

Abstract
Background:
Reactive oxygen species (ROS) contribute to platelet hyperactivation during aging. Several oxidative pathways and antioxidant enzymes have been implicated, however, their mechanistic contributions during aging remain elusive. We hypothesized that mitochondria are an important source of platelet ROS, and that mitochondrial superoxide dismutase (SOD2) protects against mitochondrial ROS-driven platelet activation and thrombosis during aging.
Methods:
We studied littermates of platelet specific SOD2-knockout (SOD2fl/flPf4Cre, pSOD2-KO) and control (SOD2fl/fl) mice at young (4–5 months) or old (18–20 months) ages. We examined agonist induced platelet activation, platelet-dependent thrombin generation potential and susceptibility to in vivo thrombosis.
Results:
Platelet αIIbβ3 activation, aggregation, and adhesion were increased to similar extents in aged mice of both genotypes compared with young mice. In contrast, the age-dependent increases in mitochondrial and total cellular ROS, calcium elevation, and phosphatidylserine exposure were augmented in platelets from pSOD2-KO mice compared with control mice. Aged pSOD2-KO mice showed increased platelet-dependent thrombin generation compared with aged control mice. In vivo, aged pSOD2-KO mice exhibited enhanced susceptibility to carotid artery and pulmonary thrombosis compared to aged control mice. Adoptive transfer of platelets from aged pSOD2-KO but not aged control mice increased thrombotic susceptibility in aged host mice, suggesting a prothrombotic effect of platelet pSOD2 deficiency. Treatment with avasopasem manganese (GC4419), a SOD mimetic, decreased platelet mitochondrial pro-oxidants, cellular ROS levels, and inhibited procoagulant platelet formation and arterial thrombosis in aged mice.
Conclusions:
Platelet mitochondrial ROS contributes to age-related thrombosis and endogenous SOD2 protects from platelet-dependent thrombin generation and thrombosis during aging.
Introduction
Aging is an established risk factor for thromboembolic events such as myocardial infarction and stroke.1 Recent studies have reported hyperactivation of platelets as an important mechanism of aging related thrombosis,2–4 however the molecular pathways that regulate platelet activation and thrombosis remain elusive.
Elevated reactive oxygen species (ROS) are important mediators of physiological platelet activation5,6 and age-induced activation of pro-oxidant pathways intrinsic to platelets could potentially contribute to platelet hyperactivity. Recent studies have described diverse sources of platelet ROS, including NADPH oxidase7 cyclooxygenase8 and xanthine oxidase,9 but controversy exists for their precise role particularly for the NADPH oxidase Nox2.10–12 Moreover, mechanistic contributions for most pro-oxidant pathways in platelet activation during aging remain to be determined. Mitochondria are an important source of ROS that contribute to excessive platelet activation in pathological conditions such as diabetes and sickle cell disease.13–15 Increased platelet mitochondrial respiration has also been observed during aging,3,16 suggesting a potential link between mitochondrial ROS production and platelet hyperactivation.
Mitochondrial redox homeostasis is regulated by antioxidant enzymes such as superoxide dismutase 2 (SOD2), a major mitochondrial antioxidant enzyme that converts superoxide to H2O2. We previously showed that overexpression of glutathione peroxidase 1 (Gpx1) that reduces H2O2 to water, protects from platelet activation and thrombosis in aged mice, but the mechanism of increased ROS generation was not identified.2 An association of platelet activation in aged humans (up to 80 yrs. of age) with markedly decreased activity of several antioxidant enzymes including SOD activity within platelets was recently described.17 Our own group previously reported that platelet SOD2 is dispensable for platelet function in young mice, suggesting that low basal production of mitochondrial superoxide within platelets at young age is insufficient to induce pathologic changes even with concomitant deficiency of SOD2. However, given that mitochondrial dysfunction3,16 and overproduction of ROS within platelets2 occur during aging, SOD2 may have a mechanistic role in modulating levels of superoxide or its ROS derivatives, thereby regulating platelet activation and thrombosis during aging. We tested the hypothesis that deficiency of intra-platelet SOD2 increases mitochondrial/cellular ROS and exacerbates platelet activation and thrombosis during aging. Using mice with platelet-specific deletion of SOD2 and complementary pharmacological and adoptive platelet transfer approaches, we report that platelet SOD2 is dispensable for age-induced increases in platelet aggregation and adhesion but is an important regulator of platelet ROS, Ca2+ elevation, phosphatidylserine exposure, thrombin generation and in vivo thrombosis during aging.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Mice
All animal studies were approved by the institutional animal care and use committees at the University of Iowa. Platelet specific SOD2-deficient mice used were generated as previously described.18 Mice harboring homozygous SOD2 floxed alleles were bred to mice expressing a Pf4 driven cre recombinase (Pf4 cre) from JAX. The Pf4 cre mouse may harbor alleles from C57BL6/N and C57BL6/J. Littermates containing floxed SOD2 (SOD2fl/fl i.e., control mice) or Pf4Cre floxed SOD2 (SOD2fl/flPf4Cre i.e., pSOD2-KO mice) were studied. We also studied transgenic mice expressing a chimera of human IL4Rα and mutated murine GP1bα in a platelet specific manner (hIL4Rα/GP1bα Tg mice) and these chimeric mice were generated on a C57BL6/J background.19 TheC57BL6/J mice were obtained from Jackson laboratory. All three mouse colonies were bred and maintained in the animal facility at the University of Iowa and fed a NIH-31 Modified Open Formula diet (7013, Teklad). Some mice were treated with 10 mg/Kg of avasopasem (GC4419),20 a superoxide dismutase mimetic or vehicle control (Na-bicarbonate buffer) for 2 weeks (IP injection once per day). All experiments were conducted in mice between the ages of 4–5 months (young cohort) or between 18–20 months (aged cohort). Both male and female mice were included in the study design, but we did not stratify groups by sex since the primary goal of the study was not to test the sex-specific effects. Therefore, sex difference and interaction were not determined in the study and the data for males and females in each group were combined for analysis.
The reported numbers within each group refer to biological replicates. In vivo studies were performed according to the current Animal Research: Reporting of In Vivo Experiment guidelines (https://arriveguidelines.org/).
Measurement of intra-platelet calcium
Intra-platelet calcium levels (mobilization and uptake) were measured in washed platelets using a previously described method with minor modifications.21 Briefly, washed platelets (2 ×108/mL) were incubated with 4.5 μM Fluo-4 for 30 min in the dark at 37 °C, washed and resuspended in Tyrode buffer containing 1 mM CaCl2 and a baseline signal was acquired using a BD Accuri C6 flow cytometer for 45 seconds, followed by addition of thrombin (0.02 U/mL) to activate platelets and the fluorescent signals were acquired continually up to 240 seconds. The cumulative signals over time were compared between the groups.
Experimental thrombosis
We used two methods to measure susceptibility to thrombosis in vivo. Carotid artery thrombosis was induced by photochemical injury as described previously.10,22 Briefly, the right common carotid artery was transilluminated with a 1.5-mV, 540 nm green laser (Melles Griot). Rose Bengal (25 mg/kg) was injected via a femoral vein catheter. Carotid artery blood flow was monitored with a Doppler flow probe for up to 90 min after injury or until a stable occlusion developed. Stable occlusion was defined as the time at which blood flow remained absent for ≥ 10 min. To assess the susceptibility to pulmonary thrombosis, we used a previously described method with minor modifications.23 Mice were infused with 0.5 μg/g collagen (Chronolog) retro-orbitally which induces platelet activation-dependent pulmonary thrombosis leading to death. After infusion of collagen, mice were monitored for up to 10 minutes for symptoms of thrombotic shock, including bradycardia, irregular breathing, and death (defined as the absence of a heartbeat for greater than 60 seconds). If death did not occur within 10 minutes, the experiment was terminated at that time.
Statistical analysis
GraphPad Prism software, version 8 was used for statistical analysis. For sample size more than 5 per group, a normality testing was performed using Shapiro-Wilk test and homogeneity was tested using Bartletts test. After confirming homogeneous variances and normality, statistical significance was assessed for means using 2-sided unpaired Student t test for 2-group comparisons, and multi-group comparisons were performed using either one-way analysis of variance (ANOVA), two-way ANOVA or two-way ANOVA with repeated measure followed by Tukey’s multiple comparisons test. For the data set not showing normal distribution, or if the sample size per group was 5 or less, Kruskal-Wallis test with Dunn’s post hoc test for multiple group comparisons was performed. Survival curves were analyzed using Log-rank (Mantel-Cox) test. Statistical significance was defined as P < 0.05. Values are reported as mean ± SE for data showing normal distribution and as median with 95% CI for those not showing normal distribution. Specific details on how many mice were included in an experiment are given in the corresponding figure legends.
Detailed information on the methods for complete blood count, platelet preparation, flowcytometric analysis for platelet ROS measurements and activation, platelet aggregation and secretion, spreading, clot retraction, platelet-dependent thrombin generation, adoptive platelet transfer, real time PCR and immunoblotting is available in the supplemental Materials and methods. Please also see the Major Resources Table in the Supplemental Materials.
