siRNA transfection. Bioconjugate Chem. 2019;30:2165–2182. doi: 10.1021/acs.bioconjchem.9b00403. [DOI] [PubMed] [Google Scholar]
- 59.Mynar J.L., Goodwin A.P., Cohen J.A., Ma Y., Fleming G.R., Fréchet J.M.J. Two-photon degradable supramolecular assemblies of linear-dendritic copolymers. Chem. Commun. 2007:2081–2082. doi: 10.1039/B701681F. [DOI] [PubMed] [Google Scholar]
- 60.Selsted M.E., Novotny M.J., Morris W.L., Tang Y.Q., Smith W., Cullor J.S. Indolicidin, a novel bactericidal tridecapeptide amide from neutrophils. J. Biol. Chem. 1992;267:4292–4295. [PubMed] [Google Scholar]
- 61.Falla T.J., Karunaratne D.N., Hancock R.E. Mode of action of the antimicrobial peptide indolicidin. J. Biol. Chem. 1996;271:19298–19303. doi: 10.1074/jbc.271.32.19298. [DOI] [PubMed] [Google Scholar]
- 62.Sitaram N., Subbalakshmi C., Nagaraj R. Indolicidin, a 13-residue basic antimicrobial peptide rich in tryptophan and proline, interacts with Ca2+-calmodulin. Biochem. Biophys. Res. Commun. 2003;309:879–884. doi: 10.1016/j.bbrc.2003.08.095. [DOI] [PubMed] [Google Scholar]
- 63.Hsu C.-H., Chen C., Jou M.-L., Lee A.Y.-L., Lin Y.-C., Yu Y.-P., Huang W.-T., Wu S.-H. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005;33:4053–4064. doi: 10.1093/nar/gki725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shai Y., Oren Z. Diastereomers of cytolysins, a novel class of potent antibacterial peptides (∗) J. Biol. Chem. 1996;271:7305–7308. doi: 10.1074/jbc.271.13.7305. [DOI] [PubMed] [Google Scholar]
- 65.Pag U., Oedenkoven M., Papo N., Oren Z., Shai Y., Sahl H.-G. In vitro activity and mode of action of diastereomeric antimicrobial peptides against bacterial clinical isolates. J. Antimicrob. Chemother. 2004;53:230–239. doi: 10.1093/jac/dkh083. [DOI] [PubMed] [Google Scholar]
- 66.Ben Hur D., Kapach G., Wani N.A., Kiper E., Ashkenazi M., Smollan G., Keller N., Efrati O., Shai Y. Antimicrobial peptides against multidrug-resistant Pseudomonas aeruginosa biofilm from cystic fibrosis patients. J. Med. Chem. 2022;65:9050–9062. doi: 10.1021/acs.jmedchem.2c00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data mentioned in this study will be made available upon request from the research community. The source code and dataset used for this study are available at GitHub: https://github.com/reymond-group/T25_analogs.