Results
Baseline Hematology:
No alterations in hemoglobin, hematocrit, white blood cell count, or platelet count were observed between control and pSOD2-KO mice (Table S1). Aging did not alter levels of hemoglobin, hematocrit, or white blood cell count, but aged mice of both genotypes exhibited significantly increased platelet count compared to young mice, consistent with a previous report.2
Platelet SOD2 mRNA and protein levels, and SOD2 activity
Levels of SOD2 mRNA and protein, and SOD2 activity were quantified in bead purified platelets. The mRNA was increased in aged control mice compared to young control mice (P < 0.01, Supplementary Figure S1A). Young or aged pSOD2-KO mice showed negligible expression of Sod2 mRNA compared to age-matched control mice (P < 0.0001). Platelet SOD2 protein levels did not increase during aging in control mice (P = 0.2 vs young control), and minimal expression of SOD2 was detected in young or aged pSOD2-KO mice (Supplementary Figure S1B & S1C). Similarly, SOD2 activity was significantly less in both young and aged pSOD2-KO mice (P < 0.0001 vs control mice, Supplementary Figure S1D), and only a modest decrease was observed in aged control mice (P = 0.07 vs young control mice). There was modest heterogeneity in the protein expression and activity in aged control mice.
Platelet SOD2-deficiency increases mitochondrial pro-oxidants and total cellular ROS in aged mice
Three approaches were used to measure intra-platelet pro-oxidant levels. Mitochondria-derived pro-oxidants in platelets were measured using MitoSox, a mitochondria-targeted oxidation sensitive probe derived from dihydroethidium (DHE). The fluorescent signals were measured under resting conditions and following activation with increasing doses of thrombin and convulxin in combination (Figure 1A & Supplementary Figure S2A - S2D). Fluorescent signal increased across the groups with increasing doses of thrombin and convulxin, however, only at the highest doses of agonists, did both aged control and aged pSOD2-KO mice exhibit significantly increased fluorescence relative to younger mice of the respective genotype (P < 0.01). Importantly, aged pSOD2-KO mice had significantly higher fluorescent signal relative to aged control mice (P < 0.05).
Figure 1. Platelet SOD2 deficiency increases mitochondrial oxidants as well as total cellular ROS within platelets in aged mice.
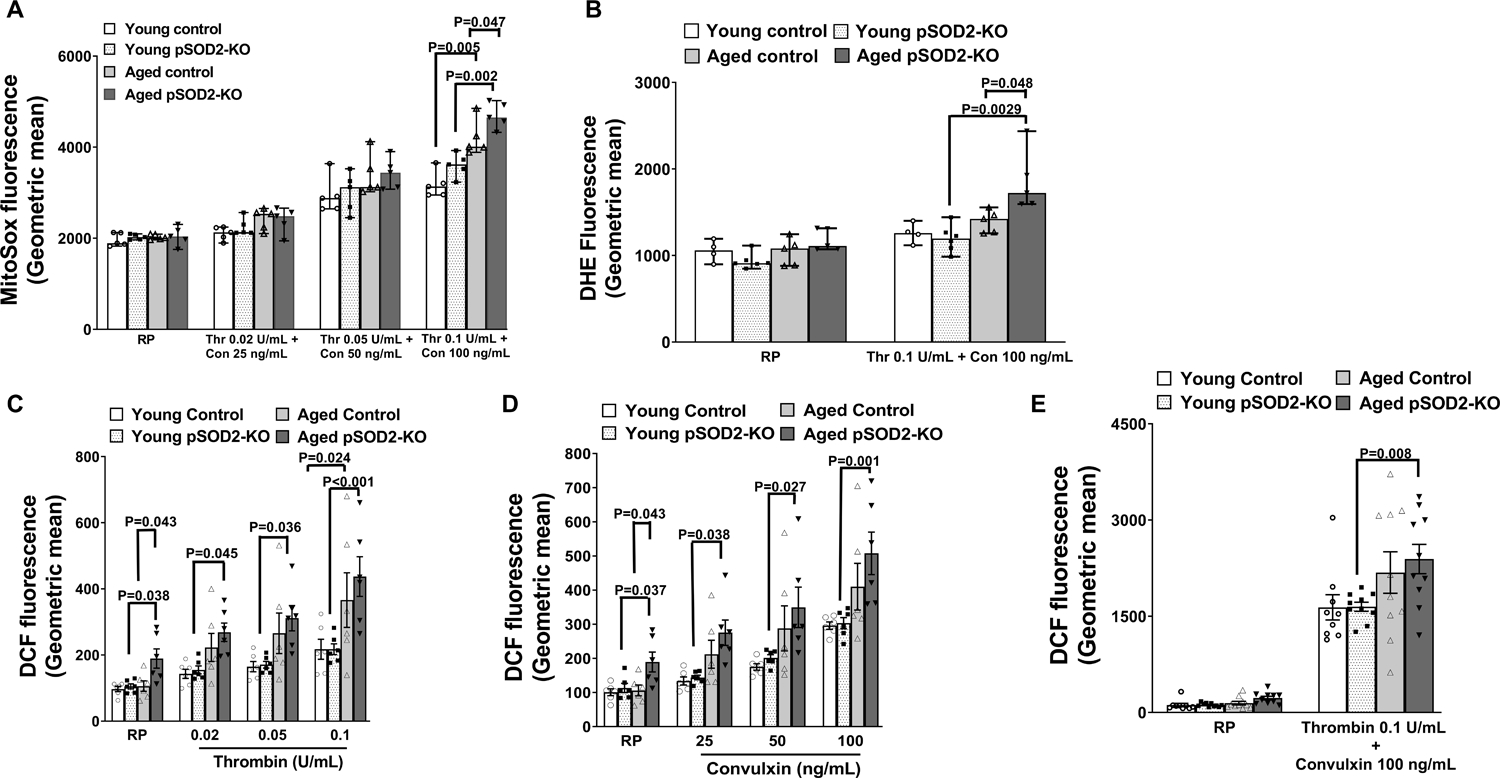
Washed platelets were prepared from young or aged mice deficient in platelet SOD2 (pSOD2-KO) or littermate controls. A. Mitochondrial pro-oxidant was detected by incubating platelets with 10 μM of MitoSox either at resting condition (RP) or in the presence of thrombin (Thr) and convulxin (Con). B. Levels of intracellular prooxidant detected by oxidation of dihydroethidium (DHE Fluorescence) measured in platelets at resting state (RP) or upon stimulation with thrombin and convulxin. C-E. Levels of intracellular ROS detected by oxidation of CM-H2DCF (DCF Fluorescence) was measured in platelets at resting state (RP) or upon stimulation with thrombin or convulxin or both. All the samples were analyzed by flow cytometry. Data for A and B are presented as median with 95% CI and analyzed by Kruskal-Wallis test with Dunn’s post hoc test for multiple group comparisons within each treatment group. Data for C-E are presented as mean ± SE and analyzed using two-way ANOVA with Tukey’s analysis for multiple group comparisons within each treatment. All the comparisons shown are for equivalent doses of agonists. N = 4–10 per group.
To measure total cellular ROS in platelets, two redox-sensitive dyes, DHE which is highly specific for superoxide and CM-H2DCF-DA a non-specific fluorescent probe for pro-oxidants were used. While in resting or unstimulated platelets there were no differences for DHE fluorescence between the 4 groups of mice, upon activation with thrombin and convulxin a significant increase was observed in aged pSOD2-KO mice (P < 0.05 vs aged control and P < 0.01 vs young pSOD2-KO mice, Figure 1B & Supplementary Figure S2). The DCF-DA probe did not detect any differences in oxidation of the probe between young control and young pSOD2-KO or aged control mice under resting condition, but a significant increase in fluorescent signal was observed in aged pSOD2-KO mice (P < 0.05 vs young pSOD2-KO or aged control mice, Figure 1C–1E, & Supplementary Figure S3). Upon stimulation with thrombin or convulxin at several doses, an age-dependent increase in fluorescence was observed. Control mice showed a modest effect of age where a significant increase compared to young control mice was observed only with the highest dose of thrombin used (0.1 U/mL) (Figure 1C Supplementary Figure S2B–S2D). In contrast, platelets from aged pSOD2-KO mice showed significant increases compared to young pSOD2-KO mice at all doses of thrombin or convulxin (Figure 1C, 1D & Supplementary Figure S3B–S3G). Similarly, although both groups of aged mice showed increased fluorescence when stimulated with the highest doses of thrombin and convulxin in combination, only aged pSOD2-KO mice exhibited significant increases relative to young pSOD2-KO mice (Figure 1E & S3H).
Together, these data indicate that aging increases mitochondrial pro-oxidants (presumably derived from superoxide) and total cellular pro-oxidant levels within platelets and this phenomenon is amplified in platelets from aged pSOD2-KO mice.
Platelets from aged pSOD2-KO mice exhibit early and sustained elevation in intra-platelet calcium
Since Ca2+ mobilization and uptake are important upstream signaling mediators of platelet activation, we sought to determine if SOD2 expression modulated intra-platelet calcium at baseline or upon thrombin activation. Quiescent platelets from aged control and aged pSOD2-KO mice had significantly higher levels of calcium (P < 0.001 vs young control or young pSOD2-KO mice respectively, Figure 2A & 2B). Upon stimulation with thrombin, all the groups showed elevation in calcium over time and age-dependent increases were observed in both control and pSOD2-KO mice (P < 0.05 vs young control or young pSOD2-KO mice respectively). Interestingly, aged pSOD2-KO mice exhibited an early elevation in calcium where the fluorescent intensity during the initial 120 seconds after addition of thrombin rose to a significantly higher level in aged pSOD2-KO mice (P < 0.001 or P < 0.01 compared to young pSOD2-KO or aged control mice, Figure 2C).
Figure 2: Platelets from aged pSOD2-KO mice exhibit early and sustained increases in calcium.
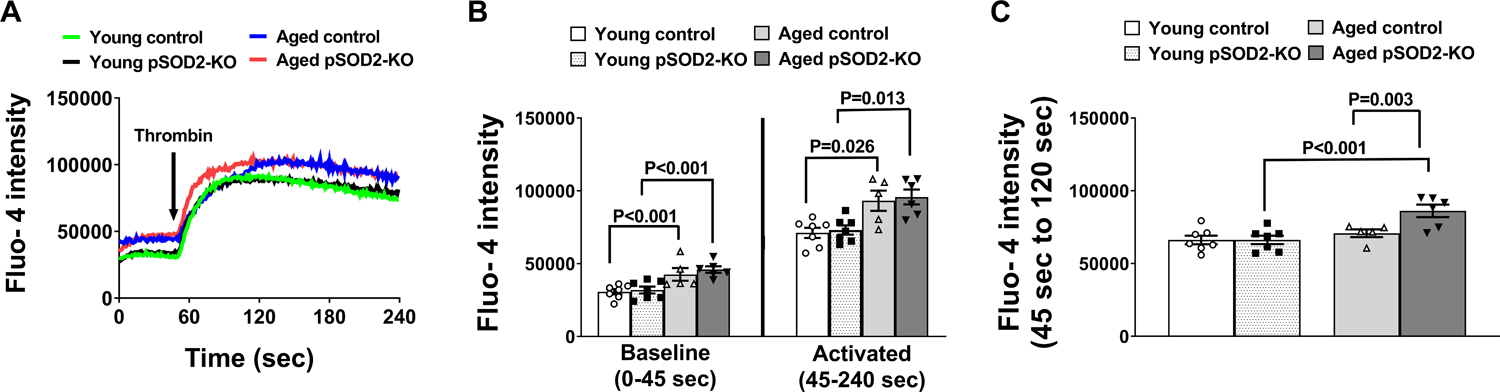
Washed platelets from young or aged mice deficient in platelet SOD2 (pSOD2-KO) or littermate control mice were loaded with Fluo-4 dye, resuspended in m-Tyrode buffer containing 1 mM calcium chloride and analyzed on BD Accuri C6 flow cytometer at baseline (0 to 45 sec) and upon activation with thrombin (45 to 240 sec) in real time. A. Cumulative tracings of calcium levels (0–240 sec). B. Quantification of cumulative fluorescent signals at baseline (0–45 sec) and during thrombin activation (from 45–240 sec). C. Quantification of cumulative fluorescent signals (45 sec to 120 sec). N = Cumulative data from 5–7 mouse in each group. Data are presented as Mean ± SE and analyzed for cumulative signal using two-way ANOVA with Tukey’s multiple comparison test.
Platelets from aged control and pSOD2-KO mice show similar increases in inside-out signaling, dense granule secretion, aggregation, and adhesion
We next determined the role of SOD2 in modulating early and late activation responses in platelets during aging. First, we examined agonist-induced activation of integrin αIIbβ3 in platelets (inside-out signaling). Both control and SOD2-KO platelets displayed age-dependent increases in αIIbβ3 activation in response to thrombin alone and there was no further increase when thrombin and convulxin was used simultaneously (Supplementary Figure S4A), The increase in αIIbβ3 activation was similar between aged control and aged pSOD2-KO mice. Next, we examined platelet aggregation and secretion in response to thrombin (Supplementary Figure S4B & S4C). Platelet aggregation and dense granule secretion were similar in both groups of young mice but were increased significantly in aged control and aged pSOD2-KO mice (P < 0.05 vs young control and P < 0.01 or 0.001 vs young pSOD2-KO mice respectively). Aggregation and secretion were increased to a similar extent in aged control or aged pSOD2-KO mice. Furthermore, platelet adhesion to collagen under arterial shear stress was also increased to a similar extent in aged control and aged pSOD2-KO mice (P < 0.05 vs young control or P < 0.01 vs young pSOD2-KO mice respectively (Supplementary Figure S4D–S4F). Together, these findings suggest that platelet SOD2 does not modulate age-associated increases in early platelet activation responses, including inside-out activation of αIIbβ3, aggregation, dense granule secretion, and adhesion.
We next measured platelet spreading and clot retraction, which are late platelet activation responses mediated by αIIbβ3 outside-in signaling. We quantified mean fluorescence and average cell area of platelets that adhered to a fibrinogen matrix after thrombin activation. No significant differences were observed between the four groups of mice (Supplementary Figure S5A–S5F). Clot retraction, measured as reduction in clot volume over time, also was similar in all four groups (Supplementary Figure S6A & S6B). These findings suggest that late platelet activation responses are not altered by age or platelet SOD2 deficiency.
Aged pSOD2-KO mice exhibit platelet mitochondrial hyperpolarization, increased surface phosphatidylserine exposure and augmented platelet-dependent thrombin generation
Since elevated calcium and superoxide-derived ROS can influence maintenance of the electrochemical gradient across mitochondrial membranes and subsequent exposure of phosphatidylserine on the platelet surface,24 we quantified membrane potential using a cationic fluorescent probe, TMRM, that accumulates in mitochondria in proportion to this gradient. Resting platelets from aged control and aged pSOD2-KO mice showed significantly higher staining for TMRM (Figure 3A & Supplementary Figure S7A, P < 0.05 vs young control or young pSOD2-KO mice, respectively). In the presence of FCCP, which chemically uncouples mitochondria, a major loss in fluorescence was observed in all groups. However, aged pSOD2-KO mice retained higher fluorescence at the submaximal dose of FCCP, compared to all other groups (P < 0.01 vs young pSOD2-KO mice, and P < 0.05 vs aged control mice Figure 3A, & Supplementary Figure S7B & S7C). The aged control mice also had relatively higher fluorescence compared to young control mice in presence of submaximal dose of FCCP (P < 0.05). These findings suggest that mitochondrial hyperpolarization increases with age in resting platelets and is retained at higher level in the absence of SOD2. We next examined platelet surface annexin V binding and observed that resting platelets showed similar bindings in all 4 groups. However, upon activation with thrombin and convulxin, platelets from aged pSOD2-KO showed increased exposure of phosphatidylserine (P < 0.01 vs young pSOD2-KO and P < 0.05 vs aged control mice (Figure 3B, & Supplementary Figure S7D & S7E).
Figure 3. Platelets from aged pSOD2-KO mice show exacerbated mitochondrial hyperpolarization and enhanced annexin V binding.
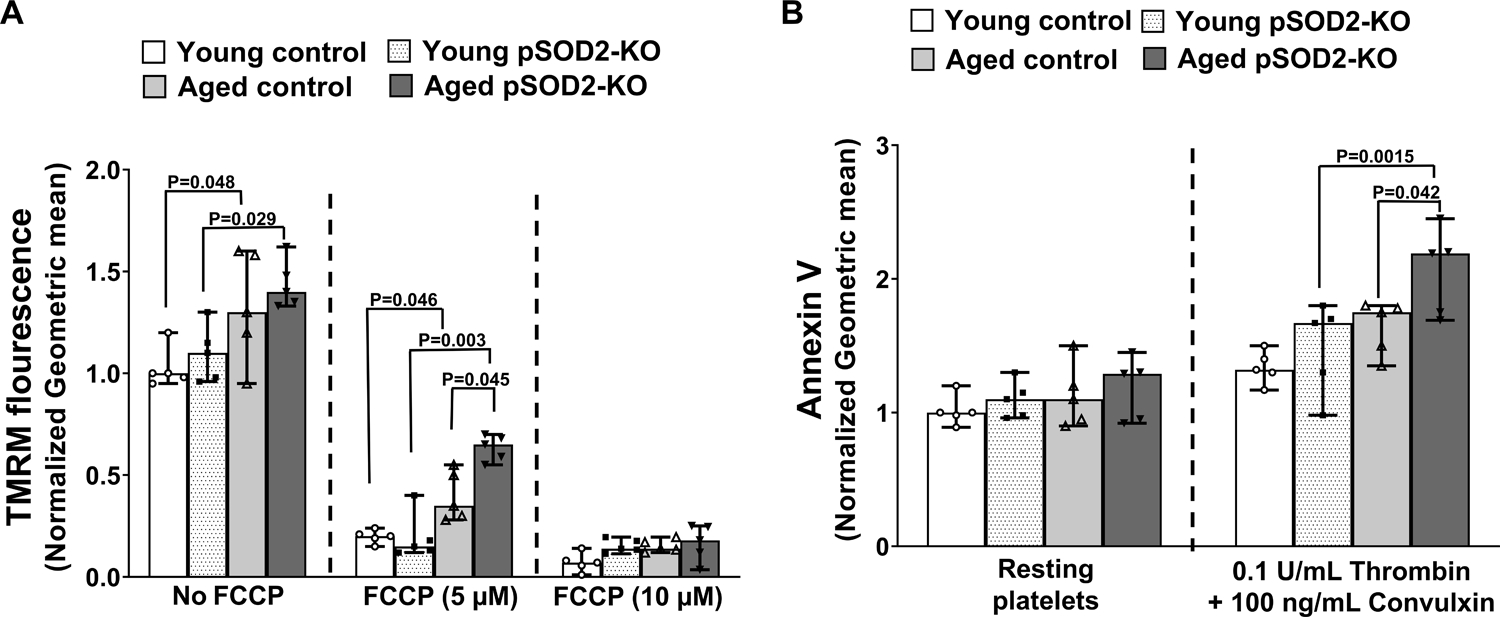
Washed platelets were prepared from young or aged mice deficient in platelet SOD2 (pSOD2-KO) or littermate controls and respective fluorescence upon treatment was measured by flow cytometry and normalized to respective young control without treatment. A. TMRM fluorescence was measured in resting platelets in the presence and absence of FCCP. B. Annexin V binding was quantified in resting and thrombin and convulxin activated platelets. Data are presented as median with 95% CI and analyzed by Kruskal-Wallis test with Dunn’s post hoc test for multiple group comparisons within each treatment group. N = 5 per group.
Since anionic phospholipid exposure on the platelet surface can fuel thrombin generation, we measured platelet-dependent thrombin generation triggered with exogenous tissue factor (Figure 4). The peak of thrombin generation was significantly higher in aged pSOD2-KO mice (P < 0.01 vs young pSOD2-KO mice) but not in aged control mice (P = 0.1 vs young control mice). Endogenous thrombin potential (ETP, measured as area under the thrombin generation curve) was increased significantly in aged control mice compared to young control mice (P < 0.05) and was further increased in aged pSOD2-KO mice (P < 0.05 vs aged control, and P < 0.0001 vs young pSOD2-KO mice). These findings suggest that amongst all the groups studied, platelets from aged pSOD2-KO mice have the highest potential to generate thrombin ex vivo, and the effect of age in control mice is more modest, where ETP is increased but not thrombin peak.
Figure 4: Platelet dependent thrombin generation is increased in aged pSOD2-KO mice.
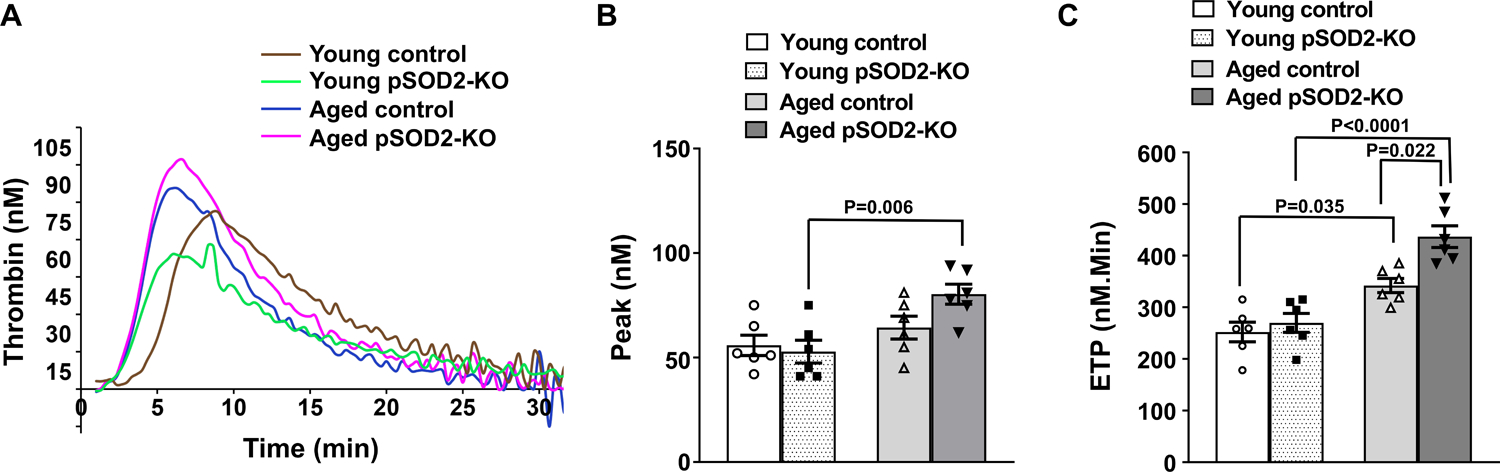
Platelet rich plasma was prepared from young and aged platelet-specific pSOD2-KO mice or control littermates, triggered with tissue factor and thrombin generation was analyzed using Calibrated Automated Thrombogram (CAT, Diagnostica Stago). A. Representative thrombin generation curves, B. thrombin peak and C. endogenous thrombin potential (ETP). Data are presented as mean ± SE and analyzed with two-way ANOVA with Tukey’s test for multiple comparisons. N = 6 per group.
Deficiency of platelet SOD2 increases susceptibility to arterial thrombosis in aged mice
We next examined whether platelet specific SOD2 deficiency in aged mice is sufficient to increase susceptibility to arterial thrombosis in vivo. After photochemical injury of the carotid artery, both groups of young mice exhibited similar times to first or stable occlusion (Figure 5A and 5B). Aged control mice demonstrated only a modest shortening in time to first or stable occlusion compared to young control mice (P = 0.1 for both). In contrast, times to stable occlusion were significantly shortened in aged pSOD2-KO mice relative to aged control mice or young pSOD2-KO mice (P < 0.05 for both), suggesting that lack of platelet SOD2 increases susceptibility to arterial thrombosis in vivo in aged mice. In an alternative model of platelet activation dependent pulmonary thrombosis induced by collagen infusion, we observed significantly shortened time to death (Figure 5C) or reduced percent survival (Figure 5D) in aged pSOD2-KO mice compared to either young pSOD2-KO or aged control mice (P < 0.05 for both). The time to death or survival in aged control mice was similar to young control mice.
Figure 5: Deficiency of platelet SOD2 increases susceptibility to thrombosis in aged mice.
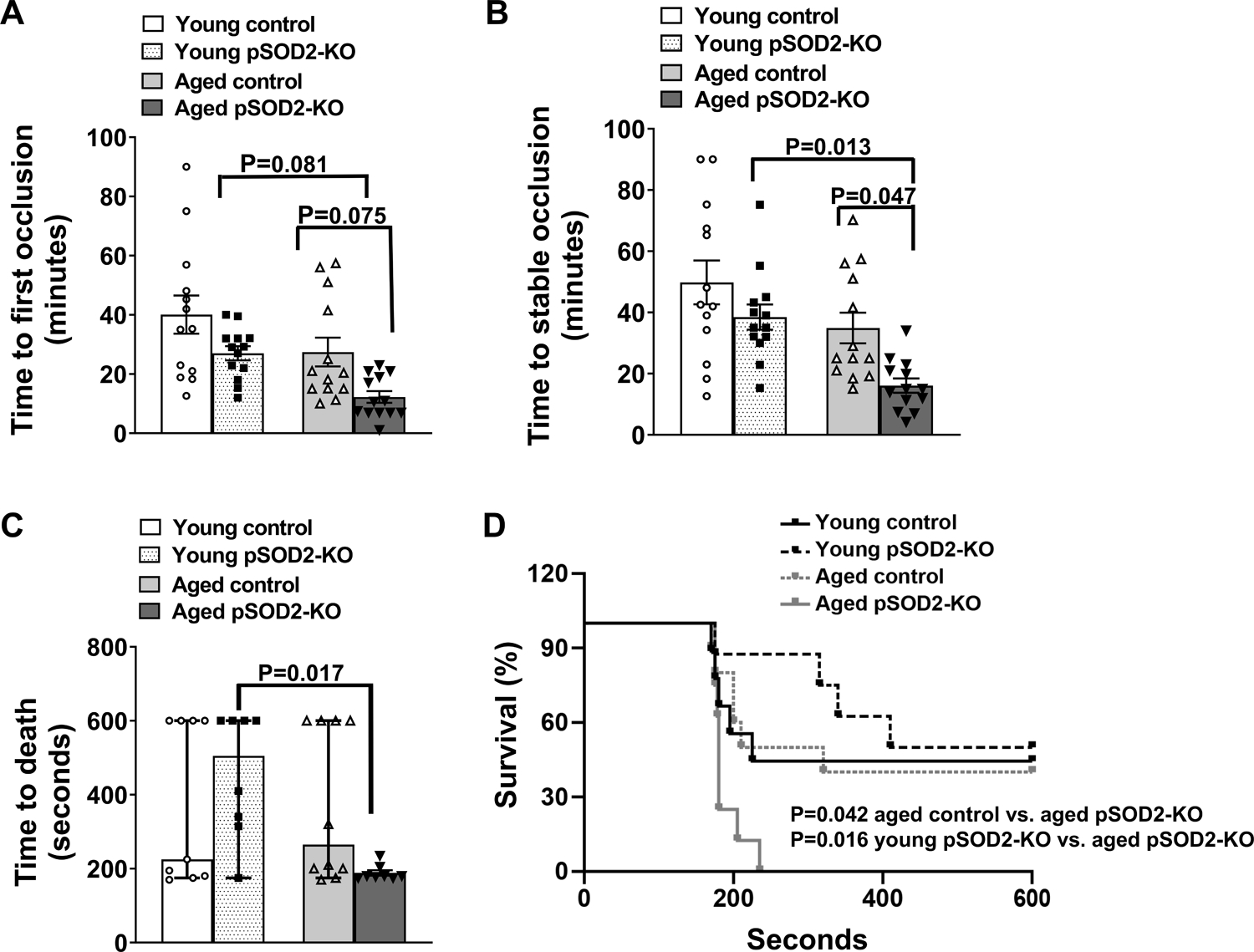
The time to first (A) and stable occlusion (B) of the carotid artery following photochemical injury, and (C) time to death after collagen infusion, was measured in young and aged platelet-specific pSOD2-KO mice or control littermates. Data for A and B are presented as mean ± SE and analyzed using two-way ANOVA with Tukey’s analysis for multiple group comparisons. Data for C is presented as median with 95% CI and analyzed by Kruskal-Wallis test with Dunn’s post hoc test for multiple group comparisons. D. Survival curve after collagen infusion and is analyzed using Log-rank (Mantel-Cox) test. N = 8–13 per group.
To determine whether the impact of platelet SOD2 deficiency on aging-associated thrombosis is independent of other age-related vascular factors, we performed adoptive platelet transfer experiments in young or aged hIL4Rα/GP1bα transgenic host mice. These mice express a chimeric human IL4Rα molecule on the surface of platelets, allowing for efficient depletion of circulating platelets using anti-human IL4R antibody followed by adoptive transfer of non-chimeric platelets from donor mice.19 In a dose response study, we first established that 0.5 mg/Kg of anti-hIL4R could effectively deplete more than 90% of endogenous circulating platelets and that infusion of donor platelets (2.5 × 106 /g host mouse) could replete the platelet count to > 200 × 103/μL, which is sufficient to develop a stable occlusion (Supplementary Figure S8). In experimental thrombosis, adoptive transfer of platelets from aged pSOD2-KO mice but no other donor groups tended to accelerate the time to occlusions in young hIL4Rα/GP1bα Tg hosts (Figure 6A), and significantly accelerated time to first and stable occlusions in aged hIL4Rα/GP1bα Tg hosts (Figure 6B). Further, we observed that the main effect of host age was significant (P < 0.01 for 1st occlusion and P < 0.0001 for stable occlusion). These data suggest a possible interaction between platelets from aged pSOD2-KO mice and the aged vasculature in increasing susceptibility to arterial thrombosis.
Figure 6: Adoptive transfer of platelets from aged pSOD2-KO mice increase susceptibility to arterial thrombosis in an aged host milieu.
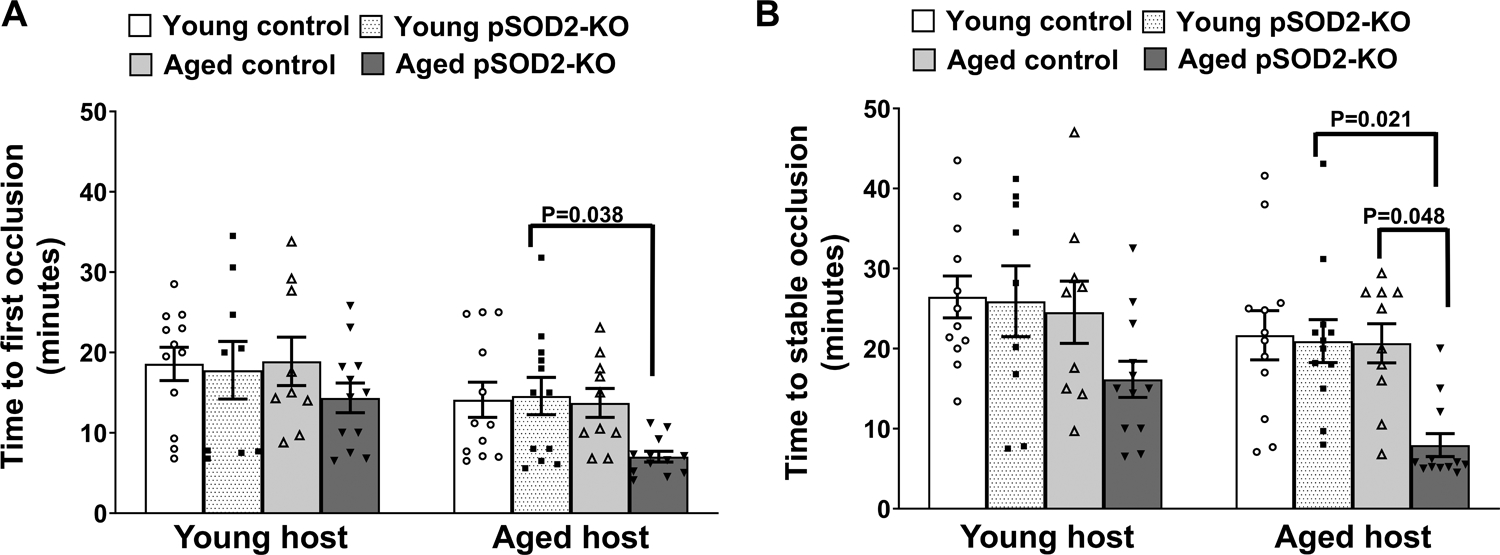
The time to first (A) and stable occlusion (B) of the carotid artery following photochemical injury was measured in young or aged hIL4Rα/GP1bα Tg hosts receiving donor platelets from young or aged control or pSOD2-KO mice. Data are presented as mean ± SE and analyzed using two-way ANOVA with Tukey’s analysis for multiple group comparisons within a host group. N = 9–12 per group.
The SOD2 mimetic, GC4419, protects from age-associated platelet pro-oxidant generation, procoagulant platelet formation, and in vivo arterial thrombosis
We next tested whether a small molecule SOD mimetic (GC4419, which is taken up by mitochondria and mimics the activity of SOD2) prevents increased generation of pro-oxidants, formation of procoagulant platelets and lowers susceptibility to experimental thrombosis characteristic of aged pSOD2-KO mice. Washed platelets were incubated with either GC4419 or vehicle buffer for 10 minutes prior to platelet activation. Incubation of platelets with vehicle buffer showed increased fluorescence for MitoSox, DCF, or annexin V binding in only aged pSOD2-KO mice (Supplementary Figure S9A–S9C). GC4419 lowered the levels of fluorescence in platelets from aged pSOD2-KO mice to levels similar to those in platelets from young mice or aged control mice. In the platelet dependent thrombin generation assay, GC4419 also lowered the ETP in aged pSOD2-KO platelets (P < 0.05 vs vehicle treated aged pSOD2-KO platelets). To assess protective effect on thrombosis, we treated pSOD2-KO mice intraperitoneally for 2 weeks with daily injection of 10 mg/Kg GC4419 or vehicle buffer. In the experimental thrombosis assay with collagen infusion, treatment with GC4419 did not influence survival time in young pSOD2-KO mice, but aged pSOD2-KO mice showed improved survival (Figure S10).
Since the prothrombotic effect of age in control mice was modest, to assess the antithrombotic effect of GC4419 in a validated model of age-dependent increase in thrombotic susceptibility, we treated aged C57BL6/J mice,.2,3,17 Aged C57BL6/J mice had significantly decreased platelet SOD2 activity despite no alterations in SOD2 protein (Figure S11). Compared to platelets from vehicle-treated mice, platelets harvested from GC4419-treated aged C57BL6/J mice, showed decreased staining for MitoSox and DCF fluorescence upon activation with thrombin and convulxin or thrombin respectively (P < 0.05 vs vehicle buffer, Figure 7A & 7B). Binding to annexin V was also decreased (P < 0.05 vs vehicle buffer, Figure 7C) in platelets from GC4419-treated aged C57BL6/J mice. Finally, in the carotid artery photochemical injury model, aged C57BL6/J mice treated with vehicle buffer occluded more rapidly compared to young mice (P < 0.05, Figure 7D) and treatment with GC4419 prolonged the time to occlusion compared to aged vehicle-treated mice (P < 0.05). Similarly, a reduced time to death or survival time following collagen induced pulmonary thrombosis was observed in aged mice (P < 0.01 vs young C57BL6/J mice) that was rescued by GC4419 treatment (Figure 7E & 7F). These data suggest that treatment with a SOD2 mimetic protects aged C57BL6/J mice from generation of procoagulant platelets and arterial thrombosis.
Figure 7: In vivo treatment with GC4419 protects aged C57BL6/J mice from agonist induced generation of ROS, phosphatidylserine exposure, and arterial thrombosis.
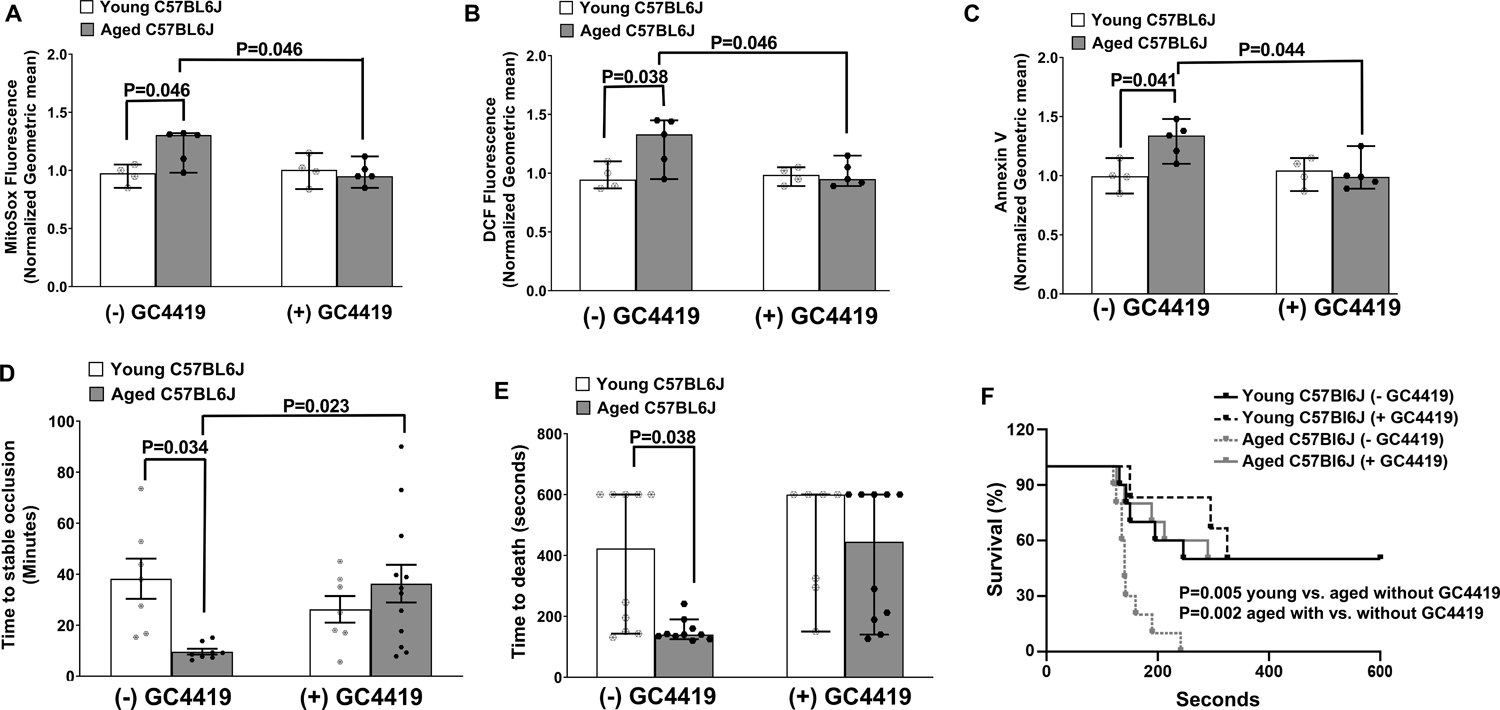
Young or aged C57BL6/J mice were treated with (+) GC4419 (10 mg/Kg daily, IP) or vehicle buffer (− GC4419) for 2 weeks. Washed platelets were prepared and activated with 0.1 U/mL thrombin and 100 ng/mL convulxin and analyzed via FACS. A. Mitochondrial pro-oxidants detection using MitoSox. B. Levels of intracellular ROS detected by oxidation of CM-H2DCF (DCF Fluorescence). C. Annexin V binding. The fluorescent signals for A-C were normalized to respective young control mice receiving vehicle buffer. D. Time to stable occlusion of the carotid artery following photochemical injury. E. Time to death after infusion with 0.5 μg/g collagen. F. Survival curve of E. Data for A-C and E are presented as median with 95% CI and analyzed by Kruskal-Wallis test with Dunn’s post hoc test for multiple group comparisons. Data for D are presented as mean ± SE and analyzed using two-way ANOVA with Tukey’s analysis for multiple group comparisons. Data for F is analyzed using Log-rank (Mantel-Cox) test. N = 4–12 per group.
Discussion
The antioxidant enzyme SOD2 is critical for maintaining mitochondrial redox homeostasis. In this study we utilized platelet specific SOD2-deficient mice to determine the contribution of SOD2 in platelet hyperactivation and arterial thrombosis during aging. Our study using two different aged mouse models reveals several novel findings. First, we observed that platelet SOD2 is dispensable for age-dependent increases in inside-out integrin αIIbβ3 activation, aggregation, dense granule secretion, and adhesion to collagen. Second, we found that deficiency of platelet SOD2 exacerbated the effects of aging on intra-platelet ROS, calcium elevation, mitochondrial hyperpolarization, annexin V binding, and platelet-dependent thrombin generation. Third, SOD2 deficiency in platelets increased susceptibility to arterial thrombosis in aged mice, an effect that was reproduced by adoptive transfer of platelets from aged pSOD2-KO but not aged control mice into aged host mice. Finally, we demonstrated that the mitochondrially targeted dismutase mimetic GC4419, decreased levels of mitochondrial and total pro-oxidants in platelets from aged mice and inhibited procoagulant platelet formation and susceptibility to arterial thrombosis in vivo.
To date the studies of the role of antioxidant enzymes in platelet activation and thrombosis in aging are limited and findings are inconsistent. This may be due in part to the inclusion of a range of age groups, species/strain differences and/or genomic effects of loxP site insertion sequences in genetically engineered mice which are of variable relevance to humans, or the presence of comorbidities in human studies. For example, Jain et al17 suggested that a decline in the activity of several cellular antioxidant enzymes such as SOD, peroxiredoxin, catalase and Gpx1 occurs within platelets in middle aged and older individuals up to 80 yrs of age with cardiovascular comorbidities and may adversely impact platelet activity. They also observed that expression of catalase and Gpx1 but not SOD1 or SOD2 decline in 12-month-old mice. In contrast, we previously reported that expression of intra-platelet catalase is increased in 18 month old mice,2 and despite no loss in Gpx1, additional overexpression of Gpx1 offered protection from elevation in platelet pro-oxidants, platelet activation and thrombosis. This study suggested that overproduction of platelet pro-oxidants occur in aging and may require supplemental antioxidants providing the rationale for identifying targetable sources of ROS.
Tight regulation of the balance between pro-oxidant generation and scavenging is essential for organellar and cellular homeostasis. Mice with complete absence of SOD2 succumb to early lethality,25 underscoring the critical protective role of endogenous SOD2-mediated ROS scavenging on cellular survival. Given the recognized role of mitochondria in regulating platelet activation,13,14 and emerging evidence that platelet activation and thrombosis during aging is mediated through a ROS-dependent mechanism,2,17 we considered a prothrombotic role for platelet mitochondria derived ROS in aging and sought to determine if deficiency of platelet SOD2 would exacerbate effects in aged mice. We first examined modulation of ROS levels by SOD2 in platelets. There was no increase in mitochondrial pro-oxidants in platelets from young pSOD2-KO mice, which is consistent with an earlier report,18 confirming that platelet SOD2 is dispensable in young mice. Mitochondrial and total cellular pro-oxidants in platelets increased with age and was exacerbated in pSOD2-KO mice. The larger impact of deficiency of mitochondrial-SOD2 on total cellular ROS in aged mice likely reflects the highly reactive nature of superoxide, which in the absence of SOD2 can quickly generate superoxide-derived pro-oxidants such as peroxynitrite or undergo spontaneous dismutation to generate H2O2 that can cross mitochondrial membranes and generate lipid peroxides within or outside mitochondria. Our study in two different models revealed that Sod2 mRNA and protein levels do not necessarily correlate with SOD2 activity and the pro-oxidant effects of aging more closely correlate with changes in SOD2 activity. Our findings also implicate additional post-translational regulatory mechanisms that might be regulating SOD2. Posttranslational modifications that modulate SOD2 activity have been described in other tissues during aging,26,27 and future studies should determine specific mechanisms that may reduce platelet SOD2 activity within platelets in aging. Despite the differences in SOD2 activity or levels of ROS in the two models in this study, our findings suggest that mitochondria are an important source of pro-oxidant stress in platelets during aging and supplemental SOD2 could reduce platelet oxidative stress.
Aging has been reported to increase early platelet activation responses,2–4,17 although the contribution of mitochondrial pro-oxidants to this phenomenon is incompletely understood. In the present study we observed age-dependent increases in platelet integrin activation, aggregation, dense granule secretion, and adhesion which corroborates previous reports.3,17,18,28 Absence of platelet SOD2 in aged mice did not worsen these responses, which suggests that platelet SOD2 and mitochondrial pro-oxidant generation are dispensable for age-dependent enhancement of early platelet activation responses in this mouse model. We also did not observe any significant effects of aging or SOD2 deficiency on platelet spreading or clot retraction. In contrast, we observed increases in platelet calcium levels, mitochondrial membrane hyperpolarization, and annexin V binding in aged pSOD2-KO mice. These results are concordant with prior work demonstrating that a sustained increase in calcium within platelets has the potential to alter mitochondrial membrane potential and mediate phosphatidylserine exposure,29–31 whereas mitochondrial calcium uptake does not appear to influence integrin activation.24 Our data reveal that the distribution of annexin V fluorescence was quite broad, with two distinct populations of annexin V-positive platelets existed in the aged groups. These observations suggest that aging may predispose a subset of activated platelets to become highly prothrombotic and this effect appeared to be more pronounced in aged pSOD2-KO platelets. Since surface exposure of phosphatidyl serine mediates platelet procoagulant activity, we measured platelet-dependent thrombin generation and observed a significant increase in aged pSOD2-KO mice relative to aged control or young mice. These findings support a model in which deficiency of platelet SOD2 during aging selectively increases platelet procoagulant activity. An alternative explanation for our findings could be that since integrin activation and aggregation are sensitive to lower doses of agonists, a threshold may have been achieved in aged control platelets limiting the ability to detect any additive effects of deficiency of pSOD2-KO.
To determine if platelet SOD2 deficiency and the subsequent changes in platelet function would affect arterial thrombosis in aged mice, we first evaluated susceptibility to develop stable occlusion after photochemical injury to the carotid artery. In agreement with our previous study,18 young pSOD2-KO mice tended to have somewhat shorter occlusion times relative to young control mice. Aged control mice also showed only a modest shortening of occlusion time. However, aged pSOD2-KO mice exhibited significant acceleration in time to occlusion compared to aged control or young pSOD2-KO mice, indicating a critical role of endogenous platelet SOD2 in mitigating age-related arterial thrombosis. A limitation of the photochemical injury model is its dependence on ROS-mediated thrombus formation which may confound findings in the setting of SOD2-deficiency in our aged mice. To overcome this limitation, we utilized an alternative model of pulmonary thrombosis where thrombus is developed by collagen-induced platelet activation and time to death is recorded as an indicator of the rate of thrombosis. Consistent with increased carotid artery occlusion there was rapid death in aged pSOD2-KO mice relative to other groups. This increased susceptibility to thrombosis in aged pSOD2-KO mice is consistent with the observed increases in ROS, calcium levels, phosphatidylserine exposure, and platelet-dependent thrombin generation. Overall, a modest or partial prothrombotic effects of age observed in our control mice with reference to platelet activation, procoagulant platelet formation or in vivo thrombosis compared to a more robust effect observed in C57BL6/J mice in this study or in others2,3,17 could be related to the inherent differences between mouse colonies, housing conditions and/or genetic effects secondary to the presence of loxP sequences. The modest prothrombotic effect seen in aged control mice could also be related to the heterogeneity in SOD2 expression specially in the face of increased mitochondrial pro-oxidants. These findings do support the notion that there is an overburden of mitochondrial pro-oxidants during aging and the physiological SOD2 levels may not be sufficient to overcome all the adverse effects of pro-oxidants.
To examine the impact of platelet SOD2 deficiency on thrombosis in aged mice independently from effects of aging on the vasculature, we undertook a rigorous adoptive platelet transfer approach to assess the effects of control or SOD2-deficient platelets on thrombosis in young or aged host hIL4Rα/GP1bα Tg mice. Both the age of the host mice and the transfer of platelets from aged pSOD2-KO mice appear to influence the time to occlusion of the carotid artery. However, significant acceleration in carotid occlusion occurred when platelets from aged pSOD2-KO mice were transferred to aged hIL4R Tg hosts. Together with the findings of procoagulant platelet formation, these observations support the notion that though aged pSOD2-KO platelets confer a prothrombotic effects, the in vivo thrombotic susceptibility is increased significantly in the presence of aged host vasculature. Since other components of vasculature may also play an important role in age-associated thrombosis, future studies should examine independent effects of these components such as the vessel wall and other cellular or non-cellular components of the blood and vasculature.
Finally, we observed that in vitro use of a SOD2 mimetic lowered platelet pro-oxidants and inhibited formation of procoagulant platelets and in vivo treatment with SOD2 mimetic protected aged pSOD2-KO mice from enhanced thrombosis. Further, in vivo treatment with SOD2 mimetic in aged C57BL6/J mice also lowered platelet pro-oxidants, inhibited formation of procoagulant platelets and decreased susceptibility to thrombosis. The potential for dismutase mimetics in lowering thrombotic susceptibility was previously reported where an alternative analogue of GC4419 (SC-52608) limited the platelet dependent growth of thrombus in carotid artery in a cyclic flow model.32 Together, these findings suggest that approaches targeted to increase SOD2 activity could lower the prothrombotic burden associated with age. These findings are novel and clinically important given that GC4419 is being tested in early phase clinical trials (NCT03340974 and NCT02508389) and a phase 3 clinical trial (NCT03689712) to mitigate radiation-induced mucositis in head and neck cancer. The use of GC4419 could potentially be extended into future trials in aged humans at high risk of thrombotic complications. Despite some differences in the phenotypes between the two models we studied, the use of a comprehensive approach spanning pharmacological, genetic, and adoptive platelet transfer methods, demonstrates that endogenous platelet SOD2 limits pathways that promote platelet-dependent thrombin generation and thrombosis in aged mice.
We conclude that platelet SOD2 plays a distinct role in modulating procoagulant platelet response during aging. Platelet SOD2 appears to be an important regulator of calcium elevation, mitochondrial hyperpolarization, surface phosphatidylserine exposure, thrombin generation, and arterial thrombosis. While SOD2 expression/activity in platelets may vary between mouse models, given that older humans have decreased total SOD activity, our findings may have important clinical implications. A missense variant in SOD2 (valine to alanine at position 16) reduces SOD2 activity.33 This polymorphism is present at high frequency among certain ethnic groups, including 45% of people with African ancestry where it is associated with increased vascular complications in sickle cell disease.34,35 This polymorphism and some others related to decreased SOD2 activity also have been linked to age-associated cardiovascular disease in several other ethnic groups,36–39 therefore, these findings could also be relevant to cardiovascular diseases in the general aging population. Hence, targeted therapies to increase platelet mitochondrial antioxidant capacity may have the potential to protect from aging-associated platelet activation, thrombosis, and their cardiovascular consequences.
Supplementary Material
{kind=link}
Novelty and Significance:
SOD2 maintains mitochondrial redox homeostasis but its contribution to platelet activation and thrombosis in aging is unknown. In a rigorous study design that included a mouse model with platelet-specific deletion of the SOD2, adoptive platelet transfer approaches and use of a pharmacological agent, we made the novel observation that endogenous platelet SOD2 protects from age-associated platelet activation and thrombosis. These findings are clinically informative since a missense variant in SOD2 in several ethnic group correlates with age-associated cardiovascular disease; hence these findings could be relevant to the cardiovascular consequences in the general aging population. Therefore, our work advances the field significantly and suggests that targeted therapies to increase platelet mitochondrial antioxidant capacity may have the potential to protect from aging-associated platelet activation and thrombosis, particularly in those at increased risk.
Highlight:
Platelet SOD2 deficiency in aged mice amplifies intra-platelet ROS generation, calcium elevation and phosphatidylserine exposure.
Endogenous platelet SOD2 protects from increased thrombin generation and thrombosis in aging.
Targeted therapy that increases platelet mitochondrial antioxidant capacity could potentially protect from aging-associated platelet procoagulant platelet formation and thrombosis.
Acknowledgements
We acknowledge Galera Therapeutics, Inc. for providing GC4419 and critical reading of the manuscript under a sponsored research agreement with the University of Iowa. We also acknowledge the Diagnostica Stago Inc., (Parsippany NJ) for loaning the Fluoroskan Ascent instrument for the thrombin generation assay and the support from the Flow Cytometry Facility, which is a Carver College of Medicine / Holden Comprehensive Cancer Center core research facility at the University of Iowa. The facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center, and Iowa City Veteran’s Administration Medical Center. Lastly, the research reported in this publication was also supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR002537.
Sources of Funding
This work was possible through funding from National Institutes of Health AG049784, Office of Research and Development and Department of Veterans Affairs I01CX001932 to S.D., National Institutes of Health T32 HL007344 to A.S.E. and M.V.S, American Heart Association 19POST34380100 to R.K, National Institutes of Health P01CA217797 to DRS and BGA, National Institutes of Health U54 HL112311 and R61HL141783 to E.D.A.
Disclosures
Drs. Spitz and Allen have Sponsored Research Agreements supported by Galera Therapeutics, Inc. to study GC4419 in preclinical and clinical studies of cancer therapy. No other author has competing financial interests in this work.
Non-standard Abbreviations and Acronyms
- ANOVA
Analysis of variance
- CM-H2DCF-DA
Chloromethyl Dichloro-dihydro-fluorescein diacetate
- DCF
Dichloro-dihydro-fluorescein
- DHE
Di hydro ethidium
- ETP
Endogenous thrombin potential
- GC4419
avasopasem manganese
- Glutathione peroxidase
Glutathione Peroxidase
- GP1bα
Glycoprotein 1bα
- hIL4Rα
Human Interleukin 4 receptor α
- KO
Knockout
- NADPH
Nicotinamide adenine dinucleotide phosphate
- ROS
Reactive oxygen species
- SOD2
superoxide dismutase
- Tg
Transgenic
Footnotes
Supplemental Material
References
- 1.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019;139:e56–e528. doi: 10.1161/CIR.0000000000000659 [DOI] [PubMed] [Google Scholar]
- 2.Dayal S, Wilson KM, Motto DG, Miller FJ Jr., Chauhan AK, Lentz SR. Hydrogen peroxide promotes aging-related platelet hyperactivation and thrombosis. Circulation. 2013;127:1308–1316. doi: 10.1161/CIRCULATIONAHA.112.000966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davizon-Castillo P, McMahon B, Aguila S, Bark D, Ashworth K, Allawzi A, Campbell RA, Montenont E, Nemkov T, D’Alessandro A, et al. TNF-alpha driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood. 2019. doi: 10.1182/blood.2019000200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang J, Zhou X, Fan X, Xiao M, Yang D, Liang B, Dai M, Shan L, Lu J, Lin Z, et al. mTORC1 promotes aging-related venous thrombosis in mice via elevation of platelet volume and activation. Blood. 2016;128:615–624. doi: 10.1182/blood-2015-10-672964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pignatelli P, Pulcinelli FM, Lenti L, Gazzaniga PP, Violi F. Hydrogen peroxide is involved in collagen-induced platelet activation. Blood. 1998;91:484–490. doi: [PubMed] [Google Scholar]
- 6.Begonja AJ, Gambaryan S, Geiger J, Aktas B, Pozgajova M, Nieswandt B, Walter U. Platelet NAD(P)H-oxidase-generated ROS production regulates alphaIIbbeta3-integrin activation independent of the NO/cGMP pathway. Blood. 2005;106:2757–2760. doi: 10.1182/blood-2005-03-1047 [DOI] [PubMed] [Google Scholar]
- 7.Delaney MK, Kim K, Estevez B, Xu Z, Stojanovic-Terpo A, Shen B, Ushio-Fukai M, Cho J, Du X. Differential Roles of the NADPH-Oxidase 1 and 2 in Platelet Activation and Thrombosis. Arterioscler Thromb Vasc Biol. 2016;36:846–854. doi: 10.1161/ATVBAHA.116.307308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson SJ, Cavanagh CC, Lesher AM, Frey AJ, Russell SE, Smyth EM. Activation-dependent stabilization of the human thromboxane receptor: role of reactive oxygen species. J Lipid Res. 2009;50:1047–1056. doi: 10.1194/jlr.M800447-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wachowicz B, Olas B, Zbikowska HM, Buczynski A. Generation of reactive oxygen species in blood platelets. Platelets. 2002;13:175–182. doi: 10.1080/09533710022149395 [DOI] [PubMed] [Google Scholar]
- 10.Sonkar VK, Kumar R, Jensen M, Wagner BA, Sharathkumar AA, Miller FJ Jr., Fasano M, Lentz SR, Buettner GR, Dayal S. Nox2 NADPH oxidase is dispensable for platelet activation or arterial thrombosis in mice. Blood Adv. 2019;3:1272–1284. doi: 10.1182/bloodadvances.2018025569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dharmarajah J, Arthur JF, Sobey CG, Drummond GR. The anti-platelet effects of apocynin in mice are not mediated by inhibition of NADPH oxidase activity. Naunyn Schmiedebergs Arch Pharmacol. 2010;382:377–384. doi: 10.1007/s00210-010-0552-3 [DOI] [PubMed] [Google Scholar]
- 12.Walsh TG, Berndt MC, Carrim N, Cowman J, Kenny D, Metharom P. The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol. 2014;2:178–186. doi: 10.1016/j.redox.2013.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cardenes N, Corey C, Geary L, Jain S, Zharikov S, Barge S, Novelli EM, Shiva S. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood. 2014;123:2864–2872. doi: 10.1182/blood-2013-09-529420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang WH, Stitham J, Jin Y, Liu R, Lee SH, Du J, Atteya G, Gleim S, Spollett G, Martin K, et al. Aldose reductase-mediated phosphorylation of p53 leads to mitochondrial dysfunction and damage in diabetic platelets. Circulation. 2014;129:1598–1609. doi: 10.1161/CIRCULATIONAHA.113.005224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamagishi SI, Edelstein D, Du XL, Brownlee M. Hyperglycemia potentiates collagen-induced platelet activation through mitochondrial superoxide overproduction. Diabetes. 2001;50:1491–1494. doi: [DOI] [PubMed] [Google Scholar]
- 16.Braganza A, Corey CG, Santanasto AJ, Distefano G, Coen PM, Glynn NW, Nouraie SM, Goodpaster BH, Newman AB, Shiva S. Platelet bioenergetics correlate with muscle energetics and are altered in older adults. JCI Insight. 2019;5. doi: 10.1172/jci.insight.128248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jain K, Tyagi T, Patell K, Xie Y, Kadado AJ, Lee SH, Yarovinsky T, Du J, Hwang J, Martin KA, et al. Age associated non-linear regulation of redox homeostasis in the anucleate platelet: Implications for CVD risk patients. EBioMedicine. 2019;44:28–40. doi: 10.1016/j.ebiom.2019.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fidler TP, Rowley JW, Araujo C, Boudreau LH, Marti A, Souvenir R, Dale K, Boilard E, Weyrich AS, Abel ED. Superoxide Dismutase 2 is dispensable for platelet function. Thromb Haemost. 2017;117:1859–1867. doi: 10.1160/TH17-03-0174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanaji T, Russell S, Ware J. Amelioration of the macrothrombocytopenia associated with the murine Bernard-Soulier syndrome. Blood. 2002;100:2102–2107. doi: 10.1182/blood-2002-03-0997 [DOI] [PubMed] [Google Scholar]
- 20.Sishc BJ, Ding L, Nam TK, Heer CD, Rodman SN, Schoenfeld JD, Fath MA, Saha D, Pulliam CF, Langen B, et al. Avasopasem manganese synergizes with hypofractionated radiation to ablate tumors through the generation of hydrogen peroxide. Sci Transl Med. 2021;13. doi: 10.1126/scitranslmed.abb3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mallick RL, Kumari S, Singh N, Sonkar VK, Dash D. Prion protein fragment (106–126) induces prothrombotic state by raising platelet intracellular calcium and microparticle release. Cell Calcium. 2015;57:300–311. doi: 10.1016/j.ceca.2015.02.002 [DOI] [PubMed] [Google Scholar]
- 22.Wilson KM, Lynch CM, Faraci FM, Lentz SR. Effect of mechanical ventilation on carotid artery thrombosis induced by photochemical injury in mice. J Thromb Haemost. 2003;1:2669–2674. doi: [DOI] [PubMed] [Google Scholar]
- 23.Lockyer S, Okuyama K, Begum S, Le S, Sun B, Watanabe T, Matsumoto Y, Yoshitake M, Kambayashi J, Tandon NN. GPVI-deficient mice lack collagen responses and are protected against experimentally induced pulmonary thromboembolism. Thromb Res. 2006;118:371–380. doi: 10.1016/j.thromres.2005.08.001 [DOI] [PubMed] [Google Scholar]
- 24.Choo HJ, Saafir TB, Mkumba L, Wagner MB, Jobe SM. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler Thromb Vasc Biol. 2012;32:2946–2955. doi: 10.1161/ATVBAHA.112.300433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376 [DOI] [PubMed] [Google Scholar]
- 26.Marzetti E, Csiszar A, Dutta D, Balagopal G, Calvani R, Leeuwenburgh C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: from mechanisms to therapeutics. Am J Physiol Heart Circ Physiol. 2013;305:H459–476. doi: 10.1152/ajpheart.00936.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bause AS, Haigis MC. SIRT3 regulation of mitochondrial oxidative stress. Exp Gerontol. 2013;48:634–639. doi: 10.1016/j.exger.2012.08.007 [DOI] [PubMed] [Google Scholar]
- 28.Yang J, Zheng Y, Gou X, Pu K, Chen Z, Guo Q, Ji R, Wang H, Wang Y, Zhou Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int J Infect Dis. 2020;94:91–95. doi: 10.1016/j.ijid.2020.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Obydennyy SI, Sveshnikova AN, Ataullakhanov FI, Panteleev MA. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J Thromb Haemost. 2016;14:1867–1881. doi: 10.1111/jth.13395 [DOI] [PubMed] [Google Scholar]
- 30.Kholmukhamedov A, Janecke R, Choo HJ, Jobe SM. The mitochondrial calcium uniporter regulates procoagulant platelet formation. J Thromb Haemost. 2018;16:2315–2321. doi: 10.1111/jth.14284 [DOI] [PubMed] [Google Scholar]
- 31.Abbasian N, Millington-Burgess SL, Chabra S, Malcor JD, Harper MT. Supramaximal calcium signaling triggers procoagulant platelet formation. Blood Adv. 2020;4:154–164. doi: 10.1182/bloodadvances.2019000182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meng YY, Trachtenburg J, Ryan US, Abendschein DR. Potentiation of endogenous nitric oxide with superoxide dismutase inhibits platelet-mediated thrombosis in injured and stenotic arteries. J Am Coll Cardiol. 1995;25:269–275. doi: 10.1016/0735-1097(94)00349-u [DOI] [PubMed] [Google Scholar]
- 33.Bastaki M, Huen K, Manzanillo P, Chande N, Chen C, Balmes JR, Tager IB, Holland N. Genotype-activity relationship for Mn-superoxide dismutase, glutathione peroxidase 1 and catalase in humans. Pharmacogenet Genomics. 2006;16:279–286. doi: 10.1097/01.fpc.0000199498.08725.9c [DOI] [PubMed] [Google Scholar]
- 34.Dosunmu-Ogunbi AM, Wood KC, Novelli EM, Straub AC. Decoding the role of SOD2 in sickle cell disease. Blood Adv. 2019;3:2679–2687. doi: 10.1182/bloodadvances.2019000527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dosunmu-Ogunbi A, Yuan S, Reynolds M, Giordano L, Sanker S, Sullivan M, Stolz DB, Kaufman BA, Wood KC, Zhang Y, et al. SOD2 V16A amplifies vascular dysfunction in sickle cell patients by curtailing mitochondria complex IV activity. Blood. 2022;139:1760–1765. doi: 10.1182/blood.2021013350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Forkan M, Wali FB, Khaleda L, Alam MJ, Chowdhury RH, Datta A, Rahman MZ, Hosain N, Maruf MF, Chowdhury MAQ, et al. Association of arsenic-induced cardiovascular disease susceptibility with genetic polymorphisms. Sci Rep. 2021;11:6263. doi: 10.1038/s41598-021-85780-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujimoto H, Taguchi J, Imai Y, Ayabe S, Hashimoto H, Kobayashi H, Ogasawara K, Aizawa T, Yamakado M, Nagai R, et al. Manganese superoxide dismutase polymorphism affects the oxidized low-density lipoprotein-induced apoptosis of macrophages and coronary artery disease. Eur Heart J. 2008;29:1267–1274. doi: 10.1093/eurheartj/ehm500 [DOI] [PubMed] [Google Scholar]
- 38.Yeh HL, Kuo LT, Sung FC, Yeh CC. Association between Polymorphisms of Antioxidant Gene (MnSOD, CAT, and GPx1) and Risk of Coronary Artery Disease. Biomed Res Int. 2018;2018:5086869. doi: 10.1155/2018/5086869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flekac M, Skrha J, Hilgertova J, Lacinova Z, Jarolimkova M. Gene polymorphisms of superoxide dismutases and catalase in diabetes mellitus. BMC Med Genet. 2008;9:30. doi: 10.1186/1471-2350-9-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.